
STUDY OF CHIRAL AZA-MACROCYCLIC LIGANDS INVOLVED IN IMPORTANT BIOLOGICAL PROCESSES · STUDY OF...
84
STUDY OF CHIRAL AZA-MACROCYCLIC LIGANDS INVOLVED IN IMPORTANT BIOLOGICAL PROCESSES Susanna Sampaolesi Dissertação para obteção do Grau de mestre em Química Orientadores: Prof. Enrico Marcantoni Doutora Alexandra Maria Moita Antunes Júri Presidente: Prof. a Maria Matilde Soares Duarte Marques Vogais: Prof. Corrado Bacchiocchi Dr. Alexandra Maria Moita Antunes Prof. Pedro Paulo de Lacerta e Oliveira Santos Novembro de 2014
Transcript of STUDY OF CHIRAL AZA-MACROCYCLIC LIGANDS INVOLVED IN IMPORTANT BIOLOGICAL PROCESSES · STUDY OF...

STUDY OF CHIRAL AZA-MACROCYCLIC LIGANDS INVOLVED
IN IMPORTANT BIOLOGICAL PROCESSES
Susanna Sampaolesi
Dissertação para obteção do Grau de mestre em
Química
Orientadores: Prof. Enrico Marcantoni
Doutora Alexandra Maria Moita Antunes
Júri
Presidente: Prof.a Maria Matilde Soares Duarte Marques
Vogais: Prof. Corrado Bacchiocchi
Dr. Alexandra Maria Moita Antunes
Prof. Pedro Paulo de Lacerta e Oliveira Santos
Novembro de 2014

1
ABSTRACT
Azamacrocyclic ligands able to coordinate metal ions in a selective way are used in a
wide variety of applications, such as in metal extraction or as models of protein
binding sites. Keeping in mind the importance of this family of compounds, we studied
synthetic methods to obtain an enantiomerically pure hexaaza tetramine macrocycle
M1, resulting from the condensation of two units of pyridine-2,6-dicarbaldehyde and
(R,R)-1,2-diaminocyclohexane and the subsequent in situ reduction. The condensation
conducted in the presence of Ba(II), which acts as a templating agent, represents the
more efficient way of synthesis, in terms of yield and selectivity. The need to get the
macrocycle of interest was dictated by the birth of a collaboration with the University
"La Sapienza" of Rome. In fact, the target macromolecule is the first of many structural
variations to be carried out on a starting prototype M, which proved extraordinarily
affinity to potassium ion, in presence of an acidic species in gas phase. Knowing the
important role of this ion at physiological level, it was decided to determine the
structural features of the macrocycle, in order to investigate possible useful
applications.
Keywords.
Chiral macrocycles; Schiff bases; Polyamines; Template synthesis.
RESUMO
Os ligandos azamacrocíclicos com capacidade de coordenação selectiva são usados
numa grande variedade de aplicações, como por exemplo na extracção de metais ou
como modelos de sítios de ligação em proteínas. Tendo em conta a importância desta
família de compostos, foram estudados métodos sintéticos para obter um hexaaza
tetramino macrociclo, M1, resultante da condensação de duas unidades de piridina-
2,6-dicarbaldeído e (R,R)-1,2-diaminociclo-hexano, com redução in situ subsequente.
A condensação na presença de Ba (II), que actua como molde, representa a

2
metodologia sintética mais eficiente, em termos de rendimento e selectividade. A
necessidade de obter o macrociclo de interesse resultou do início de uma colaboração
com a Universidade "La Sapienza" de Roma. De facto, a molécula alvo é a primeira de
muitas variantes estruturais a sintetizar a partir de um protótipo M que evidenciou
um extraordinária afinidade para o ião potássio na presença de uma espécie ácida em
fase gasosa. Considerando o importante papel deste ião a nível fisiológico, foi decidido
determinar quais as características estruturais do macrociclo, de modo a investigar
possíveis aplicações úteis.
Palavras-chave
Macrociclos quirais; Bases de Schiff; Poliaminas; Síntese a partir de um molde

3
Index of figures
Figure 1. Charles J. Pedersen.........................................................................................................................9
Figure 2. Example of saturated macrocycles……………………………………………………..............10
Figure 3. Examples of Calixerene and Cyclodextrin.........................................................................10
Figure 4. First synthesized imine ligands..............................................................................................11
Figure 5. Examples of imine ligands........................................................................................................13
Figure 6. Examples of trianglimine ligands.........................................................................................14
Figure 7. Enantiomers of 1,2-diphenylethylendiamine and trans-1,2-
diaminocyclohexane........................................................................................................................................19
Figure 8. Other examples of imine macrocycles.................................................................................27
Figure 9. Resonance structures of Schiff base derived from condensation of 2-
formylpyridine and primary amine…………………………………………………………………………….31
Figure 10. N-Pyridine orbital representation………………………………………………………........31
Figure 11. Examples of first imine macrocycles incorporating pyridine units....................32
Figure 12. Antisense therapeutic agent mechanism of action....................................................33
Figure 13. Macrocyclic ligand s involved in cleavage of DNA phosphodiester linkage...35
Figure 14. Structure of the dimeric tetracation [{Y(33)(OH)(H2O)2}2]4+ in the hydrated
nitrate salt............................................................................................................................................................36
Figure 15. Magnetic Resonance of rat abdomen before (A), 16 (B), 56 (C) and 182 (D)
minuts after administration of a contrast agent………………………………………………………...37
Figure 16. Diethylentriaminopentacetic acid (DTPA) and Ethylendiaminotetracetic acid
(EDTA)……………………………………………………………………………………………………………………..37
Figure 17. Examples of chiral macrocyclic imines...........................................................................39
Figure 18. Stereoselective receptor of biologically important dicarboxylates under
Physiological Conditions................................................................................................................................40
Figure 19. Schematic representation of equilibrium interactions of macrocycle 38 with
two enantiomers of dicarboxylate species………………………………………………………………….41

4
Figure 20. Hexazamacrocycle synthesized by condensation of 2,6-diformylpyridine with
racemic (±)-trans-1,2- diaminocyclohexane………………………………………………………………..41
Figure 21. ESI-QIT spectrometer interfaced with laser source...................................................43
Figure 22. Vibrational Energy levels for (a) a fictional species void of anharmonicity
effects and (b) a species showing the effect of anharmonicity on vibrational spacings…44
Figure 23. Model for IR multiphoton absorption from the ground state to the
quasicontinuum and to the dissociative continuum……………………………………………………45
Figure 24. Different views of [39·K·H2O]⁺complex (bond length expressed in
angstrom)…………………………………………………………………………………………………………………48
Figure 25. Different views of [39·K·HF]⁺complex (bond length expressed in
angstrom)…………………………………………………………………………………………………………………48
Figure 26. Different views of [39·K·HCl]⁺complex (bond length expressed in
angstrom)…………………………………………………………………………………………………………………49
Figure 27. A cartoon schematic of CID fragmentation...................................................................50
Figure 28. First structural variation planned.....................................................................................53
Figure 29. Stereoisomer conformations (in twist conformation cyclohexyl groups are
omitted for clarity)………………………………………………………………………………………………….....54
Figure 30. ESI-MS spectrum of reaction mixture after a) 7 hours b) 23 hours and c) 28
hours of reflux…………………………………………………………………………………………………………60
Figure 31. ESI-MS spectrum of enantiopure hexaaza tetramine macrocycle 38……….....64
Figure 32. 1H NMR spectrum of macrocycle 38 recorded in CDCl3..........................................65
Figure 33. 13C NMR spectrum of macrocycle 38 recorded in CDCl3.……………………………65
Figure 34. Hypothetic structures responsible of signal m/z=453 (reported calculated
isotopic abundances)…………………………………………………………………………………………………67
Figure 35. Hypothetic structures responsible of signal m/z=497 (reported calculated
isotopic abundances)…………………………………………………………………………………………………67
Figure 36. Hypothetic structure responsible of signal m/z=357 (reported calculated
isotopic abundances)…………………………………………………………………………………………………69

5
Figure 37. Hypothetic structure responsible of signal m/z=274 (reported calculated
isotopic abundances)…………………………………………………………………………………………………71
Figure 38. Proposed starting materials for the synthesis of new azamacrocycle [2+2]..74

6
Table of Contents
1. INTRODUCTION ................................................................................................................................. 8
1.1 Macrocycles .................................................................................................................................... 8
1.2 Imine macrocycles .................................................................................................................... 10
1.2.1 Metal-free synthesis of imine macrocycles ...................................................... 14
1.2.2 Metal-template synthesis of imine macrocycles ............................................ 24
1.3 Hexadentate imine macrocycles [2+2] deriving from pyridine units ................... 30
1.3.1 Complexes and their applications ......................................................................... 32
1.4 Chiral hexadentate macrocycles [2+2] deriving from pyridine units ................... 38
1.5 Gas phase studies ....................................................................................................................... 41
1.5.1 Infrared Multiphoton Dissociation (IR-MPD) ................................................... 42
1.5.2 Computational studies ............................................................................................... 46
1.5.3 Collitional Induced Dissociation (CID) ................................................................ 48
2. RESULTS AND DISCUSSION ................................................................................................ 53
2.1 Aim of our project .................................................................................................................................. 53
2.2 Metal-free synthesis of enantiomerically pure azamacrocycle ................................... 54
2.3 Metal-template synthesis of enantiomerically pure azamacrocycle ........................ 61
3. CONCLUSIONS ................................................................................................................................... 73
4. EXPERIMENTAL SECTION .................................................................................................... 75
4.1 Instrumentation ...................................................................................................................................... 75
4.2 Synthesis of 2,6-diformylpyridine ................................................................................................ 76

7
4.3 Synthesis of chiral hexaaza macrocycle .................................................................................... 77
4.4.1 Metal-free synthesis of [2+2] chiral hexaaza macrocycle ....................................... 77
4.4.2 Metal-template synthesis of [2+2] chiral hexaaza macrocycle with BaCl2 .. 78
REFERENCES ....................................................................................................................................... 80

8
-1-
INTRODUCTION
1.1 MACROCYCLES
When we are talking about macrocycles, first thing should be done is to define the
term “macrocycle”, although it could seem trivial. According to the IUPAC’s definition,
a macrocycle could be simply described as a cyclic macromolecule; so this definition
focuses just on the geometric properties of the molecule, such as shape and size.
However, recently, this term has a wider meaning, that goes over the trivial molecule
appearance and considers its real potential abilities.
In supramolecular chemistry, for instance, a macrocycle isn’t just a “circular”
macromolecule, but it is seen also as a compound that is able to act as an amphitryon
towards some chemical species. In fact, according to its composition and the type of
interactions that it is able to establish, a macrocycle could host ionic species, both
negative and positive ones, but also neutral species. Furthermore, in coordination
chemistry, the term macrocycle could refer to all the heterocyclic compounds able to
coordinate metal centers, that are hosted inside their cavity.
This particular conception of the term macrocycle was born after the discovery and
the evolution of the first crown-ethers identified by Charles J. Pedersen in 1968, in U.S.
DuPont laboratories.

9
Fig. 1 Charles J. Pedersen.
He was able to synthesize a new type of organic compounds; moreover he also
discovered novel properties of Cation-binding, correlated to those of natural
molecules, such as the so-called ionophores. The latter play an important role: in fact,
they has the ability to complex ions inside the body, acting as regulators of biological
processes.
As is usually the case, the resemblance of ownership between a natural compound and
a new class of synthetic chemical compounds allows a considerable increase in studies
the latter, and the improvement of their physical-chemical properties toward specific
applications. That’s why, over the past 40 years, several classes of macrocycles have
been synthesized, containing various combinations of aza- (N), oxa- (O), phospho (P),
and sulfo- (S) ligands, suitable to accommodate metal ions, or other chemical species,
inside their cavity. The simpler compounds that could be found, for example, are the
polioxa- (1, Fig. 2), poliaza- (2), or polioxazamacrocycles (3) saturates, all deriving
from the basic structures of Pedersen crown ethers. However, we could observe an
evolution of such systems in macrocycles with better complexing properties such as
cryptands (4), bicyclic systems containing nitrogen and oxygen binding sites.

