
Brain–heartinteractions: physiologyandclinical...
22
rsta.royalsocietypublishing.org Review Cite this article: Silvani A, Calandra-Buonaura G, Dampney RAL, Cortelli P. 2016 Brain–heart interactions: physiology and clinical implications. Phil. Trans. R. Soc. A 374: 20150181. http://dx.doi.org/10.1098/rsta.2015.0181 Accepted: 19 January 2016 One contribution of 16 to a theme issue ‘Uncovering brain–heart information through advanced signal and image processing’. Subject Areas: biomedical engineering Keywords: brain–heart interactions, autonomic nervous system, physiology, human disease Author for correspondence: Pietro Cortelli e-mail: [email protected] Brain–heart interactions: physiology and clinical implications Alessandro Silvani 1 , Giovanna Calandra-Buonaura 2,3 , Roger A. L. Dampney 4 and Pietro Cortelli 2,3 1 PRISM Lab, and 2 Autonomic Unit, Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy 3 IRCCS, Institute of Neurological Sciences of Bologna, Bellaria University Hospital, Block G, Via Altura 3, 40139 Bologna, Italy 4 School of Medical Sciences (Physiology) and Bosch Institute for Biomedical Research, University of Sydney, Sidney, New South Wales, Australia AS, 0000-0003-3992-3892; PC, 0000-0002-3633-8818 The brain controls the heart directly through the sympathetic and parasympathetic branches of the autonomic nervous system, which consists of multi- synaptic pathways from myocardial cells back to peripheral ganglionic neurons and further to central preganglionic and premotor neurons. Cardiac function can be profoundly altered by the reflex activation of cardiac autonomic nerves in response to inputs from baro-, chemo-, nasopharyngeal and other receptors as well as by central autonomic commands, including those associated with stress, physical activity, arousal and sleep. In the clinical setting, slowly progressive autonomic failure frequently results from neurodegenerative disorders, whereas autonomic hyperactivity may result from vascular, inflammatory or traumatic lesions of the autonomic nervous system, adverse effects of drugs and chronic neurological disorders. Both acute and chronic manifestations of an imbalanced brain–heart interaction have a negative impact on health. Simple, widely available and reliable cardiovascular markers of the sympathetic tone and of the sympathetic– parasympathetic balance are lacking. A deeper understanding of the connections between autonomic cardiac control and brain dynamics through advanced signal and neuroimage processing may lead to invaluable tools for the early detection and treatment of pathological changes in the brain–heart interaction. 2016 The Author(s) Published by the Royal Society. All rights reserved. on August 25, 2018 http://rsta.royalsocietypublishing.org/ Downloaded from
Transcript of Brain–heartinteractions: physiologyandclinical...

rsta.royalsocietypublishing.org
ReviewCite this article: Silvani A,Calandra-Buonaura G, Dampney RAL, CortelliP. 2016 Brain–heart interactions: physiologyand clinical implications. Phil. Trans. R. Soc. A374: 20150181.http://dx.doi.org/10.1098/rsta.2015.0181
Accepted: 19 January 2016
One contribution of 16 to a theme issue‘Uncovering brain–heart information throughadvanced signal and image processing’.
Subject Areas:biomedical engineering
Keywords:brain–heart interactions, autonomic nervoussystem, physiology, human disease
Author for correspondence:Pietro Cortellie-mail: [email protected]
Brain–heart interactions:physiology and clinicalimplicationsAlessandro Silvani1, Giovanna Calandra-Buonaura2,3,
Roger A. L. Dampney4 and Pietro Cortelli2,3
1PRISM Lab, and 2Autonomic Unit, Department of Biomedical andNeuromotor Sciences, University of Bologna, Bologna, Italy3IRCCS, Institute of Neurological Sciences of Bologna, BellariaUniversity Hospital, Block G, Via Altura 3, 40139 Bologna, Italy4School of Medical Sciences (Physiology) and Bosch Institute forBiomedical Research, University of Sydney, Sidney, New SouthWales, Australia
AS, 0000-0003-3992-3892; PC, 0000-0002-3633-8818
The brain controls the heart directly through thesympathetic and parasympathetic branches of theautonomic nervous system, which consists of multi-synaptic pathways from myocardial cells backto peripheral ganglionic neurons and further tocentral preganglionic and premotor neurons. Cardiacfunction can be profoundly altered by the reflexactivation of cardiac autonomic nerves in response toinputs from baro-, chemo-, nasopharyngeal and otherreceptors as well as by central autonomic commands,including those associated with stress, physicalactivity, arousal and sleep. In the clinical setting,slowly progressive autonomic failure frequentlyresults from neurodegenerative disorders, whereasautonomic hyperactivity may result from vascular,inflammatory or traumatic lesions of the autonomicnervous system, adverse effects of drugs andchronic neurological disorders. Both acute andchronic manifestations of an imbalanced brain–heartinteraction have a negative impact on health. Simple,widely available and reliable cardiovascular markersof the sympathetic tone and of the sympathetic–parasympathetic balance are lacking. A deeperunderstanding of the connections between autonomiccardiac control and brain dynamics through advancedsignal and neuroimage processing may lead toinvaluable tools for the early detection and treatmentof pathological changes in the brain–heart interaction.
2016 The Author(s) Published by the Royal Society. All rights reserved.
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

2
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
1. Physiology of the brain–heart interactions
(a) Physiological effects of sympathetic and parasympathetic nerve activities on the heart(i) Effects of sympathetic activity
The cardiac sympathetic innervation originates from sympathetic preganglionic neurons (S1N)in the upper thoracic segments of the spinal cord, which synapse with neurons (S2N) inthe cervical and upper thoracic ganglia. Sympathetic postganglionic fibres innervate thecardiac conduction system more prominently than the working myocardium [1], releasingnoradrenaline (norepinephrine) and neuropeptide Y. Noradrenaline binds to cardiac beta-1 adrenergic receptors, increasing intracellular levels of cyclic adenosine monophosphate(cAMP). In myocytes of the sinoatrial node, which is the physiological cardiac pacemaker,increased cAMP levels hasten diastolic depolarization by increasing the inward ‘funny’ (If)cation current gated by hyperpolarization-activated cyclic nucleotide-gated channels [2]. Asa result, the heart period (HP) shortens almost linearly with the frequency of sympatheticpostganglionic discharge [3]. This response is delayed by 1.7 s and the frequency responseessentially filters out the fluctuations of sympathetic activity faster than 0.15 Hz [4], whichare typically associated with breathing [5]. By itself, decreases in HP raise ventricularcontractility [6] and relaxation rate [7]. These effects are further enhanced by the increasedcAMP levels, which increase sarcoplasmic reticulum calcium reuptake because of proteinkinase A-dependent phosphorylation of phospholamban [8]. The decrease in HP also increasesatrioventricular conduction time, but this effect is counteracted by sympathetic activity [9].Sympathetic innervation of cardiac automatic, conduction and contractile tissue follows parallelyet distinct intrapericardial pathways [10], which are the basis for selective effects [11].Sympathetic activity is also associated with ventricular repolarization heterogeneity, as indexedby T-wave alternans in subjects with coronary artery disease [12] and by QT intervalvariability in dogs with experimental heart failure [13]. The sympathetic nervous system alsocontrols the heart by promoting adrenal medullary release of adrenaline (epinephrine), whoseplasma threshold for decreasing HP is in the range of plasma levels attained during activestanding [14].
(ii) Effects of parasympathetic activity
The parasympathetic preganglionic neurons (P1N) involved in the control of cardiac functionare located in the medulla oblongata within and ventrolateral to the nucleus ambiguus(NAmb), and to a much lesser extent in the dorsal motor nucleus of the vagus nerve (DMNX)and in the reticular formation between these two nuclei [15]. All of these neurons projectthrough the vagus nerves to a complex set of epicardial ganglionated plexi [16], whichdisplay functional selectivity for the effects on HP, conduction time and contractility [17], andreceive synaptic contacts by local sensory neurons [18]. Parasympathetic postganglionic fibreswidely innervate the cardiac conduction system as well as the atrial and ventricular workingmyocardium [1,16], releasing acetylcholine and vasoactive intestinal peptide. Acetylcholinedecreases cAMP levels in myocardial cells by binding to M2 muscarinic receptors [8]. Thesereceptors also open specific potassium channels that gate a hyperpolarizing current, particularlyin conditions of marked parasympathetic activity [19]. As a result, HP lengthens almost linearlywith parasympathetic preganglionic discharge rate [3]. Resting HP values are higher thanthe intrinsic HP in the absence of autonomic control [20], highlighting the prominence ofthe cardiac parasympathetic tone over the cardiac sympathetic tone. Sinoatrial responses toacetylcholine occur with minimal delay and keep up with modulations at least up to 0.4 Hz [4].Parasympathetic activity also lengthens atrioventricular conduction time [9] and decreasesatrial and ventricular contractility [16] and ventricular relaxation rate [21], but does not affectventricular repolarization heterogeneity as indexed by T-wave alternans [12] or QT intervalvariability [13].
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

3
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
(iii) Sympathetic–parasympathetic interactions
The sympathetic and parasympathetic cardiac controls are antagonistic in that they exertopposite effects on myocardial cAMP levels, and thereby on HP, atrioventricular conductiontime, ventricular contractility and relaxation rate. Evidence that sympathetic noradrenalinerelease is decreased by ongoing parasympathetic activity also suggests an inhibitory presynapticinteraction [22]. The autonomic control of the mean values of heart rate (i.e. 1/HP) displaysaccentuated nonlinear antagonism in that even high levels of sympathetic activity exertnegligible effects when parasympathetic activity is also high [23]. This nonlinear interactionis much less prominent with respect to values of atrioventricular conduction time [23] andeven of the mean values of HP [3], which are well explained by a linear summation ofsympathetic and parasympathetic effects [24]. As far as autonomic modulations are concerned,activity of each autonomic branch enhances the fluctuations of heart rate resulting fromthe modulation of the other branch [25]. This synergistic interaction suggests that the high-frequency fluctuations of heart rate that constitute respiratory sinus arrhythmia are enhancedby sympathetic activity and, therefore, do not result solely from parasympathetic modulation,as would be expected from the frequency response properties detailed previously [25]. Incontrast to this suggestion, however, analysis of HP fluctuations has shown that beta-adrenergicblockade increases respiratory sinus arrhythmia in human subjects [26]. Taken together, thesediscrepancies highlight that the interactions between sympathetic and parasympathetic activitieson cardiac function are complex and still incompletely understood, and that differences betweenanalyses based on HP and analyses based on heart rate may contribute to the variability in thereported results.
(b) Central pathways regulating the autonomic outflow to the heart(i) The central autonomic network
The autonomic outflow to the heart is regulated by a central autonomic network (CAN) ofinterconnected brain structures, which includes the medial prefrontal cortex (MPFC) and insularcortex, the amygdala and the bed nucleus of the stria terminalis (BNST), the lateral region ofthe hypothalamus and the paraventricular nucleus (PVN) and dorsomedial hypothalamic (DMH)nucleus, the periaqueductal grey (PAG) matter of the midbrain, the parabrachial Kölliker–Fuseregion of the lateral pons, as well as several regions of the medulla, which partly overlap withthose involved in respiratory control [27]. The MPFC comprises the anterior cingulate cortexand the prelimbic and infralimbic areas and is involved in both cognitive and visceromotorfunctions, thus being of potential great relevance for psychosomatic medicine [28]. The insulais a viscerosensory and visceromotor region [29] and plays a key role in physiological andpathological cardiovascular control [30]. In humans, the right (non-dominant) anterior insularcortex is involved in the generation of the mental image of one’s physical state, which underliesbasic emotional states [31]. The amygdala is primarily involved in the information processingrelated to negative emotions, whereas positive emotions tend to reduce amygdala activation [32].The insular cortex, the central nucleus of the amygdala and the BNST constitute a cortico-striatal–pallidal circuit that processes emotional information with autonomic responses [27]and projects to the hypothalamic behaviour control column [33]. The PVN is a mastercontroller of the autonomic nervous system, providing specialized innervation to all autonomicrelay centres [27], and together with the DMH it integrates neuroendocrine, homeostatic andstress responses [34]. The PAG takes part in regulating autonomic responses to physical andpsychological stressors [35]. The medullary nucleus of the tractus solitarius (NTS) and thepontine parabrachial and Kölliker–Fuse nuclei are reciprocally connected and relay visceralafferent information to other CAN structures [36]. In the medulla, the CAN includes the NAmband DMNX, the rostral (RVLM) and caudal portions of the ventrolateral medulla, and therostral ventromedial medulla (RVMM), which comprises the midline medullary raphe andthe parapyramidal area [36].
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

