
Surface chemical reactions studied via ab initio-derived molecular ...

Length and Time scale issues in Molecular
simulation
Prabal K Maiti
Center for Condensed Matter Theory, Department of
Physics
http://www.physics.iisc.ernet.in/~maiti

Ref:
Computer simulation of Liquids: M. P. Allen and D. J.
Tildesley, Oxford (1987)
Understanding Molecular simulation: Daan Frenkel and B
Smit (2nd ed)
Molecular Modelling Principles And Applications: Andrew
Leach, Prentice Hall (2001)
The art of Molecular dynamics: D. C.Rappaport
Molecular Modeling and Simulation: Tamar Schlick

What size is too big? What times are too long?
• QM (ab initio molecular dynamics)
electrons / N basis sets, speed ~ N3 or N4 (may be linear with very high prefactor)
Time steps ~10-2 fs.
Example of big and long: 64 water molecules during 10 ps.
• Classical atomistic MDAtoms/ N atoms, speed ~N2 (may be reduced with efficient algorithms, periodic coulomb is
most expensive)
Time steps ~0.5-2 fs.
Example of big and long (1 processor): 50,000 atoms for 1 ns.
(parallel simulations, can improve this much. Parallel codes available free or almost free: LAMMPS, AMBER, GROMACS, NAMD)

Short history of Molecular Simulations
• Metropolis, Rosenbluth, Teller (1953) Monte Carlo
Simulation of hard disks.
• Fermi, Pasta Ulam (1954) experiment on ergodicity
• Alder & Wainwright (1958) liquid-solid transition in hard
spheres. “long time tails” (1970)
• Vineyard (1960) Radiation damage using MD
• Rahman (1964) liquid argon, water(1971)
• Verlet (1967) Correlation functions, ...
• Andersen, Rahman, Parrinello (1980) constant pressure
MD
• Nose, Hoover, (1983) constant temperature thermostats.
• Car, Parrinello (1985) ab initio MD.

The examples for each period are
representative. The first five
systems are modeled in vacuum
and the others in solvent.
The 38 µµµµs ββββ-hairpin simulation
in 2001 represents an ensemble
(or aggregate dynamics)
simulation, as accumulated over
several short runs, rather than a
long simulation.
The table is taken from the book
by Tamar Schlick


NSF Peta scale machine



What are the forces?
• Crucial since V(q) determines the quality of result.
• Semi-empirical potentials: potential is constructed on
theoretical grounds but using some experimental data.
• Common examples are Lennard-Jones, Coulomb,
embedded atom potentials. They are only good for simple
materials. The ab initio philosophy is that potentials are to
be determined directly from quantum mechanics as
needed.
• But computer power is not yet adequate in general.
• A powerful approach is to use simulations at one level to
determine parameters at the next level.
)(2
2rV
dt
rd ρρ
∇−=

Calculation of Forces• Force is the gradient of the potential
2 1 12
12 12
1 1
12 12 12
12 1 1
1212 12
12
( )
( ) ( )
( )
( )
x y
x y
x y
u r
u r u r
x y
du r r r
dr x y
f rx y
r
→ = −∇
∂ ∂= − −
∂ ∂
∂ ∂= − + ∂ ∂
= − +
F
e e
e e
e e
2
1
r12
x12
y12
4
2
0
-2
2.52.01.51.0
Separation, r/σ
Energy Force
1/ 22 2
12 2 1 2 1( ) ( )r x x y y = − + −
Force on 1,
due to 2
2 1 1 2→ →= −F F
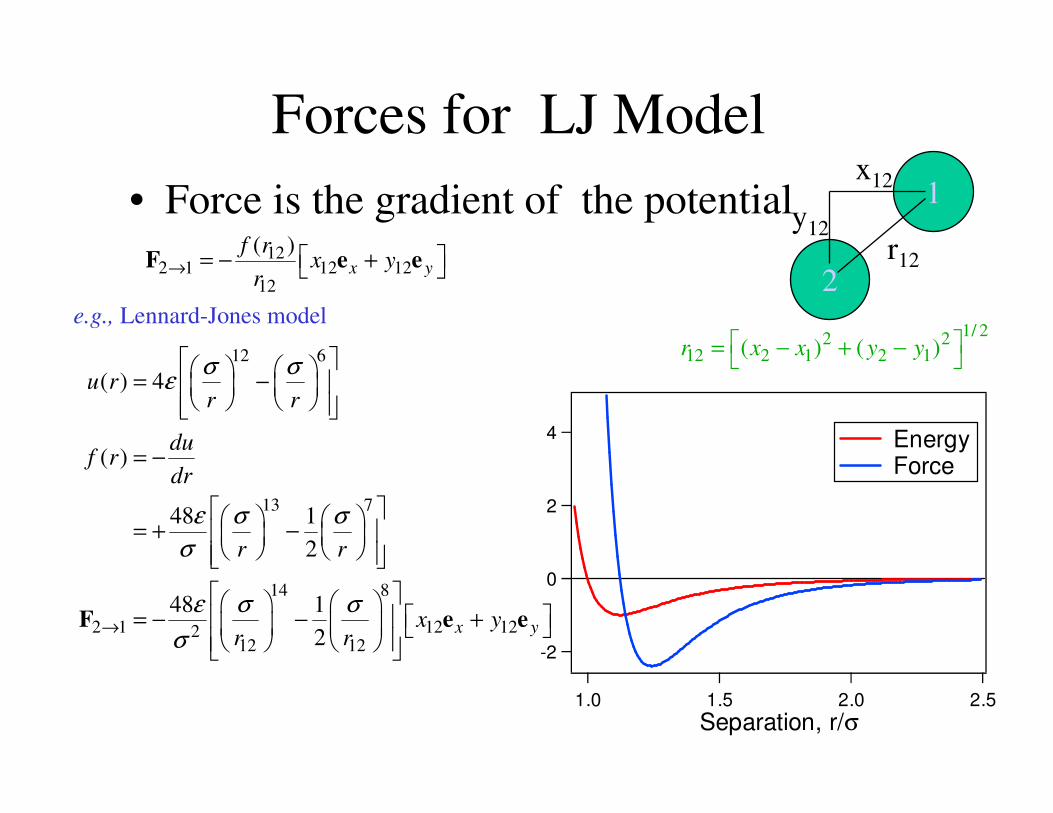
Forces for LJ Model
• Force is the gradient of the potential12
2 1 12 1212
( )x y
f rx y
r→ = − + F e e
2
1
r12
x12
y12
4
2
0
-2
2.52.01.51.0
Separation, r/σ
Energy Force
1/ 22 2
12 2 1 2 1( ) ( )r x x y y = − + −
e.g., Lennard-Jones model
12 6
13 7
14 8
2 1 12 12212 12
( ) 4
( )
48 1
2
48 1
2x y
u rr r
duf r
dr
r r
x yr r
σ σε
ε σ σ
σ
ε σ σ
σ→
= −
= −
= + −
= − − +
F e e

