
Molecular Photophysics under Shock Compression: Ab Initio ... · Molecular Photophysics under Shock...
8
Molecular Photophysics under Shock Compression: Ab Initio Nonadiabatic Molecular Dynamics of Rhodamine Dye Xin Zhou, †,‡ Linqiu Li, ‡ Dana D. Dlott, § and Oleg V. Prezhdo* ,‡,∥ † College of Environment and Chemical Engineering, Dalian University, Dalian, 116622, People’s Republic of China ‡ Department of Chemistry, University of Southern California, Los Angeles, California 90089, United States § School of Chemical Sciences and Fredrick Seitz Materials Research Laboratory, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States ∥ Department of Physics and Astronomy, University of Southern California, Los Angeles, California 90089, United States * S Supporting Information ABSTRACT: Photoluminescent chromophores can be used as probes for shock-wave propagation, provided that luminescence wavelength and/or lifetime depend on the increased pressure and temperature generated by the propagating shock-wave. Recent experiments have observed such effects with organic dyes and inorganic semiconductor quantum dots. We employ ab initio density functional theory and nonadiabatic molecular dynamics to study the effects of pressure and temperature on fluorescence properties of rhodamine-6G encapsulated in silica. Increase of pressure alone already decreases the luminescence wavelength and lifetime. The combined effect of enhanced pressure and temperature, representing shock conditions, is even stronger with temperature growth having a particularly strong influence on the nonradiative lifetime. The excitation wavelength is red-shifted due to changes in the highest occupied molecular orbital and lowest unoccupied molecular orbital energies caused by mechanical distortions of the π-electron plane of the organic chromophore. The nonradiative relaxation accelerates due to enhancement in the nonadiabatic electron- vibrational coupling, that is particularly sensitive to temperature. The difference in the sensitivity of luminescence wavelength and lifetime to pressure and temperature suggests that it may be possible to determine the thermodynamic properties separately on the basis of the two measured quantities. A broad spectrum of vibrational modes couples to the electronic transition with lower frequency modes exhibiting stronger coupling. Higher-frequency modes become more important under the shock conditions. The simulations show excellent agreement with experiment, characterize how temperature and pressure influence electron−hole recombination in rhodamine-6G encapsulated in a silica matrix, and provide important details on the mechanism of nonequilibrium dynamics in the fluorescent chromophore. 1. INTRODUCTION Insights into the pressure, volume, and temperature of materials under shock compression are important, particularly, in deriving the equation of states of materials and predicting materials response under transient extreme conditions of high pressure, high temperature, and high-strain deformation. 1−3 Various properties under shock compression have been investigated in recent years, including mechanical properties of shocked materials, 4 optical properties of crystals and semiconductor quantum dots, 5−7 phase transitions of metal oxides, 8 shock-induced changes inside polymers, 9,10 and shock dynamics of emissive organic dyes and self-assembled monolayers. 3,11,12 Strain, pressure, temperature, and composi- tions are complex functions of space and time in a shocked microstructured medium. 13−15 Applications of fluorescent materials play an important role in a growing number of disciplines, including biotechnology, medicine, photonics, and optoelectronics. 16−19 A fluorescent dye can be used to probe shock compression dynamics by measuring shock-induced changes in emission wavelength, line width, intensity, and lifetime. 20−22 Huston et al. found that the crystal-violet lifetime increased from the 100 ps at atmospheric pressure to 200 ps at 2 GPa. 11 The effect was attributed to shock-induced increases in glycerol viscosity. Taguchi et al. investigated the emission lifetimes of rhodamine-6G (R6G) in solution at high static pressures up to 0.9 GPa and attributed the observed changes to chromophore aggregation. 23 Dreger et al. studied emission lifetimes of a related dye, rhodamine-B (RhB), in a solid polyacrylic acid matrix under high static pressures. Their results showed that the RhB lifetime decreased from 6 to 3 ns with the increase of pressure from ambient to 7 Special Issue: Prashant V. Kamat Festschrift Received: December 28, 2017 Revised: March 3, 2018 Published: March 5, 2018 Article pubs.acs.org/JPCC Cite This: J. Phys. Chem. C 2018, 122, 13600-13607 © 2018 American Chemical Society 13600 DOI: 10.1021/acs.jpcc.7b12768 J. Phys. Chem. C 2018, 122, 13600−13607 Downloaded via UNIV OF SOUTHERN CALIFORNIA on November 7, 2019 at 16:51:55 (UTC). See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles.
Transcript of Molecular Photophysics under Shock Compression: Ab Initio ... · Molecular Photophysics under Shock...
Molecular Photophysics under Shock Compression: Ab InitioNonadiabatic Molecular Dynamics of Rhodamine DyeXin Zhou,†,‡ Linqiu Li,‡ Dana D. Dlott,§ and Oleg V. Prezhdo*,‡,∥
†College of Environment and Chemical Engineering, Dalian University, Dalian, 116622, People’s Republic of China‡Department of Chemistry, University of Southern California, Los Angeles, California 90089, United States§School of Chemical Sciences and Fredrick Seitz Materials Research Laboratory, University of Illinois at Urbana−Champaign, Urbana,Illinois 61801, United States∥Department of Physics and Astronomy, University of Southern California, Los Angeles, California 90089, United States
*S Supporting Information
ABSTRACT: Photoluminescent chromophores can be used as probes forshock-wave propagation, provided that luminescence wavelength and/orlifetime depend on the increased pressure and temperature generated by thepropagating shock-wave. Recent experiments have observed such effectswith organic dyes and inorganic semiconductor quantum dots. We employab initio density functional theory and nonadiabatic molecular dynamics tostudy the effects of pressure and temperature on fluorescence properties ofrhodamine-6G encapsulated in silica. Increase of pressure alone alreadydecreases the luminescence wavelength and lifetime. The combined effect ofenhanced pressure and temperature, representing shock conditions, is evenstronger with temperature growth having a particularly strong influence onthe nonradiative lifetime. The excitation wavelength is red-shifted due tochanges in the highest occupied molecular orbital and lowest unoccupiedmolecular orbital energies caused by mechanical distortions of the π-electronplane of the organic chromophore. The nonradiative relaxation accelerates due to enhancement in the nonadiabatic electron-vibrational coupling, that is particularly sensitive to temperature. The difference in the sensitivity of luminescence wavelength andlifetime to pressure and temperature suggests that it may be possible to determine the thermodynamic properties separately onthe basis of the two measured quantities. A broad spectrum of vibrational modes couples to the electronic transition with lowerfrequency modes exhibiting stronger coupling. Higher-frequency modes become more important under the shock conditions.The simulations show excellent agreement with experiment, characterize how temperature and pressure influence electron−holerecombination in rhodamine-6G encapsulated in a silica matrix, and provide important details on the mechanism ofnonequilibrium dynamics in the fluorescent chromophore.