10
Fig. 2 Example of saturated macrocycles.
During the following years, more complex systems (Fig. 3) were synthesized, which
were able to host not only ionic species but also organic molecules, forming stable
product. Calixarenes (5), for instance, are able to host molecules that present aromatic
moieties and so that can establish π-π interaction. Cyclodextrins (6), instead, have a
lipophile cavity and a hydrophile external structure, so they can interact both with
polar and apolar molecules, in different manners.
Fig. 3 Example of Calixerene and Cyclodextrin.
These systems have stimulated researchers interest thanks to the wide plethora of
applications related to them. They, for example, could be used as a model in
determining biological processes, such as the transport and the recognition of metal
ions or, furthermore, the functioning of the so-called metal-protein.1 In some cases,

11
they could act as antibiotics, after complexion of metal ions,2 but also as asymmetric
catalysts in some synthetic pathway or, moreover, as contrast agent in NMR
experiments.
1.2 IMINE MACROCYCLES
Although the macrocycles study started after the Pedersen discovery, the real starting
point of these macromolecules chemistry is bound to Schiff bases. In fact the first
example of imine macrocycle (7) was synthesized in 1961, through the condensation
of acetone and ethyldiamine in presence of Ni2+, followed by the synthesis of
compound 8 in 1964.3
Fig. 4 First synthesized imine ligands.
The evolution of this Schiff base type compounds allowed the synthesis of a great
number of new macrocyclic systems. In particular, in coordination chemistry, they
represent the most important class of heteropolidentate cyclic ligands, able to form
mono- or polinuclear complexes, both with transition metals and other metals, such as
Lanthanides and Actinides.
The importance of Schiff bases as structural feature in macrocycles derives from many
factors,1 such as:
They can be synthesized in mild condition through a cyclocondensation
between diamine and dicarbonyl compounds.
Their size, and so the size of the cavity, could be controlled carrying out a
template synthesis, that is the growing of the macrocycle around a particular
metal ion.

12
If they undergo reduction, they easily produce the relative poliamines, that
have different chemical properties and so different potential applications.
Their metal complexes are excellent candidates to simulate biological systems.4
Relative to the latter, it is possible to find analogies between these metal
complexes and some enzymatic systems. This concept represents the base of a novel
type of chemistry: the Biomimetic chemistry.
[Zn(9)]2+ complex, for example, is a good candidate as a mimetic system of
many hydrolytic enzymes containing Zinc ion. In fact, it catalyzes the hydrolysis of
methyl trifluoroacetate (Eq. 1) and also the hydratation of acetaldehyde to give the
alcohol (Eq. 2).
CF3COOCH3 + -OH CF3COO- + CH3OH
(Eq. 1)
CH3CHO + H2O CH3CH(OH)2
(Eq. 2)
Also the [Cu(9)]2+ is able to act as catalyst, in particular it catalyses the CO2
hydration, generally catalyzed by the carbonic anhydrase (Eq. 3).
CO2 + H2O H+ + HCO3-
(Eq. 3)
Cobalt complex with the macrocycle 7 is useful as catalyst in the
electrochemical reduction of O2 to give hydrogen peroxide, and, together with
[Co(9)]2+ and [Co(10)]2+, it could be used in CO2 reduction processes, generally
catalyzed by Ni2+ porphyrin complexes in biochemistry.

13
Fig. 5 Examples of imine ligands.
Another example of the usefulness of macrocycle imine complexes is represented by
Cu+ and Cu2+ complexes with 11 ligand. They show strong catalytic effect during the
ascorbic acid oxidation, in aerobic conditions.
While, as mentioned above, molecular recognition using non-natural receptors of
metal cations has reached a high level of maturity and sophistication, and as a
consequence is successfully applied on an industrial scale,5,6,7 on the contrary,
molecular recognition of small-to-medium sized organic molecules is somehow
lagging behind, mainly due to the complexity of the scientific challenge. Also in this
case macrocyclic imine systems are thought as good candidates. In particular N.
Kunhert et al.8 proposed new trianglimines (Fig. 6), that is believed to fulfill all the
desiderable requirements to bind small organic molecules. Of these, the most
important are the easy of synthesis on a large scale and in high yields, high solubility
in a wide range of solvents and the overall control of size and topology. As well as, it is
possible to obtain molecular skeletons in which to bind functional groups suitable for
molecular recognition processes for a variety of guest molecules. The availability,
finally, of various stereoisomeric forms with the judicious choice of functionality to be
incorporated, allows to increase the versatility of its application of these macrocyclic
imine ligands.

14
Fig. 6 Examples of trianglimine ligands.
From these examples it is possible to understand how the synthesis of innovative
macrocyclic structures, their complexes with different types of metals, and the study
of their properties can give a strong contribution to the growth and the development
in the scientific field of chemistry and biology. In this the organic synthesis has a
fundamental role and the various synthesis techniques involved in the formation of
imine macrocycles will be discussed.
1.2.1 METAL-FREE SYNTHESIS OF IMINE MACROCYCLES
As mentioned, the method most used for the synthesis of Schiff bases is the
condensation of carbonyl compounds with primary amines. In fact, historically,
condensation of carbonyl compounds with primary amines was discovered in 1864 by

15
Hugo Schiff.9 This acid-catalyzed reaction is universal and it allows to obtain a broad
variety of azomethines. The classical Schiff condensation using monocarbonyl
compounds and amines as the starting compounds occurs with high yields. Its
mechanism is well understood (Scheme 1).
Scheme 1 Mechanism of condensation of Carbonyl Compounds with Amines.
All steps in this reaction sequence are reversible. Therefore, the Schiff condensation
under thermodynamically controlled conditions can be used for generating dynamic
combinatorial libraries if several different amines or carbonyl compounds are used as
starting compounds simultaneously.10 So, this reaction is quite simple to make when
the precursors systems are monofunctional and it leads to the formation of a minimal
amount of byproducts, but, on the contrary, when dicarbonyl systems are put to react
with diamines, in the absence of a metal that acts as a templating agent, the
condensation is considerably more complex, by the wide spectrum of products that
may be generated.
In metal-free conditions, the reaction between diamine and dicarbonyl compound, in
fact, could lead to the formation of several products (Scheme 2).
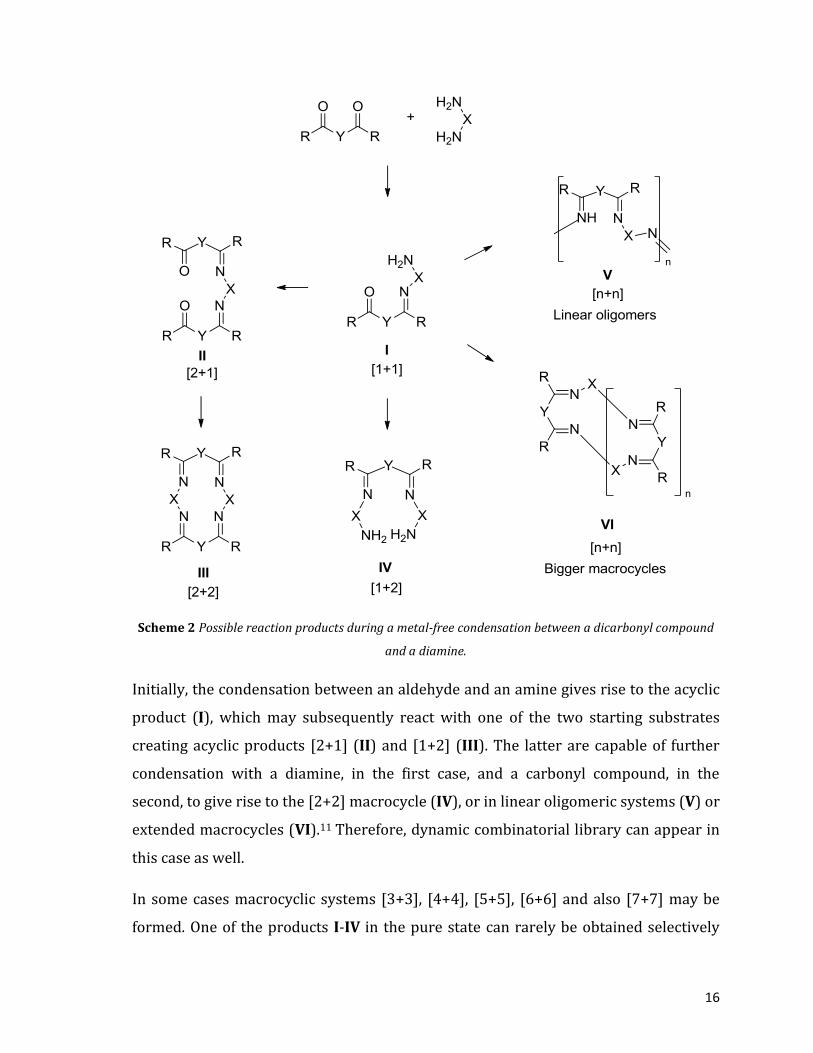
16
Scheme 2 Possible reaction products during a metal-free condensation between a dicarbonyl compound
and a diamine.
Initially, the condensation between an aldehyde and an amine gives rise to the acyclic
product (I), which may subsequently react with one of the two starting substrates
creating acyclic products [2+1] (II) and [1+2] (III). The latter are capable of further
condensation with a diamine, in the first case, and a carbonyl compound, in the
second, to give rise to the [2+2] macrocycle (IV), or in linear oligomeric systems (V) or
extended macrocycles (VI).11 Therefore, dynamic combinatorial library can appear in
this case as well.
In some cases macrocyclic systems [3+3], [4+4], [5+5], [6+6] and also [7+7] may be
formed. One of the products I-IV in the pure state can rarely be obtained selectively

17
under standard reaction conditions. Usually a mixture of these compounds contains
oligomeric products (oligomers V) of polycondensation.
Without controlling the reaction conditions, therefore, it is extremely difficult to
design a synthesis of these systems and create one of the products I-IV in a
regioselective manner.
Given that all the reactions involved in the scheme are reversible reactions, it is
possible to study the factors that influence the position of involved equilibria, in order
to increase the selectivity of the process and make predictions on the final product.
These factors may be divided into three classes:
1. The stoichiometric ratio of the starting substrates, the main factor
that governs the outcome of the reaction. The macrocyclic systems
(regardless that they are [2+2], [3+3], [4+4]... etc.) are favored by a
1:1 ratio diamine-dicarbonyl. If this ratio, however, was not equal to
the unit, one of the two substrates is in excess, favoring the acyclic
products II and III, and oligomeric V.
2. The solvent and the concentration of the substrates, which can
greatly affect the course of the reaction. When a macrocyclic system
is desired, in fact, intramolecular interactions should be favoured
with respect to intermolecular ones. The latter, leading to the
formation of oligomers, may be limited by working under conditions
of high dilution, thanks to the different kinetics of the two processes.
The ring closure reactions are generally first order with respect to
the concentration of the substrates, while intermolecular ones are of
the second order. At higher dilutions, the rate of the intramolecular
processes results, therefore, greater than intermolecular ones.
Regarding the solvent, however, it must be chosen with a very high
polarity, so it can completely solvate the starting substrates and, in
smaller entity, the final products, that are less polar than their

18
precursors. That’s the reason why methanol, ethanol and acetonitrile
are often used as solvents.
3. The nature of the starting substrates, which influences both the
stability and size of the product. Considering the dicarbonyl
substrates, most of the examples found in literature involve aromatic
compounds. The latter, in fact, are able to stabilize Schiff bases thanks
to the delocalization of π electrons over the entire aromatic system.
On the contrary, the amine substrates are used in all their forms, and
it was found that their nature will greatly influence the type of
condensation product obtainable. These chemical species could be
divided into three main groups with different reactivity: a) aliphatic
amines, which are more nucleophilic and more flexible, b) rigid
aliphatic amines, and c) aromatic amines, which are rigid and much
less nucleophilic.
A particular attention deserves the amines that can be used and therefore:
a) aliphatic diamines
The diamines of the aliphatic series are the most reactive towards the condensation to
give a Schiff base and, this is why they are the most difficult to control. The synthesis
of macrocycles of precise dimensions, therefore, is generally made through a template
synthesis, in order to conduct, kinetically and thermodynamically, the reaction
towards a single product (paragraph 1.2.2). In the absence of a metal salt, however,
the right choice of solvent and the concentrations of reagents could increase the
regioselectivity of the process, although yields couldn’t reach high values. In addition
to the high reactivity of these amines, also flexibility of the aliphatic compound has to
be considered. When two amino groups are linked by a long aliphatic spacer, in fact,
they can react independently, greatly lowering the regioselectivity of the process.
When, however, the two amino groups are located in the immediate proximity (α,β-
diamines and α,γ-diamine) the selectivity increases.

19
b) aliphatic diamines with rigid spatial arrangement of amino groups
Unlike common aliphatic amines, systems with rigidly fixed conformation, such as
(S,S)- or (R,R)-1,2-diphenylethylendiamine or trans-1,2-diaminocyclohexanes, show a
very low flexibility and precise directionality of amino groups.
Fig. 7 Enantiomers of 1,2-diphenylethylendiamine and trans-1,2-diaminocyclohexane.
Using one of these reagents, they greatly limit the geometries of the possible end
products. In particular, in trans-1,2-diaminocyclohexane, this is due to the amino
groups that occupy the equatorial positions and the dihedral angle between two C-
NH2 bonds is about 60°. Moreover, the outcomes heavily depend on the dihedral angle
in the dicarbonyl system. In fact, if (R,R)-trans-1,2-diaminocyclohexane (16a, Scheme
3), is placed to react with a dicarbonyl system with a dihedral angle of 180°, there is
the formation especially of the [3+3] trianglimine macrocycle.12,11 Of course, the
stronger steric hindrance of the substituents on the dicarbonyl compound the lower
the yield, as in the case of the product 17d (Scheme 3).