Dealing length scale: Boundary Conditions
• Small system is simulated to study/mimic the bulk properties
• Surface effect: a large fraction of the small sample lie on the surface. For example for 1000 molecules arranged in 10x10x10 cube, almost 400 molecules appear on the cube faces. Surface molecules/atoms experience different force than in the bulk.
• Impractical to contain system with a real boundary
– Enhances finite-size effects
– Artificial influence of boundary on system properties

Solution: Periodic boundary condition
•The problem of surface effect can be overcome by using. “Periodic Boundary Conditions” (PBC). The bounding box is replicated throughout space to form an infinite lattice. In the course of simulation as a molecule moves in the original box, its periodic images in the each of the neighboring boxes
•As the molecule leaves the central box, one of its image will enter through the opposite face.
•No physical wall at the boundary and so no surface molecules.

Issues with Periodic Boundary Conditions
• Suppress fluctuation that have wavelength greater than the
length of the simulation cell. This could be problem near critical
point where fluctuation plays dominant role.
• new artificial correlations
• It can also affect the rate at which a simulated liquid nucleates
and forms a solid or glass when rapidly cooled (Honeycutt and
Andersen 1984)
• Artifact of PBC can be determined by performing simulations
using a variety of cell sizes and shape

Issues with Periodic Boundary Conditions
• Other issues arise when dealing with longer-range potentials
– accounting for long-range interactions
– nearest image not always most energetic
– splitting of molecules (charges)
– discuss details later
• Other geometries possible
– any space-filling unit cell
• hexagonal in 2D
• truncated octahedron in 3D
• rhombic dodecahedron in 3D
– surface of a (hyper)sphere
– variable aspect ratio useful for solids
• relieves artificial stresses


Implementing Cubic Periodic Boundaries
• How do we handle PBC and the minimum image convention
– Box origin
• center of box, coordinates range from -L/2 to +L/2. When a molecule
leaves a box crossing one of the boundary, its image enters the box and is
accomplished either by adding or subtracting L to the particle coordinate.
If (drx[I] > L/2) rx[I] = rx[I] – L
If (drx[I] > -L/2) rx[I] = rx[I] +L
A more convenient way to handle PBC as well as
minimum image convention is to use reduce
(scaled coordinates) in the range (-1/2,1/2)
– Box size
• unit box, coordinates scaled by box length
• sep[0] = sep[0] – NINT(sep[0])
• define NINT(x) ((x) < 0.0 ? (int) ((x) - 0.5) : (int) ((x)
+ 0.5))
-0.5 +0.5+0.5
-0.5
(0,0)

