
NANO266 - Lecture 13 - Ab initio molecular dyanmics
23
Ab initio molecular dynamics Shyue Ping Ong
-
Upload
shyue-ping-ong -
Category
Education
-
view
401 -
download
2
Transcript of NANO266 - Lecture 13 - Ab initio molecular dyanmics

Ab initio molecular dynamics
Shyue Ping Ong

What is molecular dynamics (MD)?
Moving atomic nuclei by solving Newton’s equation of motion
For N nuclei, we have 3N positions and 3N velocities, i.e., a 6N dimensional phase space.
NANO266 2
M!!RI = −∂E∂RI
= FI[{RJ}]
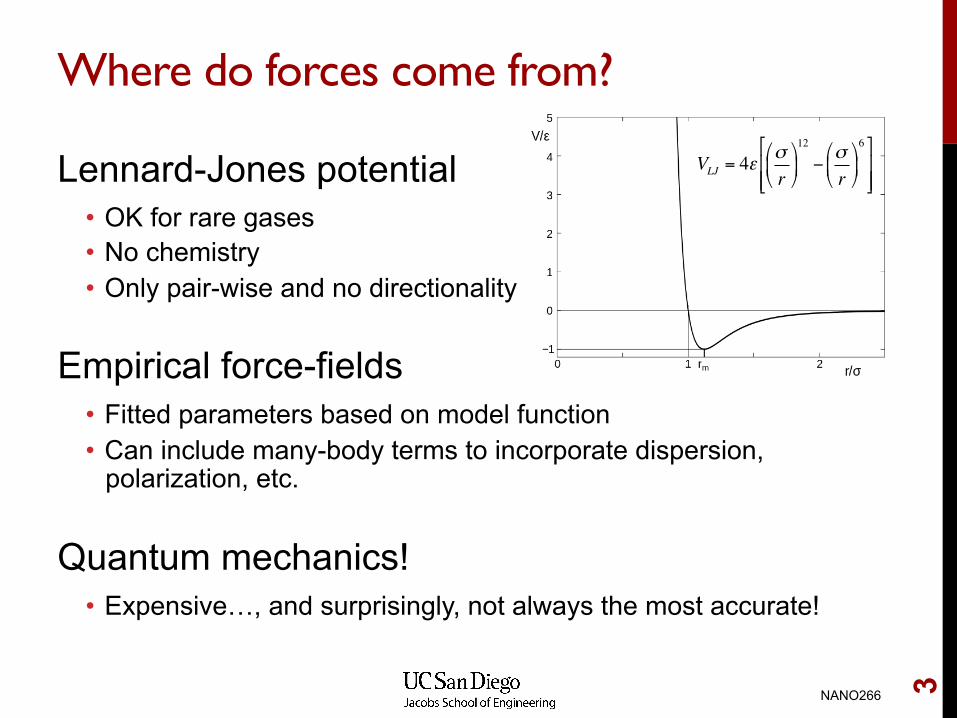
Where do forces come from?
Lennard-Jones potential • OK for rare gases • No chemistry • Only pair-wise and no directionality
Empirical force-fields • Fitted parameters based on model function • Can include many-body terms to incorporate dispersion,
polarization, etc.
Quantum mechanics! • Expensive…, and surprisingly, not always the most accurate!
NANO266 3
VLJ = 4εσr
!
"#
$
%&12
−σr
!
"#
$
%&6(
)**
+
,--

2013 Nobel Prize in Chemistry
NANO266 4

Computational Experiment
NANO266 5
Initialize positions and velocities
Compute forces
Determine new positions
Calculate thermodynamic
averages
Repeat until time is reached