20
Dicarbonyl Compounds Yield Product
90% 17a
97% 17b
90% 17c
32% 17d
99% 17e
67% 17f
80% 17g
99% 17h
68% 17i
Scheme 3 Formation of [3+3] Macrocycles condensing dialdehydes with trans-1,2-diaminocyclohexanes.
A large number of linear dicarbonyl compounds leads to the formation of [3+3]
trianglimine macrocyclic products, such as 17e-17i (Scheme 3).11 From these studies,
therefore, it could understand as the condensation reaction is guided by a structural
predisposition of substrates and acyclic intermediates to form a triangular product.
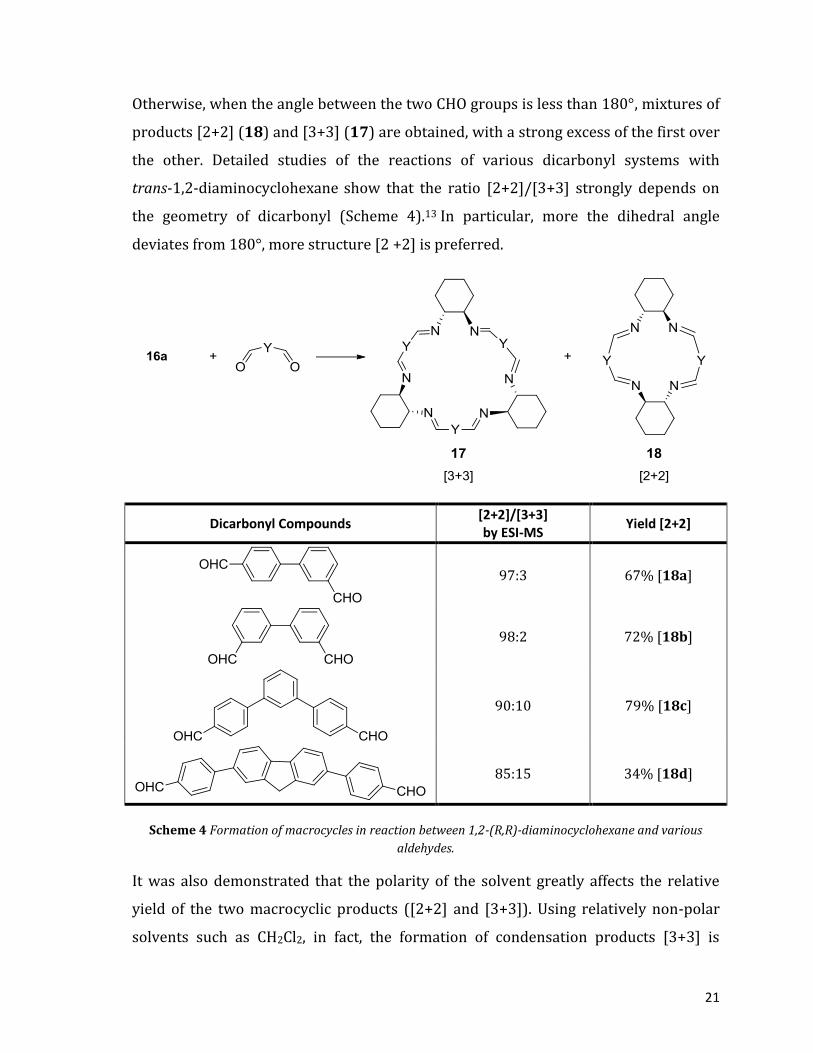
21
Otherwise, when the angle between the two CHO groups is less than 180°, mixtures of
products [2+2] (18) and [3+3] (17) are obtained, with a strong excess of the first over
the other. Detailed studies of the reactions of various dicarbonyl systems with
trans-1,2-diaminocyclohexane show that the ratio [2+2]/[3+3] strongly depends on
the geometry of dicarbonyl (Scheme 4).13 In particular, more the dihedral angle
deviates from 180°, more structure [2 +2] is preferred.
Dicarbonyl Compounds [2+2]/[3+3] by ESI-MS
Yield [2+2]
97:3 67% [18a]
98:2 72% [18b]
90:10 79% [18c]
85:15 34% [18d]
Scheme 4 Formation of macrocycles in reaction between 1,2-(R,R)-diaminocyclohexane and various
aldehydes.
It was also demonstrated that the polarity of the solvent greatly affects the relative
yield of the two macrocyclic products ([2+2] and [3+3]). Using relatively non-polar
solvents such as CH2Cl2, in fact, the formation of condensation products [3+3] is

22
promoted, while with more polar solvents, the product mixture is enriched with
products of the type [2+2]. This can be well understood, since the stability of larger
macrocycles is greater in less polar solvents. In support of this assumption, the
Gawronski group has experienced that the condensation between terephthalic
aldehyde and trans-1,2-diaminocyclohexane (17a, Scheme 3) leads to the formation of
the [3+3] macrocycle only in slightly polar solvents such as benzene and CH2Cl2. On
the other hand, using more polar solvents such as MeOH, a small percentage of the
[2+2] product is observed.14 When the angle between the two groups CHO, instead,
approaching to 120°, the selectivity of the ring sizes change and can generate
condensation products [2+2], [3+3] and also [4+4].11
c) aromatic diamines
Aromatic diamines possess a lower nucleophilicity if compared to those aliphatic. In
particular, recently, attention have turned towards the reactivity of the
ortho-phenylenediamine (19, scheme 5) and its derivatives. When both amino groups
are involved in the direct condensation with a carbonyl compound, the reactivity of
the second amino group strongly decreases after the condensation of the first.
Similiarly, if the carbonyl compound which reacts with o-phenylenediamine also has
an aromatic structure, its reactivity would drop significantly as a result of the effect of
delocalization of π orbitals. For this reason, the Schiff base 20 is able to give further
condensations only if subjected to a template synthesis (paragraph 1.2.2), where the
metal templating agent also acts as a Lewis acid, or a very strong acid catalysis.
Generally, it is used a mineral acid such as H2SO4 or HCl.
Scheme 5 Condensation between ortho-phenylenediamine and dialdehydes.

23
Factors that influence the outcome in this reaction are the same ones involved in
reaction with conformationally rigid amines. Even in this case, in fact, the
condensation reaction will be guided by a structural predisposition of starting
systems to form a particular geometry.
The examples given in the whole paragraph show that the appropriate choice of the
reaction conditions (solvent, concentration, ratio of reagents, acid catalysis) and the
use of starting materials with the right geometrical characteristics, allows to shift the
equilibrium toward the target product, and to obtain macrocycles with high selectivity
without the use of metals such as templating agents.
The study of the factors that influence these reactions are of great importance,
because it determines two advantages of metal-free synthesis with respect to the
template one. First of all, free macrocycle systems could be obtained, that is they are
not coordinated to the guest metal, with high selectivity and in high yields. Secondly,
the possibility to generate non-symmetrical macrocyclic imine. In fact, till now only
symmetrical macrocycles synthesis have been shown. Metal-free synthesis, however,
also allows to produce asymmetric systems through a strategy of Fragment-Fragment
assembly.11 Taking into account the geometric principles, the reaction conditions, and
stoichiometric requirements seen previously, this technique involves the formation of
the preliminary systems [2+1] or [1+2] (Scheme 2). Subsequently, by entering into the
system a different difunctional reagent, it will be possible to complete the cyclization,
resulting in a non-symmetric macrocyclic product. Of course, to have a high efficiency,
the requirements of complementarity of the reactants must be satisfied, so it must be
observed the ability of a reagent to adopt a particular conformation rather than
another. For example, following it could be observed (Scheme 6) how species a and b
have a similar geometry, although they are different. This allows to generate the
addition product [1+1+2] without excessive by-products observed.

24
Scheme 6 Example of non-symmetric macrocycle synthesis.
1.2.2 METAL-TEMPLATE SYNTHESIS OF IMINE MACROCYCLES
To synthesize macrocyclic systems, the metal-free method does not always allow to
achieve acceptable yields and selectivity. So an alternative approach is necessary for
the synthesis of these macromolecules, especially when the control of the reaction
conditions and the geometry of the components isn’t enough to guide to a specific
product.
The most used method, in these cases, involves the in situ action of a metal center,
which plays a very important role in the formation of the macrocycle. This ion, in fact,
may be able to direct the course of the reaction and the effect that emerges is called

25
"metal-template effect". The ability of metal ions to affect the course of some organic
reactions has been systematically studied at the beginning of the sixties. Since then,
various and amazing molecular architectures have been created as result of
employing this effect. Two classes of chemical templates have been recognized:
1. Kinetic templates: the ability of the metal to control the steric course of the
reaction, directing toward the condensing of a cyclic product rather than
oligomeric one. This is due to the coordination of the preliminary starting
materials with the ion, in order to form C=N bonds with the right orientation.
In other words, the macrocycle closure occurs on the coordination sphere of
the metal. This effect provides routes to products which are not formed in the
absence of the metal ion. Kinetic templates influence the mechanistic pathway.
Experimental data lead to the following types of kinetic metal template
reactions:15
-the molecules are coordinated and assembled around a metal cation in a
single step (Scheme 7)
Scheme 7
-the coordinated substrates react with an external molecule which bridges the
ends of the ligated ones (Scheme 8).

26
Scheme 8
It must be noticed that the most frequent products are cyclic ligands, but,
however, linear ligands have also been obtained, mostly when the reaction is
stopped along half a building stage.
2. Thermodynamic templates: the ability of the metal to stabilize the
macrocyclic system, once it has formed. In fact the metal-macrocycle
complexation, going to affect the Gibbs free energy of the system, leads to the
formation of the more stable one (see Scheme 9, pag. 25). In this case, the
metal ion promotes formation of the desired product by removing it from the
equilibrium. In other words, metal ions select and bind certain complementary
structure among an equilibrating mixture of structures. This is a
“sequestration” phenomenon.
This classification says nothing about the importance of the structural factors
affecting the template reactions. However, it is easy to understand that, designing a
metal-template synthesis, it is also necessary to choose carefully the metal ion.
The coordination of ligands to metal ions involves electronic factors as well as
geometric relationship between these two parts. Regarding to the electronic factors,
the preference of alkaline and alkaline earth metal ions for oxygen donors and their
template activity to form crown ethers can be understood through the HSAB theory

27
elaborated by Pearson, as well as the preference of the transition metal ions toward
nitrogen donors. In addition the templating potential of a metal ion depends also on
two geometrical factors: the coordination sphere geometry (octahedral, tetragonal,
square planar, square-based pyramid, etc. ...) and the ionic radius. The coordination
numbers and geometries allowed by a given metal, for example, can be a disadvantage
or favor the formation of a macrocycle. In fact the metal ion exerts a preference for a
particular kind of environment and some ligands are better able to conform to that
environment than others. On the other hand, each ligand has its own preference for a
particular geometrical rearrangement. The organization of metal ions with specific
electronic properties within a potentially template ligand system can lead to the
appropriate orientation of substrate molecules, (the preexisting ligands in the
precursor molecules) required for a particular reactions.
This can be understood by observing that neither Cu2+ nor Ni2+ act as templating
synthesis of pentadentate macrocycles 21 and 22 (Fig. 8).16 In contrast to Cd2+, Co2+,
Hg2+, which can assume a pyramidal geometry with a pentagonal base, allowing a
better overview of the products 21 and 22.2
Fig. 8 Other examples of imine macrocycles.
Among general factors influencing a metal-ion-controlled synthesis of macrocyclic
ligands, the relationship between the size of the metal ion and the opening in the
middle of the ring clearly is important. So, the dimension of the ion and its radius are
perhaps the most important features of the metal center. Natural complexes
demonstrate this statement. Iron porphyrins involve a 16-membered ring while the
cobalt in vitamin B12 occupies a 15-membered ring.

28
The compatibility between the radius of the cation and the cavity of the macrocycle
contributes to direct the synthetic pathway and to control the geometry of the
resulting complex. The matching ion-cavity has been heavily studied in the mid-
seventies by Fenton and his colleagues, synthesizing oxazamacrocyclic imines through
a template methodology (Scheme 9).17,18Among the alkaline earth metals, for instance,
only Mg2+ is able to promote the synthesis of the macrocycle [1+1] pentadentate 25,
but it is inefficient in the synthesis of the macrocycle 24, which has a cavity slightly
larger. The latter, however, is easily obtainable through a template synthesis using of
Ca2+, Ba2+, Pb2+, which have larger radii than that of magnesium (II). Also the
preferential formation of the macrocycle [1+1] 25 or [2+2] 26, from the same
substrates depends on the size of the ion. If it is too large for the cavity, then a
macrocyclic system [2+2] is generated; in fact the ions Ba2+, Pb2+, Sr2+ form the
macrocycle 26, while the Mg2+ the 25 one.

