
CHAPTER-5A AN IMPROVED PROCESS FOR THE PREPARATION...
35
176 Chapter-5 CHAPTER-5A AN IMPROVED PROCESS FOR THE PREPARATION OF ZAFIRLUKAST - AN ANTI ASTHMA DRUG
Transcript of CHAPTER-5A AN IMPROVED PROCESS FOR THE PREPARATION...

176 Chapter-5
CHAPTER-5A
AN IMPROVED PROCESS FOR THE PREPARATION OF
ZAFIRLUKAST - AN ANTI ASTHMA DRUG

177 Chapter-5
AN IMPROVED PROCESS FOR THE PREPARATION OF
ZAFIRLUKAST: AN ANTI ASTHMA DRUG
5. 1.1 INTRODUCTION
Asthma is a disease that causes swelling and narrowing the
airways of the lungs. Airways are air carriers to and from lungs. Swollen
and narrower airways affect the air flow to and from the lungs and this
lead to tightness of chest, wheezing, shortness of breath and cough.
These symptoms are often occurs in early morning and in night. Asthma
is caused by genetic and environmental factors, it was not curable
completely but this can be controlled with good medical care.
Leukotriene antagonists also known as leukast are the
medicaments that are used to reduce leukotrienes, which are produced
by several types of cells and causes inflammation in asthma and
bronchitis. Leukotriene antagonists that are available in market are
Montelukast, Zafirlukast and Pranlukast. Zafirlukast is the first leukast
compound approved for management of Asthma. US FDA approved
zafirlukast in the form of 10 mg and 20 mg tablet with the brand name
of Accolate®.1 Subsequently this was approved and launched by
innovator in few other countries.
Importance of Leukotriene antagonists for treating asthma are
motivated us to develop an improved and cost effective process for the
preparation of Zafirlukast.

178 Chapter-5
5. 1.1.1 PRODUCT PROFILE
1. Generic name : Zafirlukast
2. Chemical structure :
3. Chemical name : [3-[[2-Methoxy-4-[[[(2-methylphenyl)
sulfonyl] amino] carbonyl] phenyl]
methyl]-1- methyl-1H-indole-5-yl]
carbamic acid cyclopentyl ester
4. Molecular formula : C31H33N3O6S
5. Molecular weight : 575.69
6. CAS No : 107753-78-6
7. Therapeutic category : Anti asthmatic
5. 1.1.2 PHYSICAL CHARACTERISTICS
1. Description : Off white to pale yellow powder
2. Solubility : Tetrahydrofuran (Rx-list)
3. Melting piont : 137 – 140°C
5. 1.1.3 MARKET INFORMATION
1. Applicant : AstraZeneca
2. Patentee : ICI
3. Brand name : ACCOLATE

179 Chapter-5
5. 1.2 LITERATURE REVIEW
There are many synthetic routes for the preparation of Zafirlukast 4 is
well documented in literature. Some of the key approaches are discussed
here under. Scientists from ICI Americas Inc2 have reported process for
the synthesis of 4, which starts with esterification of
3-methoxy-4-methyl benzoic acid 53 using methanol in presence of
acetyl chloride (Scheme 5.1).
OCH3
OCH3
H3C
O
OCH3
OCH3
O
Br
OH
OCH3
H3C
O
NH
O2N
NH
O2N
53 54 55
124
125
Acetyl chloride
Methanol
OCH3
O
OCH3
N
O2N
57
OCH3
O
OCH3
CH3
N
H2N
58
OCH3
O
OCH3
CH3
N
HN
59
OCH3
O
OCH3
CH3
O
O
N
HN
60
OCH3
O
OH
CH3
O
O
N
HN
4
OCH3
O
NH
CH3
O
O
S
O O CH3
Bromine
1,4-dioxane
Silver oxide
NaH, CH3I
Tetrahydro furan
Pd/C, H2
Methanol
CPC, NMM Lithium hydroxide
THF, Methanol
OTSA, DMAPEC
CCl4
DMAP
Scheme 5.1: Synthesis of zafirlukast 4 (product patent route)

180 Chapter-5
Allylic bromination of methyl ester 54 using bromine in presence
of CCl4 resulted bromo compound 55, which was reacted with 5-nitro
indole 124 using silver oxide as catalyst to obtain condensed compound
125. N-methylation of 125 utilizing methyl iodide in presence of NaH
afforded N-methyl indole derivative 57. Thus obtained 57 was subjected
to reduction using palladium carbon (Pd/C) in methanol followed by
reacted with cyclopentyl chloroformate to obtain compound 59.
Hydrolysis of 59 using LiOH.H2O subsequently reaction with o-toluene
sulfonamide (OTSA) in presence of 1-[3-(dimethylamino)propyl]-3-ethyl
carbodiimide hydrochloride (DMAPEC) and DMAP furnished zafirlukast
4. Matassa et al3 also reported similar procedure for the synthesis of
Zafirlukast 4.
Gutman et al4 reported process for synthesis of zafirlukast 4
commenced with hydrolysis of 125 as depicted in Scheme 5.2. Basic
hydrolysis of 125 using NaOH provided 126 in the form of sodium salt,
which was converted into acid 127 by treating 126 in aqueous HCl.
Simultaneous N-methylation and methyl esterification of acid 127 using
dimethyl sulfate followed by reduction of nitro compound 57 in presence
of 10% Pd/C provided amine 58. The resulted 58 was reacted with
cyclopentyl chloro formate to obtain cyclopentyl carbamate 59, which
was subjected to hydrolysis under basic conditions (NaOH) to obtain
acid compound in the form of its sodium salt 128. Thus obtained 128

