
UNIVERSIDADE FEDERAL DE UBERLÂNDIA INSTITUTO DE FÍSICA ... · ... como requisito parcial para a...
48
UNIVERSIDADE FEDERAL DE UBERLÂNDIA INSTITUTO DE FÍSICA RENATO NEIVA SAMPAIO ESTUDOS ESPECTROSCÓPICOS EM MOLÉCULAS DE TETRAPIRIDIL PORFIRINAS TETRARUTENADAS UBERLÂNDIA 2008
Transcript of UNIVERSIDADE FEDERAL DE UBERLÂNDIA INSTITUTO DE FÍSICA ... · ... como requisito parcial para a...

UNIVERSIDADE FEDERAL DE UBERLÂNDIA
INSTITUTO DE FÍSICA
RENATO NEIVA SAMPAIO
ESTUDOS ESPECTROSCÓPICOS EM MOLÉCULAS DE TETRAPIRIDIL PORFIRINAS
TETRARUTENADAS
UBERLÂNDIA
2008

RENATO NEIVA SAMPAIO
ESTUDOS ESPECTROSCÓPICOS EM MOLÉCULAS DE TETRAPIRIDIL PORFIRINAS
TETRARUTENADAS
Orientador: Prof. Dr. Newton Martins Barbosa Neto
UBERLÂNDIA
2008
Trabalho de Conclusão de Curso apresentado
ao Instituto de Física da Universidade Federal
de Uberlândia, como requisito parcial para a
obtenção do título de bacharel em Física de
Materiais.

RENATO NEIVA SAMPAIO
ESTUDOS ESPECTROSCÓPICOS EM MOLÉCULAS DE TETRAPIRIDIL PORFIRINAS
TETRARUTENADAS
Prof. Dr. Newton Martins Barbosa Neto
Prof. Dr. Augusto Miguel Alcalde Milla
Prof. Dr. Djalmir Nestor Messias
UBERLÂNDIA
2008
Monografia apresentada como pré-requisito para obtenção do
título de Bacharel em Física de Materiais da Universidade
Federal de Uberlândia, submetida à aprovação da banca
examinadora composta pelos seguintes membros.

RESUMO
Neste trabalho, é apresentado o estudo espectroscópico das propriedades fotofísicas da
tetrapiridil porfirina base livre (H2TPyP) com a inserção de grupos periféricos de rutênio,
visando compreender a influência destes grupos periféricos sobre as propriedades fotosícas da
H2TPyP. As moléculas foram estudadas dissolvidas em clorofórmio. Através de técnicas
espectroscópicas como a absorção na região do UV-Vis e fluorescência, levantamos as
características fotofísicas das moléculas analisando a intensidade e largura das bandas
fornecidas pelos seus gráficos de absorção e emissão. A eficiência quântica de emissão e a
força do oscilador foram calculados para as moléculas afim de sistematizar ainda mais o seu
processo de caracterização. As medidas de absorção juntamente com o calculo da força do
oscilador revelam que a inserção dos grupos de rutênio favorece processos de absorção da
molécula enquanto que as medidas de fluorescência e o cálculo da eficiência quântica
demonstram o aparecimento de decaimento não radiativo, diminuindo a quantidade de luz
emitida, possivelmente via transferência de cargas.
Palavras-chaves: Tetrapiridil porifina, Absorção de UV-Vis, Lei de Beer-Lambert,
Fluorescência, Eficiência Quântica, Força do Oscilador.

4
Sumário
1. INTRODUÇÃO .............................................................................................................................. 5
2. FUNDAMENTOS TEÓRICOS ...................................................................................................... 6
2.1. Transições Eletrônicas ............................................................................................................. 6
2.2. Espectroscopia de Absorção e Lei de Beer-Lambert .............................................................. 9
2.3. Fluorescência ......................................................................................................................... 14
2.4. Eficiência Quântica ............................................................................................................... 16
2.5. Força do Oscilador ................................................................................................................ 18
3. MATERIAL E MÉTODOS .......................................................................................................... 28
3.1. Material ................................................................................................................................. 28
3.1.1. Porfirina ......................................................................................................................... 28
3.1.2. Rodamina ...................................................................................................................... 30
3.2. Métodos ................................................................................................................................. 33
4. RESULTADOS E DISCUÇÕES .................................................................................................. 35
4.1. Espectro de Absorbância ....................................................................................................... 35
4.2. Espectro de Fluorescência ..................................................................................................... 41
5. CONCLUSÕES ............................................................................................................................. 44
6. REFERÊNCIAS BIBLIOGRÁFICAS .......................................................................................... 45

5
1. INTRODUÇÃO
O estudo envolvendo materiais orgânicos com o objetivo de analisar suas propriedades
fotofísicas vem despertando bastante interesse no ramo científico e tecnológico, devido às
suas potenciais aplicações tecnológicas na indústria, tais como de telecomunicação, de laser, e
principalmente nas indústrias de que desenvolvem dispositivos fotônicos como led (light-
emitting-diode), display, limitadores ópticos, entre outros.
Este plano de trabalho faz parte de um projeto de caracterização óptica, tanto linear
quanto não linear, de moléculas orgânicas, visando o entendimento dos fatores que
influenciam em suas propriedades fotofísicas. Esse conhecimento servirá de suporte para o
desenvolvimento de novos materiais, via engenharia molecular, para aplicações tanto em
fotônica (chaves e limitadores ópticos) quanto em foto-medicina (desenvolvimento de drogas
fotosensibilizadoras para fototerapia dinâmica). Dentro deste plano de trabalho visamos o
estudo de propriedades fotofísicas da tetrapiridil porfirinas (TPyP), em solução, modificadas
pela adição de grupos periféricos de rutênio. Para isto empregamos, ao longo do projeto,
técnicas experimentais como a espectroscopia de absorção óptica, fotoluminescência e
cálculos envolvendo a eficiência quântica de fluorescência do material e a força do oscilador.
Estas moléculas se destacam devido à sua fácil manipulação estrutural e interessantes
propriedades ópticas. Além disso, estas formam o modelo molecular básico para drogas
fotosensibilizadoras em fototerapia dinâmica contra o câncer.

6
2. FUNDAMENTOS TEÓRICOS
2.1. Transições Eletrônicas
Dizemos que uma molécula está em um estado excitado quando esta, por exemplo,
absorve um fóton de energia apropriada promovendo um elétron de um orbital molecular de
menor energia para um orbital de maior energia.
Existem os orbitais σ e π. O orbital σ pode ser formado por dois orbitais atômicos s,
por um orbital atômico s e um p, ou também por dois orbitais atômicos p tendo um eixo
colinear de simetria, conforme a figura 1. Os elétrons deste orbital são chamados de elétrons σ
e quando excitados, são promovidos para um orbital mais energético antiligante σ*,
caracterizando a transição σ → σ*.
Figura 1: Esquema ilustrativo da sobreposição de orbitais atômicos formando a ligação σ e conseqüentemente o
orbital σ.
O orbital π é formado por dois orbitais atômicos sobrepostos lateralmente, de acordo
com a figura 2. Os elétrons presentes no orbital π são denominados elétrons π e quando estes
absorvem um fóton de energia específica, são promovidos para um orbital antiligante π*. Esta
transição é chamada π → π*.

7
Figura 2: Ilustração da formação da ligação π proveniente da sobreposição lateral de orbitais atômicos.
Existem também elétrons na molécula que não pertencem aos orbitais moleculares,
estes estão localizados nos heteroatomos, porém é convencionado pertencerem ao orbital
denominado orbital molecular n. Estes elétrons sofrem transições do tipo n → π* e n → σ*.
A energia envolvida nas transições citadas acima pode ser disposta em ordem
crescente da seguinte forma: ∗π→n < ∗→ ππ < *n σ→ < *σ→σ .
A figura 3 abaixo ilustra o diagrama das energias dos orbitais moleculares e das
transições eletrônicas possíveis entre eles.
Figura 3: Diagrama de energia dos orbitais moleculares e das possíveis transições eletrônicas.
Ainda em relação aos orbitais moleculares, estes podem ser classificados em relação à
sua ocupação. Podem ser HOMO (orbital atômico mais alto ocupado) e LUMO (orbital
atômico mais baixo ocupado). O HOMO refere-se ao estado fundamental, ou seja, pode ser σ,
π*
HOMO
LUMO
σ*
n(p) π
σ Estado Fundamental
σ→σ*
Ene
rgia
n→σ* π→π* n→π* σ→π*

8
π e n. Enquanto que o LUMO se refere ao orbitais mais energéticos, ou seja, os antiligantes
σ*, π*.
Podemos classificar os estados eletrônicos conforme sua configuração de spin em
tripleto e singleto. Tripleto, se a molécula possui par de spins emparelhados (dois spins up ou
dois spins down), já o singleto ocorre quando a molécula possui um spin up e um spin down
na sua camada de valência.
A absorção da radiação ultravioleta ou visível resulta geralmente da excitação dos
elétrons de ligação. Pode-se então relacionar os tipos de ligação da amostra em estudo com os
comprimentos de onda dos picos de absorção. Estes elétrons que participam do processo de
absorção de radiação eletromagnética de uma molécula orgânica são os envolvidos
diretamente na formação das ligações entre os átomos. No caso das porfirinas em particular,
os elétrons envolvidos diretamente no processo de absorção de radiação ultravioleta são os
elétrons π responsáveis pela formação das ligações duplas entre os carbonos (C=C ).
Todos os compostos orgânicos são capazes de absorver radiação eletromagnética
porque possuem elétrons nas camadas de valências que podem ser excitados a níveis
eletrônicos mais energéticos. A energia associada a excitações eletrônicas de ligações simples,
ou seja, σ, é mais alta (λ < 185 nm) enquanto que para as ligações duplas (π) essa encontra-se
em maiores comprimentos de onda (λ > 200 nm), de modo que a maioria das investigações
espectroscópicas de compostos orgânicos esta associada a comprimentos de ondas maiores
que 185 nm. A absorção de radiação UV-Vis com maiores comprimentos de onda está restrita
aos cromóforos, os quais possuem elétrons de valência com energias de excitação mais
baixas. Os espectros de absorção de compostos orgânicos contendo cromóforos são
relativamente complexos, pois resultam da superposição de transições vibracionais com
transições eletrônicas dando origem a uma combinação de linhas superpostas denominada de
banda.

