
The lipid composition of autophagic vacuoles regulates ... composition.pdf · surfactant protein B...
13
Introduction Multilamellar bodies are lysosomal organelles containing multiple concentric membrane layers. MLBs vary in size from 100 to 2400 nm and are found in various cell types where their principal functions are storage and secretion of lipids (Schmitz and Muller, 1991). In lung alveolar type II cells, MLBs are responsible for the secretion of the surfactant film that prevents alveolae from collapse during respiration (Hatasa and Nakamura, 1965). Deficient expression of the hydrophobic surfactant protein B (SP-B) results in the formation of immature MLBs and secretion of non-functional surfactant (Foster et al., 2003). Abnormal MLBs have also been observed in familial desquamative and non-specific interstitial pneumonitis associated with mutations in surfactant protein C (SP-C) gene (Thomas et al., 2002). Another protein, the ATP- binding cassette transporter A3 (ABCA3) is also localized to lamellar bodies of alveolar type II cells and is critical for the formation of MLBs and production of surfactant (Mulugeta et al., 2002; Shulenin et al., 2004). MLB formation in type II alveolar cells is therefore critically dependent on the protein composition of the organelle. The lipid composition of MLBs is 95% dipalmitoyl phosphatidylcholine (DPPC), a neutral phospholipid that represents the major active component of surfactant (Schmitz and Muller, 1991). Lung surfactant is composed of 80% glycerophospholipid, 10% cholesterol and 10% protein; the amount of cholesterol within the surfactant can increase relative to the phospholipid content under certain conditions, including exercise (Doyle et al., 1994) and hyperpnea (Orgeig et al., 2003). In lung MLBs, cholesterol is localized primarily to the limiting membrane of the organelle (Orgeig, 2001) and whether MLBs are the source of surfactant cholesterol remains uncertain (Hass and Longmore, 1979; Orgeig et al., 1995). Augmentation of cellular cholesterol stimulates MLB expression, the accumulation of cholesterol within MLBs and the uptake of palmitic acid, the precursor of DPPC, in alveolar type II cells (Kolleck et al., 2002). Extracellular cholesterol is therefore internalized and stored in the MLBs of type II alveolar cells but whether it impacts on their biogenesis remains poorly understood. The presence of MLBs is also associated with various lysosomal storage diseases, including gangliosidosis, Tay- Sachs, Fabry’s and Niemann-Pick, associated with deficiencies in various lysosomal degradative enzymes and aberrant lysosomal accumulation of lipids (Blanchette-Mackie, 2000; Gieselmann, 1995; Pentchev et al., 1987; Platt et al., 1997). Cholesterol accumulation is closely related to the expression of MLBs in the Niemann-Pick lysosomal storage diseases. 1991 Multilamellar bodies (MLBs) are responsible for surfactant secretion in type II alveolar cells but also accumulate in other cell types under pathological conditions, including cancer and lysosomal storage diseases such as Niemann- Pick C (NPC), a congenital disease where defective cholesterol transport leads to its accumulation in lysosomes. Mv1Lu type II alveolar cells transfected with Golgi β1,6 N-acetylglucosaminyltransferase V (Mgat5), enhancing the polylactosamine content of complex-type N- glycans, exhibit stable expression of MLBs whose formation requires lysosomal proteolysis within dense autophagic vacuoles. MLBs of Mgat5-transfected Mv1Lu cells are rich in phospholipids and have low levels of cholesterol. In Mv1Lu cells treated with the NPC- mimicking drug U18666A, cholesterol-rich MLBs accumulate independently of both Mgat5 expression and lysosomal proteolysis. Inhibition of autophagy by blocking the PI 3-kinase pathway with 3-methyladenine prevents MLB formation and results in the accumulation of non-lamellar, acidic lysosomal vacuoles. Treatment with 3-methyladenine inhibited the accumulation of monodansylcadaverine, a phospholipid-specific marker for autophagic vacuoles, but did not block endocytic access to the lysosomal vacuoles. Induction