1. INTRODUCTION
Insights into the pressure, volume, and temperature of materialsunder shock compression are important, particularly, inderiving the equation of states of materials and predictingmaterials response under transient extreme conditions of highpressure, high temperature, and high-strain deformation.1−3
Various properties under shock compression have beeninvestigated in recent years, including mechanical propertiesof shocked materials,4 optical properties of crystals andsemiconductor quantum dots,5−7 phase transitions of metaloxides,8 shock-induced changes inside polymers,9,10 and shockdynamics of emissive organic dyes and self-assembledmonolayers.3,11,12 Strain, pressure, temperature, and composi-tions are complex functions of space and time in a shockedmicrostructured medium.13−15
Applications of fluorescent materials play an important rolein a growing number of disciplines, including biotechnology,medicine, photonics, and optoelectronics.16−19 A fluorescent
dye can be used to probe shock compression dynamics bymeasuring shock-induced changes in emission wavelength, linewidth, intensity, and lifetime.20−22 Huston et al. found that thecrystal-violet lifetime increased from the 100 ps at atmosphericpressure to 200 ps at 2 GPa.11 The effect was attributed toshock-induced increases in glycerol viscosity. Taguchi et al.investigated the emission lifetimes of rhodamine-6G (R6G) insolution at high static pressures up to 0.9 GPa and attributedthe observed changes to chromophore aggregation.23 Dreger etal. studied emission lifetimes of a related dye, rhodamine-B(RhB), in a solid polyacrylic acid matrix under high staticpressures. Their results showed that the RhB lifetime decreasedfrom 6 to 3 ns with the increase of pressure from ambient to 7
Special Issue: Prashant V. Kamat Festschrift
Received: December 28, 2017Revised: March 3, 2018Published: March 5, 2018
Article
pubs.acs.org/JPCCCite This: J. Phys. Chem. C 2018, 122, 13600−13607
© 2018 American Chemical Society 13600 DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
Dow
nloa
ded
via
UN
IV O
F SO
UT
HE
RN
CA
LIF
OR
NIA
on
Nov
embe
r 7,
201
9 at
16:
51:5
5 (U
TC
).Se
e ht
tps:
//pub
s.ac
s.or
g/sh
arin
ggui
delin
es f
or o
ptio
ns o
n ho
w to
legi
timat
ely
shar
e pu
blis
hed
artic
les.

GPa.24 Because the duration of the shock-wave is in the tens ofnanoseconds, which is several times longer than theluminescence lifetime, the chromophore response is fasterthan the shock duration, and chromophore luminescence canbe used as an indicator for shock wave evolution.Fluorescence lifetime imaging has several advantages over
measurements of fluorescence intensity and frequency shift. Onthe one hand, lifetime provides information on dynamiccoupling between the chromophore and the shock environ-ment, not available from time-independent measurements. Onthe other hand, fluorescence lifetimes are more robust touncertainties. Fluorescence intensity can change due to manyeffects occurring during a shock-wave experiments. Forinstance, the sample can move into or away from the line offocus, or some of the chromophore species can be photo-chemically damaged. A frequency shift is insensitive to intensitychanges. However, it is hard to measure because one requiresinformation on the entire spectrum. Unlike emission wave-length, which can be obtained nearly instantaneously, lifetimemeasurements require a time window of several lifetimes.Recently, Liu et al. explored emission of R6G under shock
compression up to 9.1 GPa to understand molecular photo-physics in extreme environments.12 The lifetime decreasedfrom 3.25 to 1.25 ns as a linear function of shock pressure from0 to 9 GPa. The observed lifetime decrease was attributed tothe shock-induced enhancement of the R6G nonradiativerelaxation. R6G was studied as a free dye dissolved inpoly(methyl methacrylate) (PMMA), and as a dye encapsu-lated in silica microparticles suspended in PMMA. It has beenshown that fluorescence quantum efficiency and chromophorebrightness can be enhanced by encapsulating covalently singleor multiple dyes in silica-based materials and making smallchanges to the internal architecture of the formed par-ticles.25−27 Although many experimental works have focusedon the effect of shock compression on the lifetime offluorescent chromophores, it is unclear what particular pressureand temperature induced changes in the geometric andelectronic structure of the chromophore and the matrix,modifications of the chromophore-matrix coupling, and specificvibrational motions are responsible for the inelastic and elasticelectron-vibrational scattering that leads to the luminescencedecay. It is very challenging to explore experimentally themicroscopic properties at extreme conditions. Materialscomputer simulations are particularly useful and powerful forproblems that may be inaccessible to direct experimentalstudies.Motivated by the experimental work, and particularly ref 12,
we report an ab initio atomistic time-domain investigation ofthe influence of pressure and temperature used in shockcompression on nonradiative relaxation in R6G encapsulated ina silica matrix. We apply the nonadiabatic molecular dynamics(NAMD) approach28 formulated within the framework of real-time time-dependent density functional theory (TDDFT) inthe Kohn−Sham (KS) representation.29−32 Theory andSimulation Methods describes the essential theoretical back-ground and computational details underlying the NAMDsimulation. Results and Discussion is separated into three parts,focusing on the geometric and electronic structure, electron-vibrational interactions, and electron−hole recombinationdynamics. We investigate whether the luminescence wavelengthchanges due to distortion of chromophore geometry under theinfluence of pressure, or increased thermal fluctuations causingdeviations from the optimal geometry, or chromophore
physical or chemical interaction with the matrix. Thenonradiative channel for luminescence decay can be affectedby changes in the electronic excitation energy, the nonadiabaticcoupling, and the quantum coherence time. We study howthese three characteristics vary due to chromophore deforma-tion under pressure, faster nuclear motions at increasedtemperature, and interaction with the matrix. The simulationresults are compared with the available experimental data. Thekey findings are summarized in Conclusions.