4
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
PVN
MPFC
MDHNAmb/DMNX
NTS
l,vl PAG
amygdala
insularcortex cortex
subcorticalforebrain
midbrain
baroreceptors,chemoreceptors,
group III/IV muscleafferents
centralinspiratory
neurons medulla
receptorsnasopharyngeal
cardiac parasympatheticoutflow
Figure 1. Schematic diagram showing the major central pathways regulating the cardiac parasympathetic outflow. Nodistinction is made between excitatory and inhibitory connections. DMNX, dorsal motor nucleus of the vagus nerve; l, lateral;MDH, medullary dorsal horn of the trigeminal nucleus; MPFC, medial prefrontal cortex; NAmb, nucleus ambiguus, NTS, nucleusof the tractus solitarius; PAG, periaqueductal grey; PVN, hypothalamic paraventricular nucleus; vl, ventrolateral.
(ii) Cardiac parasympathetic preganglionic neurons
Each cardiac P2N receives inputs from only a single active P1N [37]. Accordingly, the functionalspecificity of cardiac P2N is matched by that of the P1N in the NAmb, separate groups ofwhich selectively and independently control HP, atrioventricular conduction and ventricularcontractility [38]. The P1N in the NAmb have myelinated B-fibre axons, while those in the DMNXhave unmyelinated slow-conducting C-fibre axons. The P1N in the DMNX may regulate to someextent coronary blood flow or cardiac contractility, but, in contrast to P1N in the NAmb, they haverelatively little effect on HP [39]. The P1N in the DMNX have a low level of ongoing activity thatis not related to cardiac or respiratory activity, and are unaffected by baroreceptor inputs [40],although they are activated by stimulation of pulmonary C-fibre afferents [41]. Conversely, theP1N in the NAmb have a relatively high level of activity that shows a distinct cardiac andrespiratory modulation [42]. Inputs from baroreceptors, chemoreceptors and nasopharyngealreceptors converge on individual P1N in the NAmb [37] and are gated by inspiratory-relatedinputs, with the result that the reflex bradycardia normally evoked by stimulation of thesereceptors is greatly attenuated during inspiration [43] (figure 1). The inspiratory gating of reflexbradycardic responses is one of the mechanisms responsible for respiratory sinus arrhythmia andis believed to have the effect of improving the ventilation–perfusion relationship in the lungs [44].Apart from inputs from peripheral receptors and central inspiratory neurons, the P1N in theNAmb or DMNX also receive inputs from other sources at all levels of the brain, including directprojections from the hypothalamic PVN [45] and the lateral (l) and ventrolateral (vl) parts of themidbrain PAG (figure 1) [46]. The main cortical areas that regulate HP are the MPFC, which isthought to promote cardiac parasympathetic activity [28,47], and the insular cortex, which canincrease or decrease HP depending on the stimulated site [30]. The insular cortex sites wherestimulation decreases HP and increases arterial blood pressure densely innervate the MPFC andamygdala [48]. In most of these studies, it is not completely clear if the changes in HP are dueto sympathetic or parasympathetic effects. Studies using the retrograde trans-synaptic transportof viral vectors revealed indirect descending pathways by which the MPFC and insular cortexmay regulate the activity of P1N in the NAmb or DMNX, involving the amygdala, PAG andNTS [49] (figure 1).
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

5
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
somatosensoryreceptors
cortex
subcorticalforebrain
midbrain
medulla
spinal cord
osmoreceptors
PVN DMH
MPFC insular cortex
amygdala
NTS
CVLM
l,vl PAG
RVLMRVMM
cardiac sympathetic outflow
baroreceptors,chemoreceptors,
group III/IVmuscle afferents
Figure 2. Schematic diagram showing the major central pathways regulating the cardiac sympathetic outflow. No distinctionismade between excitatory and inhibitory connections. CVLM, caudal ventrolateralmedulla; DMH, dorsomedial hypothalamus;RVLM, rostral ventrolateral medulla; RVMM, rostral ventromedial medulla (for more abbreviations, refer to figure 1).
(iii) Cardiac sympathetic premotor neurons
The major input to the cardiac S1N arises from sympathetic premotor neurons (S0N) in theRVMM, RVLM, A5 area in the pons and hypothalamic PVN [50] (figure 2). These cell groups differwith respect to their anatomical connections and functional properties. As will be discussed inmore detail below, S0N in the RVMM are activated during arousal and stress but do not generatecardiac responses to baroreceptor or chemoreceptor inputs. By contrast, S0N in the RVLM arecritical for the expression of cardiac reflex responses to baroreceptor, chemoreceptor and otherinputs [36], but do not appear to have a major role in the generation of cardiac responses instress and arousal [51]. Much less information is available about the precise functional role ofS0N in the A5 area and PVN, although in regard to the latter it is likely that they are essential forgenerating cardiac sympathetic responses to inputs associated with changes in blood volume orplasma osmolality [52]. The chemical properties of the S0N in the different regions are also highlyvaried. For example, many of the S0N in the RVMM contain serotonin, while those in the RVLMare mainly adrenergic neurons of the C1 group. The S0N in the A5 group in the pons synthesizenoradrenaline, and in the PVN many of the S0N contain oxytocin, corticotropin-releasing factor,vasopressin or angiotensin [50]. Neurons at all levels of the brain regulate the activity of the S0N,but many of the descending pathways to the S0N in the medulla include synapses in other nuclei(figure 2). As described above, the MPFC and insular cortex may regulate HP through changes incardiac sympathetic or parasympathetic activity. Activation of neurons in the MPFC may alsodecrease vasomotor sympathetic activity [53]. The descending pathways from the MPFC andinsular cortex to the S0N in the medulla are not clearly defined. Anatomical studies in animalshave revealed direct descending projections to the RVLM as well as potential indirect projectionsto both the RVMM and RVLM that include synaptic connections in the amygdala, PAG andNTS [35,53] (figure 2).
(c) Reflex control of the autonomic outflow to the heartCardiac function can be profoundly altered by the reflex activation of cardiac autonomic nervesin response to inputs from receptors including baroreceptors, chemoreceptors, receptors fromskeletal muscles and nasopharyngeal receptors. Cardiac sympathetic and parasympathetic nerve
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

6
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
activities are reciprocally altered in response to baroreceptor stimulation, whereas in responseto stimulation of chemoreceptor and nasopharyngeal inputs, both cardiac sympathetic andparasympathetic activities are increased [54].
(i) Baroreceptor reflex
The physiological advantage of reciprocal baroreflex control of the heart by sympatheticand parasympathetic nerves is that it allows for rapid and large compensatory responses toperturbations in blood pressure [54]. The essential central circuits that mediate the baroreceptorreflex control of the heart have been well defined [36]. Primary baroreceptor and chemoreceptorafferent fibres terminate in the NTS on separate and spatially distinct subgroups of the second-order neurons. From the NTS, baroreceptor signals are transmitted via excitatory glutamatergicprojection neurons directly to cardiac P1N in the NAmb (figure 1) or to the caudal ventrolateralmedulla, where they synapse with GABAergic neurons. In turn, these neurons project to andinhibit S0N in the RVLM, which control the sympathetic outflow to the heart and differentvascular beds (figure 2). There is good evidence that the S0N in the RVLM that control thesympathetic outflow to different vascular beds form separate subgroups [55]. It also seems likelythat cardiac S0N in the RVLM are distinct from sympathetic vasomotor S0N, given that the cardiacand vasomotor sympathetic activities are reflexly regulated in a differentiated manner, accordingto the afferent input [36].
(ii) Chemoreceptor reflex
The physiological cardiac response to the chemoreceptor reflex has the advantage of maximizingoxygen conservation, while at the same time maintaining an adequate perfusion pressure for thebrain and the heart. This is achieved largely by a vagally evoked bradycardia (which reducescardiac oxygen consumption), while co-activation of cardiac sympathetic outflow maintains anoptimum stroke volume and cardiac output to meet the essential metabolic needs of the brainand heart [54]. Chemoreceptor signals are transmitted from the NTS via direct projections to P1Nin the NAmb (figure 1) and to S0N in the RVLM [56,57] (figure 2). Given that chemoreceptorstimulation evokes parasympathetic and sympathetic co-activation, it seems likely that both ofthese projections are excitatory, presumably glutamatergic. In addition, chemoreceptor signalsare also transmitted indirectly to the RVLM via the Kölliker–Fuse nucleus in the pons [56].
(iii) Exercise pressor reflex
The mechanical distortion and interstitial accumulation of metabolites produced by skeletalmuscle contraction stimulate group III/IV thin fibre afferents from skeletal muscles, whichelicit the exercise pressor reflex. This reflex supports muscle perfusion by raising arterial bloodpressure as a result of increased sympathetic outflow to the heart and systemic vasculature and ofdecreased parasympathetic outflow to the heart [58]. The neural pathway of the exercise pressorreflex is not known with certainty, except for the site of the first synapse, which is in the spinalcord dorsal horn [58]. However, neurons responsive to group III muscle afferents have beendemonstrated in the NTS [59], suggesting that the NTS is a key relay also for this reflex.
(iv) Diving reflex
The diving reflex is perhaps the most powerful autonomic reflex known, as it is associatedwith intense sympathetically mediated vasoconstriction and profound vagally mediatedbradycardia [60]. Like the chemoreceptor reflex, it has the effect of conserving oxygen duringsubmersion. In non-diving animals such as rabbits, a very similar reflex is evoked by irritantvapours [61]. In both cases, nasopharyngeal receptors innervated by trigeminal afferents triggerthe reflex responses. There is evidence for both a direct projection from trigeminal afferents to theNAmb region that contains cardiac P1N, as well as indirect projections relayed via the medullarydorsal horn of the trigeminal nerve [60] (figure 1).
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