of autophagy via serum starvation resulted in an increased size of cholesterol-rich MLBs. Although expression of MLBs in the Mv1Lu cell line can be induced by modulating lysosomal cholesterol or protein glycosylation, an autophagic contribution of phospholipids is critical for the formation of concentric membrane lamellae within late lysosomal organelles. Key words: Multilamellar bodies, Cholesterol, Lysosomes, Phosphatidylinositol 3-kinase, Autophagy, Autophagic vacuoles Summary The lipid composition of autophagic vacuoles regulates expression of multilamellar bodies Patrick Lajoie 1,2 , Ginette Guay 2 , James W. Dennis 3 and Ivan R. Nabi 1,2, * 1 Department of Cellular and Physiological Sciences, University of British Columbia, 2177 Wesbrook Mall, Vancouver V6T 1Z3, British Columbia, Canada 2 Département de Pathologie et Biologie Cellulaire, Université de Montréal, CP6128 Succursale Centre-Ville, Montréal H3C 3J7, Québec, Canada 3 Samuel Lunenfeld Research Institute, Mount Sinai Hospital, University of Toronto, 600 University Avenue, Toronto M5G 1X5, Ontario, Canada *Author for correspondence (e-mail: [email protected]) Accepted 11 February 2005 Journal of Cell Science 118, 1991-2003 Published by The Company of Biologists 2005 doi:10.1242/jcs.02324 Research Article Journal of Cell Science
Transcript of The lipid composition of autophagic vacuoles regulates ... composition.pdf · surfactant protein B...

IntroductionMultilamellar bodies are lysosomal organelles containingmultiple concentric membrane layers. MLBs vary in size from100 to 2400 nm and are found in various cell types where theirprincipal functions are storage and secretion of lipids (Schmitzand Muller, 1991). In lung alveolar type II cells, MLBs areresponsible for the secretion of the surfactant film that preventsalveolae from collapse during respiration (Hatasa andNakamura, 1965). Deficient expression of the hydrophobicsurfactant protein B (SP-B) results in the formation ofimmature MLBs and secretion of non-functional surfactant(Foster et al., 2003). Abnormal MLBs have also been observedin familial desquamative and non-specific interstitialpneumonitis associated with mutations in surfactant protein C(SP-C) gene (Thomas et al., 2002). Another protein, the ATP-binding cassette transporter A3 (ABCA3) is also localized tolamellar bodies of alveolar type II cells and is critical for theformation of MLBs and production of surfactant (Mulugeta etal., 2002; Shulenin et al., 2004). MLB formation in type IIalveolar cells is therefore critically dependent on the proteincomposition of the organelle.
The lipid composition of MLBs is 95% dipalmitoylphosphatidylcholine (DPPC), a neutral phospholipid thatrepresents the major active component of surfactant (Schmitz
and Muller, 1991). Lung surfactant is composed of 80%glycerophospholipid, 10% cholesterol and 10% protein; theamount of cholesterol within the surfactant can increaserelative to the phospholipid content under certain conditions,including exercise (Doyle et al., 1994) and hyperpnea (Orgeiget al., 2003). In lung MLBs, cholesterol is localized primarilyto the limiting membrane of the organelle (Orgeig, 2001) andwhether MLBs are the source of surfactant cholesterol remainsuncertain (Hass and Longmore, 1979; Orgeig et al., 1995).Augmentation of cellular cholesterol stimulates MLBexpression, the accumulation of cholesterol within MLBs andthe uptake of palmitic acid, the precursor of DPPC, in alveolartype II cells (Kolleck et al., 2002). Extracellular cholesterol istherefore internalized and stored in the MLBs of type IIalveolar cells but whether it impacts on their biogenesisremains poorly understood.
The presence of MLBs is also associated with variouslysosomal storage diseases, including gangliosidosis, Tay-Sachs, Fabry’s and Niemann-Pick, associated with deficienciesin various lysosomal degradative enzymes and aberrantlysosomal accumulation of lipids (Blanchette-Mackie, 2000;Gieselmann, 1995; Pentchev et al., 1987; Platt et al., 1997).Cholesterol accumulation is closely related to the expressionof MLBs in the Niemann-Pick lysosomal storage diseases.