2. THEORY AND SIMULATION METHODSThe NAMD simulation is carried out using a mixed quantum-semiclassical framework by implementing the decoherence-induced surface hopping (DISH) technique33 in the time-domain KS theory.34 In this approach, the lighter and fasterelectrons are treated quantum mechanically, while the heavierand slower nuclei are described semiclassically. While fewestswitching surface hopping (FSSH) is the most commonNAMD methodology,35,36 it requires decoherence correc-tions37−39 to accurately model slow quantum transitions acrosslarge energy gaps, such as the nonradiative relaxation of theR6G chromophore to the ground state. Instead of introducingdecoherence as a correction, DISH interprets decoherenceevents as wave function collapse giving rise to surface hops. Theapproach employed in the current simulation provides adetailed ab initio picture of the coupled electron-vibrationaldynamics on the atomic scale and in the time domain. It hasapplied successfully to many systems and processes.40−47 Thefundamentals of the NAMD methodology are presented inSupporting Information, and a detailed description of themethod, as implemented in the PYXAID package, can be foundin the original literature.31,32,48
The current simulation model has been generated as follows.The silica matrix is simulated by a 2 × 2 × 2 supercell of β-cristobalite SiO2, subject to periodic boundary conditions inthree dimensions. The initial positions of the simulationsupercell were taken from the experimentally determined X-raystructure.49,50 The R6G dye contains a xanthene ringsubstituted with two methyl groups, two ethylamino groups,and a carboxyphenyl group. In vacuum, the phenyl ring is foundalmost perpendicular to the xanthene plane, and therefore thetwo fragments are not conjugated. Previous theoretical worksshowed that the lowest singlet excited state of R6G (S1) isassociated with a transition from the highest occupiedmolecular orbital (HOMO) to the lowest unoccupiedmolecular orbital (LUMO).51 These two orbitals have littlecontribution from the carboxyphenyl group. Considering thehigh cost of the NAMD simulation, the two methyl groups andthe carboxyphenyl group in R6G were replaced by hydrogenatoms, and the two ethylamino groups were replaced by aminogroups. Then, the simplified R6G molecule was inserted intothe SiO2 matrix along the diagonal of SiO2 simulation cell inorder to maximize the separation between the periodic imagesof the molecule. All atoms of the SiO2 matrix overlapping withthe molecule were removed in a stoichiometric way, that is, ingroups of SiO2. Care was taken to minimize the number ofdangling bonds of the SiO2 atoms facing the cavity. The cavitywas sufficiently large to allow some flexibility in the orientationof the molecule inside the cavity. The final model includes acubic supercell with the side length of 14.24 Å, containing 42 Si,85 O, 13 C, 2 N, and 10 H atoms.Geometry optimization, adiabatic MD, and NA coupling
calculations are carried using Vienna ab initio simulation
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13601
package (VASP).52 The exchange-correlation interactions aretreated using the Pedrew−Burke−Ernzerhof (PBE) func-tional.53 The interaction between the ionic cores and thevalence electrons are described by the projector-augmentedwave (PAW) approach.54 The van der Waals interactions aretreated by the Grimme DFT-D2 method.55 The kinetic cutoffenergy for the plane-wave expansion was set to 400 eV. For thestructural optimization, adiabatic MD, and density of states(DOS) calculations, a uniform Monkhorst−Pack mesh of 3 × 3× 3 k-points are used, while in the NA coupling and dynamicscalculations comprised the Γ-point only. In order to investigatethe effect of shock temperature and pressure on the structureand properties of R6G encapsulated in SiO2, we choose thefollowing three sets of conditions: 300 K-0.0001 GPa, 300 K-7GPa, and 525 K-7 GPa. The first set represents ambientconditions. The last set represents a typical shock-wave, whichincreases both temperature and pressure, according to theHugoniot plot shown in Figure 2a of ref 12. It important tonote that a uniform pressure and temperature increase may notfully represent a shock-wave; for example, a shear stress can bepreserved for a long time, especially in solid matrices such assilica. The middle set isolates the effect of increased pressure,while keep temperature low. Because the NA coupling dependson nuclear velocity which depends on temperature, the effect oftemperature on nonradiative relaxation is well-known.56−59
Therefore, we focus on the pressure effect.After geometry optimization of the whole model, uniform
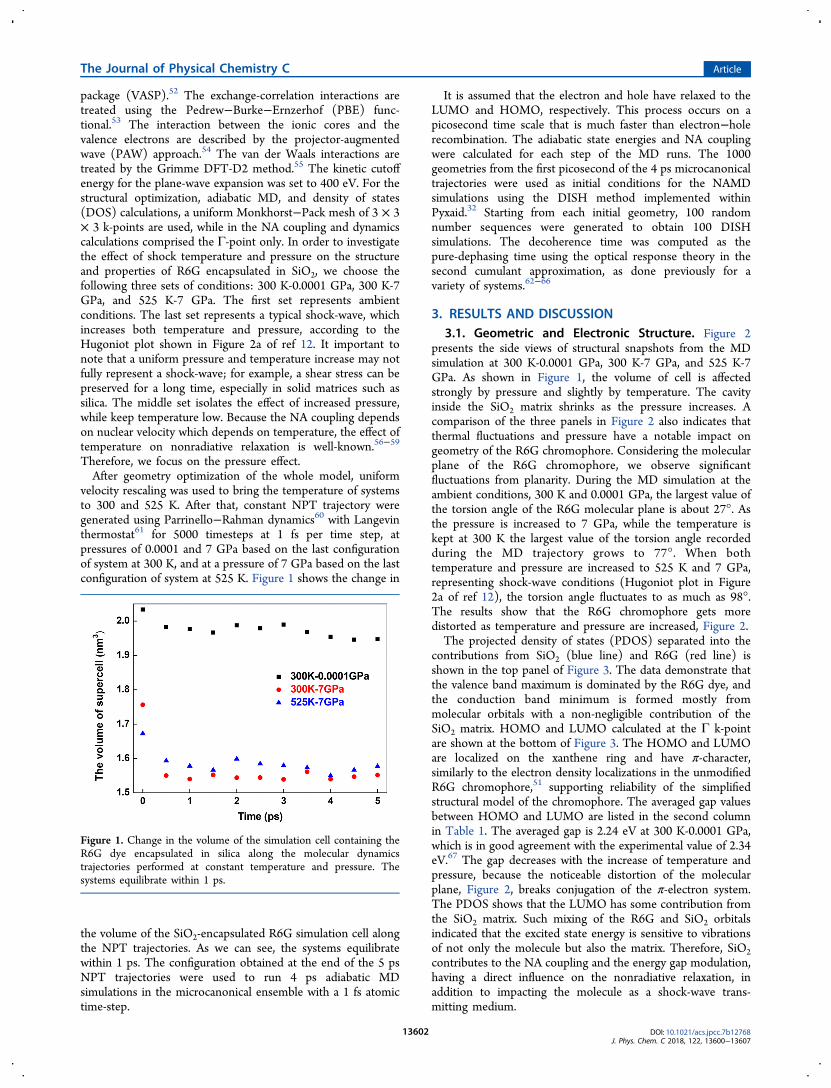
velocity rescaling was used to bring the temperature of systemsto 300 and 525 K. After that, constant NPT trajectory weregenerated using Parrinello−Rahman dynamics60 with Langevinthermostat61 for 5000 timesteps at 1 fs per time step, atpressures of 0.0001 and 7 GPa based on the last configurationof system at 300 K, and at a pressure of 7 GPa based on the lastconfiguration of system at 525 K. Figure 1 shows the change in
the volume of the SiO2-encapsulated R6G simulation cell alongthe NPT trajectories. As we can see, the systems equilibratewithin 1 ps. The configuration obtained at the end of the 5 psNPT trajectories were used to run 4 ps adiabatic MDsimulations in the microcanonical ensemble with a 1 fs atomictime-step.
