

Protein Structure Prediction David Wild Keck Graduate Institute of Applied Life Sciences...
58
Protein Structure Prediction David Wild Keck Graduate Institute of Applied Life Sciences [email protected]
-
Upload
ursula-houston -
Category
Documents
-
view
216 -
download
0
Transcript of Protein Structure Prediction David Wild Keck Graduate Institute of Applied Life Sciences...

Summary
• Motivation
• Secondary Structure Prediction
• Tertiary Structure Prediction
• Sequence/Structure Approaches
• 3D profile
• Threading
• Ab-initio Approaches
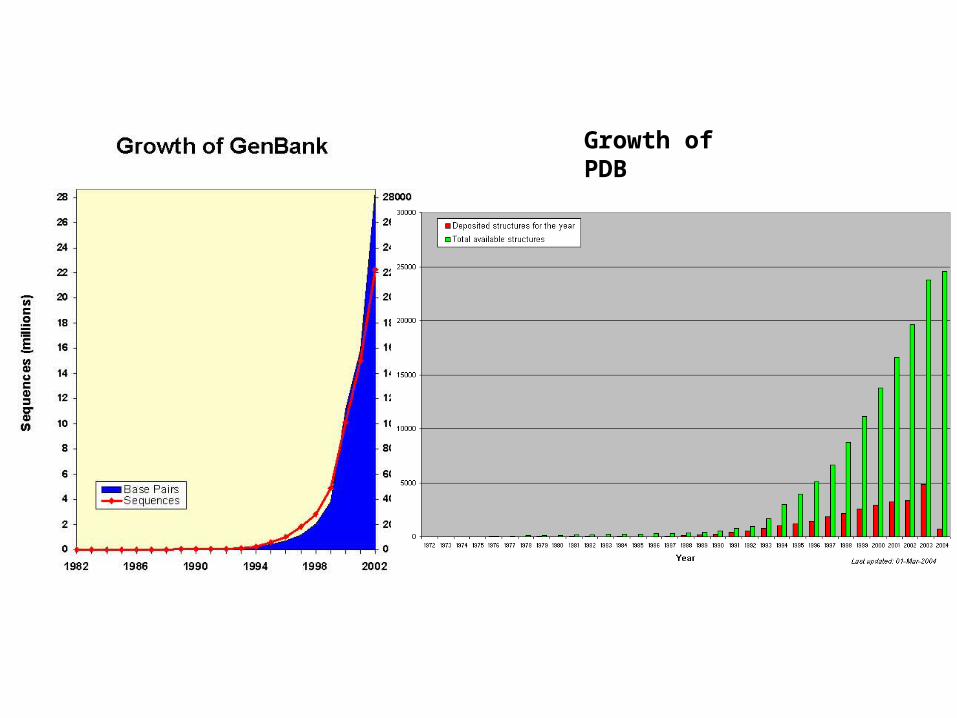
Growth of PDB

Functional assignment by homology: the function-homology gap
yeast data analyzed by GeneQuiz

Russell et al. J. Mol. Biol (1997) 269, 423-439

enterotoxin
homolog: cholera toxin80% ID 98/103 residues with rmsd 0.6A
remote homolog: toxic shock syndrome toxin; no sequence similarity but 35/95 residues with rmsd 2.4A
analog:tRNA synthetase; no sequence similarity but 41/103 residues with rmsd 2.2A no known functional similarity

From Hegyi and Gerstein(1999)

• Active site formed by loops between the carboxy end of the -strands and the amino end of the -helices at one end of the barrel
From Branden and Tooze (1999)

From Hegyi and Gerstein (1999)


Baker and Sali (2000)

Central Dogma
“The 3D structure of a protein is determined by its sequence and its environment without the obligatory
role of extrinsic factors”
• Anfinsen (1973) - renaturation of ribonuclease
• Ignores role of chaperones, disulfide interchange enzymes etc


Dominant Effects in Protein Folding
• Net protein stability - diverse chemical properties of main and side chain atoms give rise to interplay of non-covalent and entropic effects
• Hydrophobic effect - non-polar core• Atomic packing - van der Waals interactions favor
close packing• Conformational entropy - freezing of rotamers• Electrostatic effects - ion pairs and H-bonds• Disulfide bridges

Primary Secondary Tertiary

Secondary Structure Prediction
• History and Context
• Chou & Fasman
• Lim
• Garnier-Osguthorpe-Robson
• Comparison of Methods
• Newer Approaches

Secondary Structure Prediction by Eye
• Position of insertions and deletions probable loop
• Conserved Gly/Pro probable loop
• Short runs of conserved hydrophobics buried -strand
• i, i+2, i+4 pattern of conserved residues surface -strand
• i, i+3,i+4,i+7 conserved pattern surface helix
Helix
Edge strand
Buried strand

From Branden and Tooze (1999)

Single Sequence Methods
Chou & Fasman 1974
• Propensities of formation based upon frequency of occurrence
• Generate tables for , , turn & random coil
• Strong/weak/indifferent formers & breakers
• Rules for nucleation, propagation & termination
• 15 protein database - 50% accuracy!