1991
Multilamellar bodies (MLBs) are responsible for surfactantsecretion in type II alveolar cells but also accumulate inother cell types under pathological conditions, includingcancer and lysosomal storage diseases such as Niemann-Pick C (NPC), a congenital disease where defectivecholesterol transport leads to its accumulation inlysosomes. Mv1Lu type II alveolar cells transfected withGolgi β1,6 N-acetylglucosaminyltransferase V (Mgat5),enhancing the polylactosamine content of complex-type N-glycans, exhibit stable expression of MLBs whoseformation requires lysosomal proteolysis within denseautophagic vacuoles. MLBs of Mgat5-transfected Mv1Lucells are rich in phospholipids and have low levels ofcholesterol. In Mv1Lu cells treated with the NPC-mimicking drug U18666A, cholesterol-rich MLBsaccumulate independently of both Mgat5 expression andlysosomal proteolysis. Inhibition of autophagy by blocking
the PI 3-kinase pathway with 3-methyladenine preventsMLB formation and results in the accumulation ofnon-lamellar, acidic lysosomal vacuoles. Treatmentwith 3-methyladenine inhibited the accumulation ofmonodansylcadaverine, a phospholipid-specific marker forautophagic vacuoles, but did not block endocytic access tothe lysosomal vacuoles. Induction of autophagy via serumstarvation resulted in an increased size of cholesterol-richMLBs. Although expression of MLBs in the Mv1Lu cellline can be induced by modulating lysosomal cholesterolor protein glycosylation, an autophagic contribution ofphospholipids is critical for the formation of concentricmembrane lamellae within late lysosomal organelles.
Key words: Multilamellar bodies, Cholesterol, Lysosomes,Phosphatidylinositol 3-kinase, Autophagy, Autophagic vacuoles
Summary
The lipid composition of autophagic vacuolesregulates expression of multilamellar bodiesPatrick Lajoie1,2, Ginette Guay2, James W. Dennis3 and Ivan R. Nabi1,2,*1Department of Cellular and Physiological Sciences, University of British Columbia, 2177 Wesbrook Mall, Vancouver V6T 1Z3, British Columbia,Canada2Département de Pathologie et Biologie Cellulaire, Université de Montréal, CP6128 Succursale Centre-Ville, Montréal H3C 3J7, Québec, Canada3Samuel Lunenfeld Research Institute, Mount Sinai Hospital, University of Toronto, 600 University Avenue, Toronto M5G 1X5, Ontario, Canada*Author for correspondence (e-mail: [email protected])
Accepted 11 February 2005Journal of Cell Science 118, 1991-2003 Published by The Company of Biologists 2005doi:10.1242/jcs.02324
Research Article
Jour
nal o
f Cel
l Sci
ence

1992
Although Niemann-Pick A and B are associated withsphingomyelinase deficiency, Niemann-Pick C and D arecaused by impaired intracellular cholesterol trafficking(Kolodny, 2000; Pagano et al., 2000). Patients with Niemann-Pick C disease are deficient for expression of the NPC1 proteinimplicated in the regulation of intracellular cholesterol traffic(Blanchette-Mackie, 2000; Ory, 2000; Pentchev et al., 1987).A class of drugs (class 2 amphiphiles such as U18666A)impairs cellular cholesterol traffic and results in theaccumulation of cholesterol in late endosomes, lysosomes andMLBs mimicking Niemann-Pick C disease (Butler et al., 1992;Lange et al., 1998; Liscum et al., 1989).
