
UCSC MS bioinformatics report 2010
20
Introduction Sanger sequencing revolutionized biological and medical research and next‐ generation sequencing is revolutionizing it again. A single biological sample, that is sequenced by a next‐generation sequencing platform, such as SOLiD, produces 30 million oligonucleotides or reads. But to make sequencing efficient, dozens of samples are sequenced simultaneously, producing over a billion reads in a single run of the sequencing equipment. Thus, two questions arise: One is how to store all the data and the other is what to do with all the data? How to store all the data is a problem that is being tackled by those who host large computer clusters, whether stationary or in a cloud. Yet analyzing the millions of reads and designing state‐of‐ the‐art bioinformatics tools with which to analyze these data is becoming quite a challenge for bioinformaticists, clinicians and biologists alike. For instance, who is asking what biological and clinical questions and who is answering them creates new roles for researchers. For my rotation project, I built a website and analyzed data that began to address these issues. The website uploaded SOLiD sequencer read files associated with the picoeukaryote organism micromonas, strain RCC299 and a reference genome for the organism and analyzed the data for a given number of samples. I used the unmasked reference assembly genome from the Department of Energy’s Joint Genome Institute. The RCC299 strain’s genome has 17 chromosomes plus a chloroplast chromosome. I analyzed the reads corresponding to the 17 chromosomes. The envisioned output on the website was a multi‐read visualizer of the 24 samples of micromonas. Materials and Methods I established a pipeline of computer programs in order to derive the results. Figure 1 illustrates this pipeline. A suite of software programs created by others was also used to derive the results. The first software program used was BWA (Barrows‐ Wheeler Alignment), a program that came from the James Durbin laboratory, Cambridge University, UK. This “short read alignment to a large reference genome” program allowed for mismatches and gaps [Li, 2009]. Bowtie, produced by S. Salzberg’s group at the University of Maryland, was another software program that was used to map the SOLiD reads to a reference genome [Langmead, 2009]. One shortcoming with both BWA and Bowtie that they did not report all reads: “Whenever bowtie reports a subset of the valid alignments that exist, it makes an effort to sample them randomly” (Bowtie manual, [Langmead, 2009]). In other words, while BWA simply reports only the first read that maps to a specific genomic position, throwing away the other reads, Bowtie randomly selects one read among all reads that map to a specific genomic position. There are options, however, to have Bowtie report aligned reads but this subset of reads does not have the genomic position given with them. The final mapping software tool to be used was Bfast, produced at UC Los Angeles, CA in the laboratory of Stanley Nelson [Homer, 2009]. Bfast output contained all reads, mapped or not.
-
Upload
elinor-velasquez -
Category
Documents
-
view
16 -
download
2
Transcript of UCSC MS bioinformatics report 2010

Introduction
Sanger sequencing revolutionized biological and medical research and next‐generation sequencing is revolutionizing it again. A single biological sample, that is
sequenced by a next‐generation sequencing platform, such as SOLiD, produces 30 million oligonucleotides or reads. But to make sequencing efficient, dozens of
samples are sequenced simultaneously, producing over a billion reads in a single
run of the sequencing equipment. Thus, two questions arise: One is how to store all the data and the other is what to do with all the data? How to store all the data is a
problem that is being tackled by those who host large computer clusters, whether
stationary or in a cloud. Yet analyzing the millions of reads and designing state‐of‐the‐art bioinformatics tools with which to analyze these data is becoming quite a
challenge for bioinformaticists, clinicians and biologists alike. For instance, who is asking what biological and clinical questions and who is answering them creates
new roles for researchers.
For my rotation project, I built a website and analyzed data that began to address
these issues. The website uploaded SOLiD sequencer read files associated with the
picoeukaryote organism micromonas, strain RCC299 and a reference genome for the
organism and analyzed the data for a given number of samples. I used the unmasked
reference assembly genome from the Department of Energy’s Joint Genome Institute. The RCC299 strain’s genome has 17 chromosomes plus a chloroplast
chromosome. I analyzed the reads corresponding to the 17 chromosomes. The
envisioned output on the website was a multi‐read visualizer of the 24 samples of
micromonas.
Materials and Methods
I established a pipeline of computer programs in order to derive the results. Figure
1 illustrates this pipeline. A suite of software programs created by others was also used to derive the results. The first software program used was BWA (Barrows‐
Wheeler Alignment), a program that came from the James Durbin laboratory,
Cambridge University, UK. This “short read alignment to a large reference genome”
program allowed for mismatches and gaps [Li, 2009]. Bowtie, produced by S.
Salzberg’s group at the University of Maryland, was another software program that was used to map the SOLiD reads to a reference genome [Langmead, 2009]. One
shortcoming with both BWA and Bowtie that they did not report all reads:
“Whenever bowtie reports a subset of the valid alignments that exist, it makes an effort to sample them randomly” (Bowtie manual, [Langmead, 2009]). In other
words, while BWA simply reports only the first read that maps to a specific genomic position, throwing away the other reads, Bowtie randomly selects one read among
all reads that map to a specific genomic position. There are options, however, to
have Bowtie report aligned reads but this subset of reads does not have the genomic position given with them. The final mapping software tool to be used was Bfast,
produced at UC Los Angeles, CA in the laboratory of Stanley Nelson [Homer, 2009].