Initialization
2nd order PDF -> Initial positions and velocities Positions
• Reasonable guess based on structure • No overlap / short atomic distances
Velocities
• Small initial velocity • Steadily increase temperature (velocity scaling)
NANO266 6
M!!RI = −∂E∂RI
= FI[{RI}]
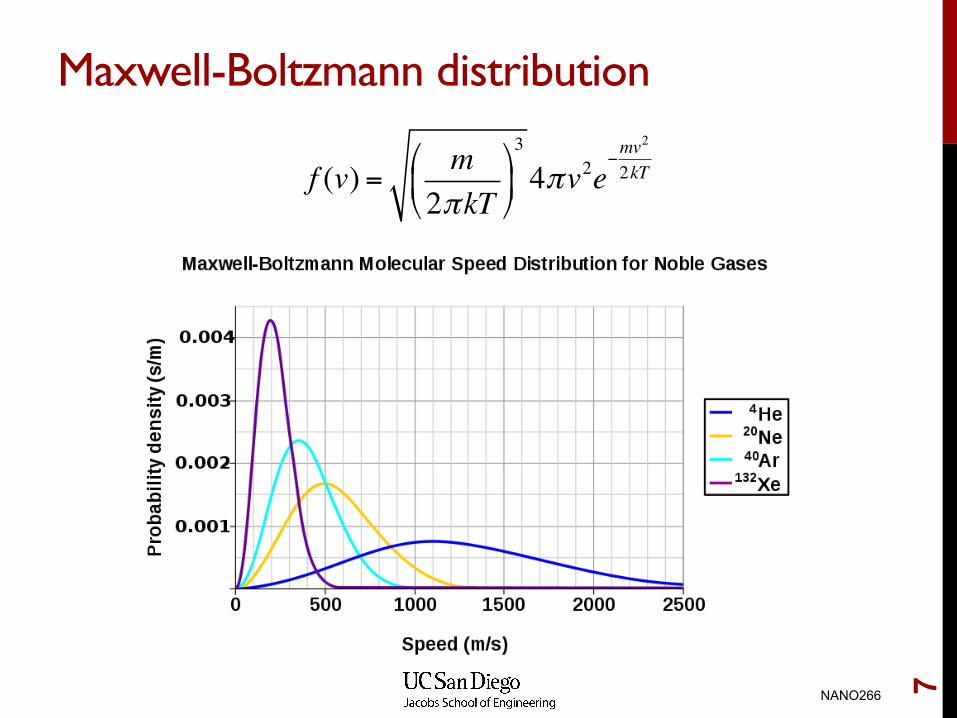
Maxwell-Boltzmann distribution
NANO266 7
f (v) = m2πkT!
"#
$
%&3
4πv2e−mv2
2kT

Ensembles
Microcanonical – N, V, E Canonical – N, V, T
• Temperature control achieved via thermostats • Examples: velocity rescaling, Andersen thermostat, Nose-Hoover
thermostat, Langevin dynamics, etc.
NANO266 8

Integrating equations of motion – Verlet algorithm
Key properties of Verlet algorithm • Errors do not accumulate • Energy is conserved • Simulations are stable
NANO266 9
M!!RI = FI[{RJ}]
!!RI =1MI
FI[{RJ}]
[RI(t +Δt)−RI(t)]−[RI(t)−RI(t −Δt)](Δt)2
=1MI
FI[{RJ}]
RI(t +Δt) = 2RI(t)+RI(t −Δt)+(Δt)2
MI
FI[{RJ(t)}]

Flavors of quantum MD
Born-Oppenheimer • Electrons stay in instantaneous ground state as nuclei move • Perform electronic SCF at each time step • Compute forces (Hellman-Feynman theorem) • Move nuclei
Car-Parrinello • Treat ions and electrons as one unified system • Accomplished with fictitious kinetic energy for electrons
NANO266 10
Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory, Phys. Rev. Lett., 1985, 55, 2471–2474, doi:10.1103/PhysRevLett.55.2471.

Coupled equations of motion
The CP Lagrangian
Equations of motion
NANO266 11
L = 12(2µ)
i=1
N
∑ dr !ψi (r)2
∫ + MII=1
NI
∑ !!RI −E[ψi,r]+ Λij drψi*(r)ψ j (r)−δij∫%& '
(ij∑
Imposes orthonormality of electronic states
µ !!ψi (r, t) = −Hψ j (r)+ Λikψk (r, t)k∑
MI!!RI = −
∂E∂RI

Comparison of BO vs CP MD
NANO266 12
CP Avoids need for
costly SCF iteration of electrons at each
step
Time step required is smaller
BO Solves Schrodinger
equation at each step
Time step can be longer

General procedure for MD simulation
NANO266 13

Applications of MD
Thermodynamics from ensemble averages Real-time evolution
• Reactions • Interaction of molecules on surfaces • Trajectories of ions
Ground-state structures
• Geometry optimization is in fact a form of MD. • For complex structures (e.g., low symmetry systems, interfaces,
liquids, amorphous solids, etc.), MD can be used to determine low-energy structures
NANO266 14
Nattino, F.; Ueta, H.; Chadwick, H.; Reijzen, M. E. Van; Beck, R. D.; Jackson, B.; Hemert, M. C. Van; Kroes, G. Ab Initio Molecular Dynamics Calculations versus Quantum-State- Resolved Experiments on CHD 3 + Pt(111): New Insights into a Prototypical Gas − Surface Reaction, J. Phys. Chem. Lett., 2014, 5, 1294–1299, doi:10.1021/jz500233n.