Fibroblasts from patients with sphingolipid storage diseasesshow defects in lipid transport and sorting (Pagano et al.,2000). In many of these diseases, altered cholesterolhomeostasis leads to perturbations in lipid traffic (Puri et al.,1999). Increased cellular cholesterol alters sphingolipidtrafficking, resulting in its delivery not to the Golgi from theplasma membrane but rather to endolysosomal compartmentssuch that altered trafficking of sphingolipids can be considereda diagnostic tool for the identification of sphingolipid storagediseases (Chen et al., 1999). U18666A-mediated cholesterolaccumulation can be reduced by overexpression of Rab 7 and9 GTPases suggesting that the modulation of endosomal lipidcomposition can impact on the delivery of material tolysosomes (Choudhury et al., 2002; Kobayashi et al., 1999;Pagano et al., 2000). Cholesterol accumulation in lysosomalorganelles has further been proposed to result in the trappingof lipid raft components in late endocytic structures that couldresult in the formation of MLBs (Lusa et al., 2001; Simons andGruenberg, 2000).
Transfection of the Mv1Lu mink lung alveolar type II cellline, which does not express MLBs, with β1-6-N-acetyl-glucosaminyl-transferase V (Mgat5) results in the stableexpression of cytoplasmic MLBs implicating glycosylation oflysosomal glycoproteins in MLB expression (Hariri et al.,2000). Interestingly, defects in galactosidases (galactosidosis)and sialidases (sialidosis) as well as accumulation ofpolylactosamine are associated with MLB accumulation inlysosomal storage diseases (Allegranza et al., 1989; Amano etal., 1983; Berra et al., 1986; DeGasperi et al., 1990). L-PHAlabelling of Mgat5 generated β1-6 GlcNAc-branched N-glycans in MLBs has also been described in melanomas andother cancers (Handerson and Pawelek, 2003).
Multilamellar bodies are therefore ubiquitous organellesexpressed under various physiological and pathologicalconditions. The similar morphology and lysosomal nature ofthese organelles in various cell types argues that commonmechanisms must necessarily regulate their formation. Studiesof MLB biogenesis in the Mgat5-transfected type II alveolarMv1Lu cells showed that MLB formation could be preventedby treatment with the protease inhibitor, leupeptin, resulting inthe accumulation of dense autophagic vacuoles, and thatinhibition of autophagy with 3-methyladenine (3-MA) alsoblocked MLB expression (Hariri et al., 2000). Similarly,lysosomal protein degradation is required for MLB formationin primary human lung type II alveolar cells (Guttentag et al.,2003). Early electron microscopic studies of fetal lungdescribing the coordinated loss of glycogen with theaccumulation of multilamellar bodies (Campiche et al., 1963;O’Hare and Sheridan, 1970) and the presence of cytoplasmic
glycogen in lamellar bodies in type II epithelial cells in fetallung, further support a role for autophagy in MLB formation(Stahlman et al., 2000; Weaver et al., 2002). However, despitethe known ability of cholesterol accumulation to stimulateMLB formation in lysosomal storage disease models, theimpact of lysosomal cholesterol on the biogenetic mechanismsunderlying MLB formation remains undetermined.
MLBs in Mgat5-transfected Mv1Lu cells are rich inphospholipids and low in cholesterol and we have used theNPC-mimicking amphiphilic drug U18666A [3β-(2-diethylaminoethoxy)-androstenone] to study the role oflysosomal cholesterol accumulation on the biogenesis ofMLBs in the Mv1Lu type II alveolar cell line. U18666Astimulates MLB formation in both parental and Mgat5-transfected Mv1Lu cells. It further transforms the denseautophagic vacuoles that accumulate following extendedtreatment with the lysosomal protease inhibitor leupeptin intoMLBs. Cholesterol therefore induces MLB formation inMv1Lu type II alveolar cells independently of both Mgat5expression and lysosomal degradation. Although inhibition ofautophagy with the PI 3-kinase inhibitor 3-methyladenineprevents the expression of concentric lamella in the U18666A-induced cholesterol-rich lysosomal vacuoles, stimulation ofautophagy by serum starvation results in an increased size ofMLBs presenting the concentric lamellar morphology typicalof MLBs. Phospholipid-rich MLBs as well as U18666A-induced cholesterol-rich MLBs are accessible to fluid-phaseuptake after 4 hours and labelled with Lysotracker Red,identifying them as late lysosomal organelles orautolysosomes. Similarly, the non-lamellar vacuoles induced inthe presence of 3-MA are late lysosomal organelles but exhibitsignificantly reduced labelling for the autolysosomal markermonodansylcadaverine. Multiple factors, including cholesterolcontent, glycosylation and autophagy, can therefore contributeto the formation of concentric lamellae within secondarylysosomes and autolysosomes.
