
Modular Synthesis of Chiral -Aminophosphine PN Ligands and ... · Modular Synthesis of Chiral...
177
Modular Synthesis of Chiral -Aminophosphine P,N- Ligands and Their Applications in Asymmetric Catalysis by Yixiong Song A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Chemistry University of Toronto © Copyright by Yixiong Song 2015
Transcript of Modular Synthesis of Chiral -Aminophosphine PN Ligands and ... · Modular Synthesis of Chiral...

Modular Synthesis of Chiral -Aminophosphine P,N-Ligands and Their Applications in Asymmetric
Catalysis
by
Yixiong Song
A thesis submitted in conformity with the requirements for the degree of Master of Science
Graduate Department of Chemistry University of Toronto
© Copyright by Yixiong Song 2015

ii
Modular Synthesis of Chiral -Aminophosphine P,N-Ligands and Their
Applications in Asymmetric Catalysis
Yixiong Song
Master of Science
Graduate Department of Chemistry
University of Toronto
2015
Abstract
A series of chiral -aminophosphine ligands bearing different carbon backbones and
electronically differentiated diarylphosphino groups were prepared using a modular approach.
These P,N-ligands were found to induce a modest level of enantioselectivities in the Pd-
catalyzed asymmetric decarboxylative allylation reaction. Thiourea-phosphine bifunctional
catalysts derived from the chiral -aminophosphine building blocks were prepared and applied to
the asymmetric Morita-Baylis-Hillman (MBH) reaction of methyl acrylate and 4-
nitrobenzaldehyde. The electronically unmodified diarylphosphino-thiourea was found to be
optimal for achieving high activity and enantioselectivity in this particular MBH reaction. We
also reported the synthesis of a P-chiral C2-symmetric bisphosphine ligand. However, the utility
of this Trost-type ligand remains to be explored in the future.

iii
Acknowledgments

iv
Table of Contents
Chapter 1. Modular Synthesis of -Aminophosphine P,N-Ligands and Their Applications in
the Pd-Catalyzed Asymmetric Decarboxylative Allylation Reaction ..................................... 1
1 Introduction ..................................................................................................................... 1
1.1 Overview of P,N-ligands in Asymmetric Catalysis .............................................. 1
1.2 Decarboxylative Allylation Reaction .................................................................10
2 Objectives .......................................................................................................................18
3 Results and Discussion ....................................................................................................19
3.1 Synthesis of -Substituted -Aminophosphines and Their appplications in the
Pd-Catalyzed Asymmetric Decarboxylative Allylation Reaction ........................19
3.2 Attempted Synthesis of -Disubstituted -Aminophosphines ...........................26
3.3 Attempted C–H Activation of (R)-2-Phenylglycinol ............................................31
4 Experimental ...................................................................................................................36
4.1 Procedures and Compounds................................................................................36
4.1 1H NMR,
13C NMR and
31P NMR Spectra ..........................................................53
Chapter 2. Synthesis of Bifunctional Thiourea-Phosphine Organocatalysts and Their
Applications in the Asymmetric Morita-Baylis-Hillman (MBH) Reaction ............................83
1 Introduction ....................................................................................................................83
1.1 Mechanism of the MBH Reaction ......................................................................83
1.2 The Asymmetric MBH Reaction........................................................................87
1.3 The Asymmetric aza-MBH Reaction ..................................................................91
1.4 Asymmetric Transformations Related to the MBH Reaction ................................95
2 Objectives .......................................................................................................................98
3 Results and Discussion ....................................................................................................98
4 Conclusion and Future Work ......................................................................................... 105

v
5 Experimental ................................................................................................................. 106
5.1 Procedures and Compounds.............................................................................. 106
5.2 1H NMR,
13C NMR and
31P NMR Spectra ........................................................ 116
5.3 X-Ray Crystallography Data ............................................................................ 139

vi
List of Abbreviations
(m) Medium
(s) Strong (w) Weak
°C Degrees Celsius
Ac Acetate
acac Acetylacetone
Ar Aryl
Boc tert-Butyloxycarbonyl
Bn Benzyl
Bz Benzoyl
cod 1,5-Cyclooctadiene
DART Direct Analysis in Real Time dba Dibenzylideneacetone
DCC Dicyclohexylcarbodiimide
DMAP 4-Dimethylaminopyridine
DMF Diastereomeric ratio
ee Enantiomeric excess
er Enantiomeric ratio
eq. Equivalent
ESI Electro-spray ionisation
Et Ethyl
EWG Electron withdrawing group
g Grams HRMS High resolution mass spectrometry
i-Pr iso-Propyl
IR Infra red
L Ligand
Leu Leucine
M Molar
m- meta m/z Mass/charge ratio
Me Methyl
MeCN Acetonitrile mg Milligrams
MHz Mega hertz
min. Minutes
mL Milliliters
n-Bu normal-Butyl
nbd Norbornadiene
NMR Nuclear magnetic resonance
Nu Nucleophile
o- ortho p- para PMP para-methoxyphenyl

vii
TBAB Tetrabutylammonium bromide
TBAB Tetrabutylammonium chloride
TBS tert-Butyldimethylsilyl
t-Bu tert-Butyl
Tf Triflate
THF Tetrahydrofuran Ts Tosyl
v/v Volume per volume

viii
List of Tables
Table 1. Scope for secondary phosphine oxides .......................................................................24
Table 2. Scope for -substituted--aminophosphine oxides .....................................................24
Table 3. Scope for -substituted--aminophosphines...............................................................25
Table 4. Screening of P,N-ligands for decarboxylative allylic alkylation ..................................26
Table 5. Decomposition products isolated in the reduction of aminophosphine oxide ...............30
Table 6. Scope for thiourea-phosphine catalysts ......................................................................98
Table 7. Screening of bifunctional catalysts in the MBH reaction ............................................98
Table 8. Screening of P-chiral bifunctional catalysts in the MBH reaction ............................. 102
Table 9. Screening of P-chiral C2-symmetrical bisphosphine ligand in Pd-catalyzed AAA
reaction ................................................................................................................................ 103

ix
List of Figures
Figure 1. X-ray crystal data Pd -allyl complex ....................................................................... 2
Figure 2. The exo-endo equilibrium of the Pd-allyl complex ..................................................... 3
Figure 3. 31
P NMR spectra of ring-opening reaction using KOtBu and NaOtBu .......................23
Figure 4. X-ray crystal structure of thiourea-phosphine-borane 2.28a .................................... 100

x
List of Schemes
Scheme 1. Asymmetric allylic substitution reactions of 1,3-dialkylallyl acetates using PHOX
ligand ...................................................................................................................................... 2
Scheme 2. Asymmetric allylic substitution of 1,3-dimethylallyl acetate with dimethyl malonate 4
Scheme 3. Asymmetric allylic substitution of cyclic substrates with dimethyl malonate............. 4
Scheme 4. Intermolecular asymmetric Heck reaction using PHOX and BINAP ligands ............. 5
Scheme 5. Ir-catalyzed asymmetric hydrogenation of non-functionalized olefins ....................... 5
Scheme 6. Asymmetric hydroboration-oxidation of arylalkenes using Rh-QUINAP .................. 5
Scheme 7. Asymmetric diboration-oxidation of olefins using Rh-QUINAP ............................... 6
Scheme 8. Asymmetric three component condensation employing QUINAP and its analog ....... 6
Scheme 9. Asymmetric Kumada coupling reaction using -aminophosphine ............................. 7
Scheme 10. Cu-catalyzed conjugated addition of diethylzinc to enones using iminophosphine ... 8
Scheme 11. Ru-catalyzed asymmetric tandem Michael addition/hydrogenation of cyclic enone . 8
Scheme 12. Fe-catalyzed hydrogenation of esters and N-heterocycles ....................................... 9
Scheme 13. Fe-catalyzed asymmetric hydrogenation of ketones and activated imines ................ 9
Scheme 14. The Tsuji reaction ................................................................................................10
Scheme 15. Proposed catalytic cycle of the decarboxylative allylic alkylation of allyl enol
carbonates and allyl β-keto esters ............................................................................................11
Scheme 16. The initial report of enantioselective Tsuji reaction using Trost P,P-ligand and
PHOX P,N-ligand ...................................................................................................................11
Scheme 17. Asymmetric decarboxylative allylic alkylation using Trost ligands ........................12
Scheme 18. Asymmetric decarboxylative allylic alkylation using PHOX 1.4 and 1.16 ..............13

xi
Scheme 19. Synthesis of chiral -fluoroketones through decarboxylative allylic alkylation ......14
Scheme 20. Enantioselective decarboxylative enolate alkylation cascade .................................14
Scheme 21. Ir-catalyzed allylic alkylation of -substituted β-ketoesters ...................................15
Scheme 22. Sequential allylic alkylation catalyzed by Ir and Pd complexes ..............................16
Scheme 23. Proposed mechanism by Stoltz and Goddard for asymmetric decarboxylative allylic
alkylation using PHOX ..........................................................................................................17
Scheme 24. Proposed mechanism by Trost for asymmetric decarboxylative allylic alkylation
using Trost ligand ...................................................................................................................17
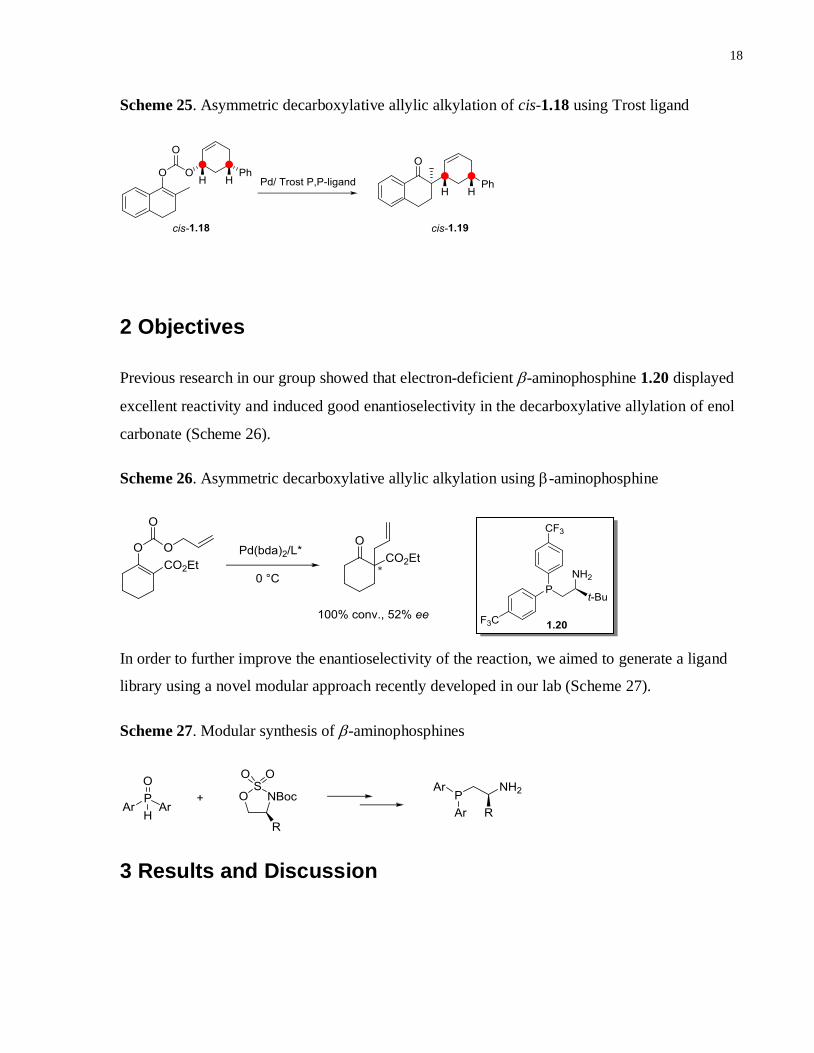
Scheme 25. Asymmetric decarboxylative allylic alkylation of cis-1.18 using Trost ligand ........18
Scheme 26. Asymmetric decarboxylative allylic alkylation using -aminophosphin .................18
Scheme 27. Modular synthesis of -aminophosphines .............................................................18
Scheme 28. Conventional synthesis of chiral -aminophosphines ............................................19
Scheme 29. Previous literature reports on synthesis of chiral -aminophosphines via tosyl
displacement...........................................................................................................................20
Scheme 30. Synthesis of chiral -aminophosphines via cyclic sulfamidates..............................21
Scheme 31. The first successful ring-opening of cyclic sulfamidate with secondary phosphine
oxide ......................................................................................................................................22
Scheme 32. Tautomerism of secondary phosphine oxides ........................................................22
Scheme 33. Competing reaction pathways in the ring-opening of cyclic sulfamidate with
secondary phosphine oxide......................................................................................................23
Scheme 34. Reductive amination of P,N-ligand .......................................................................26

xii
Scheme 35. Attempted ring-opening of 1,2-disubstituted cyclic sulfamidate with secondary
phosphine oxide ......................................................................................................................27
Scheme 36. Decomposition pathway of benzylic tosylate ........................................................27
Scheme 37. Ring-opening of cyclic sulfamidates with fluoride ................................................28
Scheme 38. Ring-opening and P-alkylation of cyclic sulfamidates ...........................................29
Scheme 39. Attempted synthesis of -disubstituted -aminophosphine derived from 1-amino-
2-indanol 1.42.........................................................................................................................31
Scheme 40. -C−H arylation of alanine ..................................................................................32
Scheme 41. Hypothetical divergent synthesis of -substituted -aminophosphines via C–H
activation ................................................................................................................................33
Scheme 42. Known C–H activation of primary benzylamines .................................................34
Scheme 43. Synthesis of starting materials for C–H activation .................................................34
Scheme 44. Attempted C–H iodination using I2 .......................................................................35
Scheme 45. Regeneration of Pd(OAc)2 from PdI2 ....................................................................35
Scheme 46. Attempted C–H iodination using IOAc .................................................................36
Scheme 47. The Morita-Baylis-Hillman reaction .....................................................................83
Scheme 48. Mechanism of MBH reaction proposed in 1980s ...................................................84
Scheme 49. Kinetic study of MBH reaction by Hill and Issac ..................................................84
Scheme 50. Kinetic study performed by McQuade et al. ..........................................................85
Scheme 51. Mechanism of MBH reaction revised by McQuade and Aggarwal .........................85
Scheme 52. Asymmetric MBH reaction using a quinidine derivative as a bifunctional catalyst .87

xiii
Scheme 53. Asymmetric MBH reaction using quinidine binaphthyl thiourea-tertiary amine 2.2 88
Scheme 54. Asymmetric MBH reaction using cyclohexyl thiourea-phosphine 2.3 ....................88
Scheme 55. Asymmetric MBH reaction using threonine derived thiourea-phosphine 2.4 ..........89
Scheme 56. Asymmetric MBH reaction using acid-base co-catalyst systems ...........................90
Scheme 57. Asymmetric aza-MBH reaction using quinidine derivatives ..................................91
Scheme 58. Asymmetric aza-MBH reaction using BINOL derived bifunctional catalyst ...........92
Scheme 59. Asymmetric aza-MBH reaction using thiourea/DABCO cocatalyst system ............93
Scheme 60. Asymmetric aza-MBH reaction using phosphine-sulfonamide catalyst 2.12 ...........93
Scheme 61. Enantioselective [3+2] annulation of allenoate with acrylate..................................94
Scheme 62. Mechanism of [3+2] annulation of allenoate with acrylate .....................................95
Scheme 63. Asymmetric Michael addition of oxindoles catalyzed by chiral phosphine .............95
Scheme 64. Mechanism of asymmetric Michael addition catalyzed by chiral phosphine ...........96
Scheme 65. Asymmetric Michael addition reaction and aza-Henry reaction catalyzed by chiral
phosphines..............................................................................................................................96
Scheme 66. Asymmetric MBH reaction catalyzed by amino acid based thiourea-phosphines ....97
Scheme 67. P-chiral phosphino-thiourea catalysts ..................................................................99
Scheme 68. Synthesis of unsymmetrical secondary phosphine oxide ........................................99
Scheme 69. Synthesis of P-chiral phosphine-thiourea catalysts .............................................. 101
Scheme 70. Synthesis of P-chiral C2-symmetrical bisphosphine ligands ................................. 102
Scheme 71. AAA reactions using Trost ligand ...................................................................... 102

xiv
Scheme 72. Rh-catalyzed asymmetric hydrosilylation of ketones ........................................... 105

1
Chapter 1
Modular Synthesis of -Aminophosphine P,N-Ligands and Their Applications in the Pd-Catalyzed
Asymmetric Decarboxylative Allylation Reaction
1 Introduction
1.1 Overview of P,N-Ligands in Asymmetric Catalysis
P,N-ligands are an important class of ligands for asymmetric catalysis. P,N-ligands are highly
effective in promoting a variety of metal-catalyzed enantioseletive transformations, including
asymmetric allylic alkylation, asymmetric hydrogenation, and enantioselective diboration of
alkenes.1 P,N-ligands contain two electronically distinct donor atoms: a “hard” nitrogen atom
with -donor property and a “soft” phosphorus atom that can act as botha -donor and a -
acceptor. This combination of mixed donors exerts regiocontrol when applied to the context of
-allyl metal complexes where the allylic terminus trans to the phosphorus donor is
preferentially attacked by the incoming nucleophile.1c
The thermodynamic trans effect of P,N-
ligands is reflected by the different Pd–C distances in the X-ray crystal structure (Figure 1).1a
PHOX ligands developed independently by the groups of Helmchen, Pfaltz and Williams,2 give
rise to reactive palladium complexes in the asymmetric allylic substitution reactions of
symmetric allylic acetates, providing up to 99% ee for 1,3-diphenylallyl acetate with dimethyl
1 a) Pfaltz, A.; Drury, W. J. Proc. Natl. Acad. Sci. USA. 2004, 101, 5723–5726. b) ern nde , E.; Guiry, P.
2 a) Helmchen, G.; Kudis, S.; Sennhenn, P.; Steinhagen, H. Pure Appl. Chem. 1997, 69, 513−518. b)
Pfaltz, A. Acta Chem. Scand. B 1996, 50, 189−194. c) Williams, J. M. J. Synlett 1996, 705−710.

2
malonate. However, diminishing enantioselectivities were observed for smaller alkyl groups.
Interestingly, there was no selectivity for cyclohexene-2-yl acetate using this ligand (Scheme 1).3
Figure 1. X-ray crystal data Pd -allyl complex
3 Helmchen, G.; Pfaltz, A. Acc. Chem. Res.2000, 33, 336−345.
3
Scheme 1. Asymmetric allylic substitution reactions of 1,3-dialkylallyl acetates using PHOX
ligand
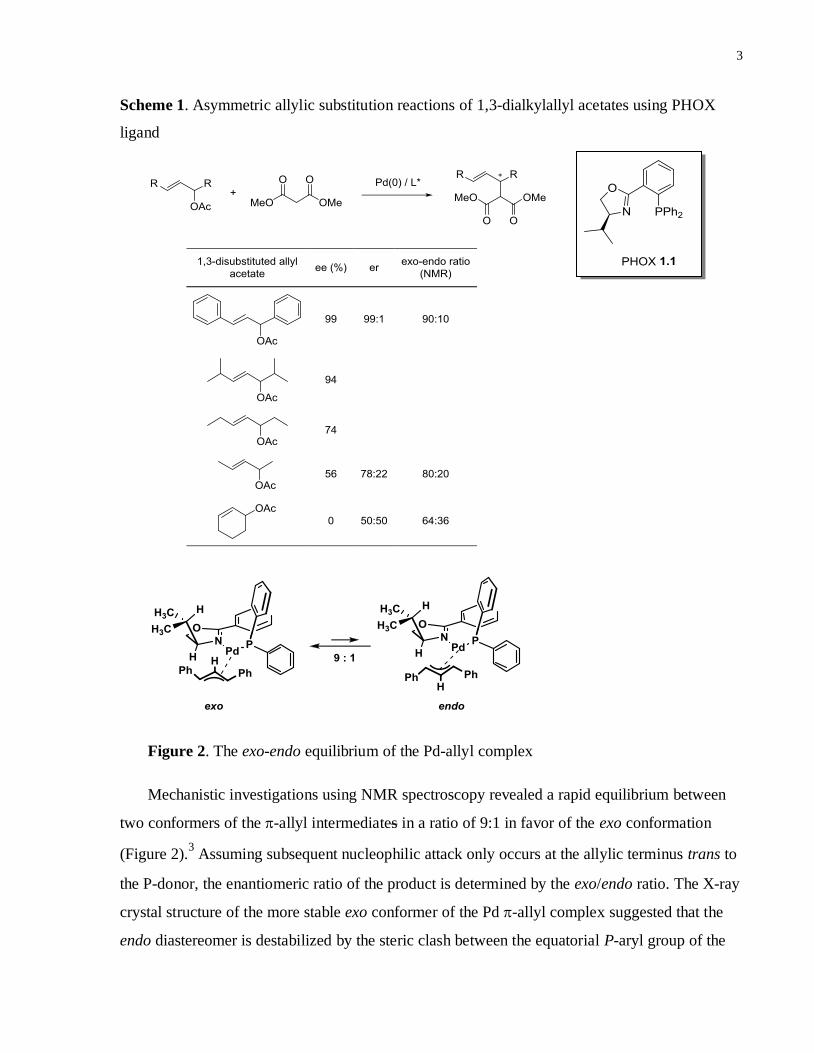
Figure 2. The exo-endo equilibrium of the Pd-allyl complex
Mechanistic investigations using NMR spectroscopy revealed a rapid equilibrium between
two conformers of the -allyl intermediates in a ratio of 9:1 in favor of the exo conformation
(Figure 2).3 Assuming subsequent nucleophilic attack only occurs at the allylic terminus trans to
the P-donor, the enantiomeric ratio of the product is determined by the exo/endo ratio. The X-ray
crystal structure of the more stable exo conformer of the Pd -allyl complex suggested that the
endo diastereomer is destabilized by the steric clash between the equatorial P-aryl group of the

4
PHOX ligand and the phenyl group of the allyl substrate.5 The energy difference between the
two diastereomers diminishes as the steric demand of the allyl substituent decreases and thus
results in the erosion of exo/endo ratio and the enantioselectivity. The modified ligand 1.2 gave
90% ee for 1,3-dimethylallyl acetate (Scheme 2), while 1.3 provided excellent ee for cyclic
substrates (Scheme 3).3
Scheme 2. Asymmetric allylic substitution of 1,3-dimethylallyl acetate with dimethyl malonate
Scheme 3. Asymmetric allylic substitution of cyclic substrates with dimethyl malonate
In addition to their application in the asymmetric allylic substitution reactions, PHOX ligands
have shown great potential in intermolecular asymmetric Heck reaction, providing significant
improvement in yields and/or enantioselectivities compared to bisphosphine ligands (Scheme
4).4 Lastly, the Ir-PHOX system is uniquely effective in the asymmetric hydrogenation of non-
functionalized olefins. Ir-PHOX system can be complementary to Rh- and Ru-bisphosphine
catalyst systems that are selective toward functionalized olefins containing coordinating groups
such as amides and carboxylic acids in close proximity to the double bond (Scheme 5).5
4 McCartney, D.; Guiry, P. J. Chem. Soc. Rev. 2011, 40, 5122−5150.
5 a) Roseblade, S. J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402−1411. b) erendel mies
i gue , M.; Andersson, P. G. Chem. Rev. 2014, 114, 2130−2169.

5
Scheme 4. Intermolecular asymmetric Heck reaction using PHOX and BINAP ligands
Scheme 5. Ir-catalyzed asymmetric hydrogenation of non-functionalized olefins
The axially chiral P,N-ligand, QUINAP, that was introduced concurrently with the PHOX
ligands in 1993, was successfully employed in the asymmetric hydroboration-oxidation of
styrene derivatives using catecholborane, achieving good to excellent enantioselectivity for a
wider range of alkenes compared to the initial Rh-BINAP system (Scheme 6).1a
Scheme 6. Asymmetric hydroboration-oxidation of arylalkenes using Rh-QUINAP
In 2003, Morken’s group reported a highly enantioselective syn-addition of
bis(catecholato)diboron across trans-disubstituted alkenes.6 However, the enantioselectivity was
6 Morgan, J. B.; Miller, S. P.; Morken, J. P. J. Am. Chem. Soc. 2003, 125, 8702−8703.
6
less satisfactory for other classes of alkene substrates (Scheme 7). In the same year, Knochel’s
group developed a powerful three-component coupling reaction between an aldehyde, a
secondary amine, and an alkyne using Cu-QUINAP as the catalyst system, giving good yields
and ees when dibenzylamines were coupled with a wide range of aliphatic aldehydes and alkynes
(Scheme 8). More recently, another variant of QUINAP 1.8 was developed to address some of
the challenging aldehyde substrates reported previously (Scheme 8). 7
Scheme 7. Asymmetric diboration-oxidation of olefins using Rh-QUINAP
Scheme 8. Asymmetric three component condensation employing QUINAP and its analog 1.8
7 a) Gommermann, N.; Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem. Int. Ed. 2003, 42, 5763−
5766. b) Gommermann, N.; Knochel, P. Chem. Eur. J. 2006, 12, 4380−4392. c) Cardoso, F. S. P.;
Abboud, K. A.; Aponick, A. J. Am. Chem. Soc. 2013, 135, 14548−14551.
7
-Aminophosphines, which can be readily prepared from chiral amino acids, were first used by
Hayashi and Kumada in the asymmetric Kumada coupling reaction (Scheme 9). The hemilabile
property of the amine group proved to be critical for obtaining high enantioselectivity. During
the enantio-discriminatig step, the dimethylamino group on the ligand is believed to dissociate
from the nickel centre and bind preferentially with one of the enantiomers of Grignard reagent.
Subsequent transmetallation and reductive elimination give enantio-enriched products in up to
94% ee (Scheme 9).8
Scheme 9. Asymmetric Kumada coupling reaction using -aminophosphine ligand 1.9
Iminophosphine 1.10 was shown to be a competent chiral ligand for the Cu-catalyzed conjugated
addition of diethylzinc to enones (Scheme 10).9
8 a) Hayashi, T.; Fukushima, M.; Konishi, M.; Kumada, M. Tetrahedron Lett. 1980, 21, 79−82. b)
Grushin, V. V. Chem. Rev. 2004, 104, 1629−1662.
9 Saitoh, A.; Morimoto, T.; Achiwa, K. Tetrahedron: Asymmetry 1997, 8, 3567.

8
Scheme 10. Cu-catalyzed conjugated addition of diethylzinc to enones using iminophosphine
1.10
Morris and coworkers developed a ruthenium hydride borohydride complex 1.11 containing -
aminophosphine ligand and demonstrated its utility in the highly enantioselective tandem
Michael addition /H2-hydrogenation of the cyclic enone (Scheme 11).10
Scheme 11. Ru-catalyzed asymmetric tandem Michael addition/hydrogenation of cyclic enone
More recently, Guan et al. and Jones et al. reported the use of an earth-abundant and nontoxic
iron-based catalyst bearing a PNP-pincer ligand in the hydrogenation of esters and N-
heterocycles (Scheme 12). 11
10 a) Guo, R.; Morris, R. H.; Song, D. J. Am. Chem. Soc. 2005, 127, 516−517. b) Guo, R.; Lough, A. J.;
Morris, R. H.; Song, D. Organometallics 2004, 23, 5524−5529. c) Abdur-Rashid, K.; Guo, R.; Lough, A.
J.; Morris, R. H.; Song. Adv. Synth. Catal. 2005, 347, 571−579.
11 Chakraborty, S.; Dai, H.; Bhattacharya, P.; Fairweather, N. T.; Gibson, M. S.; Krause, J. A.; Guan, H. J.
Am. Chem. Soc. 2014, 136, 7869−7872. b) Chakraborty, S.; Brennessel, W. W.; Jones, W. D. J. Am.
Chem. Soc. 2014, 136, 8564−8567.
9
Scheme 12. Fe-catalyzed hydrogenation of esters and N-heterocycles
Morris et al. achieved good to excellent enantioselectivities in the asymmetric hydrogenation of
ketones and activated imines employing an iron precatalyst 1.13 containing unsymmetrical N ’
pincer ligand (Scheme 13).12
Scheme 13. Fe-catalyzed asymmetric hydrogenation of ketones and activated imines
12 a) Lagaditis, P. O.; Sues, P. E.; Sonnenberg, J. F.; Wan, K. Y.; Lough, A. J.; Morris, R. H. J. Am.
Chem. Soc. 2014, 136, 1367−1380. b) Sonnenberg, J. F.; Lough, A. J.; Morris, R. H. Organometallics
2014, 33, 6452−6465.

10
In addition to their role as ligands for transition metals, -aminophosphines are receiving new
attention as the building blocks for chiral bifunctional catalysts in the field of organocatalysis,
which will be discussed in Chapter 2.
1.2 Decarboxylative Allylation Reaction
The decarboxylative allylic alkylation reaction, discovered by Tsuji and co-workers in the 1980s,
allows the construction of all-carbon quaternary centres from allyl enol carbonates or allyl β-keto
esters precursors under mild and neutral conditions (Scheme 14).13
The preparation of these α-
quaternary ketones would otherwise have been difficult under basic conditions, due to the
regioselectivity issue caused by two enolizable protons with very similar pKa values.
Scheme 14. The Tsuji reaction
The proposed mechanism is shown in Scheme 15. The coordination and ionization of the allyl
moiety by the Pd(0) complex takes place to generate a palladium carbonate intermediate, and
subsequent decarboxylation of the carbonate leads to the palladium enolate intermediate. This in
13 a) Tsuji, J.; Minami, I.; Shimizu, I. Tetrahedron Lett. 1983, 24, 1793–1796. b) Shimizu, I.; Yamada, T.;
Tsuji, J. Tetrahedron Lett. 1980, 21, 3199–3202.

11
situ generated enolate nucleophile then recombines with the palladium -allyl electrophile to
give the product and regenerates the catalyst.14
Scheme 15. Proposed catalytic cycle of the decarboxylative allylic alkylation of allyl enol
carbonates and allyl β-keto esters
The first enantioselective variant of the Tsuji reaction was reported independently by the Trost
group and the Stoltz group employing different ligands (Scheme 16).15,16
Scheme 16. The initial report of enantioselective Tsuji reaction using Trost P,P-ligand and
PHOX P,N-ligand
14 a) Tsuda, T.; Chujo, Y.; Nishi, S.; Tawara, K.; Saegusa, T. J. Am. Chem. Soc. 1980, 102, 6381–6384. b)
Tsuji, J.; Minami, I. Acc. Chem. Res. 1987, 20, 140–145. 15
Behenna, D. C.; Stoltz, B. M. J. Am. Chem. Soc. 2004, 126, 15044–15045.
16 Trost, B. M.; Xu, J. J. Am. Chem. Soc. 2005, 127, 2846–2847.
12
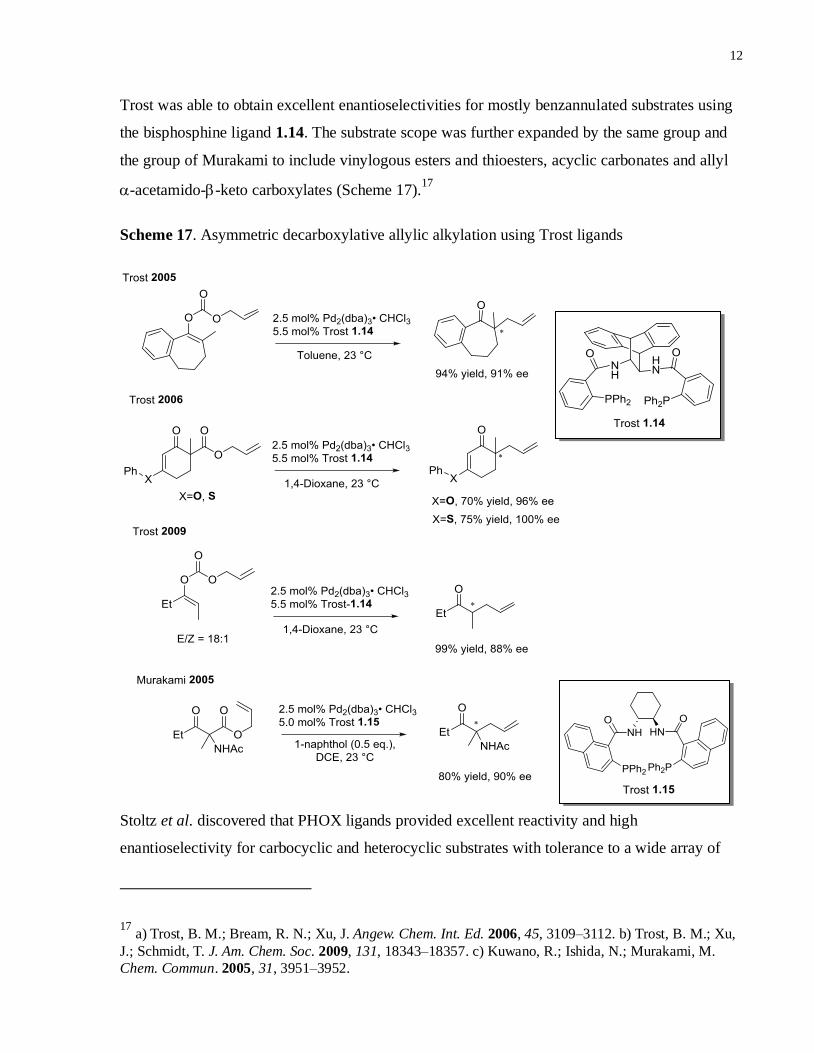
Trost was able to obtain excellent enantioselectivities for mostly benzannulated substrates using
the bisphosphine ligand 1.14. The substrate scope was further expanded by the same group and
the group of Murakami to include vinylogous esters and thioesters, acyclic carbonates and allyl
-acetamido--keto carboxylates (Scheme 17).17
Scheme 17. Asymmetric decarboxylative allylic alkylation using Trost ligands
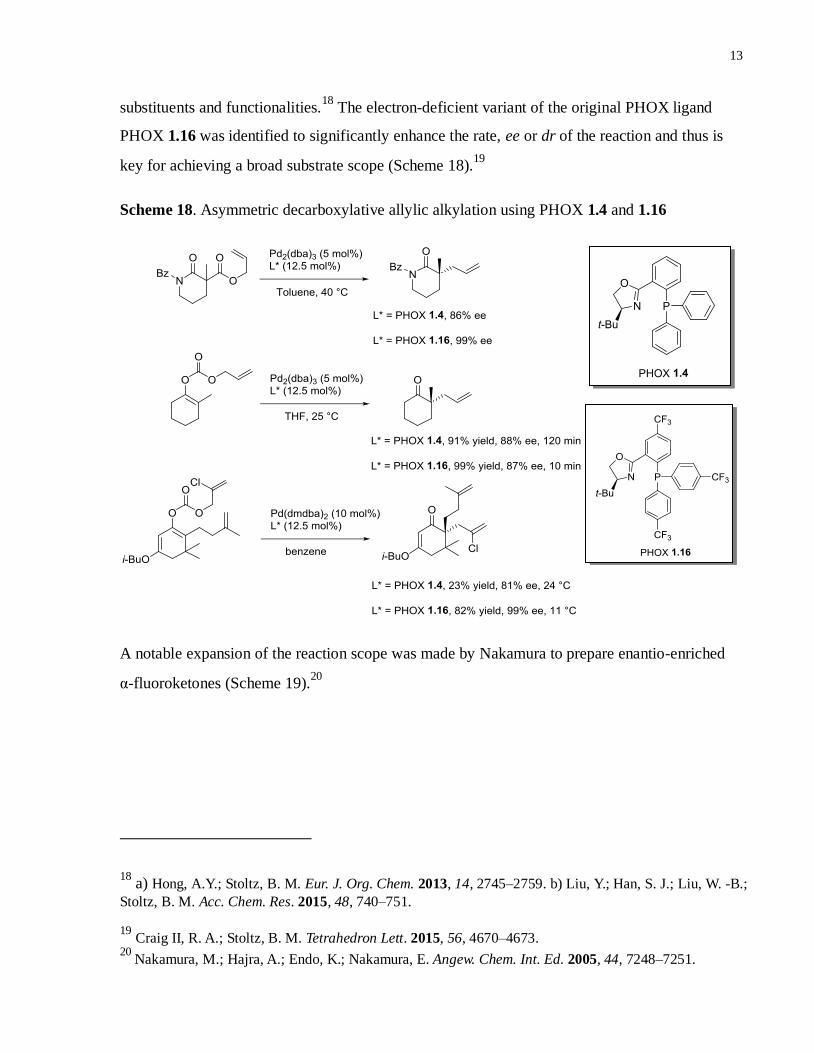
Stoltz et al. discovered that PHOX ligands provided excellent reactivity and high
enantioselectivity for carbocyclic and heterocyclic substrates with tolerance to a wide array of
17 a) Trost, B. M.; Bream, R. N.; Xu, J. Angew. Chem. Int. Ed. 2006, 45, 3109–3112. b) Trost, B. M.; Xu,
J.; Schmidt, T. J. Am. Chem. Soc. 2009, 131, 18343–18357. c) Kuwano, R.; Ishida, N.; Murakami, M.
Chem. Commun. 2005, 31, 3951–3952.
13
substituents and functionalities.18
The electron-deficient variant of the original PHOX ligand
PHOX 1.16 was identified to significantly enhance the rate, ee or dr of the reaction and thus is
key for achieving a broad substrate scope (Scheme 18).19
Scheme 18. Asymmetric decarboxylative allylic alkylation using PHOX 1.4 and 1.16
A notable expansion of the reaction scope was made by Nakamura to prepare enantio-enriched
α-fluoroketones (Scheme 19).20
18 a) Hong, A.Y.; Stoltz, B. M. Eur. J. Org. Chem. 2013, 14, 2745–2759. b) Liu, Y.; Han, S. J.; Liu, W. -B.;
Stoltz, B. M. Acc. Chem. Res. 2015, 48, 740–751.
19 Craig II, R. A.; Stoltz, B. M. Tetrahedron Lett. 2015, 56, 4670–4673.
20 Nakamura, M.; Hajra, A.; Endo, K.; Nakamura, E. Angew. Chem. Int. Ed. 2005, 44, 7248–7251.
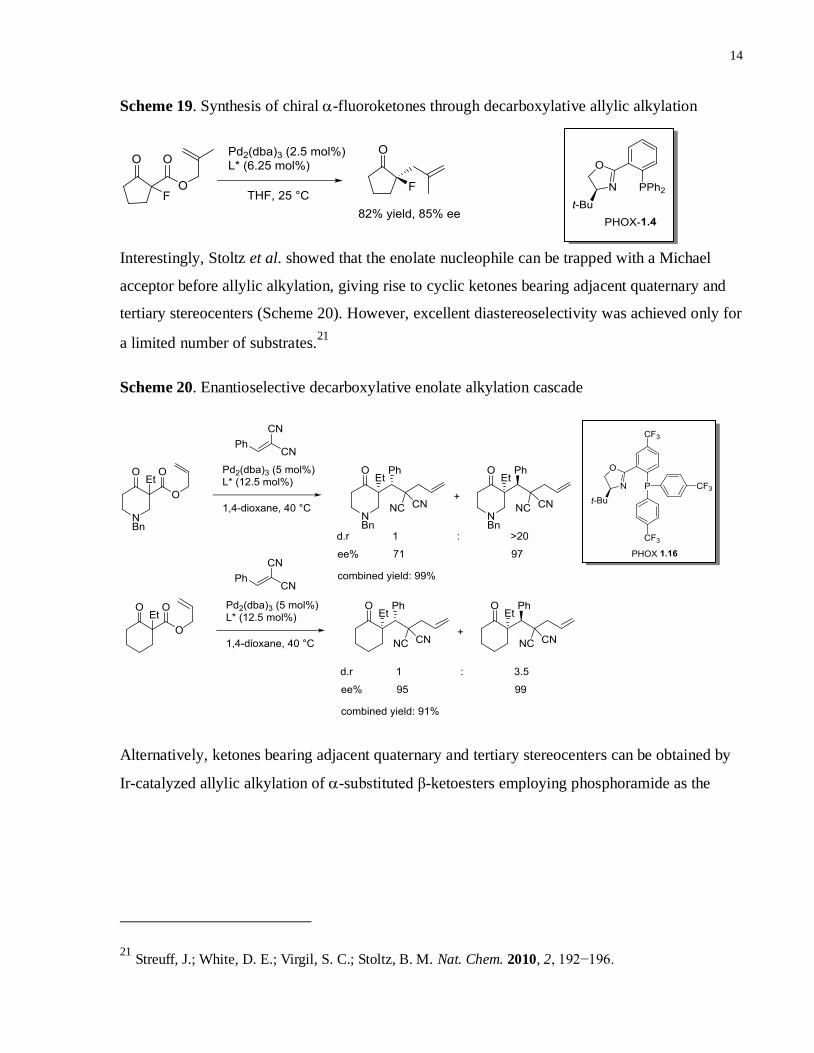
14
Scheme 19. Synthesis of chiral -fluoroketones through decarboxylative allylic alkylation
Interestingly, Stoltz et al. showed that the enolate nucleophile can be trapped with a Michael
acceptor before allylic alkylation, giving rise to cyclic ketones bearing adjacent quaternary and
tertiary stereocenters (Scheme 20). However, excellent diastereoselectivity was achieved only for
a limited number of substrates.21
Scheme 20. Enantioselective decarboxylative enolate alkylation cascade
Alternatively, ketones bearing adjacent quaternary and tertiary stereocenters can be obtained by
Ir-catalyzed allylic alkylation of -substituted β-ketoesters employing phosphoramide as the
21 Streuff, J.; White, D. E.; Virgil, S. C.; Stoltz, B. M. Nat. Chem. 2010, 2 192−196

15
chiral ligand (Scheme 21). Excellent regio‑, diastereo‑, and enantioselectivities were achieved
for both the cyclic and acyclic substrates using different lithium salt additives.22
Scheme 21. Ir-catalyzed allylic alkylation of -substituted β-ketoesters
To add another level of sophistication to the system, Stoltz demonstrated that using a cleverly
devised 2-(trimethylsilyl)ethyl -ketoester can lead to sequential double allylic alkylation using
the Ir-phosphoramide/Pd-PHOX dual catalyst system. Treatment with fluoride triggered a
cascade degradation of the 2-(trimethylsilyl)ethyl group and exposed the prochiral enolate to Pd-
allyl electrophile. The consecutive allylic alkylation reaction furnished the products with
excellent diastereo‑ and enantioselectivities (Scheme 22).19b
22 a) Liu, W.-B.; Reeves, C. M.; Stoltz, B. M. J. Am. Chem. Soc. 2013, 135, 17298–17301. b) Liu, W.-B.;
Reeves, C. M.; Virgil, S. C.; Stoltz, B. M. J. Am. Chem. Soc. 2013, 135, 10626–10629.