Bfast output contained all reads, mapped or not.

The other software tools that were used were SamTools, a set of tools used to
analyze reads that have been mapped to a reference genome, BEDTools, a set of tools also used to produce analyses of reads that have a been mapped to a reference
genome, and PicardTools, a set of tools that change the file format of read files, among other possibilities.
Figure 1. Flowchart of methodology for analysis of reads: The pre‐filter steps are
applied sequentially to raw reads. The pre‐filtering technique applied to the raw
reads resulted in a very high quality set of reads that were then mapped to the reference genome of micromonas. The mapping steps applied to the pre‐filtered
reads resulted in a careful read count for exons, introns, intergenic regions and rRNA, along with unmapped reads.
I wrote a number of software programs in order to filter and prepare read files for their analysis. The reads were pre‐filtered before mapping to a reference genome.
First, I removed reads with more than one ‘wildcard’ position. That meant that if a
read had an unknown value at any base, that read was discarded. Next, a ‘floating
window’ was prepared to assess the quality values in each read. If in a window of
five reads, the quality values for each read averaged to 10 or less, that read was trimmed at the base. Thus low quality reads were trimmed. Next the P2 adapter was
matched against each read to see if any of the reads had P2 adapter bases. If so, that
read was removed. Lastly, if a read was 35 bases or less, that read was discarded.

Next, the set of filtered reads for each sample was converted from two files (the colorspace file and the quality values file) into a single file with fastq file format.
Then, the reads were mapped to a set of poly‐T, C, G, A reads and all SOLiD adapters. If a read mapped to this set, it was discarded. The reads were then mapped to a set
of rRNA for the micromonas. The reads that mapped to the set of rRNA were
counted and then removed from the larger set of reads. Finally, BWA/Bowtie/Bfast was applied to the set of reads in order to map them to the micromonas genome. I
used BWA to map the reads of all 24 samples to the micromonas genome. Finding
that these results were unsatisfactory, I used Bowtie and Bfast to map to the micromonas genome. For BWA, the output was a set of mapped reads in SAM file
format. I converted the SAM files to BAM files using the SamTools and then converted BAM files to BED files using the BEDTools. For Bfast output, I had two
sets: One with mapped reads and one with non‐mapped reads. For the set of
unmapped reads, the output file format was in BAF file format. I used one tool from
the suite of Bfast tools to convert the BAF file format to SAM file format. I then used
a tool from PicardTools to convert the SAM file to a fastq file format. This fastq file of
unmapped reads was mapped against the micromonas genome for the second time
and the output was used for counting mapped reads and unmapped reads. I fed the
set of second unmapped reads into Bfast for a third time and used those counts of mapped and unmapped reads. The Bfast mapped reads were in SAM format. Using
the suite of tools from SamTools and BEDTools, the Bfast mapped reads were
converted to BAM and then BED file format for each sample.
For the BWA set of mapped reads, I was able to use the BEDTools software tool “intersectBed” which examines two files and determines the intersection of those
two files and counts the number of times a read from one set intersects with the
second set. A BED file is a collection of genomic positions. Thus each mapped read in each sample was converted into genomic coordinates. Next, I downloaded each
chromosome GenBank file for micromonas. I created a set of all exons from the
GenBank file of CDS coordinates for micromonas exons, by writing a computer
program that isolated each exon’s start and stop genomic position and converting
that into a BED file. Then, I intersected the BED file of mapped reads with the BED file of exons to get a count of number of reads which intersected with micromonas
exons. This means that I counted the number of reads which overlapped with the
exon genomic regions in the micromonas genome.
To create a set of micromonas introns, I used the GenBank files again. I wrote a program that found the set of introns associated to a genomic consecutive pair of
exons in each gene in each chromosome. I intersected the set of introns with the set
of mapped reads and recorded the counts. I used the GenBank files to create a set of intergenic regions for the BWA mapped reads. However, after discussion with
Marcus Breese from Indiana University, I decided that the counts for the intergenic
regions were those counts of mapped reads that remained after subtracting the counts for the exons and the introns. This technique I used for the Bfast mapped
reads. I was able to compute the exons and introns for each Bfast file for seven of the

samples (See Figure 3). I also mapped the total reads against the chloroplast
genome of micromonas as well as the genome of another strain of micromonas and the E. Coli genome in order to test for contamination.
To create Figures 4 – 28, I wrote a computer program that counted the number of
mapped reads at a given base for a specified chromosome of a reference genome.
The program’s output was the number of counts of mapped reads and the given genomic position of the base. Only nonzero counts were outputted. I created Figures
2 – 28 using the R graphics package.
Additionally, I built a website, http://inspired.soe.ucsc.edu, in which a user could
perform the above calculations for SOLiD reads for the micromonas organism, namely, mapping SOLiD reads to a reference genome and additionally output those
reads in an R software program computed graph which plotted counts against
genomic coordinates for the mapped reads. The user simply uploaded files of
colorspace data along with quality values for each read from the SOLiD sequencer,
along with an uploaded reference genome. The analysis was created through a
series of webpages that allowed the user to choose which mapping tool to a
reference genome they wanted to use and what they wanted to do once they had
mapped the SOLiD reads. The R software package was linked to the website so that the graphs could be produced.