Materials and MethodsChemicalsFilipin complex, leupeptin, 3-MA, MDC and gelvatol were purchasedfrom Sigma (St Louis, MO) and Alexa 488 goat anti-mouse IgG,Alexa 568 goat anti-mouse IgG, Alexa 647 goat anti-mouse IgGsecondary antibodies, Lysotracker Red, Sytox Green and FITC-dextran (molecular weight 10,000; lysine-fixable) were fromMolecular Probes (Eugene, OR). U18666A was purchased fromBiomol (Plymouth Meeting, PA), G418 from Invitrogen (Burlington,ON) and Nile Red from ICN Biomedicals (Costa Mesa, CA).
Cell cultureNormal Mv1Lu mink lung epithelial cells, the M9 clone of Mv1Lucells transfected with Mgat5 (Demetriou et al., 1995) were grown inDulbecco’s modified Eagle’s medium (DMEM) supplemented withglutamine, vitamins, non-essential amino acids (Invitrogen,Burlington, ON) and 10% FBS (Medicorp, Laval, Qc) in an air/5%CO2 atmosphere at constant humidity at 37°C. To maintain thephenotype of Mgat5-transfected Mv1Lu cells, the medium wassupplemented with G418 at a final concentration of 600 µg/ml (Haririet al., 2000). Cells were plated at a density of 40,000 cells/cm2 andthe medium was replaced every 2 days. To induce formation ofcholesterol-rich MLBs, medium was supplemented with U18666A(1:10,000 of a 10 mg/ml stock solution in ethanol) for 24 hours.
Journal of Cell Science 118 (9)
Jour
nal o
f Cel
l Sci
ence

1993Biogenesis of multilamellar bodies
Leupeptin was added to the medium at a concentration of 2 µg/ml. 3-MA, at a final concentration of 10 mM, was dissolved directly in themedium, which was then sterilized by filtering (Hariri et al., 2000).
ImmunofluorescenceNormal and Mgat5-transfected Mv1Lu cells were grown on glasscoverslips for 6 days and then processed for immunofluorescence aspreviously described (Hariri et al., 2000). Cells were fixed with 3%paraformaldehyde for 15 minutes, washed with PBS supplementedwith 0.1 mM Ca2+ and 1 mM Mg2+ (PBS-CM) and then incubated for15 minutes with PBS-CM supplemented with 0.2% BSA to reducenon-specific binding and 0.07% saponin to permeabilize cellularmembranes. AC17 anti-LAMP-2 antibody (Nabi et al., 1991; Nabiand Rodriguez-Boulan, 1993) was used to determine the cellulardistribution of LAMP-2 using Alexa 488 goat anti-mouse IgG assecondary antibody. Intracellular cholesterol was visualized by filipinlabelling (1:25 of a stock solution of 10 mg/ml in DMSO).Phospholipid content was determined by mounting coverslips ingelvatol containing Nile Red (1:1000 of a saturated solution).
To follow fluid-phase endocytosis, cells were incubated for variousperiods of time with lysine-fixable FITC-Dextran (5 mg/ml) in cellmedia. Cells were then washed with PBS-CM and processed forimmunofluorescence as described above. Visualisation of acidicorganelles was performed by incubation of cells for 10 minutes with0.1 µM Lysotracker Red added to the medium prior to fixation andlabelling with filipin. Labelled cells were viewed with a 100�NeoFluor objective of a Zeiss Axiophot fluorescent microscopeequipped with UV, FITC and rhodamine filter sets, a QImaging RetigaCCD camera and Northern Eclipse imaging software (EmpixImaging, Mississuaga, ON) or the 100� planapochromat objective ofa Leica TCS-SP1 confocal microscope equipped with Argon (488),Krypton (568) and HeNe (633) lasers. Quantification of endocytosiswas performed by counting the percentage of swollen LAMP-2/NileRed-positive vacuoles labelled for FITC-dextran from ten confocalimages per condition. Presented data were compiled from threeindependent experiments.