Implementing Cubic Periodic Boundaries 3
void periodic_boundary_conditions(int n_atoms, double **h,
double **scaled_atom_coords, double **atom_coords)
int i, j;
for (i = 0; i < n_atoms; ++i)
for (j = 0; j < NDIM; ++j)
scaled_atom_coords[i][j] -= NINT(scaled_atom_coords[i][j]);
atom_coords[i][0] = h[0][0] * scaled_atom_coords[i][0]
+ h[0][1] * scaled_atom_coords[i][1]
+ h[0][2] * scaled_atom_coords[i][2];
atom_coords[i][1] = h[1][1] * scaled_atom_coords[i][1]
+ h[1][2] * scaled_atom_coords[i][2];
atom_coords[i][2] = h[2][2] * scaled_atom_coords[i][2];

Implementing Cubic Periodic Boundaries
void scaled_atomic_coords(int n_atoms, double **h_inv, double **atom_coords,
double **scaled_atom_coords)
int i;
for (i = 0; i < n_atoms; ++i)
scaled_atom_coords[i][0] = h_inv[0][0] * atom_coords[i][0]
+ h_inv[0][1] * atom_coords[i][1]
+ h_inv[0][2] * atom_coords[i][2];
scaled_atom_coords[i][1] = h_inv[1][1] * atom_coords[i][1]
+ h_inv[1][2] * atom_coords[i][2];
scaled_atom_coords[i][2] = h_inv[2][2] * atom_coords[i][2];

Truncating the Potential
• For system with pair wise additive interactions force on a particle I
is computed by all its neighbors. This means for a system of N
particles we have to evaluate N(N-1)/2 pair interactions. So the time
needed for evaluation of energy/force scales as N2. This is one of the
main bottleneck in the simulation field.
• Bulk system modeled via periodic boundary condition
– not feasible to include interactions with all images
– must truncate potential at half the box length (at most) to have all
separations treated consistently
• Contributions from distant separations may be important

Minimum image convention and Truncating the Potential
These two are same
distance from central
atom, yet:
Black atom interacts
Blue atom does not
How do we evaluate force on the red atom in
the simulation box. Assuming pair wise
interaction we should include interaction of
the red atom with all other atoms in the
simulation box. There are N-1 such term.
However, we must also include interaction
coming from images lying in the surrounding
boxes. That is an infinite number of terms.
Minimum image convention
Construct a simulation box of same size as
the the original box with the red atom at its
center. Now minimum image convention
says that the red atom interact with those
atoms which lie in this region, that is with
the closest periodic images of the other N-1
atoms.

Truncating the Potential
Only interactions
considered
With the minimum image convention energy/force
computation involves 1/2N(N-1) terms.This is
significant for large system size.
For short range interaction major contribution
comes from the neighbors close to the atom of
interest. So use a spherical cutoff to truncate the
interaction.
So the red atom interact only with the atoms lying
inside the cutoff region (2 black and one blue)
The cutoff distance should be smaller than L/2 to be consistent with the minimum
image convention
Thermodynamic properties are different for a truncated potential compared to a non-
truncated case. However, we can apply long range correction to get back
approximately the non-truncated properties.
Cutoff introduces discontinuity in the force and energy computation. This has
serious consequences on the energy conservation and stability of the simulation
Points to remember

Truncating the Potential contd
Potential truncation introduces discontinuity
– Corresponds to an infinite force
– Problematic for MD simulations
• ruins energy conservation
• Shifted potentials: a constant term is subtracted from the potential at all values
– Removes infinite force
– Still discontinuity in force: at the cutoff distance, the force will have a finite value which drops to zero just beyond the cutoff
( ) ( )( )
0
c cs
c
u r u r r ru r
r r
− ≤=
>