181 Chapter-5
was isolated and further treated with HCl to get acid 60. Reaction of 60
with OTSA in presence of DMAPEC and DMAP furnished Zafirlukast 4.
NH
O2N
125
OCH3
O
OCH3
N
O2N
57
OCH3
O
OCH3
CH3
N
H2N
58
OCH3
O
OCH3
CH3
N
HN
59
OCH3
O
OCH3
CH3
O
O
N
HN
60
OCH3
O
OH
CH3
O
O
N
HN
4
OCH3
O
NH
CH3
O
O
S
O O CH3
DMAP, DCM
OTSA, DMAPEC
NH
O2N
126
OCH3
O
ONa
NH
O2N
127
OCH3
O
OH
N
HN
128
OCH3
O
ONa
CH3
O
O
NaOH
Methanol
Water
DMS, K2CO3
2-Butanone
NH2-NH2. H2O
10% Pd/C
Ethanol
CPC, NMM
DCM
NaOH
Methanol
HCl
Water
MeOH
Scheme 5.2: Synthesis of zafirlukast 4
Claire and co-workers5 have reported an alternate synthesis for
preparation of 4, which starts with hydrolysis of ester 57 as described in
Scheme 5.3. Hydrolysis of 57 in presence of hydrochloric acid provided
acid 129, which was converted into acid chloride 130 by treating with
thionyl chloride followed by reacted with o-toluene sulfonamide in
presence of DMAP to obtain amide 131. Thus obtained amide 131 was

182 Chapter-5
subjected to reduction using Pd/C followed by reacted with cyclopentyl
chloroformate to obtain Zafirlukast 4.
N
HNO
O
O
NH
S
CH3OO
CH3
4
57 129130
131132
N
O2N
O
OCH3
CH3
N
O2N
O
OH
CH3
N
O2N
O
Cl
CH3
N
O2N
O
NH
S
CH3OO
CH3
N
H2N
O
NH
S
CH3OO
CH3
Water, HCl Thionyl chloride
DMF
OTSA
DMAP
Pd/C, H2
NaOH
NaOH
OCH3 OCH3 OCH3
OCH3 OCH3
OCH3
Cyclopentyl chloroformate
Scheme 5.3: Synthesis of zafirlukast 4
Keesari et al6 have reported synthesis of 4 from benzoic acid
derivative 133 and N-methyl nitro indole 56 as described in Scheme 5.4.
Reaction of 133 with 56 in presence of ZnBr2 offered nitro acid 129,
which was hydrogenated using Raney nickel to obtain amine 134. The
resulted amine 134 was treated with OTSA in presence DCC as coupling
agent followed by reaction with cyclopentyl chloroformate in presence of
NMM furnished zafirlukast 4.

183 Chapter-5
OH
OCH3
O
Br N
O2N
+
CH3
N
HNO
O
O
NH
S
CH3OO
CH3
4
N
O2N
O
OH
CH3
N
H2N
O
OH
CH3
N
H2N
O
NH
S
CH3OO
CH3
135
ZnBr2
1,4-Dioxane
Raney nickel, H2
THF
OTSA, DCC
DMAP
133 56 129
134
CPC, NMM
DCM
OCH3
OCH3OCH3
OCH3
Scheme 5.4: Synthesis of zafirlukast
The reported synthetic routes for preparation of zafirlukast have
certain disadvantages, (i) possibility for formation of N-alkylated by-
products are high when unprotected indole compound was used for the
condensation reaction (synthetic routes 5.1, 5.2 and 5.3),5 (ii) selectivity
of alkylation at C-3 position depends on the substitutients on indole
nitrogen atom. Usage of unsubstituted indole for condensation reaction
leads to formation of C-2 alkylated by product rather than alkylation at
C-3 position, 7 (iii) formation of amide linkage via acid chloride is not
suitable for commercial level since acid chloride intermediate is non
solid, unstable and moister sensitive intermediates (scheme 5.3), (iv)
preparation of sulfonamide 135 via DCC mediated coupling reaction

184 Chapter-5
leads to formation of dimer compounds because unprotected primary
amine 134 underwent intermolecular coupling with acid group and (v)
removal of dicyclohexyl urea (obtained as by product in DCC mediated
coupling reactions) from the sulfonamide compounds needed
purification, which makes process expensive.
Apart from these drawbacks, the reported synthetic processes for
preparation of zafirlukast 4 involves usage of commercially unsafe
reagents, such as sodium hydride, acetyl chloride, Br2, CH3I and
hydrazine hydrate, expensive reagents palladium carbon, 1-[3-
(dimethylamino) propyl]-3-ethylcarbodiimide hydrochloride (DMAPEC)
and usage of column chromatography techniques purifications of crude
compounds.
5. 1.3 PRESENT WORK
5. 1.3.1 OBJECTIVE
The drawbacks associated with the reported processes motivated us
to develop an efficient, cost-effective and large-scale process for the
synthesis of zafirlukast. As per retro-synthetic path way, synthesis of
zafirlukast involves majorly four reactions (Figure 5.1).
(i) Sulfonamide linkage.
(ii) Carbamate linkage.
(iii) C-C bond formation.
(iv) N-Methylation.

185 Chapter-5
Figure 5.1: Retro-synthetic analysis of zafirlukast
All the raw materials resulted from retero-synthetic path way are
commercially available.
5. 1.3.2 RESULT AND DISCUSSION
In this context, an improved synthetic approach (scheme 2.5) for
preparation of 4 has been designed, which commenced with the
esterification of commercially available benzoic acid 53. The major
drawback associated with the esterification reaction is longer reaction
time (about 36 hours). At our end after screening the key reaction
parameters, reaction time was significantly reduced to 3 hours from 36
hours by replacing the earlier reported acetyl chloride2 with thiony
chloride. This modification is allowed us to isolate ester 54 with about
98% yield and more than 99 % of purity. Further, workup and isolation
process was simplified by quenching the reaction mixture with water.