9
2.2. Espectroscopia de Absorção e Lei de Beer-Lambert
A palavra absorção se refere ao processo em que uma molécula absorve um fóton de
energia apropriada, sendo promovida para um estado de maior energia a partir do estado
fundamental ou excitado. Em particular, neste último caso temos o que se denomina absorção
de estados excitados. O estado fundamental é definido como o estado onde a molécula se
encontra quando não interage com fatores externos.
A grandeza física que mede a eficiência de absorção para um comprimento de onda, λ,
é denominado de absorbância, A(λ), sendo definida como:
( ) ( )( )λ
λ=λ
I
IlogA
o
(1.1)
onde ( )λ0I é a intensidade de luz que entra na amostra e ( )λI a intensidade de luz que sai da
amostra. É importante frisar que na equação 1.1 estamos desprezando a perda de energia
luminosa por espalhamento e reflexão. Do ponto de vista prático isto é obtido quando
trabalhamos com amostras homogêneas e quando na medida de absorção a linha de base é
feita.
Outra grandeza usada para medir á eficiência de absorção é a transmitância T(λ). Esta
mede a quantidade de luz que é transmitida através da amostra, sendo matematicamente
expressa por:

10
( ) ( )( )λ
λ=λ
0I
IT (1.2)
onde a transmitância se relaciona com a absorbância através da relação:
( ) ( )λ−=λ TlogA ( 1.3)
Uma das maneiras de mensurarmos a capacidade de absorção de um dado material é
através da definição do coeficiente de absorção, a(λ). Este é definido como á razão da
absorbância por unidade de comprimento:
( ) ( )L
Aa
λ=λ (1.4)
onde L é o caminho óptico percorrido pela luz dentro do material. Como a absorbância é
adimensional, a(λ) é expresso em unidade de comprimento a menos um (m-1, cm-1 etc).
É comum também usarmos o coeficiente de absorção neperiano, α(λ), que é definido
como:
( ))λ(I
)λ(Iln
L
1=λα
0
(1.5)
Uma grandeza de importante valia no estudo de absorção pode ser obtida,
matematicamente, pela razão do coeficiente de absorção comum ou Neperiano pelo número
de moléculas absorventes por unidade de volume n, cujo nome é seção de choque de absorção
molecular σ(λ). Ela caracteriza a área de captura de fótons da molécula, cuja unidade de
medida é cm2.

11
( ) ( )n
λα=λσ (1.6)
Fenomenologicamente o processo de absorção em materiais é descrito pela lei de
Beer-Lambert (BL) a qual correlaciona matematicamente as grandezas já definidas. Para
deduzirmos a lei BL vamos considerar uma fina camada de material com espessura dL
contendo dN moléculas. Supondo que a concentração molar dessa solução seja igual a c e que
a frente de onda da luz possui uma seção de choque S podemos afirmar que o número de
moléculas no volume SdL é de:
cSdLNdN a= (1.7)
onde Na é o número de Avogrado.
A seção de choque de absorção total no caminho óptico dL é dada pela soma das
seções de choque de absorção molecular, isto é, σ.dN. A probabilidade de um fóton ser
absorvido é σ.dN / S que é igual a fração de luz absorvida pela camada fina (-dI/I); Logo:
dLcNS
dN
I
dIa σ=
σ=− (1.8)
integrando temos:
∫∫ σ=−L
0
a
I
I
dLcNI
dI
0
(1.9)

12
LcNI
Iln a
0 σ= (1.10)
que pode ser reescrita como:
LcN303,2
1
I
Ilog a
0 σ= (1.11)
Denominamos, ( )λε=σaN3,2
1, de coeficiente de absorção molar, temos:
( )Lc)(A λε=λ (1.12)
onde, ( )λε é dado em L mol . cm-1.
A equação (1.12) é conhecida como lei de Beer-Lambert, e mostra a relação linear da
absorbância em função da concentração molar. No entanto quando há formação de agregado
ou presença de outras substâncias absorventes essa relação não é mais valida.
Para medirmos a absorção de uma amostra utilizamos dispositivos denominados de
espectrofotômetros. Estes são instrumentos compostos, em geral, por um conjunto de
componentes do seguinte tipo: uma fonte de luz, componentes ópticos que guiam a luz até a
amostra, compartimento de amostra, e um ou mais detectores que medem a intensidade dessa
radiação, veja figura 4.

13
Figura 4: Esquema de um espectrofotômetro.
A fonte emissora (lâmpada) é responsável por emitir o feixe de luz. A lâmpada
utilizada foi de tungstênio que emite um espectro que cobre a região espectral que vai de 200
nm a 800 nm.
A segunda estrutura física presente no espectrofotômetro são seus componentes
ópticos. Como estamos trabalhando na região espectral do UV-Vis, estes instrumentos são
sempre dispersivos, o qual se trata de uma grade de difração. A finalidade deste instrumento é
o de difratar a luz de modo que diferentes comprimentos de onda incidam sobre a amostra
permitindo que se determine sua absorbância em cada um destes comprimentos. Este conjunto
de dados é o que chamamos de espectro de absorção.
Figura 5: Espectro visível de uma fonte de luz branca difratada por uma grade de difração.
Os detectores utilizados no espectrofotômetro podem ser desde fotomultiplicadoras até
arranjo de diodos. Este arranjo consiste em uma série de detectores fotodiodos posicionados
lado a lado em um cristal de silício, de modo que cada comprimento de onde difratado pela
grade atinja um ponto deste arranjo, e conseqüentemente um detector. A radiação que
atravessa a amostra é total e instantaneamente analisada pelo detector determinando-se a
absorbância em todos os comprimentos de onda.

14
Neste trabalho, utilizamos a espectroscopia de absorção na região UV-Vis, por
apresentarem as moléculas orgânicas, nesta região, bandas de absorção eletrônica intensas.
Tal fato é responsável por uma série de propriedades fotofísicas que fazem das porfirinas
sistemas moleculares extremamente interessantes. Especificamente, empregamos a
espectroscopia de UV-Vis para obtermos informações de como os grupos laterais de rutênio
alteram a absorção da TPyPs.
2.3. Fluorescência
Processos de relaxação do estado excitado para o estado fundamental via emissão de
fótons dão origem ao fenômeno da luminescência. Estes podem ser de dois tipos: 1)
fluorescência ou 2) fosforescência. No primeiro a luz é emitida a partir da relaxação entre dois
estados de mesma multiplicidade (em geral, singleto-singleto), já no segundo a emissão de
fótons ocorre a partir da relaxação entre estados de multiplicidade diferente (tripleto-singleto).
Neste trabalho estudamos a fluorescência emitida por moléculas de H2TPyP em solução de
clorofórmio.
A fluorescência estudada aqui é causada da relaxação S1 – S0 da molécula de porfirina.
Uma propriedade geral da fluorescência, a menos de algumas exceções, é que sua emissão
ocorre a partir do estado S1, e suas características não dependem do comprimento de onda de
excitação.