Thermodynamic averages
Under ergodic hypothesis
Examples of averages
• Energy (potential, kinetic, total) • Temperature • Pressure • Mean square displacements (diffusion) • Radial distribution function
NANO266 15
A =Aie
−βEi (r,p)
i∑
e−βEi (r,p)i∑
= limT→∞
1T
A(t)dt0
T
∫

Correlation Functions from MD
Pair distribution function
Time correlation function
NANO266 16
g(r) = VN 2 δ(r− rij)
j≠i∑
i∑
Einstein relation (valid at large t)
D =13
dt v(t)v(0)0
∞
∫
Example: Diffusion coefficient
D =12dt
(r(t)− r(0))22tγ = (A(t)− A(0))2
γ = dt !A(t) !A(0)0
∞
∫

Structure of Amorphous InP
NANO266 17
Lewis, L.; De Vita, A.; Car, R. Structure and electronic properties of amorphous indium phosphide from first principles, Phys. Rev. B, 1998, 57, 1594–1606, doi:10.1103/PhysRevB.57.1594.
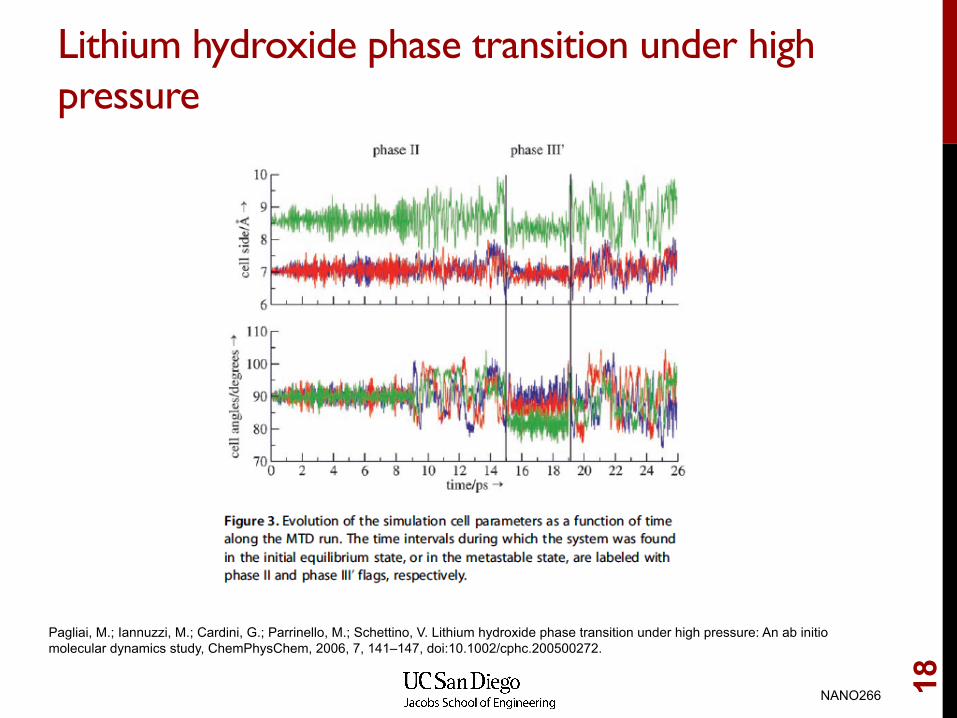
Lithium hydroxide phase transition under high���pressure
NANO266 18
Pagliai, M.; Iannuzzi, M.; Cardini, G.; Parrinello, M.; Schettino, V. Lithium hydroxide phase transition under high pressure: An ab initio molecular dynamics study, ChemPhysChem, 2006, 7, 141–147, doi:10.1002/cphc.200500272.