Results and Discussion
I produced a website which could analyze and display the samples. Figures 2 – 28
were produced on a Mac computer. Figure 4 was partially produced after a number of attempts: The Mac computer froze and the figure crashed midway through the
figure’s production. Thus a snapshot of the figure was taken before the figure
crashed.
The goal of the project was to create a visualization tool for viewing the
micromonas’ samples. The resulting Figures 2 – 3 were created to show the
percentage of exons, introns, rRNA, intergenic regions and unmapped rRNA
computed using the SOLiD data of micromonas. Figures 4 – 28 were created in order to show how the website’s viewer looked when the data was analyzed using the
website. The idea was that the user could click on which samples to display or have
all the samples display. Figures 4 – 28 are known as “bedgraphs,” nomenclature used in discussions of the UCSC genome browser. Viewing the Figures 4 – 28 is most
interesting. The reader can see that the genomic patterns change according to the sample displayed. It is clear that there are different conditions that the organism
underwent to create these diverse patterns.
Another useful visualization was “pileups,” namely displaying reads against the
genome. Since the resulting analysis of the data produced files in Bed formats, the
files could in theory be visualized using the micromonas browser. However after a discussion with Larry Meyer, of UC Santa Cruz, it was determined that these Bed
formatted files would overload the micromonas browser as well as the UCSC

browser (if the UCSC browser contained a reference genome for micromonas). A
Bed formatted file had annotations for each genomic region that results from the analysis. It would be possible to place in the annotation the number of reads for a
given genomic region, thus permitting a type of pileup. This remains future work for the website.
One crucial point for the analysis of the SOLiD data: I will use the A15_01 sample to illustrate the point. The total number of raw reads is 12,422,404. After the pre‐
filtering, the number of reads was 9,400,465. Bfast mapped 3,936,114 reads to the
micromonas reference genome. After running through Bfast twice, a total of 3,938,207 reads were produced. However, the preliminary Bed formatted file
contained only 700,606 unique genomic regions or mapped reads (prior to use of the intersectBed program). Thus, a number of mapped reads was not being counted
or were being combined to create unique reads. I could have adjusted the number of
exons, introns, rRNA and intergenic regions by multiplying the numbers by a
common factor of 3938207/700606. This would assume a uniform distribution for
each reported read. That is, each mapped read could be assumed to have
approximately 5.5 copies of that identical read. Doing the multiplication, the number
of exons (876,813 x 5.5) equaled 4,822,471, which was greater than the 3,938,207
mapped reads. Thus, there was a non‐uniform distribution of reads. The best way around this problem would be to map the pre‐filtered reads against the set of exons
(and, also introns) rather than the whole genome in order to get an accurate count
of the exon (and introns) reads.
Bfast worked in the following way. It identified CALs or candidate alignment locations, known as genomic regions in our nomenclature, for each read. If no CAL
was found for a read then that read was unmapped. Another issue was the
possibility that some unmapped reads may have been copies of mapped reads.
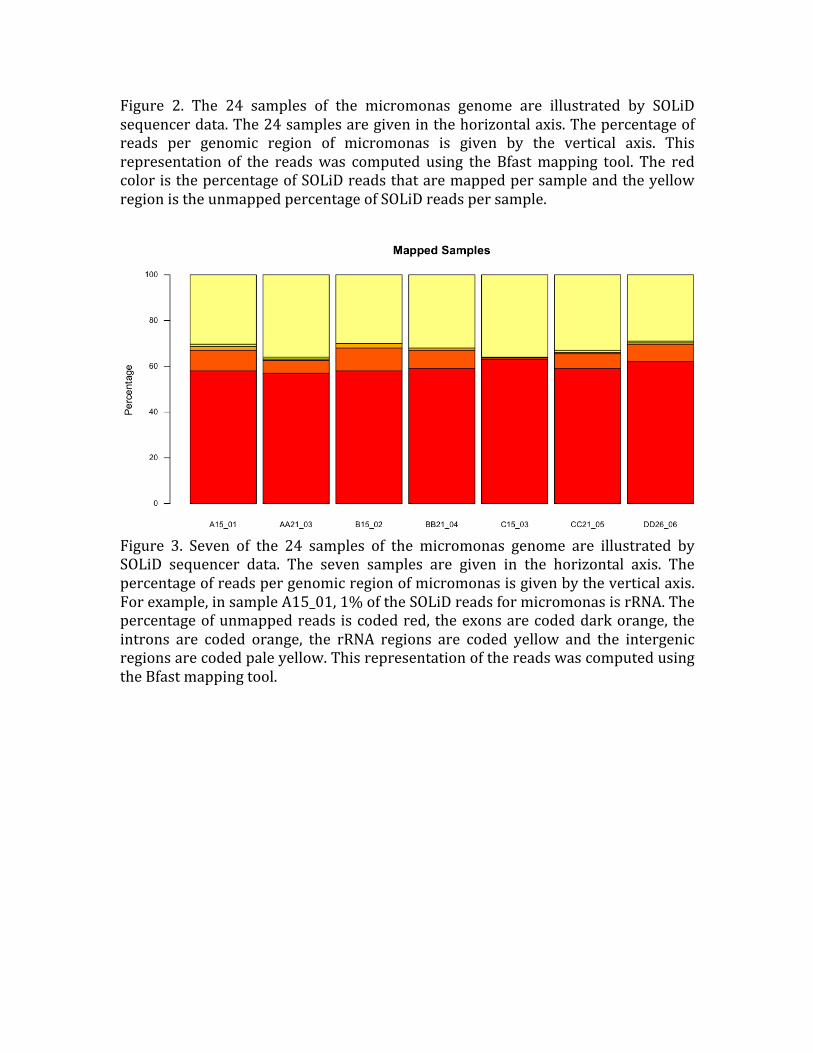
Figure 2. The 24 samples of the micromonas genome are illustrated by SOLiD
sequencer data. The 24 samples are given in the horizontal axis. The percentage of reads per genomic region of micromonas is given by the vertical axis. This
representation of the reads was computed using the Bfast mapping tool. The red color is the percentage of SOLiD reads that are mapped per sample and the yellow
region is the unmapped percentage of SOLiD reads per sample.