7
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
(d) Integrative regulation of the autonomic outflow to the heart(i) Physical activity
Physical exercise, either dynamic or static (isometric), entails a decrease in HP, which is abruptin onset, maintained or even enhanced while exercise occurs, and progressively reversed afterexercise is over [62]. The changes in HP at the onset and offset of exercise result primarilyfrom parasympathetic withdrawal and reactivation, respectively [62,63]. During steady-statedynamic exercise, the decrease in HP results from a continuum of balanced sympatho-vagalcontrol, with parasympathetic withdrawal playing the greater role the lower the workload [64].These autonomic changes result from the integration of the baroreceptor and exercise pressorreflexes with central autonomic commands [65]. These commands are classically defined as feed-forward modulation of P1N and S1N exerted by the descending somatic motor pathways, butalso incorporate feedback components associated with the perception of effort [66]. The neuralstructures that issue the central autonomic commands associated with physical exercise arestill uncertain, and have been suggested to overlap with those of the CAN at the level of theNTS, PVN, PAG, insular cortex and MPFC [62,65]. In this last respect, a remarkable study thatcoupled magnetic resonance neuroimaging with measurements of HP, muscle sympathetic nerveactivity and arterial blood pressure in human subjects provided evidence that decreased activityof the ventral MPFC is involved in the central commands that cause parasympathetic cardiacwithdrawal during static handgrip exercise [67]. During exercise, both central commands and theexercise pressor reflex reset the operating point of the arterial baroreceptor reflex towards highervalues of blood pressure and heart rate [65]. There is evidence that this resetting is mediatedby neuronal circuitry in the NTS [59]. Chronotropic incompetence, i.e. the failure to increaseheart rate to a level commensurate with increased workload, may occur in patients with heartfailure because sympathetic overactivation desensitizes myocardial beta-adrenergic receptors,and portends a poor prognosis [68]. Aerobic exercise training may decrease sympatheticactivation and ameliorate chronotropic incompetence in patients with heart failure [68,69]. Themechanisms of this beneficial effect are thought to involve inhibition of the NFκB-dependentinflammatory cascade, angiotensin II receptor expression and oxidative stress in RVLM and PVNneurons [69].
(ii) Stress and arousal
Stress may be defined as a condition in which expectations, whether genetically programmed,learned or deduced, do not match perceptions of the internal or the external environment, thisdiscrepancy eliciting compensatory responses [70]. The concept of arousal is state-dependent,and, during wakefulness, it refers to the cerebral, autonomic and behavioural activation inresponse to internal and environmental stimuli [71]. In the immediate response to a novelpsychological stress (e.g. air-puff stress), the changes in HP are often biphasic, due to co-activationof cardiac sympathetic and parasympathetic nerves, consistent with an orienting response to thearousing stimulus [54,72]. With repeated exposures, however, there is a decrease in HP whichis due predominantly to cardiac sympathetic activation [54]. There is good evidence that S0Nlocated in the RVMM (especially the midline raphe) generate stress-evoked increases in cardiacsympathetic activity [73]. The midline raphe receives a direct projection from the DMH, a regionthat is critical for the expression of the cardiovascular response to stress [72,73], and from thelPAG [74], which as mentioned above is a region that generates cardiovascular responses tophysical stressors [35,72]. In addition, both the DMH and lPAG receive direct inputs from theamygdala and direct or indirect inputs from the insular cortex (figure 2), both of which arealso involved in generating responses to physical stressors [35,72]. Furthermore, air-puff stressinduces an increase in c-Fos expression (a marker of neuronal activation) in the RVMM, butnot in the RVLM, which, as mentioned above, is activated when cardiac sympathetic activityis reflexly increased by inputs from peripheral receptors [51]. The RVMM contains more S0Nthan any other brain region [50]. The descending pathway from the DMH to the RVMM is
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

8
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
functionally asymmetric, such that disinhibition of the right DMH induces significantly largerdecreases in HP and increases in cardiac contractility and number of ectopic beats than thoseevoked from the left DMH [75]. Thus, the pathway from the right DMH to the RVMM maycontribute to stress-related increases in cardiac sympathetic activity [75], which is a main cause ofmalignant tachyarrhythmias [76], and further explains the fact that intense emotional stress cancause sudden death (see, for example, [77]), as also discussed in §2d(iv). Finally, vagally mediatedheart rate variability may also decrease with stress, and according to a remarkable meta-analysisof neuroimaging studies may index the extent to which threat representations encoded in theamygdala are inhibited by the ventral MPFC based on the external and internal perceptions ofsafe contexts [47].
(iii) Sleep
The transition from wakefulness to non-rapid eye movement (non-REM) sleep brings aboutan increase in HP [78], which is attributed to an increase in parasympathetic activity [79]. Onpassing from non-REM sleep to REM sleep, which accounts for approximately 20% of totalsleep time, HP generally shows a mild tonic decrease, which is superimposed on large transientchanges in HP associated with bursts of rapid eye movements, phasic increases in the activity ofthe pyramidal tract, ponto-geniculo-occipital waves, and acceleration of the hippocampal thetarhythm (reviewed in [78]). These sleep-related changes in HP are best explained by sleep-relatedcentral autonomic commands, which override or reset baroreflex function [80]. The hypothalamicsuprachiasmatic nucleus (SCN) is the master pacemaker responsible for the circadian rhythms ofwake–sleep states [81] and of blood pressure and HP [82]. Neurons in the SCN may issue circadianautonomic commands involving sympathetic and parasympathetic outflows by projecting to thePVN [83], which, as mentioned previously, is a master autonomic controller in the CAN [27].However, the main SCN contribution to the circadian cardiovascular rhythms is thought tocome indirectly from the circadian rhythms of rest and activity [84] or, more precisely, ofwakefulness, non-REM sleep and REM sleep [85], each of which appears associated with specificcentral autonomic commands [86]. The central autonomic commands during non-REM sleepmay involve inhibitory projections from sleep-active neurons of the hypothalamic ventrolateralpreoptic nucleus to the S0N of the PVN [87], central thermoregulatory pathways includingthe RVMM, central baroreflex pathways including the NTS, the parabrachial and Kölliker–Fusenuclei, and command neurons in the pedunculopontine tegmental (PPT) nucleus [86]. DuringREM sleep, central autonomic commands may involve the PAG, the pontine sublaterodorsal andPPT nuclei, and the vestibular and raphe obscurus medullary nuclei [86].
2. Clinical implications of the brain–heart interactions
(a) Autonomic hyperactivity versus autonomic failureNeurodegenerative disorders frequently cause slowly progressive failure of the autonomiccontrol, mainly concerning its sympathetic component, resulting in multiple clinicalmanifestations (orthostatic hypotension, impaired sweating, neurogenic bladder with erectiledysfunction in men, gastrointestinal dysmotility), which may be subtle due to compensatorymechanisms [88]. Conversely, vascular, inflammatory or traumatic lesions of the autonomicnervous system and drug adverse effects most often manifest acutely with signs of autonomichyperactivity, including abnormal excessive control of the cardiovascular system. Sustained andchronic autonomic hyperactivity may also appear in association with other chronic neurologicaldisorders, in particular sleep disorders [89]. Both acute and chronic manifestations of animbalanced brain–heart interaction represent a risk factor for the development of cardiovasculardiseases or acute cardiovascular events, which can also lead to sudden cardiac death. It may bequite difficult to separate out the cardiac component in these clinical conditions. By contrast,viewing derangements of the brain–heart interaction from the perspective of the underlying
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from
9
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
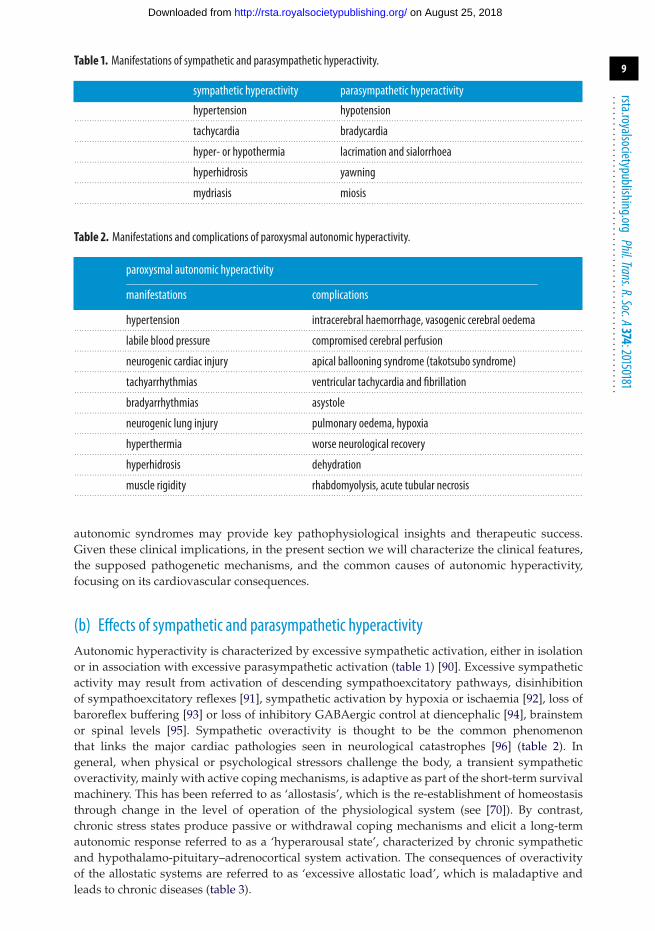
Table 1. Manifestations of sympathetic and parasympathetic hyperactivity.
sympathetic hyperactivity parasympathetic hyperactivity
hypertension hypotension. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
tachycardia bradycardia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
hyper- or hypothermia lacrimation and sialorrhoea. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
hyperhidrosis yawning. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
mydriasis miosis. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 2. Manifestations and complications of paroxysmal autonomic hyperactivity.
paroxysmal autonomic hyperactivity
manifestations complications
hypertension intracerebral haemorrhage, vasogenic cerebral oedema. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
labile blood pressure compromised cerebral perfusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
neurogenic cardiac injury apical ballooning syndrome (takotsubo syndrome). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
tachyarrhythmias ventricular tachycardia and fibrillation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
bradyarrhythmias asystole. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
neurogenic lung injury pulmonary oedema, hypoxia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
hyperthermia worse neurological recovery. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
hyperhidrosis dehydration. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
muscle rigidity rhabdomyolysis, acute tubular necrosis. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
autonomic syndromes may provide key pathophysiological insights and therapeutic success.Given these clinical implications, in the present section we will characterize the clinical features,the supposed pathogenetic mechanisms, and the common causes of autonomic hyperactivity,focusing on its cardiovascular consequences.
(b) Effects of sympathetic and parasympathetic hyperactivityAutonomic hyperactivity is characterized by excessive sympathetic activation, either in isolationor in association with excessive parasympathetic activation (table 1) [90]. Excessive sympatheticactivity may result from activation of descending sympathoexcitatory pathways, disinhibitionof sympathoexcitatory reflexes [91], sympathetic activation by hypoxia or ischaemia [92], loss ofbaroreflex buffering [93] or loss of inhibitory GABAergic control at diencephalic [94], brainstemor spinal levels [95]. Sympathetic overactivity is thought to be the common phenomenonthat links the major cardiac pathologies seen in neurological catastrophes [96] (table 2). Ingeneral, when physical or psychological stressors challenge the body, a transient sympatheticoveractivity, mainly with active coping mechanisms, is adaptive as part of the short-term survivalmachinery. This has been referred to as ‘allostasis’, which is the re-establishment of homeostasisthrough change in the level of operation of the physiological system (see [70]). By contrast,chronic stress states produce passive or withdrawal coping mechanisms and elicit a long-termautonomic response referred to as a ‘hyperarousal state’, characterized by chronic sympatheticand hypothalamo-pituitary–adrenocortical system activation. The consequences of overactivityof the allostatic systems are referred to as ‘excessive allostatic load’, which is maladaptive andleads to chronic diseases (table 3).
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