186 Chapter-5
Modification of solvent system (chloroform with cyclohexane) in
subsequent allylic bromination reaction is avoided the tedious workup
process. Targeted bromo compound 55 was isolated with about 85%
yield and more than 99% purity by adopted simple crystallization at
lower temperature (about 4 ºC). In addition to the above modifications
both esterfication reaction and allylic bromination reactions were
performed in one pot.
To enhance the selectivity at indole C-3 position and to reduce the
formation of N-alkylated by products, N-Methyl indole 56 was chosen for
condensation reaction at indole C-3 position. N-Methyl indole 56 was
synthesized with about 99.0 % yield and more than 99.5 % purity by
reacting 5-nitroindole 124 with dimethyl sulfate (DMS) in presence of
NaOH and DMF, where as similar reaction was performed earlier using
expensive catalysts and reagents such as DABCO, CH3I and (CH3)2CO3
at elevated temperature (90 oC).7
Expensive reagents like Ag2CO3, ZnBr2 and Ag2O are previously
used2-6 for the reaction of N-methyl indole 56 and bromo compound 55.
In order to improve the process efficiency and to reduce the
manufacturing cost, commercially available and less expensive reagents
such as Cu2O, CuCl, ZnO, AlCl3 and Al2O3 are examined along with the
reported reagents for synthesis of 57.
Table 5.1: Alkylation of compound 56 using different reagents

187 Chapter-5
S. No. Reagent Yield (%) 57 (%) 136 (%) 56 (%)
1 Ag2O 65 51.4 16.8 1.2
2 CuCl 30 33.4 7.5 38.3
3 ZnO 60 49.1 4.3 1.8
4 Al2O3 95 6.7 ND 35.0
5 AlCl3* --- --- --- ---
6 ZnBr2 62 53.2 9.5 4.8
7 Cu2O 85 85.3 3.6 1.0
* indicates product formation not observed; ND = not detected.
Experimental results are indicated that lower amount of
dialkylated (C-2 and C-3) compound 136 (figure 5.4) and better yield
and purity was observed in Cu2O (Table 5.1, entry 7) when compared
with other reagents.
N
O2N O
OCH3
CH3
OOCH3
136
H3CO
Figure 5.2: Chemical structures of 136
The resulted crude compound 57 was subjected to purification for
removal of the related substances. Various purification techniques and
different solvents were screened for the purification of 57, but did not

188 Chapter-5
succeed. Hence it was decided to proceed to subsequent stage rather
than the purification of crude compound 57. In subsequent step,
previously reported expensive and pyrophoric reducing reagents like
Raney nickel and Pd/C for reduction of 57 was replaced with plant-
friendly and low toxic reducing agent sodium dithionite.
OCH3
OCH3
H3C
O
OCH3
OCH3
O
Br
OH
OCH3
H3C
O
N
O2N
53 54 55
56
Methanol
N
O2N
57
OCH3
O
OCH3
CH3
N
H2N
58
OCH3
O
OCH3
CH3
N
HN
59
OCH3
O
OCH3
CH3
O
O
N
HN
60
OCH3
O
OH
CH3
O
O
N
HN
4
OCH3
O
NH
CH3
O
O
S
O O CH3
3 h, 98 % 84 %
85 %
1,4-dioxane
Cu2O
Sodium dithionate
Methanol, water
45 %
CPC, NMM
Toluene, 97 %
LiOH. H2O
Methanol
98 %
OTSA
DCM
CH3
SOCl2 DBDMH, AIBN
Cyclohexane
T3P, DMAP
86 %
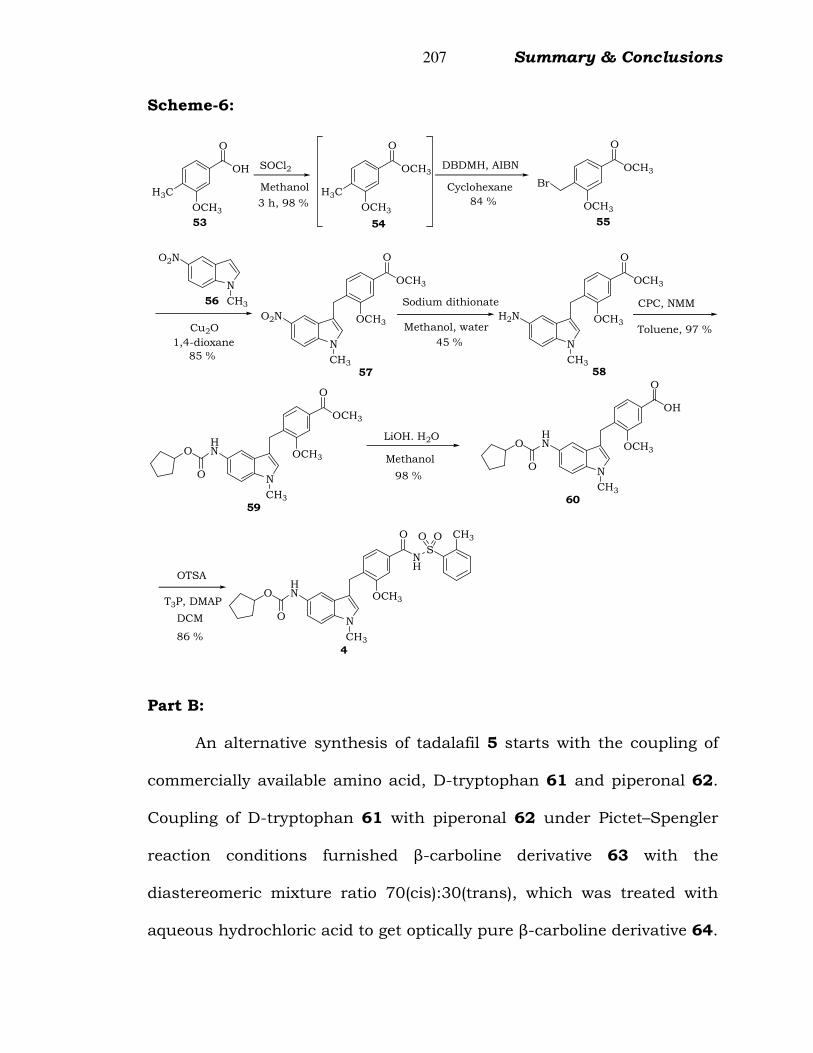
Scheme 5.5: Improved synthesis of zafirlukast
The resulted amine 58 was subjected to purification for
elimination of related compounds. After screening the various