Figure 3. Seven of the 24 samples of the micromonas genome are illustrated by SOLiD sequencer data. The seven samples are given in the horizontal axis. The
percentage of reads per genomic region of micromonas is given by the vertical axis.
For example, in sample A15_01, 1% of the SOLiD reads for micromonas is rRNA. The percentage of unmapped reads is coded red, the exons are coded dark orange, the
introns are coded orange, the rRNA regions are coded yellow and the intergenic
regions are coded pale yellow. This representation of the reads was computed using
the Bfast mapping tool.
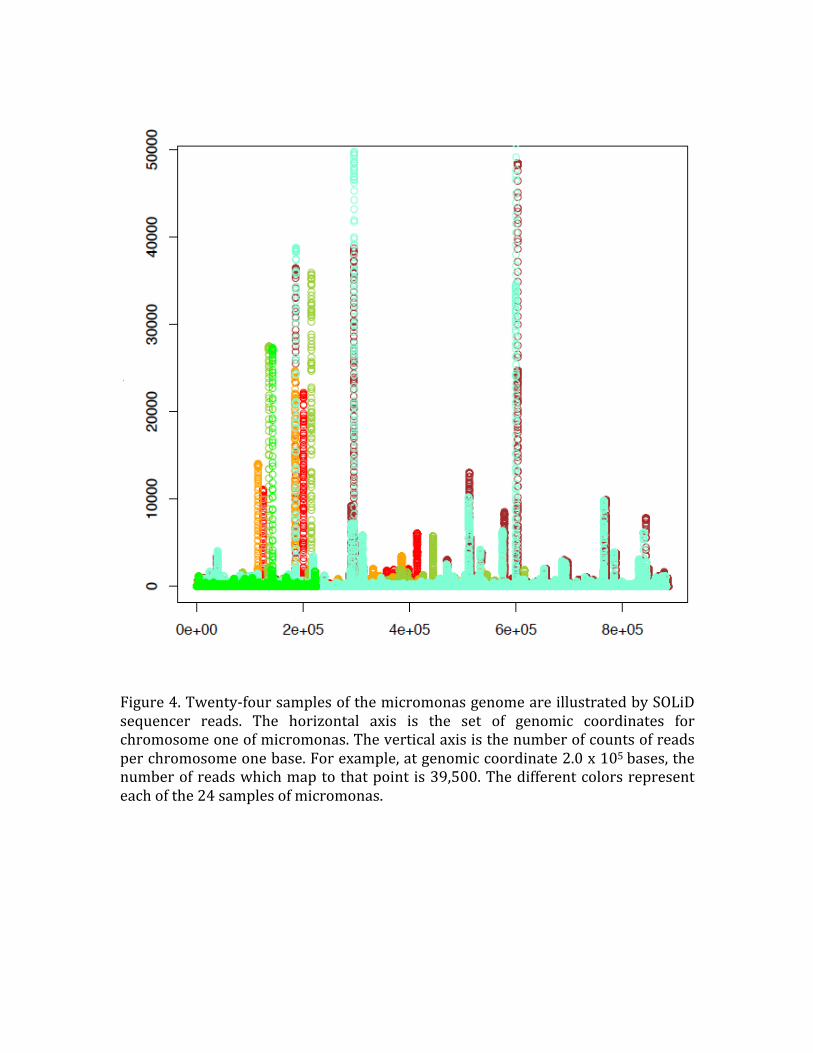
Figure 4. Twenty‐four samples of the micromonas genome are illustrated by SOLiD
sequencer reads. The horizontal axis is the set of genomic coordinates for chromosome one of micromonas. The vertical axis is the number of counts of reads
per chromosome one base. For example, at genomic coordinate 2.0 x 105 bases, the number of reads which map to that point is 39,500. The different colors represent
each of the 24 samples of micromonas.