Truncating the Potential
Shifted-force potentials : a linear term can be added to the potential, making the derivative to be zero at the cutoff
– The discontinuity now appears in the gradient, not in the force itself. The force goes smoothly to zero at cutoff.
– The shift makes the potential deviate from the true potential, so the calculated thermodynamic properties will be changed
– True values can be retrieved but is difficult to do so, so rarely used in simulation
Eliminate discontinuities in the energy and force by a switching function
( ) ( ) ( )( )
0
c c csf
c
duu r u r r r r r
u r dr
r r
− − − ≤
= >
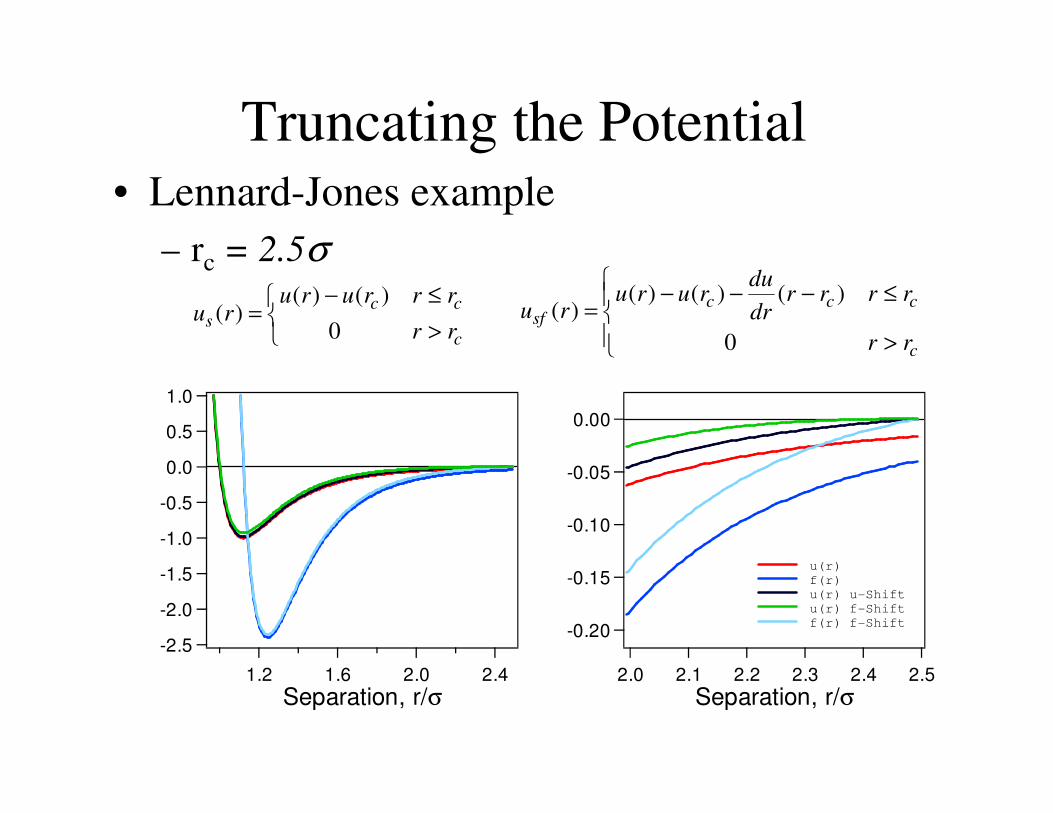
Truncating the Potential
( ) ( )( )
0
c cs
c
u r u r r ru r
r r
− ≤=
>
( ) ( ) ( )( )
0
c c csf
c
duu r u r r r r r
u r dr
r r
− − − ≤
= >
• Lennard-Jones example
– rc = 2.5σ
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
2.42.01.61.2
Separation, r/σ
-0.20
-0.15
-0.10
-0.05
0.00
2.52.42.32.22.12.0
Separation, r/σ
u(r)
f(r)
u(r) u-Shift
u(r) f-Shift
f(r) f-Shift

Potential function is multiplied by a polynomial which is a function of
distance
Switching function
)()()( rSrursu =
S(r) gradually taper the potential between two cutoff values : it smoothly
changes its value of 1 to a value of 0 between two cutoff : rl (lower
cutoff) and ru (upper cutoff) and satisfies the following criteria
02
200.1
02
200.1
=
=
==
==
=
=
==
==
urrdr
Sd
urrdr
dS
urrS
lrrdr
Sd
lrrdr
dS
lrrS

Zero first derivative ensures that the force approaches to zero smoothly
at the cutoffs. A continuous second derivative ensures the stability of the
integration algorithm.
Switching function contd.
0)(
1)(
=
=
urSl
rS
3)22(2
2)22(1
)22(0
22)]22(2
3[
3
2)22()(
rurcrurcrurc
lrurwhererurrurrS
−+−+−=
−=−−−= γγ
γ

/* Calculate pair interaction. The LJ pair potential is switched off smoothly between r_on and
r_off. */
one_over_r2_sep = 1.0 / r2_sep;
rho_2 = comb_par[id_0][id_1][2] * one_over_r2_sep;
rho_6 = CUBE(rho_2);
rho_12 = SQR(rho_6);
*u_vdw = rho_12 - rho_6;
*f_vdw = rho_12 + *u_vdw;
four_epsilon = comb_par[id_0][id_1][3];
(*u_vdw) *= four_epsilon;
(*f_vdw) *= 6.0 * one_over_r2_sep * four_epsilon;
if (r2_sep > r2_on)
sw1 = r2_off - r2_sep;
sw2 = two_over_gamma_cubed * sw1;
sw3 = sw1 * sw2 * (three_gamma_over_two - sw1);
sw4 = 6.0 * sw2 * (gamma - sw1);
*f_vdw = sw4 * (*u_vdw) + sw3 * (*f_vdw);
(*u_vdw) *= sw3;

Switching function contd.
We can use higher order polynomial also
5
5
4
4
3
3
2
210)(
−
−+
−
−+
−
−+
−
−+
−
−+=
lrur
lrr
c
lrur
lrr
c
lrur
lrr
c
lrur
lrr
c
lrur
lrr
ccrS
With the coefficients satisfying the previous criteria and that gives
C0=1, c1=0, c2=0, c3=-10, c4=15, c5=-6