10
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
Table 3. Disorders associated with chronic autonomic hyperactivity.
chronic autonomic hyperactivity-
associated disorders
obesity. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
diabetes, insulin resistance. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
hypertension. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
insomnia and anxiety. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
hyperthermia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
high energy expenditure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
muscle wasting. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
increased susceptibility to infection. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
impairment of memory. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(c) Common causes of paroxysmal sympathetic hyperactivity(i) Severe head injury
Episodes of paroxysmal sympathetic hyperactivity frequently occur in the setting of severe diffuseaxonal injury and in the post-resuscitation period after severe anoxic–ischaemic brain insults [97].These paroxysms usually start 5–7 days after injury and follow a regular pattern (1–3 times perday, lasting 1–10 h). Over time, the episodes tend to become less frequent and more prolongedand are associated with poor outcome. Release of hypothalamic CAN structures from corticalinhibitory control may explain acute episodes of autonomic hyperactivity of diencephalic origin,which may occur after closed-head injury associated with a decorticate state and widespreadaxonal damage [27].
(ii) Subarachnoid haemorrhage
Subarachnoid haemorrhage is associated with acute and massive sympathetic hyperactivity [98].This may produce electrocardiographic changes indicative of cardiac injury [99,100] anddifferent arrhythmias, particularly supraventricular tachyarrhythmias such as atrial fibrillation.Acute hydrocephalus after subarachnoid haemorrhage may cause episodes of acute autonomichyperactivity linked to disinhibition of hypothalamic CAN structures such as the PVN [27].Mechanical and chemical stimulation of the insular cortex has also been postulated as amechanism for autonomic derangements caused by subarachnoid haemorrhage in the Sylvianfissure [30].
(iii) Insular stroke
Acute stroke can impair central autonomic control, resulting in myocardial injury,electrocardiographic abnormalities, cardiac arrhythmias and ultimately sudden death. Cardiacand renal diseases, diabetes and arrhythmogenic medications increase the risk of cardiacmorbidity and mortality after stroke [100]. Autonomic imbalance is more frequent afterinfarcts involving the insular cortex, which is part of the CAN [30]. Despite evidenceindicating a lateralization of autonomic control by the insular cortex [27], there is noobvious laterality in the impact of insular cortex stroke on cardiovascular response or strokeprognosis [30].
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

11
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
(iv) Seizures
Focal seizures involving CAN structures such as the MPFC, insular cortex and amygdalaentail various autonomic manifestations [27] including sympathetic hyperactivity and cardiacarrhythmias [101]. The most common cardiac manifestation is sinus tachycardia but paroxysmalatrial fibrillation, supraventricular and ventricular tachycardia or ventricular fibrillation mayalso occur. Other autonomic manifestations of temporal lobe seizures include paroxysmalhypertension, ictal piloerection, sweating and facial flushing or pallor [102]. Temporal lobeseizures, particularly originating from the left hemisphere, may also manifest with excessiveparasympathetic activity leading to ictal bradycardia and asystole which can cause syncope [103].Seizure-related cardiac arrhythmias have been implicated as a potential pathogenetic mechanismof sudden unexpected death in epilepsy. However, ictal asystole usually has a self-limitingcourse, and postictal rather than ictal arrhythmias together with respiratory abnormalities seemof greater importance to the pathophysiology of sudden unexpected death in epilepsy [104]. Inseizures occurring during sleep, as in nocturnal frontal lobe epilepsy, an increase in sympathetic–parasympathetic balance, similar to that associated with physiological arousal from sleep, usuallyprecedes the onset of the motor manifestations. This suggests a role of autonomic activation,which is part of the arousal response, in triggering seizures [105].
(v) Brainstem lesions
Sympathetically mediated hypertension, cardiac arrhythmias or myocardial injury may occur asa consequence of brainstem lesions, particularly those involving the lateral medulla [106]. Rarely,paroxysmal sympathetic activity may be the presentation of baroreflex failure due to bilateralinvolvement of the NTS [107].
(vi) Baroreflex failure syndrome
The baroreflex failure syndrome refers to the cardiovascular manifestations resulting frominterruption of the afferent limb of the baroreflex at the level of the carotid sinus, baroreceptorafferents or medulla. The main clinical manifestations of baroreflex failure are acute hypertensionor fluctuating hypertension with or without orthostatic hypotension or orthostatic tachycardia.Episodes of severe bradycardia and hypotension, referred to as malignant vagotonia, may alsooccur [108].
(vii) Autonomic dysreflexia in spinal cord injury
Autonomic dysreflexia refers to episodes of massive sympathetic hyperactivity triggered byreflex stimuli below the lesion in patients with cervical or thoracic spinal cord injury above T5.These episodes occur upon recovery from the acute spinal shock [109] and are characterizedby severe hypertension that may result in hypertensive encephalopathy, intracranial or retinalhaemorrhage, seizures or even sudden death [110]. Headache, anxiety, facial flushing andextensive diaphoresis above the level of the lesion often precede the onset of the hypertensivecrisis. The most common stimuli originate from the bladder or rectum.
(viii) Guillain–Barré syndrome
Sympathetic or parasympathetic hyperactivity or hypofunction in different combinationsmay be detected in about two-thirds of patients with Guillain–Barré syndrome [111,112].Autonomic hyperactivity may manifest with sinus tachycardia, sustained or paroxysmalhypertension, episodes of flushing, orthostatic hypotension and cardiac arrhythmias includingcardiac arrest. The main mechanism of cardiovascular instability in Guillain–Barré syndrome isimpaired baroreflex modulation of the sympathetic cardiovascular output due to demyelinationof baroreceptor afferents [112]. Episodes of severe paroxysmal hypertension may resultin subarachnoid haemorrhage or posterior leukoencephalopathy syndrome or takotsubo
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

12
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
syndrome [113], whereas some patients are at risk of severe vagally mediated bradycardia andasystole in response to reflex stimuli such as tracheal suction [112].
(ix) Sepsis
The systemic inflammatory response to infection includes tachycardia in a context of sympatheticoveractivity and myocardial contractile dysfunction, which converts into autonomic failurepreceding shock in severe cases [114]. Inflammatory mediators act directly on the heart to causetachycardia in experimental sepsis [115]. However, the sympathetic outflow to the heart alsomarkedly increases in these conditions [116]. Cardiac sympathetic activation during sepsis mayresult from disinhibition of the DMH and the RVMM caused by prostaglandin E2 binding toneurons in the medial preoptic hypothalamus [116,117]. This is a dramatic example of the complexand still poorly understood interactions between the brain, autonomic activity and the immunesystem, which are a burgeoning area of research [118].
(x) Iatrogenic causes
Several drugs can trigger syndromes characterized by mental status changes, increase of muscletone, hyperthermia and autonomic hyperactivity. These include the neuroleptic malignantsyndrome, which may be elicited by dopamine D2 receptor blocking antipsychotics [119];malignant hyperthermia, which is a hypermetabolic response to volatile anaesthetics or tosuccinylcholine driven by excessive calcium release in the skeletal muscle [120]; the serotoninsyndrome, which results from overactivation of central and peripheral serotonin receptors,often by antidepressant drugs [121]; and the anticholinergic syndrome, which results from theinhibition of muscarinic cholinergic neurotransmission [122]. The brain structures responsible forautonomic hyperactivity associated with these iatrogenic causes are still unclear.
(d) Causes of chronic and sustained sympathetic hyperactivity(i) Fatal familial insomnia and agrypnia excitata
Fatal familial insomnia (FFI) is a rare familial prion disease whose clinical hallmark is theagrypnia excitata syndrome [123]. Somatomotor abnormalities (e.g. pyramidal signs, myoclonus,dysarthria/dysphagia and gait dysfunctions) also occur with variable latency and degree duringthe disease course. The term agrypnia (from the Greek expression for ‘to chase sleep’), asproposed by Lugaresi & Provini [124], describes an organic insomnia, which is characterizedby severe or complete lack of sleep, especially deep sleep. The sleep disorder is associatedwith sympathetic and motor hyperactivation (excitata) and with episodes of a peculiar oneiricbehaviour (oneiric stupor) [125]. Agrypnia excitata is typical of but not specific for FFI [94]. Atrest, FFI patients show from the onset of agrypnia a progressive worsening of hypertension,tachycardia and hyperthermia sustained for 24 h resulting in a progressive decline in theamplitude of circadian oscillations of the autonomic parameters [126]. FFI is characterized byunbalanced autonomic control with preserved parasympathetic activity but higher backgroundand stimulated sympathetic activity [127]. Clinico-pathological relations and functional imagingin FFI implicate the anterior ventral and mediodorsal nuclei of the thalamus in the regulationof the wake–sleep cycle and other autonomic functions, with sparing of hypothalamic andbrainstem structures [128]. The mediodorsal thalamic nucleus is an integral part of the circuitsthat connect the hypothalamus with the amygdala and the MPFC components of the CAN [33].The preferential thalamic lesions in FFI would act by disconnecting the limbic cortical areasinvolved in the control of instinctive behaviour and the cortical and subcortical regions thatpromote sleep and regulate autonomic functions. Such a disconnection syndrome results in ashift to persistent wakefulness behaviour and sympathetic hyperactivation, which makes FFI theparadigm of what can occur in humans if the arousal system cannot be shut off. Dysfunctions of
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