Dihydrogen oxide
NANO266 19
Izvekov, S.; Voth, G. a. Car-Parrinello molecular dynamics simulation of liquid water: New results, J. Chem. Phys., 2002, 116, 10372–10376, doi:10.1063/1.1473659.

Diffusion in Lithium Superionic Conductors
NANO266 20
ac
ab
1D conductors would be highly sensitive to blocking defects!
X
Trace of Li motion at 900K
Overall Ea (meV)
σ@ 300 K (mS/cm)
First principles1
210 13
Experimental2
240 12
1 S. P. Ong Y. Mo, W. Richards, L. Miara, H. S. Lee, G. Ceder. Energy & Environ. Sci., 2013, 6, 148–156. 2 N. Kamaya et al. Nat. Mater. 2011, 10, 682-686

Cation has a small effect on diffusivity of Li10MP2S12
Isovalent
Aliovalent
Ge Si Sn P Al σ @ 300 K (mS/Cm) 13 23 6 4 33
Ea (meV) 210 200 240 260 180
(Aliovalent substitutions are Li+ compensated)
S. P. Ong, Y. Mo, W. D. Richards, L. Miara, H. S. Lee, G. Ceder, Phase stability, electrochemical stability and ionic conductivity in the Li10±1MP2X12 family of superionic conductors. Energy Environ. Sci., 2012, doi: 10.1039/C2EE23355J

Phonons from MD
NANO266 22
Kim, J.; Yeh, M. L.; Khan, F. S.; Wilkins, J. W. Surface phonons of the Si(111)-7x7 reconstructed surface, Phys. Rev. B, 1995, 52, 14709–14718, doi:10.1103/PhysRevB.52.14709.
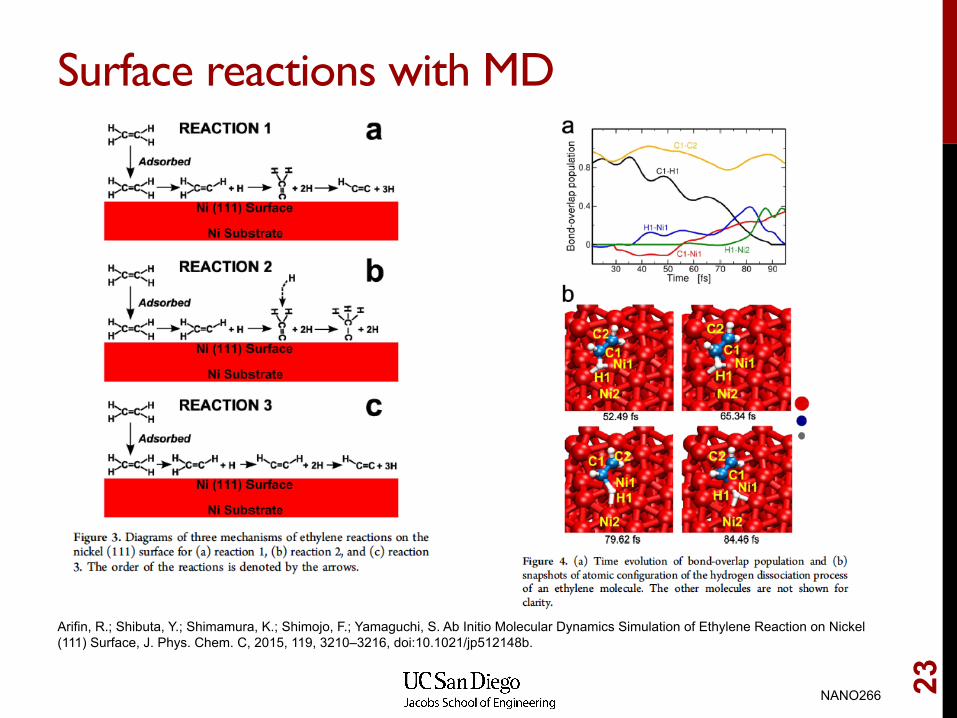
Surface reactions with MD
NANO266 23
Arifin, R.; Shibuta, Y.; Shimamura, K.; Shimojo, F.; Yamaguchi, S. Ab Initio Molecular Dynamics Simulation of Ethylene Reaction on Nickel (111) Surface, J. Phys. Chem. C, 2015, 119, 3210–3216, doi:10.1021/jp512148b.


