Verlet List
i
rl ru
Particles outside cutoff do not
contribute to the energy of the
particle I.
Exclude those particles from the
energy computation
Verlet List: Introduce a second
cutoff radius ru >rl
Make a list of all the particles
within a radius of ru of particle i
If the maximum displacement of
the particles is less than (ru -rl) we
have to consider only the particles
in this list for the energy/force
computation
Update the Verlet list as soon as
one of the particle is displaced
more than (ru -rl)
For MD it is sufficient to have a Verlet list with half the number of
particles for each particle as long as interaction i-j is accounted for in
either the list of particle i or that of j

1
2
34
5
6
7
rc
7’
ru
The cutoff sphere and its skin around
an atom 1. Atoms 2,3,4,5 and 6 are on
the list of atom 1, but 7 is not. Among
these only 2,3,4 are within the rc when
the list is made
The skin is thick enough such that
when reconstructing an atom 7,
which was not in the original list of
atom 1can not penetrate the rc sphere.
Atom 3, 4 can move in and out but
their interaction get counted as they
are in the original list until the list get
updated.
From Allen and Tildesley

For a periodic system Verlet list has an elegant implementation due to
Bekker et. al. (Mol. Sim. 14, 137-151, 1995)
)(1
13
13
/
jkiF
N
j ki
F ∑=
∑−=
=
In a periodic system total force on particle i can be written as
Where the prime denotes that summation is performed over the nearest
image of the particle j in the central box (k=0) or in one of its 26
periodic images (or 8 in 2-d). (j.k) denotes the periodic image of
particle j in box k. Box k is defined by the integer numbers nx, ny, nz
k = 9nx+3 ny + nz

As the size of the system increases neighbor list becomes too large to
store easily
Also the testing of every pair separation is also very inefficient
Make the neighbor list using Cell list
Problem with Verlet List

Cell Lists or Linked-list
The simulation box is divided into cells with size equal to or slightly
larger than the cutoff rc
Distribute the particle to the cell according to their position
Each particle in a given cell interacts with only those particles in the
same or neighboring cells
This scales as N rc
The simulation cell is divided into
cells of size rc x rc, a particle i
interacts with those particles in the
same cell or neighboring cell (in 2D
9 cells; in 3D 27 cells)
i

The Cell list is created using the linked list methods:
Sorting of atoms to their respective cells
Two arrays are created (HEAD AND LIST)
HEAD (head of the chain) array has one element for each cell. This
contains the identification number of one of the molecules sorted into
that cell
LIST (linked list array) contains the number of next molecules in
that cell.
HEAD (head of the chain) array element is used to address the LIST
array element.
If we follow the trail of linked-list we will eventually reach an
element of LIST which is zero. This indicates that there are no more
molecules in that cell and we move on to the head of chain of the next
cell.
See Allen and Tildesley for details

void neighbor_lists_period()
/* Purge neighbor lists. */
for (i = 0; i < n_atoms; ++i)
nl_tail[i] = NULL;
/* Update cell lists. */
update_cells(period_switch, n_atoms, h, scaled_atom_coords, atom_coords,
atom_cells, first, last, phantoms, kc, phantom_skip, nc, nc_p);
/* Update positions of phantom atoms. */
update_phantoms(n_atoms, atom_cells, phantom_skip, phantoms, atom_coords);
/* Loop over central atoms. */
for (i = 0; i < n_atoms; ++i)
/* Get attributes of atom i. */
for (k = 0; k < NDIM; ++k)

Radial Distribution Function
• Radial distribution function, g(r)
– key quantity in statistical mechanics
– quantifies correlation between atom pairs
• Definition
( )( )
( )id
r dg r
r d
ρ
ρ=
r
r
Number of atoms at
r for ideal gas
Number of atoms at
r in actual system
( )id Nr d d
Vρ =r r
dr
4
3
2
1
0
543210
Hard-sphere g(r) Low density High density

),(),(2
1),(
jr
ira
jr
irg
jdr
idr
Vj
ri
ra ∫=
Various thermodynamic quantities in terms of g(r)
Ensemble average of any pair function can be expressed in terms
of g(r) as follows
For example we can write the energy as (assuming pair-wise
additivity
)()(242
)( rgru
crrN
ijrcuE ∫
∞+∑= πρ

Simple Long-Range Correction
• Approximate distant interactions by assuming
uniform distribution beyond cutoff: g(r) = 1 r > rcut
• Corrections to thermodynamic properties
– Internal energy
– Virial
– Chemical potential
2( )42
cut
lrc
r
NU u r r drρ π
∞
= ∫Expression for Lennard-Jones model
2 214
6cut
lrc
r
duP r r dr
drρ π
∞
= ∫
2( )4 2
cut
lrclrc
r
Uu r r dr
Nµ ρ π
∞
= =∫
9 3
383
9
LJlrc
c c
U Nr r
σ σπ ρσ ε
= −
9 3
2 332 3
9 2
LJlrc
c c
Pr r
σ σπρ σ ε
= −
For rc/σ = 2.5, these are about
5-10% of the total values

Overcoming time issues
• Ewlad method O(N3/2)
• Particle Mesh Ewald O(NlogN)
• Cell Multipole Methods O(N)