16
Scheme 22. Sequential allylic alkylation catalyzed by Ir and Pd complexes
The mechanism of the Pd-catalyzed decarboxylative allylic alkylation reaction of enol
carbonates has been investigated by both the Stoltz and the Trost groups. Based on theoretical
calculations, Stoltz and Goddard proposed that a relatively stable pentacoordinated-
allylpalladium enolate complex exo-5 is formed following the decarboxylation (Scheme 23).
This pentacoordinate species rearranges to the tetracoordinate -allylpalladium enolate complex
(Re/Si)-8. Furthermore, the Re/Si faces of the enolate interact differently with PHOX ligand
during the transition state. Therefore, the formation of (Re/Si)-8 could be the enantio-
determining step, provided that the rotation barrier (currently unknown) about Pd-O bond is large
enough to prevent racemization of (Re/Si)-8. (Re/Si)-8 then undergoes an uncommon pericyclic
reductive rearrangement transition state that is different from the traditional three-centered
reductive elimination. Hence, the final product is believed to be generated via an “inner sphere”
mechanism.23
23 a) Keith, J. A.; Behenna, D. C.; Mohr, J. T.; Ma, S.; Marinescu, S. C.; Oxgaard, J.; Stoltz, B. M.;
Goddard, W. A. J. Am. Chem. Soc. 2007, 129, 11876–11877. b) Keith, J. A.; Behenna, D. C.; Mohr, J. T.; Ma, S.; Marinescu, S. C.; Nielsen, R. J.; Oxgaard, J.; Stoltz, B. M.; Goddard, W. A. J. Am. Chem. Soc.
2012, 131, 19050–19060.
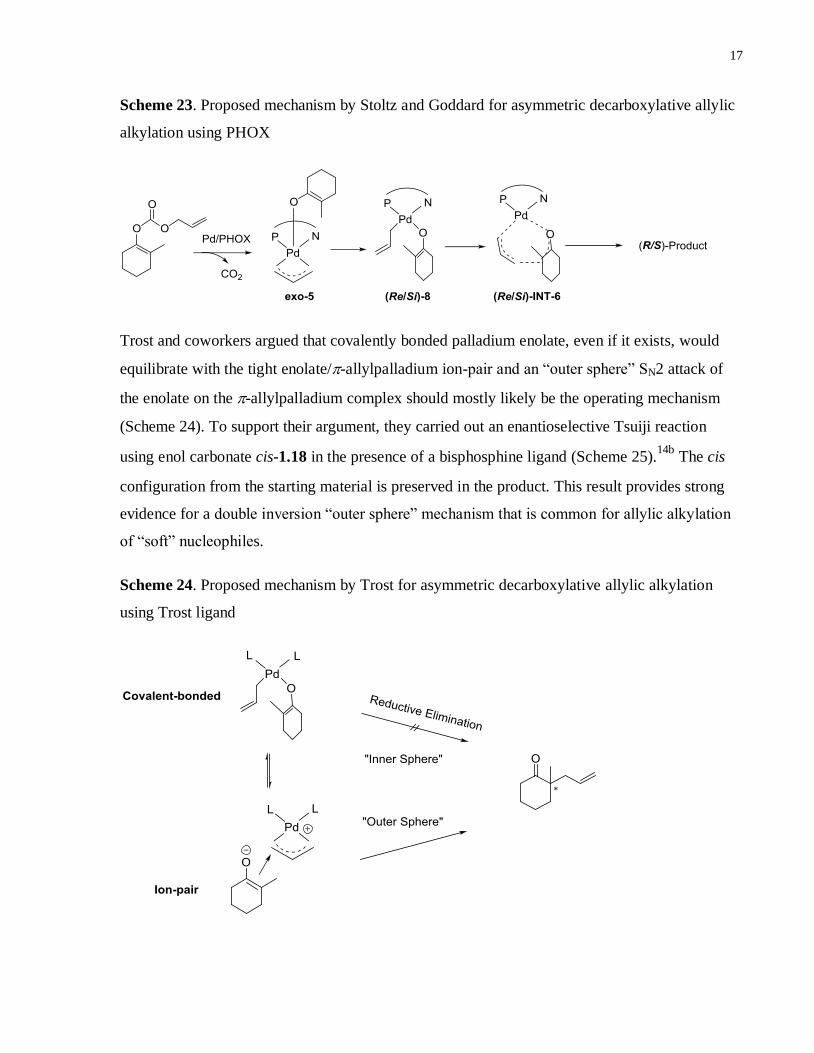
17
Scheme 23. Proposed mechanism by Stoltz and Goddard for asymmetric decarboxylative allylic
alkylation using PHOX
Trost and coworkers argued that covalently bonded palladium enolate, even if it exists, would
equilibrate with the tight enolate/-allylpalladium ion-pair and an “outer sphere” SN2 attack of
the enolate on the -allylpalladium complex should mostly likely be the operating mechanism
(Scheme 24). To support their argument, they carried out an enantioselective Tsuiji reaction
using enol carbonate cis-1.18 in the presence of a bisphosphine ligand (Scheme 25).14b
The cis
configuration from the starting material is preserved in the product. This result provides strong
evidence for a double inversion “outer sphere” mechanism that is common for allylic alkylation
of “soft” nucleophiles.
Scheme 24. Proposed mechanism by Trost for asymmetric decarboxylative allylic alkylation
using Trost ligand
18
Scheme 25. Asymmetric decarboxylative allylic alkylation of cis-1.18 using Trost ligand
2 Objectives
Previous research in our group showed that electron-deficient -aminophosphine 1.20 displayed
excellent reactivity and induced good enantioselectivity in the decarboxylative allylation of enol
carbonate (Scheme 26).
Scheme 26. Asymmetric decarboxylative allylic alkylation using -aminophosphine
In order to further improve the enantioselectivity of the reaction, we aimed to generate a ligand
library using a novel modular approach recently developed in our lab (Scheme 27).
Scheme 27. Modular synthesis of -aminophosphines
3 Results and Discussion

19
3.1 Synthesis of -Substituted -Aminophosphines and Their
Applications in Decarboxylative Allylation Reaction
Traditionally, chiral -aminophosphines are prepared from chiral 1,2-aminoalcohols in four
synthetic steps: Boc-protection of the primary amine, tosylation of the alcohol followed by
displacement of the tosyl group with potassium diphenylphosphide, and, finally, deprotection of
the Boc group (Scheme 28).24
Scheme 28. Conventional synthesis of chiral -aminophosphines
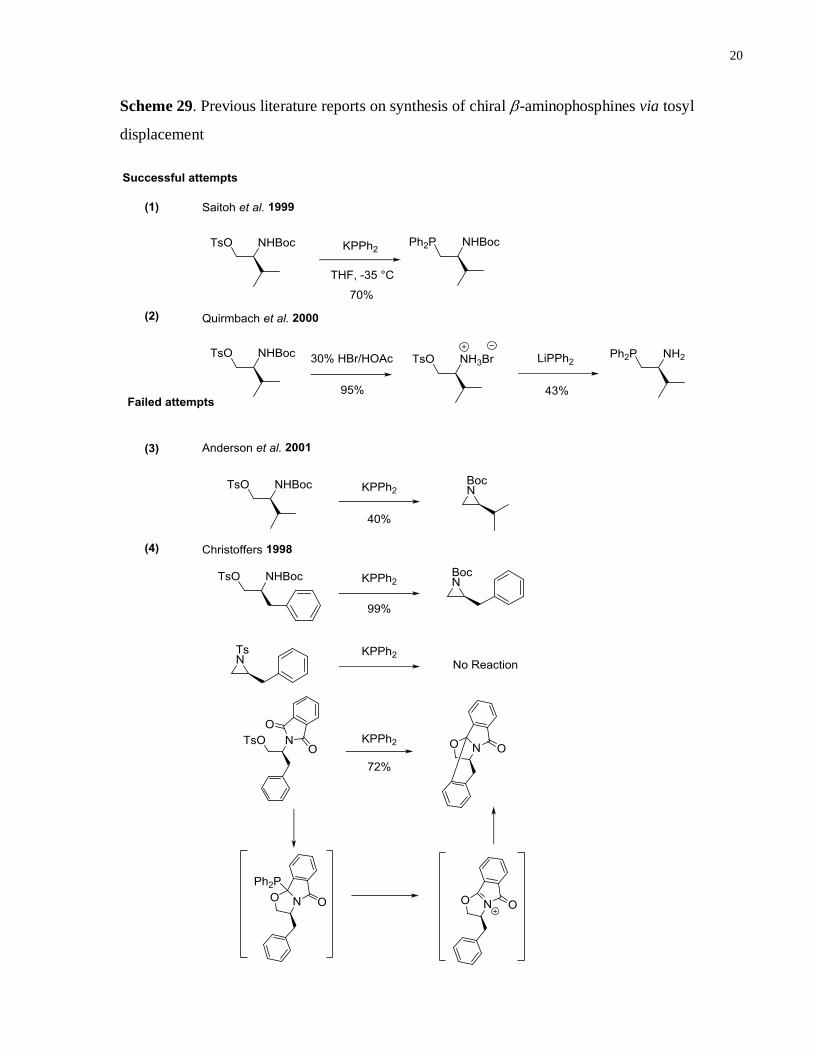
There are reports in the literature that describe the successful syntheses of these -
aminophosphines from the corresponding tosylate precursors.(Scheme 29 (1) and (2))21
There
are also findings where these -amino tosylates fall short of serving as precursor to the desired
phosphine products (Scheme 29 (3) and (4))25
A major drawback of this approach is the
formation of aziridine that cannot be re-opened by KPPh2. Changing the Boc protecting group to
phthalimide only led to the formation of the another side product via neighboring group
participation of the amide carbonyl group.
24 a) Saitoh, A.; Uda, T.; Morimoto, T. Tetrahedron: Asymmetry 1999, 10, 45014511. b) Quirmbach, M.;
Holz, J.; Tararov, V. I.; Borner, A.Tetrahedron 2000, 56, 775780.
25 a) Anderson, J. C.; Cubbon, R. J.; Harling, J. D. Tetrahedron: Asymmetry 2001, 12, 923935. b)
Christoffers, J. Helvetica Chimica Acta. 1998, 81, 845852.
20
Scheme 29. Previous literature reports on synthesis of chiral -aminophosphines via tosyl
displacement

21
To get around such these previous complications, Guo et al. demonstrated that a variety of chiral
1,2-aminoalcohols can be reliably converted to stable N-Boc protected cyclic sulfamidates. The
ring opening of these cyclic sulfamidates with potassium diphenylphosphide followed by the
deprotection of Boc affords chiral aminophosphines in good yields (Scheme 30).26
Scheme 30. Synthesis of chiral -aminophosphines via cyclic sulfamidates
However, the reaction was only demonstrated for diphenylphosphino and BINAP-phosphepine
moieties. Attempts to synthesize electron-deficient P,N-ligand by simply switching the
nucleophile to bis(4-(trifluoromethyl)phenyl)phosphanide failed to deliver any desired product.27
In light of this limitation, we set out to develop a modified version of this method that would
permit easy modification of the diaryl substituents on the phosphine group.
26 Guo, R.; Lu, S.; Chen, X.; Tsang, C.-W.; Jia, W.; Sui-Seng, C.; Amoroso, D.; Abdur-Rashid, K. J. Org.
Chem. 2010, 75, 937940.
27 J. Su, unpublished results

22
Scheme 31. The first successful ring-opening of cyclic sulfamidate with secondary phosphine
oxide
It was identified that the combination of potassium tert-butoxide and bis(4-
(trifluoromethyl)phenyl) phosphine oxides was able to open the cyclic sulfamidate (Scheme
31).24
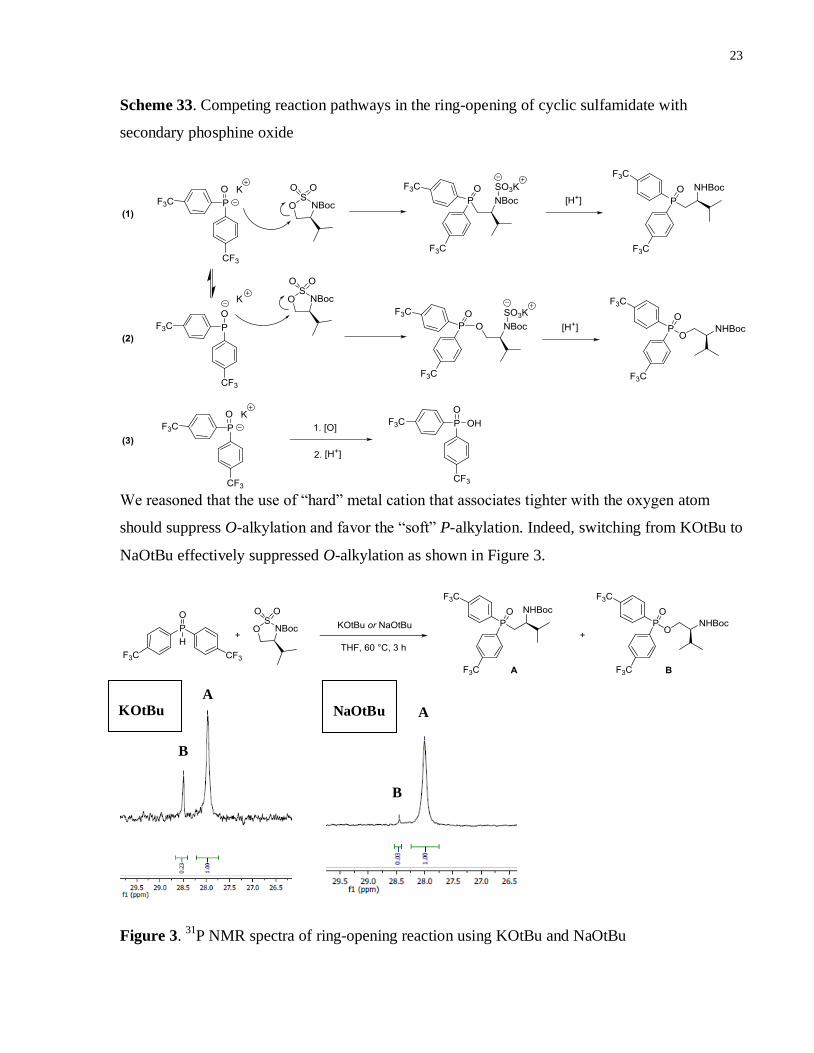
Efforts towards optimizing the reaction conditions revealed two other competing pathways.
Firstly, it is known that penta-valent secondary phosphine oxides can tautomerize in solution to
trivalent phosphinous acids under neutral condition (Scheme 32).28
Under basic conditions, the
oxygen atom can potentially compete with the phosphorus atom that would otherwise give rise to
the desired product (Scheme 33 (1) and (2)). 24
Another side reaction is the oxidation of
secondary phosphine oxides (phosphinous acids) to phosphinic acids (Scheme 33 (3)) which is
hydrophilic and dissolves in the aqueous layer during workup.29
Scheme 32. Tautomerism of secondary phosphine oxides
28 Christiansen, A.; Li, C.; Garland, M.; Selent, D.; Ludwig, R.; Spannenberg, A.; Baumann, W.; Franke,
R.; Börner, A. Eur. J. Org. Chem. 2010, 2010, 27332741.
29 Tsvetkov, E. N.; Bondarenko, N. A.; Malakhova, I. G.; Kabachnik, M. I. Synthesis, 1986, 3, 198208.
23
Scheme 33. Competing reaction pathways in the ring-opening of cyclic sulfamidate with
secondary phosphine oxide
We reasoned that the use of “hard” metal cation that associates tighter with the oxygen atom
should suppress O-alkylation and favor the “soft” P-alkylation. Indeed, switching from KOtBu to
NaOtBu effectively suppressed O-alkylation as shown in Figure 3.
Figure 3. 31
P NMR spectra of ring-opening reaction using KOtBu and NaOtBu
B
A
B
A KOtBu NaOtBu

24
In addition, we carried out these ring-opening reactions in degassed THF under inert atmosphere
to minimize the oxidation of secondary phosphine oxides. Various secondary phosphine oxide
precursors were prepared by reacting the in situ generated Grignard reagents with diethyl
phosphite (Table 1). With the optimized reaction conditions, several sterically and electronically
differentiated aryl groups were installed on the phosphorus atom (Table 2).
Table 1. Scope for secondary phosphine oxides
Table 2. Scope for -substituted--aminophosphine oxides

25
The -aminophosphine oxides were then successfully reduced using diphenylsilane at 140 °C
(Table 3).30
Due to the air-sensitive nature of organophosphines, the final P,N-ligands are
purified through flash column chromatography using degassed eluent under argon.
Table 3. Scope for -substituted--aminophosphines
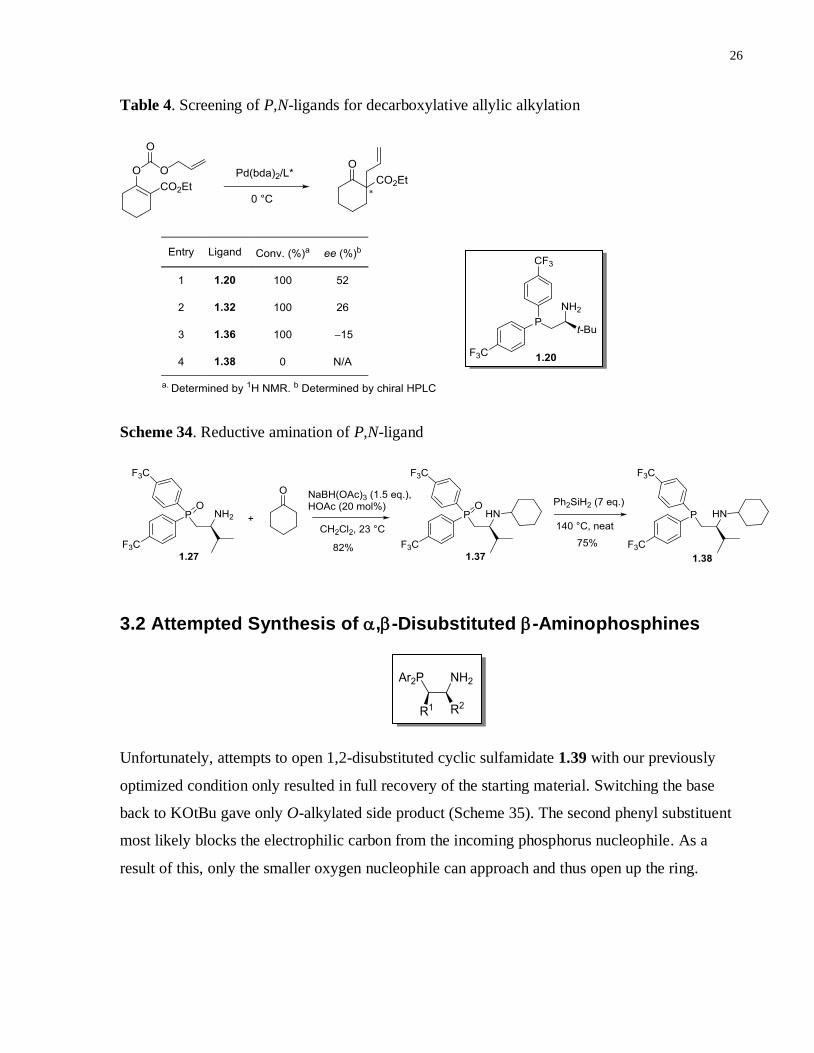
Electron-deficient P,N-ligands 1.32 and 1.36 were tested in the decarboxylative allylation
reaction of enol carbonate (Table 4). Both ligands promoted the reaction to full conversion.
However, the ee values were lower than what has been achieved using 1.20. To answer whether
or not functionalizing the primary amine group would improve the enantioselectivity, ligand 1.38
was prepared by reductive amination of ligand 1.27 followed by reduction of 1.37 (Scheme 34).
Unfortunately, ligand 1.38 did not display any catalytic activity. Considering the fact that
electron-withdrawing groups such as 4-trifluoromethylphenyl group on the phosphine has been
found to be optimal for these decarboxylative allylation reactions, we turned our attention toward
optimizing the enantioselectivity of the reaction further by introducing a second chirality centre
onto the carbon backbone of the P,N-ligand.
30 McDougal, N. T.; Streuff, J.; Mukherjee, H.; Virgil, S. C.; Stoltz, B. M. Tetrahedron Lett. 2010, 51,
55505554.
26
Table 4. Screening of P,N-ligands for decarboxylative allylic alkylation
Scheme 34. Reductive amination of P,N-ligand
3.2 Attempted Synthesis of ,-Disubstituted -Aminophosphines
Unfortunately, attempts to open 1,2-disubstituted cyclic sulfamidate 1.39 with our previously
optimized condition only resulted in full recovery of the starting material. Switching the base
back to KOtBu gave only O-alkylated side product (Scheme 35). The second phenyl substituent
most likely blocks the electrophilic carbon from the incoming phosphorus nucleophile. As a
result of this, only the smaller oxygen nucleophile can approach and thus open up the ring.

27
Scheme 35. Attempted ring-opening of 1,2-disubstituted cyclic sulfamidate with secondary
phosphine oxide
We were then prompted to reconsider the possibility of using an open-chain, conformationally
less rigid electrophile that could permit easier access for the incoming phosphorus nucleophile
through carbon-carbon bond rotation. However, the isolation of the benzylic tosylate
intermediate was not possible according to Guo et al., due to neighboring group participation of
the Boc group, which rapidly displaces the tosyl group to form oxazolidinone as the only product
(Scheme 36).23
Scheme 36. Decomposition pathway of benzylic tosylate

28
We considered whether we could install a less labile leaving group so that the intermediate
would be more resistant against the intramolecular cyclization of the Boc group, but at the same
time be adequately reactive towards intermolecular SN2 attack of the secondary phosphine oxide
nucleophile. Commonly employed leaving groups such as chloride and bromide naturally came
to mind. Although the required benzylic chloride and bromide had not been known in the
literature, the ring-opening of cyclic sulfamidates with a fluoride is precedented as a means to
prepare 18
F-labeled compounds for imaging through Positron Emission Tomography (PET)
(Scheme 37). 31
Scheme 37. Ring-opening of cyclic sulfamidates with fluoride
Knowing this, we decided to try opening the cyclic sulfamidate with tetra-n-butylammonium
chloride and bromide. Gratifyingly, the reactions proceeded to full conversion after stirring at
room temperature overnight and the products were isolated in 87 and 33 % yield, respectively
(Scheme 38). Next, to test whether the halides could be displaced by a phosphorus nucleophile,
the two intermediates were treated with secondary phosphine oxides under basic conditions. We
found that while the benzylic chloride was unreactive, the benzylic bromide afforded the product
in 40% yield. After deprotection of the Boc group, however, all efforts to reduce the
aminophosphine oxide 1.41 to the aminophosphine failed (Table 5). Heating 1.41 neat with
diphenylsilane at 140 °C resulted in the liberation of secondary phosphine oxide 1.21 through C–
P bond cleavage. Treatment of 1.41 with BH3▪SMe at 70 °C, led to the cleavage of C–C bond.32
The use of 1,1,3,3-tetramethyldisiloxane (TMDS) with catalytic amount of Ti(OiPr)4 also
31 VanDort, M. E.; Jung, Y.-W.; Sherman, P. S.; Kilbourn, M. R.; Wieland, D. M. J. Med. Chem. 1995, 38,
810815.
32 Hérault, D.; Nguyen, D. H.; Nuela, D.; Buono, G. Chem. Soc. Rev. 2015, 44, 25082528.

29
resulted in C–C bond cleavage.33
It seems that the phosphine oxide starting material is prone to
decomposition in the presence of a Lewis acid.
Scheme 38. Ring-opening and P-alkylation of cyclic sulfamidates
33 M. Berthod, A. Favre-Réguillon, J. Mohamad, G. Mignani, G. Docherty, M. Lemaire, Synlett, 2007,
15451548.

30
Table 5. Decomposition products isolated in the reduction of aminophosphine oxide
Another synthetic target that we examined was derived from 1-amino-2-indanol 1.42 (Scheme
39). The tosylate intermediate 1.43 was expected to be stable from ring-closure because the tosyl
group and the Boc group are locked in a cis configuration. The two known decomposition
pathways - aziridination and oxazolidinone formation - require the neighboring group to
approach from the opposite side. Surprisingly, treatment of tosylate 1.43 with secondary
phosphine oxide and base only resulted in the full recovery of the starting material. Switching to
a more reactive triflate group afforded the product 1.45 in modest yield. The attempt to remove
the Boc group with trifluoroacetic acid led to P–C bond cleavage, suggesting that the product is
unstable towards acidic conditions. Instead, stirring 1.45 with zinc bromide overnight
successfully hydrolyzed the Boc group and furnished the product 1.46 in good purity after work
up. It should be noted that, eluting the crude product through a silica gel column resulted in
complete decomposition of 1.46. Unfortunately, the desired phosphine product was not observed

31
by 31
P spectroscopy even with attempted reduction of the crude phosphine oxide mixture using
either diphenylsilane or BH3.
Scheme 39. Attempted synthesis of -disubstituted -aminophosphine derived from 1-amino-
2-indanol 1.42
3.3 Attempted C–H activation of (R)-2-phenylglycinol
Given the daunting synthetic challenge of -disubstituted -aminophosphines, we turned our
attention back to the ligand scaffold bearing a single chiral centre. By this time, we had almost
exhausted the small pool of commercially available chiral 1,2-aminoalcohols that could be
relevant to our research. We were therefore drawn to the idea of modifying the existing chiral
backbone through C–H activation. The power of this approach was demonstrated recently by Yu
and his collaborators in Bristol-Myers Squibb, where simple alanine was transformed into a
diverse range of non-natural amino acids through sequential sp3 C–H arylation. The resulting
32
amino acids were readily reduced to amino alcohols which were used to make novel BOX and
PyBOX ligands (Scheme 40).34
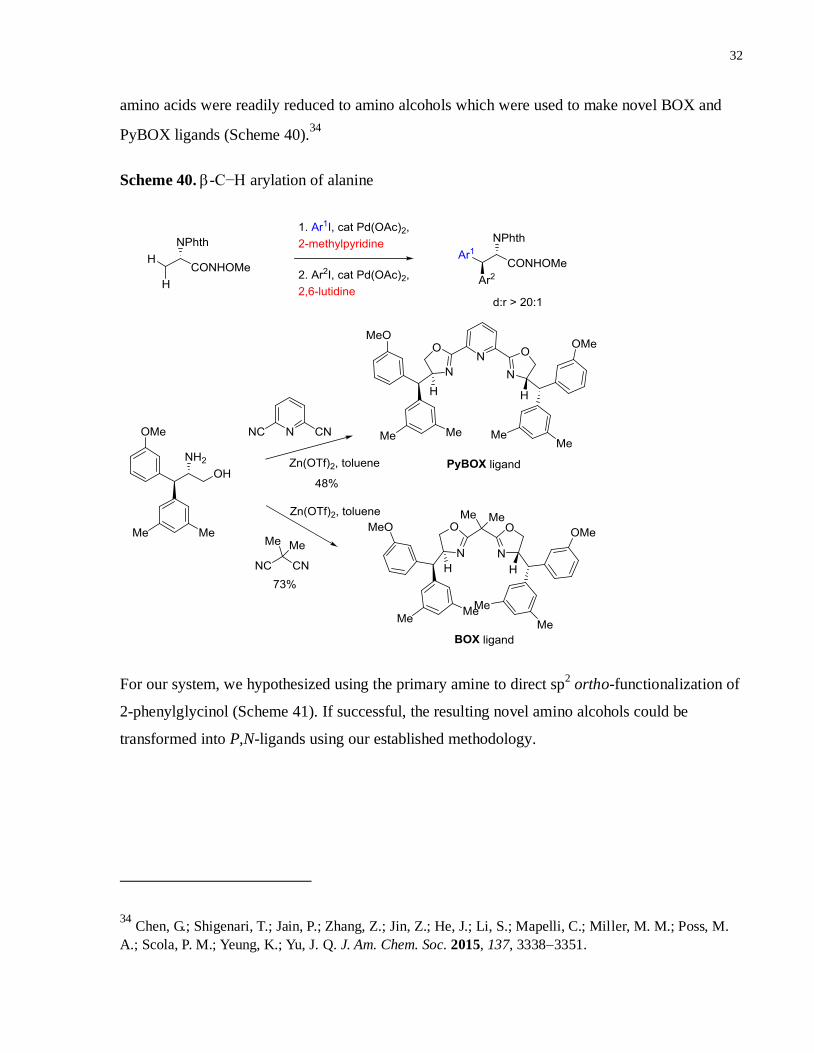
Scheme 40. -C−H arylation of alanine
For our system, we hypothesized using the primary amine to direct sp2 ortho-functionalization of
2-phenylglycinol (Scheme 41). If successful, the resulting novel amino alcohols could be
transformed into P,N-ligands using our established methodology.
34 Chen, G.; Shigenari, T.; Jain, P.; Zhang, Z.; Jin, Z.; He, J.; Li, S.; Mapelli, C.; Miller, M. M.; Poss, M.
A.; Scola, P. M.; Yeung, K.; Yu, J. Q. J. Am. Chem. Soc. 2015, 137, 33383351.

33
Scheme 41. Hypothetical divergent synthesis of -substituted -aminophosphines via C–H
activation
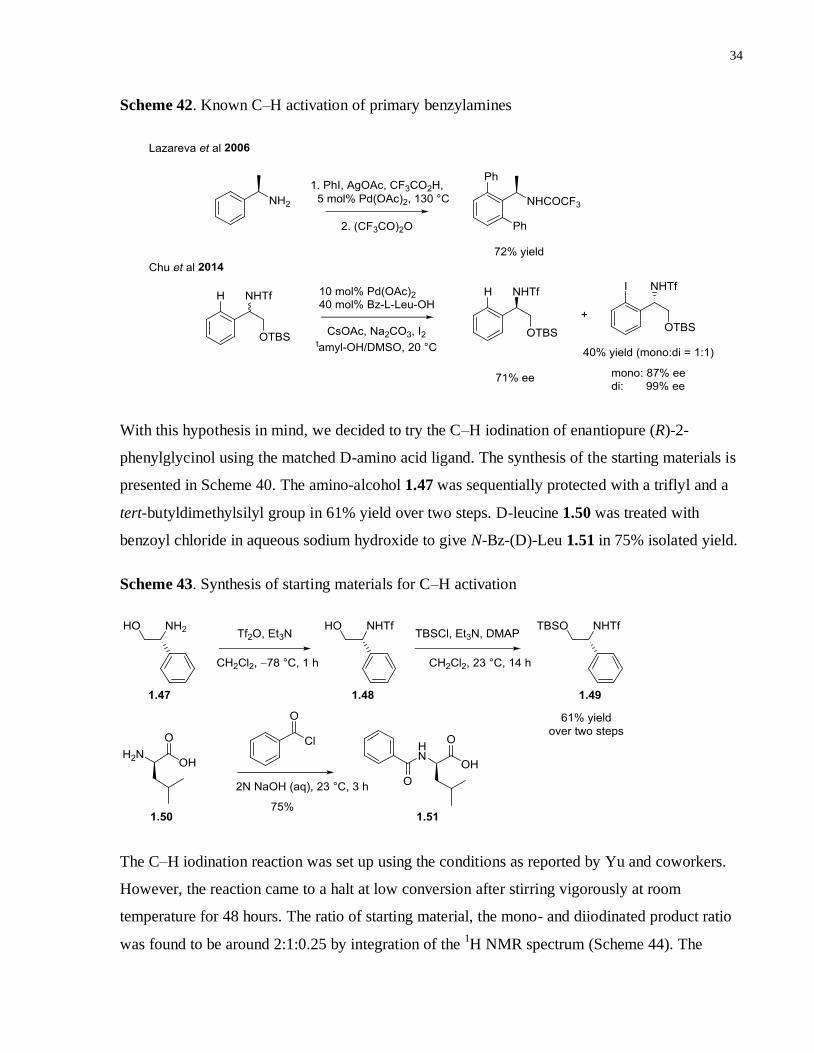
Although ortho-palladation of N,N-dialkylbenzylamines is well-known, the first synthetically
relevant C–H activation using primary benzylamine as the directing group was not reported until
2006 (Scheme 42).35
Under rather harsh reaction conditions, the ortho-diarylation of
benzylamines possessing only simple hydrocarbon backbones was realized and the products
were isolated as trifluoroacetamides in decent yields. More recently, Yu and coworkers reported
an enantioselective C–H iodination of benzyamines via kinetic resolution employing I2 as the
sole oxidant (Scheme 42).36
The C–H activation of one of the enantiomers is selectively
accelerated by the addition of matched ligand – either L or D mono-protected amino acid. The
drawback of this approach was the generation of a 1:1 mixture of mono- and di-iodinated
products. However, this limitation may not necessarily be a pitfall for an initial screening as it
effectively increases the chemical space covered by the ligand library.
35 Lazareva, A.; Daugulis, O. Org. Lett. 2006, 8, 52115213.
36 Chu, L.; Xiao, K. J.; Yu, J. Q. Science. 2014, 346, 451455.
34
Scheme 42. Known C–H activation of primary benzylamines
With this hypothesis in mind, we decided to try the C–H iodination of enantiopure (R)-2-
phenylglycinol using the matched D-amino acid ligand. The synthesis of the starting materials is
presented in Scheme 40. The amino-alcohol 1.47 was sequentially protected with a triflyl and a
tert-butyldimethylsilyl group in 61% yield over two steps. D-leucine 1.50 was treated with
benzoyl chloride in aqueous sodium hydroxide to give N-Bz-(D)-Leu 1.51 in 75% isolated yield.
Scheme 43. Synthesis of starting materials for C–H activation
The C–H iodination reaction was set up using the conditions as reported by Yu and coworkers.
However, the reaction came to a halt at low conversion after stirring vigorously at room
temperature for 48 hours. The ratio of starting material, the mono- and diiodinated product ratio
was found to be around 2:1:0.25 by integration of the 1H NMR spectrum (Scheme 44). The

35
turnover number of the palladium catalyst was calculated to be around 4. Several attempts to
improve the conversion, including increasing the temperature, varying the palladium-to-ligand
ratio, and adjusting the solvent ratio, proved fruitless. The only way to improve the conversion
was by increasing the palladium loading. Based on these results, we suspected that the inactive
PdI2 formed after two catalytic cycles was not being effectively converted back to the reactive
Pd(OAc)2 by CsOAc, Na2CO3 and DMSO, a combination of reagents that was previously shown
to regenerate the active catalyst in a different reaction (Scheme 45).37
Scheme 44. Attempted C–H iodination using I2
Scheme 45. Regeneration of Pd(OAc)2 from PdI2
We wondered whether we could prevent the formation of PdI2 in the first place by using IOAc as
the oxidant. IOAc, generated by mixing I2 with AgOAc or PhI(OAc)2, had been employed by Yu
37 Wang, X.; Hu, Y.; Bonacorsi, S.; Hong, Y.; Burrell, R.; Yu, J. Q. J. Am. Chem. Soc. 2013, 135,
1032610329.

36
and coworkers as the C–H iodination reagent before they discovered the conditions (Scheme 45)
that enabled them to adopt the much more practical I2 for C–H iodination.38
The result we
obtained from the use of IOAc is presented in Scheme 46. We observed predominantly the di-
iodinated product along with a negligible amount of mono-iodinated product, and the turnover
number was improved to around 7. However, full conversion of the starting material was not
attained even with a 20 mol% Pd loading. From a practical perspective, no further C–H
iodination reaction was attempted beyond this point.
Scheme 46. Attempted C–H iodination using IOAc
4 Experimental
4.1 Procedures and Compounds
General procedure for preparing diaryl secondary phosphine oxides 1.21–1.24.
Bis(4-(trifluoromethyl)phenyl)phosphine oxide (1.21)
Mg turnings (0.56 g, 23 mmol) were activated with iodine in diethyl ether (12 mL) and 1-bromo-
4-(trifluoromethyl)benzene (5 g, 22 mmol) was added at 0 °C. The mixture was stirred under
gentle reflux for 90 min. Diethyl phosphite (1.02 g, 7.4 mmol) was then added dropwise at 0 ºC.
38 Giri, R.; Chen, X.; Yu, J. Q. Angew. Chem. Int. Ed. 2005, 44, 21122115.

37
The reaction mixture was stirred at room temperature for 14 h. 2N Hydrochloric acid (12 mL)
was added dropwise at 0 ºC and the mixture was stirred at 23 ºC for 5 min. Extraction with
diethyl ether followed by purification by column chromatography (Hexanes /EtOAc, 3:1 v/v)
afforded phosphine oxide 1 (1.5 g, 61%) as a yellow solid.
1H NMR (300 MHz, CDCl3) δ 8 20 (d, J (P-H) = 492 Hz, 1H), 7.96 – 7.74 (m, 10H).
13C NMR
(101 MHz, CDCl3) δ 134 84 (dd J = 33.0, 3.0 Hz), 134.82 (d, J = 100.0 Hz), 131.19 (d, J = 12.0
Hz), 126.04 (dq, J = 13.2, 3.8 Hz), 123.30 (dd, J = 272.9, 1.1 Hz).31
P NMR (121 MHz, CDCl3) δ
17.81.
Di-o-tolylphosphine oxide (1.22)
Synthesized according to general procedure using 1-bromo-2-methylbenzene (5.68 g, 33.2
mmol), Mg turning (834 mg, 34.3 mmol) and diethyl phosphite (1.53 g, 11 mmol). Purified by
flash chromatography on silica gel (EtOAc /CH2Cl2, 1:4 v/v) to afford 1.22 (1.38 g, 55%) as a
white solid.
1H NMR (400 MHz, CDCl3) δ 8 19 (d J = 477.6 Hz, 1H), 7.70 (m, 2H), 7.45 (m, 2H), 7.31 (m,
2H), 7.23 (m, 1H), 2.36 (s, 6H). 13
C NMR (101 MHz, CDCl3) δ 141 08 (d J = 9.9 Hz), 132.52
(d, J = 2.7 Hz), 132.42 (d, J = 11.8 Hz), 131.22 (d, J = 10.3 Hz), 129.29 (d, J = 100.2 Hz),
126.04 (d, J = 12.8 Hz), 20.15 (d, J = 7.0 Hz). 31
P NMR (121 MHz, CDCl3) δ 17 64
Bis(4-methoxyphenyl)phosphine oxide (1.23)
Synthesized according to general procedure using 1-bromo-4-methoxybenzene (5.98 g, 32.0
mmol), Mg turning (802 mg, 33.0 mmol) and diethyl phosphite (1.47 g, 10.7 mmol). Purified by
flash chromatography on silica gel (100% EtOAc) to afford 1.23 (1.08 g, 39%) as a white solid.

38
1H NMR (400 MHz, CDCl3) δ 8 01 (d = 478 0 H 1H) 7 71 – 7.52 (m, 4H), 7.07 – 6.94 (m,
4H), 3.83 (s, 6H). 13
C NMR (101 MHz, CDCl3) δ 162 92 (d J = 2.9 Hz), 132.67 (d, J = 13.0 Hz),
122.92 (d, J = 108.0 Hz), 114.46 (d, J = 13.9 Hz), 55.39. 31
P NMR (121 MHz, CDCl3) δ 20 50
Diphenylphosphine oxide (1.24)
Synthesized according to general procedure using bromobenzene (5.98 g, 38.1 mmol), Mg
turning (957 mg, 39.4 mmol) and diethyl phosphite (1.75 g, 12.7 mmol). Purified by flash
chromatography on silica gel (100% EtOAc) to afford 1.24 (1.48 g, 57%) as a white solid.
1H NMR (300 MHz, CDCl3) δ 8 08 (d J = 480.2 Hz, 1H), 7.71 (m, 4H), 7.63 – 7.45 (m, 6H).
13C NMR (101 MHz, CDCl3) δ 131 26 (d J = 10.4 Hz), 130.74 (d, J = 11.4 Hz), 128.94 (d, J =
12.9 Hz), 128.29 (d, J = 13.3 Hz). 31
P NMR (121 MHz, CDCl3) δ 21 38
General procedure for preparing cyclic sulfamidates 1.25, 1.26, 1.39.
tert-Butyl (S)-4-isopropyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide (1.25)
A solution of (Boc)2O (520 mg, 2.4 mmol) in THF (4 mL) was added to a mixture of (S)-2-
amino-3-methyl-1-butanol (220 mg, 2.1 mmol) and sodium carbonate (0.5 g, 4.7 mmol) in
THF/H2O (1/1, 8 mL) at 0 ºC. The mixture was stirred at room temperature for 3 h. Water was
added to the mixture and was extracted with ethyl acetate. The combined organic layer was
washed with brine, dried over anhydrous MgSO4, and concentrated to afford the crude product in
quantitative yield as a colorless viscous liquid, which was used in the next step without
purification.