MDC labelling was performed as previously described (Biederbicket al., 1995; Munafo and Colombo, 2001). Images were obtained withthe 60� PlanApo objective of an Olympus-PTI microscope systemequipped with DeltaRam V monochromator and Cascade BF CCDcamera. Equivalent acquisition settings and display parameters wereused for all samples from a single experiment. Quantification wasperformed by measuring the average intensity of MDC labellingwithin large vacuoles (1.4 to 8 µm), labelled in a second fluorescentchannel for Nile Red, using Image Master software. Presented data,compiled from three independent experiments, were normalized to themean MDC intensity values of MLBs from control M9 cells and atwo-tailed Student’s t-test was performed.
Induction of autophagy by serum starvation was performed on cellsgrown on coverslips for 5 days and then treated with U18666A for 24hours. Cells were washed twice with serum-containing or serum-free
DMEM containing U18666A and incubated for 0.5, 1, 3 or 6 hoursbefore fixation and labelling with filipin and Sytox Green. Imageswere obtained with the 60� PlanApo objective of an OlympusFV1000 confocal microscope. The surface area of filipin-positivestructures, with diameter 0.5 to 5 µm that corresponded to MLB size(Table 1), was determined using Image Master software. Overlappingvacuoles were manually segmented where possible and only single,isolated filipin-positive structures were quantified. The number ofcells present in each field was determined by counting Sytox Green-labelled nuclei. Equivalent acquisition settings and display parameterswere used for all samples from a single experiment and data fromthree different experiments were compiled and a two-tailed Student’st-test performed.
Electron microscopyCells were rinsed with 0.1 mM sodium cacodylate, pH 7.3 and thenfixed for 1 hour with 2% glutaraldehyde at 4°C. After fixation, thecells were rinsed with cacodylate buffer, scraped from the Petri dish,pelleted and post-fixed with 2% osmium tetroxide at 4°C. The cellswere dehydrated and then embedded in LR-White resin. Ultra-thinsections were prepared and treated with uranyl acetate and lead citrateto enhance contrast. The sections were visualized with a ZeissCEM902 electron microscope. Quantification of the expression ofMLBs and of swollen lysosomal vacuoles in the 3-MA experimentswas determined by circumscribing the cytoplasm (excluding thenucleus) and the MLBs and non-lamellar lysosomal vacuoles fromsix images at 3000� magnification and determining the area ofthe circumscribed regions. MLBs were defined as membrane-bound cytoplasmic organelles presenting at least three distinctcircumferential concentric membrane lamellae. MLBs werecomposed either completely of concentric lamellae or of concentriclamellae surrounding a single dense core (Hariri et al., 2000). Swollenlysosomal vacuoles were defined by the presence of multiple internalstructures surrounded by a limiting membrane and could bemorphologically distinguished from MLBs.