• Newton’s equation of motion are time reversible and so should be
our algorithm.
• Hamiltonian dynamics preserve the magnitude of volume element in
phase space and so our algorithm should have this area preserving
property
• simplicity (How long does it take to write and debug?)
• efficiency (How fast to advance a given system?)
• stability (what is the long-term energy conservation?)
• reliability (Can it handle a variety of temperatures, densities,
potentials?)
Criteria for an Integrator

The nearly universal choice for an integrator is the Verlet (leapfrog) algorithm
r(t+δt) = r(t) + v(t) δt + 1/2 a(t) δt 2 + b(t) δt 3 + O(δt 4) Taylor expand
r(t- δt) = r(t) - v(t) δt + 1/2 a(t) δt 2 - b(t) δt 3 + O(δt 4) Reverse time
r(t+ δt) = 2 r(t) - r(t- δt) + a(t) δt 2 + O(δt 4) Add
v(t) = (r(t+ δt) - r(t- δt))/(2 δt) + O(δt 2) estimate velocities
Time reversal invariance is built in the energy does not drift.
Velocity is not required to compute the new position.
Once the new position is computed using position at t- δt, discard the old
Position. The current position become the old positions and the new position
become the current position.
Note that velocity is used only to compute the kinetic energy and hence the
temperature of the system
Time reversible and area preserving
Tuckerman, Berne, Martyna, JCP, 97, 1990 1992

Linear Stability analysis for Harmonic oscillator
xxF 2)( ω−=
=
+
+
)(
)(
)(
)(
tv
txS
ttv
ttx ω
δ
δω
−−
−−=
2/1
)4/1(2/1
2
22
εε
εεεS
Position Verlet scheme can be written as
where tωδε =and
42 <ε
πωδ
2//2
pTt<< ωπ /2=pT
Powers of S is bonded if
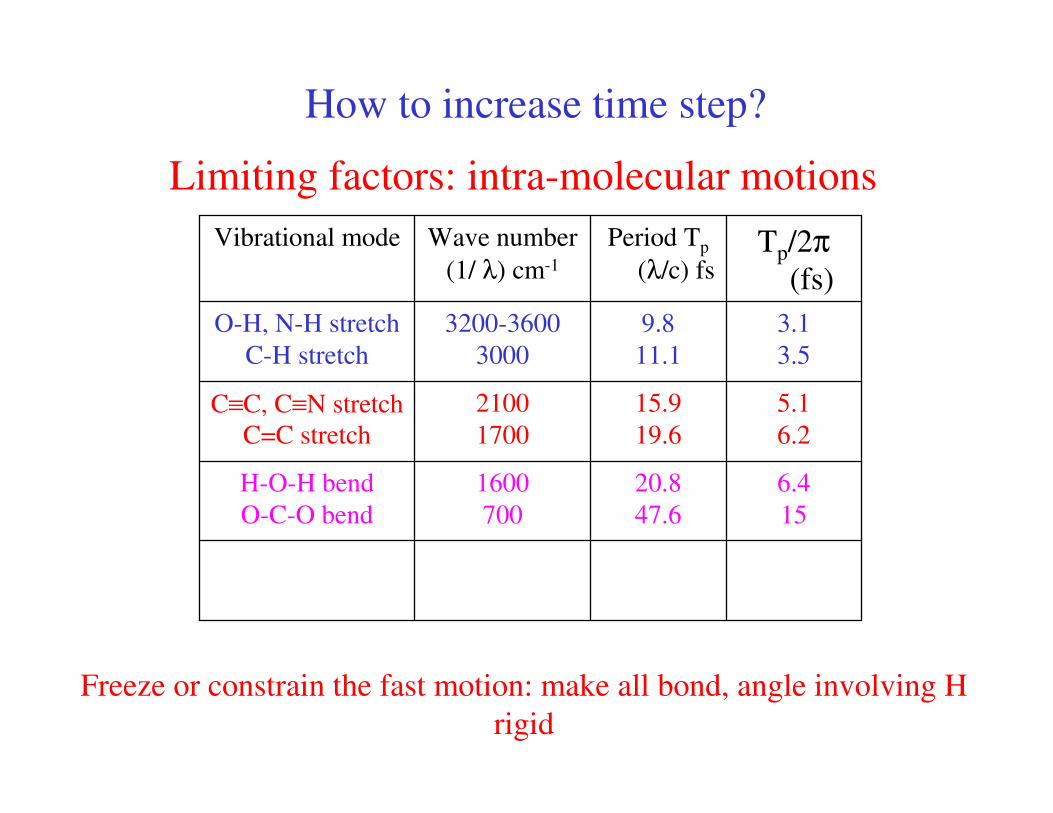
How to increase time step?
Limiting factors: intra-molecular motions
6.4
15
5.1
6.2
3.1
3.5
Tp/2π(fs)
1600
700
2100
1700
3200-3600
3000
Wave number
(1/ λ) cm-1
20.8
47.6
H-O-H bend
O-C-O bend
15.9
19.6
C≡C, C≡N stretch
C=C stretch
9.8
11.1
O-H, N-H stretch
C-H stretch
Period Tp
(λ/c) fs
Vibrational mode
Freeze or constrain the fast motion: make all bond, angle involving H
rigid