39
Pyridine (1 g, 10 mmol) was added dropwise to a solution of N-Boc-protected (S)-2-amino-3-
methyl-1-butanol (426mg, 2.10 mmol) and SOCl2 (621 mg, 5.2 mmol) in MeCN (6 mL) at -40
ºC. The reaction mixture was allowed to warm up and stirred at room temperature for 3 h. Water
was added to quench the reaction at 0 ºC. The aqueous layer was extracted with ethyl acetate.
The organic phase was dried over anhydrous MgSO4 and concentrated to afford an orange color
oil. The residual was azeotroped three times with toluene and was dried on high vacuum to
remove pyridine. The crude product was used for the next step without purification.
RuCl3∙3H2O (30 mg, 0.146 mmol) was added to a solution of cyclic sulfamidite (0.45 g) in
CH3CN/H2O (1:1, 10 mL) at 0 ºC. NaIO4 (0.67 g, 3.13 mmol) was added in one portion. The
reaction was stirred at room temperature for 14 h. The aqueous layer was extracted with diethyl
ether. The combined organic layer was washed with brine, dried over MgSO4 and concentrated.
The residual was re-dissolved in CH2Cl2 and was filtered through a short plug of silica to remove
Ru catalyst. The solvent was removed in vacuo to afford the final product (293mg, 53% over
three steps) as a white or slightly yellow crystalline solid.
1H NMR (400 MHz, CDCl3) δ 4 57 (dd J = 9.5, 6.4 Hz, 1H), 4.39 (dd, J = 9.5, 1.8 Hz, 1H), 4.19
(ddd, J = 6.4, 5.2, 1.8 Hz, 1H), 2.27 (pd, J = 6.9, 5.2 Hz, 1H), 1.55 (s, 9H), 1.02 (d, J = 6.9 Hz,
3H), 0.97 (d, J = 7.0 Hz, 3H). 13
C NMR (101 MHz, CDCl3) δ 149 10 85 34 67 02 62 01, 30.04,
27.89, 18.00, 16.46.
tert-Butyl (R)-4-phenyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide (1.26)
Synthesized according to general procedure using (R)-2-amino-2-phenylethan-1-ol (364 mg, 2.65
mmol). The final product 1.26 (380 mg, 48% over three steps) was obtained as a white
crystalline solid.

40
1H NMR (300 MHz, CDCl3) δ 7 56 – 7.34 (m, 5H), 5.44 – 5.19 (dd, J = 6.7, 4.2 Hz, 1H), 4.87
(dd, J = 9.2, 6.7 Hz, 1H), 4.41 (dd, J = 9.2, 4.2 Hz, 1H), 1.43 (s, 9H). 13
C NMR (101 MHz,
CDCl3) δ 148 28 136 95, 129.26, 129.15, 126.17, 85.59, 71.79, 60.77, 27.83.
tert-Butyl (4R,5S)-4,5-diphenyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide (1.39)
Synthesized according to general procedure using (1S,2R)-2-amino-1,2-diphenylethan-1-ol (521
mg, 2.44 mmol). The final product 1.39 (666 mg, 72% over three steps) was obtained as a white
crystalline solid.
1H NMR (399 MHz, CDCl3) δ 7 16 (m 6H) 7 10 – 7.01 (m, 2H), 6.96 – 6.88 (m, 2H), 6.16 (d, J
= 5.6 Hz, 1H), 5.42 (d, J = 5.6 Hz, 1H), 1.49 (s, 9H). 13
C NMR (101 MHz, CDCl3) δ 148 24
133.53, 130.57, 129.31, 128.53, 128.42, 128.37, 128.27, 127.21, 126.27, 85.67, 83.36, 66.60,
27.89.
General procedure for preparing aminophosphine oxides 1.27–1.31.
(S)-(2-Amino-3-methylbutyl)bis(4-(trifluoromethyl)phenyl)phosphine oxide (1.27)
Bis(4-(trifluoromethyl)phenyl)phosphine oxide 1.21 (186 mg, 0.55 mmol) was added to a
solution of sodium tert-butoxide (53 mg, 0.55 mmol) in degassed THF (5 mL) under inert
atmosphere. The mixture was stirred for 5 min. tert-Butyl (S)-4-isopropyl-1,2,3-oxathiazolidine-
3-carboxylate 2,2-dioxide 1.25 solid (146 mg, 0.55 mmol) was added and the reaction mixture
was stirred at 60 for 3 h. The reaction was quenched with 2N H2SO4 (4 mL) at room

41
temperature and the mixture was stirred for another 20 min. Extraction with diethyl ether
followed by purification by flash chromatography on silica gel (EtOAc /CH2Cl2, 1:4 v/v).
Trifluoroacetic acid (1.1 mL, 14.3 mmol) was added to a solution of the purified product in dry
CH2Cl2 (5 mL) at 0 °C. The reaction was allowed to warm up and stirred at room temperature for
3 h. Saturated Na2CO3 was added dropwise to the solution at 0 °C. The aqueous layer was
extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated to
afford the final product 1.27 (160 mg, 69%) as colorless oil.
1H NMR (300 MHz, CDCl3) δ 7 98 – 7.82 (m, 4H), 7.79 – 7.68 (m, 4H), 3.07 (m, 1H), 2.67 –
2.18 (m, 2H), 1.91 (s, 2H), 1.67 (m, 1H), 0.88 (app dd, J = 6.8, 5.6 Hz, 6H). 13
C NMR (101
MHz, CDCl3) δ 137 69 (d J = 95.4 Hz), 136.53 (d, J = 96.3 Hz), 134.48 – 134.00 (m), 134.00 –
133.58 (m), 131.36 (d, J = 9.5 Hz), 131.03 (d, J = 9.8 Hz), 126.26 – 125.46 (m), 124.78, 122.06,
52.01 (d, J = 4.6 Hz), 34.71 (d, J = 12.7 Hz), 33.75 (d, J = 72.9 Hz), 18.29, 17.51. 31
P NMR
(121 MHz, CDCl3) δ 30 79 IR (neat, cm-1
): 3044 (w), 2961 (w), 1684 (w), 1400 (m), 1317 (s),
1183 (m), 1163 (s), 1117 (s), 1100 (s), 1060 (s), 1017 (s), 840 (m), 790 (m), 764 (m), 708 (s),
670 (m). Optical rotation: []D20
(c 1.15, CHCl3) = +26.8. HRMS (DART-TOF) m/z: [M + H]+
calcd for C19H21F6NOP 424.12649; found 424.12666.
(S)-(2-Amino-3-methylbutyl)di-o-tolylphosphine oxide (1.28)
Synthesized according to general procedure using di-o-tolylphosphine oxide 1.22 (115 mg, 0.5
mmol), sodium tert-butoxide (48 mg, 0.5 mmol) and tert-Butyl (S)-4-isopropyl-1,2,3-
oxathiazolidine-3-carboxylate 2,2-dioxide 1.25 (133 mg, 0.5 mmol). The intermediate was
purified by flash chromatography on silica gel (EtOAc /CH2Cl2, 1:3 v/v) before treatment of
trifluoroacetic acid. The final product 1.28 (67 mg, 42%) was isolated as a colorless oil.
1H NMR (300 MHz, CDCl3) δ 7 81 (ddd J = 12.5, 7.7, 1.5 Hz, 1H), 7.64 (ddd, J = 12.9, 7.7, 1.4
Hz, 1H), 7.46 – 7.34 (m, 2H), 7.34 – 7.24 (m, 2H), 7.24 – 7.12 (m, 2H), 3.21 – 2.89 (m, 1H),
2.49 – 2.45 (m, 2H), 2.46 – 2.34 (m, 2H), 2.31 (s, 3H), 2.22 (s, 3H), 1.79 – 1.61 (m, 1H), 0.87

42
(app t, 6H). 13
C NMR (101 MHz, CDCl3) δ 141 68 (d J = 8.4 Hz), 141.21 (d, J = 8.8 Hz),
132.43 (d, J = 9.6 Hz), 132.05 (d, J = 96.4 Hz), 132.01 , 131.91 , 131.89 , 131.87 , 131.30 (d, J =
11.1 Hz), 130.65 (d, J = 95.3 Hz), 125.79 (d, J = 6.6 Hz), 125.68 (d, J = 7.0 Hz), 52.16 (d, J =
4.0 Hz), 34.52 (d, J = 12.8 Hz), 32.37 (d, J = 71.9 Hz), 21.26 (d, J = 4.0 Hz), 21.15 (d, J = 4.4
Hz), 18.22, 17.88. 31
P NMR (121 MHz, CDCl3) δ 35 29 IR (neat, cm-1
): 2957 (m), 2871 (w),
1593 (m), 1568 (m), 1452 (s), 1385 (m), 1283 (m), 1174 (s), 1138 (s), 1084 (w), 1071 (w), 927
(m), 805 (m), 751 (s), 730 (s), 688 (m). Optical rotation: []D20
(c 1.45, CHCl3) = +68.7.
HRMS (DART-TOF) m/z: [M + H]+
calcd for C19H27F6NOP 316.18303, found 316.18342.
(S)-(2-Amino-3-methylbutyl)bis(4-methoxyphenyl)phosphine oxide (1.29)
Synthesized according to general procedure using bis(4-methoxyphenyl)phosphine oxide 1.23
(105 mg, 0.4 mmol), sodium tert-butoxide (39 mg, 0.4 mmol) and tert-Butyl (S)-4-isopropyl-
1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide 1.25 (106 mg, 0.4 mmol). The intermediate was
purified by flash chromatography on silica gel (MeOH /CH2Cl2, 1:50 v/v) before treatment of
trifluoroacetic acid. The final product 1.29 (34 mg, 25%) was isolated as a colorless oil.
1H NMR (399 MHz, CDCl3) δ 7 69 – 7.54 (m, 4H), 6.93 (m, 4H), 3.80 (s, 3H), 3.77 (s, 3H), 3.00
(dddd, J = 11.7, 9.6, 4.9, 2.6 Hz, 1H), 2.69 (s, 3H), 2.36 – 2.09 (m, 2H), 1.65 (pd, J = 6.8, 4.8 Hz,
1H), 0.83 (app t, J = 6.7 Hz, 6H). 13
C NMR (100 MHz, CDCl3) δ 162 28 (d J = 2.8 Hz), 162.23
(d, J = 2.9 Hz), 132.73 (d, J = 10.5 Hz), 132.30 (d, J = 10.8 Hz), 125.41 (d, J = 105.3 Hz),
123.37 (d, J = 104.8 Hz), 55.31 (d, J = 1.6 Hz), 55.27 (d, J = 1.7 Hz), 52.00 (d, J = 4.3 Hz),
34.41 (d, J = 12.7 Hz), 33.91 (d, J = 72.8 Hz), 18.16, 17.74. 31
P NMR (121 MHz, CDCl3) δ
33.14. IR (neat, cm-1
): 2956 (m), 2839 (w), 1596 (s), 1570 (m), 1503 (s), 1463 (m), 1408 (m),
1292 (s), 1253 (s), 1173 (s), 1119 (s), 1026 (s), 930 (m), 829 (s), 802 (s), 762 (m), 729 (m), 660
(m). Optical rotation: []D20
(c 3.5, CHCl3) = +36.1. HRMS (DART-TOF) m/z: [M + H]+ calcd
for C19H27NO3P 348.17285, found 348.17327.

43
(S)-(2-Amino-3-methylbutyl)diphenylphosphine oxide (1.30)
Synthesized according to general procedure using diphenylphosphine oxide 1.24 (162 mg, 0.8
mmol), sodium tert-butoxide (77 mg, 0.8 mmol) and tert-Butyl (S)-4-isopropyl-1,2,3-
oxathiazolidine-3-carboxylate 2,2-dioxide 1.25 (212 mg, 0.8 mmol). The intermediate was
purified by flash chromatography on silica gel (MeOH /CH2Cl2, 1:50 v/v) before treatment of
trifluoroacetic acid. The final product 1.30 (123 mg, 54%) was isolated as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 7 73 (m 4H) 7 57 – 7.35 (m, 6H), 3.04 (dddd, J = 12.0, 10.0, 4.9,
2.3 Hz, 1H), 2.48 (br s, 2H), 2.37 (ddd, J = 15.0, 9.2, 2.3 Hz, 1H), 2.26 (ddd, J = 15.0, 12.0, 10.0
Hz, 1H), 1.66 (m, 1H), 0.85 (app t, J = 7.1 Hz, 6H). 13
C NMR (101 MHz, CDCl3) δ 134.00 (d, J
= 98.7 Hz), 132.33 (d, J = 98.3 Hz), 131.77 (app t, J = 2.8 Hz), 130.93 (d, J = 9.1 Hz), 130.47 (d,
J = 9.3 Hz), 128.75 (d, J = 1.5 Hz), 128.63 (d, J = 1.6 Hz), 51.95 (d, J = 4.3 Hz), 34.57 (d, J =
12.7 Hz), 33.79 (d, J = 71.9 Hz), 18.20 , 17.66 . 31
P NMR (162 MHz, CDCl3) δ 32 83 IR (neat,
cm-1
): 2958 (m), 2871 (w), 1401 (m), 1591 (w), 1465 (w), 1437 (s), 1387 (s), 1179 (s), 1119 (s),
1028 (m), 997 (m), 930 (m), 744 (s), 719 (s). Optical rotation: []D20
(c 1.3, CHCl3) = +41.7.
HRMS (DART-TOF) m/z: [M + H]+ calcd for C17H23NOP 288.15173, found 288.15217.
(R)-(2-Amino-2-phenylethyl)bis(4-(trifluoromethyl)phenyl)phosphine oxide (1.31)
Synthesized according to general procedure using bis(4-(trifluoromethyl)phenyl)phosphine oxide
1.21 (244 mg, 0.72 mmol), potassium tert-butoxide (81 mg, 0.72 mmol) and tert-Butyl (R)-4-
phenyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide 1.26 (180 mg, 0.6 mmol). The
intermediate was purified by flash chromatography on silica gel (EtOAc /CH2Cl2, 1:9 v/v) before

44
treatment of trifluoroacetic acid. The final product 1.31 (100 mg, 35%) was isolated as a white
solid.
1H NMR (300 MHz, CDCl3) δ 7 93 – 7.77 (m, 4H), 7.77 – 7.65 (m, 4H), 7.39 – 7.11 (m, 5H),
4.51 (ddd, J = 10.5, 9.0, 4.0 Hz, 1H), 2.90 – 2.60 (m, 2H), 1.94 (s, 2H). 13
C NMR (101 MHz,
CDCl3) δ 144 99 (s) 144 87 (s) 131 29 (d), 130.93 (d), 128.76, 127.78, 126.10, 125.71 (dt, J =
11.9, 3.8 Hz), 51.27 (d, J = 3.4 Hz), 39.52 (d, J = 69.9 Hz). 31
P NMR (121 MHz, CDCl3) δ 28 52
IR (neat, cm-1
): 2900 (w), 1496 (w), 1456 (w), 1400 (m), 1321 (s), 1165 (s), 1120 (s), 1100 (s),
1061 (s), 1017 (s), 835 (m), 789 (m), 768 (m), 742 (m), 708 (m), 696 (s). Optical rotation:
[]D20
(c 0.3, CHCl3) = –18.7. HRMS (DART-TOF) m/z: [M + H]+ calcd for C22H19F6NOP
458.11084, found 458.11092.
General procedures for preparing aminophosphines 1.32–1.36 through silane reduction
(S)-1-(Bis(4-(trifluoromethyl)phenyl)phosphanyl)-3-methylbutan-2-amine (1.32)
Diphenylsilane (0.29 g, 1.58 mmol) was added into a 2 dram vial containing (S)-(2-amino-3-
methylbutyl)bis(4-(trifluoromethyl)phenyl)phosphine oxide 1.27 (96 mg, 0.23 mmol). The
reaction was heated at 140 under inert atmosphere for 14 h. The reaction was allowed to cool
down to room temperature and the crude mixture was directly loaded onto silica gel column. The
eluent was degassed in advance through sparging with argon for 20 min. The crude product was
purified through silica gel flash column chromatography using 50:1 v/v CH2Cl2/MeOH under a
positive pressure of argon. The final product 1.32 (53 mg, 57%) was isolated as a colorless oil.
1H NMR (300 MHz, CDCl3) δ 7 78 – 7.41 (m, 8H), 2.63 (m, 1H), 2.33 (m, 1H), 2.04 (m, 1H),
1.72 (m, 1H), 0.90 (app dd, J = 7.9, 6.8 Hz, 6H). 13
C NMR (101 MHz, CDCl3) δ 143 81 (d J =
15.5 Hz), 142.57 (d, J = 16.8 Hz), 133.50 (d, J = 19.8 Hz), 132.64 (d, J = 18.4 Hz), 131.23 (d, J
= 32.4 Hz), 130.69 (d, J = 32.6 Hz), 125.89 – 124.91 (m), 54.19 (d, J = 13.1 Hz), 34.58 (d, J =

45
7.4 Hz), 34.37 (d, J = 12.3 Hz), 18.85, 17.05. 31
P NMR (121 MHz, Chloroform-d) δ -20.39.
HRMS (DART-TOF) m/z: [M + H]+ calcd for C19H21F6NP 408.13158, found 408.13147.
(S)-1-(Di-o-tolylphosphanyl)-3-methylbutan-2-amine (1.33)
Synthesized according to general procedure using diphenylsilane (0.26 g, 1.4 mmol) and (S)-(2-
amino-3-methylbutyl)di-o-tolylphosphine oxide 1.28 (63 mg, 0.20 mmol). Purified by flash
chromatography on silica gel (MeOH /CH2Cl2, 1:50 v/v) to afford 1.33 (36 mg, 61%) was
isolated as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 7 25 – 7.10 (m, 8H), 2.81 – 2.63 (m, 1H), 2.45 (s, 3H), 2.43 (s,
3H), 2.25 (ddd, J = 13.8, 4.4, 1.6 Hz, 1H), 1.94 (ddd, J = 13.8, 9.2, 2.9 Hz, 1H), 1.83 (m, 1H),
0.95 (d, J = 6.8 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H). 13
C NMR (101 MHz, CDCl3) δ 142 74 (d J =
25.7 Hz), 142.00 (d, J = 25.6 Hz), 137.18 (d, J = 12.3 Hz), 136.38 (d, J = 13.4 Hz), 131.22 (d, J
= 7.7 Hz), 130.12 (d, J = 2.2 Hz), 130.07 (d, J = 2.0 Hz), 128.44 (d, J = 11.7 Hz), 126.07 (d, J =
0.8 Hz), 126.04(d, J = 0.8 Hz), 54.05 (d, J = 14.0 Hz), 34.28 (d, J = 7.4 Hz), 33.37 (d, J = 12.1
Hz), 21.40 (d, J = 7.7 Hz), 21.18 (d, J = 7.4 Hz), 19.02 , 17.04. 31
P NMR (162 MHz, CDCl3) δ -
44.02. IR (neat, cm-1
): 3055 (w), 2956 (m), 1589 (w), 1466 (m), 1451 (m), 1378 (m), 1270 (w),
1200 (w), 1130 (m), 1032 (m), 910 (w), 747 (s), 719 (m). Optical rotation: []D20
(c 1.25,
CHCl3) = +82.4. HRMS (DART-TOF) m/z: [M + H]+ calcd for C19H27NP 300.18811, found
300.18886.
(S)-1-(Bis(4-methoxyphenyl)phosphanyl)-3-methylbutan-2-amine (1.34)

46
Synthesized according to general procedure using diphenylsilane (0.30 g, 1.61 mmol) and (S)-(2-
amino-3-methylbutyl)bis(4-methoxyphenyl)phosphine oxide 1.29 (79 mg, 0.23 mmol). Purified
by flash chromatography on silica gel (MeOH /CH2Cl2, 1:30 v/v) to afford 1.34 (30 mg, 40%)
was isolated as a colorless oil.
1H NMR (300 MHz, CDCl3) δ 7 44 – 7.31 (m, 4H), 6.93 – 6.80 (m, 4H), 3.80 (s, 3H), 3.78 (s,
3H), 2.66 (m, 1H), 2.25 (m, 1H), 1.99 (m, 1H), 1.75 (m, 1H), 1.26 (br s, 2H), 0.91 (d, J = 9.1 Hz,
3H), 0.88 (d, J = 9.0 Hz, 3H). 13
C NMR (101 MHz, CDCl3) δ 160 28, 159.93, 134.50 (d, J =
20.6 Hz), 133.78 (d, J = 19.5 Hz), 130.20 (d, J = 9.3 Hz), 129.07 (d, J = 10.4 Hz), 114.20 (d, J =
4.7 Hz), 114.12 (d, J = 4.1 Hz), 55.18, 54.22, 54.08, 35.06 (d, J = 11.1 Hz), 34.19 (d, J = 7.6 Hz),
18.90, 17.17. 31
P NMR (121 MHz, CDCl3) δ -25.59. IR (neat, cm-1
): 2956 (m), 2835 (w), 1593
(s), 1568 (m), 1497 (s), 1462 (m), 1441 (m), 1401 (w), 1282 (s), 1245 (s), 1176 (s), 1094 (s),
1030 (s), 910 (w), 825 (s), 797 (m), 732 (m). Optical rotation: []D20
(c 2.65, CHCl3) = +71.4.
HRMS (DART-TOF) m/z: [M + H]+ calcd for C19H27NO2P 332.17794, found 332.17812.
(S)-1-(Diphenylphosphanyl)-3-methylbutan-2-amine (1.35)
Synthesized according to general procedure using diphenylsilane (0.36 g, 1.95 mmol) and (S)-(2-
amino-3-methylbutyl)diphenylphosphine oxide 1.30 (80 mg, 0.28 mmol). Purified by flash
chromatography on silica gel (MeOH /CH2Cl2, 1:30 v/v) to afford 1.35 (35 mg, 46%) was
isolated as a yellow oil.
1H NMR (400 MHz, CDCl3) δ 7 48 – 7.20 (m, 10H), 2.60 (m, 1H), 2.25 (m, 1H), 1.94 (m, 1H),
1.68 (m, 1H), 0.82 (app dd, 6H). 13
C NMR (101 MHz, CDCl3) δ 133 31 133 12 132 57 132 38
128.88, 128.54, 128.48, 128.46, 128.40, 128.38, 54.27, 54.13, 34.53, 34.41, 34.17, 34.09, 18.89,
17.11. 31
P NMR (162 MHz, CDCl3) δ -21.62.
(R)-2-(Bis(4-(trifluoromethyl)phenyl)phosphanyl)-1-phenylethan-1-amine (1.36)

47
Synthesized according to general procedure using diphenylsilane (0.69 g, 3.72 mmol) and (R)-(2-
amino-2-phenylethyl)bis(4-(trifluoromethyl)phenyl)phosphine oxide 1.31 (243 mg, 0.53 mmol).
Purified by flash chromatography on silica gel (MeOH /CH2Cl2, 1:50 v/v) to afford 1.36 (119 mg,
51%) was isolated as a slightly yellow solid.
1H NMR (300 MHz, CDCl3) δ 7 63 – 7.55 (m, 4H), 7.55 – 7.44 (m, 4H), 7.41 – 7.16 (m, 5H),
4.03 (m, 1H), 2.67 – 2.41 (m, 2H), 1.82 (s, 2H). 13
C NMR (100 MHz, CDCl3) δ 133 31 (d J =
19.6 Hz), 132.81 (d, J = 18.9 Hz), 128.68 (s), 127.53(s), 126.05(s), 125.32 (m), 53.87 (d, J =
16.5 Hz), 39.43 (d, J = 14.7 Hz). 31
P NMR (121 MHz, CDCl3) δ -21.05. IR (neat, cm-1
): 2935
(w), 2870 (w), 1606 (m), 1454 (w), 1398 (m), 1321 (s), 1165 (s), 1119 (s), 1104 (s), 1059 (s),
1015 (s), 952 (m), 879 (m), 828 (s), 769 (m), 724 (m), 699 (s). Optical rotation: []D20
(c 0.8,
CHCl3) = -26.0. HRMS (DART-TOF) m/z: [M + H]+ calcd for C22H19F6NP 442.11593, found
442.11624.
(S)-N-(1-(bis(4-(trifluoromethyl)phenyl)phosphanyl)-3-methylbutan-2-yl)cyclohexanamine
(1.38)
Cyclohexanone (46 mg, 0.47 mmol), sodium triacetoxyborohydride (125 mg, 0.59 mmol) and
acetic acid (4.8 mg, 0.08 mmol) were added sequentially to a solution of (S)-(2-amino-3-
methylbutyl)bis(4-(trifluoromethyl)phenyl)phosphine oxide 1.27 (168 mg, 0.39 mmol) in CH2Cl2
(2 mL). The reaction was stirred at room temperature for 2 h. Saturated Na2CO3 was added to the
reaction. The aqueous layer was extracted with CH2Cl2. The combined organic layer was dried
over anhydrous MgSO4, concentrated and purified through flash column chromatography on

48
silica gel (MeOH /CH2Cl2, 1:50 v/v) to afford 1.37 as a white solid (169 mg, 82%). 1.37 was
reduced with diphenylsilane (0.41 g, 2.23 mmol) using the general procedure to afford 1.38 (116
mg, 75%) as a colorless oil after column (MeOH /CH2Cl2, 1:50 v/v).
1H NMR (400 MHz, CDCl3) δ 7 65 – 7.54 (m, 6H), 7.54 – 7.47 (m, 2H), 2.63 – 2.48 (m, 1H),
2.35 (tt, J = 10.1, 3.3 Hz, 1H), 2.25 (ddd, J = 13.8, 4.7, 2.0 Hz, 1H), 2.03 (ddd, J = 14.0, 8.6, 1.9
Hz, 1H), 1.99 – 1.89 (m, 1H), 1.70 – 1.58 (m, 4H), 1.59 – 1.44 (m, 1H), 1.22 – 1.04 (m, 3H),
1.04 – 0.90 (m, 2H), 0.89 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.9 Hz, 3H). 13
C NMR (101 MHz,
CDCl3) δ 144 39 (d = 16 6 H ) 143 24 (d = 16 9 H ) 133 44 (d = 19 4 H ) 132 79 (d =
18.4 Hz), 131.25 (d, J = 42.9 Hz), 130.98 (d, J = 32.4 Hz), 130.55 (d, J = 32.5 Hz), 130.28 (d, J =
43.0 Hz), 125.79 – 124.60 (m), 56.42 (d, J = 12.8 Hz), 53.84, 34.31, 33.64, 30.80 (d, J = 13.0
Hz), 30.54 (d, J = 7.2 Hz), 26.07, 24.99 (d, J = 4.8 Hz), 18.41, 16.95. 31
P NMR (162 MHz,
CDCl3) δ -20.14.
tert-Butyl ((1R,2R)-2-bromo-1,2-diphenylethyl)carbamate (1.40)
Method A.
Tetra-n-butylammonium bromide (129 mg, 0.4 mmol) was added to a solution of tert-butyl
(4R,5S)-4,5-diphenyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide 1.39 (75 mg, 0.2 mmol) in
THF (1.5 mL). The reaction was stirred at room temperature for 10 h. 2N H2SO4 (1 mL) was
added to the solution and stirred for 20 min. Extraction with diethyl ether followed by
purification by column chromatography (Hexanes /EtOAc, 9:1 v/v) afforded 1.40 (22 mg, 30%)
as a white solid.
Method B.
18-Crown-6 (590 mg, 2.23 mmol) was added to a suspension of potassium bromide (664 mg,
5.58 mmol) and tert-butyl (4R,5S)-4,5-diphenyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide
1.39 (419 mg, 1.12 mmol) in THF (6 mL). The reaction was stirred at 60 °C for 5 h. Water was
added to the reaction and the aqueous layer was extracted with diethyl ether. The combined

49
organic layer was dried over anhydrous MgSO4, concentrated in vacuo to afford 1.40 (289 mg,
70%) as a white solid. The product was used in the next step without purification.
1H NMR (400 MHz, CDCl3) δ 7 42 – 7.34 (m, 2H), 7.33 – 7.24 (m, 6H), 7.24 – 7.18 (m, 2H),
5.54 (d, J = 8.6 Hz, 1H), 5.24 (br, 2H), 1.43 (s, 9H). 13
C NMR (100 MHz, CDCl3) δ 154 97
138.89, 128.79, 128.50, 128.42, 128.35, 128.28, 128.13, 127.70, 126.92, 80.05, 60.24, 28.35.
HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C19H22BrNNaO2 398.0726, found 398.0722.
((1S,2R)-2-Amino-1,2-diphenylethyl)bis(4-(trifluoromethyl)phenyl)phosphine oxide (1.41)
Bis(4-(trifluoromethyl)phenyl)phosphine oxide 1.21 (253 mg, 0.75 mmol) was added to a
solution of sodium tert-butoxide (72 mg, 0.75 mmol) in degassed THF (5 mL) under inert
atmosphere. The mixture was stirred for 5 min. tert-Butyl ((1R,2R)-2-bromo-1,2-
diphenylethyl)carbamate 1.40 solid (281 mg, 0.75 mmol) was added and the reaction mixture
was stirred at 60 °C for 3 h. The reaction was quenched with 2N H2SO4 (5 mL) at room
temperature and the mixture was stirred for another 20 min. The aqueous layer was extracted
with diethyl ether. The combined organic layer was dried over anhydrous MgSO4, concentrated
in vacuo to afford a white solid (460 mg). Trifluoroacetic acid (1.48 mL, 19.4 mmol) was added
to a solution of the crude product in dry CH2Cl2 (10 mL) at 0 °C . The reaction was allowed to
warm up and stirred at room temperature for 3 h. Saturated Na2CO3 was added dropwise to the
solution at 0 °C . Extraction with CH2Cl2 followed by purification by flash chromatography on
silica gel (MeOH /CH2Cl2, 1:40 v/v) afford the final product 1.41 (140 mg, 35%) as a white solid.
1H NMR (300 MHz, CDCl3) δ 7 81 (dd J = 10.6, 8.1 Hz, 2H), 7.63 – 7.51 (m, 4H), 7.42 (dd, J =
8.4, 2.5 Hz, 2H), 7.38 – 7.30 (m, 2H), 7.17 (dd, J = 4.9, 1.9 Hz, 3H), 7.12 – 7.06 (m, 2H), 7.02
(m, 3H), 4.85 (t, J = 6.6 Hz, 1H), 3.78 (t, J = 6.4 Hz, 1H), 1.71 (s, 2H). 13
C NMR (100 MHz,
CDCl3) δ 141 86 (d J = 6.9 Hz), 137.14 (d, J = 22.2 Hz), 136.20 (d, J = 18.3 Hz), 133.57 –

50
132.45 (m), 131.15 (d, J = 9.0 Hz), 130.94 (d, J = 8.8 Hz), 130.66 (d, J = 7.0 Hz), 128.53 (d, J =
1.3 Hz), 128.12 , 127.82 , 127.75 (d, J = 1.9 Hz), 127.41 , 125.47 – 125.11 (m), 125.10 – 124.70
(m), 56.16 , 54.46 (d, J = 69.2 Hz). 31
P NMR (121 MHz, CDCl3) δ 29 26 IR (neat, cm-1
): 3065
(w), 3025 (w), 1401 (m), 1321 (s), 1169 (s), 1123 (s), 1062 (s), 1369 (w), 1018 (m), 837 (m), 791
(m), 756 (m), 710 (s), 670 (s). Optical rotation: []D20
(c 0.79, CHCl3) = +13.9. HRMS (ESI-
TOF) m/z: [M + H]+ calcd for C28H23F6NOP 534.1416, found 534.1420.
(1R,2S)-1-((tert-Butoxycarbonyl)amino)-indan-2-yl trifluoromethanesulfonate (1.44)
A solution of (Boc)2O (321 mg, 1.47 mmol) in THF (2 mL) was added to a mixture of (1R,2S)-
(+)-cis-1-amino-2-indanol 1.42 (200 mg, 1.34 mmol) and sodium carbonate (0.31 g, 2.9 mmol)
in THF/H2O (1/1, 4 mL) at 0 ºC. The mixture was stirred at room temperature for 3 h. Water was
added to the mixture and was extracted with ethyl acetate. The combined organic layer was
washed with brine, dried over anhydrous MgSO4, and concentrated to afford the crude product in
quantitative yield as a white solid, which was used in the next step without purification.
Trifluoromethanesulfonic anhydride (56 mg, 0.2 mmol) was added dropwise to a solution of
pyridine (19 mg, 0.24 mmol) and N-Boc protected (1R,2S)-(+)-cis-1-amino-2-indanol (50 mg,
0.2 mmol) in dry CH2Cl2 (1 mL) at –40 ºC. The reaction was stirred at –40 ºC for 1 h. Saturated
NH4Cl was added to quench the reaction. The aqueous layer was extracted with CH2Cl2. The
combined organic layer was dried over anhydrous MgSO4, and concentrated to afford the
product 1.44 (67mg, 89%) as a yellow or orange solid.
1H NMR (300 MHz, CDCl3) δ 7 31 (m 4H) 5 67 (br s 1H) 5 46 (dd J = 9.3, 4.5 Hz, 1H), 5.04
(d, J = 9.3 Hz, 1H), 3.45 – 3.14 (m, 2H), 1.51 (s, 9H). 19
F NMR (282 MHz, CDCl3) δ -75.13.

51
((1R,2R)-1-Amino-indan-2-yl)bis(4-(trifluoromethyl)phenyl)phosphine oxide (1.45)
Bis(4-(trifluoromethyl)phenyl)phosphine oxide 1.21 (494 mg, 1.46 mmol) was added to a
solution of sodium tert-butoxide (140 mg, 1.46 mmol) in degassed THF (8 mL) under inert
atmosphere. The mixture was stirred for 5 min. (1R,2S)-1-((tert-Butoxycarbonyl)amino)-indan-
2-yl trifluoromethanesulfonate 1.44 solid (556 mg, 1.46 mmol) was added and the reaction
mixture was stirred at 60 ºC for 3 h. The reaction was quenched with 2N H2SO4 (8 mL) at room
temperature and the mixture was stirred for another 10 min. Extraction with diethyl ether
followed by purification by flash chromatography on silica gel (EtOAc /CH2Cl2, 1:9 v/v) afford
the final product 1.45 (216 mg, 26%) as a white solid.
1H NMR (300 MHz, CDCl3) δ 7 82 (t J = 9.1 Hz, 2H), 7.76 – 7.64 (m, 4H), 7.61 (dd, J = 8.5,
2.7 Hz, 2H), 7.30 – 7.20 (m, 1H), 7.15 (t, J = 7.4 Hz, 1H), 7.07 (t, J = 8.5 Hz, 2H), 5.84 (d, J =
6.2 Hz, 1H), 3.04 – 2.68 (m, 3H), 2.07 (dt, J = 17.4, 9.0 Hz, 1H), 1.55 – 1.39 (m, 2H), 1.25 (s,
9H). 31
P NMR (121 MHz, CDCl3) δ 32 83 HRMS (ESI-TOF) m/z: [M + H]+ calcd for
C28H27F6NO3P 570.1627, found 570.1614.
(R)-N-(2-((tert-Butyldimethylsilyl)oxy)-1-phenylethyl)-1,1,1-trifluoromethanesulfonamide
(1.49)
To a stirred solution of the (R)-2-amino-2-phenylethan-1-ol (685 mg, 5 mmol) in CH2Cl2 (10 mL)
was added triethylamine (556 mg, 5.5 mmol) at -78 °C under inert atmosphere. After stirring for
10 min at -78 °C, trifluoromethanesulfonic anhydride (1.41 g, 5 mmol) was added dropwise and
the mixture was stirred for 1 h at that temperature before being quenched by water (20 mL). The
organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined

52
organic phase was washed with brine, dried over anhydrous MgSO4, and concentrated to give the
trifluoromethanesulfonamide without purification.
To a 50 mL round bottle was added the crude product from the previous step, tert-
butyldimethylsilyl chloride (825 mg, 5.5 mmol), DMAP (60 mg, 0.5 mmol) and CH2Cl2 (12 mL).
The mixture was cooled to 0 °C and Et3N (560 mg, 5.5 mmol) was added dropwise. The solution
was warmed to room temperature and stirred for 12 h. Then the reaction was quenched with
water. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The
combined organic layer was washed with brine and concentrated under reduced pressure to give
the crude product, which was purified by column chromatography on silica gel (EtOAc/hexanes,
1:20 v/v) to afford 1.49 (1.19 g, 62% over 2 steps) as a colorless oil.
1H NMR (300 MHz, CDCl3) δ 7 43 – 7.27 (m, 5H), 5.91 (d, J = 6.5 Hz, 1H), 4.72 (m, 1H), 3.97
(dd, J = 10.4, 4.2 Hz, 1H), 3.79 (dd, J = 10.4, 4.7 Hz, 1H), 0.86 (s, 9H), -0.03 (s, 3H), -0.04 (s,
3H).
Benzoyl-D-leucine (1.51)
Benzoyl chloride (281 mg, 2 mmol) was added dropwise to a suspension of D-leucine (262 mg, 2
mmol) in aqueous 2N NaOH (5 mL) at 0 °C. The reaction was stirred at room temperature for 3h.
Then the reaction was acidified with 1N HCl until pH<2. Extraction with EtOAc followed by
column chromatography on silica gel (MeOH/ CH2Cl2, 1:50 v/v) afforded product 1. 51 (176 mg,
75%) as a white solid.
1H NMR (399 MHz, CDCl3) δ 10 79 (br s, 1H), 7.77 (d, J = 7.3 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H),
7.38 (t, J = 7.5 Hz, 2H), 6.88 (d, J = 8.0 Hz, 1H), 4.82 (td, J = 8.1, 4.2 Hz, 1H), 2.02 – 1.57 (m,
2H), 0.96 (d, J = 2.1 Hz, 3H), 0.95 (d, J = 2.2 Hz, 3H).
4.2 1H NMR, 13C NMR and 31P NMR Spectra
53
54
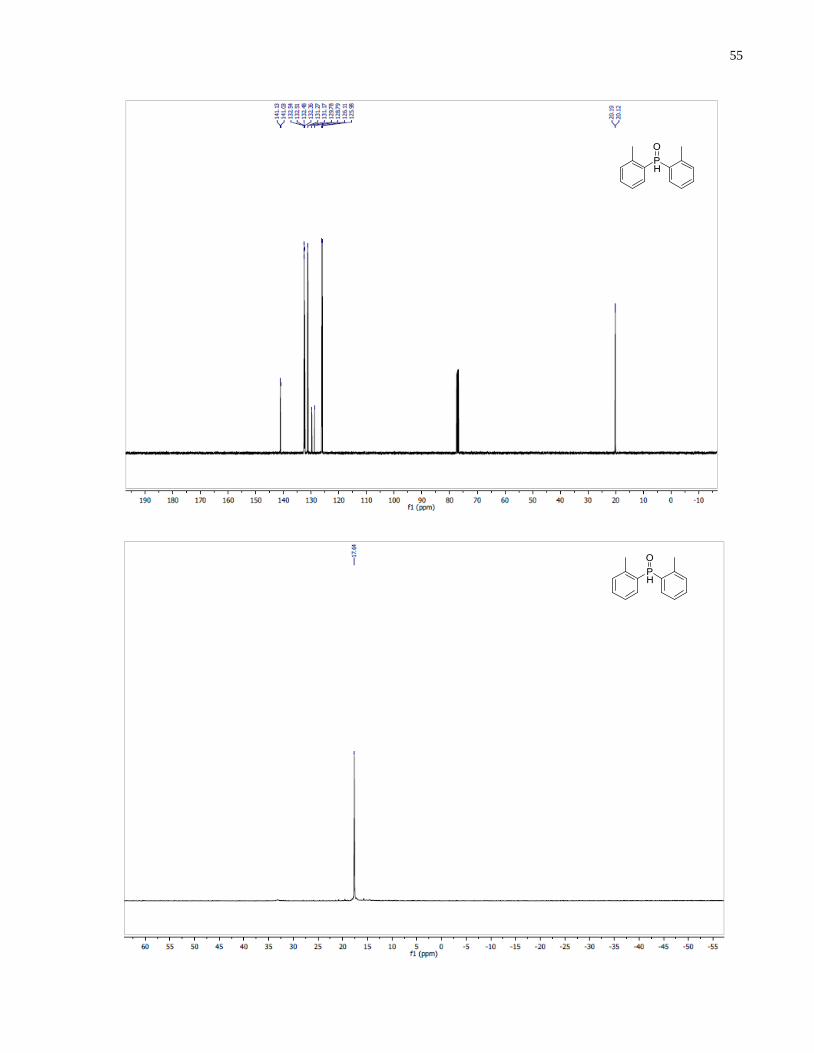
55
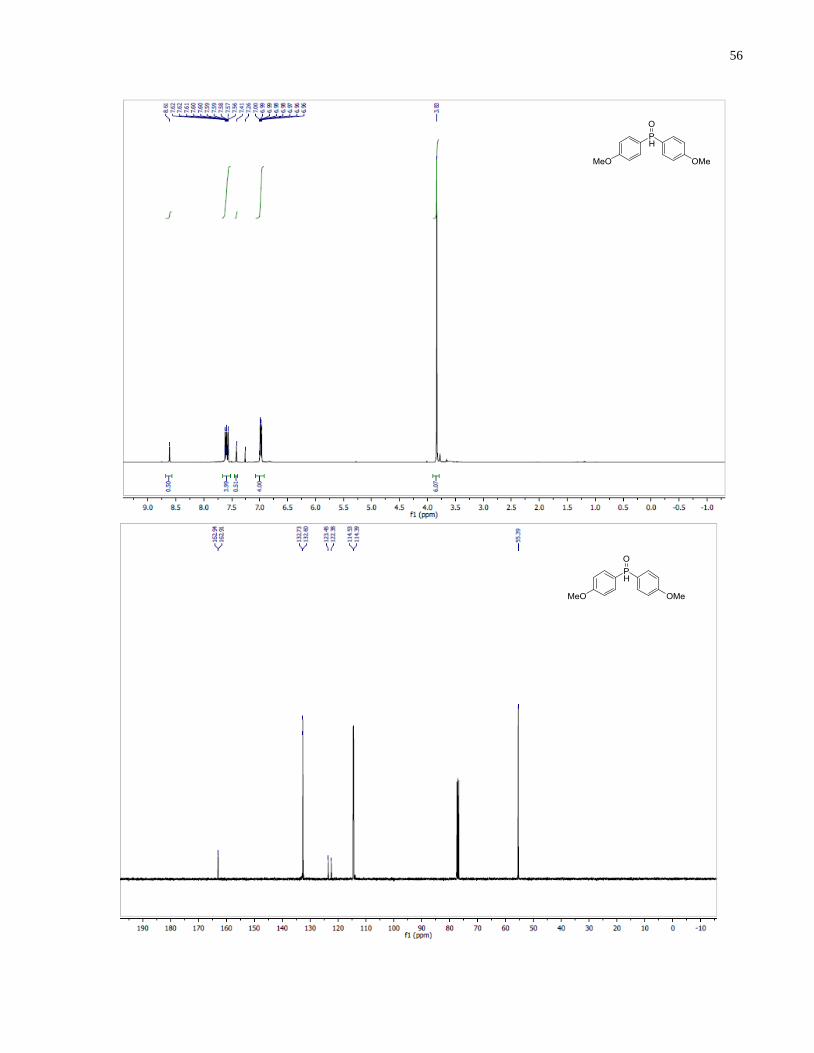
56
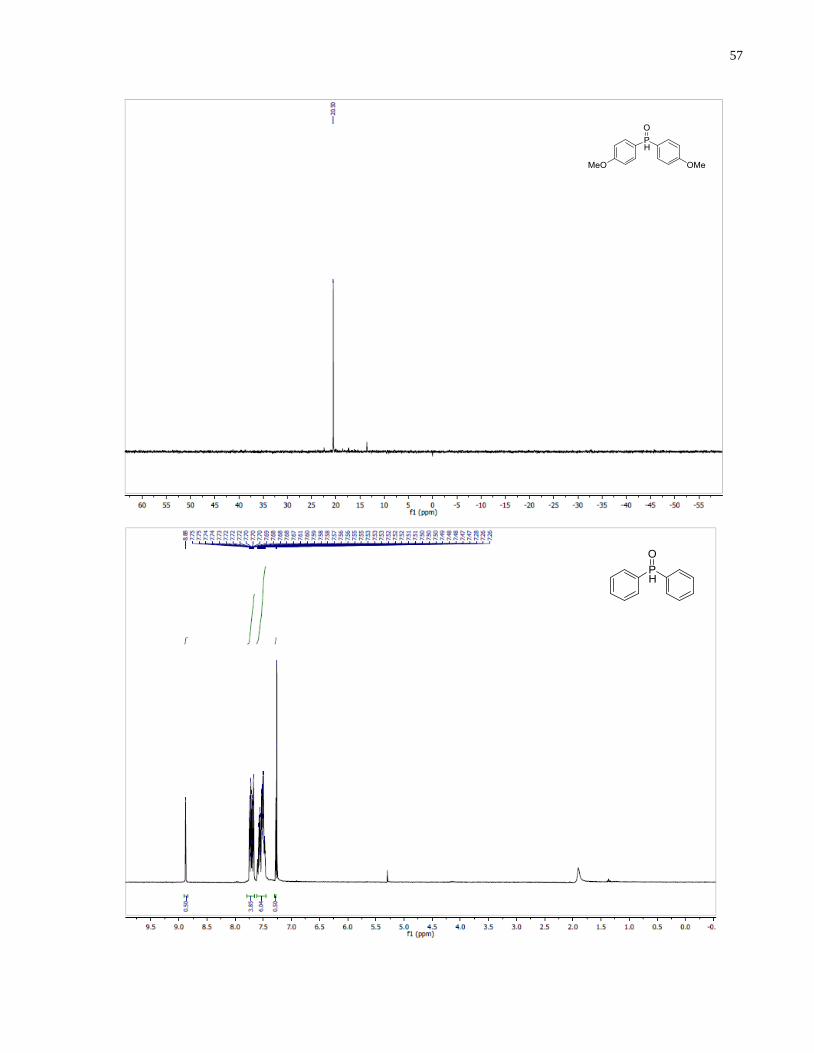
57
58
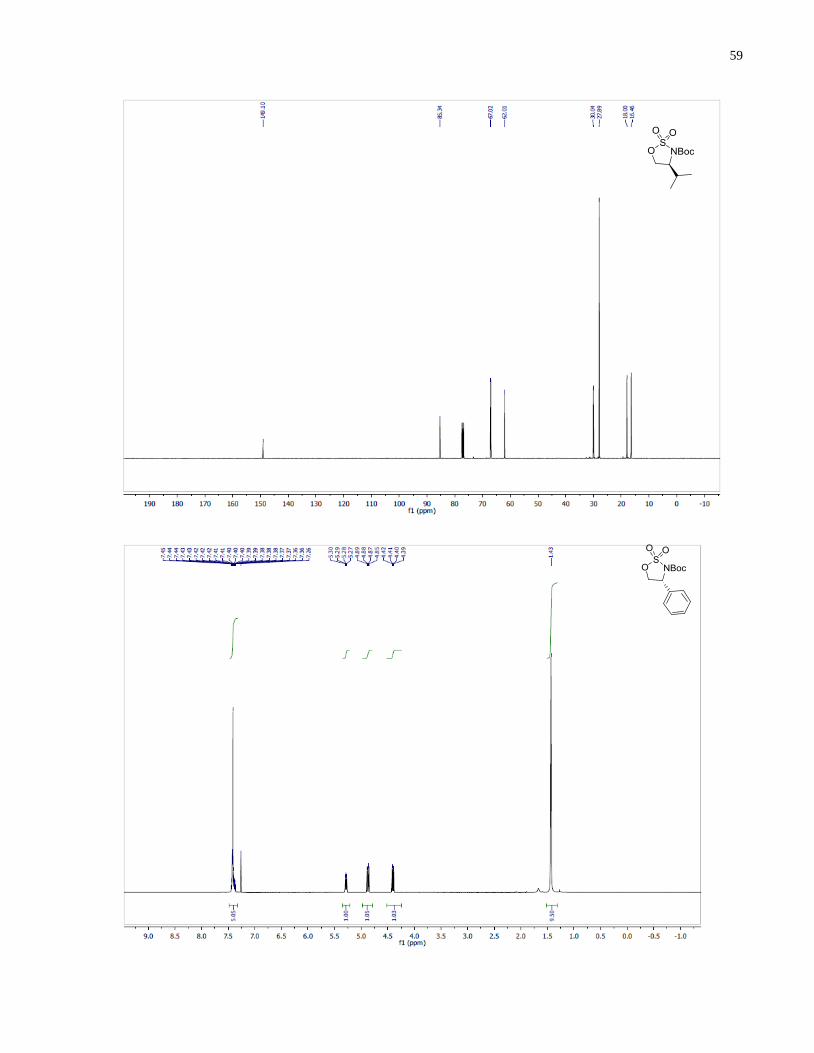
59
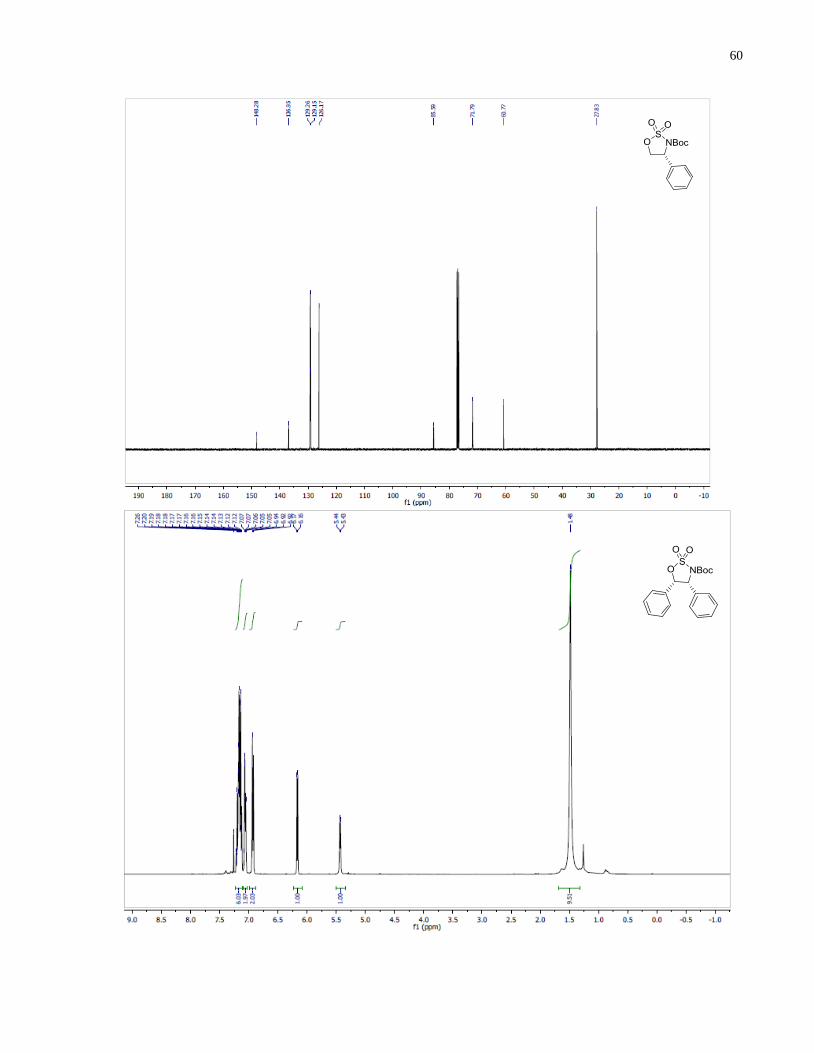
60
61
62
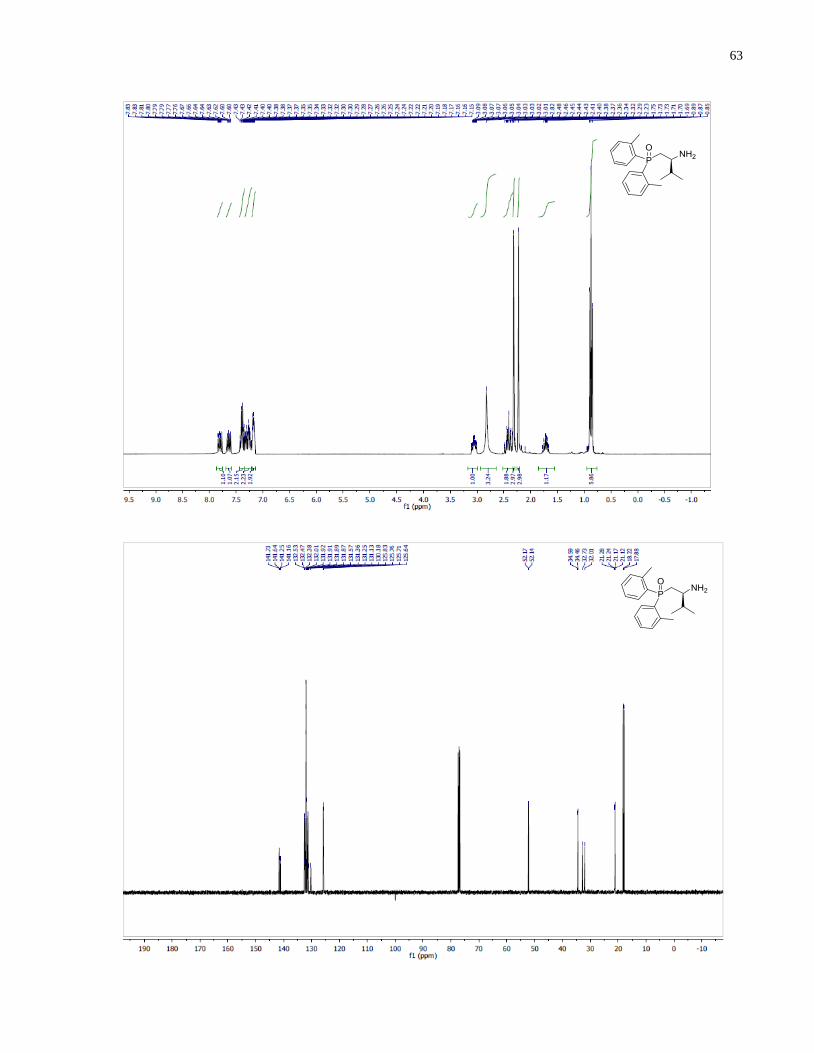
63
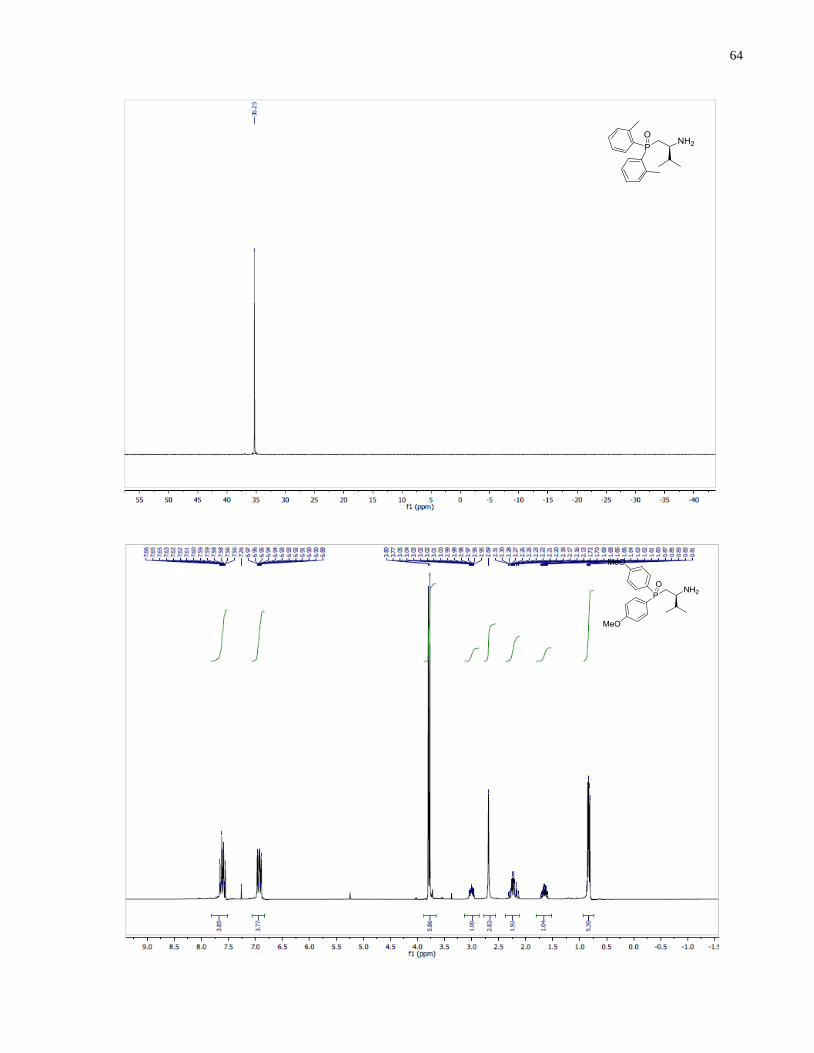
64
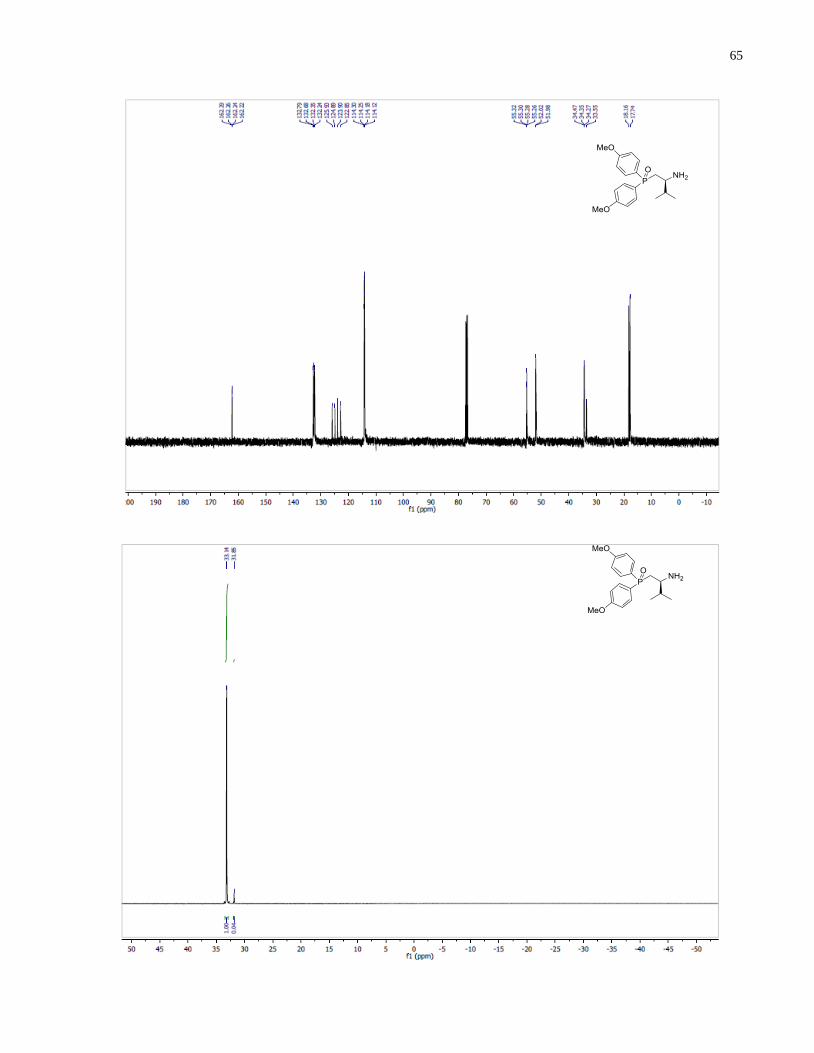
65
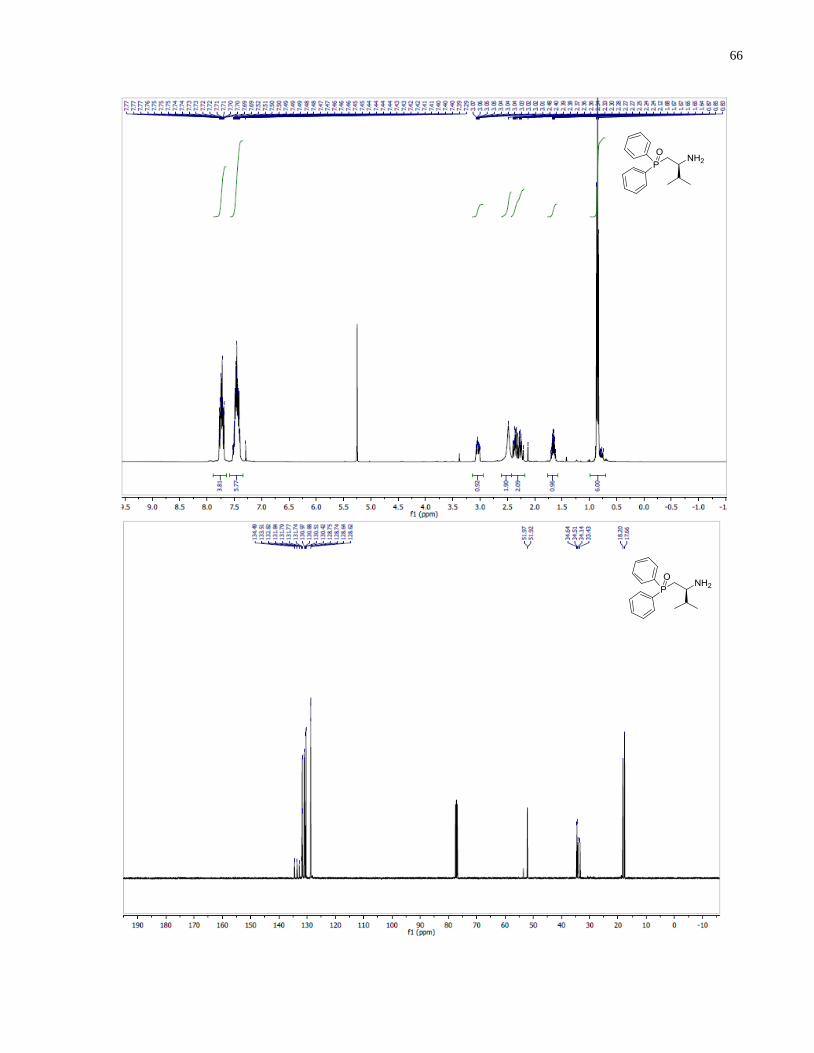
66
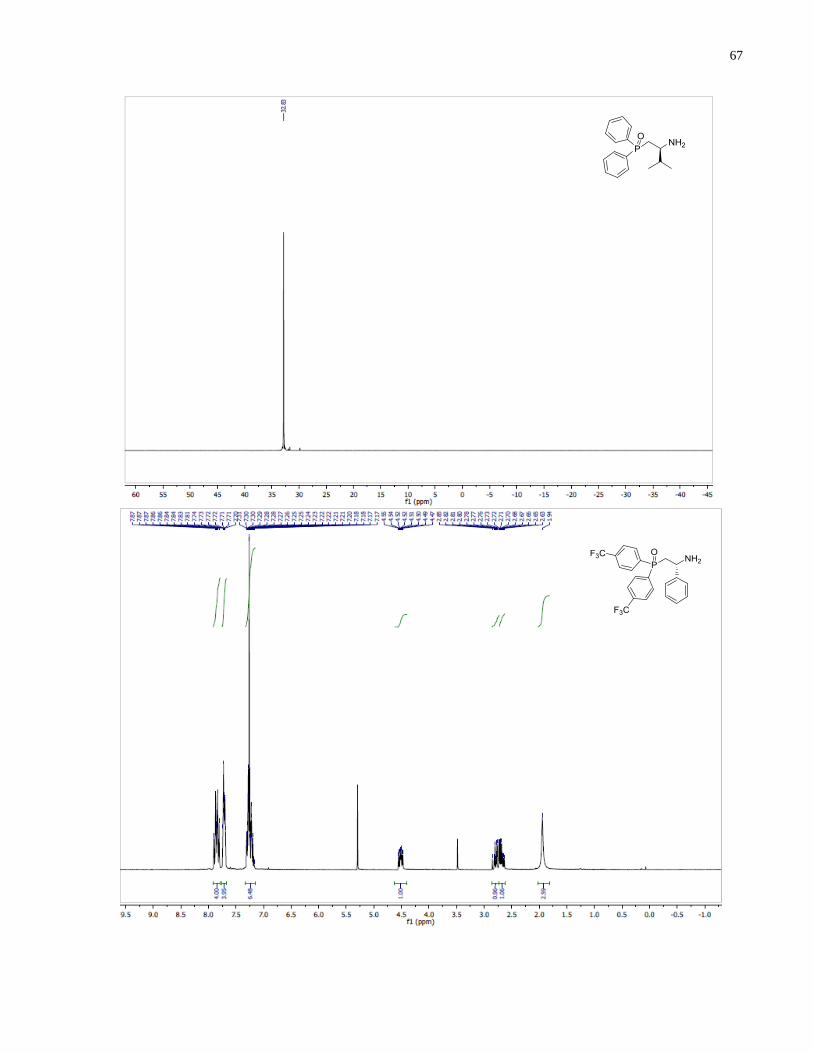
67
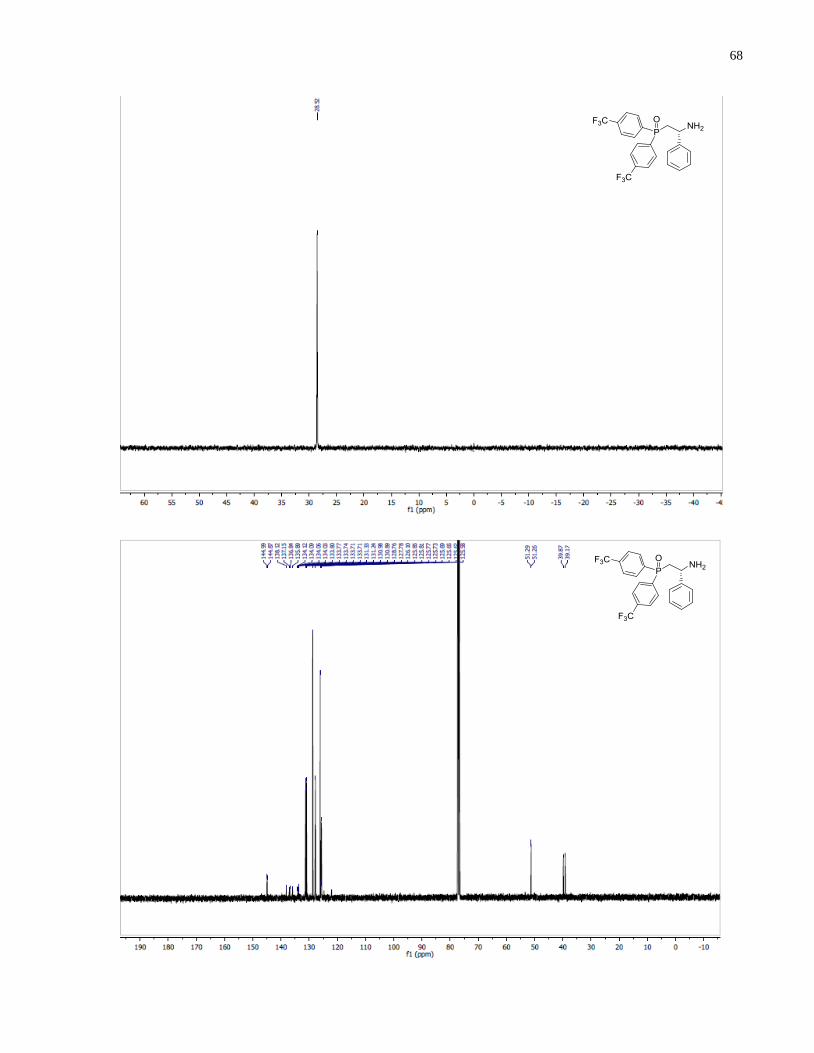
68
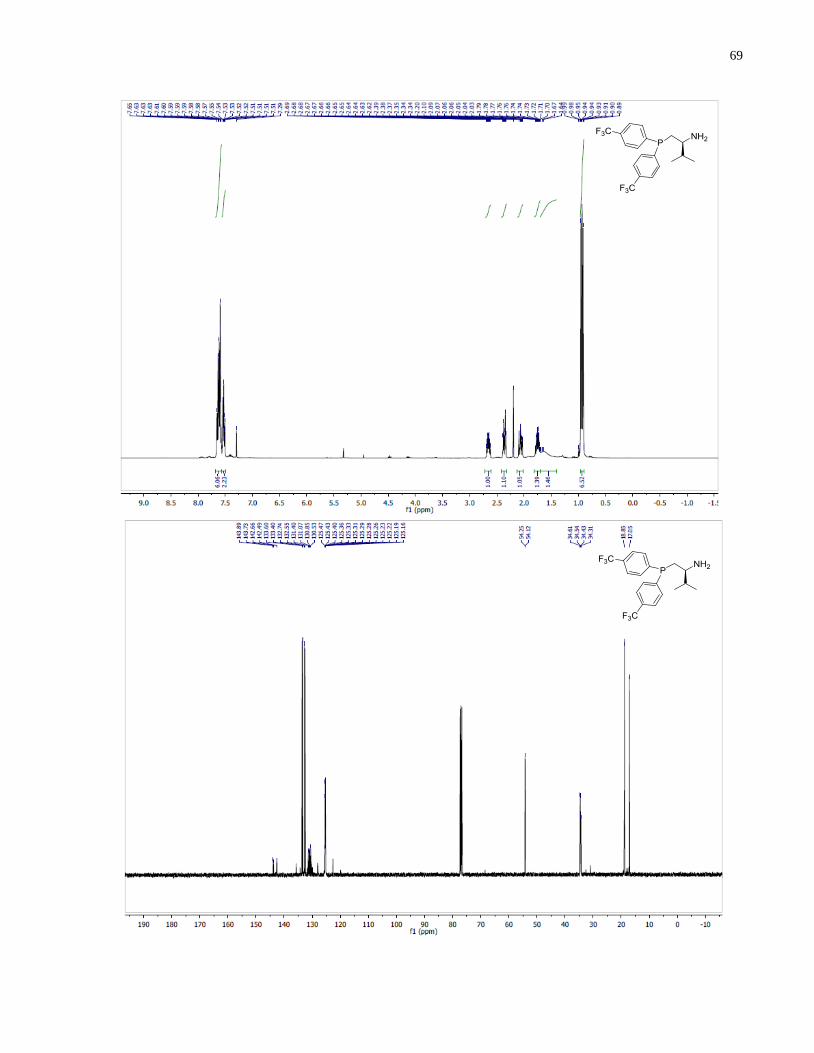
69
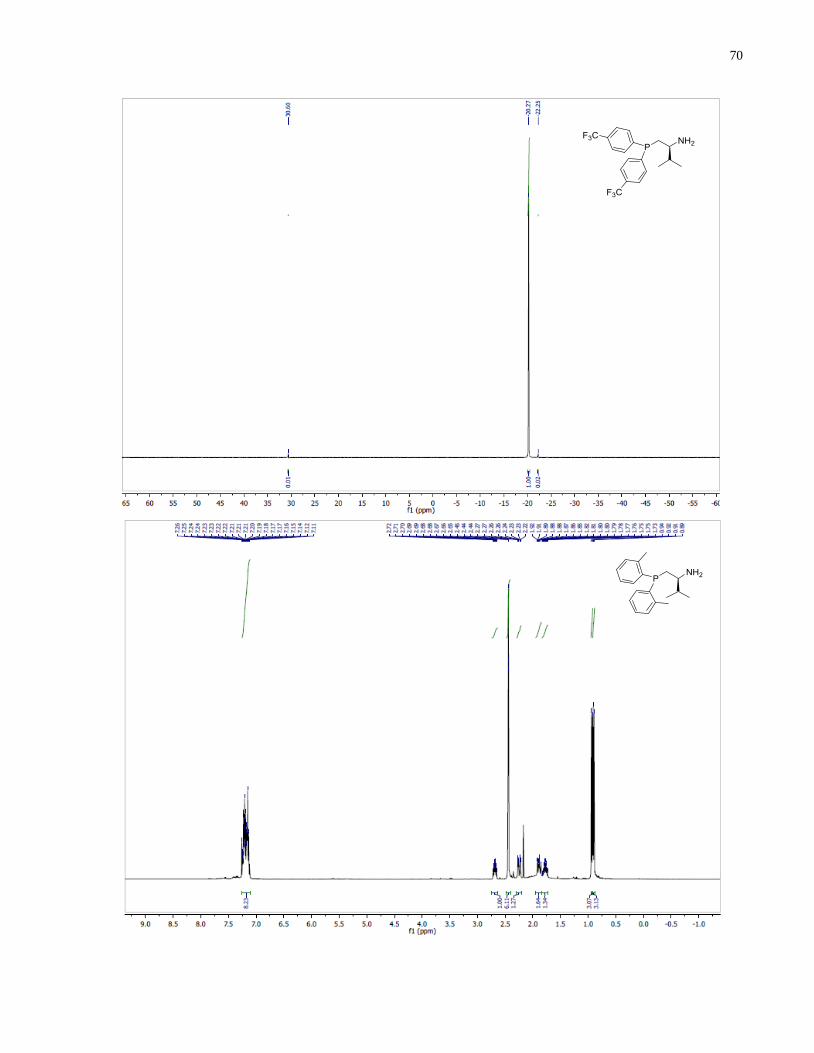
70
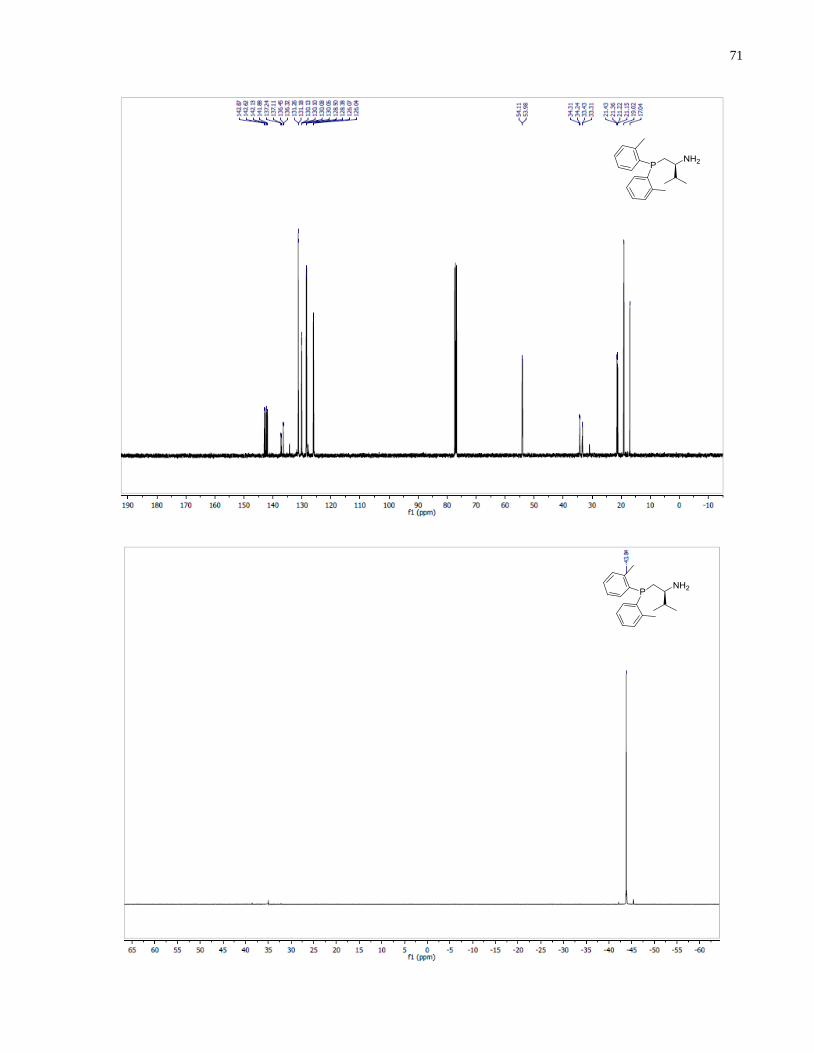
71
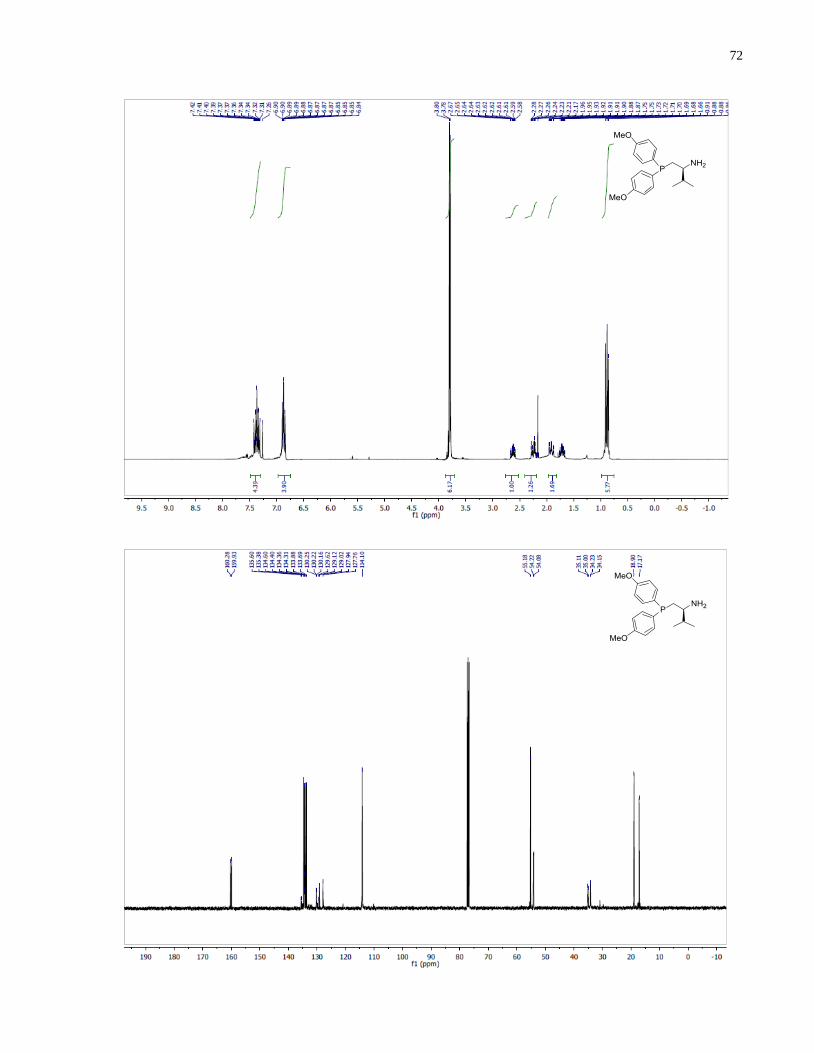
72
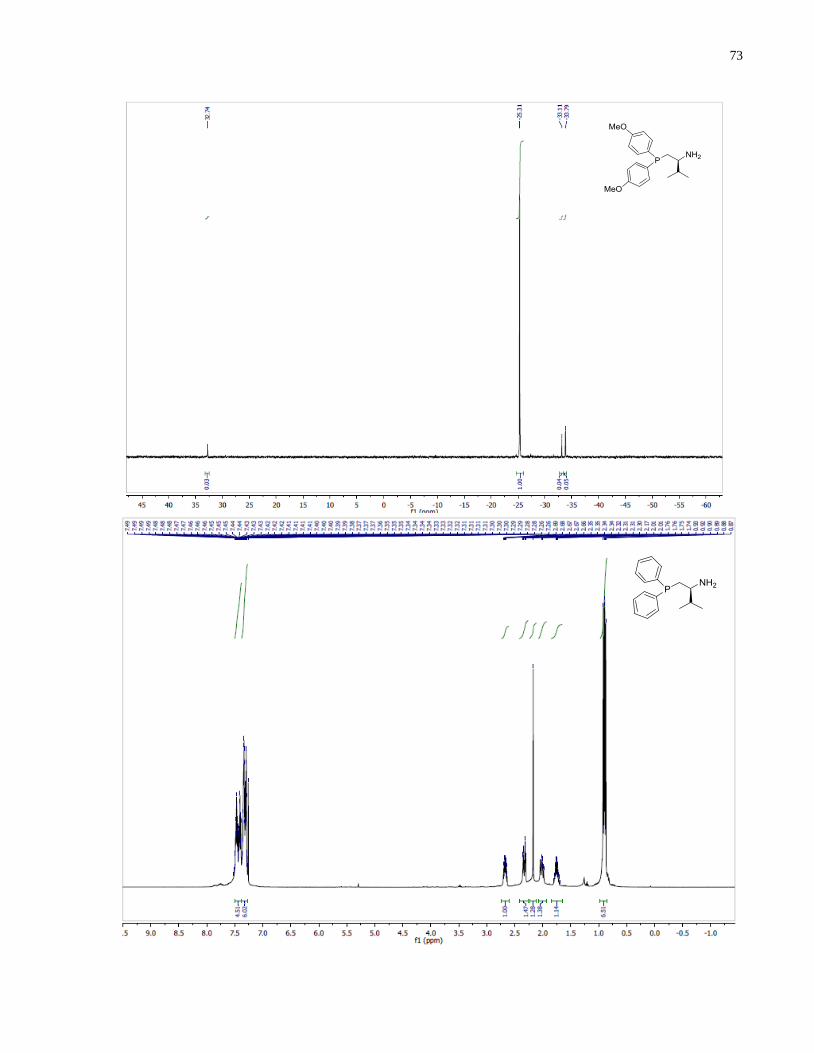
73
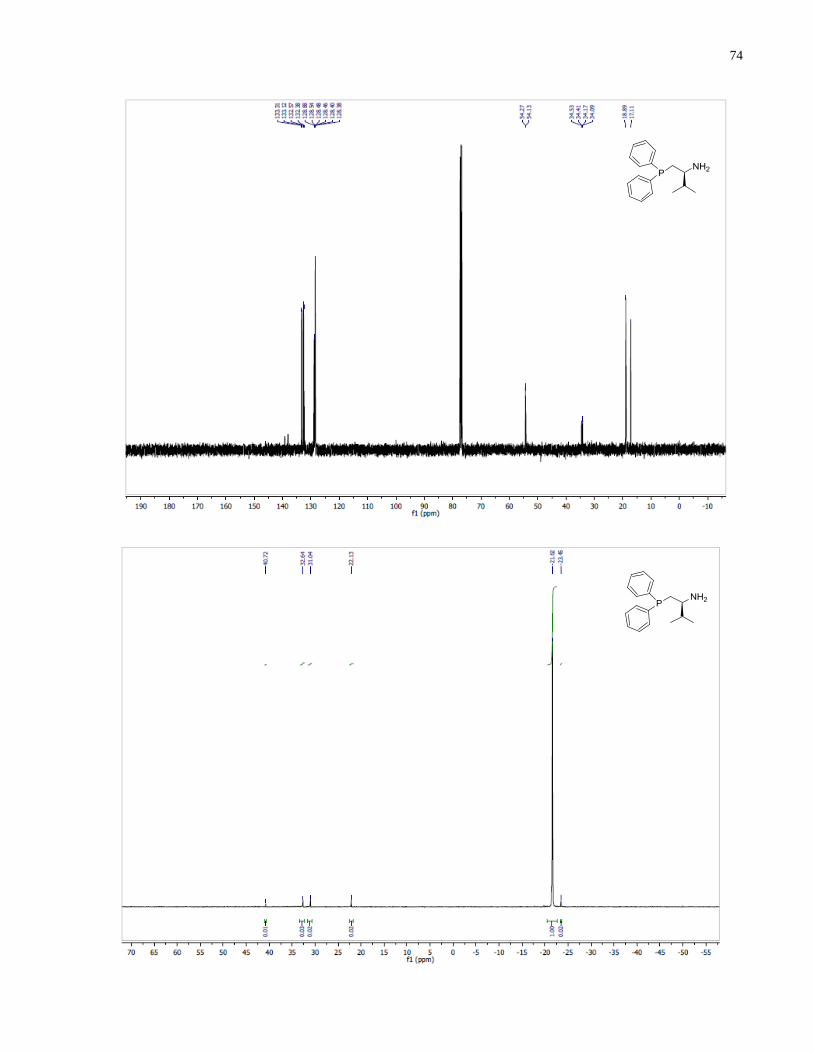
74
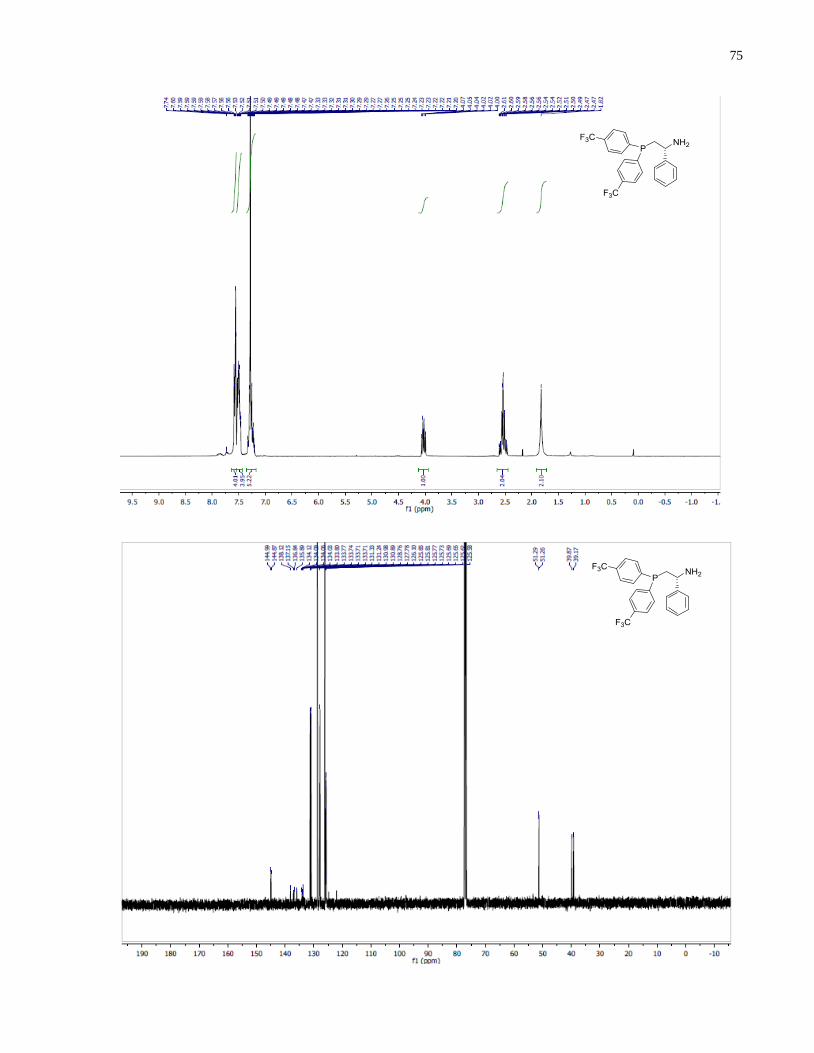
75
76
77
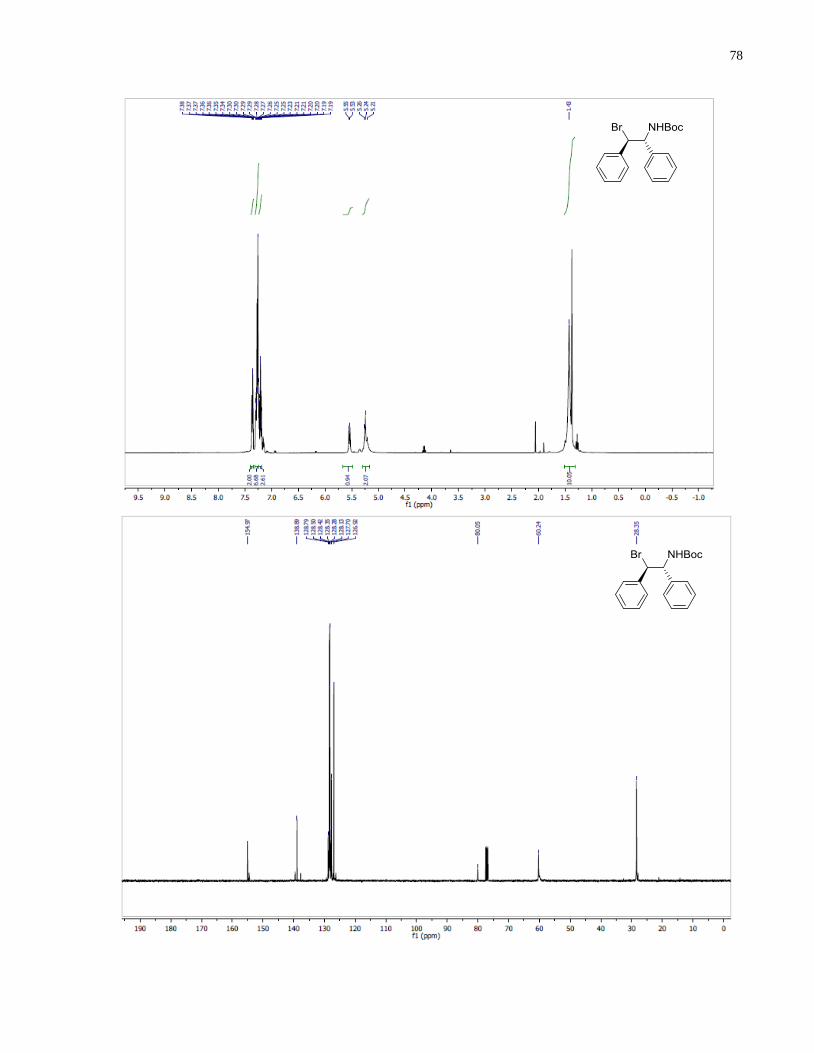
78
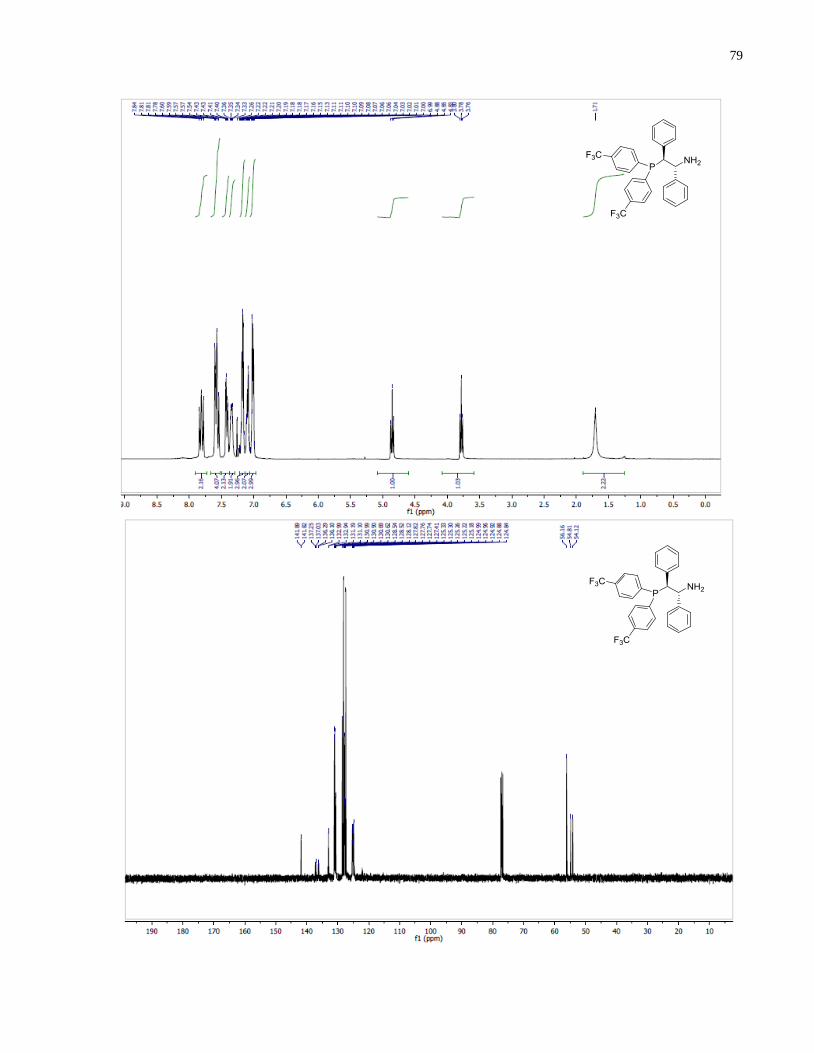
79
80
81
82