The Lim Method (1974)
• Theory based on packing of polypeptide chains
e.g.: -helices that make contact with the main protein body need a hydrophobic side
• Hydrophobic residues must face internally and pack closely together
• Method defines hydrophobics/hydrophilics and passageway residues
• Advantage: rules have a clear basis in protein chemistry theory
• Disadvatange: rules complex & difficult to understand

-helix -strand
strong former glu, ala, leu met, val, ile
former his, met, gln, trp, val, phe cys, tyr, phe, gln, leu, thr, trp
weak former leu, ile ala
indifferent former asp, thr, ser, arg, cys arg, gly, asp
breaker asn, tyr lys, ser, his, asn, pro
strong breaker pro, gly glu

Single Sequence Methods
Garnier, Osguthorpe, Robson (GOR), 1978
• Window of 17 residues (i-8 i i+8)
• 4 states - predicted structure is highest value summed over window
• “Information theoretic” approach
• Single sequence GORI - 55% accuracy
• GORIII - pair information - correlate the type of residues in a window with the residue to be predicted
• Sensitive to database size - getting better all the time

GOR I Example
• For alanine
240 in helix, 150 not in helix, total 390 residues
• For all residues
780 in helix (H), 1050 not in helix (~H), total 1830
P(S=H|A) = 240/390 = 0.615
P(S=~H|A) = 150/390 = 0.385
P(S=H) = 780/1830 = 0.426
P(S=~H) = 1050/1830 = 0.573
I(S=H:~H;A) = ln(0.615/0.385) - ln(0.426/0.573) (log-odds ratio)
= 0.4683 - 0.2964
= 0.7647

Neural networks applied to SS prediction
• Use known structures as target function
• Single sequence methods not that successful, but better than GOR (Qian & Sejnowski, 1988 ~ 63%)
• Adding information from an alignment substantially improves accuracy
• Disadvantage: one loses sight of original problem due to ‘black box’ nature of prediction method
• Large number of parameters


Qian and Sejnowski (1988)
Input
Prediction of center residue X
13 residue window-6,-5,-4,-3,-2,-1, X, +1,+2,+3,+4,+5,+6
Input Layer13 groups, each of 21 units
(20 residues plus space)
Hidden layer
Output Layer
3 groups (H, E, C)

• Binary coding of amino acid residues
– 20 residues require 5 bits
– for instance
ala = 00001
cys = 00010
asp = 00011
…
trp = 10100
• Could alternatively encode 5 properties, e.g.: hydrophobicity, side chain size etc...

PHD Neural NetworkRost & Sander, 1993
• Uses multiple independent neural networks as prediction engine
• Balanced training - present network with one structural class at a time
• Addition of evolutionary information improves prediction quality
How…
1. Sequence to structure - input coded as a profile, trained against known structure
2. Structure to structure - predicted SS trained against known structure
3. Jury decision - numerical average over number of different level 2 networks

Profile/PSSM
• Position Specific Scoring Matrix, or weight matrix, is calculated based on observed frequencies in a column
GCGGTGATAATGGTTGCATGTTGGGTATATTTGACTATGGATGCATACACTATAGGTGTGTGCAGTAAGATACAAATGGCATGGTTATAGTATGCCCATG
Acknowledgement: Mike Gribskov

A 2 95 26 59 51 1C 9 2 14 13 20 3G 10 1 16 15 13 0T 79 3 44 13 17 96 T A T A A T
Weight Matrix Methods
• Position specific scoring matrix (PSSM)• Feature is represented as a matrix with a score for
every possible character
• A simple weight matrix for the bacterial promoter -10 region, values here are simply % frequencies
Acknowledgement: Mike Gribskov



From Baldi and Brunak (2001)

Nearest Neighbor Methods
Salamov & Solovyev, NSSP 1995
• Use database of proteins of known structure
• Match each segment of query sequence against all sequences in database
• Choose secondary structure state of the majority of its neighbors as the prediction
• Neighbors are decided upon by using amino acid substitution tables and scoring tables

Indentifying factors that affect secondary structure
King & Sternberg, DSC 1996
• Relative aa position in chain
• Treatment of insertions/deletions
• Hydrophobic moment
• %aa content
• not a ‘black box’ technique

CASP2 - Blind Prediction of Protein Secondary Structure
0 10 20 30 40 50 60 70 80
Accuracy
DSC-M
GOR-S
SSPRED-M
NNPRED-S
NNSSP-M
SSP-M
PHD-M
Method
Server Predictions M=Multiple S=Single
Zemla et al. Proteins (1997) Suppl. 1, 140-150