How to increase time step?
Multiple time steps algorithm
Short-range interactions governs the intra-molecular motion, time scale
of which are very fast. In that time scale “long range” part of the
interaction hardly changes and need not be computed at same frequency
of the short range interactions.
longshort FFF +=
longmedshort FFFF ++=
or
a time step to compute short-range interactions
another time stop to compute medium range interactions
another time step to compute long-range interactions
Use Multiple time steps

Solvation Models
in Molecular Simulations
•Explicit Solvent
Large system - CPU intensive
Long equilibration simulation times
Quality depends on forcefield used
•Implicit Solvent
Continuous: medium surrounding solute
SASA: Solvent Accessible solute interaction
GB:Treat long-range effects
AVGB: add short-range effects to GB

Water Solvation Effects
•Long Range Effects (Electrostatic)
Solute Polarization (reaction field)
Dielectric Screening
•Short Range Effects (contraction to 1st solvation shell)
Dispersion Interactions (vdW)Cavity Formation (Entropic)
Hydrogen Bonding (Charge Transfer)

The solvation free energy is the free energy change to transfer a molecule from vacuum
to solvent. The solvation free energy has three components
cavGvdw
Gelec
Gsolv
G ∆+∆+∆=∆
elecG∆ is the electrostatic component: very important component for polar and
charged solute
is the van der Walls interaction between the solute and solventvdwG∆
cavG∆ is the free energy required to form the solute cavity within the solvent. This
component is positive and comprises the entropic penalty associated with the
reorganization of the solvent molecules around the solute together with the
work done against the solvent pressure to create the cavity

Short-Range van der Walls Effects
•Only atoms in first solvation shell feel effects.
•Solvation free energy dependent on solvent accessible
surface.
•Surface tension parameters σ
•SASA:
•Fused-sphere model•Connolly Quadratic Surface
∑=
=∆+∆N
i
iicavvdW AGG1
σ
Courtesy: G. Zamanakos, Caltech

Long Range Effects
Continuum Dielectric Approximation: Poisson-Boltzman
)(4)()( rrrρρρ
πρε =Φ∇⋅∇−
)(rρ
Φ Electrostatic potential dielectric, screening, and charge density)(),(),( rrrρρρ
ρκε
The solute is treated as a body of constant low dielectric and the solvent is modeled as a
continuum of high dielectric
επρφ )(4
)(2 rr −=∇
For a set of point charges in a constant dielectric medium the Poisson equation
reduces to Coulomb’s Law
When dielectric changes with position Poisson equation becomes

)(4)(sinh2)()()( rrrrrρρρρρ
πρκε =Φ+Φ∇⋅∇−
)(rρ
κ
When mobile ions are present Poisson equation needs to be modified to take into
account for their redistribution in the solution in response to the electric potential. The
ions distribution is described by a Boltzmann distribution of the form
)/)(exp()( TB
krVNrn −= n(r) is the number density at position r
N is the bulk number density
V(r) is the energy change to bring the ion from
infinity to the position r
When the above fact is taken into consideration the Poisson equation becomes Poisson-
Boltzmann equation
is related to the inverse of the Debye-Huckel length
Expanding the hyperbolic sine function as Taylor series and keeping only the first term we
have linearised Poisson-Boltamann equation
)(4)(2)()()( rrrrrρρρρρ
πρκε =Φ+Φ∇⋅∇−

•Numerical solution, CPU intensive, scales as )( 3NO
•Lack of gradients (atomic forces) •Accuracy depends on grid resolution.
•Applications show successful predictions.
•Not parallelizable, QM implementations available

The Generalized Born Model
•Polar Solvation Energy of a charged sphere of radius R and
permittivity εin in dielectric medium εout (Born):
R
qG
outin
pol
211
2
1
−−=∆
εε
•System of N spheres of radii αi, far away from each other:
+
−−=∆ ∑∑∑
=≠ ==
N
ji
N
j ij
jiN
i i
i
outin
polr
qqqG
1 11
211
2
1
αεε
•We seek interpolation formula between the two limits:
∑∑= =
−−=∆
N
i
N
j ij
ji
outin
polf
qqG
1 1
11
2
1
εε

The Generalized Born Model
( )jiijjiijij rrf αααα 4exp 22 −+=
•Interpolation formula:
•Limit iiiij fr α=⇒= 0
i
i
outin
pol
qG
αεε
211
2
1
−−=∆ (Born Formula)
•Limit ijijij rfr →⇒∞→
ij
ji
outin
polij
r
qqG
−−→∆
εε
11
2
1(Coulomb Limit)