15
Figura 6: Dinâmica de excitação e relaxação de níveis e/ou bandas eletrônicas
Uma vez excitada para os níveis eletrônicos e vibracionais mais altos, o excesso de
energia é rapidamente dissipado deixando o fluoróforo (entidade responsável pela emissão na
molécula) no menor estado de energia vibracional de S1. A relaxação dos níveis eletrônicos
mais altos para o menor estado de energia vibracional S1 ocorre em cerca de 10-12 s, e devido a
esse curto tempo o espectro de emissão é considerado independente do comprimento de onda
de excitação.
O espectro da fluorescência está localizado em maiores comprimentos de onda, ou
seja, baixas energias, do que o espectro de absorção, por causa da perda de energia no estado
excitado devido às relaxações vibracionais.
Um ponto interessante a se observar é que para a grande maioria dos fluoróforos, o
espectro de emissão é uma imagem da absorção S0 – S1, este fato é conhecido como a regra do
espelho. A natureza simétrica desses espectros é o resultado das mesmas transições
envolvidas na absorção e emissão e as similaridades das energias vibracionais dos níveis S0 e
S1.
Quanto ao regime temporal, as medidas de fluorescência podem ser classificadas em
dois tipos: 1) estado estacionário ou 2) resolvido no tempo. A primeira forma é aquela
executada com uma iluminação e observação constantes. A amostra é iluminada com um
feixe de luz contínuo, sendo registrado seu espectro de emissão. Já na fluorescência resolvida
no tempo a amostra é submetida a um pulso de luz, onde a largura do pulso é menor do que o
tempo de decaimento da amostra. O decaimento da intensidade é registrado com um sistema
Sn
S1
S0

16
de detecção de alta resolução temporal que permite a detecção dos processos de decaimento.
No nosso caso, trabalhamos com medidas de estado estacionário.
2.4. Eficiência Quântica
Quando um fluorofóro absorve um fóton de luz, um estado excitado é ocupado. A
energia absorvida pelo fluorofóro é perdida logo em seguida, devido à instabilidade energética
dos estados excitados, e este retorna ao estado fundamental. Os principais mecanismos de
perda da energia absorvida são a fluorescência (perda de energia pela emissão de um fóton),
conversões internas, relaxações vibracionais (perda de energia não-radioativa, via geração de
calor) e cruzamento intersistemas.
A eficiência quântica da fluorescência (Φ) é a razão dos fótons emitidos pelos fótons
absorvidos. Em outras palavras a eficiência quântica nos fornece a probabilidade do estado
excitado decair emitindo fótons entre estados de mesma multiplicidade.
Um dos métodos mais empregados para se obter Φ é o método comparativo de
Williams [4], o qual se utiliza amostras de referência bem caracterizada com Φ conhecido.
Uma vez que Φ para a amostra de referência é conhecida, pode-se obter Φ para a amostra que
se quer medir; porém é necessário levar em conta certas considerações: é preciso trabalhar
cuidadosamente dentro de um dado intervalo de concentração para evitar certos efeitos como
o de filtro interno e garantir a validade da Lei de Beer-Lambert. Caso sejam usados solventes
diferentes para a referência e para a amostra, deve-se levar em consideração os índices de
refração dos solventes nos cálculos; a amostra de referência deve ser escolhida de modo que
ela absorva no comprimento de onda de excitação escolhido para o experimento e se possível,

17
que tenha emissão em uma região similar a da amostra a qual queremos medir sua eficiência
quântica.
Portanto, para processos mediados pela absorção de um único fóton e trabalhando com
concentrações que garantam a não formação de agregados moleculares, nem o surgimento de
efeitos de transferência de energia radiativa, temos que a área do espectro é proporcional a
potência da luz que excita amostra sendo escrita como:
PA β= (2.1)
onde A é a área do espectro de emissão, P a potência do laser de excitação. β é o coeficiente
angular que depende, dos seguintes parâmetros [4]: 1) do índice de refração do solvente (n), 2)
da absorbância da amostra para o comprimento de onda de excitação (Absexc), 3) da eficiência
quântica de emissão (Φ) e 4) da geometria do aparato experimental utilizado (G),
Φ⋅⋅⋅=β exc2 AbsnG (2.2)
Fazendo o gráfico da área do espectro de fluorescência em função da potência de
excitação para a amostra que se deseja medir a eficiência quântica e a solução referência
temos respectivamente:
P)AbsGn(A
amo
amoexcamo
2amoamo 444 3444 21
β
Φ= (2.3a)
P)AbsGn(A
refe
refeexcrefe
2referefe 444 3444 21
β
Φ= (2.3b)

18
onde os coeficientes angulares são obtidos a partir da regressão linear dos dados
experimentais.
Tomando a razão entre os coeficiente angulares temos:
refexcref
2ref
amoexcamo
2amo
ref
amo
Absn
Absn
Φ⋅⋅
Φ⋅⋅=
β
β (2.4)
sendo o fator geométrico é o mesmo para as duas amostras. Então a partir da equação 2.4,
temos:
refexcamo
2amo
excref
2ref
ref
amoamo Absn
AbsnΦ
⋅
⋅⋅
β
β=Φ (2.5)
a qual é empregada para determinarmos a eficiência quântica de emissão da amostra, uma vez
que todos os parâmetros do lado direito da equação 2.5 são conhecidos.
2.5. Força do Oscilador
A força do oscilador é um número adimensional introduzido para especificar as
probabilidades das transições eletrônicas e representar a fração de osciladores atômicos
efetivos para cada freqüência ressonante.
Classicamente, a força do oscilador é usada como um indicador estatístico para o
numero relativo de osciladores em cada freqüência ressonante. Quanticamente, a força do
oscilador proporciona a obtenção da “intensidade” relativa entre as transições eletrônicas em

19
sistemas atômicos e moleculares. É um importante parâmetro usado para calcular os
coeficientes A e B de Einstein, probabilidades de transições, elementos da matriz de momento
de dipolo, concentrações de impurezas e coeficientes de absorção. Além disso, é bastante
usado na validação de modelos teóricos.
Os passos a seguir referem-se ao desenvolvimento clássico para se encontrar a força
do oscilador.
Levando em consideração que a maioria das propriedades ópticas de sólidos pode ser
entendidas utilizando a teoria eletromagnética clássica, partiremos das equações de Maxwell
em um meio dielétrico.
i) ���� · ���� � 0 iii) ���� �� � � � ����
ii) ���� · ��� � 0 iv) ���� ���� � �������
onde, ���� � ���� � ���, ��� � �����, ���� � �� ��� e que � � ��.
Utilizando as equações acima podemos derivar a equação de onda. Aplicando o
rotacional na equação iii), temos.:
���� ����� ��� � ������� ��� �!
���� · ����� · ��� � ����"�� � � ��! #�
������! $
����"�� � �� �"���!"
����"�� � ��� �%&����% � 0 (3.1)
A equação 3.1 nos fornece a equação de uma onda em um meio dielétrico, a qual
possui solução do tipo ���', ! � ������)*�+,-./� e 0, � 0 � 10′ � 2���3.

20
Tratando o sistema como sendo um dipolo, onde o movimento do elétron é descrito
pela equação de um oscilador harmônico amortecido forçado, temos:
45 � 645 � 3�"4 � � 78�� (3.2)
Assumindo que o campo elétrico aplicado varia harmonicamente em função do tempo
e que o movimento do elétron também siga este comportamento, temos uma função posição
do elétron que pode ser dada por 4 � 4�).*/�. Derivando duas vezes e substituindo na
equação 3.2, obtemos:
4 � � 78 9 &��
�/:%./%�.*;/< (3.3)
Onde 6 é o parâmetro de amortecimento, 3� é a freqüência ressonante do oscilador, )
é a carga do elétron e = a massa do elétron.
Em um meio dielétrico e isotrópico temos que o elétron está sujeito a uma polarização
� � �>)4 (onde N é a densidade eletrônica) quando se encontra deslocado de uma distancia
x do equilíbrio. Deste modo,, substituindo a equação 3.3 na polarização obtemos:
� � ?7%8 9 &��
�/:%./%�.*;/ < (3.4)
Utilizando a relação ��� � �����, encontramos:
� � ?7%@:8 9 �
�/:%./%�.*;/ < (3.5)

21
No estudo da interação da radiação com a matéria, a suscetibilidade elétrica é um
parâmetro de extrema importância e a partir dele podemos encontrar �:
���� � ��� ���� � ���� � ��� ��� � ����� A � � ���1 � �
� � �� � ?7%8 9 �
�/:%./%�.*;/ < (3.6)
Substituindo a equação 3.6 na equação 3.1, temos:
����"�� � �C% D1 � ?7%
@:8 9 ��/:%./%�.*;/ <E �%&��
��% � 0 (3.7)
E tomando �� � �)*�+,-./� como solução, encontramos:
�0,�" � /%C% 91 � ?7%
@:8 F ��/:%./%�.*;/ G< (3.8)
Da relação 0, � 2���3, obtemos que:
0, � 3H 21 � �
Para gases e em geral para sistemas diluídos, � I 1, de modo que podemos expandir a
relação acima em séries de Taylor e pegando até o primeiro termo da expansão, obtemos:
0, � JH K1 � �
2M

22
Com isso é possível substituir a equação 3.5 na relação acima e obtermos:
0, � /C 91 � ?7%
"@:8 F ��/:%./%�.*;/ G< (3.9)
Separando as partes real e imaginária de 0,:
0, � /C 91 � ?7%
"@:8 F �/:%./%��/:%./%�%.�;/ % G � 1 ?7%
"@:8 F �;/ �/:%./%�%.�;/ % G< (3.10)
Sabendo que 0, � 0 � 10′, obtemos:
0 � /C 91 � ?7%
"@:8 F �/:%./%��/:%./%�%.�;/ % G< (3.11)
0 ′ � /C 9 ?7%
"@:8 F �;/ �/:%./%�%.�;/ % G< (3.12)
O problema é que na realidade não existe apenas uma freqüência ressonante 3�.
Existem diferentes osciladores com diferentes constantes de mola, de modo que passamos a
ter uma série de freqüências ressonantes 3�N. Assumimos então que uma fração ON de
osciladores no material terão uma freqüência ressonante 3�N, ou seja, ON é um peso estatístico.
Denominado de força do oscilador, o valor f é então o número efetivo de elétrons por átomo
ou molécula para uma transição particular.
0, � /C #1 � ?7%
"@:8∑ QRK/:R% ./%M.*;R/ N $ (3.13)