It is assumed that the electron and hole have relaxed to theLUMO and HOMO, respectively. This process occurs on apicosecond time scale that is much faster than electron−holerecombination. The adiabatic state energies and NA couplingwere calculated for each step of the MD runs. The 1000geometries from the first picosecond of the 4 ps microcanonicaltrajectories were used as initial conditions for the NAMDsimulations using the DISH method implemented withinPyxaid.32 Starting from each initial geometry, 100 randomnumber sequences were generated to obtain 100 DISHsimulations. The decoherence time was computed as thepure-dephasing time using the optical response theory in thesecond cumulant approximation, as done previously for avariety of systems.62−66
3. RESULTS AND DISCUSSION3.1. Geometric and Electronic Structure. Figure 2
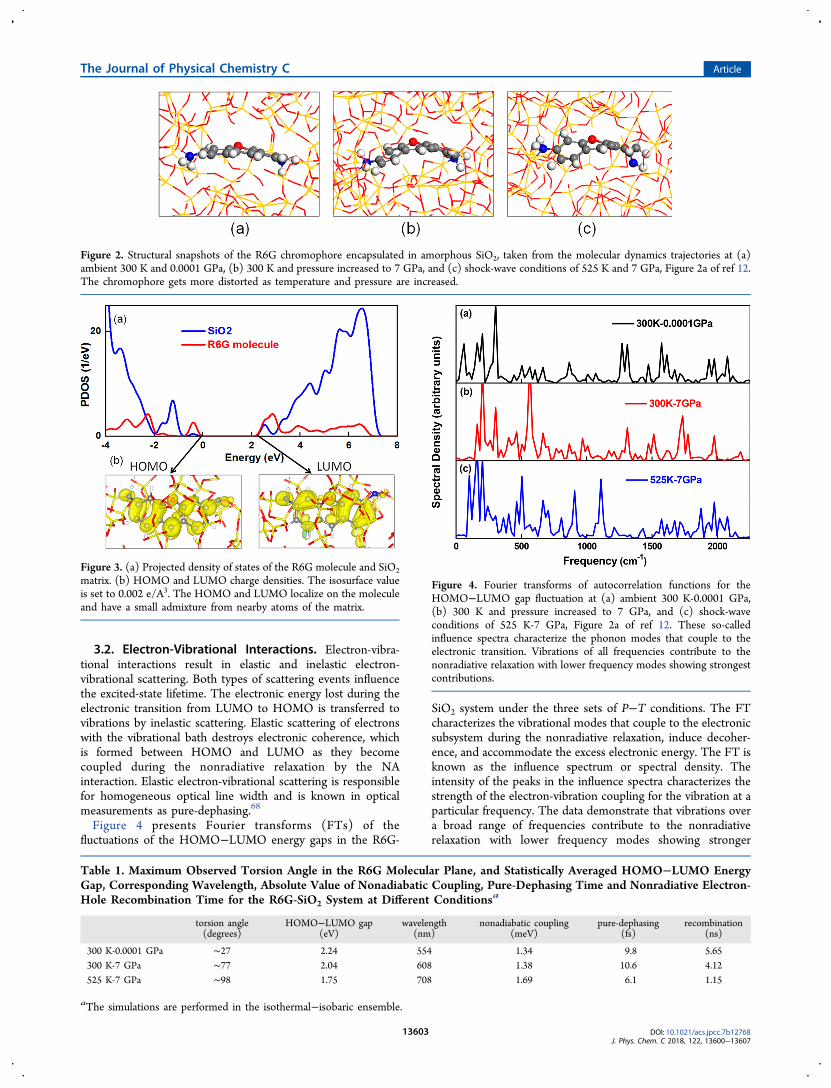
presents the side views of structural snapshots from the MDsimulation at 300 K-0.0001 GPa, 300 K-7 GPa, and 525 K-7GPa. As shown in Figure 1, the volume of cell is affectedstrongly by pressure and slightly by temperature. The cavityinside the SiO2 matrix shrinks as the pressure increases. Acomparison of the three panels in Figure 2 also indicates thatthermal fluctuations and pressure have a notable impact ongeometry of the R6G chromophore. Considering the molecularplane of the R6G chromophore, we observe significantfluctuations from planarity. During the MD simulation at theambient conditions, 300 K and 0.0001 GPa, the largest value ofthe torsion angle of the R6G molecular plane is about 27°. Asthe pressure is increased to 7 GPa, while the temperature iskept at 300 K the largest value of the torsion angle recordedduring the MD trajectory grows to 77°. When bothtemperature and pressure are increased to 525 K and 7 GPa,representing shock-wave conditions (Hugoniot plot in Figure2a of ref 12), the torsion angle fluctuates to as much as 98°.The results show that the R6G chromophore gets moredistorted as temperature and pressure are increased, Figure 2.The projected density of states (PDOS) separated into the
contributions from SiO2 (blue line) and R6G (red line) isshown in the top panel of Figure 3. The data demonstrate thatthe valence band maximum is dominated by the R6G dye, andthe conduction band minimum is formed mostly frommolecular orbitals with a non-negligible contribution of theSiO2 matrix. HOMO and LUMO calculated at the Γ k-pointare shown at the bottom of Figure 3. The HOMO and LUMOare localized on the xanthene ring and have π-character,similarly to the electron density localizations in the unmodifiedR6G chromophore,51 supporting reliability of the simplifiedstructural model of the chromophore. The averaged gap valuesbetween HOMO and LUMO are listed in the second columnin Table 1. The averaged gap is 2.24 eV at 300 K-0.0001 GPa,which is in good agreement with the experimental value of 2.34eV.67 The gap decreases with the increase of temperature andpressure, because the noticeable distortion of the molecularplane, Figure 2, breaks conjugation of the π-electron system.The PDOS shows that the LUMO has some contribution fromthe SiO2 matrix. Such mixing of the R6G and SiO2 orbitalsindicated that the excited state energy is sensitive to vibrationsof not only the molecule but also the matrix. Therefore, SiO2contributes to the NA coupling and the energy gap modulation,having a direct influence on the nonradiative relaxation, inaddition to impacting the molecule as a shock-wave trans-mitting medium.
Figure 1. Change in the volume of the simulation cell containing theR6G dye encapsulated in silica along the molecular dynamicstrajectories performed at constant temperature and pressure. Thesystems equilibrate within 1 ps.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13602
3.2. Electron-Vibrational Interactions. Electron-vibra-tional interactions result in elastic and inelastic electron-vibrational scattering. Both types of scattering events influencethe excited-state lifetime. The electronic energy lost during theelectronic transition from LUMO to HOMO is transferred tovibrations by inelastic scattering. Elastic scattering of electronswith the vibrational bath destroys electronic coherence, whichis formed between HOMO and LUMO as they becomecoupled during the nonradiative relaxation by the NAinteraction. Elastic electron-vibrational scattering is responsiblefor homogeneous optical line width and is known in opticalmeasurements as pure-dephasing.68
Figure 4 presents Fourier transforms (FTs) of thefluctuations of the HOMO−LUMO energy gaps in the R6G-
SiO2 system under the three sets of P−T conditions. The FTcharacterizes the vibrational modes that couple to the electronicsubsystem during the nonradiative relaxation, induce decoher-ence, and accommodate the excess electronic energy. The FT isknown as the influence spectrum or spectral density. Theintensity of the peaks in the influence spectra characterizes thestrength of the electron-vibration coupling for the vibration at aparticular frequency. The data demonstrate that vibrations overa broad range of frequencies contribute to the nonradiativerelaxation with lower frequency modes showing stronger
Figure 2. Structural snapshots of the R6G chromophore encapsulated in amorphous SiO2, taken from the molecular dynamics trajectories at (a)ambient 300 K and 0.0001 GPa, (b) 300 K and pressure increased to 7 GPa, and (c) shock-wave conditions of 525 K and 7 GPa, Figure 2a of ref 12.The chromophore gets more distorted as temperature and pressure are increased.
Figure 3. (a) Projected density of states of the R6G molecule and SiO2matrix. (b) HOMO and LUMO charge densities. The isosurface valueis set to 0.002 e/A3. The HOMO and LUMO localize on the moleculeand have a small admixture from nearby atoms of the matrix.
Table 1. Maximum Observed Torsion Angle in the R6G Molecular Plane, and Statistically Averaged HOMO−LUMO EnergyGap, Corresponding Wavelength, Absolute Value of Nonadiabatic Coupling, Pure-Dephasing Time and Nonradiative Electron-Hole Recombination Time for the R6G-SiO2 System at Different Conditionsa
torsion angle(degrees)
HOMO−LUMO gap(eV)
wavelength(nm)
nonadiabatic coupling(meV)
pure-dephasing(fs)
recombination(ns)
300 K-0.0001 GPa ∼27 2.24 554 1.34 9.8 5.65300 K-7 GPa ∼77 2.04 608 1.38 10.6 4.12525 K-7 GPa ∼98 1.75 708 1.69 6.1 1.15
aThe simulations are performed in the isothermal−isobaric ensemble.