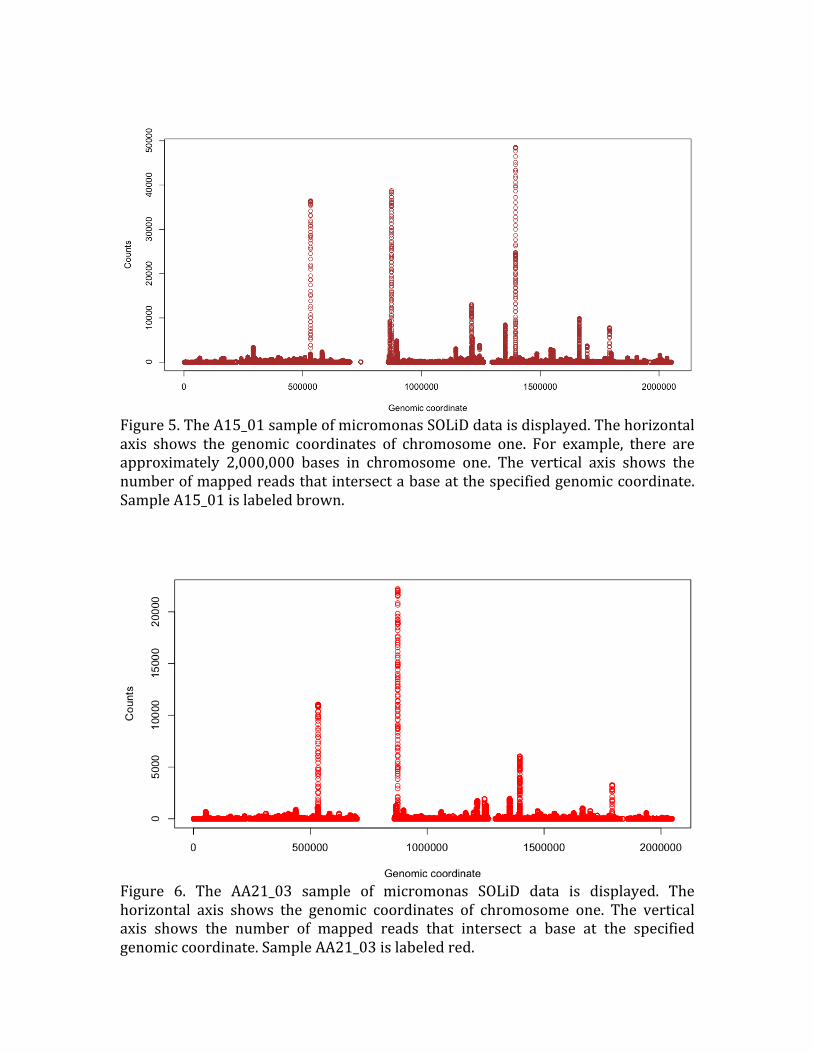
Figure 5. The A15_01 sample of micromonas SOLiD data is displayed. The horizontal
axis shows the genomic coordinates of chromosome one. For example, there are
approximately 2,000,000 bases in chromosome one. The vertical axis shows the
number of mapped reads that intersect a base at the specified genomic coordinate.
Sample A15_01 is labeled brown.
Figure 6. The AA21_03 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample AA21_03 is labeled red.

Figure 7. The B15_02 sample of micromonas SOLiD data is displayed. The horizontal
axis shows the genomic coordinates of chromosome one. The vertical axis shows the
number of mapped reads that intersect a base at the specified genomic coordinate.
Sample B15_02 is labeled orange.
Figure 8. The BB21_04 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified genomic coordinate. Sample BB21_04 is labeled yellow‐green.

Figure 9. The C15_03 sample of micromonas SOLiD data is displayed. The horizontal
axis shows the genomic coordinates of chromosome one. The vertical axis shows the
number of mapped reads that intersect a base at the specified genomic coordinate.
Sample C15_03 is labeled aquamarine.
Figure 10. The CC21_05 sample of micromonas SOLiD data is displayed. The horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample CC21_05 is labeled green.