ResultsMLB expression in type II alveolar cells followingU18666A-mediated lysosomal cholesterol accumulationTo assess the phospholipid and cholesterol distribution of theMLBs expressed upon Mgat5 transfection of Mv1Lu cells,both parental Mv1Lu and the M9 clone of Mgat5-transfectedMv1Lu cells (Demetriou et al., 1995; Hariri et al., 2000) werelabelled for Nile Red, a phospholipid-specific dye that labelsMLBs of alveolar type II cells (Gonzales et al., 2001; Guttentaget al., 2003), and for filipin, a cholesterol-specific dye.Although neither Nile Red nor filipin labelling associated withthe LAMP-2-positive lysosomes of Mv1Lu cells (Fig. 1A-D),the multiple large LAMP-2-positive lysosomal structures that
Table 1. Quantification of MLB expression in Mv1Lu cells and M9 clones transfected with Mgat5 before and aftertreatment with U18666A and 3-MA
Multilamellar bodies (MLBs) Non-lamellar lysosomal vacuoles
Size range Size range Cell type Number (median) (µm) % cell area Number (median) (µm) % cell area
Mv1Lu control 1 4.6 0.13 – – –Mv1Lu+U18666A 130 0.3-6.2 (1.6) 4.35±0.8 – – –Mv1Lu+3MA 0 0 0 62 0.5-3.5 (1.3) 1.1±0.5Mv1Lu+U18666A+3MA 7 0.2-5.4 (1.3) 0.02±0.05 86 0.8-6.0 (1.7) 3.5±0.7M9 control 107 0.1-6.7 (1.7) 5.1±1.6 – – –M9+U18666A 169 0.1-7.8 (1.9) 7.4±0.9 – – –M9+3MA 22 0.1-5.8 (1.2) 0.4±0.3 76 0.1-3.3 (1.1) 1.26±0.7M9+U18666A+3MA 9 0.1-6.1 (1.5) 0.4±0.3 100 0.1-7.4 (1.9) 6.5±0.5
Jour
nal o
f Cel
l Sci
ence

1994
correspond to MLBs in the M9 clone of Mgat5-transfectedMv1Lu alveolar cells (Hariri et al., 2000) were labelled forNile Red indicating that they are rich in phospholipids (Fig.1E-H). Labelling of cholesterol with filipin shows that thelumen of the MLBs is not labelled although a peripheral filipin-positive ring is frequently observed to circumscribe the NileRed positive MLBs (Fig. 1G,Q-T), consistent with the fact thatin type II alveolar cells, cholesterol is mainly localized to thelimiting membrane of the organelles (Orgeig and Daniels,2001; Punnonen et al., 1988).
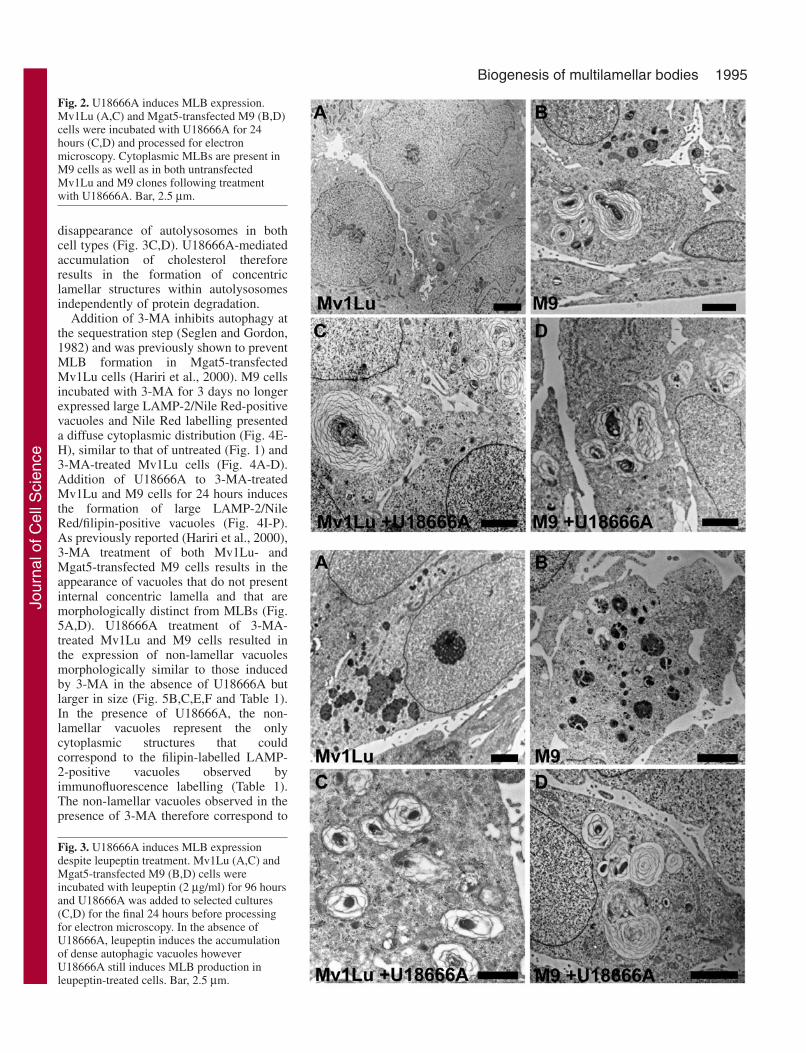
Treatment of both Mv1Lu and Mgat5-transfected M9 cellswith U18666A resulted in the expression of large LAMP-2-positive vacuoles labelled for both Nile Red and filipin,reflecting the ability of U18666A to induce cholesterolaccumulation in both lysosomes and MLBs, respectively (Fig.1I-P). MLBs are not observed in Mv1Lu cells using electronmicroscopy, whereas M9 cells present multiple cytoplasmicMLBs (Fig. 2A,B and Table 1), as previously reported (Haririet al., 2000). In both cell lines, numerous MLBs are observedupon treatment with U18666A (Fig. 2C,D and Table 1)