Figure 4. Fourier transforms of autocorrelation functions for theHOMO−LUMO gap fluctuation at (a) ambient 300 K-0.0001 GPa,(b) 300 K and pressure increased to 7 GPa, and (c) shock-waveconditions of 525 K-7 GPa, Figure 2a of ref 12. These so-calledinfluence spectra characterize the phonon modes that couple to theelectronic transition. Vibrations of all frequencies contribute to thenonradiative relaxation with lower frequency modes showing strongestcontributions.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13603
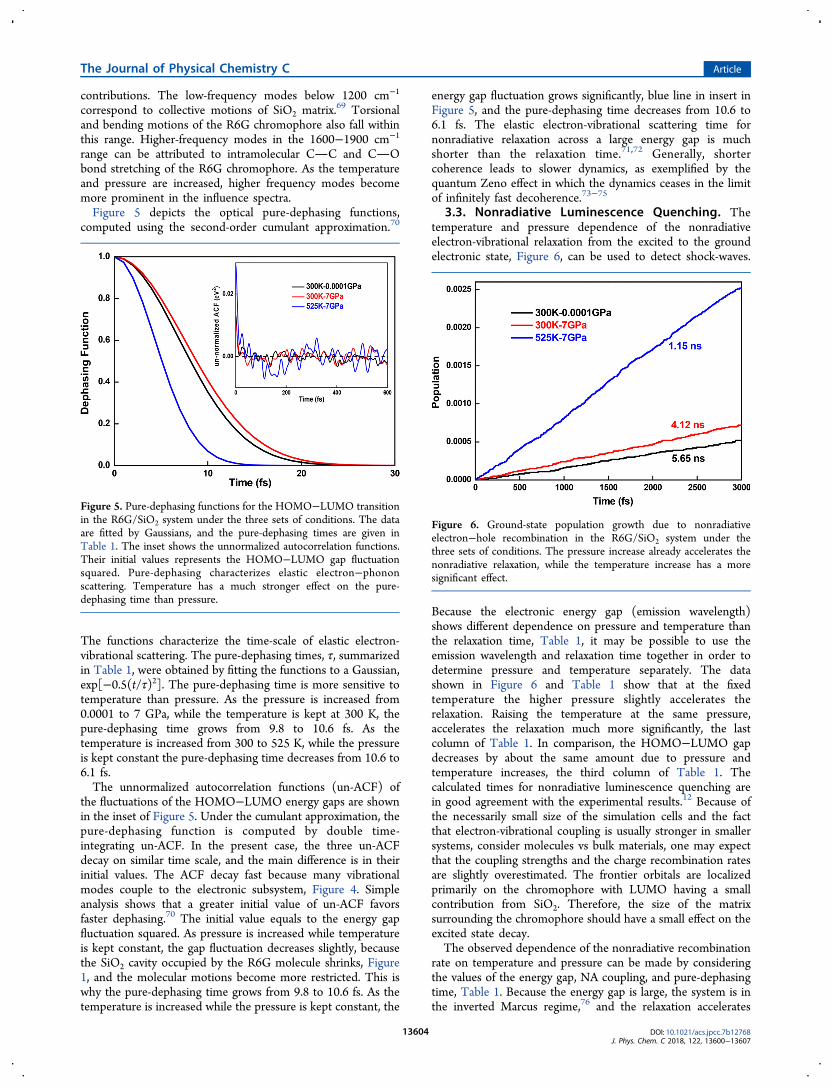
contributions. The low-frequency modes below 1200 cm−1
correspond to collective motions of SiO2 matrix.69 Torsionaland bending motions of the R6G chromophore also fall withinthis range. Higher-frequency modes in the 1600−1900 cm−1
range can be attributed to intramolecular CC and CObond stretching of the R6G chromophore. As the temperatureand pressure are increased, higher frequency modes becomemore prominent in the influence spectra.Figure 5 depicts the optical pure-dephasing functions,
computed using the second-order cumulant approximation.70
The functions characterize the time-scale of elastic electron-vibrational scattering. The pure-dephasing times, τ, summarizedin Table 1, were obtained by fitting the functions to a Gaussian,exp[−0.5(t/τ)2]. The pure-dephasing time is more sensitive totemperature than pressure. As the pressure is increased from0.0001 to 7 GPa, while the temperature is kept at 300 K, thepure-dephasing time grows from 9.8 to 10.6 fs. As thetemperature is increased from 300 to 525 K, while the pressureis kept constant the pure-dephasing time decreases from 10.6 to6.1 fs.The unnormalized autocorrelation functions (un-ACF) of
the fluctuations of the HOMO−LUMO energy gaps are shownin the inset of Figure 5. Under the cumulant approximation, thepure-dephasing function is computed by double time-integrating un-ACF. In the present case, the three un-ACFdecay on similar time scale, and the main difference is in theirinitial values. The ACF decay fast because many vibrationalmodes couple to the electronic subsystem, Figure 4. Simpleanalysis shows that a greater initial value of un-ACF favorsfaster dephasing.70 The initial value equals to the energy gapfluctuation squared. As pressure is increased while temperatureis kept constant, the gap fluctuation decreases slightly, becausethe SiO2 cavity occupied by the R6G molecule shrinks, Figure1, and the molecular motions become more restricted. This iswhy the pure-dephasing time grows from 9.8 to 10.6 fs. As thetemperature is increased while the pressure is kept constant, the
energy gap fluctuation grows significantly, blue line in insert inFigure 5, and the pure-dephasing time decreases from 10.6 to6.1 fs. The elastic electron-vibrational scattering time fornonradiative relaxation across a large energy gap is muchshorter than the relaxation time.71,72 Generally, shortercoherence leads to slower dynamics, as exemplified by thequantum Zeno effect in which the dynamics ceases in the limitof infinitely fast decoherence.73−75
3.3. Nonradiative Luminescence Quenching. Thetemperature and pressure dependence of the nonradiativeelectron-vibrational relaxation from the excited to the groundelectronic state, Figure 6, can be used to detect shock-waves.
Because the electronic energy gap (emission wavelength)shows different dependence on pressure and temperature thanthe relaxation time, Table 1, it may be possible to use theemission wavelength and relaxation time together in order todetermine pressure and temperature separately. The datashown in Figure 6 and Table 1 show that at the fixedtemperature the higher pressure slightly accelerates therelaxation. Raising the temperature at the same pressure,accelerates the relaxation much more significantly, the lastcolumn of Table 1. In comparison, the HOMO−LUMO gapdecreases by about the same amount due to pressure andtemperature increases, the third column of Table 1. Thecalculated times for nonradiative luminescence quenching arein good agreement with the experimental results.12 Because ofthe necessarily small size of the simulation cells and the factthat electron-vibrational coupling is usually stronger in smallersystems, consider molecules vs bulk materials, one may expectthat the coupling strengths and the charge recombination ratesare slightly overestimated. The frontier orbitals are localizedprimarily on the chromophore with LUMO having a smallcontribution from SiO2. Therefore, the size of the matrixsurrounding the chromophore should have a small effect on theexcited state decay.The observed dependence of the nonradiative recombination
rate on temperature and pressure can be made by consideringthe values of the energy gap, NA coupling, and pure-dephasingtime, Table 1. Because the energy gap is large, the system is inthe inverted Marcus regime,76 and the relaxation accelerates
Figure 5. Pure-dephasing functions for the HOMO−LUMO transitionin the R6G/SiO2 system under the three sets of conditions. The dataare fitted by Gaussians, and the pure-dephasing times are given inTable 1. The inset shows the unnormalized autocorrelation functions.Their initial values represents the HOMO−LUMO gap fluctuationsquared. Pure-dephasing characterizes elastic electron−phononscattering. Temperature has a much stronger effect on the pure-dephasing time than pressure.