83
Chapter 2
Synthesis of Bifunctional Thiourea-Phosphine
Organocatalysts and Their Applications in the
Asymmetric Morita-Baylis-Hillman (MBH) Reaction
1 Introduction
1.1 Mechanism of the Morita-Baylis-Hillman (MBH) Reaction
The Morita-Baylis-Hillman (MBH) reaction is a carbon-carbon bond forming reaction between
an aldehyde and an activated alkene catalyzed by tertiary amines or phosphines.39
If an activated
imine is used instead of an aldehyde, the reaction is commonly referred to as the aza-Morita-
Baylis-Hillman (aza-MBH) reaction (Scheme 47).
Scheme 47. The Morita-Baylis-Hillman reaction
The mechanism of a tertiary amine-catalyzed MBH reaction is presented in Scheme 48.40
The
conjugate addition of tertiary amine A to the Michael acceptor generates the zwitterionic
39 a) Morita, K.; Suzuki, Z.; Hirose, H. Bull. Chem. Soc. Jpn. 1968, 41, 2815. b) Morita, K. Japan Patent,
6803364, 1968. b) Baylis, A. B.; Hillman, M. E. D. German Patent 2155113, 1972.
40 Hoffmann, H. M. R.; Rabe, J. Angew. Chem. Int. Ed. 1983, 22, 795.

84
ammonium-enolate intermediate B, which then attacks the incoming aldehyde and forms
intermediate C. Intramolecular proton transfer and subsequent elimination affords the product
and regenerates the tertiary amine catalyst A.
Scheme 48. Mechanism of MBH reaction proposed in 1980s
The aldol addition step was proposed to be the rate-determining step by Hill and Issac. They
observed that the rate of reaction was first order in acetaldehyde, acrylonitrile and DABCO, and
the kinetic isotope effect (KIE) at the -position of the acrylonitrile was low (Scheme 49).41
Scheme 49. Kinetic study of MBH reaction by Hill and Issac
However, this theory has been challenged by McQuade et al. and Aggarwal et al., who proposed
that the proton transfer step is rate limiting based on both kinetic and theoretical studies.
41 J. S. Hill, N. S. Isaacs, J. Phys. Org. Chem. 1990, 3, 285288.

85
McQuade observed a large normal KIE for the -position of methyl acrylate and a large inverse
KIE for the aldehyde proton (Scheme 50). In addition, the reaction was found to be second-order
in aldehyde and first-order in DABCO and acrylate in all tested solvents (DMSO, DMF, MeCN,
THF, CHCl3).42
Scheme 50. Kinetic study performed by McQuade et al.
McQuade therefore proposed that the mechanism involves the formation of a hemiacetal adduct
with a second equivalent of aldehyde and the -proton is abstracted in the rate determining step
via a six-membered transition state (Scheme 51).40
42 a) Price, K. E.; Broadwater, S. J.; Jung, H. M.; McQuade, D. T. Org. Lett. 2005, 7, 147150. b) Price,
K. E.; Broadwater, S. J.; Walker, B. J.; McQuade, D. T. J. Org. Chem. 2005, 70, 39803987.

86
Scheme 51. Mechanism of MBH reaction revised by McQuade and Aggarwal
Aggarwal et al. suggested that protic additives can act as proton shuffles to accelerate the -
deprotonation (Scheme 51).43
The proton transfer step is rate limiting only at the beginning of
the reaction (<20% conversion). Once the allylic alcohol product, which can act as a proton
shuffle, has built up to a certain extent, the aldol addition step becomes the rate-determining step.
They observed that the normal acrylate is consumed faster than the -deuterated acrylate in up to
20% conversion.41a
In a subsequent computational study, Aggarwal and Harvey confirmed that
under aprotic conditions, the formation of hemiacetal and the proton transfer via a cyclic
transition state proposed by McQuade is the most likely operating mechanism. In the presence of
methanol however, a slightly lower energy pathway in which the alcohol serves as a shuttle to
transfer the proton from carbon to oxygen was found.41b
Most recently, Singleton refuted the
proton-shuttle mechanism and questioned the usefulness of computational study in predicting the
thermodynamics and kinetics of MBH reactions. A two-step acid-base mechanism involving
proton transfer to and from protic solvent was fully supported by their experiments. Their study
also suggested the involvement of competitive rate-limiting steps in the methanol mediated
43 a) Aggarwal, V. K.; Fulford, S. Y.; Lloyd-Jones, G. C. Angew. Chem. Int. Ed. 2005, 44, 17061708. b)
Robiette, R.; Aggarwal, V. K.; Harvey, J. N. J. Am. Chem. Soc. 2007, 129, 1551315525.

87
MBH reactions. The proton transfer of the elimination step was found to be rate-limiting at 25 °C,
while the aldol addition became the primary rate-limiting step at low temperatures.44
1.2 The Asymmetric Morita-Baylis-Hillman (MBH) Reaction
Since the seminal discovery of a highly enantioselective MBH reaction using a bifunctional
catalyst derived from quinidine in 1999, several acid-base cocatalysts and bifunctional catalyst
systems have been developed to improve the efficiency and expand the scope of the initial
reaction. In the following sections, progress in the asymmetric MBH and aza-MBH reactions
will be presented separately for clarity purposes, although there is considerable overlap between
research in both fields, as the catalyst systems initially developed for MBH reaction often found
successful application later in the aza-MBH reaction and vice versa. Each catalyst system will be
presented in the chronological order of its discovery.
In 1999, Hatakeyama et al. discovered that a hydroxylated chiral amine, -isocupreidine (ICD)
2.1, confered considerable rate acceleration and asymmetric induction in the MBH reaction.
Excellent enantioselectivities were achieved using hexafluoroisopropyl acrylate with several
aromatic and aliphatic aldehydes despite modest yields (Scheme 52).45
The phenolic hydroxyl
group was believed to stabilize oxyanion intermediate through hydrogen bonding, which not
only accelerates the aldol reaction, but also creates an asymmetric environment together with the
chiral amine scaffold to enable enantioseletive addition to the aldehyde.
44 Plata, R. E.; Singleton, D. A. J. Am. Chem. Soc. 2015, 137, 3811−3826
45 Iwabuchi, Y.; Nakatani, M.; Yokoyama, N.; Hatakeyama, S. J. Am. Chem. Soc. 1999, 121,
1021910220.

88
Scheme 52. Asymmetric MBH reaction using a quinidine derivative as a bifunctional catalyst
Guided by this principle, Wang and coworkers designed the binaphthyl thiourea-tertiary amine
catalyst 2.2 that is effective for the MBH reaction of cyclohexenone and aliphatic aldehydes.
Lower yields and enantioselectivities were obtained when aromatic aldehydes were used,
however (Scheme 53).46
Scheme 53. Asymmetric MBH reaction using quinidine binaphthyl thiourea-tertiary amine 2.2
Another thiourea-phosphine catalyst 2.3 bearing a 1,2-cyclohexane backbone developed by Wu
et al. performed well with methyl vinyl ketone as the activated alkene. Considerable rate
acceleration was achieved with 2.3, where most reactions were completed within one hour
(Scheme 54).47
46 Wang, J.; Li, H.; Yu, X.; Zu, L.; Wang, W. Org. Lett. 2005, 7, 42934296.
47 Yuan, K.; Zhang, L.; Song, H.-L.; Hu, Y. J.; Wu, X. Y. Tetrahedron Lett. 2008, 49, 62626264.

89
Scheme 54. Asymmetric MBH reaction using cyclohexyl thiourea-phosphine 2.3
Finally, a class of threonine derived thiourea-phosphine catalysts reported by Lu et al. expanded
the substrate scope to include simple acrylates. Excellent yields and very good ee’s were
achieved in the reactions of acrylates with electron-deficient aromatic aldehydes. The use of
benzaldehyde required longer reaction time and resulted in a lower yield without much erosion
of enantioselectivity (Scheme 55).48
Scheme 55. Asymmetric MBH reaction using threonine derived thiourea-phosphine 2.4
48 Han, X.; Wang, Y.; Zhong, F.; Lu, Y. Org. Biomol. Chem. 2011, 9, 67346740.

90
Parallel to the research in bifunctional catalysis, the development of acid-base cocatalysis also
led to several breakthroughs in asymmetric MBH reaction.49
Schaus pioneered the use of a
BINOL-derived chiral Brønsted acid 2.5 as the catalyst along with triethylphosphine as the
nucleophilic promoter for MBH reaction of cyclohexenone with aldehydes. Excellent yields and
enantioselectivities were achieved only for aliphatic aldehydes.47a
A complimentary cocatalyst
system discovered by Connell employing u’s chiral MA catalyst 2.6 and magnesium iodide
as Lewis acid exhibited high yields and enantioselectivities in reactions of cyclopentenone with
aromatic aldehydes.47b
Ito demonstrated for the first time that high enantioselectivities could be
obtained in the reactions of cyclohexenone with both aromatic and aliphatic aldehydes using a
bis-thiourea 2.7/DABCO cocatalyst system (Scheme 56).47c
49 a) McDougal, N. T.; Schaus, S. E. J. Am. Chem. Soc. 2003, 125, 1209412095. b) Bugarin, A.; Connell,
B. T. Chem. Commun. 2010, 46, 26442646. c) Nakayama, Y.; Gotanda, G.; Ito, K. Tetrahedron Lett. 2011,
52, 62346237.
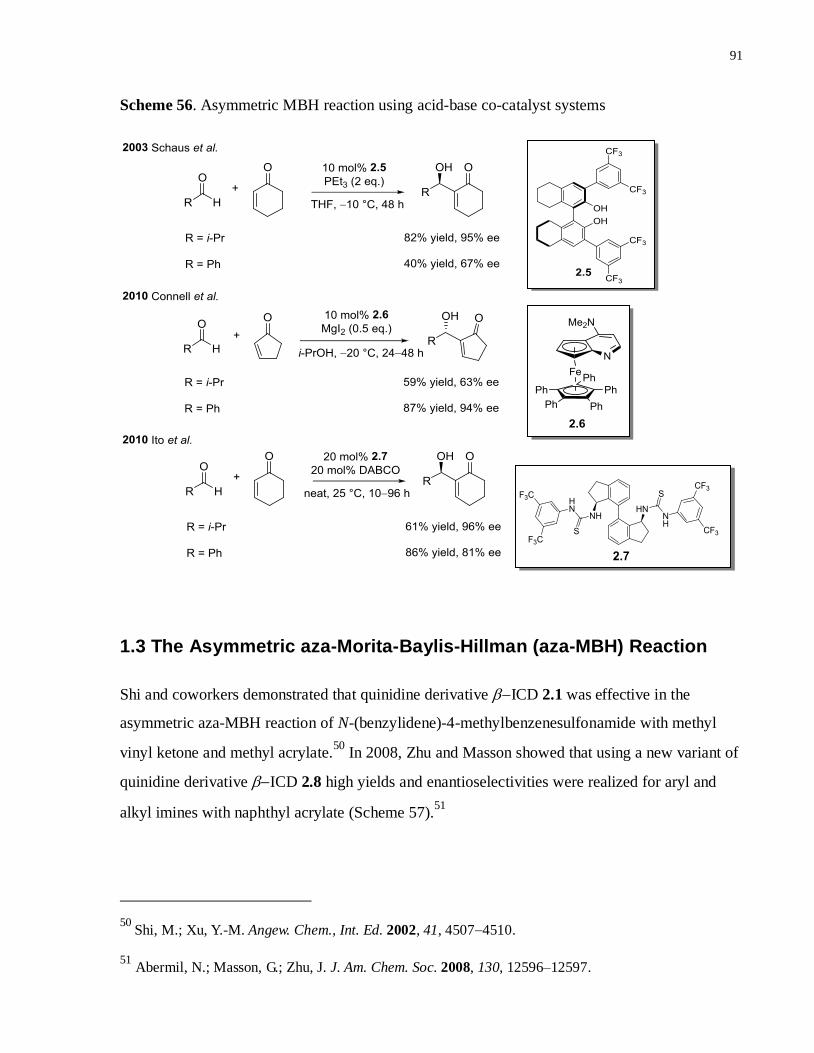
91
Scheme 56. Asymmetric MBH reaction using acid-base co-catalyst systems
1.3 The Asymmetric aza-Morita-Baylis-Hillman (aza-MBH) Reaction
Shi and coworkers demonstrated that quinidine derivative ICD 2.1 was effective in the
asymmetric aza-MBH reaction of N-(benzylidene)-4-methylbenzenesulfonamide with methyl
vinyl ketone and methyl acrylate.50
In 2008, Zhu and Masson showed that using a new variant of
quinidine derivative ICD 2.8 high yields and enantioselectivities were realized for aryl and
alkyl imines with naphthyl acrylate (Scheme 57).51
50 Shi, M.; Xu, Y.-M. Angew. Chem., Int. Ed. 2002, 41, 45074510.
51 Abermil, N.; Masson, G.; Zhu, J. J. Am. Chem. Soc. 2008, 130, 12596–12597.

92
Scheme 57. Asymmetric aza-MBH reaction using quinidine derivatives
Shi reported that in the presence of a BINOL-derived phosphine catalyst 2.8, the aza-MBH
reaction of N-sulfonylated imines with methyl vinyl ketone could be obtained in high yields and
enantioselectivities at low temperature. Using a modified catalyst 2.9, the same reaction could be
carried out at room temperature in shorter times. Although the catalyst was less effective for
acrylates, high yield and enantioselectivity were achieved for acrolein.52
Sasai independently
reported an equally effective BINOL-derived catalyst 2.10 containing a tertiary amine and a
pyridyl group, which gave even higher yields and enantioselectivities for certain substrates
(Scheme 58).53
52 Shi, M.; Chen, L.-H.; Li, C.-Q. J. Am. Chem. Soc. 2005, 127, 37903800.
53 Matsui, K.; Takizawa, S.; Sasai, H. J. Am. Chem. Soc. 2005, 127, 3680–3681.
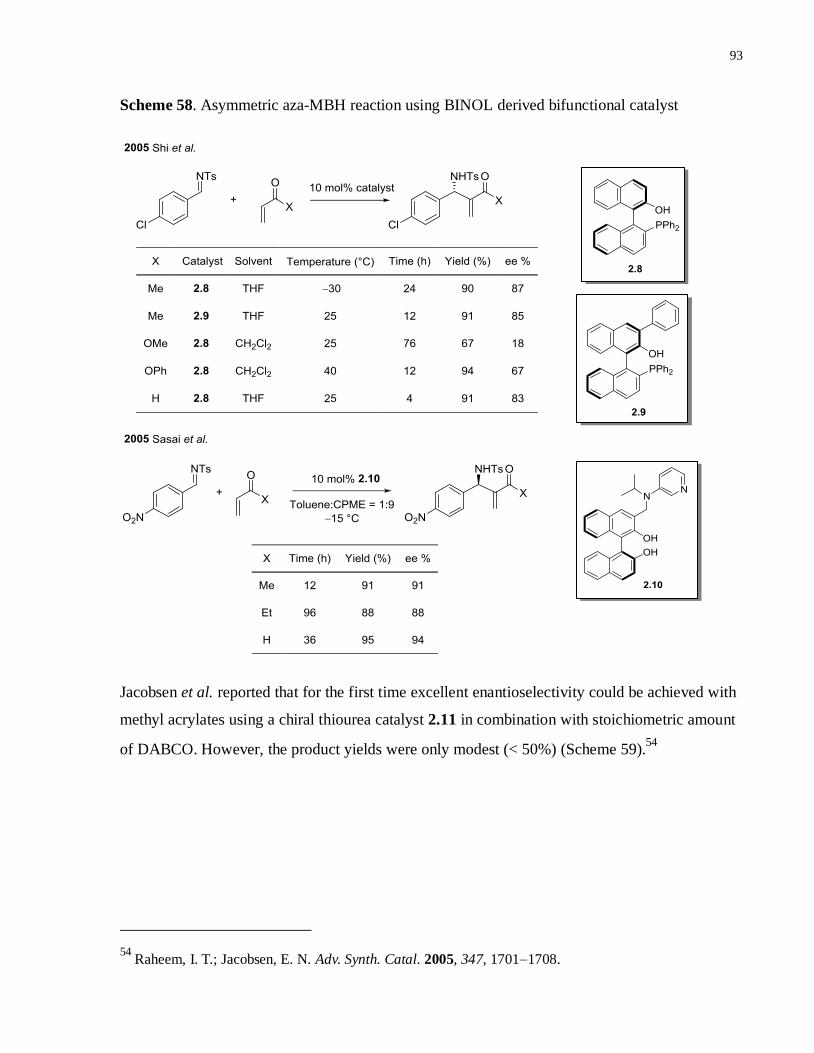
93
Scheme 58. Asymmetric aza-MBH reaction using BINOL derived bifunctional catalyst
Jacobsen et al. reported that for the first time excellent enantioselectivity could be achieved with
methyl acrylates using a chiral thiourea catalyst 2.11 in combination with stoichiometric amount
of DABCO. However, the product yields were only modest (< 50%) (Scheme 59).54
54 Raheem, I. T.; Jacobsen, E. N. Adv. Synth. Catal. 2005, 347, 17011708.
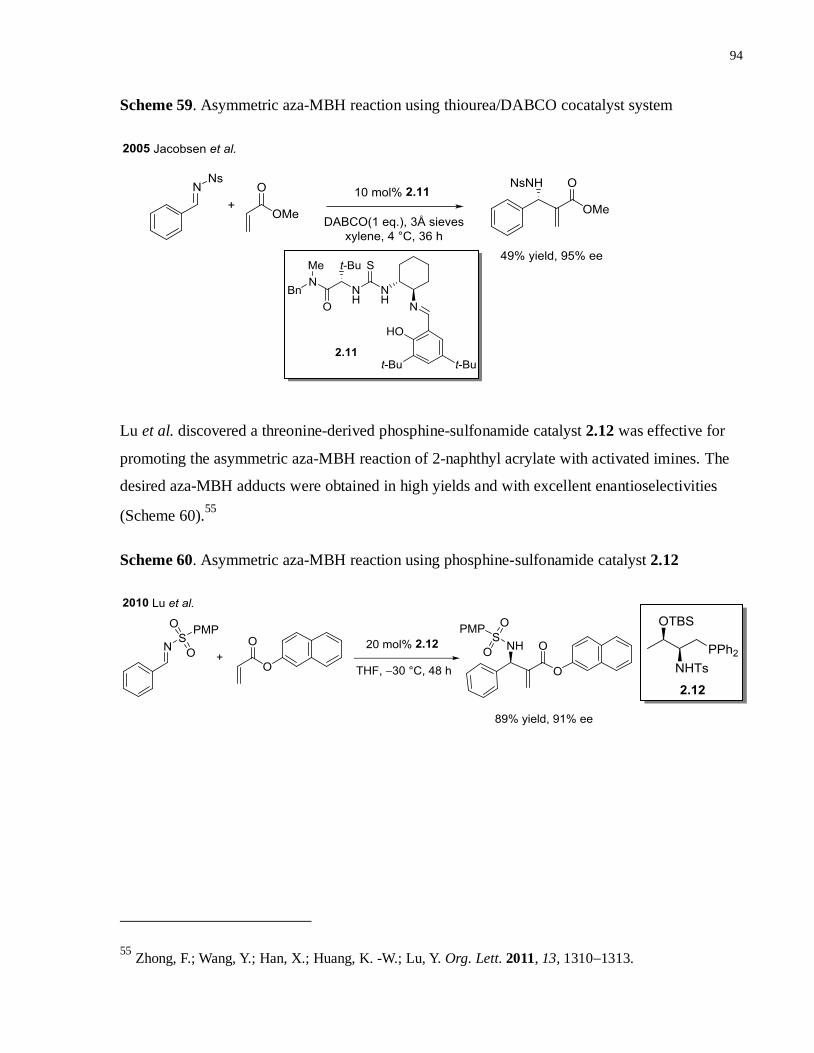
94
Scheme 59. Asymmetric aza-MBH reaction using thiourea/DABCO cocatalyst system
Lu et al. discovered a threonine-derived phosphine-sulfonamide catalyst 2.12 was effective for
promoting the asymmetric aza-MBH reaction of 2-naphthyl acrylate with activated imines. The
desired aza-MBH adducts were obtained in high yields and with excellent enantioselectivities
(Scheme 60).55
Scheme 60. Asymmetric aza-MBH reaction using phosphine-sulfonamide catalyst 2.12
55 Zhong, F.; Wang, Y.; Han, X.; Huang, K. -W.; Lu, Y. Org. Lett. 2011, 13, 13101313.

95
1.4 Asymmetric Transformations Related to the Morita-Baylis-Hillman
(MBH) Reaction
The application of bifunctional organocatalysts is not limited to asymmetric MBH/aza-MBH
reactions. Given the particular relevance of amino acid-derived phosphine catalysts to our
research, a brief survey of their utility outside MBH/aza-MBH reactions is presented in the
following section. The reactions listed below are related mechanistically to the MBH reaction.
Lu and coworkers prepared a series of five-membered carbocycles with a chiral quaternary
centre through highly enantioselective [3+2] annulations of an allenoate and an -substituted
acrylate using a novel dipeptide derived phosphine catalyst 2.13. They further demonstrated the
synthetic value of the cycloaddition product by converting it in three steps into a cytotoxic agent
containing a spirooxindole core (Scheme 61).56
Scheme 61. Enantioselective [3+2] annulation of allenoate with acrylate
The proposed mechanism is shown in Scheme 62. The phosphonium enolate intermediate A
attacks the less hindered terminus of the activated alkene B. The catalyst 2.13 presumably
positions B in a particular orientation relative to the A through favourable hydrogen-bonding
56 Han, X.; Wang, Y.; Zhong, F.; Lu, Y. J. Am. Chem. Soc. 2011, 133, 1726–1729.

96
interactions so that enolate A preferentially adds to one face of the olefin. Subsequent proton
transfer and elimination yield the product and regenerate the catalyst as in the MBH reaction.
Scheme 62. Mechanism of [3+2] annulation of allenoate with acrylate
Lu reported the first asymmetric Michael addition mediated by the chiral phosphine 2.14
(Scheme 63). They envisioned that the in situ generated phosphonium enolate A could act as a
base to deprotonate the pronucleophile and form a tight ion pair between enolate nucleophile B
and chiral phosphonium C. Efficient chirality transfer during the Michael addition step affords
the enantio-enriched adduct D, which then abstracts a proton from the pronucleophile to generate
the final product E and completes the catalytic cycle (Scheme 64).57
Scheme 63. Asymmetric Michael addition of oxindoles catalyzed by chiral phosphine
57 Zhong, F.; Dou, X.; Han, X.; Yao, W.; Zhu, Q.; Meng, Y.; Lu, Y. Angew. Chem., Int. Ed. 2013, 52, 943–
947.
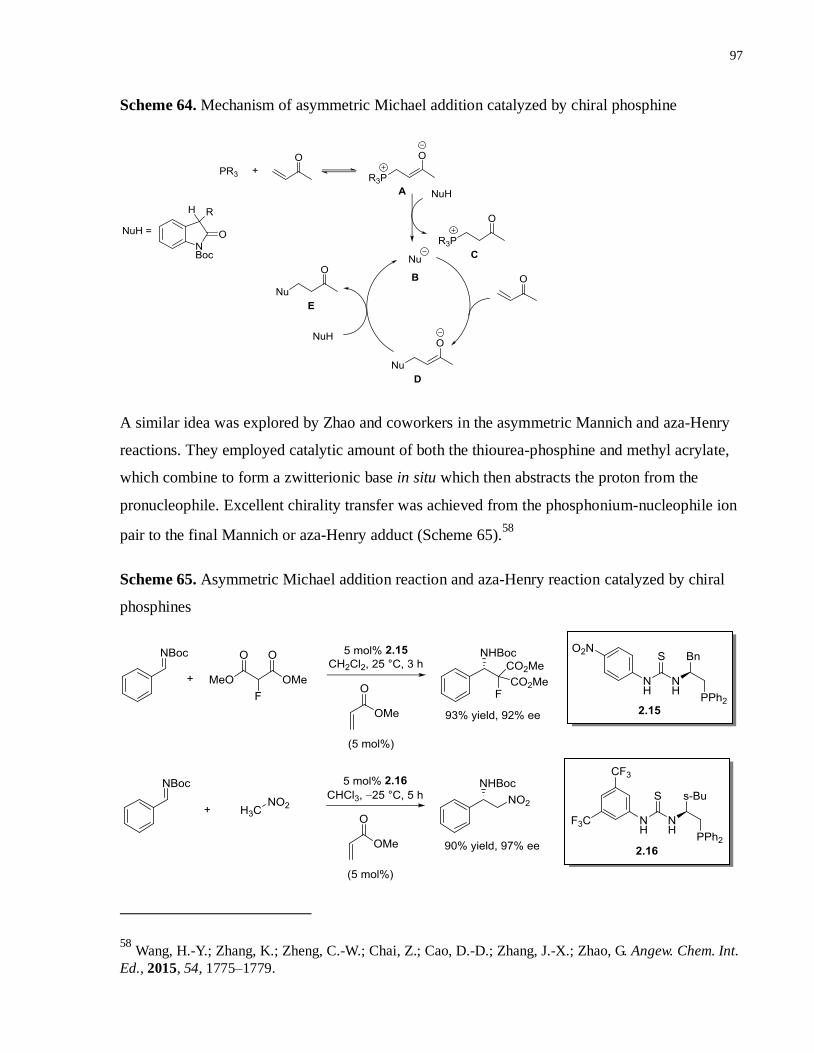
97
Scheme 64. Mechanism of asymmetric Michael addition catalyzed by chiral phosphine
A similar idea was explored by Zhao and coworkers in the asymmetric Mannich and aza-Henry
reactions. They employed catalytic amount of both the thiourea-phosphine and methyl acrylate,
which combine to form a zwitterionic base in situ which then abstracts the proton from the
pronucleophile. Excellent chirality transfer was achieved from the phosphonium-nucleophile ion
pair to the final Mannich or aza-Henry adduct (Scheme 65).58
Scheme 65. Asymmetric Michael addition reaction and aza-Henry reaction catalyzed by chiral
phosphines
58 Wang, H.-Y.; Zhang, K.; Zheng, C.-W.; Chai, Z.; Cao, D.-D.; Zhang, J.-X.; Zhao, G. Angew. Chem. Int.
Ed., 2015, 54, 1775–1779.