29
Scheme 9 Matching ion-cavity in the imine macrocycles formation.
Another effect, related to the size of the ions, is called Ring-Contraction. This effect can
be observed on systems containing side chains with additional functionality (-NH or
-OH), available for nucleophilic addition to the imine bond of the macrocycle. For
instance, in Scheme 10, the species 27 allow the contraction or expansion of the ring,
depending on the templating metal used. If the synthesis is carried out by ions of high
radius as the Ba2+, in fact, the side chain is fully extended, obtaining the 20-membered
macrocyclic complex 28.
Scheme 10 Ring-contraction induced by metal ions.
If instead, smaller ions are used such as Pb2+, the side chain is able to rearrange and
create contract complex 29, containing oxazole (Y=O) or imidazole (Y=NH) moiety. It
is interesting to observe that, if both complexes 28 and 29 are treated with much
smaller metal salts such as Cu2+, it easily generates the dinuclear complex 30, which
implies an expansion of the ring.

30
As showed, the ring expansion and contraction are reversible equilibrium reactions. If,
in fact, the condensation reaction is carried out in metal-free conditions, the presence
in solution of both the extended form and the compact one in equilibrium is observed
(Scheme 11). The presence of the metal allows to stabilize one of the two cyclic forms
28a or 29a, acting directly on the thermodynamics of the system. For this reason, this
example may help to understand the meaning of thermodynamic template effect. Big
ion stabilizes the large macrocycle 28a. In contrast, the presence of small ion
significantly lowers the Gibbs Free Energy of the macrocycle 29a, shifting the
equilibrium towards it.
Scheme 11 Ring expansion and contraction in metal-free condition in solution.
With respect to metal-free synthesis, metal template reactions offer simple ways to
obtain organic molecules which otherwise involve complicates organic routes, high
amounts of solvents, small yields and high costs. The organic molecules act as ligands
and the high stability of complexes allows reactions at the coordinated ligands
without complex destruction. For example, the coordinated imino groups can be
reduced to give coordinated amine groups using very effective agents like hydrogen,
using platinum catalyst, sodium borohydride, cathodic reduction. Other reactions
including derivatization, functionalizing and isotopic exchange reactions are also
possible. The ligands, obtained by a metal template route, can be also released and
used, both free or to obtain further metal complexes.
1.3 HEXADENTATE IMINE MACROCYCLE [2+2] DERIVING FROM
PYRIDINE UNITS

31
Imine macrocycles can be generated by many different building-blocks and, according
to the used units, systems with different donor atoms and in different sizes are
originated. Among all imine systems synthesized and studied in recent years,
however, many ones involve the use of pyridine units, such as 2,6-diformylpyridine
and 2,6-diacetylpyridine that have favorable characteristics, both from a point of view
of synthesis and application. In fact the dicarbonyl-derivatives of pyridine are
precursors for easy access, as they can be synthesized by various methodologies. The
macrocycles derived from pyridine precursors, also they have strong advantages in
stability and reactivity. The electron delocalization on the aromatic ring tends to
stabilize the Schiff base resulting from their condensation with amine functionalities
(Fig. 9).
Fig. 9 Resonance structures of Schiff base derived from condensation of 2-formylpyridine and primary
amine.
Although the aromatic ring involved in the stabilization of the double bond C=N, a
further advantage is given by the fact that the nitrogen possesses a free electron pair,
placed on an orbital sp2, which does not participate in electron delocalization of the
aromatic ring. This fact, therefore, unlike the systems pyrrolidine, allows the aromatic
ring to act as a nucleophile or Lewis base.
Fig. 10 N-Pyridine orbital representation.

32
These structures are therefore of great interest in macrocycles chemistry and their
coordination processes. Being its characteristics between Hard and Soft, the nitrogen
can coordinate to a large number of metal cations, implementing the versatility of the
binder.
Further advantages of the 2,6-dicarbonyl derivatives of pyridine arise from their
particular geometric structure. The dihedral angle of about 120° between the two
carbonyl functionalities imparts to these molecules a particular tendence to form
macrocyclic systems [2+2], greatly simplifying the processes of metal-free synthesis.
The [2+2] macrocycles, resulting from the condensation of pyridine units and α,β- or
α,γ-diamines, have also cavity size comparable to that of the heavier alkaline earth
metals (eg, Ba2+) or lighter lanthanides (eg, La3+, Nd3+, Sm3+, Eu3+) who have similar
ionic radii. For this reason, these ions have been used since 70 years as templating
agents in the synthesis of pyridine macrocyclic imines. This synthesis has led to the
formation of a large amount of mononuclear complexes of Lanthanides, which were
subsequently characterized and highly studied.19 Over the years, in fact, multiple
applications of these systems of coordination (paragraph 1.3.1) have been discovered
and even today, chemists are trying to synthesize, purify and stabilize new complexes
that might be useful in other fields.
1.3.1 COMPLEXES AND THEIR APPLICATIONS
Among the first hexadentate compounds containing pyridine units, the
complexes of the lanthanide series (Ln3+) of the macrocycles 31 and 32 (Fig. 11) are
found.20

33
Fig. 11 Example of first imine macrocycles incorporating pyridine units.
Such complexes are greatly used for biologically relevant applications, thanks to their
stability and inertia in physiological conditions. Generally, in fact, the imine systems
may decompose easily in an aqueous environment through hydrolysis, releasing metal
ions coordinated to them. From kinetic studies on decomposition of such systems,
however, a prolonged stability in physiological conditions (37°C, pH = 7.3) and in the
presence of other ligands such as EDTA, which could capture the metal ions, has been
found.21
The complex [Eu(31)]3+, for example, is an efficient catalyst for the processes of
transesterification of the RNA, and for this reason it is involved in the study of new
artificial nucleases.22 One of the most attractive applications of these catalysts of
transesterification is the possibility of generating systems that act according to the
Antisense Technology. Antisense therapeutic agents are based upon a simple and
elegant concept as illustrated in Fig. 12.
Fig. 12 Antisense therapeutic agent mechanism of action.

34
As genes are expressed to produce specific proteins, the two complementary strands
of DNA begin uncoiling within the nucleus. The “sense” strands carries nucleic acid
bases in order which specifies which amino acids should be assembled to produce the
protein. The complementary strand or “antisense” strand is used as a template for
assembling a complementary strand of messenger RNA (mRNA) in the process called
transcription. The mRNA will have the same sequence as the “sense” strand of DNA. In
eucaryotes, the mRNA is further processed in the nucleus by capping and slicing and is
transported into the cytoplasm where ribosomes translate the mRNA into proteins.
Antisense drugs are short stretches of deoxyribonucleotide analogs (antisense
oligonucleotides) which binds to specific complementary areas of the mRNA by
Watson-Crick base pairing. In doing so, they can induce a nuclease (RNase H) which
cleaves the mRNA at site of binding or can physically block translation or other steps
in mRNA processing and transport, thus stopping protein synthesis. Another class of
antisense agents, ribozymes acts catalyzing cleavage at specific sequence in a specific
mRNA substrate, preventing its translation. That’s also the case of some chemical
agents: in complex [Eu(31)]3+, for instance, the metal center has the task to perform
the transesterification of the mRNA chain (Scheme 12), while the ligand has the role of
stabilizing the metal, avoiding its release inside the body.
Scheme 12 mRNA transesterification.
In fact, this complex does not exchange easily the metal ion with other binders, and its
decomposition is very low. More precisely, in physiological conditions, and after three
days, only 8% of the complex is degraded. Furthermore, the macrocycle may be
functionalized to increase the selectivity of the catalyst and mimic the behavior of
natural nucleases.

35
Antisense drugs thus work at an early stage in the production of a disease-causing
protein and theoretically can be applied to a number of disease where the basic
pathophysiology involves an overexpression or aberrant expression of a give protein
molecule. Viral diseases, cancers, and inflammatory diseases are all examples of such
diseases that potentially can be treated via antisense mechanisms.
Also double stranded DNA hydrolysis is became of interest for treatment of
genetically based diseases. The increasing interest in using lanthanide ions or
complexes as artificial restrinction enzymes for cleaving DNA has prompted several
research groups to investigate the application of lanthanide macrocyclic complexes in
this area. Also in DNA the sites of cleavage, catalyzed by lanthanides, are at the
phosphodiester linkages, although they are more difficult to hit than those in RNA
strands. For instance, two dimeric Y(III) and Nd(III) complexes of an 18-membered
hexaaza macrocycle 33 (Fig. 13) have been prepared and characterized by Bligh
et al23.
Fig. 13 Macrocyclic ligand involved in cleavage of DNA phosphodiester linkage .
They found that all monomeric complexes of Gd3+, Y3+ and Dy3+ had no measurable
ability to cleave DNA, but the dimeric ones with Y3+ (Fig. 14) and Nd3+ showed
marked cleavage activity, demonstrating their potential for therapeutic applications in
genetically based disease.

36
Fig. 14 Structure of the dimeric tetracation [{Y(33)(OH)(H2O)2}2]4+ in the hydrated nitrate salt.
Further studies on the complex of the macrocycle 31 has also led to evaluate their
potential application as contrast agents in magnetic resonance imaging (MRI), a
technique widely used in diagnostic. This technology takes advantage of the unique
properties of certain atomic nuclei to absorb radio waves of specific energy when they
are subjected to a magnetic field. In particular, the MRI makes use of the proton
signals of the water contained in the tissues to produce sharp images of the internal
organs, like brain, liver and spleen. The intensity of this image depends strongly on
the relaxation time of the protons; the lower this time the greater the intensity of the
signal. The complexes of transition metals and paramagnetic ions of Lanthanides have
precisely the ability to lower these relaxation times of the nuclei close to them,
through the dipolar interactions (Fig. 15).24 The main feature of such contrast media
lies in the fact that they are displayed in an indirect way, and not directly as other
diagnostic means.

37
Fig. 15 Magnetic Resonance of rat abdomen before (A), 16 (B), 56 (C) and 182 (D) minuts after
administration of a contrast agent.
Various studies have been carried out on metal complexes with acyclic ligands such as
DTPA (34, Fig. 16) or EDTA (35), but their stability under physiological conditions is
extremely low, and at the same time the influence they have on relaxation time is low.
Fig. 16 Diethylentriaminopentacetic acid (DTPA) and Ethylendiaminotetracetic acid (EDTA).
On the contrary, complex [Gd(31)]3+ has a greater stability and effectiveness in
lowering of the relaxation times of H2O.25 Both the excretion of systems such as
[Eu(31)]3+ and [Gd(31)]3+ occurs within a few hours, then, the kinetics of
decomposition is slow enough to be able to be used.
Lots of others applications could be described to underling the enormous plethora of
possibilities that these complexes allow. Furthermore one can mention how the
peculiar chemical, structural, spectroscopic and magnetic properties of the trivalent
lanthanide ions associated with their 4fn configuration make them suitable for
development of novel fascinating supramolecular photonic light-converting devices

38
and sensors, potential radiopharmaceuticals, sensitizers for photodynamic therapy
and biomedical diagnostics.26
1.4 CHIRAL HEXADENTATE MACROCYCLES [2+2] DERIVING FROM
PYRIDINE UNITS
The concept of chirality of object which are related as mirror images is completely
pervasive in the living world and yet, perhaps because of this, it is often overlooked.
From the beginning of the evolutionary process right up to the present diversity of
biological forms, life has been under the constant influence of chirality, and this is
because the whole environment in which life forms developed was asymmetric.
Optically active compounds are ubiquitous in our everyday lives. They are the active
constituents of many medicines, vitamins, flavors and fragrances, herbicides and
pesticides used in crop protection. Basically, there are three methodologies to
synthesize enantiomerically pure compounds. The first method is based on getting
enantiopure starting materials from the chiral pool. It refers to inexpensive, readily
available natural products and their derivatives. These substances can be transformed
into synthetic products by chemical manipulation that may involve retention or
inversion of configuration or chirality transfer. The second one is the resolution of
racemic mixture through diastereomer crystallization or kinetic resolution, while the
third is represented by asymmetric synthesis, performed through chemocatalysis or
biocatalysis.
Optically active polyazamacrocycles are important compounds in organic, 27
supramolecular,28 medicinal29 and bioorganic30 chemistry.
Their synthesis, for instance, could be performed introducing chiral diamines in the
synthesis of hexaazamacrocycles. Among the most examples, significant are the
hexazamacrocycle 36, deriving from the condensation between 2,6-diformylpyridine
and (R,R)-1,2-diphenylethylendiamine, or the macrocycle 37, synthesized from a
biphenyl system.