Figure 6. Ground-state population growth due to nonradiativeelectron−hole recombination in the R6G/SiO2 system under thethree sets of conditions. The pressure increase already accelerates thenonradiative relaxation, while the temperature increase has a moresignificant effect.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13604

when the gap decreases. The relaxation also accelerates forlarger NA coupling and longer pure-dephasing time. Thepressure growth leads to a decrease of the energy gap, anincrease in the NA coupling, and an increase in the pure-dephasing times. All factors favor faster relaxation. Because theof changes in the gap, NA coupling, and pure-dephasing aresmall, the relaxation time changes little. The growth intemperature decreases the energy gap by a larger amount. Itincreases the NA coupling significantly, because the NAcoupling is directly proportion to nuclear velocity, whichgrows with temperature. The pure-dephasing time becomesshorter. The increase in the NA coupling is key to the reducedrelaxation time.
4. CONCLUSIONS
We have used NAMD in combination with real-time TDDFTto simulate nonradiative electron-vibrational relaxation respon-sible for luminescence quenching of the R6G chromophoreencapsulated in a silica matrix under shock-wave conditions.Showing good agreement with the experimental data, thereported study characterizes the effects of temperature andpressure on luminescence wavelength and lifetime andgenerates an atomistic understanding of the observed effects.The excited state energy and lifetime exhibit differentdependencies on pressure and temperature, and thereforetogether they may be used to determine independently thepressure and temperature in a propagating shock-wave. Thesimulations indicate that increases in pressure and temperaturehave similar effects on the luminescence wavelength, whilenonradiative quenching of luminescence depends much morestrongly on temperature than pressure. The energy gap isinfluenced by geometric distortions to the π-electron plane ofthe conjugated R6G chromophore. Both increased pressure andtemperature perturb the chromophore structure and reduce thegap. The nonradiative relaxation depends much more stronglyon temperature than pressure, because the NA electron-vibrational coupling is proportional to nuclear velocity. A broadrange of vibrational modes couple to the electronic subsystemwith lower frequency modes having a larger contribution. Theweak NA electron-vibrational coupling combined with a fastloss of coherence in the electronic subsystem result innanosecond luminescence lifetimes, which are sufficientlylong to facilitate experimental detection, and sufficiently shortcompared to shock-wave duration, allowing one to track shock-wave propagation. The research presented in this paperadvances our understanding of the key factors influencing andcontrolling luminescence properties of organic chromophoresunder shock compression.
■ ASSOCIATED CONTENT
*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.jpcc.7b12768.
Basics of the time-dependent density functional theoryand nonadiabatic molecular dynamics, snapshots of theR6G chromophore encapsulated in an amorphous SiO2
matrix, representative system coordinates and densities ofstates along the MD trajectories, and NAMD con-vergence tests (PDF)
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
ORCID
Xin Zhou: 0000-0002-4385-8379Oleg V. Prezhdo: 0000-0002-5140-7500NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
The research described in this study was based on worksupported by the US Army Research Office under awardnumber W911NF-16-1-0406 (MURI). X.Z. acknowledgessupport of the National Natural Science Foundation ofChina, Grant 21473183.
■ REFERENCES(1) Franken, J.; Hambir, S. A.; Hare, D. E.; Dlott, D. D. Shock Wavesin Molecular Solids: Ultrafast Vibrational Spectroscopy of the FirstNanosecond. Shock Waves 1997, 7, 135−145.(2) Dlott, D. D. New Developments in the Physical Chemistry ofShock Compression. Annu. Rev. Phys. Chem. 2011, 62, 575−597.(3) Lagutchev, A. S.; Patterson, J. E.; Huang, W.; Dlott, D. D.Ultrafast Dynamics of Self-Assembled Monolayers under ShockCompression: Effects of Molecular and Substrate Structure. J. Phys.Chem. B 2005, 109, 5033−5044.(4) Forbes, J. W. Shock Wave Compression of Condensed Matter: APrimer; Springer: New York, 2012.(5) Liu, Q. C.; Zhou, X. M.; Luo, S. N. Optical Absorbances ofGd3ga5o12 Single Crystals under Shock Compression to 211 GPa. J.Appl. Phys. 2017, 121, 145901.(6) Kang, Z.; et al. Exploration of Cdte Quantum Dots as MesoscalePressure Sensors Via Time-Resolved Shock-Compression Photo-luminescent Emission Spectroscopy. J. Appl. Phys. 2016, 120, 043107.(7) Xiao, P.; Kang, Z.; Bansihev, A. A.; Breidenich, J.; Scripka, D. A.;Christensen, J. M.; Summers, C. J.; Dlott, D. D.; Thadhani, N. N.;Zhou, M. Laser-Excited Optical Emission Response of Cdte QuantumDot/Polymer Nanocomposite under Shock Compression. Appl. Phys.Lett. 2016, 108, 011908.