189 Chapter-5
purification techniques, purity of the 58 was significantly improved
(more than 99.5%) by converting crude compound 58 as its HCl salt.
Having developed advantageous process for synthesis of 58, our focus
was moved towards subsequent reactions. Reaction of amine 58 with
cyclopentyl chloroformate using N-Methyl morpholine as base in toluene
medium afforded carbamate 59 with 98 % yield and about 99.7 %
purity. Thereafter, 59 was converted into 60 using lithium hydroxide
monohydrate as base in mixture of methanol and water with 98% yield
and more than 99% of purity. These processes are advantageous over
the processes reported in literature with respect to yield, cycle time and
purity.
Good number of references available in literature for the
condensation of acid 60 with o-toluene sulfonamide and these processes
involves the usage dicyclocarbodiimide (DCC) as coupling agent. Usage
of DCC as coupling reagent for formation of amide linkage facilitates
formation of dicyclohexylurea (DCU) as by-product. As per the ICH
guidelines limit of DCU in final drug compound is less than or
equivalent to 0.1%. To meet the regulatory requirement, DCU present in
crude zafirlukast 4 need to be removed by employing purification.
Multiple purifications lead to loss of yield and this makes process
expensive. In view of this, DCC is replaced with non toxic and
commercially available coupling reagent propylphosphonic anhydride
also known as T3P®. By-products obtained during the T3P mediated

190 Chapter-5
amide formation are water soluble and these can be removed with simple
water washings. By implement all the process improvements, zafirlukast
was obtained with more than 99.5% purity and all other known and
unknown impurities are less than 0.1%.
An improved synthetic method depicted in Scheme 5.5 for the
preparation of Zafirlukast 4 has certain advantages over the earlier
reported processes that are mentioned below.
(i) Time cycle for esterification reaction was reduced from 36 hours to
3 hours.
(ii) Tedious work-up processes were avoided.
(iii) Formation of N-alkylated by products was controlled by using N-
methyl indole 56 as starting material for alkylation reaction.
(iv) Formation of dialkylated compound 136 during alkylation reaction
is significantly reduced to below 4.0%.
(v) Expensive and pyrophoric reducing reagents like Raney nickel and
Pd/C are replaced with plant-friendly and low toxic reducing agent
like sodium dithionite.
(vi) Purifications for removal of DCU (obtained as by-product in DCC
mediated coupling reactions) in crude Zafirlukast 4 is avoided using
commercially available less toxic coupling reagent T3P® instead of
DCC.
(vii) Usage of toxic reagents like bromine and acetyl chloride is avoided.

191 Chapter-5
5. 1. 4 CONCLUSION
In conclusion, an efficient, robust and large scale manufacturing
process for zafirlukast 4 was developed. The product obtained by this
process fulfills all the regulatory needs.
5. 1. 5 EXPERIMENTAL SECTION
5. 1. 5.1 Process description
5. 1.5.1.1 Methyl 4-bromomethyl-3-methoxybenzoate (55)
Thionyl chloride (50 mL, 0.689 mol) was slowly added to the
stirring solution of acid 53 (100 g, 0.602 mol) & methanol (150 mL).
Reaction mass was heated to 60 °C and stirred at same temperature for
3 hours, quenched with precooled water at below 15 °C. Separated solid
was filtered, washed with 10 % aqueous Na2CO3 solution (200 mL) to
afford 108 g of ester compound 54. To the stirring solution of ester 54 in
cyclohexane (600 mL), DBDMH (80 g, 0.28 mol) and AIBN (0.6 g, 3.6
mmol) were charged and the resultant reaction mixture was heated to 80
°C and stirred for 4 hours. AIBN (0.1 g, 0.601 mmol) and DBDMH (20 g,
0.072 mol) charged into reaction mixture at ambient temperature then
heated to 80 °C and stirred for 2 hours. Reaction mass was quenched
with water (400 mL) at 55 °C and stirred for 45–60 min. Organic phase

192 Chapter-5
and aqueous phase was separated at about 55 °C, organic phase was
cooled to 10 °C and stirred at same temperature for 1 hour. The
separated solid was collected by filtration, washed with cyclohexane (50
mL) and dried under reduced pressure 50 °C for 4 hours to give 84 %
(130 g) of title compound 55 with 99.1 % purity.
IR (KBr, cm–1): 2951 (Ali, CH),1717 (ester, C═O), 1276 (Ether, C-O-C).
1H NMR (200 MHz, CDCl3): δ 7.60 (d, 1H, Ar-H), 7.54 (s, 1H, Ar-H),
7.39 (d, 1H, Ar-H), 4.55 (s, 2H, CH2), 3.95 (s, 3H, CH3), 3.92 (s, 3H,
CH3).
MS (m/z): 283.2 (M+ + Na).
5. 1.5.1. 2 1-Methyl-5-nitro-1H-indole (56)
N
CH3
O2N
Dimethyl sulfate (91 g, 0.722 mol) was slowly added to the
suspension of NaOH (52 g, 1.3 mol), DMF (400 mL) and indole
compound 124 (100 g, 0.617 mol) and stirred at 28 °C for 4 hours.
Water (1.0 L) was added to the reaction mass and stirred for 1 hour. The
separated solid was filtered, washed with water (500 mL) and dried
under reduced pressure at 55 °C for 4 hours to afford 99 % of title
compound 56 with 99.6 % purity.
IR (KBr, cm–1): 1579 & 1397 (NO2, asym. and sym.).