39
Fig. 17 Examples of chiral macrocyclic imines.
In addition to the applicability as asymmetric catalysts, the interest towards these
systems deals with two other chiral property. One is the ability of their complexes
with the lanthanides, in particular europium (III) and terbium (III), to produce
circularly polarized luminescence, useful in various technological fields.31 The second
one is the possibility of using such complexes as chiral derivatizing agents (CDA) in
spectroscopic analysis of mixtures of enantiomers. From studies made by the research
group of Lisowski, in fact, these complexes exhibit good interactions with amino acids
of the D series and the L series. In the presence of a racemic mixture, the
enantiomerically pure metal complexes form diastereoisomeric adducts which can be
identified, for example, with NMR techniques.32
The interesting chemistry of chiral azamacrocyclic molecules is not limited to imine
compounds, also the corresponding amines and their relative chemistry deserve
attention. For instance, macrocycle 38 (Fig. 18) has been found to have anion binding
abilities, useful for a new mechanism for the enantiomeric recognition of anion in
aqueous solution.33

40
Fig. 18 Stereoselective Receptor of biologically Important Dicarboxylates under Physiological Conditions.
The interaction of a synthetic enantiopure azamacrocyclic receptor (38) with
biologically important chiral dicarboxylates has been studied by means of
potentiometric titrations in 0.15 M NaCl aqueous solution in a wide pH range. This
macrocycle forms strong complexes of the type [Hn38A](n-2) (with n=0-5). As a general
trend, the binding is much tighter at basic or neutral pH than in acidic medium.
Interestingly, nonprotected excitatory amino acids (Asp and Glu) are strongly bound
even at acidic pH. Regarding selectivity, the receptor showed stereoselective binding
toward those substrates bearing an H-bonding donor at Cα, being S-selective in most
of the cases, except for glutamic acid.
Fig. 19 Schematic representation of equilibrium interactions of macrocycle 38 with two enantiomers of
dicarboxylate species.

41
1.5 GAS PHASE STUDIES
In order to justify, just in a qualitative way, the starting point of our project, this
section is dedicated to the gas phase studies carried out on a prototype (39, Fig. 18),
which demonstrated peculiar characteristics, deserving of attention and exhaustive
analysis.
Fig. 20 Hexazamacrocycle synthesized by condensation of 2,6-diformylpyridine with racemic (±)-trans-
1,2-diaminocyclohexane.
The hexaza tetramine macrocycle 39 was synthesized in Prof. Marcantoni research
group few years ago, through the macrocyclization between a racemic
(±)-trans-1,2-diaminocyclohexane and 2,6-diformylpyridine and successive reduction
of imine moieties, performed with NaBH4. This compound, that is proved to be the
meso compound, was studied at the Prof. Filippi and Prof. Speranza research group,
showed a particular affinity towards potassium ions, in presence of an acidic species,
in gas phase analyses, such as mass spectrometry. In fact the ESI-MS spectrum of the
macrocycle 39 only shows an important intensity of the [MH+], with a less extent the
[MNa+], while the [MK+] signal is almost not observable. Surprisingly this trend is
reverse when inside the sample an acidic species, such as HCl, HF and amino acids, is
put: the [MK+] signal increases a lot its intensity whilst [MH+] and [MNa+] intensities
are lower. So infrared multiphoton dissociation and collisional induced dissociation
were then used to try to measure the entity of affinity towards potassium ions.

42
Knowing the extremely important role of this ion from a physiologically point of view,
both research groups decided to collaborate in order to determine the reason of this
strong affinity and to eventually investigate the usefulness in possible applications.
1.5.1 INFRARED MULTIPHOTON DISSOCIATION (IR-MPD)
The interaction of gas-phase molecules with intense infrared (IR) lasers is now well
understood. Since the observation of collisionless unimolecular dissociation of
neutrals after absorption of radiation from IR laser around 1970, thousands of
publication on the topic have appeared. The breadth of research conducted using IR
multiphoton dissociation (IRMPD) extends from theoretical physicists studying the
physics of multiphoton absorption to experimental biochemists studying the
structures of large biological molecules.
It took only few years after the original IRMPD publications before mass
spectrometrists took advantage of combining the technique with trapped-ion mass
spectrometry (MS) for studying the properties of charged species.34
This is also the case of the ionic complex [39·K·A]⁺. The analysis consists in the
formation of gas phase ionic complex and in isolation of this one for enough time to be
submitted to a laser beam. So instruments that are capable to trapping ions for
extended period of time are commonly used. As showed in Fig. 21, quadrupole ion
trap (QIT) or also Ion cyclotron resonance (ICR) spectrometer are ideal for IR-MPD
studies. Often linear ion traps are used.
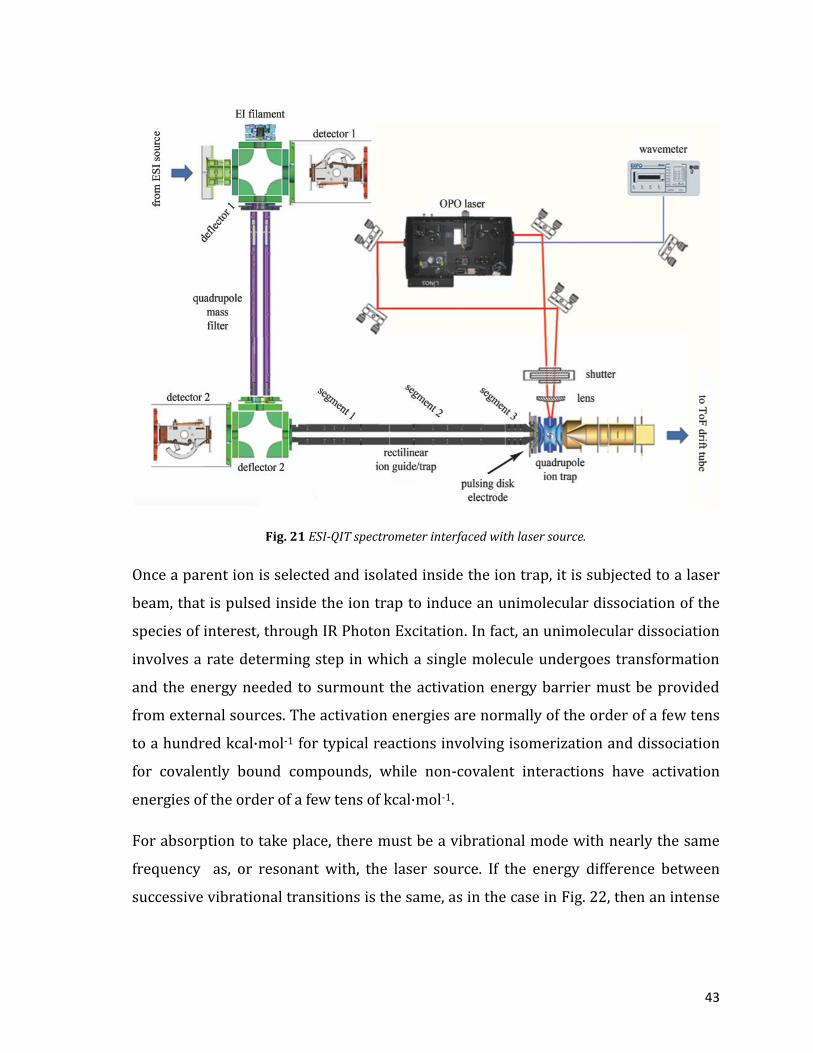
43
Fig. 21 ESI-QIT spectrometer interfaced with laser source.
Once a parent ion is selected and isolated inside the ion trap, it is subjected to a laser
beam, that is pulsed inside the ion trap to induce an unimolecular dissociation of the
species of interest, through IR Photon Excitation. In fact, an unimolecular dissociation
involves a rate determing step in which a single molecule undergoes transformation
and the energy needed to surmount the activation energy barrier must be provided
from external sources. The activation energies are normally of the order of a few tens
to a hundred kcal·mol-1 for typical reactions involving isomerization and dissociation
for covalently bound compounds, while non-covalent interactions have activation
energies of the order of a few tens of kcal·mol-1.
For absorption to take place, there must be a vibrational mode with nearly the same
frequency as, or resonant with, the laser source. If the energy difference between
successive vibrational transitions is the same, as in the case in Fig. 22, then an intense

44
source of photons at the frequency corresponding to the energy difference will take
the system to the threshold for chemical reaction.
Fig. 22 Vibrational Energy levels for (a) a fictional species void of anharmonicity effects and (b) a
species showing the effect of anharmonicity on vibrational spacings.
However anharmonicity in molecules causes a shift to lower frequency for subsequent
transitions, so that the overtone transition from υ=0 to υ=2 actually requires less
energy than twice the energy difference for υ=0 to υ=1 transition. Therefore,
absorption of the first photon is straightforward process; however, successive
absorptions are immediately out of resonance with the monochromatic laser due to
the anharmonicity. Other mechanisms must be in operation for the second and
subsequent photons by the molecule in Region I (Fig. 23), that are not discussed in
details here. However after absorption of the first few IR photons, the complex has
absorbed a many hundreds to few thousand cm-1 of energy and stored it as internal
energy. At this point the density of states in the complex become quite large and
allows the absorbing mode to mix with background modes of the complex (Region II,
Fig. 23).

45
Fig. 23 Model for IR multiphoton absorption from the ground state to the quasicontinuum and to the
dissociative continuum.
If favourable mixing of vibrational state exists, energy can be quickly randomized
throughout the vibrational modes of the complex, depopulating the absorbing modes,
allowing further absorption of photons. Further, the complex can absorb radiation by
a range of states in Region II due to the availability of suitable energy spacing.
Absorption continues until the ionic complex has acquired enough energy to reach the
true continuum above the dissociation threshold. At this point the IRMPD occurs and
it is detected by a loss in precursor ion signal and the observation of fragments ions.
In order to obtain structural information on a particular ion using this technique, it is
imperative to dissociate the ion. In fact, scanning a large range of frequencies with the
laser beam (for instance 2800-3800 cm-1, as in our case), it is possible to build an
absorption spectrum and to correlate absorption bands with the generation of
particular ionic fragment. In this way is possible to have elucidation on energies
involved in non-covalent interactions between two species and so a valuation of
affinity between them. Computational studies always support the right interpretation
of results; in fact they are useful to make prevision on less energetic conformation,
bond distances and energies involved.
Regarding the particular ionic species [39·K·A]⁺, unusual results have been obtained.
As previously said, less energy is required to break non-covalent interactions with

46
respect to covalent ones. So initially, removal of potassium or the acidic species was
supposed to happen, but raising irradiation energy leads just to uncontrolled
fragmentation. This result was surprising, so that great interest was aroused in
determining the causes of this particular behavior. On the other hand, this analysis
couldn’t furnish substantial elucidation because of missed fragmentation. So
Collitional Induced Dissociation have been performed, together with theoretical
studies.
1.5.2 COMPUTATIONAL STUDIES
Computational studies uses computer simulation to assist in solving chemical
problems. It uses methods of theoretical chemistry, incorporated into
efficient computer programs, to calculate the structures and properties
of molecules and solids. Examples of such properties are structure (i.e. the expected
positions of the constituent atoms), absolute and relative (interaction) energies,
electronic charge distributions, dipoles and higher multiple moments, vibrational
frequencies, reactivity or other spectroscopic quantities, and cross sections for
collision with other particles. The methods employed cover both static and dynamic
situations. System of interest can be a single molecule, a group of molecules, or a solid.
Computational chemistry methods range from highly accurate to very approximate;
highly accurate methods are typically feasible only for small systems. Ab
initio methods are based entirely on quantum mechanics and basic physical constants.
Other methods are called empirical or semi-empirical because they employ additional
empirical parameters. Both ab initio and semi-empirical approaches involve
approximations. These range from simplified forms of the first-principles equations
that are easier or faster to solve, to approximations limiting the size of the system (for
example, periodic boundary conditions), to fundamental approximations to the
underlying equations that are required to achieve any solution to them at all. For
example, most ab initio calculations make the Born–Oppenheimer approximation,
which greatly simplifies the underlying Schrödinger equation by assuming that the
nuclei remain in place during the calculation. In principle, ab
initio methods eventually converge to the exact solution of the underlying equations

47
as the number of approximations is reduced. In practice, however, it is impossible to
eliminate all approximations, and residual error inevitably remains. The goal of
computational chemistry is to minimize this residual error while keeping the
calculations tractable.
Regarding macrocycle 39, the theoretical studies have been performed by a
biophysicist at University of La Sapienza, in Rome. Results of preliminary calculations
have been obtained through a simulating annealing with classical force field;
simulated annealing is a strategy that is used to solve optimization problems and it is
carried out heating at high temperature (1000 K or even more) the structures of
interest for a certain time and then cool them. This was done in 1000 conformations
of these structures. This procedure allows to overcome the energies related to the
torsional barriers, in order to obtain a more detailed conformational investigation.
Moreover, classical forced field means that the molecular structure is represented by
balls (atoms) and springs (bonds), so it is not possible to simulate the creation and
breaking of bonds or proton transfer. Structures with the lowest energy (typically
chosen within a range of 5 kcal·mol-1), obtained with this procedure, were then
chosen to perform static quantum calculation, that is without considering the time
evolution and temperature. The method used is the ab intio density functional theory
(DFT).
The results of these preliminary calculations shows that the protonated host has a
major stability when protonation occurs on aliphatic nitrogen with respect to the
protonation on pyridine one. The energy difference is about 6 kcal·mol-1 and it is
easily justified by the greater basicity of aliphatic amine than that of aromatic ones.
Considering the complex [39·K·A]⁺, in which A is represented by water, the most
stable structure binds not dissociated water (Fig. 24).