(8) Tan, H.; Jain, A.; Voznyy, O.; Lan, X.; de Arquer, F. P. G.; Fan, J.Z.; Quintero-Bermudez, R.; Yuan, M.; Zhang, B.; Zhao, Y.; et al.Efficient and Stable Solution-Processed Planar Perovskite Solar CellsVia Contact Passivation. Science 2017, 355, 722−726.(9) Zheng, X.; Curtis, A. D.; Shaw, W. L.; Dlott, D. D. ShockInitiation of Nano-Al + Teflon: Time-Resolved Emission Studies. J.Phys. Chem. C 2013, 117, 4866−4875.(10) Rastogi, V.; Chaurasia, S.; Rao, U.; Sijoy, C. D.; Poswal, H. K.;Mishra, V.; Kumar, M.; Deo, M. N. In Situ Raman SpectroscopicStudies of Polyvinyl Toluene under Laser-Driven Shock Compressionand Comparison with Hydrostatic Experiments. J. Raman Spectrosc.2017, 48, 1300−1306.(11) Huston, A. L.; Justus, B. L.; Campillo, A. J. Direct Measurementof the Viscosity of Glycerol under Laser Driven Shock Compression:Fluorescence Lifetime Changes in Crystal Violet. Chem. Phys. Lett.1985, 122, 617−621.(12) Liu, W.-l.; Bassett, W. P.; Christensen, J. M.; Dlott, D. D.Emission Lifetimes of a Fluorescent Dye under Shock Compression. J.Phys. Chem. A 2015, 119, 10910−10916.(13) Turneaure, S. J.; Gupta, Y. M. Real-Time Microstructure ofShock-Compressed Single Crystals from X-Ray Diffraction LineProfiles. J. Appl. Crystallogr. 2011, 44, 574−584.(14) Barua, A.; Zhou, M. Computational Analysis of TemperatureRises in Microstructures of Hmx-Estane Pbxs. Comp. Mech. 2013, 52,151−159.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13605

(15) Barua, A.; Kim, S.; Horie, Y.; Zhou, M. Prediction ofProbabilistic Ignition Behavior of Polymer-Bonded Explosives fromMicrostructural Stochasticity. J. Appl. Phys. 2013, 113, 184907.(16) Jaiswal, J. K.; Mattoussi, H.; Mauro, J. M.; Simon, S. M. Long-Term Multiple Color Imaging of Live Cells Using Quantum DotBioconjugates. Nat. Biotechnol. 2003, 21, 47.(17) Goldman, E. R.; Clapp, A. R.; Anderson, G. P.; Uyeda, H. T.;Mauro, J. M.; Medintz, I. L.; Mattoussi, H. Multiplexed Toxin AnalysisUsing Four Colors of Quantum Dot Fluororeagents. Anal. Chem.2004, 76, 684−688.(18) Burns, A.; Ow, H.; Wiesner, U. Fluorescent Core-Shell SilicaNanoparticles: Towards “Lab on a Particle” Architectures forNanobiotechnology. Chem. Soc. Rev. 2006, 35, 1028−1042.(19) Rampazzo, E.; Bonacchi, S.; Montalti, M.; Prodi, L.; Zaccheroni,N. Self-Organizing Core−Shell Nanostructures: Spontaneous Accu-mulation of Dye in the Core of Doped Silica Nanoparticles. J. Am.Chem. Soc. 2007, 129, 14251−14256.(20) Shen, X. A.; Gupta, Y. M. Shock-Induced Fluorescence Shift ofRhodamine-6g Dye in Ethanol Solution. J. Appl. Phys. 1991, 70, 7549−7553.(21) Brown, K. E.; Fu, Y.; Shaw, W. L.; Dlott, D. D. Time-ResolvedEmission of Dye Probes in a Shock-Compressed Polymer. J. Appl.Phys. 2012, 112, 103508.(22) Brown, K. E.; Shaw, W. L.; Zheng, X.; Dlott, D. D. SimplifiedLaser-Driven Flyer Plates for Shock Compression Science. Rev. Sci.Instrum. 2012, 83, 103901.(23) Taguchi, T.; Hirayama, S.; Okamoto, M. New SpectroscopicEvidence for Molecular Aggregates of Rhodamine 6g in AqueousSolution at High Pressure. Chem. Phys. Lett. 1994, 231, 561−568.(24) Dreger, Z. A.; Yang, G.; White, J. O.; Li, Y.; Drickamer, H. G.One- and Two-Photon-Pumped Fluorescence from Rhodamine B inSolid Poly(Acrylic Acid) under High Pressure. J. Phys. Chem. B 1998,102, 4380−4385.(25) Nguyen, D. T.; Smit, M.; Dunn, B.; Zink, J. I. Stabilization ofCreatine Kinase Encapsulated in Silicate Sol−Gel Materials andUnusual Temperature Effects on Its Activity. Chem. Mater. 2002, 14,4300−4306.(26) Gilliland, J. W.; Yokoyama, K.; Yip, W. T. Solvent Effect onMobility and Photostability of Organic Dyes Embedded inside SilicaSol−Gel Thin Films. Chem. Mater. 2005, 17, 6702−6712.(27) Ow, H.; Larson, D. R.; Srivastava, M.; Baird, B. A.; Webb, W.W.; Wiesner, U. Bright and Stable Core−Shell Fluorescent SilicaNanoparticles. Nano Lett. 2005, 5, 113−117.(28) Jasper, A. W.; Nangia, S.; Zhu, C.; Truhlar, D. G. Non-Born−Oppenheimer Molecular Dynamics. Acc. Chem. Res. 2006, 39, 101−108.(29) Craig, C. F.; Duncan, W. R.; Prezhdo, O. V. Trajectory SurfaceHopping in the Time-Dependent Kohn-Sham Approach for Electron-Nuclear Dynamics. Phys. Rev. Lett. 2005, 95, 163001.(30) Fischer, S. A.; Habenicht, B. F.; Madrid, A. B.; Duncan, W. R.;Prezhdo, O. V. Regarding the Validity of the Time-Dependent Kohn−Sham Approach for Electron-Nuclear Dynamics Via Trajectory SurfaceHopping. J. Chem. Phys. 2011, 134, 024102.(31) Akimov, A. V.; Prezhdo, O. V. The Pyxaid Program for Non-Adiabatic Molecular Dynamics in Condensed Matter Systems. J. Chem.Theory Comput. 2013, 9, 4959−4972.(32) Akimov, A. V.; Prezhdo, O. V. Advanced Capabilities of thePyxaid Program: Integration Schemes, Decoherence Effects, Multi-excitonic States, and Field-Matter Interaction. J. Chem. Theory Comput.2014, 10, 789−804.(33) Jaeger, H. M.; Fischer, S.; Prezhdo, O. V. Decoherence-InducedSurface Hopping. J. Chem. Phys. 2012, 137, 22A545.(34) Marques, M. A. L.; Gross, E. K. U. Time-Dependent DensityFunctional Theory. Annu. Rev. Phys. Chem. 2004, 55, 427−455.(35) Tully, J. C. Molecular Dynamics with Electronic Transitions. J.Chem. Phys. 1990, 93, 1061−1071.(36) Parandekar, P. V.; Tully, J. C. Mixed Quantum-ClassicalEquilibrium. J. Chem. Phys. 2005, 122, 094102.