13
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
the medial thalamus and related limbic areas are believed to underlie also the agrypnia excitataassociated with delirium tremens, Morvan syndrome [123], and Whipple disease [129].
(ii) Obstructive sleep apnoea syndrome
The obstructive sleep apnoea syndrome (OSAS) is characterized by repetitive episodes ofcomplete (apnoea) or partial (hypopnoea) upper airway obstruction occurring during sleep,which usually result in blood oxygen desaturation and often terminate with brief arousal. Theapnoeic episodes are associated with sympathetic hyperactivity and blood pressure increase, andwith HP increase at onset of apnoeas followed by tachycardia on resumption of breathing [130].OSAS with this continuous repetition of obstructive respiratory events during the night is aparadigm of how a sleep breathing disorder can lead to a permanent dysregulation of theautonomic cardiovascular control resulting in sustained sympathetic hyperactivity. Recent dataon animal models highlight the key role played in this process by the NTS and PVN [131].A remarkable functional imaging study on patients suggested that the elevated sympatheticactivity associated with OSAS may be driven by changes in activity in higher cortical regions,such as the MPFC [132]. An increase in sympathetic tone is thought to underlie the cardiovascularcomplications responsible for the increased mortality rate in patients with sleep apnoea [133].In addition, inhibition of the excitatory pathway from the PVN to P1N during chronichypoxia/hypercapnia may decrease cardiac parasympathetic activity, further increasing the riskof adverse cardiac events associated with OSAS [45]. At least in women, OSAS severity issignificantly associated with incident heart failure [134], which brings about cardiopulmonaryreflex desensitization and severe cardiac and renal sympathoexcitation [135]. The OSAS isalso related to the development of daytime hypertension [136] and to excessive daytimesleepiness [137]. A reduced baroreflex sensitivity is also a well-documented feature of OSASbefore the onset of cardiovascular complications [138]. Daytime hypertension and excessivedaytime sleepiness may share a common underlying cause: the cardiovascular effects of OSASmay be due to the top-down dysfunction of the baroreflex, while the reduced diurnal vigilancemay be explained by a bottom-up dysfunction of the baroreceptor afferent effect [71]. Thiscould be better understood from an allostatic perspective where the chronic hypertensive stateassociated with OSAS might be viewed as the result of autonomic nervous system adaptation tothe episodic recurrence of sympathetic surges during the night. Therefore, the reduced baroreflexsensitivity consistently described in OSAS could be an indirect index of an as yet unknownmaladaptive mechanism resulting in a decreased baroreflex function.
(iii) Autonomic effects of sleep debt and narcolepsy with cataplexy
Sleep has important homeostatic functions. Whether sleep deprivation and fragmentation isdue to sleep disorders such as restless legs syndrome [89] or anxiety or depression or ahectic lifestyle, it may disturb many essential homeostatic mechanisms with adverse effects onautonomic, hormonal and metabolic systems [139]. There is evidence that short sleep durationis associated with cardiovascular morbidity in epidemiological surveys [140] and that sleepdeprivation profoundly affects the autonomic nervous system [141]. Acute [142] and chronic [143]sleep deprivation are associated with increased sympathetic and decreased parasympatheticcardiovascular modulation. Long-term sleep deprivation is a chronic stressor that may decreaseresting HP and increase systolic and diastolic blood pressure [144], entailing a modest butindependent increase in the risk of cardiac ischaemia, stroke and sudden cardiac death [145].The central neural mechanisms responsible for the autonomic effects of sleep debt are stillunclear. However, it seems reasonable to surmise that they consist of altered function of thosesame structures of the CAN which underlie the sleep-related central autonomic commands(see §1d(iii)). Another sleep disorder with possible autonomic involvement is narcolepsy withcataplexy, a chronic disorder of the sleep–wake behaviour associated with the loss of neuronsreleasing hypocretin/orexin peptides [146]. Preliminary evidence indicates that autonomiccontrol of cardiac variability by baroreflex and central autonomic (feed-forward) mechanisms
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

14
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
is altered in narcolepsy with cataplexy patients during spontaneous sleep–wake behaviour, andparticularly during wakefulness before sleep [147]. The extent to which narcolepsy with cataplexyentails derangements in the control of the cardiovascular system is still a matter of debate [146].Interestingly, however, hypocretin/orexin neurons project to all of the brain structures that arethought to underlie the sleep-related central autonomic commands [86].
(iv) Triggering of cardiovascular events by emotional stressors
After the pioneering work of Walter B. Cannon in 1942 [148] with an article proposing a scientificbasis for ‘voodoo’ death, several studies were performed to clarify the neurobiological basisof the link between emotional and cardiovascular events that are a major cause of morbidityand mortality in the developed world. Today we have strong evidence that cardiovascularevents can be triggered by acute mental stress caused by events such as an earthquake, atelevised high-drama soccer game, job strain or the death of a loved one [149]. Animal studiessuggest that stressors presumed to provoke acute negative emotions affect cardiovascularphysiological control and increase the risk of sudden cardiac death through haemodynamic andelectrophysiological pathways increasing sympathetic output, impairing endothelial function andcreating a hypercoagulable state [150]. Interestingly, post-traumatic stress disorder, which is ananxiety disorder initiated by exposure to a traumatic event, is also independently associated withincreased risk of incident coronary heart disease and mortality [151].
3. Conclusion and perspectivesFrom the perspective of the clinician, the analysis of heart rate variability (HRV) based onthe electrocardiogram holds promise as an attractively simple tool for detecting autonomicimpairments and for predicting the prognosis of some neurological disorders through theassessment of the brain–heart connections. Unfortunately, we are still far from the realization ofthis very important unmet need. According to the Task Force on HRV evaluation (1996) [152],there are only two clinical situations where HRV analysis should be performed: to assessmortality risk in patients after myocardial infarction and to detect early evidence of cardiacautonomic neuropathy in diabetic patients. In the light of the clinical relevance of the brain–heartconnection for so many diseases, this implies that what is really lacking to develop specific clinicalapplications of the knowledge on heart–brain interactions are simple, widely available andreliable cardiovascular markers of the sympathetic tone and of the sympathetic–parasympatheticbalance. Such markers would be invaluable for the early detection of signs of cardiovasculardysautonomia, the treatment of which can avoid the occurrence of the life-threatening paroxysmalor chronic autonomic hyperactivity.
This state of affairs highlights the need for a deeper physiological understanding of thelinks between brain dynamics and the corresponding autonomic cardiac dynamics. The keyissue at stake here is to combine different sources such as: information on animal modelsand human subjects, information on basic physiology and clinical disorders, but also, andcritically, information from different biosignals in each given setting. In the physiologylaboratory, key advances can be obtained by multi-modal recordings of electroencephalogram,electrocardiogram, electromyogram, blood pressure and respiration in animal models withcongenital or acquired (viral vectors, drug-inducible expression systems) genetic modificationof brain circuits. Multi-modal recordings including beat-to-beat blood pressure (finger volumeclamp), respiration, muscle sympathetic nerve activity (peroneal nerve microneurography) andbody temperature are also desirable when investigating brain–heart interactions in the clinicallaboratory, in order to interpret cardiac control in its true physiological context. Taking advantageof the recent developments in magnetic resonance neuroimaging, as well as in advancedprocessing techniques of electroencephalographic, electrocardiographic and cerebrovascular flowsignals, interdisciplinary and multi-modal research approaches now have the chance of bringingour understanding of the brain–heart interactions to the next level.
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

15
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
Authors’ contributions. A.S., G.C.-B., R.A.L.D. and P.C. carried out the bibliographic research, analysed the data,interpreted the results and wrote sections of the paper. R.A.L.D. constructed the figure schemes. A.S. draftedthe full manuscript, which was reviewed by G.C.-B., R.A.L.D. and P.C. P.C. coordinated the project. All authorsgave final approval for publication.Competing interests. We have no competing interests.Funding. No specific funding was dedicated to this project.
References1. Crick SJ, Wharton J, Sheppard MN, Royston D, Yacoub MH, Anderson RH, Polak JM. 1994
Innervation of the human cardiac conduction system. A quantitative immunohistochemicaland histochemical study. Circulation 89, 1697–1708. (doi:10.1161/01.CIR.89.4.1697)
2. Bucchi A, Baruscotti M, Robinson RB, DiFrancesco D. 2007 Modulation of rate by autonomicagonists in SAN cells involves changes in diastolic depolarization and the pacemakercurrent. J. Mol. Cell Cardiol. 43, 39–48. (doi:10.1016/j.yjmcc.2007.04.017)
3. Berntson GG, Cacioppo JT, Quigley KS. 1995 The metrics of cardiac chronotropism: biometricperspectives. Psychophysiology 32, 162–171. (doi:10.1111/j.1469-8986.1995.tb03308.x)
4. Berger RD, Saul JP, Cohen RJ. 1989 Transfer function analysis of autonomic regulation. I.Canine atrial rate response. Am. J. Physiol. Heart Circ. Physiol. 256, H142–H152.
5. Connelly CA, Wurster RD. 1985 Spinal pathways mediating respiratory influences onsympathetic nerves. Am. J. Physiol. Regul. Integr. Comp. Physiol. 249, R91–R99.
6. Furnival CM, Linden RJ, Snow HM. 1970 Inotropic changes in the left ventricle: the effectof changes in heart rate, aortic pressure and end-diastolic pressure. J. Physiol. 211, 359–387.(doi:10.1113/jphysiol.1970.sp009283)
7. DeSantiago J, Maier LS, Bers DM. 2002 Frequency-dependent acceleration of relaxation inthe heart depends on CaMKII, but not phospholamban. J. Mol. Cell Cardiol. 34, 975–984.(doi:10.1006/jmcc.2002.2034)
8. Bers DM, Despa S. 2009 Na/K-ATPase—an integral player in the adrenergic fight-or-flightresponse. Trends Cardiovasc. Med. 19, 111–118. (doi:10.1016/j.tcm.2009.07.001)
9. Wallick DW, Martin PJ, Masuda Y, Levy MN. 1982 Effects of autonomic activity andchanges in heart rate on atrioventricular conduction. Am. J. Physiol. Heart Circ. Physiol. 243,H523–H527.
10. Ardell JL, Randall WC, Cannon WJ, Schmacht DC, Tasdemiroglu E. 1988 Differentialsympathetic regulation of automatic, conductile, and contractile tissue in dog heart. Am. J.Physiol. Heart Circ. Physiol. 255, H1050–H1059.
11. Salo LM, Campos RR, McAllen RM. 2006 Differential control of cardiac functions by thebrain. Clin. Exp. Pharmacol. Physiol. 33, 1255–1258. (doi:10.1111/j.1440-1681.2006.04520.x)
12. Rashba EJ, Cooklin M, MacMurdy K, Kavesh N, Kirk M, Sarang S, Peters RW, ShorofskySR, Gold MR. 2002 Effects of selective autonomic blockade on T-wave alternans in humans.Circulation 105, 837–842. (doi:10.1161/hc0702.104127)
13. Piccirillo G et al. 2009 Autonomic nervous system activity measured directly and QT intervalvariability in normal and pacing-induced tachycardia heart failure dogs. J. Am. Coll. Cardiol.54, 840–850. (doi:10.1016/j.jacc.2009.06.008)
14. Clutter WE, Bier DM, Shah SD, Cryer PE. 1980 Epinephrine plasma metabolic clearance ratesand physiologic thresholds for metabolic and hemodynamic actions in man. J. Clin. Invest.66, 94–101. (doi:10.1172/JCI109840)
15. Hopkins DA, Bieger DJ, Steinbusch WM. 1996 Vagal efferent projections: viscerotopy,neurochemistry and effects of vagotomy. Prog. Brain Res. 107, 79–96. (doi:10.1016/S0079-6123(08)61859-2)
16. Coote JH. 2013 Myths and realities of the cardiac vagus. J. Physiol. 591, 4073–4085.(doi:10.1113/jphysiol.2013.257758)
17. Randall DC, Brown DR, McGuirt AS, Thompson GW, Armour JA, Ardell JL. 2003Interactions within the intrinsic cardiac nervous system contribute to chronotropicregulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R1066–R1075. (doi:10.1152/ajpregu.00167.2003)
18. Armour JA. 2008 Potential clinical relevance of the ‘little brain’ on the mammalian heart. Exp.Physiol. 93, 165–176. (doi:10.1113/expphysiol.2007.041178)
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