48
Fig. 24 Different views of [39·K·H2O]⁺complex (bond length expressed in angstrom).
Starting from [39·K·H2O]⁺complex, water is replaced by hydrogen fluoride (Fig. 25).
Fig. 25 Different views of [39·K·HF]⁺complex (bond length expressed in angstrom).

49
Also in this case, hydrogen fluoride is found as an undissociated species. Moreover, its
coordination is more stable on aromatic nitrogen with respect to that one on aliphatic
nitrogen. In particular the stabilization difference corresponds to 1 kcal·mol-1.
Starting from [39·K·HF]⁺complex, water is replaced by hydrogen chloride (Fig. 26).
Fig. 26 Different views of [39·K·HCl]⁺complex (bond length expressed in angstrom).
In this case, the situation is reverse. The most stable structure is found to bind
hydrogen chloride, that is dissociated in H+ and Cl-. Its coordination is more stable on
aliphatic nitrogen with respect to that one on aromatic nitrogen, with an extent of 1,9
kcal·mol-1. In every case however, the potassium ion is closely coordinate to the
aromatic nitrogen.

50
These initial results could be considered, remembering that the method of calculation
is based on a wide approximation of real systems. As said above, temperature has not
been taken into account initially, although it obviously influences the conformation
that the system could adopt. Ab initio molecular dynamics, in which the temperature
is considered, are work in progress.
1.5.2 COLLISIONAL INDUCED DISSOCIATION
Collisional Induced Dissociation (CID), sometimes also called collisionally activated
decomposition (CAD), is a further technique that allows partial or complete structural
determination. From IUPAC Compendium on Analytical Nomenclature, Collision-
induced Dissociation is a analytical technique consisting in an ion-neutral species
interaction, wherein the projectile ion is dissociated as a result of interaction with a
target neutral species. This is brought about by conversion of part of the translational
energy of the ion to internal energy in the ion during collision.
As in IR-MPD analysis, in CID the isolation of ionic complex under study is needed, in
order to submit it to the action of collision gas. This is carried out using a MS/MS or
MSn spectrometer. As shown in Fig. 27, the precursor ion enters the collision cell (or in
the case of ion-traps or FT-MS, the precursor ion is isolated in the trap) containing a
high pressure of an energised, chemically inert collision gas: Ar, He, N2, CO2 etc.
Fig. 27 A cartoon schematic of CID fragmentation.

51
The precursor ion undergoes repeated collisions with the collision gas, building up
potential energy in the molecule, until eventually the fragmentation threshold is
reached and the product ions are formed. The types of fragmentation vary
considerably with the type of product ion and the amount of energy involved. At lower
energies (close to the threshold), fragmentation reactions are often limited to neutral
losses (H2O, MeOH, CO, CO2, MeCN etc.) depending on the nature of the precursor ion.
These neutral losses are often not considered structurally significant, although they
can be used to obtain information about functional groups. At higher energies, retro-
synthetic type reactions are often observed. These are much more structurally
significant, and often result in cleavage of the molecule at characteristic positions. If
the energy is too high, C-C bong cleavage can occur leading to uncontrolled
fragmentation, which should be avoided. Usually it is best to work at around the
fragmentation threshold, or just above, to maintain most control over the
fragmentation processes. Ion-trap and FT-MS instruments allow for
the most control over CID, but also tend to produce less energetic reactions. Triple
quadrupole tend to produce more energetic CID with more fragmentation, but less
operator control. However ion-trap and FT-MS allow multistage fragmentation
experiments to be conducted, which is essential for structural elucidation studies.
Performing the CID analysis on complex [39·K·A]⁺, the same unexpected results have
been obtained: operating at lower energies the lack of potassium ion or the acidic
species isn’t observed, while at high collisional energy uncontrolled fragmentation
have been observed. So, the goal to determine energetic information, in particular
bond energies of non-covalent interaction, has not yet been reached.
However prof. Filippi research group is going to develop further studies using fourier-
transform ion cyclotron resonance mass spectrometry (FT-ICR), in particular for
kinetic constant determination in exchange reaction of the type:
[39·K·A]⁺ + B → [39·K·B]⁺+ A
(Eq. 4)

52
where A and B are acidic species, such as hydrogen halides or amino acids.
As mentioned above, till now gas phase studies, together with theoretical ones, gave
just preliminary results and further deepened investigation are required. However the
preamble is of great interest, so that collaboration between organic, inorganic and
computational chemists is strongly motivated, in order to shed light on the reason of
the particular affinity of macrocycle 39 toward potassium ions.
The general idea of the collaboration is to make systematic structural variations on
macrocycle 39, that represents the starting point of our project, in order to
synthesized different azamacrocycles, which will be investigate in the same manner.
In fact, in this way, the moieties and the structural features, that are responsible of the
strong binding with potassium, should be determined.

53
-2-
RESULTS AND DISCUSSION
2.1 AIM OF OUR PROJECT
Taking part of a collaboration in which different research groups buckle to solve a
problem, the role of organic chemist is to find a valuable pathway to synthesize the
azamacrocycle of interest. Moreover, also the logical planning of the structural
variation to make on the prototype 39 is of fundamental importance to determine
unequivocally the moieties and structural features of the molecule that are
responsible of the binding to potassium ion.
The first structural variation planned consists in the introduction of chirality into the
hexaaza macrocycle 39, so in the synthesis of the enantiopure macrocycle 38 (Fig. 28).
This variation could be provided using the enantiopure (R,R)-1,2-diaminocyclohexane
instead the racemic mixture of trans-diaminocyclohexane.
Fig. 28 First structural variation planned.

54
The reason of this choice resides on the fact that great conformational variation of the
internal cavity could be induced using enantiopure diamines as reagents. In fact,
observing the two imino type diastereoisomers (Fig. 29), from a structural point of
view, one could note that the spatial dispositions are different. They adopt two
different conformations, that could strongly influence the coordination to a metal ion.
The meso compound could adopt a chair conformation, while the enatiopure form
arranges itself according a twist conformation.
Fig. 29 Stereoisomer conformations (in twist conformation cyclohexyl groups are omitted for clarity).
Moreover, these ligands are able to adjust their conformation to match the size of
coordinated metal ion. This adjustment is based on combination of various degree of
bending and helical twisting of the macrocycle.1,20 Basing on the assumption that also
the corresponding hexaaza macrocycle amines have different behaviors on
coordination of metal ions, this structural variation has been approved to be the first
one to try.
2.2 METAL-FREE SYNTHESIS OF ENANTIOMERICALLY PURE
AZAMACROCYCLE
The first step was to synthesize the dialdehyde 41, which can be generated in various
ways, either starting from 2,6-dimethylpyridine and from the corresponding alcohol
42 (2,6-dihydroxymethylpyridine). The higher yields, generally, are obtained from the

55
alcohol, so the latter have been chosen as the starting material (Scheme 13). It could
be oxidazed through two hypothetical reaction pathways: a) Swern reaction,35 b)
Oxidation by SeO2.36
Scheme 13 Possible pathways of synthesis of 2,6-diformylpyridine.
The second pathway, however, represents the favored choice because of the bad smell
that diffuses carring out the Swern oxidation. The dialdehyde 42 have been obtained
as white crystals with a yield of 80%. Its formation is confirmed by GC-MS: the
gaschromatogram in fact contains only one peak (R.T.=6,93 min), whose patter of
fragmentation corresponds to that one of the desired aldehyde. The IR spectrum is
also useful to confirm the formation of the carbonyl group with the presence of a
strong band at 1711 cm-1. Also 1H NMR spectrum underlines the formation of formyl
moiety by the singlet at 10,17 ppm, relative to the -CHO proton and the lack of signal
relative to hydroxyl proton. Often the synthesis does not conduct to the pure product,
but a purification could be carried out by column chromatography. This synthesis has
been performed from time to time because of the instability of the 2,6-
diformylpyridine towards oxidation. That’s also the reason why it is not purchased
directly.
Once that the starting material has been synthesized, the condensation reaction
between aldehyde and diamine has been carried out in order to obtain the [2+2]
macrocycle of interest. Following the guidelines presented in section 1.2.1 and the
approach used to synthesize the meso form 33, this reaction was carried out under

56
conditions of high dilution and in a protic polar solvent such as methanol, using the
starting materials in a stoichiometric ratio 1:1. These measures promote the
cyclization reactions rather than those of the formation of oligomers. The use of the
highly polar solvent, in fact, allows the precipitation of the cyclized products, much
less polar of the starting substrates, shifting the equilibrium toward their formation.
The control of the progress of the reaction has been effectuated by ESI-MS analysis
because of the thermal instability of the obtained products and so the impossibility to
use other common analysis such as gas chromatography or gas chromatography
interfaced with electronic ionization mass spectrometry. At the end of reaction the
formation of two macrocycles was observed (Scheme 14).
Scheme 14 Condensation reaction of 2,6-diformylpyridine with (R,R)-1,2-diaminocyclohexane.
The ESI-MS spectrum of the reaction mixture showed a signal m/z=427, relative to the
protonated macrocycle 40, but also an intense signal with m/z=640, corresponding to
the protonated macrocycle 43, that results from the condensation of three dialdehyde
units and three diamine units. The formation of [3+3] product is confirmed by a work
of Gregolinski et al.37
Before the adoption of another strategy of synthesis, the reduction of the imine
products has been carried out in order to shelve the equilibrium between them and to
determine the relative concentration. Another reason of this attempt was to verify if,
in the reduction step, a perturbation of the equilibrium could be provoked and just
one of the macrocyclic amine formed, as in the case of the meso compound 33.

57
As mentioned, once a macrocyclic imine is formed through the condensation between
dicarbonyl compounds and diamines, it is able to undergo reduction of imine moieties,
leading to the formation of the corresponding macrocyclic amines, that are more
stable towards hydrolytic decomposition. The reduction can be carried out using
several reagents, but the most used is surely sodium borohydride, that could be
directly added to the reaction mixture after the macrocyclization step.
Scheme 15 Onepot synthesis of hexaaza macrocyclic amines.
The commercial availability, the easy handling and simpleness of synthesis make the
sodium borohydride the best choice. Because of the reaction is esothermic, the
released heat could perturb the equilibrium established in the macrocyclization step,
so in some particular cases an ice bath is used during the addition of the reagent.
Unlike in the synthesis of the meso amine 39, macrocyclic amines 38 and 44 were
obtained as a mixture, which was analyzed by 1H NMR in order to evaluate the

58
integration of signal and stabilize the relative concentration, that otherwise has not
been possible with gas chromatographic analysis. Unfortunately this strategy hadn’t a
positive response because of the similar nature of the two products: they have same
structural features and they differ only in ring size. This leads to the superimposition
of signals, preventing the right interpretation of the integrations.
Nevertheless, separation of the macrocyclic amines 38 and 42 has been tried, in order
to recover the product of interest. The first methodological approach has been
consisted in chromatography, although in literature no clear way to elute this type of
compound was found. Several mobile phases were investigate through thin layer
chromatography in order to find effective conditions for a valuable separation. The
difficulty was in finding a means to allow macrocycles 38 and 42 to run along the TLC.
In fact these species are extremely basic and has strong interaction with silica that is
acidic. These interactions led to stripes rather than precise spots. Putting
triethylamine or formic acid into the eluent or dabbing the silica did not lead to a real
improvement, so this attempt to separate the two polyamines was abandoned.
Searching another solution in litherature, a purification of [3+3] macrocyclic amine
via precipitation in dichloromethane/acetonitrile mixture was found.38 The idea was
to precipitate the [3+3] macrocycle and recovering the filtrate. Unfortunately,
although several successive precipitations were performed, a sufficient level of purity
of [2+2] macrocycle wasn’t reached.
Metal-free synthesis was then tried again, leaving the reaction mixture of the
macrocyclization step to reflux for a prolong period of time. This idea arose from a
work of Kunhert et al.39 In fact they have synthesized macrocyclic systems similar to
those of our interest, starting from (R,R)-1,2-diaminocyclohexane and 1,3-
disubstituted dialdehydes. They found that [3+3] cyclocondensation product are
formed under kinetic control, while [2+2] cyclocondensation products points towards
these macrocycles as the products of thermodynamic control (Scheme 16).

59
Scheme 16 Example of conversion of the kinetic [3+3] product to the thermodynamic one.
So, the possibility to convert totally the side product in [2+2] product of interest was
considered as a chance to try (Scheme 17).
Scheme 17 Failed attempt to convert [3+3] product into the [2+2] one.
Nevertheless, in ESI-MS spectrum, no changes in signal intensity was observed,
regarding both the [3+3] and [2+2] cyclocondensation products, after several hours of
reflux (Fig. 30).

60
Fig. 30 ESI-MS spectrum of reaction mixture after a) 7 hours b) 23 hours and c) 28 hours of reflux.
Furthermore, also some experiments were conducted with microwaves assistance
instead the classical heating, in order to investigate if the use of microwaves could
accelerate the rate of formation of [2+2] cyclocondensation product. Both
dichlorometane and methanol were used as solvents (Scheme 18): in fact different
temperatures could be reached in shorter time lapses. Moreover, methanol and
dichloromethane, having different polarity, were supposed to be able to stabilize the
two macrocycles 40 and 43 with different extent.