23
0 � /C #1 � ?7%
"@:8∑ K/:R% ./%MQRK/:R% ./%M%.�;R/�%N $ (3.14)
0 ′ � /C # ?7%
"@:8∑ �;R/�QRK/:R% ./%M%.�;R/�%N $ (3.15)
Nosso objetivo agora é derivar relações que nos permitam calcular a força do oscilador
por meio de quantidades mensuráveis como a energia e o coeficiente de absorção.
Há uma relação possível de se obter entre a Lei de Beer-Lambert e a potência
absorvida pelo oscilador, que mais tarde nos fornecerá a força do oscilador. Para isso
desejamos no momento calcular a energia e a potencia absorvida pelo oscilador.
ST � U� · S4� � �)V�C�! WXW� S! (3.16)
� � WYW� � �)V�C).*/�4Z (3.17)
Onde as equações 3.16 e 3.17 fornecem a energia e a potência instantânea absorvida
pelo oscilador respectivamente. Substituindo a equação 3.3 na equação 3.17 obtemos a
potência instantânea. Importante observar que precisamos calcular é media temporal de
potência, já que esta é o que realmente medimos experimentalmente.
[ � \� �] ^ �S!�_]
� � 7%&`ab% /%;"8cd�/; %_�/:%./%�%e (3.18)
Considerando que o campo tenha uma distribuição espectral uniforme de energia sobre
todas as freqüências próximas de 3�, podemos inserir um determinado peso espectral dado
por f/, que é constante próximo à freqüência de ressonância.

24
V�C" � gV�C" �3 f/S3
Desde que a equação 3.3 seja linear, podemos usar a superposição de cada
componente espectral de " para obtermos a potencia total absorvida sobre todas as
freqüências.
[ ����hV \� ^�/S3 � ^ 7%&`ab% /%;"8cd�/; %_�/:%./%�%eS3 � i7%&`ab% jk
"8c (3.19)
Este resultado se aplica para um oscilador clássico simples, mas estamos lidando com
transições em um conjunto de osciladores quânticos. Podemos usar do principio da
correspondência com o nosso resultado se admitirmos que um sistema quântico simples
possui diversas freqüências ressonantes com a força do oscilador distribuída entre as
transições, ou seja, um peso estatístico. Assim, cada transição age como um oscilador em
particular, portanto a soma entre elas é valida. Podemos então relacionar a absorção de uma
única transição com a absorção total.
�hlmn!o= � O �hlmopH. Hrnpp. � O s)"V�C" f/2=7
Em termos da potência absorvida por unidade de volume, temos:
tuvwx�V. � >O i7%&`ab% jk
"8c (3.20)

25
Considere a Lei de Beer-Lambert dada por:
y � y�).zX
yhlm � y��1 � ).zX
Convertendo para a forma diferencial:
Syhlm � y�).zX{S4 | W}uvwWX � y{
W}uvwWX � Wtuvw
W~ � y{
Para um sistema real, a transição terá uma faixa de freqüências a qual irá absorver,
{ � {�3 , temos então que considerar apenas a lei de Beer-Lambert em cada componente
espectral da intensidade. Podemos realizar isso utilizando uma intensidade espectral, como
visto anteriormente.
yS3 � y�S3 ).z�/ X � y�f/).z�/ X
�hlmS� � gy�3 S3 {�3 S3
Como estamos interessados apenas em um S4 sobre o qual cada oscilador experiência
a intensidade, y�3 � y��3 sobre S4.
tuvwW~ � yf/ ^{�3 S3
yf/ ^{�3 S3 �>O i7%&`ab% jk"8c (3.21)

26
Usando a relação entre intensidade e o vetor de Poyting e considerando que y� � y, temos:
�y�� � ������ � �] ^ ��� �����S! � ~@&%
"�_]�
y � C@:&%" � C@&b%
"� (3.22)
Substituindo a equação 3.22 na equação 3.21, obtemos:
^{�3 S3 �>O i7%�8cC@ d&`ab%
&b% e (3.23)
Para continuarmos, precisamos encontrar a expressão para o nosso campo local.
V�C � C � ��V � �7h� (3.24)
Onde C é o campo sobre todo o volume, ��V é o campo devido a polarização e �7h�
é o campo estático próximo aos íons. ��V é dado por ��V � � ���ε:.
�C � ��C � �C
�C � �C | �C � C�� � �� ��V � � &b�@.@:
�@: (3.25)

27
Para o efeito de íons locais, temos que saber a natureza dos íons no ponto de simetria
do oscilador. Lorentz achou que �7h� � 0. Combinando as equações 3.24 e 3.25:
V�C � C �@_"@: �@: (3.26)
Substituindo a equação 3.26 na equação 3.23, obtemos:
g{�3 S3 �>O s)"��� � 2�� "9=7H��"�
Assumindo um material não magnético, temos quem � � �� e � � ���", portanto
obtemos que:
O � �8cC@:?7%
����%_" % ^{�� S� (3.27)
Onde O é a força do oscilador, =7 e ) são a massa e a carga do elétron
respectivamente, > é a densidade eletrônica, � é o índice de refração do solvente e {�� é o
coeficiente de absorção da molécula em função da freqüência.

28
3. MATERIAL E MÉTODOS
3.1. Material
3.1.1. Porfirina
As porfirinas são moléculas tanto encontradas na natureza como sintetizadas em
laboratório em uma grande variedade de formas, sendo sua estrutura básica formada por um
anel macrocíclico conhecido como anel porfirínico. Este anel é constituindo por quatro grupos
pirróis ligados entre si por átomos de carbono (denominados mesocarbonos) através de pontes
metínicas, e estruturalmente estabilizado por um substituinte central ligado aos átomos de
nitrogênio dos grupos pirrólicos. Por possuírem alto comprimento de conjugação π, as
porfirinas apresentam uma série de propriedades ópticas não lineares, tais como absorção de
dois fótons e refração não linear, e uma larga banda de absorção na região do UV-Vis, bem
com emissão proveniente de estados excitados singleto e tripleto. Além disso, por
apresentarem fácil manipulação estrutural, as moléculas de porfirina se tornam boas
candidatas a uma série de aplicações, que vão desde dispositivos fotônicos, até drogas
fotossensibilizadoras utilizadas em tratamentos contra o câncer. No mais, estas apresentam
considerável importância biológica e espectroscópica em diversos processos como
fotossíntese, transporte de oxigênio, oxidação-redução e transporte de elétrons.
Em particular, a tetrapiridil porfirina possui quatro anéis piridínicos ligados aos
mesocarbonos e distorcidos do plano que contem o anel porfirínico, sendo sua estrutura
apresentada na figura 7.

29
Figura 7: molécula de tetrapiridil porfirina.
O estudo das propriedades fotofísicas e fotoquímicas de porfirinas desperta a atenção
da comunidade científica devido principalmente à sua forte absorção de luz na região do
visível o que permite caracterizações por uma grande variedade de técnicas espectroscópicas e
às suas propriedades ópticas de estados excitados.
Em relação a suas propriedades fotofísicas, as porfirinas podem ser definidas como
regulares ou irregulares. Porfirinas regulares têm os seus espectros de absorção e emissão
determinados essencialmente pelos elétrons π do anel porfirínico, sofrendo apenas uma
pequena perturbação devido ao substituinte central. São exemplos desse tipo de porfirina: a
porfirina base livre (2H+), e as metaloporfirinas cujo íon central possui camada externa
fechada (camadas d0 ou d10). Íons metálicos que possuem essa propriedade encontram-se nos
grupos IA, IIA, IIIA, IIB, IIIB, IVB, VB da tabela periódica. Metaloporfirinas dos grupos
IVA e VA, com altos estados de oxidação, também possuem uma configuração de camada
fechada sendo, portanto, classificadas como porfirinas regulares. São elas: Si(IV), Ge(IV),
Sn(IV), Pb(IV), P(V), As(V), Sb(V). As porfirinas irregulares são formadas por íons
metálicos de transição com camadas d e f incompletas. Nestas porfirinas os elétrons das
camadas semi-preenchidas interagem fortemente com os elétrons π do anel, através do

30
acoplamento spin-órbita, provocando uma perturbação significativa nas propriedades
fotofísicas destas, em comparação com as regulares. A absorção e emissão são alteradas e os
tempos de vida de estado excitado tornam-se mais curtos, em função do aparecimento de
níveis de transferência de carga. Além disso, estes níveis também fazem com que as porfirinas
irregulares apresentem, ao contrário das regulares, baixo sinal de fluorescência. Neste
trabalho, as porfirinas H2TPyP são classificadas como regulares.
3.1.2. Rodamina
Rodamina é um nome genérico para uma família de compostos orgânicos, corantes
chamados fluoronas (estrutura básica de várias substâncias químicas, de fórmula química
C13H10O). Sua estrutura molecular básica é mostrada na Figura 8. Devido a sua alta eficiência
quântica de emissão, estas são empregadas como meio ativo em lasers de corante, sendo
espectroscopicamente detectável ao redor de 610 nm quando usada como corante laser . Além
disso, são extensivamente usadas em aplicações biotecnologicas tais como a microscopia de
fluorescencia . Corantes do tipo rodamina são geralmente tóxicos, e são soluveis a água,
metanol e etanol. São exemplos de rodamina a Rodamina 6G e Rodamina B.