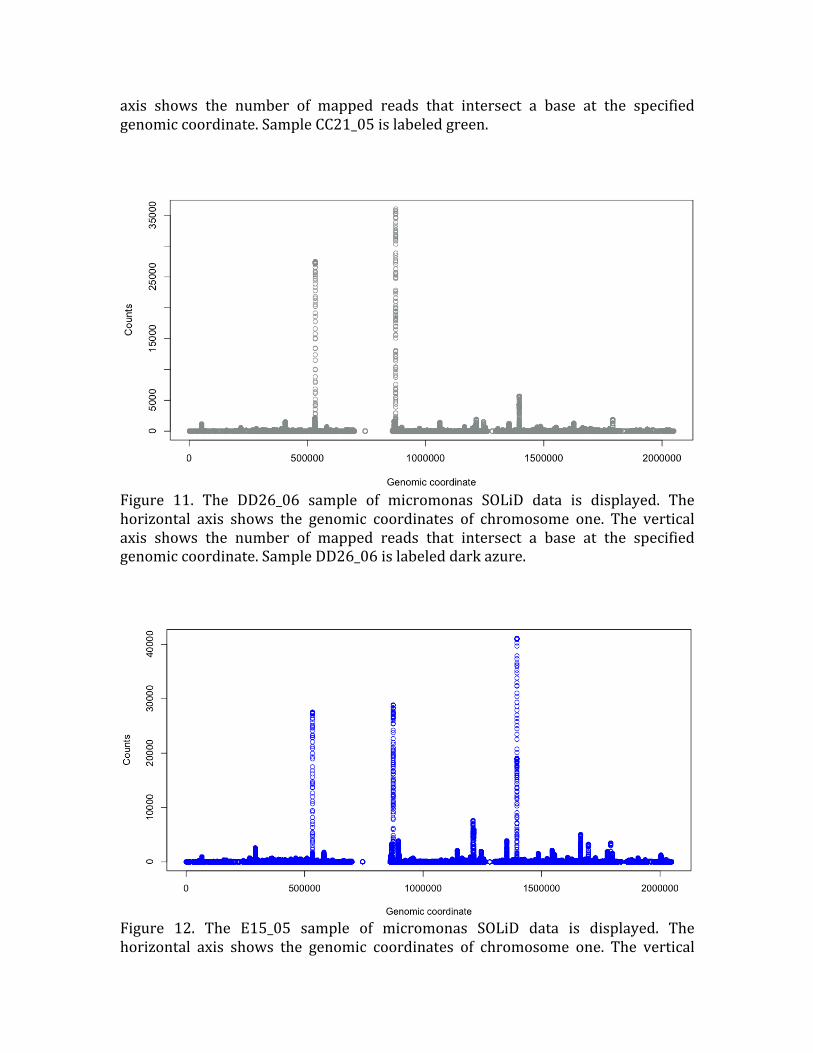
Figure 11. The DD26_06 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified genomic coordinate. Sample DD26_06 is labeled dark azure.
Figure 12. The E15_05 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample E15_05 is labeled blue.
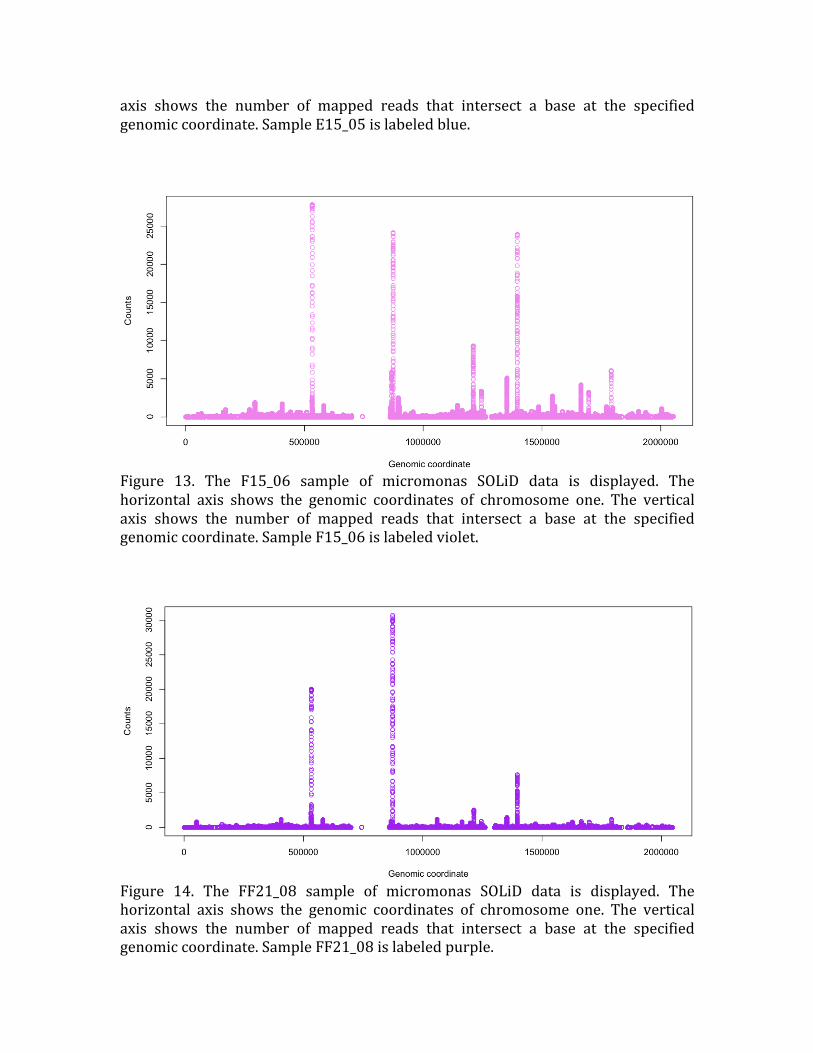
Figure 13. The F15_06 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample F15_06 is labeled violet.
Figure 14. The FF21_08 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified genomic coordinate. Sample FF21_08 is labeled purple.