16
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
19. Di Francesco D, Ducouret P, Robinson RB. 1989 Muscarinic modulation of cardiac rate at lowacetylcholine concentrations. Science 243, 669–671. (doi:10.1126/science.2916119)
20. Opthof T. 2000 The normal range and determinants of the intrinsic heart rate in man.Cardiovasc. Res. 45, 177–184. (doi:10.1016/S0008-6363(99)00322-3)
21. Henning RJ, Cheng J, Levy MN. 1989 Vagal stimulation decreases rate of left ventricularrelaxation. Am. J. Physiol. Heart Circ. Physiol. 256, H428–H433.
22. Levy MN, Blattberg B. 1976 Effect of vagal stimulation on the overflow of norepinephrineinto the coronary sinus during cardiac sympathetic nerve stimulation in the dog. Circ. Res.38, 81–84. (doi:10.1161/01.RES.38.2.81)
23. Levy MN, Zieske H. 1969 Autonomic control of cardiac pacemaker activity andatrioventricular transmission. J. Appl. Physiol. 27, 465–470.
24. Ursino M. 1998 Interaction between carotid baroregulation and the pulsating heart: amathematical model. Am. J. Physiol. Heart Circ. Physiol. 275, H1733–H1747.
25. Sunagawa K, Kawada T, Nakahara T. 1998 Dynamic nonlinear vago-sympathetic interactionin regulating heart rate. Heart Vessels 13, 157–174. (doi:10.1007/BF01745040)
26. Taylor JA, Myers CW, Halliwill JR, Seidel H, Eckberg DL. 2001 Sympathetic restraint ofrespiratory sinus arrhythmia: implications for vagal-cardiac tone assessment in humans. Am.J. Physiol. Heart Circ. Physiol. 280, H2804–H2814.
27. Benarroch EE. 1993 The central autonomic network: functional organization,dysfunction, and perspective. Mayo Clinic Proc. Mayo Clinic 68, 988–1001. (doi:10.1016/S0025-6196(12)62272-1)
28. Lane RD. 2008 Neural substrates of implicit and explicit emotional processes: a unifyingframework for psychosomatic medicine. Psychosom. Med. 70, 214–231. (doi:10.1097/PSY.0b013e3181647e44)
29. Verberne AJ, Owens NC. 1998 Cortical modulation of the cardiovascular system. Prog.Neurobiol. 54, 149–168. (doi:10.1016/S0301-0082(97)00056-7)
30. Nagai M, Hoshide S, Kario K. 2010 The insular cortex and cardiovascularsystem: a new insight into the brain-heart axis. J. Am. Soc. Hypertens. 4, 174–182.(doi:10.1016/j.jash.2010.05.001)
31. Craig DA. 2002 How do you feel? Interoception: the sense of the physiological condition ofthe body. Nat. Rev. Neurosci. 3, 655–666. (doi:10.1038/nrn894)
32. Burgdorf J, Panksepp J. 2006 The neurobiology of positive emotions. Neurosci. Biobehav. Rev.30, 173–187. (doi:10.1016/j.neubiorev.2005.06.001)
33. Swanson LW. 2000 Cerebral hemisphere regulation of motivated behavior. Brain Res. 886,113–164. (doi:10.1016/S0006-8993(00)02905-X)
34. Thompson RH, Swanson LW. 2003 Structural characterization of a hypothalamicvisceromotor pattern generator network. Brain Res. Rev. 41, 153–202. (doi:10.1016/S0165-0173(02)00232-1)
35. Bandler R, Keay KA, Floyd N, Price J. 2000 Central circuits mediating patternedautonomic activity during active vs passive emotional coping. Brain Res. Bull. 53, 95–104.(doi:10.1016/S0361-9230(00)00313-0)
36. Dampney RAL. 1994 Functional organization of central pathways regulating thecardiovascular system. Physiol. Rev. 74, 323–364.
37. McAllen RM, Salo LM, Paton JF, Pickering AE. 2011 Processing of central and reflex vagaldrives by rat cardiac ganglion neurones: an intracellular analysis. J. Physiol. 589, 5801–5818.(doi:10.1113/jphysiol.2011.214320)
38. Gatti PJ, Johnson TA, Massari VJ. 1996 Can neurons in the nucleus ambiguus selectivelyregulate cardiac rate and atrio-ventricular conduction? J. Auton. Nerv. Syst. 57, 123–127.(doi:10.1016/0165-1838(95)00104-2)
39. Geis GS, Kozelka JW, Wurster RD. 1981 Organization and reflex control of vagal cardiomotorneurons. J. Auton. Nerv. Syst. 3, 437–450. (doi:10.1016/0165-1838(81)90080-1)
40. Jordan D. 2005 Vagal control of the heart: central serotonergic (5-HT) mechanisms. Exp.Physiol. 90, 175–181. (doi:10.1113/expphysiol.2004.029058)
41. Jones JFX, Wang Y, Jordan D. 1998 Activity of C fibre cardiac vagal efferents in anaesthetizedcats and rats. J. Physiol. 507, 869–880. (doi:10.1111/j.1469-7793.1998.869bs.x)
42. McAllen RM, Spyer KM. 1978 The baroreceptor input to cardiac vagal motoneurones.J. Physiol. 282, 365–374. (doi:10.1113/jphysiol.1978.sp012469)
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

17
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
43. Gilbey MP, Jordan D, Richter DW, Spyer KM. 1984 Synaptic mechanisms involved in theinspiratory modulation of vagal cardio-inhibitory neurones in the cat. J. Physiol. 356, 65–78.(doi:10.1113/jphysiol.1984.sp015453)
44. Hayano J, Yasuma F, Okada A, Mukai S, Fujinami T. 1996 Respiratory sinus arrhythmia. Aphenomenon improving pulmonary gas exchange and circulatory efficiency. Circulation 94,842–847. (doi:10.1161/01.CIR.94.4.842)
45. Dergacheva O, Dyavanapalli J, Pinol RA, Mendelowitz D. 2014 Chronic intermittent hypoxiaand hypercapnia inhibit the hypothalamic paraventricular nucleus neurotransmissionto parasympathetic cardiac neurons in the brain stem. Hypertension 64, 597–603.(doi:10.1161/HYPERTENSIONAHA.114.03603)
46. Farkas E, Jansen AS, Loewy AD. 1997 Periaqueductal gray matter projection to vagalpreganglionic neurons and the nucleus tractus solitarius. Brain Res. 764, 257–261.(doi:10.1016/S0006-8993(97)00592-1)
47. Thayer JF, Ahs F, Fredrikson M, Sollers JJ, Wager TD. 2012 A meta-analysis of heart ratevariability and neuroimaging studies: implications for heart rate variability as a marker ofstress and health. Neurosci. Biobehav. Rev. 36, 747–756. (doi:10.1016/j.neubiorev.2011.11.009)
48. Yasui Y, Breder CD, Saper CB, Cechetto DF. 1991 Autonomic responses and efferentpathways from the insular cortex in the rat. J. Comp. Neurol. 303, 355–374. (doi:10.1002/cne.903030303)
49. Ter Horst GJ, Postema F. 1997 Forebrain parasympathetic control of heart activity: retrogradetransneuronal viral labeling in rats. Am. J. Physiol. Heart Circ. Physiol. 273, H2926–H2930.
50. Jansen AS, Wessendorf MW, Loewy AD. 1995 Transneuronal labeling of CNS neuropeptideand monoamine neurons after pseudorabies virus injections into the stellate ganglion. BrainRes. 683, 1–24. (doi:10.1016/0006-8993(95)00276-V)
51. Furlong TM, McDowall LM, Horiuchi J, Polson JW, Dampney RA. 2014 The effect ofair puff stress on c-Fos expression in rat hypothalamus and brainstem: central circuitrymediating sympathoexcitation and baroreflex resetting. Eur. J. Neurosci. 39, 1429–1438.(doi:10.1111/ejn.12521)
52. Stocker SD, Simmons JR, Stornetta RL, Toney GM, Guyenet PG. 2006 Water deprivationactivates a glutamatergic projection from the hypothalamic paraventricular nucleus to therostral ventrolateral medulla. J. Comp. Neurol. 494, 673–685. (doi:10.1002/cne.20835)
53. Verberne AJM. 2011 Modulation of autonomic function by the cerebral cortex. In Centralregulation of autonomic functions (eds IJ Llewellyn-Smith, AJM Verberne), pp. 202–219. NewYork, NY: Oxford University Press.
54. Paton JF, Boscan P, Pickering AE, Nalivaiko E. 2005 The yin and yang of cardiac autonomiccontrol: vago-sympathetic interactions revisited. Brain Res. Brain Res. Rev. 49, 555–565.(doi:10.1016/j.brainresrev.2005.02.005)
55. McAllen RM, May CN, Shafton AD. 1995 Functional anatomy of sympathetic premotor cellgroups in the medulla. Clin. Exp. Hypert. 17, 209–221. (doi:10.3109/10641969509087066)
56. Dampney RA, Coleman MJ, Fontes MA, Hirooka Y, Horiuchi J, Li YW, Polson JW, PottsPD, Tagawa T. 2002 Central mechanisms underlying short- and long-term regulation ofthe cardiovascular system. Clin. Exp. Pharmacol. Physiol. 29, 261–268. (doi:10.1046/j.1440-1681.2002.03640.x)
57. Guyenet PG. 2006 The sympathetic control of blood pressure. Nat. Rev. Neurosci. 7, 335–346.(doi:10.1038/nrn1902)
58. Murphy MN, Mizuno M, Mitchell JH, Smith SA. 2011 Cardiovascular regulation by skeletalmuscle reflexes in health and disease. Am. J. Physiol. Heart Circ. Physiol. 301, H1191–H1204.(doi:10.1152/ajpheart.00208.2011)
59. Degtyarenko AM, Kaufman MP. 2006 Barosensory cells in the nucleus tractus solitariusreceive convergent input from group III muscle afferents and central command. Neuroscience140, 1041–1050. (doi:10.1016/j.neuroscience.2006.02.050)
60. Panneton WM. 2013 The mammalian diving response: an enigmatic reflex to preserve life?Physiology 28, 284–297. (doi:10.1152/physiol.00020.2013)
61. White S, McRitchie RJ, Korner PI. 1975 Central nervous system control of cardiorespiratorynasopharyngeal reflexes in the rabbit. Am. J. Physiol. 228, 404–409.
62. Fisher JP, Young CN, Fadel PJ. 2015 Autonomic adjustments to exercise in humans. Compr.Physiol. 5, 475–512. (doi:10.1002/cphy.c140022)
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