31
Figura 8: Molécula de Rodamina B
A Rodamina B, também chamada de Rosa Rodamina B, Rodamina 610 ou Violeta
Básico (C.I. 45170) é usada em biologia como um corante fluorescente em coloração
citológica, também usada como corante básico no tingimento de papel, madeira e derivados
de celulose. Além de usada como referência no ramo cientifico para experimentos que
envolvem o cálculo de eficiência quântica de materiais. Neste trabalho estas foram
empregadas como material de referência nas medidas de eficiência quântica de fluorescência
da porfirinas tetrarutenadas. Da literatura obtivemos o valor da eficiência quântica para a
Rodamina B diluída em água, a qual é nossa referência, e é de 31% excitando a amostra em
514nm.
Em seguida, temos o espectro de absorção para a Rodamina B.
450 500 550 600 650
0,00
0,02
0,04
0,06
Ab
sorb
ancia
Comprimento de Onda (nm)
Figura 13: Espectro de absorção da Rodamina B.
32
O espectro de absorção da Rodamina B nos mostra que este material possui uma larga
banda de absorção na região do visível, tendo um pico bem definido em 555nm e um outro
em aproximadamente 514nm. Podemos observar então que a Rodamina B absorve em 514
nm, que é o comprimento de onda de excitação das amostras escolhido para a realização de
nosso experimento, condição essencial para podermos realizar os cálculos da eficiência
quântica.
550 600 650 700 750
0
100
200
300
Inte
nsid
ad
e
Comprimento de Onda (nm)
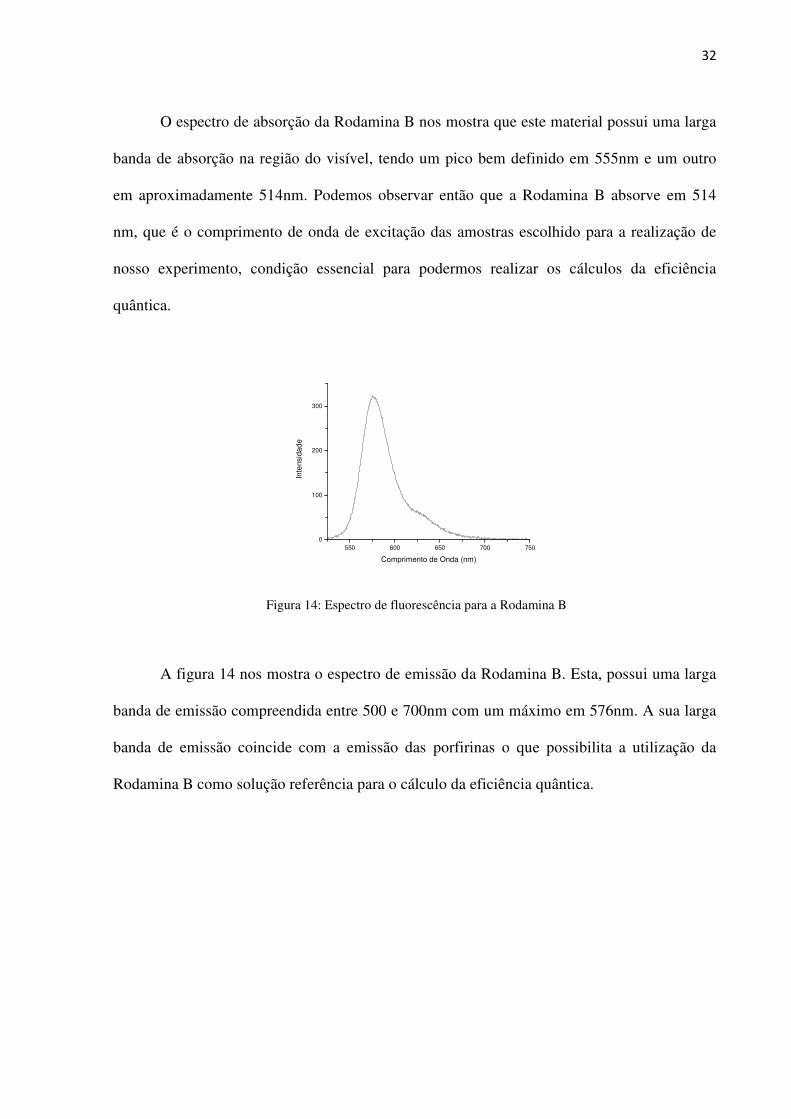
Figura 14: Espectro de fluorescência para a Rodamina B
A figura 14 nos mostra o espectro de emissão da Rodamina B. Esta, possui uma larga
banda de emissão compreendida entre 500 e 700nm com um máximo em 576nm. A sua larga
banda de emissão coincide com a emissão das porfirinas o que possibilita a utilização da
Rodamina B como solução referência para o cálculo da eficiência quântica.

3.2. Métodos
Todas as porfirinas, tanto base livre quanto as rutenadas foram sintetizadas pela equipe
do Prof. Dr. Alzir Azevedo Batista do Departamento de Química da Universidade Federal de
São Carlos de acordo com
porfirínico mais quatro moléculas de piridina ligadas aos mesocarbonos. Na Figura
mostramos a estrutura molecular das amostras estudadas neste trabalho.
Figura 9: Acima temos o anel porfirínico, onde a letra R representa os diferentes tipos de substituintes na posição
dos meso-carbonos. São eles: 1)
(3 – Ru). A amostra de H2TPyP, sem grupos de rutênio é denominada de 0
Foram estudados os efeitos de três grupos diferentes de Rutênio sobre as propriedades
fotofísicas da H2TPyP. São eles: Grupo1:
Grupo 3: RuCl2(PPh3)2CO
respectivamente. A porfirina sem grupo de rutênio será denominada de 0
As soluções foram preparadas diluindo
B em água. Todas as medidas foram realizadas a temperatura ambiente, com as soluções
Todas as porfirinas, tanto base livre quanto as rutenadas foram sintetizadas pela equipe
do Prof. Dr. Alzir Azevedo Batista do Departamento de Química da Universidade Federal de
São Carlos de acordo com a referência [16]. Sua estrutura básica é formada pelo anel
porfirínico mais quatro moléculas de piridina ligadas aos mesocarbonos. Na Figura
mostramos a estrutura molecular das amostras estudadas neste trabalho.
: Acima temos o anel porfirínico, onde a letra R representa os diferentes tipos de substituintes na posição
carbonos. São eles: 1) RuCl (dppb)(N-N) (1 – Ru); 2) RuCl2(dppb)CO (2 –
TPyP, sem grupos de rutênio é denominada de 0 – Ru.
Foram estudados os efeitos de três grupos diferentes de Rutênio sobre as propriedades
TPyP. São eles: Grupo1: RuCl (dppb)(N-N); Grupo 2: RuCl
. que serão a partir de agora denominados de 1
respectivamente. A porfirina sem grupo de rutênio será denominada de 0
As soluções foram preparadas diluindo-se as porfirinas em clorofórmio e a Rodamina
B em água. Todas as medidas foram realizadas a temperatura ambiente, com as soluções
33
Todas as porfirinas, tanto base livre quanto as rutenadas foram sintetizadas pela equipe
do Prof. Dr. Alzir Azevedo Batista do Departamento de Química da Universidade Federal de
. Sua estrutura básica é formada pelo anel
porfirínico mais quatro moléculas de piridina ligadas aos mesocarbonos. Na Figura 9
mostramos a estrutura molecular das amostras estudadas neste trabalho.
: Acima temos o anel porfirínico, onde a letra R representa os diferentes tipos de substituintes na posição
– Ru) e 3) RuCl2(PPh3)2CO
Foram estudados os efeitos de três grupos diferentes de Rutênio sobre as propriedades
; Grupo 2: RuCl2(dppb)CO e
. que serão a partir de agora denominados de 1-Ru, 2-Ru e 3-Ru
respectivamente. A porfirina sem grupo de rutênio será denominada de 0-Ru.
as em clorofórmio e a Rodamina
B em água. Todas as medidas foram realizadas a temperatura ambiente, com as soluções