Issues
• Definition of secondary structure from 3D coordinates is not exact
• Different algorithms to define secondary structure
DSSP, STRIDE, DEFINE, Author, P-Curve
give different definitions:
DSSP/Stride Stride/Define DSSP/Define
95% 74% 73%
• Definition itself is open to interpretation - there are more than 3 states defined:
H, E, G, I, T, C, B, S H, E, C

-sheets are formed by long range interactions

Generative probabilistic models(Schmidler et al. (2000); Chu et al. (2004))

Tertiary Structure Prediction
• Comparative modeling–Homology modeling• Fragment-based
–COMPOSER–SWISS-MODEL
• 3D distance constraints–MODELER
• Fold Recognition/Threading/Inverse Folding
• Proteins may have undetectable sequence similarity but striking structural similarity.• Glimmers in the twilight zone (Doolittle, 1987)

0
20
40
60
80
100
120
0 20 40 60 80 100 120
%ID
%Res-Res (in reliable regions)
Sequence Alignment Accuracy: %correctly aligned residues vs. %sequence identity
Saqi et al. Prot. Eng (1998)

Russell et al. J. Mol. Biol (1997) 269, 423-439

Fold Recognition Methods
• Sequence profile– PSI-BLAST– HMM– Environmental PSSM
• Structural profile– 3D-1D profile
• Threading– Pair potential based fold recognition

Ab initio/De Novo FoldingCombinatorial approaches
• Secondary structure prediction + Docking
Energy minimization
Monte Carlo simulation• Fragments of highly resolved protein structures are joined
together and the feasibility of the fold is evaluated with a potential function.
Lattice simulations
– Still mainly developer based usage.

From Higgins and Taylor (2000)



Bowie, Luthy and Eisenberg (1991)


Threader Jones et al 1999
• Structural role of residue described in terms of interactions
• ‘Network’ of pairwise interatomic energy terms (potentials) from a statistical analysis of proteins of known structure and inverse Boltzman equation (Sippl 1990) used as sequence-structure compatibility function

For specified atoms in a pair of residues {a,b}, with a sequence separation of k and distance interval s, the potential is given by
mab is the number of pairs ab observed at sequence separation k
is the weight of each observation
fabk(s) is the equivalent frequency of occurrence of
residue pair ab.
fk(s) is the frequency of occurrence of all residue pairs at sequence separation k and separation distance s
Potentials corresponding to short (sequence separation , k < 11), medium (11 k 22), and long (k > 30) range interactions, have been utilized.
])()(
1[)1ln(sfsf
mRTmRTEk
abk
abababk +−+=Δ

Ab-initio ApproachesLINUS (Srinivasan & Rose, 1995)
Folding by “Hierarchical Condensation”
Cstart
NΔ6(
jcycle
1 ( Generate Trial conformation C*1. Randomly choose backbone conformation2. Bump check C*3. Calculate energy of C*, U(C*)4. If U(C*)<U(C) or x < e- ΔE ,
where is x is random and 0<x<1 then C = C*
N-1Step
2{ }))Fragments of 50 residues, interaction interval 6<Δ<48
Simple potential:
• Contact energy
• H-bonding
• Main chain ‘torsional potential’ ( > 0 except for glycine)

ROSETTA Simons et al, 1997
• Metropolis Monte Carlo simulated annealing procedure
• 3 and 9 residue fragments of known structures with local sequences similar to the target sequence
•Potential function - sequence dependent terms hydrophobic burial electrostatics and disulfide bonding,
• sequence independent terms hard sphere packing, alpha-helix and beta-strand packing collection of beta-strands in beta-sheets

FRAGFOLD Jones (1997, 2001)
Library of super-secondary structures fragments
-hairpin
motif
From Branden and Tooze (1999)

Folding Proteins with Boltzmann Learning Rule
• NOT traditional ab initio folding
• Learn the potentials that maximize the probability of known native folds
• Then, use learned potential for future folding
(Ole Winter & Anders Krogh 2003)

Boltzmann Learning Rule
The probability of nativei fold given sequencei and the model parameters :
( ),sequencenative iiP
( )∑∇+=i
iioldnew P θηθθ θ ,sequencenativeln
The updating of parameters with the rate .

Potentials
• Lennard-Jones between atoms X and Y
• Hydrogen bonds
• Others
∑⎪⎭
⎪⎬⎫
⎪⎩
⎪⎨⎧
⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟⎟
⎠
⎞⎜⎜⎝
⎛=
612
65XY
XY
XY
XYXYLJ rr
Eσσ
ε
( )∑⎪⎭
⎪⎬⎫
⎪⎩
⎪⎨⎧
⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟⎟
⎠
⎞⎜⎜⎝
⎛=
1012
2anglesHO
HO
HO
HOHOhb rr
uEσσ
ε
Total of more than 1000 model parameters to learn

Assessment
• LiveBench
• CAFASP3 Servers – Evaluation Results
• CASP5– Evaluation Results


