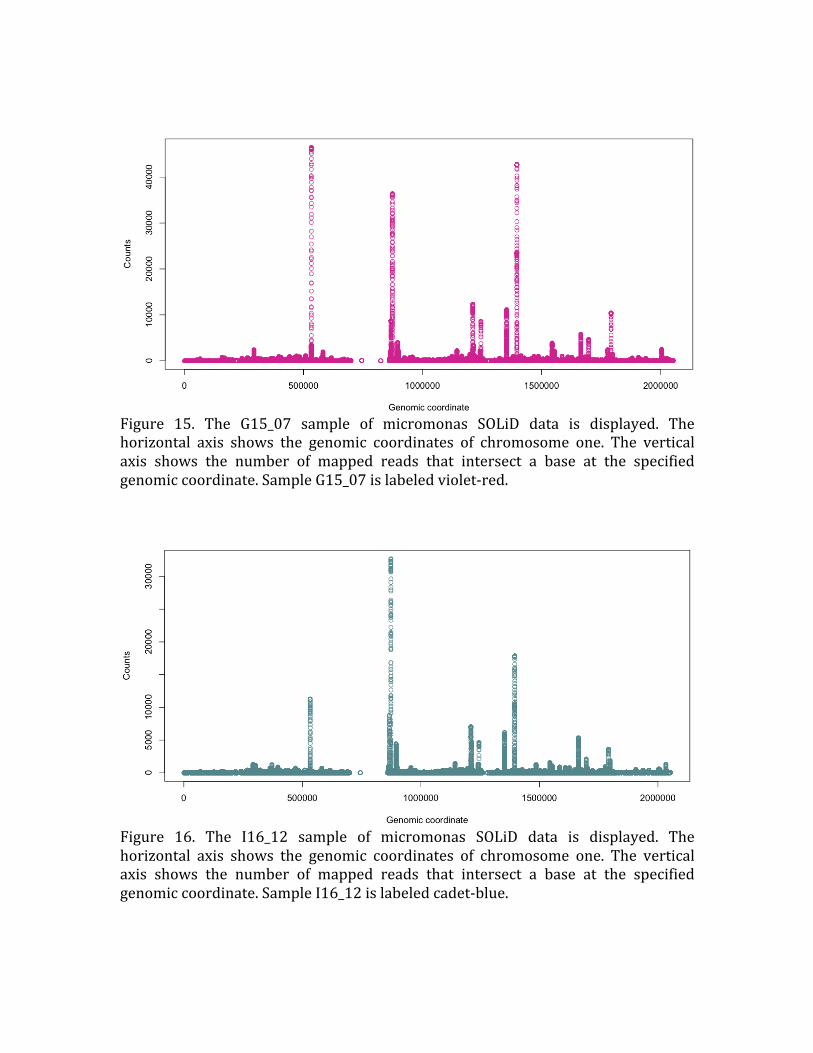
Figure 15. The G15_07 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample G15_07 is labeled violet‐red.
Figure 16. The I16_12 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample I16_12 is labeled cadet‐blue.
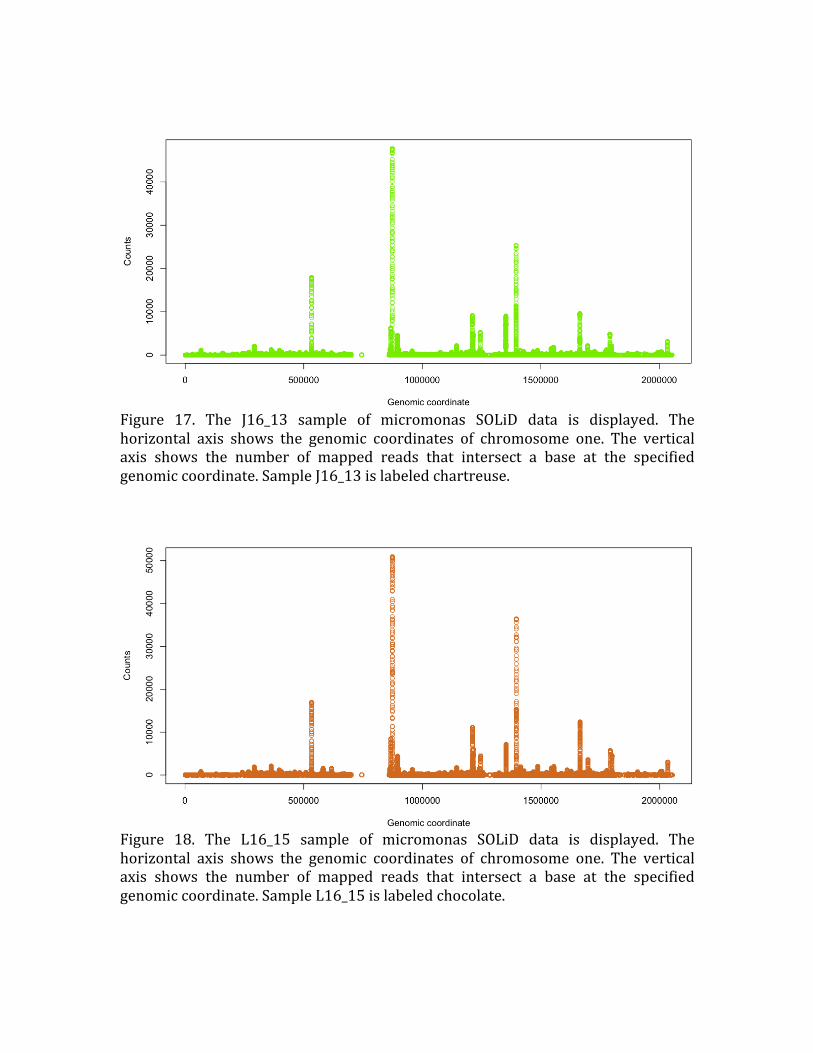
Figure 17. The J16_13 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample J16_13 is labeled chartreuse.