18
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
63. Imai K, Sato H, Hori M, Kusuoka H, Ozaki H, Yokoyama H, Takeda H, Inoue M, KamadaT. 1994 Vagally mediated heart rate recovery after exercise is accelerated in athletesbut blunted in patients with chronic heart failure. J. Am. Coll. Cardiol. 24, 1529–1535.(doi:10.1016/0735-1097(94)90150-3)
64. White DW, Raven PB. 2014 Autonomic neural control of heart rate during dynamic exercise:revisited. J. Physiol. 592, 2491–2500. (doi:10.1113/jphysiol.2014.271858)
65. Michelini LC, O’Leary DS, Raven PB, Nobrega AC. 2015 Neural control of circulation andexercise: a translational approach disclosing interactions between central command, arterialbaroreflex, and muscle metaboreflex. Am. J. Physiol. Heart Circ. Physiol. 309, H381–H392.(doi:10.1152/ajpheart.00077.2015)
66. Williamson JW. 2010 The relevance of central command for the neural cardiovascular controlof exercise. Exp. Physiol. 95, 1043–1048. (doi:10.1113/expphysiol.2009.051870)
67. Wong SW, Massé N, Kimmerly DS, Menon RS, Shoemaker JK. 2007 Ventral medialprefrontal cortex and cardiovagal control in conscious humans. Neuroimage 35, 698–708.(doi:10.1016/j.neuroimage.2006.12.027)
68. Brubaker PH, Kitzman DW. 2011 Chronotropic incompetence: causes, consequences, andmanagement. Circulation 123, 1010–1020. (doi:10.1161/CIRCULATIONAHA.110.940577)
69. Haack KKV, Zucker IH. 2015 Central mechanisms for exercise training-inducedreduction in sympatho-excitation in chronic heart failure. Auton. Neurosci. 188, 44–50.(doi:10.1016/j.autneu.2014.10.015)
70. Goldstein DS. 2013 Concepts of scientific integrative medicine applied to the physiologyand pathophysiology of catecholamine systems. Compr. Physiol. 3, 1569–1610. (doi:10.1002/cphy.c130006)
71. Silvani A, Calandra-Buonaura G, Benarroch EE, Dampney RA, Cortelli P. 2015 Bidirectionalinteractions between the baroreceptor reflex and arousal: an update. Sleep Med. 16, 210–216.(doi:10.1016/j.sleep.2014.10.011)
72. Dampney RAL. 2015 Central mechanisms regulating coordinated cardiovascular andrespiratory function during stress and arousal. Am. J. Physiol. Regul. Integr. Comp. Physiol.309, R429–R443. (doi:10.1152/ajpregu.00051.2015)
73. DiMicco JA, Zaretskaia MV, Zaretsky DV. 2006 Stress-induced cardiac stimulation andfever: common hypothalamic origins and brainstem mechanisms. Auton. Neurosci. 126–127,106–119. (doi:10.1016/j.autneu.2006.02.010)
74. Farkas E, Jansen AS, Loewy AD. 1998 Periaqueductal gray matter input to cardiac-relatedsympathetic premotor neurons. Brain Res. 792, 179–192. (doi:10.1016/S0006-8993(98)00029-8)
75. Xavier CH, Nalivaiko E, Beig MI, Menezes GB, Cara DC, Campagnole-Santos MJ, Fontes MA.2009 Functional asymmetry in the descending cardiovascular pathways from dorsomedialhypothalamic nucleus. Neuroscience 164, 1360–1368. (doi:10.1016/j.neuroscience.2009.09.018)
76. Meredith IT, Broughton A, Jennings GL, Esler MD. 1991 Evidence of a selective increase incardiac sympathetic activity in patients with sustained ventricular arrhythmias. N. Engl. J.Med. 325, 618–624. (doi:10.1056/NEJM199108293250905)
77. Leor J, Poole WK, Kloner RA. 1996 Sudden cardiac death triggered by an earthquake. N.Engl. J. Med. 334, 413–419. (doi:10.1056/NEJM199602153340701)
78. Silvani A. 2008 Physiological sleep-dependent changes in arterial blood pressure: centralautonomic commands and baroreflex control. Clin. Exp. Pharmacol. Physiol. 35, 987–994.(doi:10.1111/j.1440-1681.2008.04985.x)
79. Baust W, Bohnert B. 1969 The regulation of heart rate during sleep. Exp. Brain Res. 7, 169–180.(doi:10.1007/BF00235442)
80. Silvani A, Magosso E, Bastianini S, Lenzi P, Ursino M. 2011 Mathematical modelingof cardiovascular coupling: central autonomic commands and baroreflex control. Auton.Neurosci. 162, 66–71. (doi:10.1016/j.autneu.2011.04.003)
81. Easton A, Meerlo P, Bergmann B, Turek FW. 2004 The suprachiasmatic nucleus regulatessleep timing and amount in mice. Sleep 27, 1307–1318.
82. Janssen BJ, Tyssen CM, Duindam H, Rietveld WJ. 1994 Suprachiasmatic lesionseliminate 24-h blood pressure variability in rats. Physiol. Behav. 55, 307–311. (doi:10.1016/0031-9384(94)90138-4)
83. Buijs RM, Fleur SE, Wortel J, Van Heyningen C, Zuiddam L, Mettenleiter TC, KalsbeekA, Nagai K, Niijima A. 2003 The suprachiasmatic nucleus balances sympathetic and
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

19
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
parasympathetic output to peripheral organs through separate preautonomic neurons.J. Comp. Neurol. 464, 36–48. (doi:10.1002/cne.10765)
84. Sheward WJ et al. 2010 Circadian control of mouse heart rate and blood pressure by thesuprachiasmatic nuclei: behavioral effects are more significant than direct outputs. PLoS ONE5, e9783. (doi:10.1371/journal.pone.0009783)
85. Bastianini S, Silvani A, Berteotti C, Lo Martire V, Zoccoli G. 2012 Mice show circadianrhythms of blood pressure during each wake-sleep state. Chronobiol. Int. 29, 82–86.(doi:10.3109/07420528.2011.635231)
86. Silvani A, Dampney RA. 2013 Central control of cardiovascular function during sleep. Am. J.Physiol. Heart Circ. Physiol. 305, H1683–H1692. (doi:10.1152/ajpheart.00554.2013)
87. Uschakov A, Gong H, McGinty D, Szymusiak R. 2006 Sleep-active neurons in thepreoptic area project to the hypothalamic paraventricular nucleus and perifornical lateralhypothalamus. Eur. J. Neurosci. 23, 3284–3296. (doi:10.1111/j.1460-9568.2006.04860.x)
88. Sambati L, Calandra-Buonaura G, Doria A, Cortelli P. 2015 Diagnosis and management ofautonomic failure in neurodegenerative disorders. Eur. Neurol. 73, 126–133. (doi:10.1159/000368828)
89. Calandra-Buonaura G, Provini F, Guaraldi P, Plazzi G, Cortelli P. 2015 Cardiovascularautonomic dysfunctions and sleep disorders. Sleep Med. Rev. 26, 43–56. (doi:10.1016/j.smrv.2015.05.005)
90. Benarroch EE. 2014 Autonomic hyperactivity. In Autonomic neurology (ed. EE Benarroch),pp. 170–183. New York, NY: Oxford University Press.
91. Baguley IJ. 2008 The excitatory:inhibitory ratio model (EIR model): an integrativeexplanation of acute autonomic overactivity syndromes. Med. Hypotheses 70, 26–35.(doi:10.1016/j.mehy.2007.04.037)
92. Dickinson CJ. 1990 Reappraisal of the Cushing reflex: the most powerful neural bloodpressure stabilizing system. Clin. Sci. (Lond.) 79, 543–550. (doi:10.1042/cs0790543)
93. Norcliffe-Kaufmann L, Kaufmann H. 2012 Familial dysautonomia (Riley-Daysyndrome): when baroreceptor feedback fails. Auton. Neurosci. 172, 26–30. (doi:10.1016/j.autneu.2012.10.012)
94. Montagna P, Lugaresi E. 2002 Agrypnia excitata: a generalized overactivity syndrome anda useful concept in the neurophysiopathology of sleep. Clin. Neurophysiol. 113, 552–560.(doi:10.1016/S1388-2457(02)00022-6)
95. Mitsumoto H, Schwartzman M, Estes M, Chou S, La Franchise E, De Camilli P, Solimena M.1991 Sudden death and paroxysmal autonomic dysfunction in stiff-man syndrome. J. Neurol.238, 91–96. (doi:10.1007/BF00315688)
96. Samuels MA. 2007 The brain-heart connection. Circulation 116, 77–84. (doi:10.1161/CIRCULATIONAHA.106.678995)
97. Baguley I, Heriseanu R, Cameron I, Nott M, Slewa-Younan S. 2008 A critical review ofthe pathophysiology of dysautonomia following traumatic brain injury. Neurocrit. Care 8,293–300. (doi:10.1007/s12028-007-9021-3)
98. Di Pasquale G, Andreoli A, Lusa A, Urbinati S, Biancoli S, Cerè E, Borgatti M. 1998Cardiologic complications of subarachnoid hemorrhage. J. Neurosurg. Sci. 42, 33–36.
99. Chatterjee S. 2011 ECG changes in subarachnoid haemorrhage: a synopsis. Neth. Heart J. 19,31–34. (doi:10.1007/s12471-010-0049-1)
100. Richard C. 2011 Stress-related cardiomyopathies. Ann. Intensive Care 1, 39. (doi:10.1186/2110-5820-1-39)
101. Freeman R. 2006 Cardiovascular manifestations of autonomic epilepsy. Clin. Auton. Res. 16,12–17. (doi:10.1007/s10286-006-0278-y)
102. Lam E, Worrell G, Laughlin R. 2010 Semiology of the rare seizure subtype piloerection. Arch.Neurol. 67, 1524–1527. (doi:10.1001/archneurol.2010.304)
103. Tinuper P, Bisulli F, Cerullo A, Carcangiu R, Marini C, Pierangeli G, Cortelli P. 2001 Ictalbradycardia in partial epileptic seizures: autonomic investigation in three cases and literaturereview. Brain 124, 2361–2371. (doi:10.1093/brain/124.12.2361)
104. van der Lende M, Surges R, Sander JW, Thijs RD. 2016 Cardiac arrhythmias during or afterepileptic seizures. J. Neurol. Neurosurg. Psychiatry 87, 69–74.
105. Calandra-Buonaura G et al. 2012 Physiologic autonomic arousal heralds motormanifestations of seizures in nocturnal frontal lobe epilepsy: implications forpathophysiology. Sleep Med. 13, 252–262. (doi:10.1016/j.sleep.2011.11.007)
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