Born Radii
•If we set all charges to zero except qk, we get:
k
k
outin
pol
qG
αεε
211
2
1
−−=∆
•Parameters αk are the Born Radii.
•Physical Meaning: the effective radius of an ion of charge qk, whose solvation energy is equal to the
self-energy of polarization of atom in the molecule.
•Difficult to calculate.
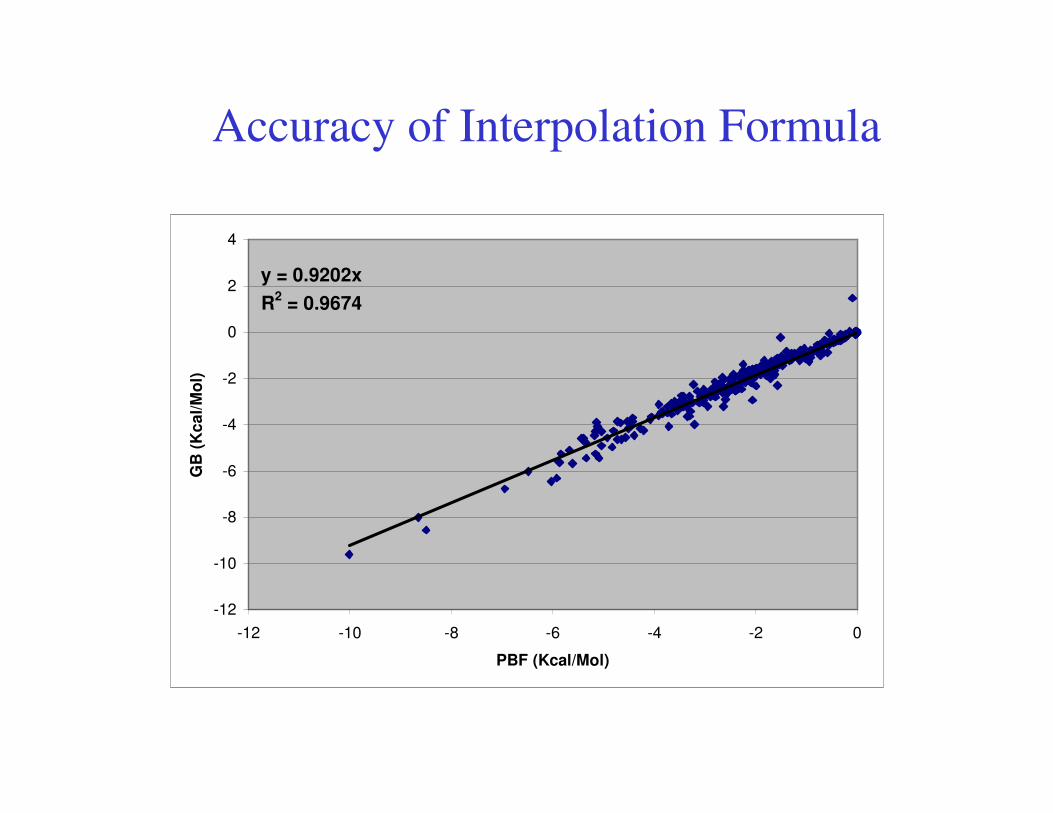
Accuracy of Interpolation Formula
y = 0.9202x
R2 = 0.9674
-12
-10
-8
-6
-4
-2
0
2
4
-12 -10 -8 -6 -4 -2 0
PBF (Kcal/Mol)
GB
(K
ca
l/M
ol)

Calculation of Born Radii
•Solute charge distribution polarizes solvent which in turn interacts with the solute (reaction field ).)(rreac
ρΦ
•Assume the reaction field is coulombic in nature. It can be proven that:
∫Ω −
−=
k
rdrrR
kkk
3
4
1
4
111ρρπα
•Pairwise approximation:
∑ ∫∫≠Ω −
=− kk V kk kk
rdrr
rdrr '
3
4
3
4
'
11ρρρρ
•Analytical solution exists for integral over sphere Need volumes of every atom.
3
4
3
πkeff
k
VR =

AVGB Solvation Energy
•Long-Range Effects:
( )∑∑= = −+
−−=∆
N
i
N
jjiijjiij
ji
outin
pol
rr
qqG
1 122 4exp
11
2
1
ααααεε
∑ ∫≠ −
−=kk V kkk
k
rdrrR '
3
4
'
1
4
111ρρπα
•Short-Range Effects:
∑=
=∆+∆N
i
iicavvdW AGG1
σ
Solvent accessible surface areas and solvent excluded volumes must be calculated fast and accurately

The Fused-Sphere Model
•Analytical calculation of volumes and areas, with
gradients
•Complicated topologies
•Robust algorithms

Area Calculation
Gauss-Bonnet Theorem
( ) ( ) ( )∑∑ ∫∫∫==
=Ω++m
i
i
n
i RC
g RdssKdllk
i11
2πχ
Geodesic curvature, gaussian curvature of surface, Euler-Poincare characteristic of surface R
External angles of surface
Zamanakos, Phd thesis (Caltech), 2002


