Figure 18. The L16_15 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample L16_15 is labeled chocolate.
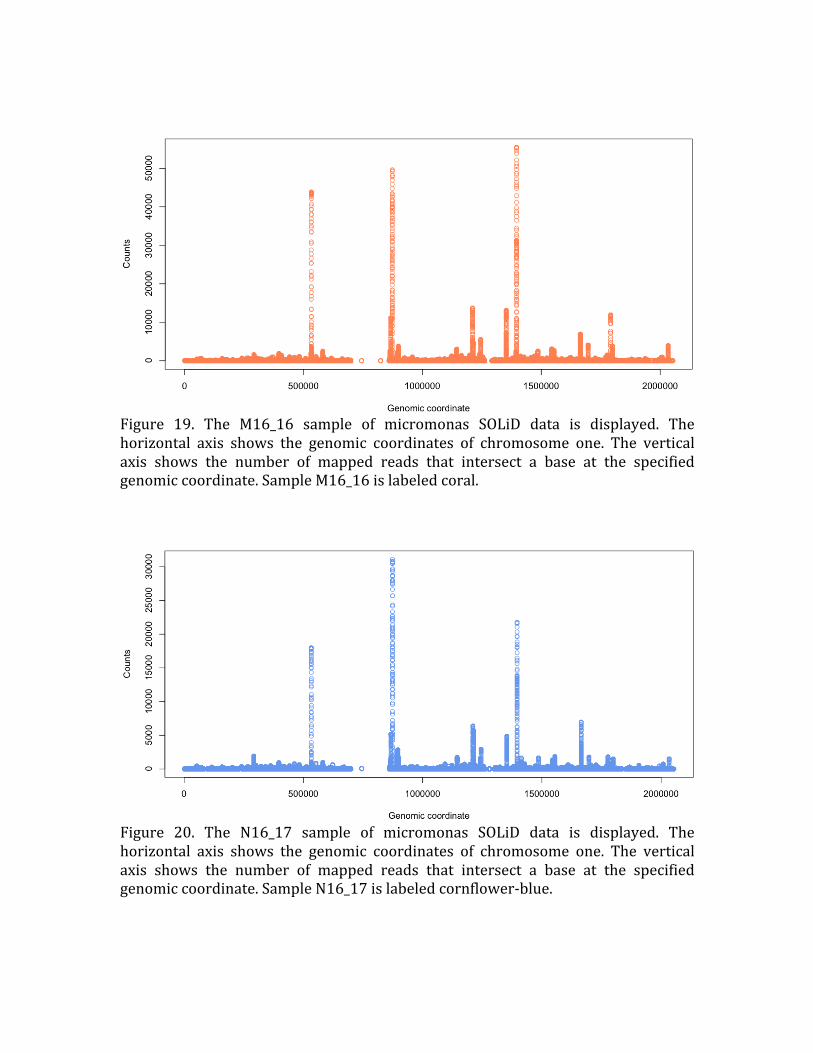
Figure 19. The M16_16 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample M16_16 is labeled coral.
Figure 20. The N16_17 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified genomic coordinate. Sample N16_17 is labeled cornflower‐blue.

Figure 21. The O16_18 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample O16_18 is labeled cyan.
Figure 22. The Q16_20 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample Q16_20 is labeled dark cyan.

Figure 23. The R16_21 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified genomic coordinate. Sample R16_21 is labeled dark goldenrod.
Figure 24. The S16_22 sample of micromonas SOLiD data is displayed. The horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified genomic coordinate. Sample S16_22 is labeled dark brown.

Figure 25. The U21_01 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample U21_01 is labeled blue‐violet.
Figure 26. The V21_02 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample V21_02 is labeled dark aquamarine.

Figure 27. The W21_03 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample W21_03 is labeled dark chocolate.
Figure 28. The Z21_02 sample of micromonas SOLiD data is displayed. The
horizontal axis shows the genomic coordinates of chromosome one. The vertical
axis shows the number of mapped reads that intersect a base at the specified
genomic coordinate. Sample Z21_02 is labeled dark antique white.

References
Li, H. et al. (2009) Fast and accurate short read alignment with Burrows‐Wheeler
transform. Bioinformatics, 25, 1754‐1760.
Langmead, B. et al. (2009) Ultrafast and memory‐efficient alignment of short DNA
sequences to the human genome. Genome Biology, 10:R25, 1‐10.
Homer, N. et al. (2009) BFAST: An alignment tool for large scale genome
resequencing. PLoS ONE, 4(11), e7767: 1‐12.


