20
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
106. Jaster JH, Fitzek S, Fitzek C, Smith TW, Becske T. 2001 Myocardial injury after hemorrhageinto the lateral medulla oblongata. Neurology 57, 1145. (doi:10.1212/WNL.57.6.1145)
107. Biaggioni I, Whetsell WO, Jobe J, Nadeau JH. 1994 Baroreflex failure in a patient with centralnervous system lesions involving the nucleus tractus solitarii. Hypertension 23, 491–495.(doi:10.1161/01.HYP.23.4.491)
108. Robertson D, Hollister AS, Biaggioni I, Netterville J, Mosqueda-Garcia R, Robertson RM.1993 The diagnosis and treatment of baroreflex failure. N. Engl. J. Med. 329, 1449–1455.(doi:10.1056/NEJM199311113292003)
109. Mathias CJ, Frankel HL. 1988 Cardiovascular control in spinal man. Annu. Rev. Physiol. 50,577–592. (doi:10.1146/annurev.ph.50.030188.003045)
110. Dolinak D, Balraj E. 2007 Autonomic dysreflexia and sudden death in people with traumaticspinal cord injury. Am. J. Forensic Med. Pathol. 28, 95–98. (doi:10.1097/PAF.0b013e3180600f99)
111. Pfeiffer G, Schiller B, Kruse J. 1999 Indicators of dysautonomia in severe Guillain-Barrésyndrome. J. Neurol. 246, 1015–1022. (doi:10.1007/s004150050506)
112. Ropper AH, Wijdicks EF. 1990 Blood pressure fluctuations in the dysautonomia of Guillain-Barré syndrome. Arch. Neurol. 47, 706–708. (doi:10.1001/archneur.1990.00530060120029)
113. Bavikatte G, Gaber T, Eshiett MU. 2010 Posterior reversible encephalopathy syndromeas a complication of Guillain-Barré syndrome. J. Clin. Neurosci. 17, 924–926. (doi:10.1016/j.jocn.2009.11.009)
114. Rudiger A, Singer M. 2007 Mechanisms of sepsis-induced cardiac dysfunction. Crit. CareMed. 35, 1599–1608. (doi:10.1097/01.CCM.0000266683.64081.02)
115. Takayama K et al. 2005 Thromboxane A2 and prostaglandin F2α mediate inflammatorytachycardia. Nat. Med. 11, 562–566. (doi:10.1038/nm1231)
116. Booth LC, Ramchandra R, Calzavacca P, May CN. 2014 Role of prostaglandins indetermining the increased cardiac sympathetic nerve activity in ovine sepsis. Am. J. Physiol.Regul. Integr. Comp. Physiol. 307, R75–R81. (doi:10.1152/ajpregu.00450.2013)
117. Nakamura K. 2011 Central circuitries for body temperature regulation and fever. Am. J.Physiol. Regul. Integr. Comp. Physiol. 301, R1207–R1228. (doi:10.1152/ajpregu.00109.2011)
118. Kenney MJ, Ganta CK. 2014 Autonomic nervous system and immune system interactions.Compr. Physiol. 4, 1177–1200. (doi:10.1002/cphy.c130051)
119. Carbone JR. 2000 The neuroleptic malignant and serotonin syndromes. Emerg. Med. Clin.North Am. 18, 317–325. (doi:10.1016/S0733-8627(05)70127-9)
120. Rosenberg H, Davis M, James D, Pollock N, Stowell K. 2007 Malignant hyperthermia.Orphanet J. Rare Dis. 2, 21. (doi:10.1186/1750-1172-2-21)
121. Boyer EW, Shannon M. 2005 The serotonin syndrome. N. Engl. J. Med. 352, 1112–1120.(doi:10.1056/NEJMra041867)
122. Cohen S, Hunter CW, Yanni B, Striker P, Hijazi RH. 2006 Central anticholinergic syndromestrikes again. J. Clin. Anesth. 18, 399–400. (doi:10.1016/j.jclinane.2005.11.007)
123. Lugaresi E, Provini F, Cortelli P. 2011 Agrypnia excitata. Sleep Med. 12, S3–S10.(doi:10.1016/j.sleep.2011.10.004)
124. Lugaresi E, Provini F. 2001 Agrypnia excitata: clinical features and pathophysiologicalimplications. Sleep Med. Rev. 5, 313–322. (doi:10.1053/smrv.2001.0166)
125. Guaraldi P, Calandra-Buonaura G, Terlizzi R, Montagna P, Lugaresi E, Tinuper P, CortelliP, Provini F. 2011 Oneiric stupor: the peculiar behaviour of agrypnia excitata. Sleep Med. 12,S64–S67. (doi:10.1016/j.sleep.2011.10.014)
126. Montagna P, Cortelli P, Gambetti P, Lugaresi E. 1995 Fatal familial insomnia: sleep,neuroendocrine and vegetative alterations. Adv. Neuroimmunol. 5, 13–21. (doi:10.1016/0960-5428(94)00042-M)
127. Cortelli P et al. 1991 Cardiovascular dysautonomia in fatal familial insomnia. Clin. Auton.Res. 1, 15–21. (doi:10.1007/BF01826053)
128. Lugaresi E, Tobler I, Gambetti P, Montagna P. 1998 The pathophysiology of fatal familialinsomnia. Brain Pathol. 8, 521–526. (doi:10.1111/j.1750-3639.1998.tb00173.x)
129. Calandra-Buonaura G, Provini F, Guaraldi P, Pizza F, Cecere A, Barletta G, Lugaresi E,Pierangeli G, Cortelli P. 2013 Oculomasticatory myorhythmia and agrypnia excitata guidethe diagnosis of Whipple disease. Sleep Med. 14, 1428–1430. (doi:10.1016/j.sleep.2013.06.022)
130. Coccagna G, Mantovani M, Brignani F, Parchi C, Lugaresi E. 1972 Continuous recording ofthe pulmonary and systemic arterial pressure during sleep in syndromes of hypersomniawith periodic breathing. Bull. Physiopathol. Respir. (Nancy). 8, 1159–1172.
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

21
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
131. Mifflin S, Cunningham JT, Toney GM. 2015 Neurogenic mechanisms underlying the rapidonset of sympathetic responses to intermittent hypoxia. J. Appl. Physiol. 119, 1441–1448.(doi:10.1152/japplphysiol.00198.2015)
132. Fatouleh RH, Hammam E, Lundblad LC, Macey PM, McKenzie DK, Henderson LA,Macefield VG. 2014 Functional and structural changes in the brain associated with theincrease in muscle sympathetic nerve activity in obstructive sleep apnoea. Neuroimage Clin.6, 275–283. (doi:10.1016/j.nicl.2014.08.021)
133. Friedman O, Logan AG. 2009 The price of obstructive sleep apnea-hypopnea: hypertensionand other ill effects. Am. J. Hypertens. 22, 474–483. (doi:10.1038/ajh.2009.43)
134. Querejeta Roca G, Redline S, Claggett B, Bello N, Ballantyne CM, Solomon SD, ShahAM. 2015 Sex-specific association of sleep apnea severity with subclinical myocardialinjury, ventricular hypertrophy, and heart failure risk in a community dwelling cohort: theAtherosclerosis Risk in Communities–Sleep Heart Health Study. Circulation 132, 1329–1337.(doi:10.1161/CIRCULATIONAHA.115.016985)
135. May CN, Yao ST, Booth LC, Ramchandra R. 2013 Cardiac sympathoexcitation in heart failure.Auton. Neurosci. 175, 76–84. (doi:10.1016/j.autneu.2012.10.017)
136. Cortelli P, Parchi P, Sforza E, Contin M, Pierangeli G, Barletta G. 1994 Cardiovascularautonomic dysfunction in normotensive awake subjects with obstructive sleep apnoeasyndrome. Clin. Auton. Res. 4, 57–62. (doi:10.1007/BF01828839)
137. Lombardi C et al. 2008 Daytime sleepiness and neural cardiac modulation in sleep-relatedbreathing disorders. J. Sleep Res. 17, 263–270. (doi:10.1111/j.1365-2869.2008.00659.x)
138. Cortelli P, Lombardi C, Montagna P, Parati G. 2012 Baroreflex modulation duringsleep and in obstructive sleep apnea syndrome. Auton Neurosci. 169, 7–11. (doi:10.1016/j.autneu.2012.02.005)
139. McEwen BS. 2006 Sleep deprivation as a neurobiologic and physiologic stressor: allostasisand allostatic load. Metabolism 55, S20–S23. (doi:10.1016/j.metabol.2006.07.008)
140. Meier-Ewert HK, Ridker PM, Rifai N. 2004 Effect of sleep loss on C-reactive protein,an inflammatory marker of cardiovascular risk. J. Am. Coll. Cardiol. 43, 678–683.(doi:10.1016/j.jacc.2003.07.050)
141. Ogawa Y, Kanbayashi T, Saito Y, Takahashi Y, Kitajima T, Takahashi K, Hishikawa Y,Shimizu T. 2003 Total sleep deprivation elevates blood pressure through arterial baroreflexresetting: a study with microneurographic technique. Sleep 26, 986–989.
142. Zhong X, Hilton HJ, Gates GJ, Jelic S, Stern Y, Bartels MN, Demeersman RE,Basner RC. 2005 Increased sympathetic and decreased parasympathetic cardiovascularmodulation in normal humans with acute sleep deprivation. J. Appl. Physiol. 98, 2024–2032.(doi:10.1152/japplphysiol.00620.2004)
143. Takase B, Akima T, Satomura K, Ohsuzu F, Mastui T, Ishihara M, Kurita A. 2004 Effects ofchronic sleep deprivation on autonomic activity by examining heart rate variability, plasmacatecholamine, and intracellular magnesium levels. Biomed. Pharmacother. 58, S35–S39.(doi:10.1016/S0753-3322(04)80007-6)
144. Lusardi P, Zoppi A, Preti P, Pesce RM, Piazza E, Fogari R. 1999 Effects of insufficientsleep on blood pressure in hypertensive patients: a 24-h study. Am. J. Hypertens. 12, 63–68.(doi:10.1016/S0895-7061(98)00200-3)
145. Ferrie JE, Shipley MJ, Cappuccio FP, Brunner E, Miller MA, Kumari M, Marmot MG. 2007 Aprospective study of change in sleep duration: associations with mortality in the WhitehallII cohort. Sleep 30, 1659–1666.
146. Grimaldi D, Silvani A, Benarroch EE, Cortelli P. 2014 Orexin/hypocretin systemand autonomic control: new insights and clinical correlations. Neurology 82, 271–278.(doi:10.1212/WNL.0000000000000045)
147. Silvani A, Grimaldi D, Barletta G, Bastianini S, Vandi S, Pierangeli G, Plazzi G, Cortelli P.2013 Cardiovascular variability as a function of sleep-wake behaviour in narcolepsy withcataplexy. J. Sleep Res. 22, 178–184. (doi:10.1111/jsr.12007)
148. Cannon WB. 1942 ‘Voodoo’ death. Am. Anthropol. 44, 169–181. (doi:10.1525/aa.1942.44.2.02a00010)
149. Schwartz BG, French WJ, Mayeda GS, Burstein S, Economides C, Bhandari AK, CannomDS, Kloner RA. 2012 Emotional stressors trigger cardiovascular events. Int. J. Clin. Pract. 66,631–639. (doi:10.1111/j.1742-1241.2012.02920.x)
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from

22
rsta.royalsocietypublishing.orgPhil.Trans.R.Soc.A374:20150181
.........................................................
150. Edmondson D, Newman JD, Whang W, Davidson KW. 2013 Emotional triggers inmyocardial infarction: do they matter? Eur. Heart J. 34, 300–306. (doi:10.1093/eurheartj/ehs398)
151. Edmondson D, Cohen BE. 2013 Posttraumatic stress disorder and cardiovascular disease.Prog. Cardiovasc. Dis. 55, 548–656. (doi:10.1016/j.pcad.2013.03.004)
152. Task Force of the European Society of Cardiology and the North American Society of Pacingand Electrophysiology. 1996 Heart variability. Standards of measurements, physiologicalinterpretation and clinical use. Circulation 93, 1043–1065. (doi:10.1161/01.CIR.93.5.1043)
on August 25, 2018http://rsta.royalsocietypublishing.org/Downloaded from


