34
colocadas em uma cubeta de vidro para fluorescência, com quatro janelas polidas, com 1cm
de caminho óptico. Os espectros de absorção no UV-Vis foram obtidos com um
espectrômetro UV 1650 PC Shimadzu. Os espectros de fluorescência foram obtidos com um
espectrofotômetro portátil Ocean Optics USB 2000, sendo a amostra excitada por um laser de
argônio refrigerado a ar (module laser da Edmund Optics), em 514 nm. As medidas foram
obtidas em configuração de espalhamento a 90o.
Para medirmos a eficiência quântica de emissão seguimos a discussão contida na seção
2.4, utilizando como padrão amostra de Rodamina B, excitando a amostra em 514 nm com
potências diferentes. Variamos a potência do laser modificando a corrente da fonte, tomando
o cuidado de esperar a cavidade estabilizar antes de obtermos cada espectro. Isto é feito para
garantirmos que não temos alteração na forma de linha do espectro de fluorescência (largura
de banda, razão entre os picos, posição dos picos etc.) devido a flutuações da fonte de
excitação.
A força do oscilador foi obtida utilizando a relação definida na seção 2.5. A partir dos
espectros de absorção fizemos uma modificação em suas unidades, transformando o
comprimento de onda em freqüência e a absorbância em coeficiente de absorção molar em
função da freqüência. Com isso é possível utilizar da relação existente entre a área do gráfico
���� 4 �� e a força do oscilador.
35
4. RESULTADOS E DISCUSSÕES
4.1. Espectro de Absorbância
O espectro de absorção da H2TPyP é caracterizado por uma forte absorção na região
UV-Vis sendo formado basicamente por duas bandas principais: a banda B ou banda de Soret,
localizada próxima a região 400 nm, e banda Q compreendida entre 470 e 700 nm. Estas
bandas são causadas por transições π- π* dos elétrons presentes no anel porfirínico. Na figura
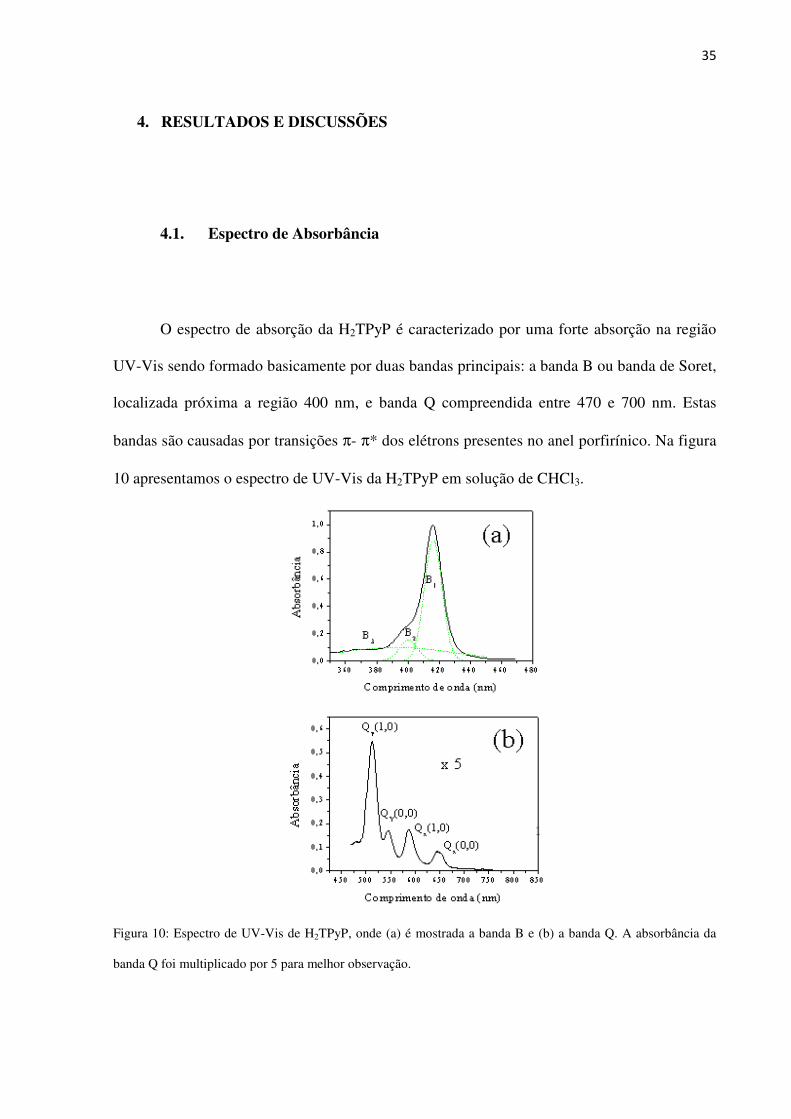
10 apresentamos o espectro de UV-Vis da H2TPyP em solução de CHCl3.
Figura 10: Espectro de UV-Vis de H2TPyP, onde (a) é mostrada a banda B e (b) a banda Q. A absorbância da
banda Q foi multiplicado por 5 para melhor observação.
36
A banda B se origina da transição S0→S2, de acordo com o digrama na figura 6, sendo
uma transição altamente permitida com alto valor de seção de choque de absorção (∼10-13
cm2). De acordo com Gouterman esta transição ocorre do orbital molecular a1u (HOMO) para
o orbital eg (LUMO) [17].
Devido a simetria do anel, as porfirinas base livre apresentam 4 sub-bandas na banda
Q, denominadas de Qy(1,0), Qy(0,0), Qx(1,0) e Qx(0,0), como indicado na figura 10b, sendo
estas da ordem de 20 vezes menos intensa do que a banda de Soret. Em ordem crescente de
absorbância estas são organizadas, para a solução de H2TPyP, como:
Qy(1,0)>Qx(1,0)>Qy(0,0)>Qx(0,0). As sub-bandas Qy são devido à transição b2g←b1u
enquanto as Qx são devido à transição b3g←b1u. A banda Q corresponde ao estado singleto de
menor energia (S1).
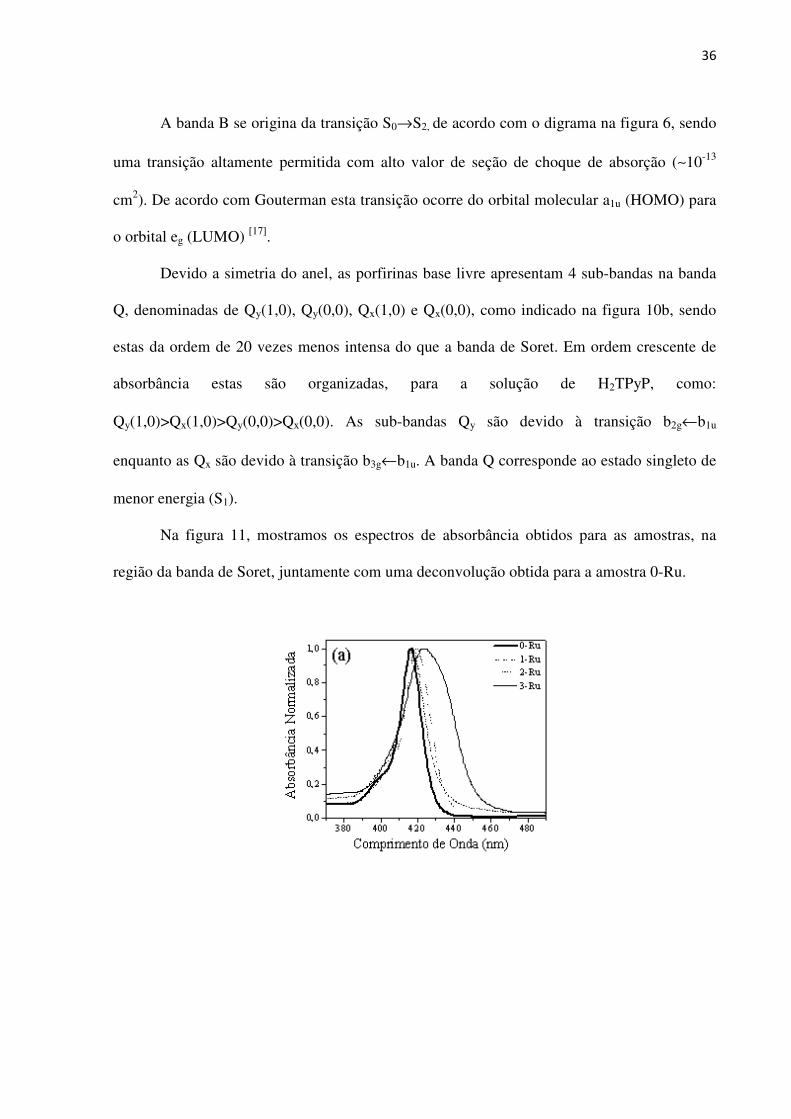
Na figura 11, mostramos os espectros de absorbância obtidos para as amostras, na
região da banda de Soret, juntamente com uma deconvolução obtida para a amostra 0-Ru.

37
Figura 11 – (a) Banda de Soret e (b) deconvolução da banda de Soret obtida para amostra 0-Ru.
Vemos da figura 11a que a adição dos grupos de Rutênio provoca tanto um
deslocamento para o vermelho quanto o alargamento da banda de Soret, em especial para a
amostra 3-Ru.
Da Figura 11b, vemos que a deconvolução da banda de Soret mostra que esta é
formada por três sub-bandas, que são associadas a transições de sub-níveis vibracionais
superpostos. Esta característica é observada para todas as amostras estudadas, independente
da adição dos grupos periféricos de rutênio. Os ajustes foram realizados pela deconvolução de
três gaussianas denominadas de B1, B2 e B3. Utilizou-se o ajuste gaussiano devido a solução
ser inomogênia. Na tabela 1, apresentamos as características das sub-bandas. Nas tabelas 1.a,
1.b, 1.c e 1.d se encontram os dados referentes aos valores para a força do oscilador para a
banda B e suas sub-bandas.