98
2 Objectives
Amino acid-derived bifunctional phosphine-thiourea catalysts were found to be very efficient in
promoting asymmetric MBH reaction of acrylates with aromatic aldehydes. Valine-based
catalyst 2.17 displayed high reactivity and enantioselectivity, although the best catalyst was
found to be the threonine-derived 2.4 (Scheme 66).43
Scheme 66. Asymmetric MBH reaction catalyzed by amino acid based thiourea-phosphines
So far, the refinement of these catalysts has been limited to modification of the carbon backbone
as well as the thiourea component. We were interested to investigate whether tuning the
electronic property of the phosphine moiety could impart any improvement on the reactivity and
enantioselectivity of the catalyst. Thus, a small library of ligands analogous to 2.17 with different
diaryl substituents on the phosphorus atom were prepared and tested in the asymmetric MBH
reaction.
3 Results and Discussion
Previously prepared P,N-ligands were coupled with 4-nitrophenyl isothiocyanate to afford
thiourea-phosphine catalysts 2.172.20 in good yields (Table 6).
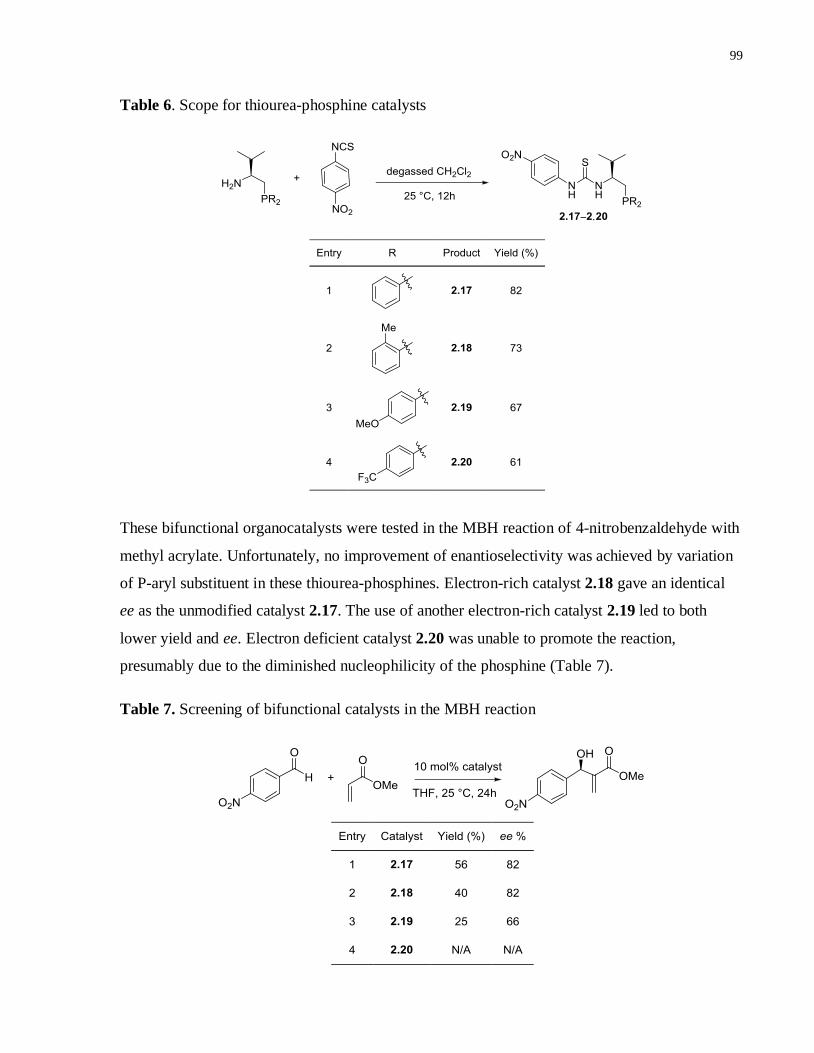
99
Table 6. Scope for thiourea-phosphine catalysts
These bifunctional organocatalysts were tested in the MBH reaction of 4-nitrobenzaldehyde with
methyl acrylate. Unfortunately, no improvement of enantioselectivity was achieved by variation
of P-aryl substituent in these thiourea-phosphines. Electron-rich catalyst 2.18 gave an identical
ee as the unmodified catalyst 2.17. The use of another electron-rich catalyst 2.19 led to both
lower yield and ee. Electron deficient catalyst 2.20 was unable to promote the reaction,
presumably due to the diminished nucleophilicity of the phosphine (Table 7).
Table 7. Screening of bifunctional catalysts in the MBH reaction

100
Around the same time, a novel P-stereogenic thiourea-phosphine catalyst 2.21 made in our lab
gave an improved enantioselectivity of 86% ee (Scheme 67).24
Encouraged by this result, we
decided to make another P-chiral catalyst 2.29, by replacing one of the aryl groups on the
phosphorus atom with a linear alkyl chain. As a result, the steric properties of the two
substituents would be further differentiated, which might impose higher level of enantio-
discrimination in the reaction. In addition, the aliphatic substituent should increase the
nucleophilicity of the catalyst, which can potentially promote reactions of less reactive substrates
that are otherwise sluggish with diarylphosphino-thiourea catalysts. Of course, we recognized the
synthetic challenges associated with making electron-rich P-chiral phosphines, mainly due to
their air sensitivity and the tendency for them to racemize at higher temperatures.
Scheme 67. P-chiral phosphino-thiourea catalysts
Addition of triethyl phosphite 2.22 to in situ generated phenyl Grignard reagent followed by
hydrolysis of the resulting phosphonite intermediate under acidic conditions afforded phenyl-H-
phosphinate 2.23. Treatment with n-butyllithium displaced the ethoxy group and generated the
unsymmetrical secondary phosphine oxide 2.24 (Scheme 68).
Scheme 68. Synthesis of unsymmetrical secondary phosphine oxide
Ring opening of the cyclic sulfamidate with 2.24 in the presence of NaOtBu afforded a 1:1
diastereomeric mixture of the products 2.25 in modest yield (Scheme 69). Separation of the
diastereomers proved to be very difficult. Using 1% THF:diethyl ether as the eluent, only a small
fraction of the less polar diastereomer could be isolated in pure form. We then carried the
mixture of diastereomers forward, hoping that the separation could be accomplished through

101
recrystallization of the HCl salts of 2.26. Unfortunately, several recrystallization attempts in
different solvent systems only resulted in the formation of oil droplets instead of crystalline
solids. The ineffective crystallization can most likely be attributed to the flexible n-butyl chain.
The diastereomeric mixture of 2.26 was then reduced and protected in situ as the phosphine-
borane complex 2.27. Gratifyingly, we were able to separate the diastereomers at this stage using
flash column chromatography on silica gel. The less polar diastereomer 2.27a was then coupled
with 4-nitrophenyl isothiocyanate to afford crystalline thiourea 2.28a. We managed to grow a
single crystal of 2.28a through liquid-liquid diffusion. Diethyl ether was slowly added into a
solution of 2.28a in dichloromethane, forming a bilayer solvent mixture. Slow diffusion of the
two solvents induces the precipitation of the solute and an X-ray quality crystal of 2.28a was
obtained after standing at room temperature for 24 hours. The relative configuration of the P-
chirality centre was determined by X-ray crystallography. Interestingly, the N-H hydrogens are
in a disordered conformation in the crystal structure, suggesting that the thiourea group does not
contribute to the crystal packing through hydrogen bonding interactions (Figure 4).
Figure 4. X-ray crystal structure of thiourea-phosphine-borane 2.28a
Attempts to deprotect the phosphine-borane compounds 2.28a/2.28b using 2.5 equivalents of
DABCO resulted in full recovery of the starting material after heating at 40 ˚C for 6 hours.
Considering that both raising the temperature and prolonged heating might risk racemizing or
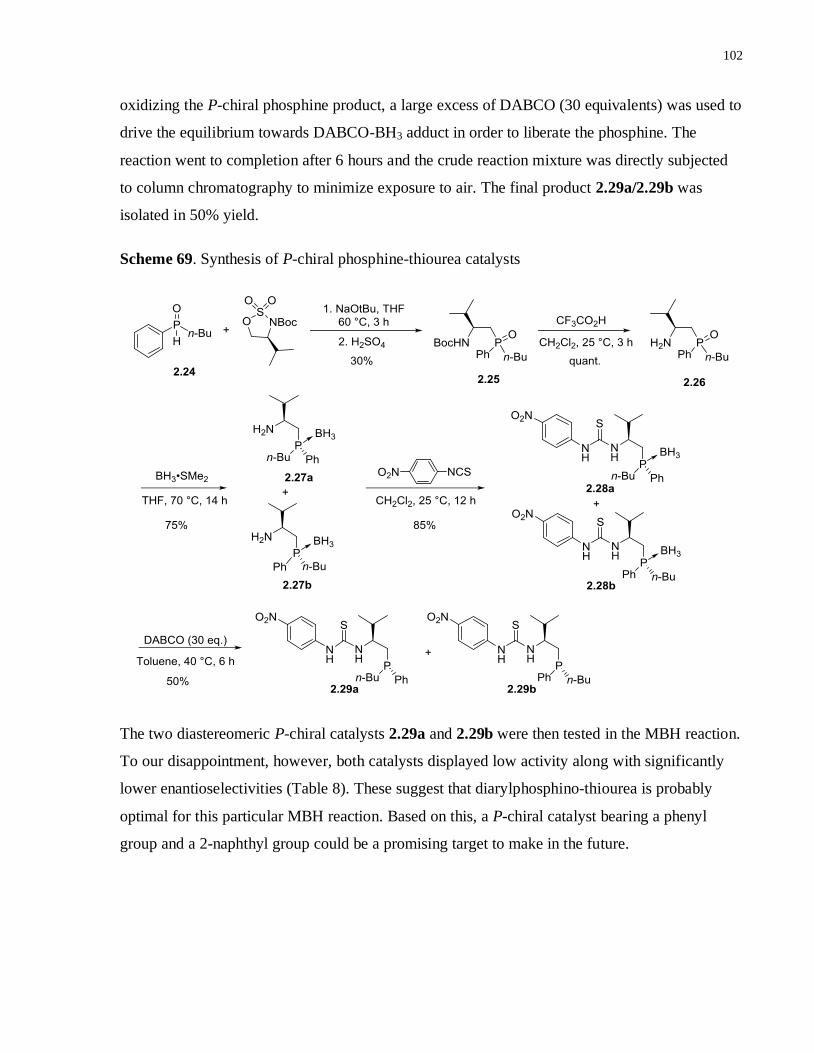
102
oxidizing the P-chiral phosphine product, a large excess of DABCO (30 equivalents) was used to
drive the equilibrium towards DABCO-BH3 adduct in order to liberate the phosphine. The
reaction went to completion after 6 hours and the crude reaction mixture was directly subjected
to column chromatography to minimize exposure to air. The final product 2.29a/2.29b was
isolated in 50% yield.
Scheme 69. Synthesis of P-chiral phosphine-thiourea catalysts
The two diastereomeric P-chiral catalysts 2.29a and 2.29b were then tested in the MBH reaction.
To our disappointment, however, both catalysts displayed low activity along with significantly
lower enantioselectivities (Table 8). These suggest that diarylphosphino-thiourea is probably
optimal for this particular MBH reaction. Based on this, a P-chiral catalyst bearing a phenyl
group and a 2-naphthyl group could be a promising target to make in the future.
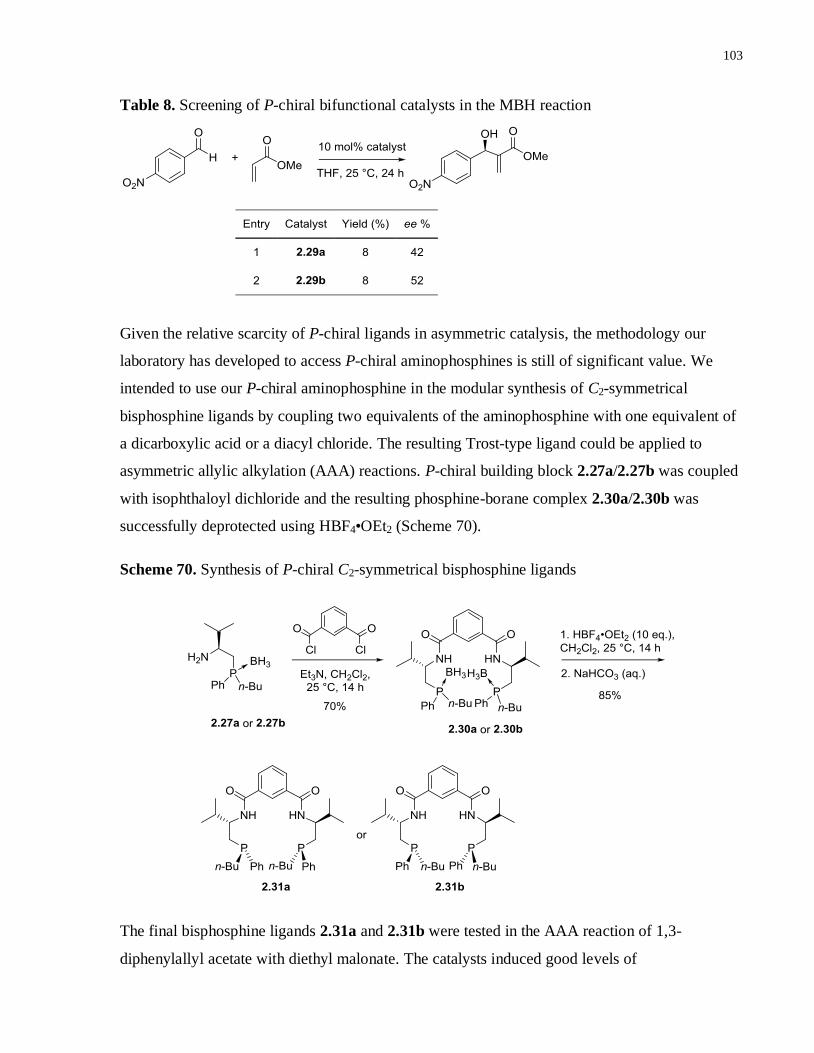
103
Table 8. Screening of P-chiral bifunctional catalysts in the MBH reaction
Given the relative scarcity of P-chiral ligands in asymmetric catalysis, the methodology our
laboratory has developed to access P-chiral aminophosphines is still of significant value. We
intended to use our P-chiral aminophosphine in the modular synthesis of C2-symmetrical
bisphosphine ligands by coupling two equivalents of the aminophosphine with one equivalent of
a dicarboxylic acid or a diacyl chloride. The resulting Trost-type ligand could be applied to
asymmetric allylic alkylation (AAA) reactions. P-chiral building block 2.27a/2.27b was coupled
with isophthaloyl dichloride and the resulting phosphine-borane complex 2.30a/2.30b was
successfully deprotected using HBF4•OEt2 (Scheme 70).
Scheme 70. Synthesis of P-chiral C2-symmetrical bisphosphine ligands
The final bisphosphine ligands 2.31a and 2.31b were tested in the AAA reaction of 1,3-
diphenylallyl acetate with diethyl malonate. The catalysts induced good levels of
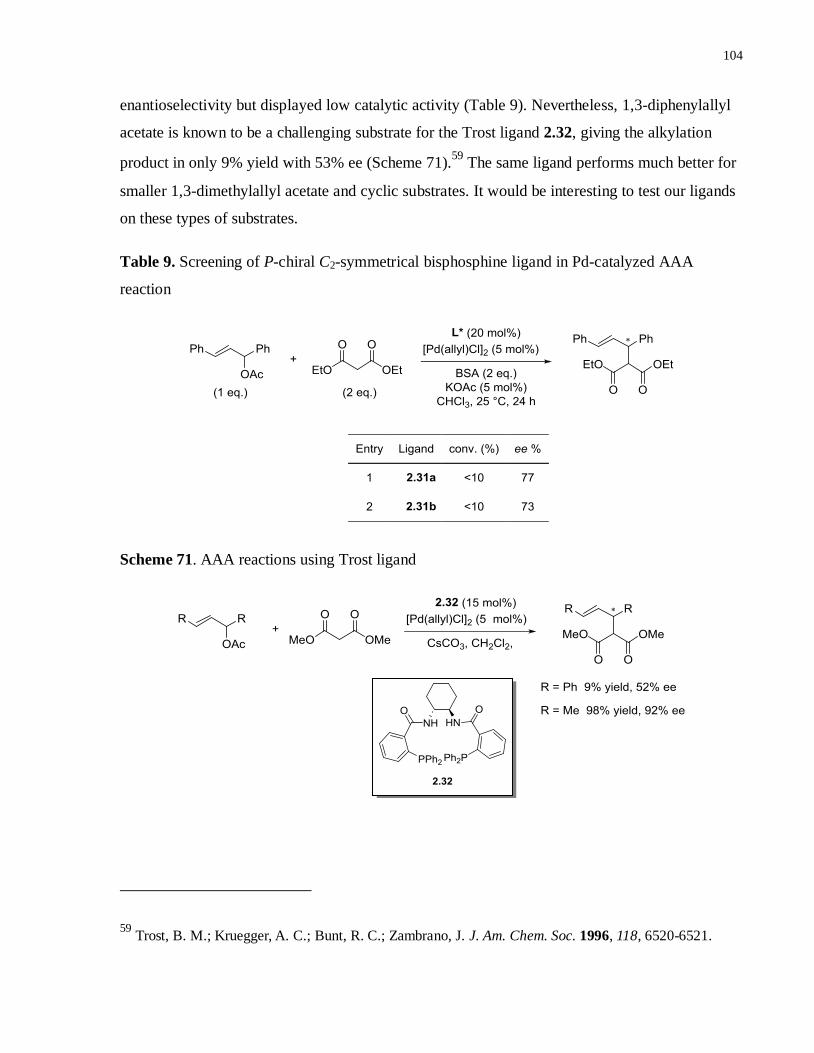
104
enantioselectivity but displayed low catalytic activity (Table 9). Nevertheless, 1,3-diphenylallyl
acetate is known to be a challenging substrate for the Trost ligand 2.32, giving the alkylation
product in only 9% yield with 53% ee (Scheme 71).59
The same ligand performs much better for
smaller 1,3-dimethylallyl acetate and cyclic substrates. It would be interesting to test our ligands
on these types of substrates.
Table 9. Screening of P-chiral C2-symmetrical bisphosphine ligand in Pd-catalyzed AAA
reaction
Scheme 71. AAA reactions using Trost ligand
59 Trost, B. M.; Kruegger, A. C.; Bunt, R. C.; Zambrano, J. J. Am. Chem. Soc. 1996, 118, 6520-6521.

105
4 Conclusion and Future Work
A small library of -aminophosphines bearing different chiral carbon backbones and different
diaryl substituents on the phosphine group was prepared through ring-opening of cyclic
sulfamidates with secondary phosphine oxides. The electron-deficient -substituted P,N-ligands
were tested in the asymmetric decarboxylative allylation reaction. Although the new ligands
derived from (S)-valinol and (R)-phenylglycinol demonstrated excellent catalytic activity, they
were less enantioselective than the original ligand derived from (S)-tert-valinol. Attempts to
prepare ,-disubstituented P,N-ligands derived from (1S,2R)-2-amino-1,2-diphenylethanol and
(1R,2S)-1-amino-2-indanol failed at the final reduction step. As for future work, it would be
worthwhile to examine the use of secondary phosphine-borane and butyllithium in the opening
of cyclic sulfamidates and displacement of halides or sulfonates. The deprotection of
aminophosphine-borane complex can be achieved under much milder condition than the
reduction of corresponding aminophosphine oxide.
A series of phosphine-thiourea bifunctional catalysts derived from -aminophosphines were
prepared and applied in the asymmetric MBH reaction. Attempts to improve the
enantioselectivity of the original catalyst by tuning the electronic properties of the phosphine
moiety were fruitless. The P-chiral catalyst bearing a phenyl group and an n-butyl group
displayed lower activity and enantioselectivity. These results suggest that electronically
unmodified diarylphosphino-thiourea is probably optimal for this particular MBH reaction. A
future direction will be to incorporate two sterically differentiated diaryl groups, such as one
phenyl group and one 2-naphthyl group, on the phosphorous donor of the P-chiral catalyst.
The P-chiral aminophosphines were employed as building blocks in the modular synthesis of C2-
symmteric bisphophine ligands. The performance of these ligands in the Pd-catalyzed AAA
reaction remains to be explored for 1,3-dimethylallyl acetate and cyclic substrates. Morimoto
reported the use of ligand in the Rh-catalyzed asymmetric hydrosilylation of ketones (Scheme
72).9 It would be interesting to see if our P-chiral catalyst can impart any improvement on the
activity and enantioselectivity of the same reaction. Finally, P-chiral P,N-ligands could be used
to construct PNP’ pincer ligands which could form active metal complexes for the asymmetric
hydrogenations of ketones and imines.

106
Scheme 72. Rh-catalyzed asymmetric hydrosilylation of ketones
5 Experimental
5.1 Procedures and Compounds
General procedures for preparing thiourea-phosphines 2.17-2.20, 2.29a, 2.29b
(S)-1-(1-(Diphenylphosphanyl)-3-methylbutan-2-yl)-3-(4-nitrophenyl)thiourea (2.17)
4-Nitrophenyl isothiocyanate (26 mg, 0.14 mmol) was added to a solution of (S)-1-
(diphenylphosphanyl)-3-methylbutan-2-amine 1.35 (35 mg, 0.13 mmol) in degassed CH2Cl2 (2
mL) under argon. The reaction was stirred at room temperature for 14 h. The reaction mixture
was directly loaded onto silica gel. The eluent was degassed in advance through sparging with
argon for 20 min. The crude product was purified through silica gel flash column
chromatography (Hexanes/EtOAc, 5:1 v/v) under a positive pressure of argon. The final product
(48 mg, 82%) was isolated as a yellow solid.
1H NMR (300 MHz, CDCl3) δ 8 65 (br s 1H) 8 21 – 8.07 (m, 2H), 7.50 – 7.39 (m, 4H), 7.38 –
7.27 (m, 8H), 6.43 (br s, 1H), 4.61 (br s, 1H), 2.51 (m, 1H), 2.31 (m, 1H), 2.18 (m, 1H), 0.95 (s,
3H), 0.92 (s, 3H). 13
C NMR (101 MHz, CDCl3) δ 179 47 144 12 143 16 133 02 132 83
132.78, 132.59, 129.14, 129.00, 128.74, 128.67, 125.42, 122.17, 58.87, 58.72, 32.08, 30.77,
18.59. 31
P NMR (121 MHz, CDCl3) δ -24.40.

107
(S)-1-(1-(di-o-tolylphosphanyl)-3-methylbutan-2-yl)-3-(4-nitrophenyl)thiourea (2.18)
Synthesized according to general procedure using 4-nitrophenyl isothiocyanate (16 mg, 0.09
mmol) and (S)-1-(di-o-tolylphosphanyl)-3-methylbutan-2-amine 1.33 (25 mg, 0.08 mmol).
Purification through silica gel flash column chromatography (Hexanes/EtOAc, 5:1 v/v) afforded
the product (29 mg, 73%) as a yellow solid.
1H NMR (399 MHz, CDCl3) δ 8 50 (br s 1H) 8 21 – 8.09 (d, 2H), 7.33 (m, 4H), 7.25 – 7.06 (m,
6H), 6.32 (br s, 1H), 4.57 (s, 1H), 2.41 (s, 6H), 2.37 – 2.33 (m, 1H), 2.32 – 2.26 (m, 1H), 2.24 –
2.15 (m, 1H), 0.95 (s, 1H), 0.93 (s, 1H). 31
P NMR (162 MHz, CDCl3) δ -46.86. IR (neat, cm-1
):
2959 (w), 1595 (m), 1505 (s), 1468 (s), 1450 (m), 1422 (m), 1325 (s), 1300 (s), 1255 (s), 1174
(s), 1111 (s), 1032 (w), 909 (w), 850 (m), 746 (s), 717 (s). Optical rotation: [D(cCHCl3)
= +18.3
(S)-1-(1-(bis(4-methoxyphenyl)phosphanyl)-3-methylbutan-2-yl)-3-(4-nitrophenyl)thiourea
(2.3)
Synthesized according to general procedure using 4-nitrophenyl isothiocyanate (28 mg, 0.16
mmol) and (S)-1-(bis(4-methoxyphenyl)phosphanyl)-3-methylbutan-2-amine 1.34 (53 mg, 0.16
mmol). Purification through silica gel flash column chromatography (Hexanes/EtOAc, 2:1 v/v)
afforded the product (55 mg, 67%) as a yellow solid.
1H NMR (400 MHz, CDCl3) δ 8 45 (s 1H) 8 23 – 8.03 (m, 2H), 7.46 – 7.22 (m, 6H), 6.90 –
6.75 (m, 4H), 6.36 (s, 1H), 4.61 (s, 1H), 3.76 (s, 3H), 3.75 (s, 3H), 2.43 (ddd, J = 14.3, 4.5, 2.4
Hz, 1H), 2.27 – 2.11 (m, 2H), 0.94 (d, J = 6.8 Hz, 6H).13
C NMR (101 MHz, CDCl3) δ 160 41
160.31 , 144.01 , 134.24 (d, J = 20.8 Hz), 133.98 (d, J = 20.6 Hz), 125.38 , 121.86 , 114.37 (d, J

108
= 7.7 Hz), 59.02 (d, J = 13.2 Hz), 55.18 , 32.08 , 30.99 , 18.70 , 18.58. 31
P NMR (121 MHz,
cdcl3) δ -28.39. IR (neat, cm-1
): 3294 (w), 2959 (w), 2836 (w), 1593 (s), 1568 (m), 1496 (s),
1462 (m), 1441 (m), 1326 (s), 1300 (s), 1283 (s), 1243 (s), 1175 (s), 1109 (s), 1094 (s), 1026 (s),
909 (m), 850 (m), 823 (s), 797 (s), 724 (s). Optical rotation: [D(cCHCl3) = +31.7.
HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H31N3O4PS 512.1767, found 512.1775.
(S)-1-(1-(bis(4-(trifluoromethyl)phenyl)phosphanyl)-3-methylbutan-2-yl)-3-(4-
nitrophenyl)thiourea (2.4)
Synthesized according to general procedure using 4-nitrophenyl isothiocyanate (24 mg, 0.13
mmol) and (S)-1-(bis(4-(trifluoromethyl)phenyl)phosphanyl)-3-methylbutan-2-amine 1.32 (50
mg, 0.12 mmol). Purification through silica gel flash column chromatography (Hexanes/EtOAc,
9:1 v/v) afforded the product (43 mg, 61%) as a yellow solid.
1H NMR (400 MHz, CDCl3) δ 8 20 – 8.12 (m, 2H), 8.10 (s, 1H), 7.68 – 7.61 (m, 2H), 7.57 (m,
2H), 7.52 – 7.40 (m, 6H), 3.19 (tdd, J = 9.5, 5.9, 3.4 Hz, 1H), 2.66 (ddd, J = 14.0, 9.5, 1.0 Hz,
1H), 2.47 (ddd, J = 14.0, 3.4, 1.1 Hz, 1H), 2.08 – 1.91 (m, 1H), 0.97 (d, J = 6.8 Hz, 3H), 0.92 (d,
J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 148 99 140 96 133 23 (d J = 10.4 Hz),
133.04 (d, J = 10.4 Hz), 128.72 , 125.33 (dd, J = 7.0, 3.6 Hz), 125.17 – 124.81 (m), 123.62 ,
75.35 (d, J = 13.3 Hz), 34.30 (d, J = 9.2 Hz), 32.28 (d, J = 12.8 Hz), 19.44 , 18.56 . 31P NMR
(162 MHz, CDCl3) δ -19.05. IR (neat, cm-1
): 2963 (w), 2873 (w), 1644 (w), 1603 (m), 1522 (m),
1396 (m), 1345 (m), 1320 (s), 1163 (s), 1121 (s), 1059 (s), 1015 (s), 951 (m), 827 (s), 748 (m),
695 (m). Optical rotation: [D(cCHCl3) = +110.0. HRMS (ESI-TOF) m/z: [M + H]
+
calcd for C26H25F6N3O2PS 588.1304, found 588.1294.
Butyl(phenyl)phosphine oxide (2.9)

109
n-Butyllithium (0.84 mL, 2.1 mmol) was added dropwise into a solution of ethyl
phenylphosphinate 2.8 (170 mg, 1.0 mmol) in dry pentane (5 mL) at 78 ºC. The reaction was
stirred at this temperature for 5 h. Saturated NH4Cl was added to quench the reaction. Extraction
with CH2Cl2 followed by flash column chromatography on silica gel (CH2Cl2/MeOH, 40:1 v/v)
afforded the product (109 mg, 60%) as colorless oil.
1H NMR (400 MHz, CDCl3) δ 7 49 (m 2H) 7 26 (d J = 463.3 Hz, 1H), 7.38 – 7.22 (m, 3H),
1.78 (m, 2H), 1.47 – 1.30 (m, 2H), 1.27 – 1.07 (m, 2H), 0.67 (t, J = 7.3 Hz, 3H). 31
P NMR (162
MHz, CDCl3) δ 27 46 13
C NMR (101 MHz, CDCl3) δ 132 16 (d J = 2.9 Hz), 130.98 (d, J =
96.0 Hz), 129.63 (d, J = 10.9 Hz), 128.68 (d, J = 12.2 Hz), 29.83 (d, J = 68.0 Hz), 23.48 (d, J =
14.6 Hz), 23.31 (d, J = 3.8 Hz), 13.38.
(S)-1-(Butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-amine (2.12)
Butyl(phenyl)phosphine oxide 2.9 (320 mg, 1.75 mmol) was added to a solution of potassium
tert-butoxide (196 mg, 1.75 mmol) in degassed THF (8 mL) under inert atmosphere. The mixture
was stirred for 5 min. tert-Butyl (S)-4-isopropyl-1,2,3-oxathiazolidine-3-carboxylate 2,2-dioxide
1.25 solid (464 mg, 1.75 mmol) was added and the reaction mixture was stirred at 60 ºC for 3 h.
The reaction was quenched with 2N H2SO4 (8 mL) at room temperature and the mixture was
stirred for another 20 min. Extraction with diethyl ether followed by purification by flash
chromatography on silica gel (EtOAc /CHCl3, 1:3 v/v) afforded diastereo-mixture 2.10 (200 mg,
31%) as a white solid. Trifluoroacetic acid (1.96 g, 17.2 mmol) was added to a solution of 2.10
(244 mg, 0.66 mmol) in dry CH2Cl2 (5 mL) at 0 ºC. The reaction was allowed to warm up and
stirred at room temperature for 3 h. Saturated Na2CO3 was added dropwise to the solution at 0
ºC. The aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over
MgSO4 and concentrated to afford the diastereo-mixture 2.11 (168 mg, 95%) as colorless oil.
BH3▪SMe was added to a solution of 2.11 (168 mg, 0.63 mmol) in dry THF (10 mL). The
reaction was stirred at 70 ºC for 14 h. Saturated NH4Cl was added to quench the reaction.

110
Extraction with CH2Cl2 followed by flash column chromatography on silica gel (CH2Cl2/MeOH,
50:1 v/v) afforded 2.12a (65 mg, 38%) and 2.12b (65 mg, 38%) as colorless oil.
(S)-1-((R)-Butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-amine (2.12a)
1H NMR (400 MHz, CDCl3) δ 7 79 (m 2H) 7 57 – 7.44 (m, 3H), 3.01 (m, 1H), 2.02 (m, 1H),
1.96 – 1.79 (m, 3H), 1.66 (m, 1H), 1.58 – 1.48 (m, 1H), 1.42 (br, 2H), 1.40 – 1.23 (m, 3H), 0.91
(d, J = 2.9 Hz, 3H), 0.90 (J = 2.9 Hz, 3H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3)
δ 131 96 (d J = 8.8 Hz), 131.30 (d, J = 2.5 Hz), 128.86 (d, J = 9.6 Hz), 52.53 (d, J = 0.9 Hz),
34.91 (d, J = 10.5 Hz), 31.51 (d, J = 35.0 Hz), 26.11 (d, J = 37.0 Hz), 24.88 (d, J = 1.1 Hz),
24.20 (d, J = 13.6 Hz), 18.45, 17.40, 13.55. 31P NMR (121 MHz, CDCl3) δ 12 83 (d J = 89.6
Hz).
(S)-1-((S)-Butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-amine (2.12b)
1H NMR (300 MHz, CDCl3) δ 7 73 (m 2H) 7 48 (m 3H) 2 79 (tdd J = 11.9, 5.3, 2.7 Hz, 1H),
2.28 (br, 2H), 2.08 (td, J = 14.5, 2.5 Hz, 1H), 1.90 (m, 2H), 1.84 – 1.73 (m, 1H), 1.64 – 1.43 (m,
2H), 1.41 – 1.19 (m, 3H), 0.98 – 0.80 (m, 6H), 0.76 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz,
CDCl3) δ 132 06 (d J = 8.6 Hz), 131.34 (d, J = 2.5 Hz), 128.79 (d, J = 9.6 Hz), 52.28 , 34.31 ,
30.95 (d, J = 35.3 Hz), 26.63 (d, J = 36.9 Hz), 24.87 (d, J = 1.2 Hz), 24.15 (d, J = 13.9 Hz),
18.19 , 17.27 , 13.52. 31P NMR (121 MHz, CDCl3) δ 13 15 (br d J = 83.1 Hz).
1-((S)-1-((R)-Butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-yl)-3-(4-
nitrophenyl)thiourea (2.13a)

111
Synthesized according to the general procedure using 4-nitrophenyl isothiocyanate (20 mg, 0.113
mmol) and 2.12a (30 mg, 0.113 mmol). Purification through silica gel flash column
chromatography (Hexanes/EtOAc, 2:1 v/v) afforded the product (42 mg, 84%) as a yellow solid.
1H NMR (400 MHz, CDCl3) δ 8 41 (br s 1H) 8 19 (d J = 9.0 Hz, 2H), 7.75 (m, 2H), 7.67 –
7.41 (m, 5H), 6.83 (br s, 1H), 4.71 (br s, 1H), 2.25 (m, 2H), 2.07 – 1.84 (m, 3H), 1.57 – 1.38 (m,
1H), 1.33 (q, J = 7.2 Hz, 2H), 1.20 (m, 1H), 0.93 (d, J = 6.7 Hz, 3H), 0.84 (t, J = 7.2 Hz, 3H),
0.80 – 0.66 (m, 3H). 13
C NMR (101 MHz, CDCl3) δ 179 61 144 17 131 97 (d J = 8.9 Hz),
131.73, 129.06 (d, J = 9.7 Hz), 125.08, 122.66, 57.05, 31.98, 28.20 (d, J = 33.0 Hz), 25.21 (d, J
= 37.5 Hz), 24.69, 24.08 (d, J = 14.0 Hz), 18.92, 18.44, 13.53. 31
P NMR (162 MHz, CDCl3) δ
9.95.
1-((S)-1-((S)-Butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-yl)-3-(4-
nitrophenyl)thiourea (2.13b)
Synthesized according to the general procedure using 4-nitrophenyl isothiocyanate (10 mg, 0.057
mmol) and 2.12b (15 mg, 0.057 mmol). Purification through silica gel flash column
chromatography (Hexanes/EtOAc, 2:1 v/v) afforded the product (22 mg, 88%) as a yellow solid.
1-((S)-1-((R)-Butyl(phenyl)phosphanyl)-3-methylbutan-2-yl)-3-(4-nitrophenyl)thiourea
(2.14a)

112
Degassed toluene (0.5 mL) was added into a Schlenk tube containing 2.13a (13 mg, 0.029 mmol)
and DABCO (98 mg, 0.87 mmol) under argon. The reaction was stirred at 40 ºC for 6 h. The
crude mixture was directly loaded onto silica gel and was purified through flash column
chromatography using degassed CH2Cl2 as eluent under a positive pressure of argon. The final
product (6 mg, 50%) was isolated as a yellow solid.
1H NMR (400 MHz, Chloroform-d) δ 8 20 (m, 3H), 7.55 (m, 2H), 7.48 – 7.29 (m, 5H), 6.28 (d,
J = 9.1 Hz, 1H), 4.66 (s, 1H), 2.23 – 1.91 (m, 3H), 1.88 – 1.70 (m, 2H), 1.48 – 1.29 (m, 2H),
1.11 – 0.71 (m, 9H). 13
C NMR (101 MHz, Chloroform-d) δ 144 28 132 59 (d J = 19.5 Hz),
129.39 , 128.65 (d, J = 7.2 Hz), 125.56 , 122.18 , 59.03 (d, J = 14.9 Hz), 32.56 , 30.94 , 28.18 (d,
J = 9.7 Hz), 27.86 (d, J = 12.9 Hz), 24.20 (d, J = 12.1 Hz), 18.53 , 13.75. 31
P NMR (162 MHz,
CDCl3) δ -31.93. MS (DART-TOF) m/z: [M + H]+calcd for C22H31N3O2PS 432.2, found 432.2.
1-((S)-1-((S)-Butyl(phenyl)phosphanyl)-3-methylbutan-2-yl)-3-(4-nitrophenyl)thiourea
(2.14b)
Degassed toluene (0.5 mL) was added into a Schlenk tube containing 2.13b (22 mg, 0.049 mmol)
and DABCO (166 mg, 1.48 mmol) under argon. The reaction was stirred at 40 ºC for 6 h. The
crude mixture was directly loaded onto silica gel and was purified through flash column
chromatography using degassed CH2Cl2 as eluent under a positive pressure of argon. The final
product (10 mg, 50%) was isolated as a yellow solid.
1H NMR (400 MHz, Chloroform-d) δ 8 25 (s 2H) 8 24 – 8.15 (m, 2H), 7.53 (m, 3H), 7.44 –
7.27 (m, 4H), 6.19 (s, 1H), 4.41 (s, 1H), 2.29 – 2.06 (m, 2H), 1.88 (ddd, J = 14.4, 8.1, 3.6 Hz,
1H), 1.74 (m, 2H), 1.47 – 1.21 (m, 4H), 0.96 (d, J = 6.7, 3H), 0.92 – 0.78 (m, 6H). 13
C NMR
(101 MHz, Chloroform-d) δ 144 18 132 68 (d J = 19.6 Hz), 129.39, 129.05 (d, J = 1.9 Hz),
128.94 , 128.62 (d, J = 7.4 Hz), 125.49 , 122.12 , 58.93 , 31.62 , 30.58 , 28.22 (d, J = 9.3 Hz),
28.14 (d, J = 13.9 Hz), 24.24 (d, J = 12.5 Hz), 18.74, 18.61, 13.77. 31
P NMR (162 MHz, CDCl3)

113
δ -34.01. HRMS (DART-TOF) m/z: [M + H]+
calcd for C22H31N3O2PS 432.18852, found
432.18746.
N1,N
3-Bis((S)-1-((R)-butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-yl)isophthalamide
(2.15a)
Et3N (9.6 mg, 0.095 mmol) was added into a solution of 2.12a (24 mg, 0.090 mmol) and
Isophthaloyl dichloride (9 mg, 0.045 mmol) in dry CH2Cl2. The reaction was stirred at room
temperature for 14 h. Then the reaction mixture was directly loaded onto silica gel without work-
up. Purification through flash column chromatography on silica gel (MeOH/CH2Cl2, 1:50 v/v)
afforded the final product 2.15a (22 mg, 74%) was isolated as a white solid.
1H NMR (400 MHz, CDCl3) δ 8 00 (t J = 1.8 Hz, 1H), 7.85 (dd, J = 7.7, 1.7 Hz, 2H), 7.78 (m,
4H), 7.48 – 7.35 (m, 7H), 6.54 (d, J = 8.9 Hz, 2H), 4.35 – 4.12 (m, 2H), 2.42 (ddd, J = 15.0, 11.2,
8.8 Hz, 2H), 2.19 – 2.07 (m, 2H), 2.06 – 1.83 (m, 6H), 1.55 – 1.41 (m, 2H), 1.35 – 1.16 (m, 6H),
0.91 (d, J = 6.7 Hz, 6H), 0.81 (t, J = 7.2 Hz, 6H), 0.77 (d, J = 6.7 Hz, 6H). 13
C NMR (101 MHz,
CDCl3) δ 165 56 134 00 132 08 (d J = 8.8 Hz), 131.41 (d, J = 2.5 Hz), 130.45 , 128.90 (d, J =
9.6 Hz), 128.43 (d, J = 52.1 Hz), 124.71 , 52.06 (d, J = 2.4 Hz), 32.64 (d, J = 7.2 Hz), 28.99 (d, J
= 33.7 Hz), 25.32 (d, J = 37.3 Hz), 24.76, 24.13 (d, J = 14.0 Hz), 18.83, 18.62, 13.50. 31
P NMR
(162 MHz, CDCl3) δ 11 30 HRMS (DART-TOF) m/z: [M – H2 + H]+
calcd for C38H59B2N2O2P2
659.42379, found 659.42462.
N1,N
3-Bis((S)-1-((S)-butyl(phenyl)phosphanyl-borane)-3-methylbutan-2-yl)isophthalamide
(2.15b)

114
Et3N (6 mg, 0.06 mmol) was added into a solution of 2.12b (15 mg, 0.056 mmol) and
Isophthaloyl dichloride (6 mg, 0.028 mmol) in dry CH2Cl2. The reaction was stirred at room
temperature for 14 h. Then the reaction mixture was directly loaded onto silica gel without work-
up. Purification through flash column chromatography on silica gel (MeOH/CH2Cl2, 1:50 v/v)
afforded the final product 2.15b (13 mg, 72%) was isolated as a white solid.
1H NMR (400 MHz, CDCl3) δ 7 87 (m 2H) 7 85 (d J = 1.7 Hz, 1H), 7.65 (m, 4H), 7.47 – 7.41
(m, 2H), 7.40 – 7.30 (m, 5H), 6.30 (d, J = 8.5 Hz, 2H), 4.03 (m, 2H), 2.37 – 2.17 (m, 4H), 2.09
(m, 2H), 2.03 – 1.91 (m, 4H), 1.61 – 1.47 (m, 2H), 1.46 – 1.28 (m, 6H), 0.95 (d, J = 6.8 Hz, 6H),
0.93 – 0.87 (m, 12H).13
C NMR (101 MHz, CDCl3) δ 165 52 133 85, 131.89 (d, J = 8.8 Hz),
131.40 (d, J = 2.5 Hz), 130.56, 128.87 (d, J = 9.9 Hz), 127.40 (d, J = 52.4 Hz), 124.91, 51.24 (d,
J = 3.3 Hz), 32.86 (d, J = 7.6 Hz), 27.83 (d, J = 34.1 Hz), 25.54 (d, J = 37.1 Hz), 24.82 , 24.25 (d,
J = 13.9 Hz), 18.49, 18.15, 13.58. 31
P NMR (162 MHz, CDCl3) δ 11 39 HRMS (DART-TOF)
m/z: [M – H2 + H]+ calcd for C38H59B2N2O2P2 659.42379, found 659.42585.
N1,N
3-Bis((S)-1-((R)-butyl(phenyl)phosphanyl)-3-methylbutan-2-yl)isophthalamide (2.16a)
HBF4• Et2 (24 mg, 0.15 mmol) was added into a solution of 2.15a (10 mg, 0.015 mmol) in
degassed CH2Cl2. The reaction was stirred at room temperature for 14 h. The reaction was
quenched with degassed sodium bicarbonate. The aqueous layer was extracted with CH2Cl2. The
combined organic layer was directly loaded onto silica gel. Purification through flash column

115
chromatography (MeOH/CH2Cl2, 1:50 v/v) afforded the final product 2.16a (7.5 mg, 80%) as a
white solid.
1H NMR (400 MHz, CDCl3) δ 7 94 – 7.90 (m, 1H), 7.62 (dd, J = 7.7, 1.8 Hz, 2H), 7.58 – 7.50
(m, 4H), 7.35 (t, J = 7.8 Hz, 1H), 7.32 – 7.27 (m, 6H), 5.94 (d, J = 9.1 Hz, 2H), 4.21 (m, 2H),
2.03 – 1.93 (m, 6H), 1.78 – 1.67 (m, 4H), 1.41 – 1.16 (m, 8H), 0.96 (d, J = 6.8 Hz, 6H), 0.89 (d,
J = 6.8 Hz, 6H), 0.82 (t, J = 7.1 Hz, 6H). 13
C NMR (101 MHz, CDCl3) δ 165 83 138 60 (d J =
15.0 Hz), 134.88 , 132.65 (d, J = 20.0 Hz), 129.55 , 129.08 , 128.59 , 128.58 , 128.52 , 53.30 (d,
J = 12.3 Hz), 32.90 (d, J = 7.4 Hz), 31.82 (d, J = 14.4 Hz), 28.28 (d, J = 10.9 Hz), 27.95 (d, J =
13.4 Hz), 24.24 (d, J = 12.0 Hz), 18.86, 18.05, 13.75. 31
P NMR (162 MHz, CDCl3) δ -31.26.
HRMS (DART-TOF) m/z: [M + H]+calcd for C38H55N2O2P2 633.37388, found 633.37428.
N1,N
3-Bis((S)-1-((S)-butyl(phenyl)phosphanyl)-3-methylbutan-2-yl)isophthalamide (2.16b)
HBF4• Et2 (54 mg, 0.33 mmol) was added into a solution of 2.15b (22 mg, 0.033 mmol) in
degassed CH2Cl2. The reaction was stirred at room temperature for 14 h. The reaction was
quenched with degassed sodium bicarbonate. The aqueous layer was extracted with CH2Cl2. The
combined organic layer was directly loaded onto silica gel. Purification through flash column
chromatography (MeOH/CH2Cl2, 1:50 v/v) afforded the final product 2.16b (18 mg, 86%) as a
white solid.
1H NMR (300 MHz, Chloroform-d) δ 7 93 (m 1H) 7 64 (dd J = 7.7, 1.8 Hz, 2H), 7.54 (td, J =
7.5, 2.1 Hz, 4H), 7.38 (t, J = 7.7 Hz, 1H), 7.34 – 7.27 (m, 6H), 5.95 (d, J = 9.0 Hz, 2H), 4.23 –
3.85 (m, 2H), 2.19 – 1.97 (m, 4H), 1.90 (m, 2H), 1.82 – 1.63 (m, 4H), 1.46 – 1.22 (m, 8H), 0.96
(d, J = 6.7 Hz, 6H), 0.92 – 0.82 (m, 12H). 13
C NMR (101 MHz, Chloroform-d) δ 165 85
138.06 (d, J = 15.1 Hz), 134.99 , 132.68 (d, J = 19.7 Hz), 129.34 (d, J = 37.0 Hz), 128.58 ,
128.58 (d, J = 7.2 Hz), 125.16 , 52.98 (d, J = 10.6 Hz), 32.33 (d, J = 7.3 Hz), 32.09 (d, J = 14.7
Hz), 28.22 (d, J = 14.6 Hz), 28.07 (d, J = 9.9 Hz), 24.30 (d, J = 12.4 Hz), 19.06, 18.17, 13.77.