193 Chapter-5
1H NMR (200 MHz, CDCl3): δ 8.56 (d, 1H, Ar-H), 8.10 (dd, 1H, Ar-H),
7.32 (d, 1H, Ar-H), 7.20 (d, 1H, Ar-H), 6.66 (dd, 1H, Ar-H), 3.85 (s, 3H,
CH3).
MS (m/z): 177.0 (M+ + H).
5. 1.5.1. 3 Methyl 3-methoxy-4-(1-methyl-5-nitro-1H-indol-3-yl
methyl) benzoate (57)
A suspension of bromo derivative 55 (95.5 g, 0.368 mol), N-methyl
indole compound 56 (50 g, 0.284 mol), Cu2O (12.2 g, 0.853 mol) and
1,4-dioxane (350 mL) was heated to 95 °C and stirred at same
temperature for 27 hours. Thus obtained reaction mass was filtered
through celite bed and washed with 1, 4-dioxane (100 mL). Solvent from
the filtrate was evaporated, methanol (450 mL) and ethyl acetate (50 mL)
was added. The resultant reaction mixture was heated to 65 °C and
stirred for 1 hour, then cooled to ambient temperature and stirred for 4
hours. The separated solid was collected by filtration and dried at 55 °C
to give 85 % of title compound 57 with 85 % purity.
IR (KBr, cm–1): 2949 (Ali, CH), 1709 (ester, C═O), 1579 & 1324 (asym.
and sym., NO2), 1291 (ether, C-O-C).

194 Chapter-5
1H NMR (200 MHz, CDCl3): δ 6.9-8.6 (m, 1H, Ar-H), 3.98 (s, 3H, CH3),
3.94 (s, 3H, CH3) and 3.80 (s, 2H, CH2).
MS (m/z): 377 (M+ + Na).
5. 1.5.1.4 Methyl 4-(5-amino-1-methyl-1H-indole-3-yl-methyl)-3
methoxybenzoate (58)
To the stirring suspension of compound 57 (100 g, 0.282 mol),
dichloromethane (500 mL), triethylamine (78.7 mL, O.565 mol) and
sodium dithionite (98.3 g, 0.565 mol) was added slowly water (100 mL)
at 28 °C and stirred for 4 hours. Thus obtained reaction mass was
acidified with 50% aqueous hydrochloride solution (100 mL) at 27 °C
and stirred for 45 minutes. The separated compound was filtered and
washed with water (50 mL). Wet compound, water (500 mL) and
dichloromethane (500 mL) was stirred at 28 °C for 4 hours. The obtained
solid was filtered, water (500 mL) was added to the resulted wet
compound and pH of the reaction mixture was adjusted to 7-8 with 10%
Na2CO3 solution. The precipitated solid was filtered to give 44 % of title
compound 58 with 99.5 % HPLC purity.

195 Chapter-5
IR (KBr, cm–1): 3442 & 3360 (Amine, NH2), 2937 (Ali, CH), 1703 (ester,
C═O), 1297 (ether, C-O-C).
1H NMR (200 MHz, CDCl3): δ 6.7−7.50 (m, 7H, Ar-H) 4.07 (s, 2H, CH2),
3.93 (s, 3H, CH3), 3.90 (s, 3H, CH3), 3.66 (s, 3H, CH3).
MS (m/z): 325 (M+ + H).
5. 1.5.1.5 4-(5-Cyclopentyloxycarbonylamino-1-methyl-1H-indol-3-
yl methyl)-3-methoxybenzoicacid methyl ester (59)
To a stirring solution of amine 58 (70 g, 0.216 mol), toluene (350
mL) and N-methyl morpholine (27 g, 0.267 mol) was slowly added
cyclopentyl chloroformate (50 g, 0.337 mol) at ambient temperature and
stirred at same temperature for 1 hour. Solvent from the resulted
reaction mixture was evaporated completely, methanol (350 mL) was
charged to the obtained crude and stirred for 30 minutes. Separated
solid was filtered, washed with methanol (70 mL) and dried at 55 °C for
3 hours to afford 98 % of title compound with more than 99.6 % purity.
IR (KBr, cm–1): 3247 (amine, NH), 1719 (ester, C═O), 1692 (carbamate,
C═O), 1232 (ether, C-O-C).

196 Chapter-5
1H NMR (400 MHz, DMSO–d6): δ 9.18 (bs, 1H, NH), 7.0-7.60 (s, 7H, Ar-
H) 5.0−5.1 (m, 1H, CH), 4.0 (s, 2H, CH2), 3.9 (s, 3H, CH3), 3.8 (s, 3H,
CH3), 3.7 (s, 3H, CH3), 1.6−1.9 (m, 8H, CH2).
MS (m/z): 437.4 (M+ + H).
5. 1.5.1.6 [4-(5-Cyclopentyloxycarbonylmethyl-1-methyl-1H-indol-
3-ylmethyl)-3-methoxybenzoic acid (60)
Mixture of carbamate compound 59 (50 g, 0.115 mol), methanol
(300 mL), LiOH.H2O (7.5 g, 0.178 mol) and water (75 mL) was heated to
65 °C and the stirred at same temperature for 2 hours. The resulted
reaction mass pH was adjusted to 1.0-2.0 with diluted hydrochloric acid
at 28 °C and stirred for 2 hours. Separated solid was filtered, washed
with water (100 mL) and dried under reduced pressure at 75 °C to
provide 98 % of title compound 60 with more than 99 % of purity.
IR (KBr, cm–1): 3288 (amine, NH), 2957 (hydroxyl, OH), 1696 (acid,
C═O), 1646 (carbamate, C═O), 1264 (ether, C-O-C).
1H NMR (400 MHz, CDCl3): δ 7.63 (s, 1H), 7.1–7.6 (m, 7H, Ar-H), 6.8 (s,
1H, CH), 5.1−5.2 (m, 1H, CH), 4.1 (s, 2H, CH2), 3.9 (s, 3H, CH3), 3.7 (s,
3H, CH3), 1.6−1.9 (m, 8H, CH2).