38
Tabela 1: Dados espectroscópicos da banda B.
Porfirina Banda B1 Banda B2 Banda B3
λc (nm) ∆ω (nm) λc (nm) ∆ω (nm) λc (nm) ∆ω (nm)
0 – Ru 404(1) 18.21 417(4.3) 11.6 427(0.4) 9.1
1 – Ru 404(1) 17.9 417(3.4) 12.4 423(1.24) 18.3
2 – Ru 405(1) 17.1 419(4.4) 13.3 428(1.05) 13.7
3 – Ru 410(1) 20.4 420(1) 12.1 430(2.85) 23.2
Os valores entre parênteses são as intensidades relativas das sub-bandas em relação a sub-banda B1.
Tabela 1.a: Força do oscilador para a 0 - Ru.
0 - Ru (Base Livre)
Banda B
0,013770855
Banda B1 Banda B2 Banda B3
0,003322509 0,005665928 0,00048087
Tabela 1.b: Força do oscilador para a 1 - Ru.
1 - Ru
Banda B
0,021674496
Banda B1 Banda B2 Banda B3
0,001218831 0,010289158 0,002786868

39
Tabela 1.c: Força do oscilador para a 2 - Ru.
2 - Ru
Banda B
0,014787141
Banda B1 Banda B2 Banda B3
0,002383449 0,002763922 0,005036467
Tabela 1.d: Força do oscilador para a 3 - Ru.
Nota-se que a adição dos grupos de rutênio favorece a transição associada à sub-banda
B3, o que pode ser verificado pelo aumento da intensidade relativa desta, em relação a
H2TPyP, além de provocar o seu alargamento, observando a largura a meia altura ∆ω notamos
que esta possui um grande aumento. Este fato explica o porque do alargamento e
deslocamento da Banda de Soret devido a adição de grupos de rutênio. A força do oscilador
apresentada para as porfirinas mostra que a inserção dos grupos de rutênio favorece as
transições envolvidas no processo de absorção da radiação eletromagnética, principalmente
em relação a sub-banda B3 da banda de Soret, a força do oscilador apresenta um aumento da
ordem de 10 quando comparada com a porfirina base livre sem grupos de rutênio, podendo
associar esse aumento quantitativo à mudanças qualitativas como aumento da intensidade e
largura de banda.
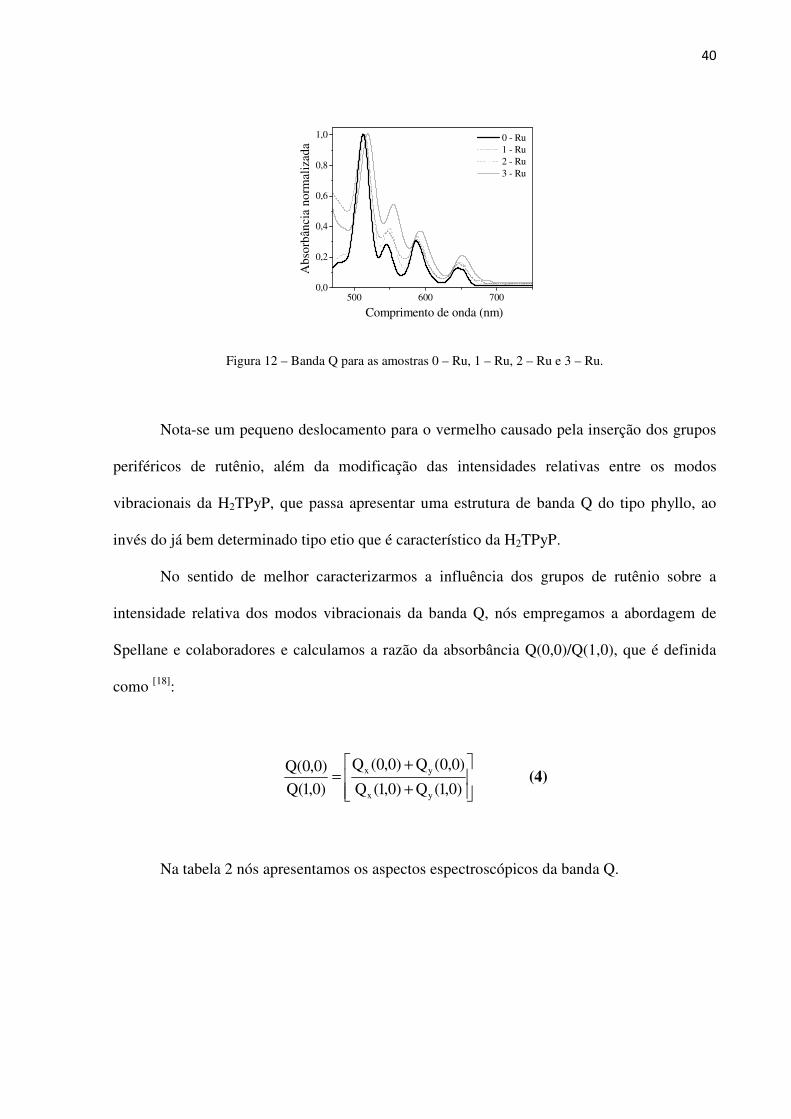
Na figura 12 apresentamos a região espectral que compreende a Banda Q das amostras
estudadas.
3 - Ru
Banda B
0,018868392
Banda B1 Banda B2 Banda B3
0,001798623 0,003888092 0,005474114
40
500 600 7000,0
0,2
0,4
0,6
0,8
1,0
Abs
orbâ
ncia
nor
mal
izad
aComprimento de onda (nm)
0 - Ru 1 - Ru 2 - Ru 3 - Ru
Figura 12 – Banda Q para as amostras 0 – Ru, 1 – Ru, 2 – Ru e 3 – Ru.
Nota-se um pequeno deslocamento para o vermelho causado pela inserção dos grupos
periféricos de rutênio, além da modificação das intensidades relativas entre os modos
vibracionais da H2TPyP, que passa apresentar uma estrutura de banda Q do tipo phyllo, ao
invés do já bem determinado tipo etio que é característico da H2TPyP.
No sentido de melhor caracterizarmos a influência dos grupos de rutênio sobre a
intensidade relativa dos modos vibracionais da banda Q, nós empregamos a abordagem de
Spellane e colaboradores e calculamos a razão da absorbância Q(0,0)/Q(1,0), que é definida
como [18]:
+
+=
)0,1(Q)0,1(Q
)0,0(Q)0,0(Q
)0,1(Q
)0,0(Q
yx
yx (4)
Na tabela 2 nós apresentamos os aspectos espectroscópicos da banda Q.

41
Tabela 2: Dados espectroscópicos da banda Q.
Porfirina Qy(1,0) Qy(0,0) Qx(1,0) Qx(0,0) [Q(0,0)]/[Q(1,0]
0 – Ru 513(1) 545(0.28) 588(0.31) 645(0.13) 0.31
1 – Ru 513(1) 547(0.35) 588(0.31) 647(0.16) 0.39
2 – Ru 515(1) 549(0.39) 589(0.33) 647(0.17) 0.42
3 – Ru 519(1) 555(0.54) 592(0.37) 650(0.21) 0.55
Os valores entre parênteses são as intensidades relativas da banda Q em relação a banda Qy(1,0).
Verificamos que a adição de rutênio interfere na banda Q aumentando de forma
substancial a razão da absorbância o que implica que estes favorecem as transições (0,0). As
intensidades relativas mostram que isto ocorre tanto para as sub-bandas x quanto para as y. É
importante frisar que os dados espectroscópicos da banda Q para a amostra 0 – Ru estão em
excelente acordo com os dados reportados anteriormente na literatura.
4.2. Espectro de Fluorescência
Abaixo, na figura 15, mostramos um espectro de emissão para a H2TPyP em
clorofórmio, enquanto que na Tabela 3 estão os dados quantitativos dos máximos do espectro
com seus respectivos comprimentos de onda, a intensidade relativa entre os dois picos e a
eficiência quântica das porfirinas.

42
600 650 700 750 800
0
100
200
300
400
Inte
nsid
ae
Comprimento de Onda (nm)
Figura 15: Espectro de emissão da H2TPyP.
Tabela 3: Dados espectroscópicos de emissão das porfirinas
Porfirina Q(0,0) Q(1,0) Φ�
0 – Ru 650(1,0) 713(0,51) 0,014
1 – Ru 653(1,0) 715(0,48) 0,007354
2 – Ru 652(1,0) 714(0,54) 0,003076
3 – Ru 656(1,0) 717(0,46) -----
Os valores entre parênteses são as intensidades relativas dos picos de emissão em relação a banda Q(0,0).
A banda de emissão da porfirina está localizada na região entre 600 nm e 800 nm
apresentando dois picos bem definidos, centrados em torno de 659 nm e 713 nm. Estes picos
são relacionados às transições ν=0 → ν=0 (659nm) e ν=0 → ν=1 (713nm). O deslocamento
das bandas não chega a ser tão sistemático quanto a diminuição da intensidade de
fluorescência ao adicionarmos grupos de rutênio ao anel porfirínico. Para a medida 3-Ru a
queda da intensidade chega a ser tão crítica, ainda que com uma potência elevada para o laser,
que foi descartado o cálculo de sua eficiência quântica pois não conseguimos desta forma
garantir a forma de linha do espectro de fluorescência (largura de banda, razão entre os picos,
posição dos picos etc.) devido a flutuações da fonte de excitação.