61
Scheme 18 Microwave assisted macrocyclization between diamine and dialdehyde in a) dichloromethane
and b) methanol.
At the end of several attempts to find a way to obtain the macrocycle 40 pure with a
metal-free synthesis, unfortunately no effective conditions have been found both for
the purification step and for the selective synthesis. This conclusion led to the
complete change of the strategy of synthesis.
2.3 METAL-TEMPLATE SYNTHESIS OF ENANTIOMERICALLY PURE
AZAMACROCYCLE
The template synthesis has been the alternative means to take into account, in order
to obtain selectively the enantiopure hexaaza macrocyclic imine 40 and successively,
with the reduction of imine moieties, the enantiomerically pure macrocyclic amine 38.

62
In fact, often metal-free synthesis offers different advantages with respect to the
template one, but, on the other hand, in some case does not allow to reach efficient
results, in terms of yield and selectivity, as in the case of our study. On the contrary,
template synthesis could permit to obtain selectively the product of interest among
the several possible products, cyclic or acyclic ones, simplifing the process of
synthesis and increasing the yield.
Taking into account the guidelines presented in section 1.2.2, a metal ions screening
has been performed in order to investigate the appropriate choice, as reported in
Scheme 19. Doing research, in literature barium (II) has been found as good
templating agent,40 together with lanthanides (III).1,26
Metal center Counter ion Cocat. Metal ionic radius (nm)
[2+2] [3+3]
Ce3+
NO3
- -------
0,114
40a Not pure
-------
Cl- CuI, I
2 ------- -------
Cl- NaI
40a Not pure
-------
Ba2+
I
-
------- 0,135
40b Not Pure
-------
Cl-
40b Pure
-------
Eu3+
Cl- ------- 0,108
40c Not pure
-------
Ag+
NO3
- ------- 0,129 ------- Not pure
Cu2+
NO3
- ------- 0,073 ------- -------

63
Scheme 19 Template syntheses of hexaaza tetraimine macrocycle 40 performed using different metal
salts.
The metal ion screening was based on the idea of using the same procedure and the
same reaction conditions for all the different trials, in which only metal ion has been
changed, in order to observe just its effect on the pathway of synthesis. Synthesis
procedures have been inspired by a work of Busto and coworkers:40 they consisted in
dissolving the two starting materials in a mixture of methanol and dichlorometane.
The reaction mixture was stirred for 15 minuts before metal salt addition. After about
15 hours the macrocyclization was complete and reduction was carried out by adding
sodium borohydride in large excess. Metal salts has been chosen also on the base of
the counter ion: in fact generally nitrate or chloride salts are found in literature in this
type of reactions.
Describing the general trend of all template reactions, surely that one carried out
using barium chloride dry as templating agent has given the best result (Scheme 20).
Scheme 20 Template synthesis of macrocycle 38 using BaCl2.
After work up, the enantiopure hexa aza tetramine macrocycle 38 has been obtained
pure, as light brown oil, without need of purification and with high yield (85%). Its
identity has been determined by ESI-MS analysis and NMR analyses. The ESI-MS
spectrum (Fig. 31) shows exclusively the protonated and diprotonated macrocycle 38
adducts and the adduct with sodium.

64
Fig. 31 ESI-MS spectrum of enantiopure hexaaza tetramine macrocycle 38.
Also 1H NMR (Fig. 32) and 13C NMR (Fig. 33), both recorded in CDCl3, give evidence of
the presence of a single macrocycle form.

65
Fig. 32 1H NMR spectrum of macrocycle 38 recorded in CDCl3.
Fig. 33 13C NMR spectrum of macrocycle 38 recorded in CDCl3.

66
Both spectrums don’t present signal relative to nuclei belonging imine moieties,
suggesting that the reduction step proceeded easily to completion. This results is
extremely important, not only because the selective formation of [2+2] macrocycle
adduct, but also because of the obtained high yield. In fact, the synthesis of chiral
polyazamacrocycles, that incorporated trans-cyclohexane-1,2-diamine, are not trivial,
due to the low yields usually associated to the key macrocyclization step.
Using BaI2·2H2O as templating agent in the same reaction conditions, the
results changed (Scheme 21).
Scheme 21 Template synthesis of macrocycle 38 using BaI2·2H2O.
ESI-MS spectrum of the crude product shows an intense signal with m/z=435 that is
referred to the protonated macrocycle 38. Unfortunately, the synthesis wasn’t
selective and two intense signals with m/z= 453 and m/z= 497 are present. Some
assumptions has been proposed to interpret the identity of chemical species
responsible of these signals. Regarding signal with m/z=453, it was hypothesized that
could represent both the protonated macrocycle 38 coordinated with a water
molecule, both the acyclic [2+2] product 47 (Fig. 34).

67
Fig. 34 Hypothetic structures responsible of signal m/z=453 (reported calculated isotopic abundances).
Regarding signal with m/z= 497, the only by-product supposed, that could be
associated to this one (Fig. 35), derives from incomplete reduction.
Fig. 35 Hypothetic structures responsible of signal m/z=497 (reported calculated isotopic abundances).
In doing these hypotheses, confirmation was search in NMR spectrum. 1H NMR
spectrum suggests the presence of more than one species. However, certain
conclusions couldn’t be develop because of the complexity of the spectrum, also due to

68
signal superimposition. Separation of all by-product should be performed in order to
identify their nature individually. The lack in efficiency of the process, with respect to
the previous one, could derive both from the nature of the counter ion or, probably,
from the presence of two water molecule of crystallization.
After barium, cerium (III) has been investigated as possible templating agent. In
particular Ce(NO3)3·6H2O and CeCl3·7H2O have been chosen. The first because it is
usually used in such kind of reactions, the latter because of several characteristics that
make it a good candidate to act as a promoter. Firstly, of course, relates to its low
toxicity and its low cost, which allows use on a large scale, and therefore also at the
industrial level. As a second feature, however, we find its good stability and activity in
the presence of both air and water. In reality, the system CeCl3·7H2O, if used as the
sole initiator of reaction, seems to have a low activation processes in which it is
involved. In fact, it has been observed that the addition of a salt of iodine in the
system, in particular NaI, is essential for its implementation and, therefore, for a good
outcome of the reaction.41
The synthesis in which Ce(NO3)3·6H2O has been used as templating agent (Scheme
22), led to the formation of the target molecule.
Scheme 22 Template synthesis of macrocycle 38 using Ce(NO3)3·6H2O.
As the previous synthesis, also in this case the ESI-MS shows a signal with m/z=453,
which origins has been discussed above, but also a more intense signal with m/z=357.

69
It has been associated to an acyclic product of the type [1+2], i.e. generated by the
condensation of one diamine unit and two dialdehyde units (Fig. 36), followed by the
reduction of both imine moieties and formyl groups.
Fig. 36 Hypothetic structure responsible of signal m/z=357 (reported calculated isotopic abundances).
The formation of acyclic by-products was also observed in synthesis using CeCl3·7H2O,
activated by the promoter NaI (Scheme 23).
Scheme 23 Template synthesis of macrocycle 38 using CeCl3·7H2O/NaI.
The recurring fail on macrocycle closure, encountered especially with Cerium salts,
led to suppose that something went wrong during the coordination of starting
materials to the ion templating agent: in fact, as said in paragraph 1.2.2, the formation
of a macrocycle happens because of the juxtaposition of diamine and dialdehyde units

70
on coordination to the metal center. So possible explanation of the reason why the
cyclization does not go to completion could be represented by the ineffective
disposition of starting materials that prevents the condensation or by the lack of
vacant coordination sites that should be available for the reactants. The latter
represents a better hypothesis: in fact in most cases macrocyclization occurs, so
effective prerequisites for the macrocycle closure are present. The explanation could
reside on the difficult coordination of starting materials, due to the presence of other
ligands, such as water molecules. This hypothesis suggested to try the template
synthesis of the macrocycle 38 using cerium trichloride dry in order to see if it was
more active, but the formation of the target molecule wasn’t observed. Other
explanations of this behavior should be searched.
Negative results have been obtained using CeCl3·7H2O, activated by CuI/I2.
Theoretically the system CuI/I2 should break the oligomer structure of cerium
trichloride and the cerium ion should act as template reagent. However, the
macrocycle 38 wasn’t formed (Scheme 24).
Scheme 24 Template synthesis attempt of macrocycle 38 using CeCl3·7H2O/CuI/I2.
The total failure of this methodologies is due probably to the presence of Cu2+ ions. In
fact also copper is able to act as templating agent, directing the reaction pathway to
the formation of other species. As reported in Scheme 18, copper (II) has a smaller
ionic radius. This could be confirmed also by the results obtained from the template
synthesis with Cu(NO3)2·3H2O, in which the formation of macrocyclic adduct 38
wasn’t observed (Scheme 25).

71
Scheme 25 Template synthesis attempt of macrocycle 38 using Cu(NO3)2·3H2O.
In both reactions that include the use of copper (II) a signal with m/z=274 has been
observed, that could be associated to a [1+1] product (Fig. 37), coordinated to a
potassium ion.
Fig. 37 Hypothetic structure responsible of signal m/z=274 (reported calculated isotopic abundances).
Furthermore, optically active [2+2] macrocycle, adopting a twisted conformation,
generates an atypical coordination sphere to host just one transition metal ion, while
it can coordinate easier Ln (III) ions. So also the preferred coordination geometry of a
metal ion and the possibility of adjustment of the macrocycle to this one influences
greatly the course of the reaction. In fact it was observed that Ag (I), although it has an
ionic radius similar to that one of Ba (II), doesn’t allow the formation of macrocycle
38, but the ESI-MS spectrum evidences the presence of the [3+3] macrocycle 44,
associated at an intense signal with m/z=652.

72
At this point, knowning that purification of macrocyclic species was extremely
difficult, a third and last attempt to isolate and determine the relative yield of the
obtained macrocyclic adduct 38, especially in all template reactions, has consisted in
finding a valuable method to adopt using HPLC technique. Working in a reverse phase,
several mobile phases were tried, such as several mixtures of water and methanol and
methanol solution of formic acid (1%). Also in this case a problem arose: the
macrocycle 38 interacts so strongly with the stationary phase of the used column that
it was not able to come out of the column. The use of a specific column for highly polar
compounds was needed in order to performed the effective separation of crude.

73
-3-
CONCLUSIONS
Through this work, we have learned the plethora of factors that influence
macrocyclization reactions and how to control them, in order to direct selectively the
synthesis to the target products. In fact, the synthesis of azamacrocycle through the
formation of Schiff bases, has been a perfect example of how many ways can be
accessed, using relative simple, difunctionalized starting materials. What is fascinating
in the synthetic chemistry is the ongoing challenge that exists between the chemist
and molecules, where the first one tries to influence the behavior of the latter and to
reach a precise goal.
We’ve also learned how chirality plays a extremely important role, particularly in
synthesis: in fact, using an enatiopure diamine as reactant instead a racemic mixture,
the results, in trying to obtain hexaaza tetramine macrocycle [2+2], are totally
different and mostly dependent on dynamic equilibria establishment and
conformational stability of final adducts.
Moreover we’ve learned that actually both macrocyclic Schiff bases and relative
amines show enormous potential. Their applicability seems to range in various
scientific fields, from pharmacology, medicine, catalysis to supramolecular chemistry,
making them of great interest.
In conclusion, the synthesis of chiral hexaaza tetramine macrocycle has been carried
out in high yields (85%) by a templated one-pot two-steps process, starting from
(R,R)-1,2-diamincyclohexane and 2,6-diformylpyridine and using BaCl2 as templating
agent. Also cerium has showed a templating potential relative to the formation of the
[2+2] product of interest, although the synthesis isn’t already optimized and needs
further investigation, while transition metal ions failed as templating agents.

74
Future prospect are directed towards the development of an HPLC method able to
allow effective purification of the target macrocycle. Also the monitoring of template
synthesis is an interesting tool to understand more deeply the process of
macrocyclization.
A useful attempt to allow the chromatographic purification could be the protection of
amine groups on the hexaaza tetramine macrocycle with BOC, for instance. In fact we
think that the protected macrocycle, being more lipophilic and sterically hidered,
could have weaker interaction with the silica stationary phase, allowing the
purification via column chromatography.
From the point of view of gas phase studies, surely the enantiopure macrocycle will be
tested as the same as the meso form, in order investigate its particular behavior and to
planned further structural variations. Among several different macrocyclic species
that could be synthesized, those that deserve attention could be prepared, starting
from the follow bulding blocks (Fig. 38).
Fig. 38 Proposed starting materials for the synthesis of new azamacrocycle [2+2].
Maintaning the (R,R)-1,2-diaminocycloheane as starting material, the use of the
dialdehydes 52a, 52b and 53 is thought to investigate if the computational studies
previsions, in which potassium ion coordinates preferentially the pyridine nitrogen,
are true and in which extent they contribute to the interaction with it. Moreover, the
use of 15a diamine could also be try because of its minor rigidity, with respect to the
cyclohexane omologue, that could influence the preferential conformation of the final
adduct.