(37) Bittner, E. R.; Rossky, P. J. Decoherent Histories andNonadiabatic Quantum Molecular Dynamics Simulations. J. Chem.Phys. 1997, 107, 8611−8618.(38) Habenicht, B. F.; Prezhdo, O. V. Nonradiative Quenching ofFluorescence in a Semiconducting Carbon Nanotube: A Time-DomainAb Initio Study. Phys. Rev. Lett. 2008, 100, 197402.(39) Wang, L. J.; Akimov, A.; Prezhdo, O. V. Recent Progress inSurface Hopping: 2011−2015. J. Phys. Chem. Lett. 2016, 7, 2100−2112.(40) Liu, J.; Neukirch, A. J.; Prezhdo, O. V. Non-Radiative Electron−Hole Recombination in Silicon Clusters: Ab Initio Non-AdiabaticMolecular Dynamics. J. Phys. Chem. C 2014, 118, 20702−20709.(41) Long, R.; Prezhdo, O. V. Instantaneous Generation of Charge-Separated State on Tio2 Surface Sensitized with Plasmonic Nano-particles. J. Am. Chem. Soc. 2014, 136, 4343−4354.(42) Li, L.; Long, R.; Prezhdo, O. V. Charge Separation andRecombination in Two-Dimensional Mos2/Ws2: Time-Domain AbInitio Modeling. Chem. Mater. 2017, 29, 2466−2473.(43) Hyeon-Deuk, K.; Prezhdo, O. V. Multiple Exciton Generationand Recombination Dynamics in Small Si and Cdse Quantum Dots:An Ab Initio Time-Domain Study. ACS Nano 2012, 6, 1239−1250.(44) Kilina, S. V.; Neukirch, A. J.; Habenicht, B. F.; Kilin, D. S.;Prezhdo, O. V. Quantum Zeno Effect Rationalizes the PhononBottleneck in Semiconductor Quantum Dots. Phys. Rev. Lett. 2013,110, 180404.(45) Akimov, A. V.; Muckerman, J. T.; Prezhdo, O. V. NonadiabaticDynamics of Positive Charge During Photocatalytic Water Splitting onGan(10−10) Surface: Charge Localization Governs Splitting Effi-ciency. J. Am. Chem. Soc. 2013, 135, 8682−8691.(46) Fischer, S. A.; Duncan, W. R.; Prezhdo, O. V. Ab InitioNonadiabatic Molecular Dynamics of Wet-Electrons on the Tio2Surface. J. Am. Chem. Soc. 2009, 131, 15483−15491.(47) Chaban, V. V.; Prezhdo, V. V.; Prezhdo, O. V. Covalent LinkingGreatly Enhances Photoinduced Electron Transfer in Fullerene-Quantum Dot Nanocomposites: Time-Domain Ab Initio Study. J.Phys. Chem. Lett. 2013, 4, 1−6.(48) Duncan, W. R.; Craig, C. F.; Prezhdo, O. V. Time-Domain AbInitio Study of Charge Relaxation and Recombination in Dye-Sensitized Tio2. J. Am. Chem. Soc. 2007, 129, 8528−8543.(49) Wyckoff, R. W. G. Crystal Structures, 2nd ed.; Interscience: NewYork, 1965.(50) Chang, E.; Thekkek, N.; Yu, W. W.; Colvin, V. L.; Drezek, R.Evaluation of Quantum Dot Cytotoxicity Based on IntracellularUptake. Small 2006, 2, 1412−1417.(51) Guthmuller, J.; Champagne, B. Resonance Raman Scattering ofRhodamine 6g as Calculated by Time-Dependent Density FunctionalTheory: Vibronic and Solvent Effects. J. Phys. Chem. A 2008, 112,3215−3223.(52) Kresse, G.; Furthmuller, J. Efficient Iterative Schemes for AbInitio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys.Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169−11186.(53) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized GradientApproximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865−3868.(54) Blochl, P. E. Projector Augmented-Wave Method. Phys. Rev. B:Condens. Matter Mater. Phys. 1994, 50, 17953−17979.(55) Grimme, S. Semiempirical Gga-Type Density FunctionalConstructed with a Long-Range Dispersion Correction. J. Comput.Chem. 2006, 27, 1787−1799.(56) Zhou, X.; Li, L. Q.; Dong, H.; Giri, A.; Hopkins, P. E.; Prezhdo,O. V. Temperature Dependence of Electron-Phonon Interactions inGold Films Rationalized by Time-Domain Ab Lnitio Analysis. J. Phys.Chem. C 2017, 121, 17488−17497.(57) Tafen, D.; Prezhdo, O. V. Size and Temperature Dependence ofElectron Transfer between Cdse Quantum Dots and a Tio2 Nanobelt.J. Phys. Chem. C 2015, 119, 5639−5647.(58) Chen, L. L.; Bao, H.; Tan, T. Z.; Prezhdo, O. V.; Ruan, X. L.Shape and Temperature Dependence of Hot Carrier RelaxationDynamics in Spherical and Elongated Cdse Quantum Dots. J. Phys.Chem. C 2011, 115, 11400−11406.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13606

(59) Bao, H.; Habenicht, B. F.; Prezhdo, O. V.; Ruan, X. L.Temperature Dependence of Hot-Carrier Relaxation in Pbse Nano-crystals: An Ab Initio Study. Phys. Rev. B: Condens. Matter Mater. Phys.2009, 79, 235306.(60) Parrinello, M.; Rahman, A. Polymorphic Transitions in SingleCrystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52,7182−7190.(61) Allen, M. P.; Tildesley, D. J. Computer Simulation of Liquids;Oxford University Press, 2017.(62) Nelson, T. R.; Prezhdo, O. V. Extremely Long NonradiativeRelaxation of Photoexcited Graphane Is Greatly Accelerated byOxidation: Time-Domain Ab Initio Study. J. Am. Chem. Soc. 2013, 135,3702−3710.(63) Kilina, S.; Velizhanin, K. A.; Ivanov, S.; Prezhdo, O. V.; Tretiak,S. Surface Ligands Increase Photoexcitation Relaxation Rates in CdseQuantum Dots. ACS Nano 2012, 6, 6515−6524.(64) Chaban, V. V.; Prezhdo, V. V.; Prezhdo, O. V. Covalent LinkingGreatly Enhances Photoinduced Electron Transfer in Fullerene-Quantum Dot Nanocomposites: Time-Domain Ab Initio Study. J.Phys. Chem. Lett. 2013, 4, 1−6.(65) Kamisaka, H.; Kilina, S. V.; Yamashita, K.; Prezhdo, O. V.Ultrafast Vibrationally-Induced Dephasing of Electronic Excitations inPbse Quantum Dots. Nano Lett. 2006, 6, 2295−2300.(66) Madrid, A. B.; Hyeon-Deuk, K.; Habenicht, B. F.; Prezhdo, O.V. Phonon-Induced Dephasing of Excitons in SemiconductorQuantum Dots: Multiple Exciton Generation, Fission, and Lumines-cence. ACS Nano 2009, 3, 2487−2494.(67) Du, H.; Fuh, R.-C. A.; Li, J.; Corkan, L. A.; Lindsey, J. S.Photochemcad‡: A Computer-Aided Design and Research Tool inPhotochemistry. Photochem. Photobiol. 1998, 68, 141−142.(68) Mukamel, S. Principles of Nonlinear Optical Spectroscopy; OxfordUniversity Press: New York, 1995.(69) Coh, S.; Vanderbilt, D. Structural Stability and Lattice Dynamicsof ${\Text{Sio}}_{2}$ Cristobalite. Phys. Rev. B: Condens. MatterMater. Phys. 2008, 78, 054117.(70) Akimov, A. V.; Prezhdo, O. V. Persistent Electronic CoherenceDespite Rapid Loss of Electron−Nuclear Correlation. J. Phys. Chem.Lett. 2013, 4, 3857−3864.(71) Marchioro, A.; Teuscher, J.; Friedrich, D.; Kunst, M.; van deKrol, R.; Moehl, T.; Gratzel, M.; Moser, J.-E. Unravelling theMechanism of Photoinduced Charge Transfer Processes in LeadIodide Perovskite Solar Cells. Nat. Photonics 2014, 8, 250.(72) Stamplecoskie, K. G.; Manser, J. S.; Kamat, P. V. Dual Nature ofthe Excited State in Organic-Inorganic Lead Halide Perovskites. EnergyEnviron. Sci. 2015, 8, 208−215.(73) Kilina, S. V.; Neukirch, A. J.; Habenicht, B. F.; Kilin, D. S.;Prezhdo, O. V. Quantum Zeno Effect Rationalizes the PhononBottleneck in Semiconductor Quantum Dots. Phys. Rev. Lett. 2013,110, 180404.(74) Bray, A. J.; Moore, M. A. Influence of Dissipation on QuantumCoherence. Phys. Rev. Lett. 1982, 49, 1545−1549.(75) Prezhdo, O. V. Quantum Anti-Zeno Acceleration of a ChemicalReaction. Phys. Rev. Lett. 2000, 85, 4413−4417.(76) Akimov, A. V.; Prezhdo, O. V. Large-Scale Computations inChemistry: A Bird’s Eye View of a Vibrant Field. Chem. Rev. 2015, 115,5797−5890.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.7b12768J. Phys. Chem. C 2018, 122, 13600−13607
13607


