43
Os espectros de fluorescência das quatro porfirinas são visivelmente similares
possuindo pequenos deslocamentos das bandas conforme a adição dos grupos de rutênio. A
intensidade relativo entre os picos pode ser disposta na seguinte ordem 2-Ru > 0-Ru > 1-Ru >
3-Ru.
Como foi dito anteriormente, na seção 2, para o cálculo da eficiência quântica
precisamos obter o parâmetro � presente no gráfico A x P dos espectros de fluorescência das
porfirinas e da Rodamina B. Abaixo segue um gráfico demonstrando este procedimento,
lembrando que não pegamos uma medida em particular para a Rodamina B, cada medida com
as porfirinas foi seguida com uma para a Rodamina B, assim temos 3 padrões para o
parâmetro �, cada uma correspondente a uma porfirina em particular. A figura 16 demonstra
quantitativamente este processo.
Pode-se observar que para nas porfirinas Base-livre, 1-Ru, 2-Ru a disposição dos
pontos para o ajuste linear são bem melhores quando comparado à 3-Ru. Isso é devido ao
sinal detectado para a fluorescência. O procedimento de variação da potência com a 3-Ru não
foi satisfatório, pois mesmo atingindo a potência máxima do laser não obtivemos uma forma
da linha do espectro de emissão confiável, sujeito a muitas variações como largura de banda e
posição dos picos, provavelmente devido a flutuações da fonte de excitação de modo que a
ocorrência de erros no cálculo da eficiência quântica torna-se razoavelmente grande,
inviabilizando o cálculo. A concentração das soluções já se encontrava próxima ao limite
aceitável pela Lei de Beer-Lambert sem possibilidade então de ampliarmos o sinal de
fluorescência, com isso foi descartada as medidas para a porfirina 3-Ru.
Resta-nos então apenas as porfirinas base livre, 1-Ru e 2-Ru para calcularmos a
eficiência quântica. Os índices de refração do clorofórmio e da água são 1,4459 e 1,332988
respectivamente e o valor da eficiência quântica para a Rodamina B segundo a literatura é de
31% (0,31).
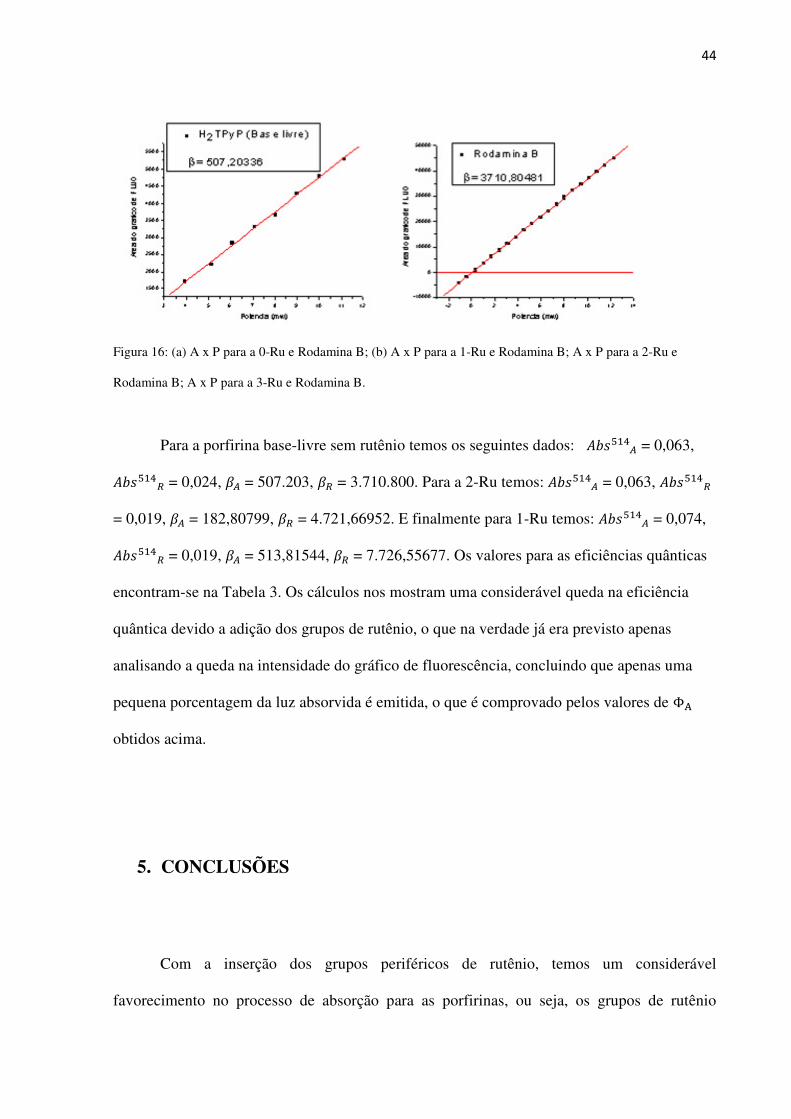
44
Figura 16: (a) A x P para a 0-Ru e Rodamina B; (b) A x P para a 1-Ru e Rodamina B; A x P para a 2-Ru e
Rodamina B; A x P para a 3-Ru e Rodamina B.
Para a porfirina base-livre sem rutênio temos os seguintes dados: ��p���� = 0,063,
��p���� = 0,024, �� = 507.203, �� = 3.710.800. Para a 2-Ru temos: ��p���� = 0,063, ��p����
= 0,019, �� = 182,80799, �� = 4.721,66952. E finalmente para 1-Ru temos: ��p���� = 0,074,
��p���� = 0,019, �� = 513,81544, �� = 7.726,55677. Os valores para as eficiências quânticas
encontram-se na Tabela 3. Os cálculos nos mostram uma considerável queda na eficiência
quântica devido a adição dos grupos de rutênio, o que na verdade já era previsto apenas
analisando a queda na intensidade do gráfico de fluorescência, concluindo que apenas uma
pequena porcentagem da luz absorvida é emitida, o que é comprovado pelos valores de Φ�
obtidos acima.
5. CONCLUSÕES
Com a inserção dos grupos periféricos de rutênio, temos um considerável
favorecimento no processo de absorção para as porfirinas, ou seja, os grupos de rutênio

45
modificam a estrutura do anel porfirínico de modo que suas transições eletrônicas envolvidas
no processo de absorção também são alteradas, gerando canais de maior absorção. Enquanto
que no processo de relaxação pela perda de energia por fluorescência, inserção de grupos
periféricos de rutênio no anel porfirínico cria um fluxo de carga retirando parte da energia,
proveniente do processo de absorção, do anel porfirínico para os grupos periféricos, gerando
assim canais de decaimento não radioativos. Com isso a proporção entre a luz absorvida e
emitida é reduzida, o que explica a redução da eficiência quântica das porfirinas rutenadas
quando comparadas com a base livre.
6. REFERÊNCIAS BIBLIOGRÁFICAS
[1] J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum
Press, New York, 1999, Second Edition.
[2] J. C. Scaiano (Ed.), Handbook of Organic Photochemistry, CRC Press, 1989.
[3] J. R. Lakowicz, Topics in Fluorescence Spectroscopy Vol.1, Kluwer Academic/ Plenum
Publishers, 2002.
[4] D. F. Eaton, Reference Materials for Fluorescence Measurement, Pure & Appl. Chem.,
Vol. 60, No. 7, pp. 1107-1114, 1998.

46
[5] K. Kalyanasundaram, Photochemistry of Water-Soluble Porphyrins: Comparative Study of
Isomeric Tetrapyridyl- and Tetrakis (N-Methylpyridiniumyl) porphyrins, Inorg. Chem. 1984,
23, 2453-2459.
[6] Xuezhong He, Guangming Xia, Yalin Zhou, Manhua Zhang, Tao Shen, Comparative
study of photophsical properties of isomeric tetrapyridyl- and tetra- (N-
hexadecylpyridiniumyl) porphyrins, Spectrochimica Acta Part A 55 (1999) 873-880.
[7] B. Valeur, “Molecular Fluorescence: principles and applications”, Wiley-VHC, New
York, (2002)
[8] P. W. Atikins, “Physical Chemistry”, Oxford University Press, 6o Edition, Oxford.
[11] H. H. PerKampus, “UV-Vis spectroscopy and its applications”, Springer Laboratory,
New York (1992).
[12] N. M. Barbosa Neto, L. De Boni, J. J. Rodrigues Jr., L. Misoguti, C. R. Mendonça, L. R.
Dinelli, A. A. Batista, S. C. Zílio, “Dynamic saturable optical nonlinearities in free base
tetrapyridylporphyrin”, J. Porphyr. Phthalocya. 7, 452-456 (2003).
[13] K. Kalyanasundaram, “Photochemistry of polypyridine and porphyrin complexes”,
Academic Press, San Diego (1992).
[14] C. Thiel, “A Discussion of Oscillator Strenghts and Related Issues”, Cone Laboratory –
Physics Department, Montana State University.

47
[15] M. A. Linne, “Spectroscopy Measurement – An Introduction to the Fundamentals”,
Academic Press (2002)
[16] L. R. Dinelli, “Estudo das Propriedades Estruturais e Desenvolvimento de Eletrodos
Modificados de Novas Porfirinas Polimetaladas”, Tese de Doutorado apresentada ao
Departamento de Química de Universidade Federal de São Carlos (UFSCar).
[17] M. Gouterman, “Spectra of Porphyrins”, Journal of Molecular Spectroscopy, Volume 6 ,
p. 138-163.
[18] P.J. Sepllane, M. Gouterman, A. Antipas, S. Kim, and Y.C. Lin, Inorg. Chem., 19, 386
(1980)


