116
31P NMR (121 MHz, cdcl3) δ -33.16. HRMS (DART-TOF) m/z: [M + H]
+ calcd for
C38H55N2O2P2 633.37388, found 633.37324.
5.2 1H NMR, 13C NMR and 31P NMR spectra
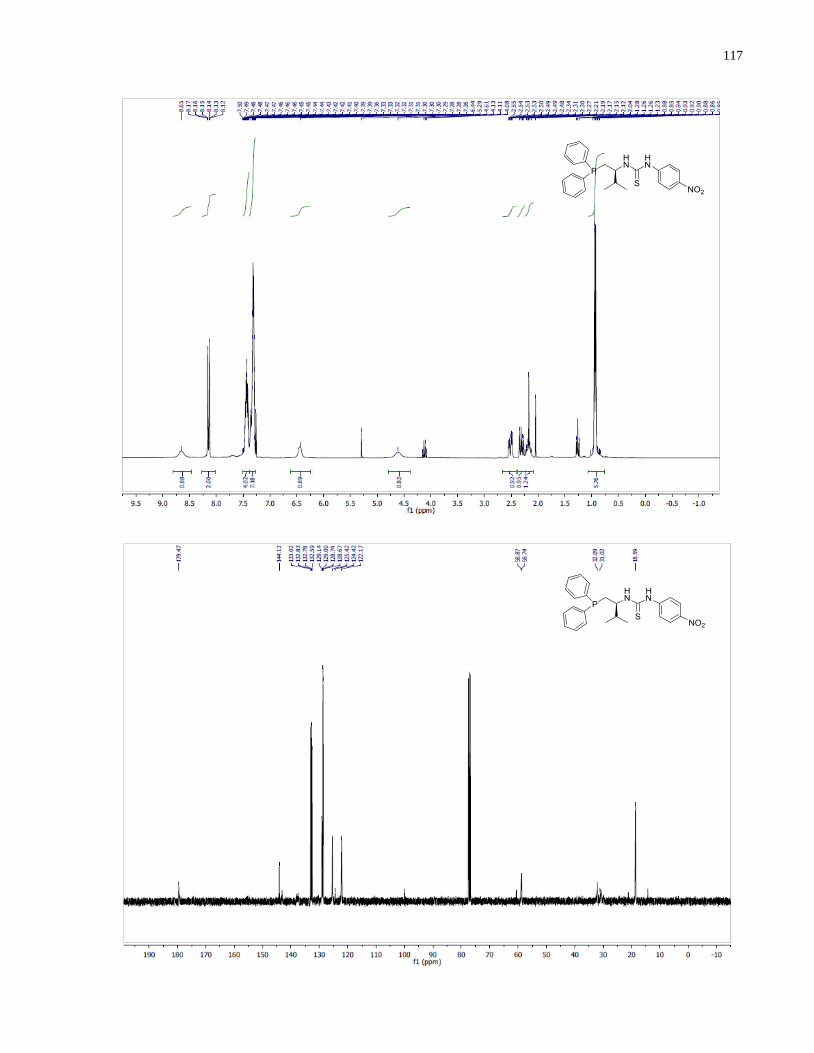
117
118
119
120
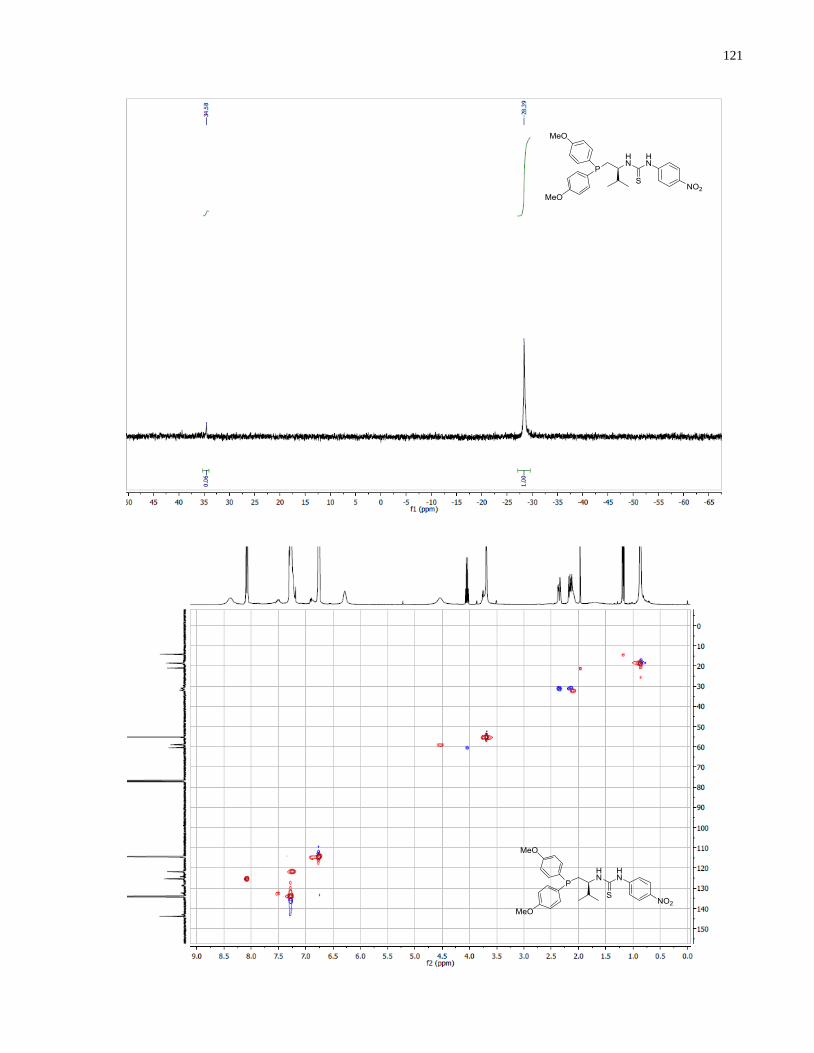
121
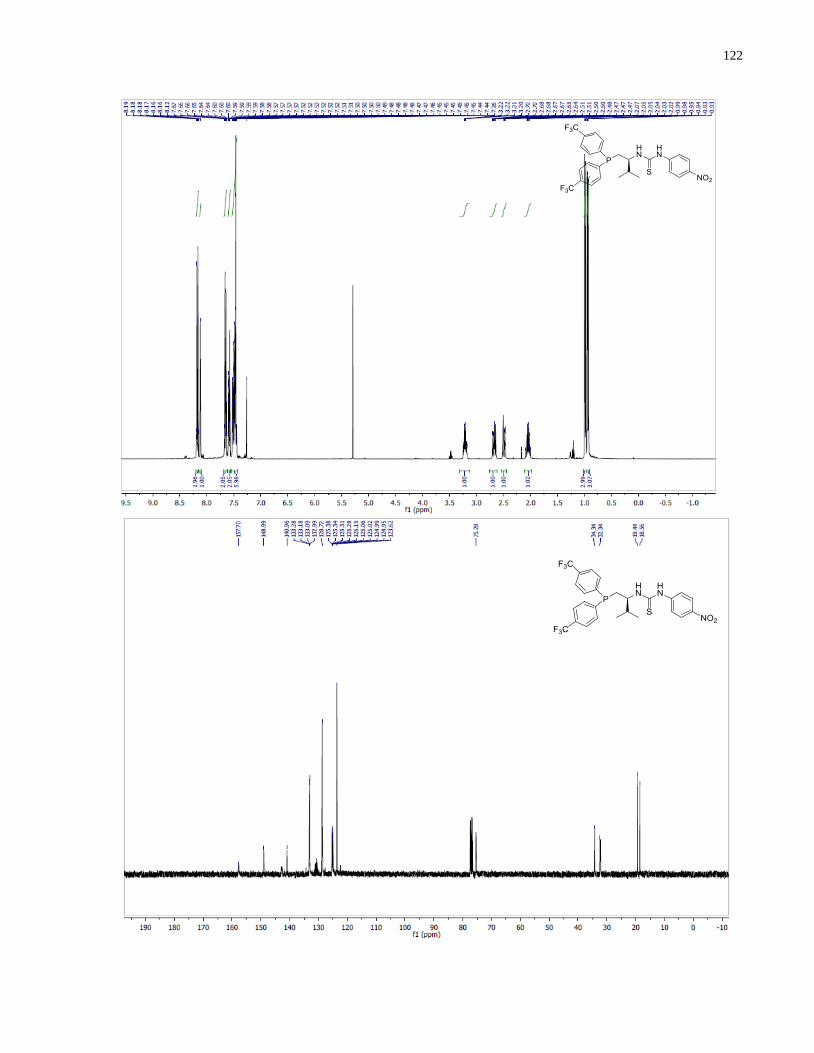
122
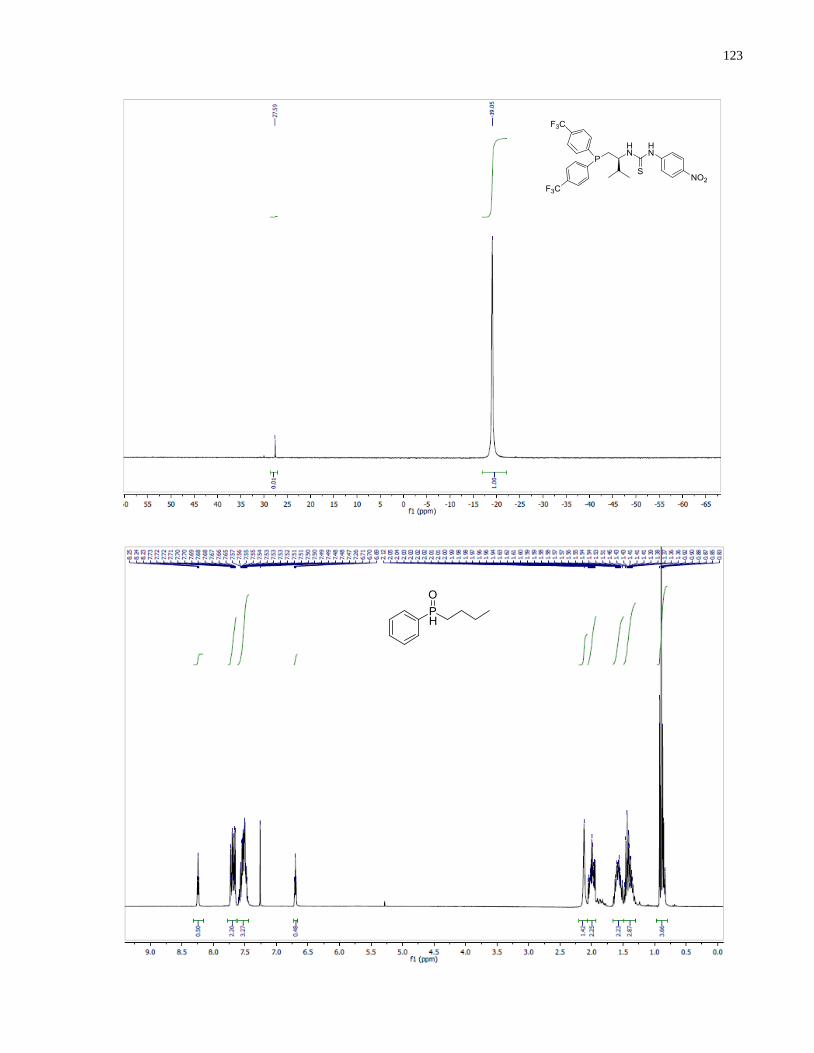
123
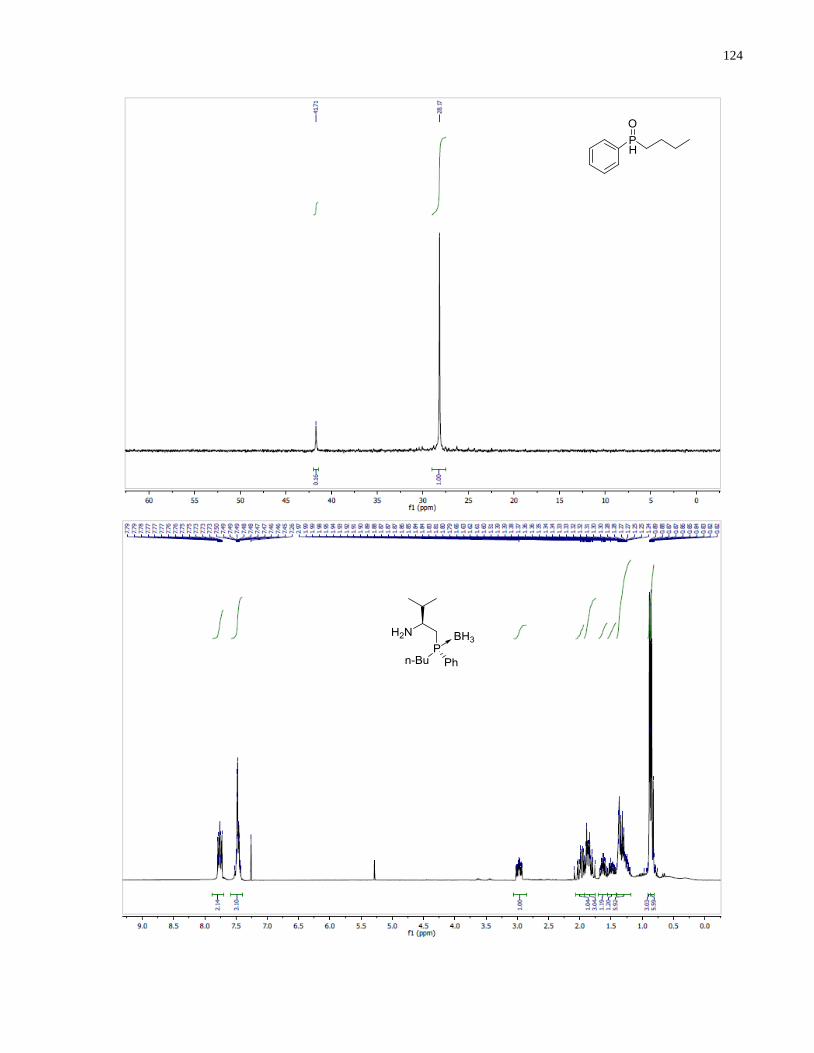
124
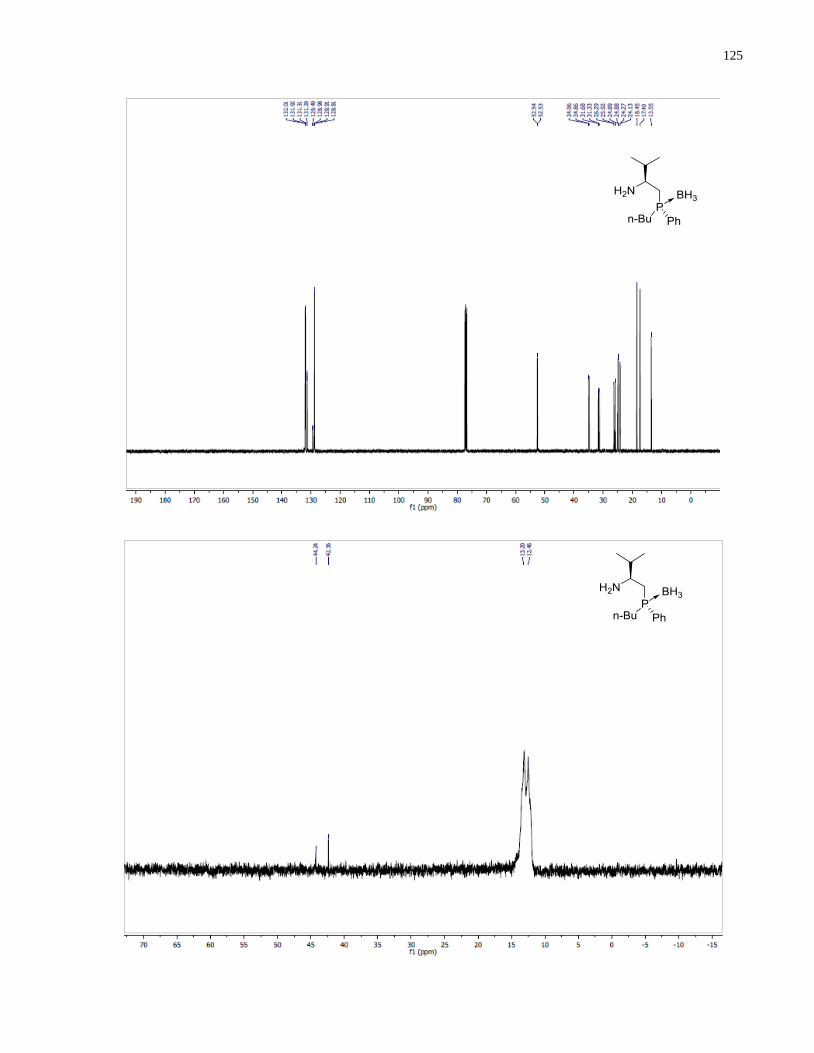
125
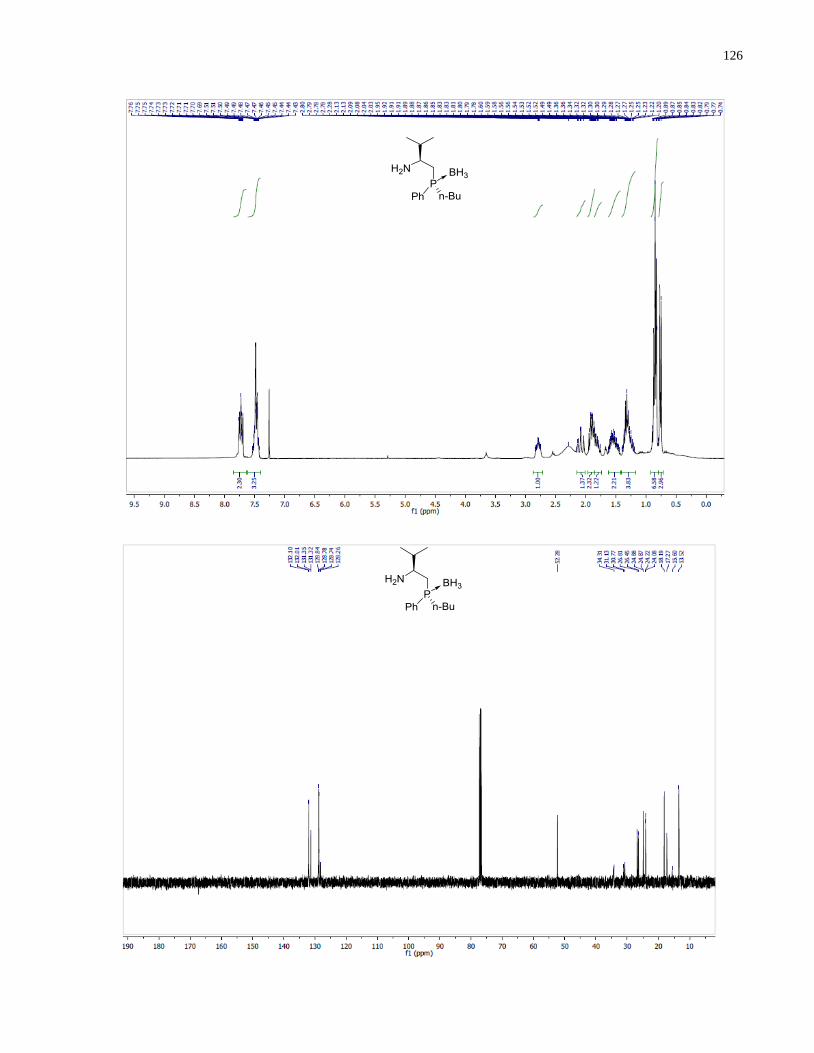
126
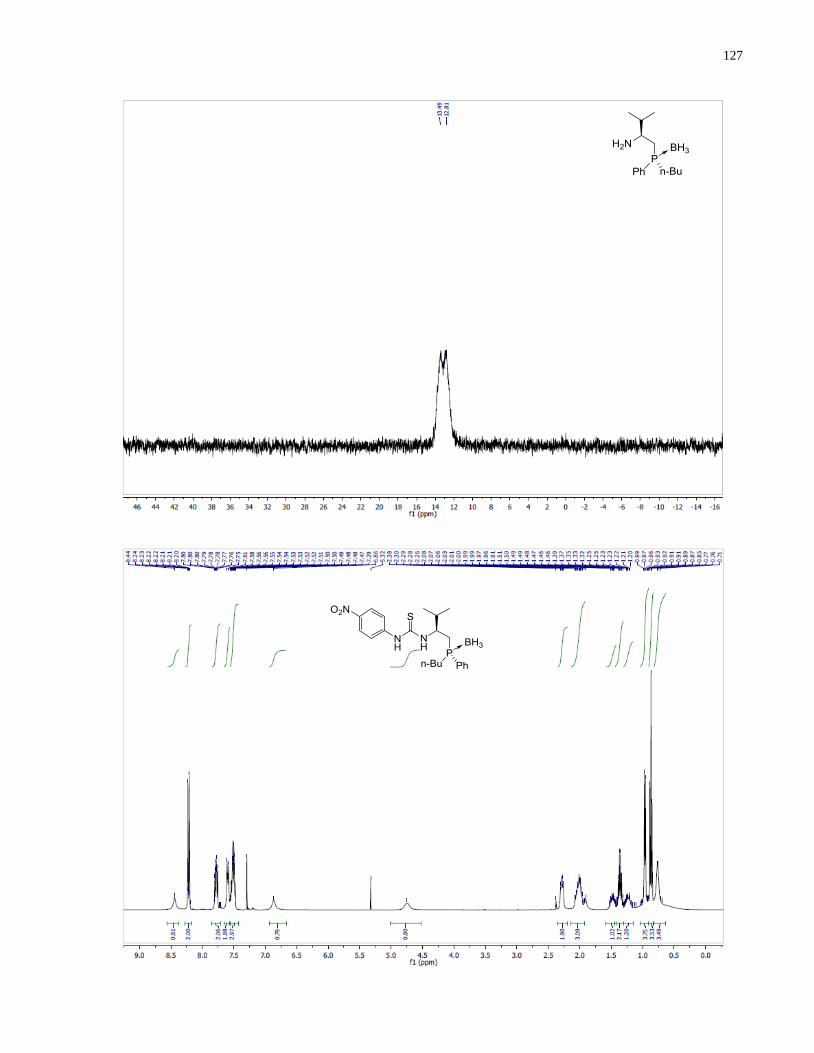
127
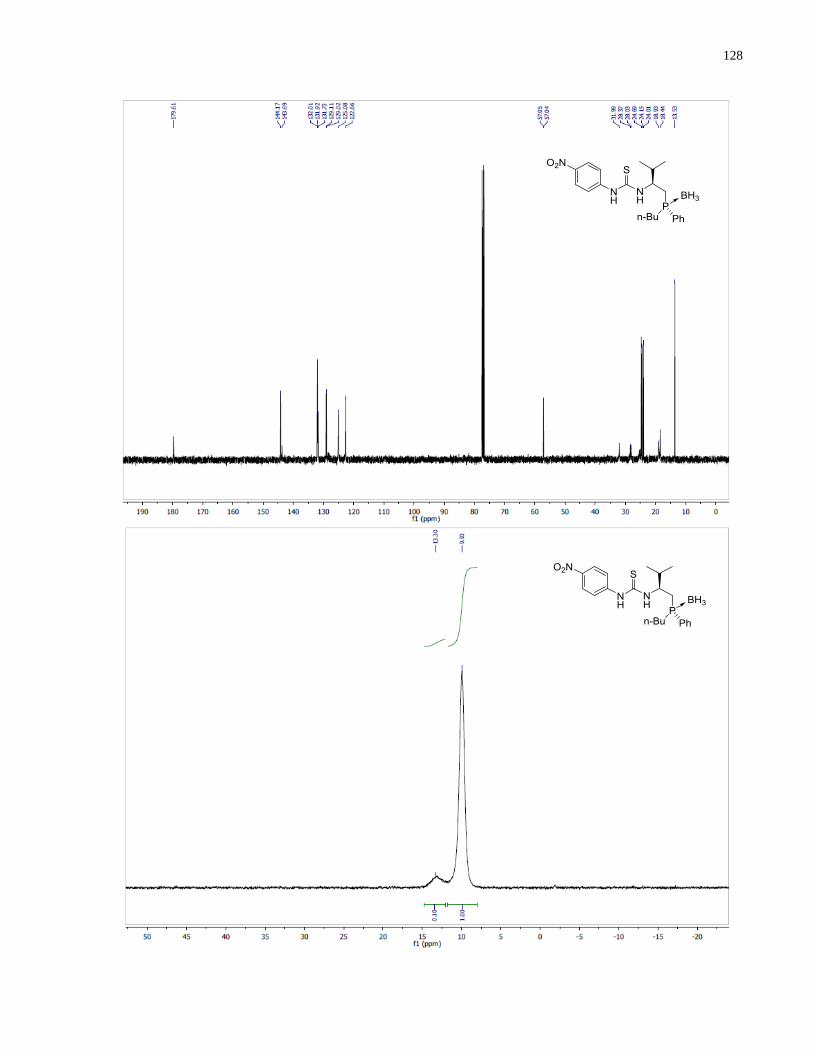
128
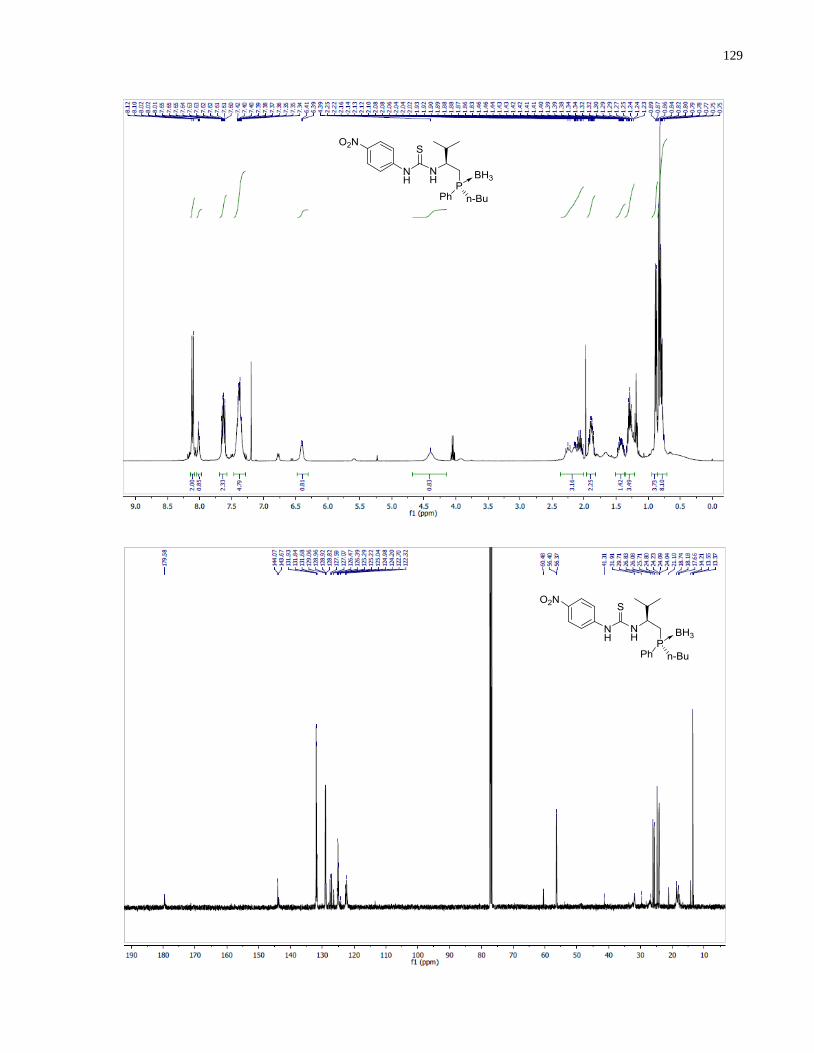
129

130
131
132
133
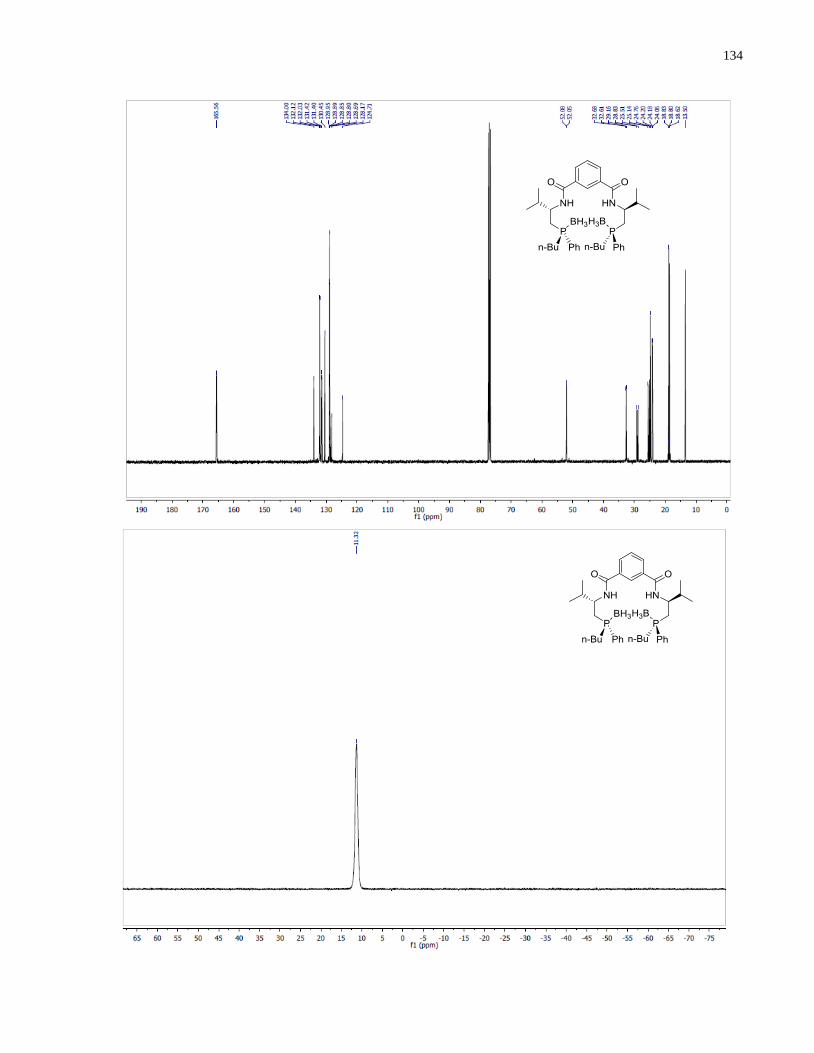
134
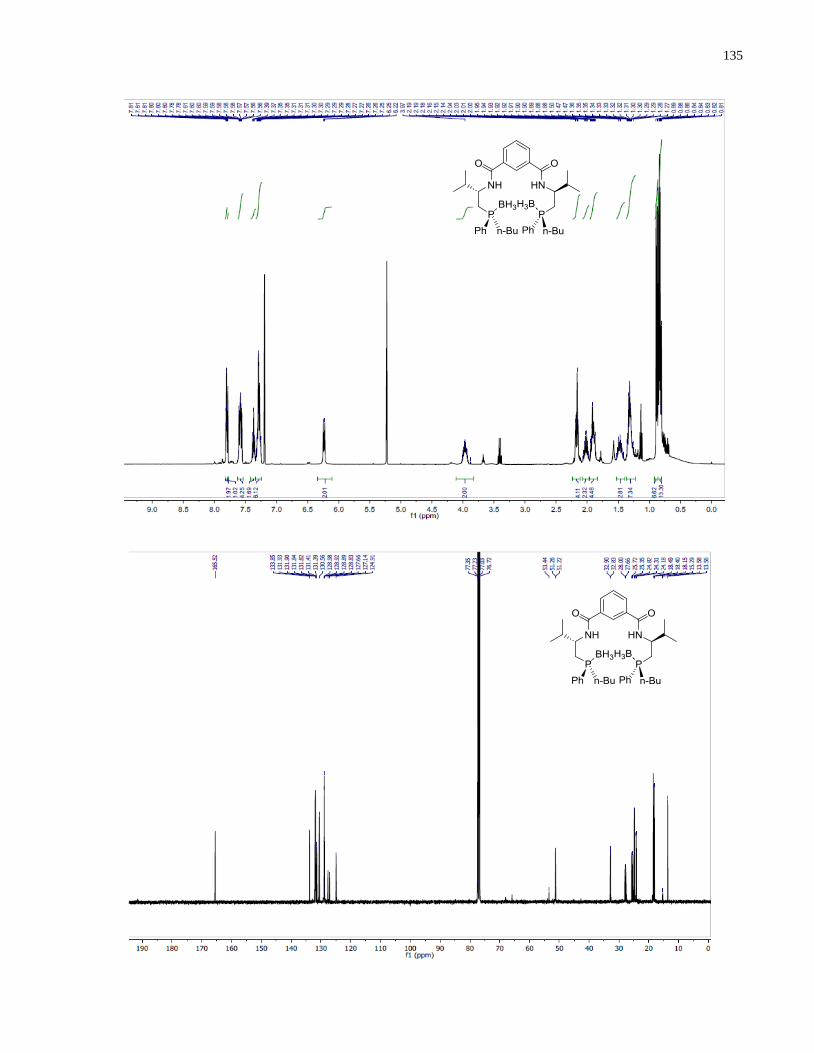
135
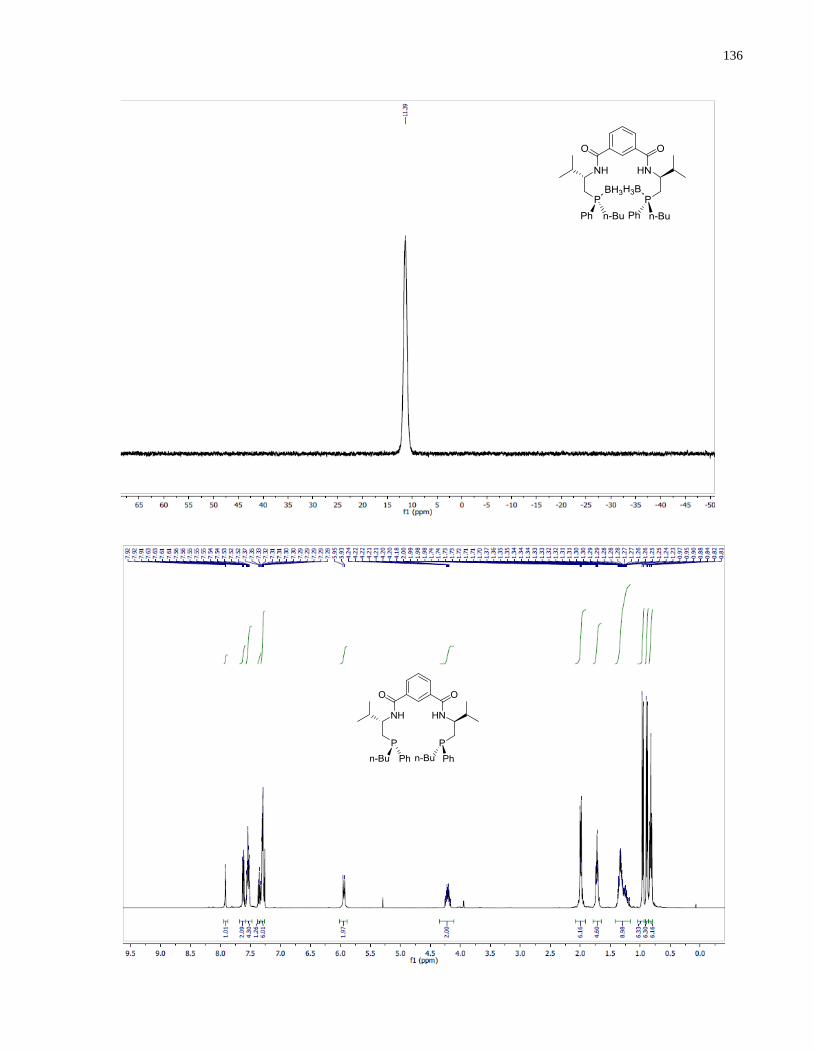
136
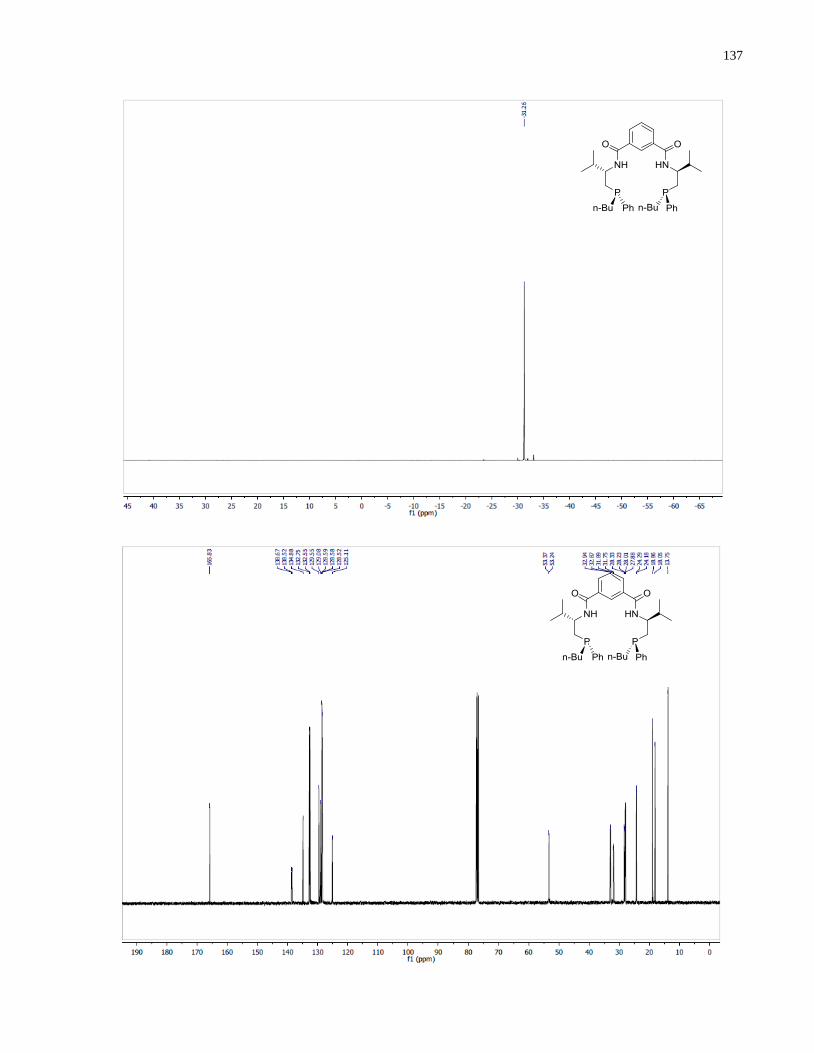
137
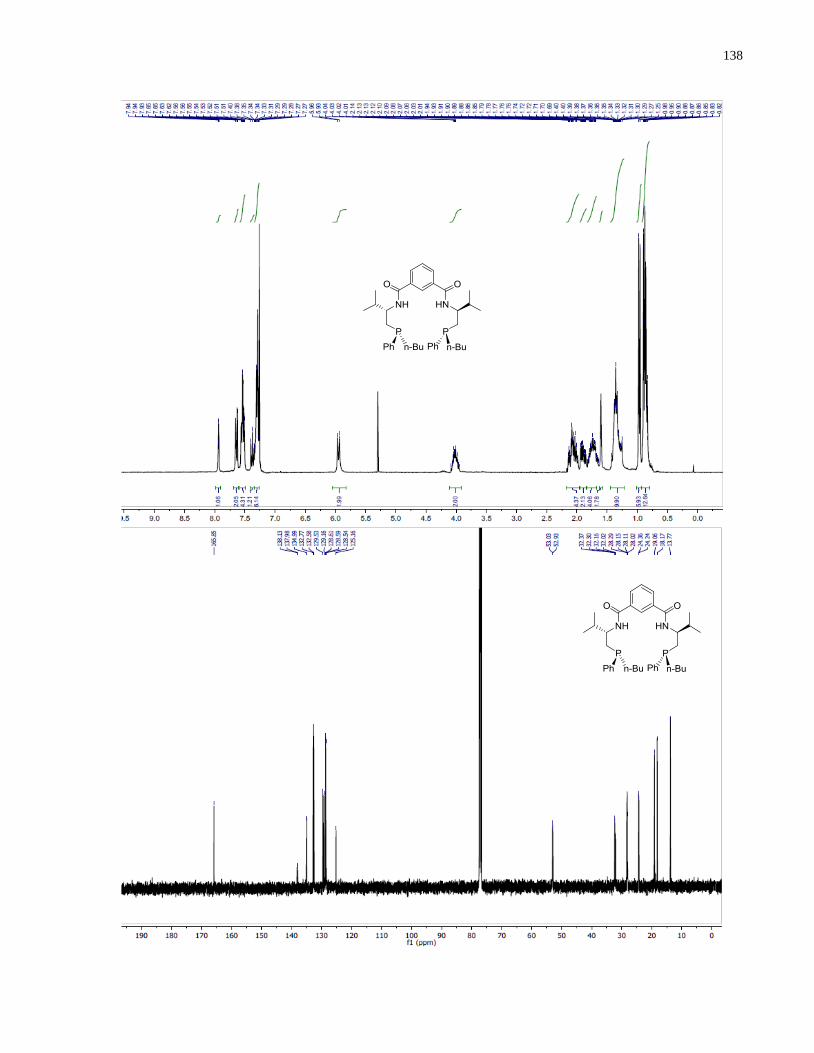
138
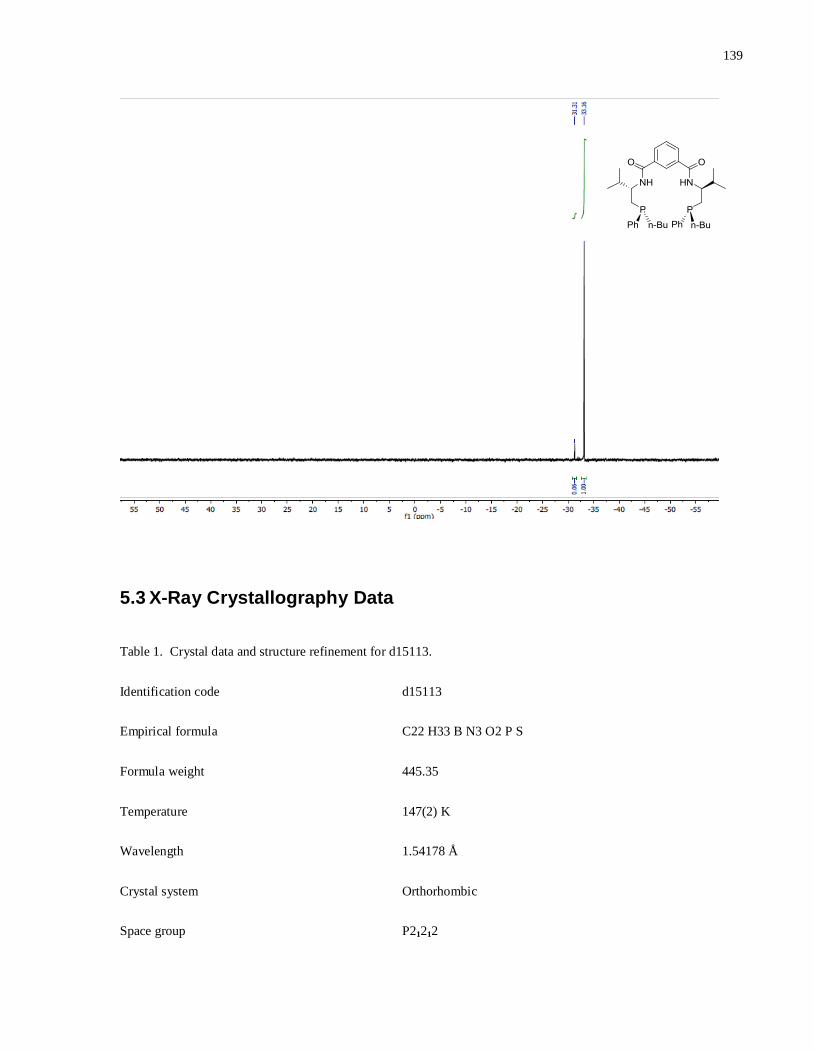
139
5.3 X-Ray Crystallography Data
Table 1. Crystal data and structure refinement for d15113.
Identification code d15113
Empirical formula C22 H33 B N3 O2 P S
Formula weight 445.35
Temperature 147(2) K
Wavelength 1.54178 Å
Crystal system Orthorhombic
Space group P21212