197 Chapter-5
MS (m/z): 423.3 (M+ + H).
5. 1.5.1.7 {3-[2-Methoxy-4-(toluene-2-sulfonylaminocarbonyl)
benzyl]-1-methyl-1H-indol-5-yl} acetic acid cyclopentyl ester (4)
Propylphosphonic anhydride in dichloromethane solution (170
mL, 0.355 mol) was slowly added to the stirring suspension of
compound 60 (50 g, 0.118 mol), diisopropyl ethylamine (41.3 mL, 0.236
mol), o-toluene sulfonamide (24.2 g, 0.141 mol) and dichloromethane
(400 mL) at 28 °C and stirred at same temperature for 4 hours. The
resultant reaction mixture was quenched with water (500 mL), organic
phase and aqueous phase was separated. Solvent from organic phase
was evaporated; acetonitrile (200 mL) was added to the crude compound
and stirred at 27 °C for 45 minutes. Separated solid was filtered,
washed with methanol (50 mL) and dried under reduced pressure at 75
°C to afford 85 % of crude 4. Suspension of crude zafirlukast (20 g),
silica gel (40 g) and DCM (300 mL) were stirred at ambient temperature
for I hour and filtered the reaction mixture. Filtrate solvent was
evaporated, acetonitrile (240 mL) charged into crude compound and
stirred at 80 °C for 45 minutes. The resultant reaction mass was cooled

198 Chapter-5
to 28 °C and stirred at same temperature for 45 minutes. Separated
solid was filtered, washed with acetonitrile (60 mL) and dried under
reduced pressure at 75 °C to afford pure zafirlukast with more than
99.5% purity.
IR (KBr, cm–1): 3371 (amine, NH), 2960 (Ali, CH), 1690 (amide, C═O),
1340 & 1162 (asym. and sym, SO2).
1H NMR (400 MHz, CDCl3): δ 9.35 (br, 1H), 7.0-8.2 (m, 11H, Ar-H), 6.7
(s, 1H), 6.5 (s, 1H), 5.1−5.2 (m, 1H), 4.00 (s, 2H, CH2), 3.8 (s, 3H, CH3),
3.68 (s, 3H, CH3), 2.7 (s, 3H, CH3), 1.5−1.9 (m, 8H, CH2).
13C NMR (100 MHz, CDCl3): δC 20.0, 23.4, 23.4, 24.8, 32.3, 32.5, 55.2,
77.5, 109.1, 109.4, 109.7, 111.7, 115.2, 119.8, 126.0, 127.7, 128.0,
129.4, 129.7, 131.1, 132.1, 133.5, 134.0, 136.0, 136.7, 137.4, 154.3,
157.1, 164.8.
MS (m/z): 576 (M+ + H).

199 Chapter-5
5.7 REFERENCES
1. Phipatanakul, W.; Eggleston, P. A.; Conover-Walker, M. K.;
Kesavanathan, J.; Sweitzer, D.; Wood, R. A. Journal of Allergy and
Clinical Immunol., 2000, 105 (4), 704–710.
2. Bernstein, P. R.; Brown, F. J.; Matassa, V. G.; Yee, Y. K. US
4859692, 1989.
3. Matassa, V. G.; Thomas, P. M,; Jr., Howard, S.; Shapiro, Barrie,
H.; David, W. S.; David, A.; Robert, D. K.; Richard, A. K. J. Med.
Chem., 1990, 33, 1781–1790.
4. Arie, G.; Genndy, N.; Igor, Z.; Victor, P.; Maxim, S. WO 02/46153
A2, 2002.
5. Claire, L. A.; Ian, D.; Jonathan, D. M.; Jeffery, A. S. Org. Proc. Res.
& Dev., 2004, 8, 808–813.
6. Srinivas, K.; Srinivasan, N.; Krishna, M. R.; Reddy, C. R.;
Muthulingam. A.; Lalitha, R.; Reddy, K. S. R.; Reddy, B. S.; Reddy,
G. M.; Reddy, P. P.; Kumar, M. K.; Reddy M. S. N. Org. Proc. Res.
& Dev., 2004, 8, 952–954.
7. (a). Jiang, X.; Tiwari, A.; Thompson, M.; Chen, Z.; Cleary, T. P.;
Lee, T. B. K. Org. Process Res. Dev., 2001, 5, 604–608; (b). Laurila,
L. N.; Magnus, A. N.; Staszak, A. M.; Org. Process Res. Dev., 2009,
13, 1199–1201.
8. Anu, M.; Howard, S.; Mario, G.; John, C. M.; Tetrahedron Lett.,
2003, 44, 4589–4591.

200 Chapter-5
9. http://www.Rxlist.com
10. Robert, J.; Timko, R. J.; Bradway, A. C. US 5993859, 1999.
11. James, J. Holohan, I. J.; Edwards, R. J.; Timko, R. J.; Bradway, A.
C. US 5482963, 1996.
12. Holohan, J. J.; Edwards, I. J. US 5319097, 1994.
13. International Conference on Harmonisation of Technical
Requirements for Registration of Pharmaceuticals for Human Use
(ICH). Q3C (R3): Impurities: Guideline for Residual Solvents,
1997.