75
-4-
EXPERIMENTAL SECTION
4.1 INSTRUMENTATION
Reactions are monitored through thin layer chromatography on Merck silica gel plates
Kieselgel 60 F254, through gaschromatography on a gaschromatograph 6850 Agilent
Technologies, with capillary column (0,32 mm x 30 m) and stationary phase OV1
Agilent of 0,40-0,45 μm and through mass spectrometry. Mass spectrum are obtained
by a gaschromatograph interfaced with a mass spectrometer Hewlett-Packard GC/MS
6890N that works with the EI method (70eV), or by an HPLC-MS HEWLETT PACKARD
1100MSD series model G1946A, with a column C18 Lichrospher 100 and mass
spectrometer API-ES, in positive mode.
Characterization of products is effectuated through mass spectrometry, infrared
spectroscopy and 1H and 13C nuclear magnetic resonance. IR spectrum are obtained
with an IR spectrophotometer Perkin-Elmer 1310 in the 4000-600 cm-1 range. NMR
spectrum are acquired with a spectrometer Varian Mercury Plus 400, operating at 400
MHz, using various deuterated solvents. Chemical shifts are expressed in δ (ppm)
regard to the not deuterated solvent. The following abbreviation are used: s = singlet,
d = doublet, t = triplet, q = quartet, quint. = quintet, bs = broaded singlet, dd = double
doublet, dt = double triplet, tt = triple triplet, m = multiplet. Reactions under
microwave irradiations were performed using BIOTAGE INITIATOR MICROWAVE
REACTOR with the follow technical feature: temperature range (40–250°C), heating
rate (2-5°C/sec), pressure range (0-20 bar), power range (0-400W) with magnetron

76
(2.4 GHz), and variable magnetic stirrer. Substrates, reactants and solvents are
acquired from common commercial sources and used as received.
4.2 SYNTHESIS OF 2,6-DIFORMYLPYRIDINE
A 50mL two neck flask equipped with a cooling system is treated with a nitrogen flow
and the entire system is dried. 2,6-bis(hydromethyl)pyridine (0,400g, 2,9mmol) is
added to dry dioxane (15mL), creating a suspension. SeO2 (0,322g, 2,9mmol) is added
to this suspension and the mixture is refluxed. When the reaction is complete (control
with TLC, CHCl3 : MeOH (9 : 1) as eluent), the reaction mixture is filtred using celite
and washing with dioxane. The product is purified by chromatographic column CHCl3 :
EtOAc 8 : 2. White crystals are obtained. Yield : 80%.
Characterization of 2,6-diformylpyridine (42):
C7H5NO2
IR (cm-1): 3084m, 3018w (υ C-H); 2861m (υ C-H, formyl); 1711s (υ C=O).
GC analysis (RT, min): 6,93.
MS-EI m/z (%): 135(M+), 107 (100%), 86, 78, 52, 44, 38, 29.

77
1H-NMR (400MHz, CDCl3): δ 8.04-8.08 (m, 1H, γ-pyridine); δ 8.18 (d, 2H, J=7.27,
β-pyridine); δ 10.17 (s, 2H, -CHO).
4.3 SYNTHESIS OF CHIRAL HEXAAZA MACROCYCLE
4.3.1 METAL-FREE SYNTHESIS OF [2+2] CHIRAL HEXAAZA MACROCYCLE
(R,R)-1,2-diaminocyclohexane (1,1mmol, 127mg) is dissolved in methanol (10mL)
inside two neck round bottom flask. Then, a solution of 2,6-diformylpyridine
(1,1mmol, 150mg) in methanol (1,2mL) is dropped inside the flask. The reaction
mixture is refluxed for 3 hours. The reaction mixture is cooled to room temperature
and toluene (11,2mL) is added to form a mixture MeOH : Tol 1:1. NaBH4 (3×0,6mmol,
3×23mg) is added to the reaction mixture, that is stirred for 24 hours at room
temperature. Then the reaction mixture is dried under reduced pressure and the
residue is dissolved in water. Then NaOH 5% is added to the solution, till pH becomes
13. Extraction with CHCl3 is done and the combined organic phases are dried over
Na2SO4. Filtration of organic phase, which is subsequently dried under reduced
pressure gives a mixture of [2+2] and [3+3] products.

78
4.3.2 METAL-TEMPLATE SYNTHESIS OF [2+2] CHIRAL HEXAAZA
MACROCYCLE WITH BaCl2
(R,R)-1,2-diaminocyclohexane (0,6mmol, 68mg) is dissolved in a mixture of methanol
(5mL) and dichloromethane (5mL). Then 2,6-diformylpyridine is added to the
reaction mixture that is stirred for 15 minutes. Then BaCl2 (1,2mmol, 250mg) is added
and the reaction mixture is stirred for 15 hours at room temperature. Then NaBH4
(3×0,8mmol, 3×31mg) is added to the reaction mixture, that is stirred for 7 hours at
room temperature. The reaction is quenched with HCl conc. (0,75mL). Then NaOH 4N
(10mL) is added. Extraction with CH2Cl2 is done and the combined organic phases are
dried over Na2SO4. Filtration of organic phase, which is subsequently dried under
reduced pressure gives the pure [2+2] product. Yield: 85%.
Characterization of polyamine macrocycle 38:
C26H38N6
ESI-MS m/z (%): 435 (MH+), 457 (MNa+), 218 (M+2H+).

79
1H-NMR (400MHz, CDCl3): δ 0,97-1,09 (m, β-cyclohexyl); δ 1,58 (m, α-cyclohexyl); δ
2,06-1,97 (m, NCH); δ 3,64 (s, br, NH); δ 3,74 (d, 4H, NCH2); δ 4,02 (d, 4H, NCH2); δ
6,99 (d, 4H, β-pyridine); δ 7,49 (t, 2H, γ-pyridine).
13C NMR (400MHz, CDCl3): δ 24,86 (β-cyclohexyl); δ 32,814 (α-cyclohexyl); δ 51,51
(NCH2); δ 59,46 (NCH); δ 121,38 (β-pyridine); δ 136,95 (γ-pyridine); δ 160,26 (α-
pyridine).

80
REFERENCES
1Vigato, P.A.; Tamburini, S. Coord. Che. Rev. 2004, 248, 1717-2128.
2Alexander, V. Chem. Rev. 1995, 95, 273-342.
3Collinson, S.R.; Fenton, D.E. Coord. Chem. Rev. 1996, 148, 19-40.
4Yatsimirskii, K.B. Russian Chemical Reviews. 1990, 59, 12, 1150-1156.
5Cation Binding by Macrocycles, eds.Y. Inoue and G. W. Gokel, Marcel Dekker, Inc., New
York/Basel, 1990.
6A. F. Danil de Namor;R. M. Cleverley;M. L. Zapata-Ormachea, Chem. Rev. 1998, 98,
2495.
7P. D. Beer, P. A. Gale and A. Philip, Angew. Chem., Int. Ed. 2001, 40, 486–516.
8Kuhnert, N.; Lopez-Periago, A.; Rossignolo, G. M. Org. Biomol. Chem. 2005, 3, 524–
537.
9Schiff, H. Annalen 1864, 131, 118.
10Godoy-Alcantar, C.; Yatsimirsky, A. K.; Lehn, J.-M. J. Phys. Org. Chem. 2005, 18, 979.
11Borisova, N.E.; Reshetova, M.D.; Ustynyuk, Y.A. Chem. Rev. 2007, 107, 46-79.
12Saczewski, F.; Dziemidowicz-Borys, E.; Bednarski, P.; Grunert, R.; Gnadiec, M.; Tabin,
P. J. Inorg. Biochem. 2006, 100, 1389-1398.
13Kuhnert, N.; Patel, C.; Jami, F. Tetrahedron Lett. 2005, 46, 7575-7579.
14Gawronski, J.; Gawronska, K.; Grajewski, J.; Kwit, M.; Plutecka, A.; Rychlewska,U.
Chem. Eur. J. 2006, 12, 1807-1817.

81
15Metal Mediated Template Synthesis of Ligands, Costisor, O.; Linert,W. World Scientific
Pub Co Inc 2004.
16Cairns, C.; McFall, S.G.; Nelson, S.M.; Drew, M.G.B. J. Chem. Soc., Dalton Trans. 1979,
446.
17Cook, D.H.; Fenton, D.E.; Drew, M.G.B.; Rodgers, A.; McCann, A.; Nelson, S.M. J. Chem.
Soc., Dalton Trans. 1979, 414.
18Cook, D.H.; Fenton, D.E. J. Chem. Soc., Dalton Trans. 1979, 266.
19Gregolinski, J.; Kochel, A.; Lisowski, J. Polyhedron. 2006, 25, 2745-2754.
20Radecka-Paryzek, W.; Patroniak, V.; Lisowski, J. Coord. Chem. Rev. 2005, 249, 2156-
2175.
21Voss Jr., D.A.; Buttrey-Thomas, L.A.; Janik, T.S.; Churchill, M.R.; Morrow, J.R. Inorg.
Chim. Acta. 2001, 317, 149-156.
22Morrow, J.R.; Buttrey, L.A.; Shelton, V.M.; Berback, K.A. J. Am. Chem. Soc. 1992, 114,
1903-1905.
23Bligh, S. W. A.; Choi, N.; Evagorou, E. G.; McPartlin, M.; White, K. N. J. Chem. Soc.,
Dalton Trans. 2001, 3169-3172.
24Lauffer, R.B. Chem. Rev. 1987, 87, 901-927.
25Smith, P.H.; Brainard, J.R.; Morris, D.E.; Jarvinen, G.D.; Ryan, R.R. J. Am. Chem. Soc.
1989, 111, 7437-7443.
26Radecka-Paryzek, W.; Patroniak, V.; Lisowski,J. Coord. Chem. Rev. 2005, 249, 2156–
2175
27(a) Savoia, D.; Gualandi, A. Curr. Org. Synth. 2009, 6, 102-118; (b) Savoia, D.;
Gualandi, A. Curr. Org. Synth. 2009, 6, 119-142.

82
28(a) Choi, K.; Hamilton, A. D. J. Am. Chem. Soc. 2001, 123, 2456-2457; (b) Choi, K.;
Hamilton, A. D. J. Am. Chem. Soc. 2003, 125, 10241-10249; (c) Choi, K.;Hamilton, A. D.
Coord. Chem. Rev. 2003, 240, 101-110; (d) Hembury, G. A.; Borovkov, V. V.; Inoue, Y.
Chem. Rev. 2008, 108, 1-73.
29(a) Reid, R. C.; Abbenante, G.; Taylor, S. M.; Fairlie, D. P. J. Org. Chem. 2003, 68, 4464-
4471; (b) Loughlin, W. A.; Tyndall, J. D. A.; Glenn, M. P. Chem. Rev. 2004, 104, 6085-
6117.
30(a) Fernández-López, S.; Kim, H.-S.; Choi, E. C.; Delgado, M.; Granja, J. R.; Khasanov,
A.; Kraehenbuehl, K.; Long, G.; Weinberger, D. A.; Wilcoxen, K. M.; Ghadiri, M. R. Nature
2001, 412, 452-455; (b) Horne,W. S.; Stout, C. D.; Ghadiri, M. R. J. Am. Chem. Soc. 2003,
125, 9372-9376.
31Tsobomura, T.; Yasaku, K.; Sato, T.; Morita, M. Inorg. Chem. 1992, 31, 447.
32Lisowski, J. Magn. Reson. Chem. 1999, 37, 287.
33González-Álvarez, A.; Alfonso, I.; Díaz, P.; García-España, E.; Gotor-Fernández, V.;
Gotor, V. J. Org. Chem. 2008, 73, 374-382.
34Gross, M.L.; Caprioli R. Encyclopedia of Mass Spectrometry: Fundamentals of and
Application to Organic (Organometallic) Compounds 2005, 4, Elsevier, Oxford, 327-
337.
35 Hicks, R.G.; Koivisto, B.D.; Lemaire, M.T. Org. Lett. 2004, 6, 12, 1887-1890.
36 Luning, U.; Baumstark, R.; Peters, K.; von Schnering, H.G. Liebigs Ann. Chem. 1990,
129-143.
37 Gregolinski, J.; Lisowski, J.; Lis, T. Org. Biomol. Chem. 2005, 3, 3162-3166.
38 Gregolinski, J.; Starynowicz, P.; Hua, K.; Lunkley, J.L.; Muller, G.; Lisowski, J. J. Am.
Chem. Soc. 2008, 130(52), 17761–17773.

83
39 Kunhert, N.; Rossignolo, G.M.; Lopez-Periago, A. Org. Biomol. Chem. 2003, 1, 1157–
1170.
40 Busto, E.; González-Álvarez, A.; Gotor-Fernández, V.; Alfonso, I.; Gotor, V.
Tetrahedron 2010, 66, 6070-6077.
41 Bartoli, G; Marcantoni, E.; Marcolini, M.; Sambri, L. Chem. Rev. 2010, 110,6140-6143.


