140
Unit cell dimensions a = 15.0286(5) Å = 90°.
b = 22.2881(8) Å = 90°.
c = 7.3704(3) Å = 90°.
Volume 2468.78(16) Å3
Z 4
Density (calculated) 1.198 Mg/m3
Absorption coefficient 1.947 mm-1
F(000) 952
Crystal size 0.200 x 0.200 x 0.100 mm3
Theta range for data collection 3.547 to 67.154°.
Index ranges -17<=h<=17, -26<=k<=26, -8<=l<=8
Reflections collected 42609
Independent reflections 4363 [R(int) = 0.0344]
Completeness to theta = 67.154° 99.2 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.7529 and 0.6642
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 4363 / 5 / 310
Goodness-of-fit on F2 1.049
Final R indices [I>2sigma(I)] R1 = 0.0252, wR2 = 0.0666
R indices (all data) R1 = 0.0257, wR2 = 0.0670
Absolute structure parameter 0.011(16)

141
Extinction coefficient n/a
Largest diff. peak and hole 0.317 and -0.196 e.Å-3
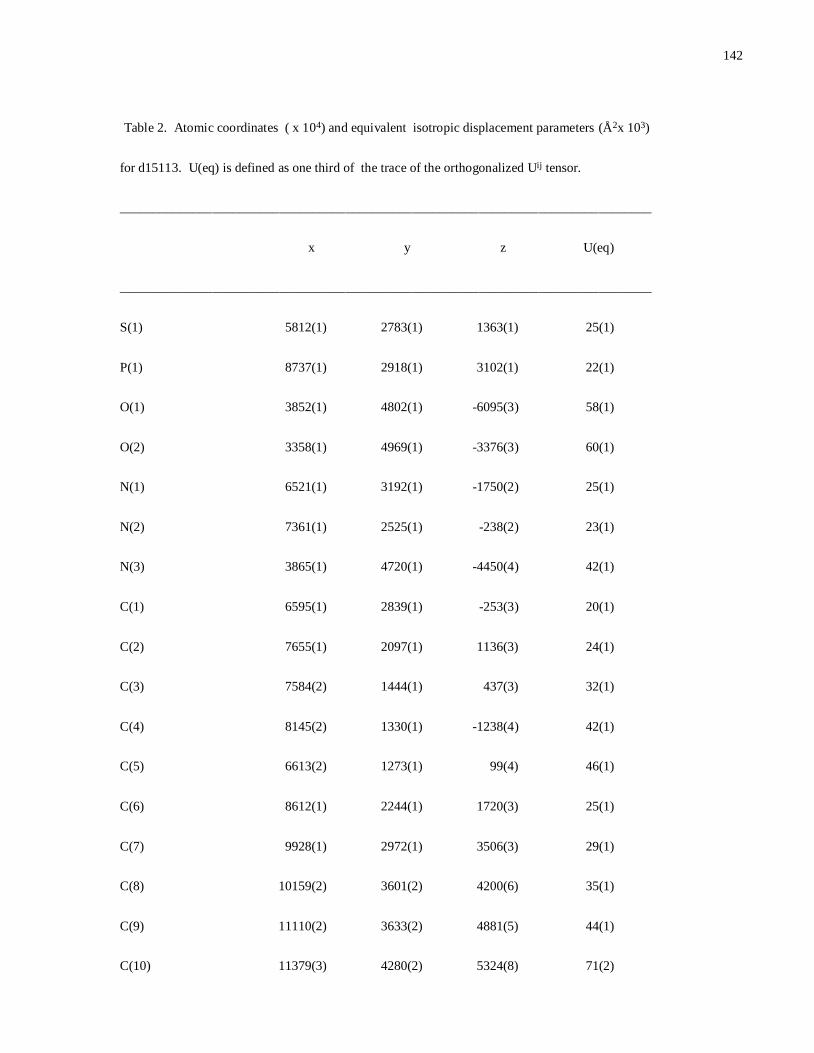
142
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for d15113. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
S(1) 5812(1) 2783(1) 1363(1) 25(1)
P(1) 8737(1) 2918(1) 3102(1) 22(1)
O(1) 3852(1) 4802(1) -6095(3) 58(1)
O(2) 3358(1) 4969(1) -3376(3) 60(1)
N(1) 6521(1) 3192(1) -1750(2) 25(1)
N(2) 7361(1) 2525(1) -238(2) 23(1)
N(3) 3865(1) 4720(1) -4450(4) 42(1)
C(1) 6595(1) 2839(1) -253(3) 20(1)
C(2) 7655(1) 2097(1) 1136(3) 24(1)
C(3) 7584(2) 1444(1) 437(3) 32(1)
C(4) 8145(2) 1330(1) -1238(4) 42(1)
C(5) 6613(2) 1273(1) 99(4) 46(1)
C(6) 8612(1) 2244(1) 1720(3) 25(1)
C(7) 9928(1) 2972(1) 3506(3) 29(1)
C(8) 10159(2) 3601(2) 4200(6) 35(1)
C(9) 11110(2) 3633(2) 4881(5) 44(1)
C(10) 11379(3) 4280(2) 5324(8) 71(2)

143
C(7A) 9928(1) 2972(1) 3506(3) 29(1)
C(8A) 10326(6) 3459(3) 4719(10) 30(2)
C(9A) 10178(5) 4098(3) 4092(9) 35(2)
C(10A) 10747(6) 4546(3) 5160(12) 52(2)
C(11) 8460(1) 3552(1) 1680(3) 25(1)
C(12) 8924(2) 3650(1) 69(3) 33(1)
C(13) 8721(2) 4144(1) -988(3) 43(1)
C(14) 8060(2) 4535(1) -471(4) 47(1)
C(15) 7598(2) 4439(1) 1116(4) 46(1)
C(16) 7796(2) 3950(1) 2202(3) 34(1)
C(17) 5832(1) 3569(1) -2331(3) 22(1)
C(18) 5331(1) 3926(1) -1161(3) 25(1)
C(19) 4674(1) 4296(1) -1860(3) 29(1)
C(20) 4540(1) 4314(1) -3709(3) 30(1)
C(21) 5039(2) 3973(1) -4901(3) 31(1)
C(22) 5685(1) 3598(1) -4196(3) 26(1)
B(1) 8092(2) 2856(1) 5333(3) 32(1)
________________________________________________________________________________

144
Table 3. Bond lengths [Å] and angles [°] for d15113.
_____________________________________________________
S(1)-C(1) 1.6791(19)
P(1)-C(11) 1.807(2)
P(1)-C(7A) 1.819(2)
P(1)-C(7) 1.819(2)
P(1)-C(6) 1.824(2)
P(1)-B(1) 1.914(2)
O(1)-N(3) 1.226(3)
O(2)-N(3) 1.231(3)
N(1)-C(1) 1.360(3)
N(1)-C(17) 1.402(3)
N(1)-H(1N) 0.77(3)
N(2)-C(1) 1.347(3)
N(2)-C(2) 1.461(3)
N(2)-H(2N) 0.82(3)
N(3)-C(20) 1.464(3)
C(2)-C(6) 1.536(3)
C(2)-C(3) 1.547(3)
C(2)-H(2A) 1.0000
C(3)-C(4) 1.517(4)
C(3)-C(5) 1.528(3)

145
C(3)-H(3A) 1.0000
C(4)-H(4A) 0.9800
C(4)-H(4B) 0.9800
C(4)-H(4C) 0.9800
C(5)-H(5A) 0.9800
C(5)-H(5B) 0.9800
C(5)-H(5C) 0.9800
C(6)-H(6A) 0.9900
C(6)-H(6B) 0.9900
C(7)-C(8) 1.532(4)
C(7)-H(7A) 0.9900
C(7)-H(7B) 0.9900
C(8)-C(9) 1.517(5)
C(8)-H(8A) 0.9900
C(8)-H(8B) 0.9900
C(9)-C(10) 1.532(6)
C(9)-H(9A) 0.9900
C(9)-H(9B) 0.9900
C(10)-H(10A) 0.9800
C(10)-H(10B) 0.9800
C(10)-H(10C) 0.9800
C(7A)-C(8A) 1.528(6)

146
C(7A)-H(7A1) 0.9900
C(7A)-H(7A2) 0.9900
C(8A)-C(9A) 1.514(6)
C(8A)-H(8AA) 0.9900
C(8A)-H(8AB) 0.9900
C(9A)-C(10A) 1.533(7)
C(9A)-H(9AA) 0.9900
C(9A)-H(9AB) 0.9900
C(10A)-H(10D) 0.9800
C(10A)-H(10E) 0.9800
C(10A)-H(10F) 0.9800
C(11)-C(16) 1.389(3)
C(11)-C(12) 1.394(3)
C(12)-C(13) 1.382(4)
C(12)-H(12A) 0.9500
C(13)-C(14) 1.375(4)
C(13)-H(13A) 0.9500
C(14)-C(15) 1.377(4)
C(14)-H(14A) 0.9500
C(15)-C(16) 1.385(4)
C(15)-H(15A) 0.9500
C(16)-H(16A) 0.9500

147
C(17)-C(22) 1.394(3)
C(17)-C(18) 1.394(3)
C(18)-C(19) 1.384(3)
C(18)-H(18A) 0.9500
C(19)-C(20) 1.379(4)
C(19)-H(19A) 0.9500
C(20)-C(21) 1.383(3)
C(21)-C(22) 1.383(3)
C(21)-H(21A) 0.9500
C(22)-H(22A) 0.9500
B(1)-H(1) 1.07(3)
B(1)-H(2) 1.16(3)
B(1)-H(3) 1.06(3)
C(11)-P(1)-C(7A) 105.65(10)
C(11)-P(1)-C(7) 105.65(10)
C(11)-P(1)-C(6) 107.23(9)
C(7A)-P(1)-C(6) 104.31(9)
C(7)-P(1)-C(6) 104.31(9)
C(11)-P(1)-B(1) 115.99(11)
C(7A)-P(1)-B(1) 111.24(11)
C(7)-P(1)-B(1) 111.24(11)

148
C(6)-P(1)-B(1) 111.61(11)
C(1)-N(1)-C(17) 130.93(18)
C(1)-N(1)-H(1N) 117(2)
C(17)-N(1)-H(1N) 112(2)
C(1)-N(2)-C(2) 127.15(17)
C(1)-N(2)-H(2N) 115.8(15)
C(2)-N(2)-H(2N) 117.1(15)
O(1)-N(3)-O(2) 123.9(2)
O(1)-N(3)-C(20) 118.2(2)
O(2)-N(3)-C(20) 117.9(2)
N(2)-C(1)-N(1) 112.11(17)
N(2)-C(1)-S(1) 123.71(15)
N(1)-C(1)-S(1) 124.16(15)
N(2)-C(2)-C(6) 109.74(16)
N(2)-C(2)-C(3) 111.27(17)
C(6)-C(2)-C(3) 111.09(16)
N(2)-C(2)-H(2A) 108.2
C(6)-C(2)-H(2A) 108.2
C(3)-C(2)-H(2A) 108.2
C(4)-C(3)-C(5) 110.9(2)
C(4)-C(3)-C(2) 112.93(19)
C(5)-C(3)-C(2) 110.79(18)

149
C(4)-C(3)-H(3A) 107.3
C(5)-C(3)-H(3A) 107.3
C(2)-C(3)-H(3A) 107.3
C(3)-C(4)-H(4A) 109.5
C(3)-C(4)-H(4B) 109.5
H(4A)-C(4)-H(4B) 109.5
C(3)-C(4)-H(4C) 109.5
H(4A)-C(4)-H(4C) 109.5
H(4B)-C(4)-H(4C) 109.5
C(3)-C(5)-H(5A) 109.5
C(3)-C(5)-H(5B) 109.5
H(5A)-C(5)-H(5B) 109.5
C(3)-C(5)-H(5C) 109.5
H(5A)-C(5)-H(5C) 109.5
H(5B)-C(5)-H(5C) 109.5
C(2)-C(6)-P(1) 115.38(13)
C(2)-C(6)-H(6A) 108.4
P(1)-C(6)-H(6A) 108.4
C(2)-C(6)-H(6B) 108.4
P(1)-C(6)-H(6B) 108.4
H(6A)-C(6)-H(6B) 107.5
C(8)-C(7)-P(1) 109.76(18)

150
C(8)-C(7)-H(7A) 109.7
P(1)-C(7)-H(7A) 109.7
C(8)-C(7)-H(7B) 109.7
P(1)-C(7)-H(7B) 109.7
H(7A)-C(7)-H(7B) 108.2
C(9)-C(8)-C(7) 111.6(3)
C(9)-C(8)-H(8A) 109.3
C(7)-C(8)-H(8A) 109.3
C(9)-C(8)-H(8B) 109.3
C(7)-C(8)-H(8B) 109.3
H(8A)-C(8)-H(8B) 108.0
C(8)-C(9)-C(10) 111.4(4)
C(8)-C(9)-H(9A) 109.3
C(10)-C(9)-H(9A) 109.3
C(8)-C(9)-H(9B) 109.3
C(10)-C(9)-H(9B) 109.3
H(9A)-C(9)-H(9B) 108.0
C(9)-C(10)-H(10A) 109.5
C(9)-C(10)-H(10B) 109.5
H(10A)-C(10)-H(10B) 109.5
C(9)-C(10)-H(10C) 109.5
H(10A)-C(10)-H(10C) 109.5
151
H(10B)-C(10)-H(10C) 109.5
C(8A)-C(7A)-P(1) 121.9(4)
C(8A)-C(7A)-H(7A1) 106.9
P(1)-C(7A)-H(7A1) 106.9
C(8A)-C(7A)-H(7A2) 106.9
P(1)-C(7A)-H(7A2) 106.9
H(7A1)-C(7A)-H(7A2) 106.7
C(9A)-C(8A)-C(7A) 115.6(5)
C(9A)-C(8A)-H(8AA) 108.4
C(7A)-C(8A)-H(8AA) 108.4
C(9A)-C(8A)-H(8AB) 108.4
C(7A)-C(8A)-H(8AB) 108.4
H(8AA)-C(8A)-H(8AB) 107.4
C(8A)-C(9A)-C(10A) 112.0(5)
C(8A)-C(9A)-H(9AA) 109.2
C(10A)-C(9A)-H(9AA) 109.2
C(8A)-C(9A)-H(9AB) 109.2
C(10A)-C(9A)-H(9AB) 109.2
H(9AA)-C(9A)-H(9AB) 107.9
C(9A)-C(10A)-H(10D) 109.5
C(9A)-C(10A)-H(10E) 109.5
H(10D)-C(10A)-H(10E) 109.5

152
C(9A)-C(10A)-H(10F) 109.5
H(10D)-C(10A)-H(10F) 109.5
H(10E)-C(10A)-H(10F) 109.5
C(16)-C(11)-C(12) 119.6(2)
C(16)-C(11)-P(1) 120.24(17)
C(12)-C(11)-P(1) 120.14(16)
C(13)-C(12)-C(11) 119.7(2)
C(13)-C(12)-H(12A) 120.2
C(11)-C(12)-H(12A) 120.2
C(14)-C(13)-C(12) 120.5(2)
C(14)-C(13)-H(13A) 119.7
C(12)-C(13)-H(13A) 119.7
C(13)-C(14)-C(15) 120.0(2)
C(13)-C(14)-H(14A) 120.0
C(15)-C(14)-H(14A) 120.0
C(14)-C(15)-C(16) 120.3(2)
C(14)-C(15)-H(15A) 119.8
C(16)-C(15)-H(15A) 119.8
C(15)-C(16)-C(11) 119.8(2)
C(15)-C(16)-H(16A) 120.1
C(11)-C(16)-H(16A) 120.1
C(22)-C(17)-C(18) 119.94(19)
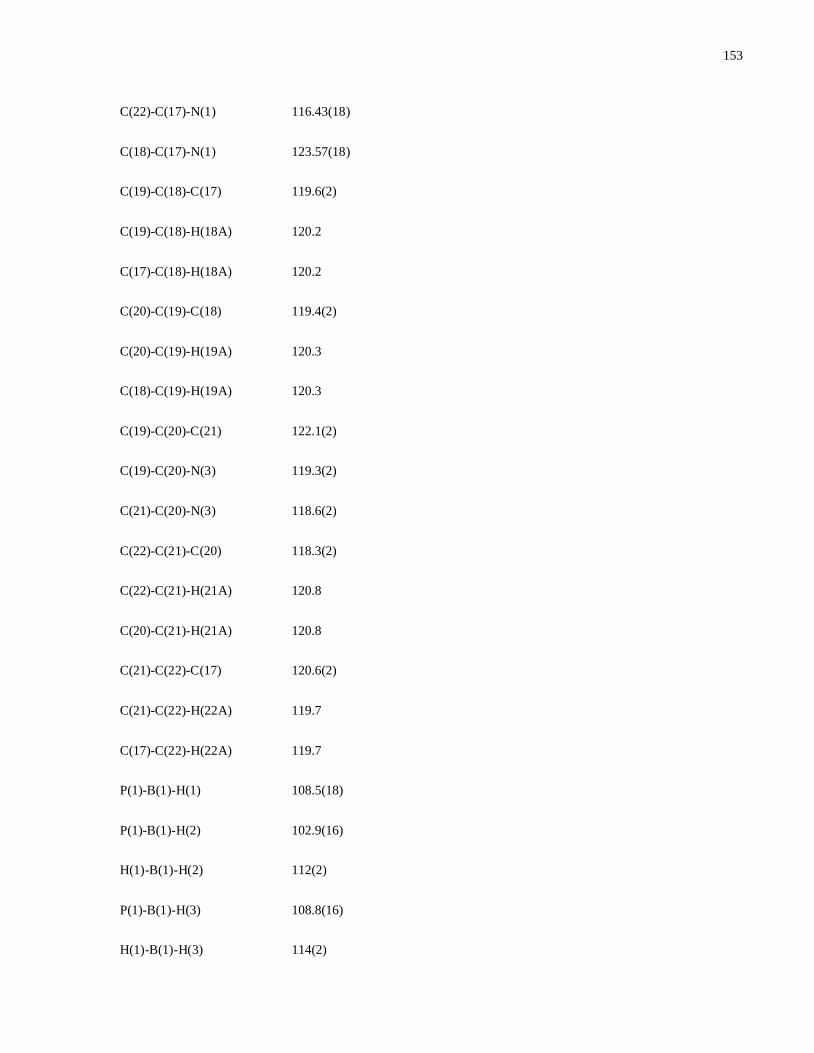
153
C(22)-C(17)-N(1) 116.43(18)
C(18)-C(17)-N(1) 123.57(18)
C(19)-C(18)-C(17) 119.6(2)
C(19)-C(18)-H(18A) 120.2
C(17)-C(18)-H(18A) 120.2
C(20)-C(19)-C(18) 119.4(2)
C(20)-C(19)-H(19A) 120.3
C(18)-C(19)-H(19A) 120.3
C(19)-C(20)-C(21) 122.1(2)
C(19)-C(20)-N(3) 119.3(2)
C(21)-C(20)-N(3) 118.6(2)
C(22)-C(21)-C(20) 118.3(2)
C(22)-C(21)-H(21A) 120.8
C(20)-C(21)-H(21A) 120.8
C(21)-C(22)-C(17) 120.6(2)
C(21)-C(22)-H(22A) 119.7
C(17)-C(22)-H(22A) 119.7
P(1)-B(1)-H(1) 108.5(18)
P(1)-B(1)-H(2) 102.9(16)
H(1)-B(1)-H(2) 112(2)
P(1)-B(1)-H(3) 108.8(16)
H(1)-B(1)-H(3) 114(2)

154
H(2)-B(1)-H(3) 110(2)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
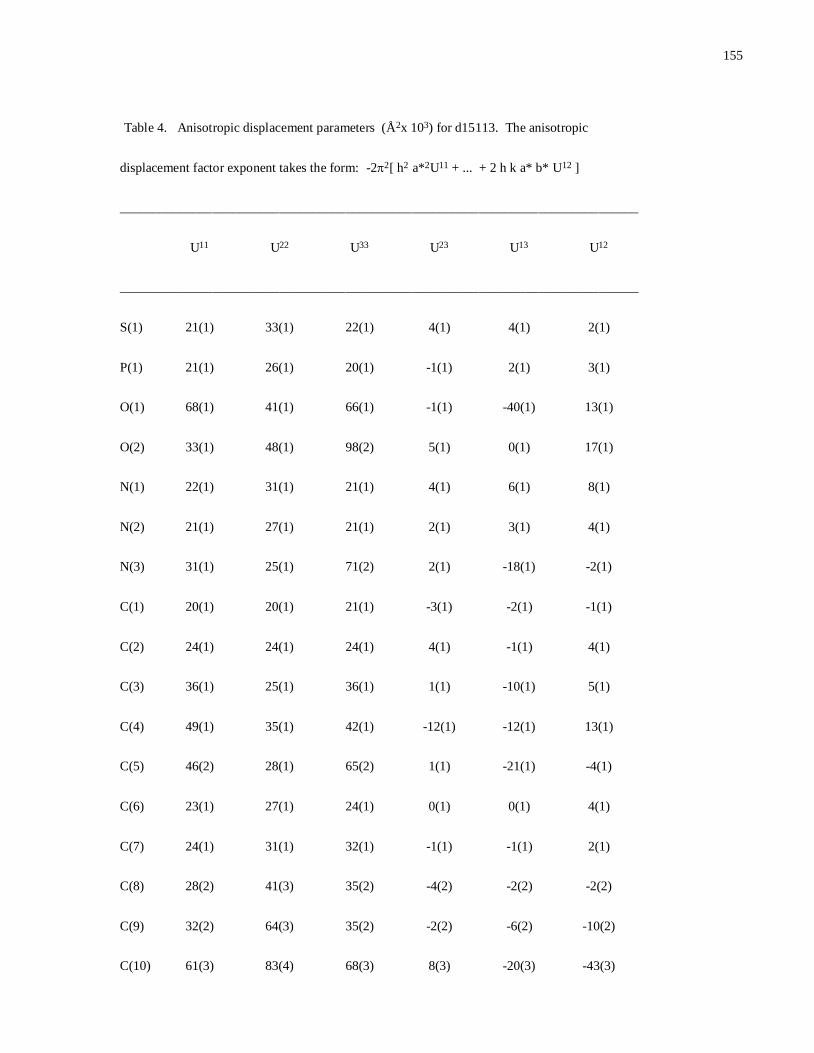
155
Table 4. Anisotropic displacement parameters (Å2x 103) for d15113. The anisotropic
displacement factor exponent takes the form: -22[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________
S(1) 21(1) 33(1) 22(1) 4(1) 4(1) 2(1)
P(1) 21(1) 26(1) 20(1) -1(1) 2(1) 3(1)
O(1) 68(1) 41(1) 66(1) -1(1) -40(1) 13(1)
O(2) 33(1) 48(1) 98(2) 5(1) 0(1) 17(1)
N(1) 22(1) 31(1) 21(1) 4(1) 6(1) 8(1)
N(2) 21(1) 27(1) 21(1) 2(1) 3(1) 4(1)
N(3) 31(1) 25(1) 71(2) 2(1) -18(1) -2(1)
C(1) 20(1) 20(1) 21(1) -3(1) -2(1) -1(1)
C(2) 24(1) 24(1) 24(1) 4(1) -1(1) 4(1)
C(3) 36(1) 25(1) 36(1) 1(1) -10(1) 5(1)
C(4) 49(1) 35(1) 42(1) -12(1) -12(1) 13(1)
C(5) 46(2) 28(1) 65(2) 1(1) -21(1) -4(1)
C(6) 23(1) 27(1) 24(1) 0(1) 0(1) 4(1)
C(7) 24(1) 31(1) 32(1) -1(1) -1(1) 2(1)
C(8) 28(2) 41(3) 35(2) -4(2) -2(2) -2(2)
C(9) 32(2) 64(3) 35(2) -2(2) -6(2) -10(2)
C(10) 61(3) 83(4) 68(3) 8(3) -20(3) -43(3)
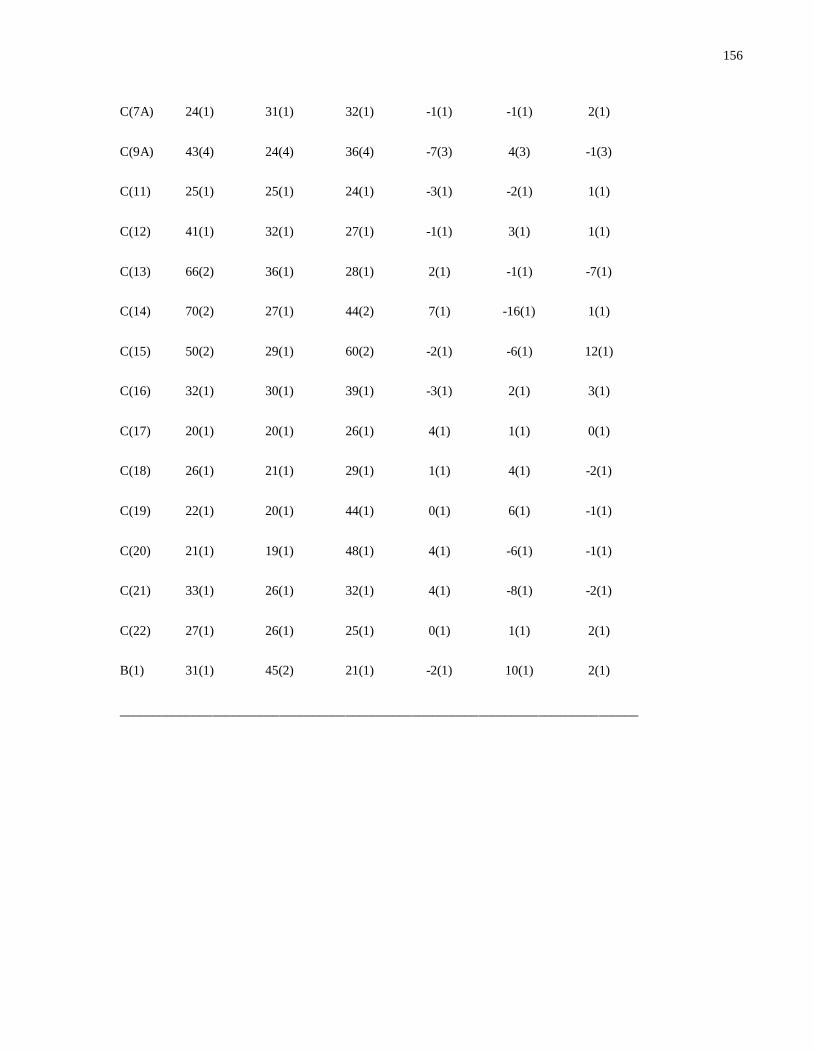
156
C(7A) 24(1) 31(1) 32(1) -1(1) -1(1) 2(1)
C(9A) 43(4) 24(4) 36(4) -7(3) 4(3) -1(3)
C(11) 25(1) 25(1) 24(1) -3(1) -2(1) 1(1)
C(12) 41(1) 32(1) 27(1) -1(1) 3(1) 1(1)
C(13) 66(2) 36(1) 28(1) 2(1) -1(1) -7(1)
C(14) 70(2) 27(1) 44(2) 7(1) -16(1) 1(1)
C(15) 50(2) 29(1) 60(2) -2(1) -6(1) 12(1)
C(16) 32(1) 30(1) 39(1) -3(1) 2(1) 3(1)
C(17) 20(1) 20(1) 26(1) 4(1) 1(1) 0(1)
C(18) 26(1) 21(1) 29(1) 1(1) 4(1) -2(1)
C(19) 22(1) 20(1) 44(1) 0(1) 6(1) -1(1)
C(20) 21(1) 19(1) 48(1) 4(1) -6(1) -1(1)
C(21) 33(1) 26(1) 32(1) 4(1) -8(1) -2(1)
C(22) 27(1) 26(1) 25(1) 0(1) 1(1) 2(1)
B(1) 31(1) 45(2) 21(1) -2(1) 10(1) 2(1)
______________________________________________________________________________
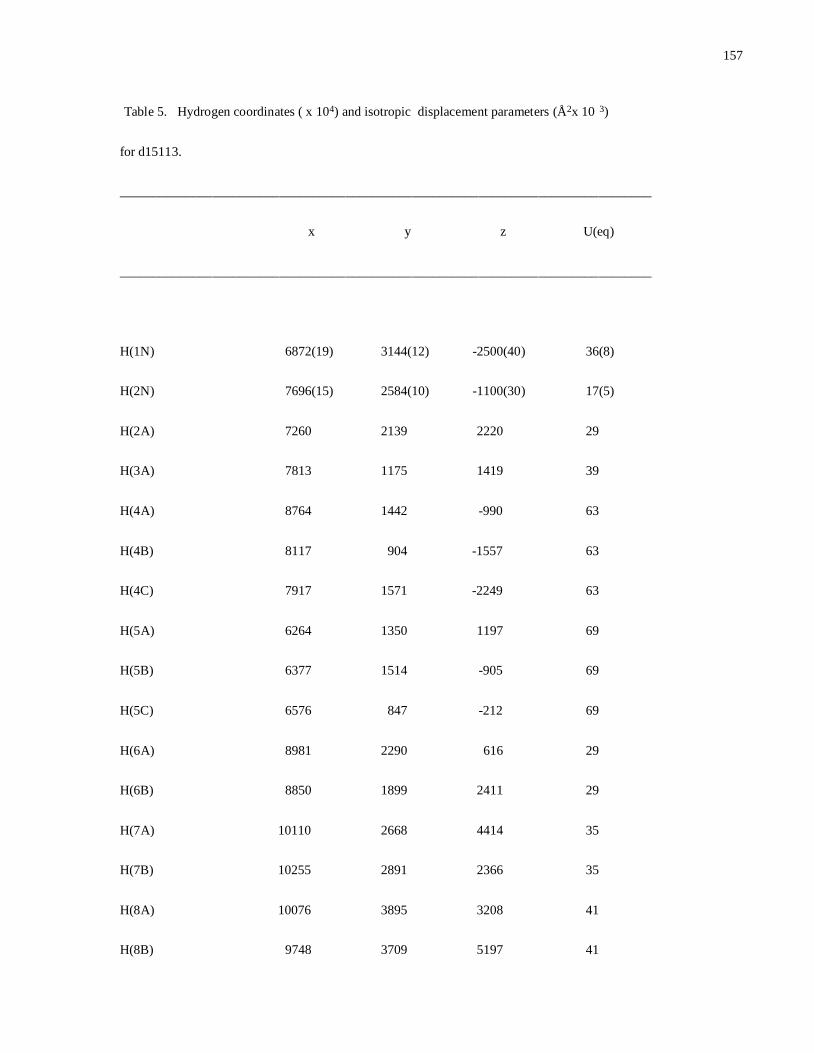
157
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3)
for d15113.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(1N) 6872(19) 3144(12) -2500(40) 36(8)
H(2N) 7696(15) 2584(10) -1100(30) 17(5)
H(2A) 7260 2139 2220 29
H(3A) 7813 1175 1419 39
H(4A) 8764 1442 -990 63
H(4B) 8117 904 -1557 63
H(4C) 7917 1571 -2249 63
H(5A) 6264 1350 1197 69
H(5B) 6377 1514 -905 69
H(5C) 6576 847 -212 69
H(6A) 8981 2290 616 29
H(6B) 8850 1899 2411 29
H(7A) 10110 2668 4414 35
H(7B) 10255 2891 2366 35
H(8A) 10076 3895 3208 41
H(8B) 9748 3709 5197 41
158
H(9A) 11515 3470 3945 53
H(9B) 11171 3383 5984 53
H(10A) 11995 4287 5759 106
H(10B) 11329 4527 4228 106
H(10C) 10985 4440 6266 106
H(7A1) 10120 2581 4012 35
H(7A2) 10216 3011 2304 35
H(8AA) 10071 3415 5951 36
H(8AB) 10975 3388 4816 36
H(9AA) 9541 4202 4241 42
H(9AB) 10325 4129 2786 42
H(10D) 10632 4953 4715 78
H(10E) 10595 4523 6451 78
H(10F) 11378 4449 4997 78
H(12A) 9377 3380 -300 40
H(13A) 9040 4213 -2080 52
H(14A) 7922 4871 -1210 56
H(15A) 7141 4709 1467 56
H(16A) 7479 3886 3301 40
H(18A) 5439 3916 109 30
H(19A) 4320 4534 -1073 34
H(21A) 4940 3995 -6173 37

159
H(22A) 6032 3357 -4990 31
H(1) 7420(20) 2717(14) 5030(50) 53(8)
H(2) 8480(20) 2492(15) 6120(50) 60(9)
H(3) 8133(19) 3270(13) 6020(40) 45(8)
________________________________________________________________________________

160
Table 6. Torsion angles [°] for d15113.
________________________________________________________________
C(2)-N(2)-C(1)-N(1) -178.97(18)
C(2)-N(2)-C(1)-S(1) -0.7(3)
C(17)-N(1)-C(1)-N(2) 177.5(2)
C(17)-N(1)-C(1)-S(1) -0.8(3)
C(1)-N(2)-C(2)-C(6) -130.5(2)
C(1)-N(2)-C(2)-C(3) 106.2(2)
N(2)-C(2)-C(3)-C(4) 60.1(2)
C(6)-C(2)-C(3)-C(4) -62.5(2)
N(2)-C(2)-C(3)-C(5) -65.0(2)
C(6)-C(2)-C(3)-C(5) 172.4(2)
N(2)-C(2)-C(6)-P(1) 71.85(19)
C(3)-C(2)-C(6)-P(1) -164.69(14)
C(11)-P(1)-C(6)-C(2) -66.98(16)
C(7A)-P(1)-C(6)-C(2) -178.73(15)
C(7)-P(1)-C(6)-C(2) -178.73(15)
B(1)-P(1)-C(6)-C(2) 61.06(18)
C(11)-P(1)-C(7)-C(8) 51.8(2)
C(6)-P(1)-C(7)-C(8) 164.7(2)
B(1)-P(1)-C(7)-C(8) -74.9(2)
P(1)-C(7)-C(8)-C(9) 169.6(3)

161
C(7)-C(8)-C(9)-C(10) 172.0(4)
C(11)-P(1)-C(7A)-C(8A) 70.9(4)
C(6)-P(1)-C(7A)-C(8A) -176.3(3)
B(1)-P(1)-C(7A)-C(8A) -55.8(4)
P(1)-C(7A)-C(8A)-C(9A) -61.9(7)
C(7A)-C(8A)-C(9A)-C(10A) -168.7(6)
C(7A)-P(1)-C(11)-C(16) -125.50(18)
C(7)-P(1)-C(11)-C(16) -125.50(18)
C(6)-P(1)-C(11)-C(16) 123.67(17)
B(1)-P(1)-C(11)-C(16) -1.8(2)
C(7A)-P(1)-C(11)-C(12) 53.23(19)
C(7)-P(1)-C(11)-C(12) 53.23(19)
C(6)-P(1)-C(11)-C(12) -57.59(19)
B(1)-P(1)-C(11)-C(12) 176.96(17)
C(16)-C(11)-C(12)-C(13) 0.2(3)
P(1)-C(11)-C(12)-C(13) -178.53(19)
C(11)-C(12)-C(13)-C(14) -0.6(4)
C(12)-C(13)-C(14)-C(15) 0.4(4)
C(13)-C(14)-C(15)-C(16) 0.1(4)
C(14)-C(15)-C(16)-C(11) -0.5(4)
C(12)-C(11)-C(16)-C(15) 0.3(3)
P(1)-C(11)-C(16)-C(15) 179.06(19)

162
C(1)-N(1)-C(17)-C(22) -144.1(2)
C(1)-N(1)-C(17)-C(18) 38.8(3)
C(22)-C(17)-C(18)-C(19) 1.6(3)
N(1)-C(17)-C(18)-C(19) 178.60(18)
C(17)-C(18)-C(19)-C(20) -1.4(3)
C(18)-C(19)-C(20)-C(21) 0.2(3)
C(18)-C(19)-C(20)-N(3) -178.00(17)
O(1)-N(3)-C(20)-C(19) 168.4(2)
O(2)-N(3)-C(20)-C(19) -9.8(3)
O(1)-N(3)-C(20)-C(21) -9.9(3)
O(2)-N(3)-C(20)-C(21) 171.9(2)
C(19)-C(20)-C(21)-C(22) 0.7(3)
N(3)-C(20)-C(21)-C(22) 178.94(18)
C(20)-C(21)-C(22)-C(17) -0.4(3)
C(18)-C(17)-C(22)-C(21) -0.7(3)
N(1)-C(17)-C(22)-C(21) -177.89(19)
________________________________________________________________
Symmetry transformations used to generate equivalent atoms:

163














![P-Chiral Phosphorus Ligands: Synthesis and Application in ...¶rn_Gschwend.pdf · [4b] Later, Knowles’ P-stereogenic diphosphine ligand DIPAMP was synthesized by oxidative coupling](https://static.fdocuments.us/doc/165x107/5fb1050757199a29b110498b/p-chiral-phosphorus-ligands-synthesis-and-application-in-rngschwendpdf.jpg)