201 Summary & Conclusions
SUMMARY & CONCLUSIONS
CHAPTER-1
We have described importance of: (i) novel anti-diabetic agent
pioglitazone hydrochloride 1 (ii) anti- anginal drug Ranolazine 2, (iii)
anti- thrombotic drug clopidogrel bisulfate 3 (iv) anti-asthma drug
zafirlukast 4 and PDE5 inhibitor tadalafil 5. We further discussed
significance of impurity profile (Related substances) in the process
development.
CHAPTER-2
An efficient, new and robust synthetic method (two alternative
routes) for the preparation of pioglitazone 1 is developed and these are
more compatible on commercial scale. Our new synthetic methods to 1
are schematically described in Scheme 1 and scheme 2.
Scheme-1:
N
EtOH
TEA, TolueneN
EtOMs
K2CO3, Toluene
OHC
OH
N
Et
O
CHO
S
HNO O
Piperidine
MethanolN
Et
OS
NH
O
O
MeOH, DMF
5% NaOHN
Et
OS
NH
O
O
MsCl
7
8
9
10
11 1
100% 83%
75%
NaBH4, DMG
Co(NO3)2 . 6H2O
HCl
WaterN
Et
OS
NH . HCl
O
O
1.HCl
6
98%
90%

202 Summary & Conclusions
Scheme-2:
N
EtOH
TEA, TolueneN
EtOMs
K2CO3, Toluene
OHC
OH
N
Et
O
CHO
S
HNO O
N
Et
OS
NH
O
O
MsCl
7
8
9
10
1
100% 83%
HCl
Water N
Et
OS
NH . HCl
O
O
1.HCl
6
NaBH4
MeOHN
Et
O
12
OH
N
Et
O
13
ClHCl
DCMLiAlH4, THF
Possible potential impurities generated during the synthesis of
pioglitazone 1 and its key intermediate 11 are identified, synthesized
and controlled in pioglitazone 1. Structures of these impurities are
depicted in Figure 1.
Figure-1:
N
Et
N
Et
OS
NH
O
O
HOS
NH
O
O
HOS
NH
O
ON
Et
OH
OH
N
Et
OS
N
O
O
N
Et
O
CHO
9
14
11
15 16
18
17
19
N
Et
N
Et
OS
N
O
O N
Et

203 Summary & Conclusions
CHAPTER-3
An efficient, commercially viable and greener process for the
synthesis of ranolazine 2 has been disclosed in this chapter.
Additionally, comprehensive study on impurity profile of crude
ranolazine and its key intermediates including identification, synthesis
and characterization is presented in this chapter.
Synthesis of ranolazine 2 started from the commercially available
starting materials 2, 6-dimethyl aniline 20 and 2-methoxy phenol 25 as
described in Scheme 3.
Scheme-3:
O
OHOCl+
O
O
NaOHWater
O
Methanol
NH
HN
24
25 26
22
NH2
ClCl
O
+
TEADCM
HN
Cl
O
2
20 21
Methanol80%
N
NO
HN O
O
OH
27
HN
N
O NH
23
96%
97%77%
Based on the molecular weight obtained from LC-MS data,
reaction conditions and reagents/reactants used for reactions,
impurities generated during synthesis of 2 and its key intermediates
(Figure 2) were identified and characterized with spectral data. All these

204 Summary & Conclusions
impurities were controlled to below 0.05% level in final ranolazine 2 and
root causes for the formation of these impurities were discussed.
Figure 2:
O
O Cl
OH O
O OH
OHO
O O
OH O
28 29 30
HN
N
O NNH
O
HN
Cl
O
Cl
31 32
O
HO
O
N
NH
O
HO
O
N
N
O
OH
O
33
34
HN
N
O N N
OH N
HN
O
HN
N
O NOH
OHN
N
O NOH
OO
O
35
36 37
CHAPTER-4
Whilst there are many synthetic routes to clopidogrel bisulfate 3,
we focused on two which have potential for industrial manufacturing
with some limitations that are addressed in this chapter. Our efficient
and large scale processes to 3 are described in Scheme 4 and Scheme 5.

205 Summary & Conclusions
Scheme-4:
+
3.0 vol. toluene
1.65 eq. TEA. HCl
N
Cl
CO2CH3
. H2SO4S
HN
Cl
CO2CH3
S
OTs
S
OH
S
SO2Cl
CH3
38 40 42
3
41
1.5 vol. toluene
3.0 eq. K2HPO4
100-110 oC, 18 h
HCl, 67%
1. HCHO
25-35 0 C, 22 h
DCM
2. Acetone
0.92 eq. H2SO4
H2N
Cl
CO2CH3
39
Scheme-5:
S
N
CN
Cl
S
NH.HClOHC
Cl
S
N
CONH2
Cl
1.0 eq.NaCN
Water
3.0 eq. KOH,
t-Butanol
DMS, H2SO4
Methanol
+
60-65 °C, 2 h
100%
67%
water, Acetone. CSA
(S)-(+)-CSA. H2O
S
N
COOCH3
Cl
NaHCO3
DCM
H2SO4
2-Butanol
.H2SO4
S
N
COOCH3
Cl
43 44 45
46 47(±) 3
80-85 °C, 3h
92%
Acetone
.H2SO4S
N
COOCH3
Cl
3
Additionally, two unknown impurities generated during the
synthesis of 3 were identified, synthesized and characterized by spectral
data. Further, synthetic process for the preparation of the three USP
listed impurities along with the two new impurities (Figure 3) is
presented in this chapter.

206 Summary & Conclusions
Figure-3:
S
N
Cl
CO2CH3
S N
Cl
CO2CH3
S
N
Cl
CO2CH3
HO
S
N
Cl
CO2H
S
HN
Cl
CO2CH3
S
N
Cl
CO2CH3
48 49 50
51 42 52
CHAPTER-5
This chapter divided into two parts. In the first part (part-A) an
improved process for the synthesis for zafirlukast 4 is described. In the
second part (part B) an alternative synthetic approach for the
preparation of PDE5 inhibitor tadalafil 5 is presented.
Part A:
An improved process for the synthesis of zafirlukast 4 is
developed. This process is commenced with the commercially available
key raw material 3-methoxy-4-methyl benzoic acid 53 and N-methyl 5-
nitro indole 56 as described in Scheme 6. Usage of expensive and
pyrophoric reducing reagents like Raney nickel and Pd/C are replaced
with plant-friendly and low toxic reducing agent like sodium dithionite.
Purifications for removal of DCU (obtained as by-product in DCC
mediated coupling reactions) in crude Zafirlukast 4 is avoided using
commercially available less toxic coupling reagent T3P® instead of DCC.
207 Summary & Conclusions
Scheme-6:
OCH3
OCH3
H3C
O
OCH3
OCH3
O
Br
OH
OCH3
H3C
O
N
O2N
53 54 55
56
Methanol
N
O2N
57
OCH3
O
OCH3
CH3
N
H2N
58
OCH3
O
OCH3
CH3
N
HN
59
OCH3
O
OCH3
CH3
O
O
N
HN
60
OCH3
O
OH
CH3
O
O
N
HN
4
OCH3
O
NH
CH3
O
O
S
O O CH3
3 h, 98 % 84 %
85 %
1,4-dioxane
Cu2O
Sodium dithionate
Methanol, water
45 %
CPC, NMM
Toluene, 97 %
LiOH. H2O
Methanol
98 %
OTSA
DCM
CH3
SOCl2 DBDMH, AIBN
Cyclohexane
T3P, DMAP
86 %
Part B:
An alternative synthesis of tadalafil 5 starts with the coupling of
commercially available amino acid, D-tryptophan 61 and piperonal 62.
Coupling of D-tryptophan 61 with piperonal 62 under Pictet–Spengler
reaction conditions furnished β-carboline derivative 63 with the
diastereomeric mixture ratio 70(cis):30(trans), which was treated with
aqueous hydrochloric acid to get optically pure β-carboline derivative 64.

208 Summary & Conclusions
Diimidation of 64 with sarcosine ethyl ester hydrochloride under DCC
and HOBt conditions provided tadalafil 5 (scheme 7).
Scheme-7:
NH
O
OOHC Trifluoro acetic acid NH
NH
OO
CO2H
DCC, HOBt, TEA
85 %
61
5
NH
N
OO
N
O
O
CH3
+
62
63 (dr= 7:3)
Sarcosine ethyl ester HCl
NH2
O
OH
90 %
NH
NH
OO
CO2H
64
.HCl
Water, 1N HCl
Dichloromethane

209 Publications
LIST OF PUBLICATIONS
1. An Improved Process for Pioglitazone and Its Pharmaceutically
Acceptable Salt.
Lokeswara Rao Madivada, Raghupathi Reddy Anumala,
Goverdhan Gilla, Sampath Alla, Kavitha Charagondla, Mukkanti
Khagga, Apurba Bhattacharya and Rakeshwar Bandichhor.
Organic Process Research & Development 2009, 13, 1190–
1194.
2. An efficient synthesis of 1-(2-methoxyphenoxy)-2, 3-epoxypropane:
Key intermediate of β-blockers.
Lokeswara Rao Madivada, Raghupathi Reddy Anumala,
Goverdhan Gilla, Mukkanti Khagga and Rakeshwar Bandichhor.
Organic Process Research & Development 2012, 16,
1660−1664.
3. Alternative Synthesis of Tadalafil: PDE-5 Inhibitor.
Raghupathi Reddy Anumula, Lokeswara Rao Madivada,
Goverdhan Gilla, V. V. N. K. Prasad Raju, Mukkanti Khagga, Padi
Pratap Reddy, Apurba Bhattacharya and Rakeshwar Bandichhor.
Synthetic communications, 2008, 38, 4265-4271
4. An efficient and large scale synthesis of Clopidogrel: Antiplatelet
drug

210 Publications
Lokeswara Rao Madivada, Raghupathi Reddy Anumala,
Goverdhan Gilla, Mukkanti Khagga and Rakeshwar Bandichhor.
Der Pharma Chemica, 2012, 4 (1): 479-488.
5. An efficient and greener synthesis of Zafirlukast: Anti Asthma
drug.
Lokeswara Rao Madivada, Goverdhan Gilla, Mukkanti Khagga
and Rakeshwar Bandichhor.
Manuscript is ready for communication
6. An efficient and high-yielding synthesis of Ranolazine: Antianginal
drug
Lokeswara Rao Madivada, Mukkanti Khagga, and Rakeshwar
Bandichhor.
Presented in National seminar on recent advances in heterocyclic
chemistry (NSRACH 2011) organized by department of chemistry,
JNTUH college of engineering during November 4th and 5th, 2011.
7. An efficient and large scale synthesis of Clopidogrel: Antiplatelet
drug
Lokeswara Rao Madivada, Mukkanti Khagga and Rakeshwar
Bandichhor.
Presented in National seminar on recent research trends in chemical
sciences (RRTCS 2011) held on 23rd December, 2011 at Sri
Venkateswara University, Tirupati.



![CLSM B SLiCeS Improved Immunohistochemical … Preparation and Staining The protocol for preparation of hippocampal slice cultures is described elsewhere [5]. After](https://static.fdocuments.us/doc/165x107/5ada0e837f8b9aee348c39ba/clsm-b-slices-improved-immunohistochemical-preparation-and-staining-the-protocol.jpg)














![Classes/The River... · PERSPECTIVES The River Continuum Concept] ROBIN L. VANNOTE Stroud Water Research Center, Academy of Natural Sciences of Philadelphia, Avondale, PA 19311, USA](https://static.fdocuments.us/doc/165x107/5b90a2f109d3f2b86e8c7126/classesthe-river-perspectives-the-river-continuum-concept-robin-l-vannote.jpg)