indicating that lysosomal accumulation of cholesterol resultsin MLB formation independently of Mgat5 expression levels.In Mv1Lu cells, lysosomal cholesterol accumulation due toU18666A treatment therefore results in de novo formation ofMLBs. In M9 cells, U18666A treatment increases the numberof MLBs and the cytoplasmic area covered by MLBs (Table 1)indicating that cholesterol accumulation occurs within pre-existing phospholipid-rich MLBs without disrupting theirlamellar morphology but also induces new MLBs.
Cholesterol accumulation overrides the role of lysosomaldegradation but not autophagy in MLB formationMLBs are lysosomal organelles and express various lysosomalhydrolases (de Vries et al., 1985; DiAugustine, 1974; Hatasaand Nakamura, 1965; Hook and Gilmore, 1982). Thepreviously demonstrated ability of leupeptin treatment toprevent MLB formation in Mgat5-transfected Mv1Lu cells(Hariri et al., 2000) led us to determine whether U18666Acould induce MLB formation in leupeptin-treated M9 cells.
After 96 hours in the presence ofleupeptin, Mv1Lu and M9 cells expresslarge LAMP-2/Nile Red-positivevacuoles and treatment with U18666Afor 24 hours results in the formation ofvacuoles strongly labelled for filipin,essentially identical to that observed incells not treated with leupeptin (datanot shown). Treatment of Mv1Lu cellswith leupeptin for 4 days induced theaccumulation of autolysosomes asviewed by electron microscopy (Fig.3A). In M9 cells, leupeptin treatmentresulted in the disappearance of MLBsand their replacement by largeautolysosomes (Fig. 3B), as previouslyreported (Hariri et al., 2000). Treatmentof leupeptin-treated cells withU18666A for the final 24 hours resultedin the induction of MLBs and
Journal of Cell Science 118 (9)
Fig. 1. U18666A treatment induces theaccumulation of swollen cholesterol-rich,LAMP-2-positive vacuoles. UntransfectedMv1Lu cells (A-D,I-L) and Mgat5-transfected M9 clones (E-H,M-T) werecultured in regular medium for 6 days(A-H,Q-T) and selected cultures incubatedwith U18666A for the final 24 hours (I-P).The cells were then fixed and triplelabelled with anti-LAMP-2 antibody(A,E,I,M,Q), Nile Red (B,F,J,N,R) andfilipin (C,G,K,O,S). Merged images(D,H,L,P,T) show LAMP-2 in green, NileRed in red, and filipin in blue. In untreatedM9 cells, filipin labelling is concentrated atthe limiting membrane of the vacuoles(Q-T). In the presence of U18666A, theLAMP-2/Nile Red-positive vacuoles(arrowheads) accumulate cholesterol andare all positive for filipin after 24 hours.Bar, 5 µm.
Jour
nal o
f Cel
l Sci
ence
1995Biogenesis of multilamellar bodies
disappearance of autolysosomes in bothcell types (Fig. 3C,D). U18666A-mediatedaccumulation of cholesterol thereforeresults in the formation of concentriclamellar structures within autolysosomesindependently of protein degradation.
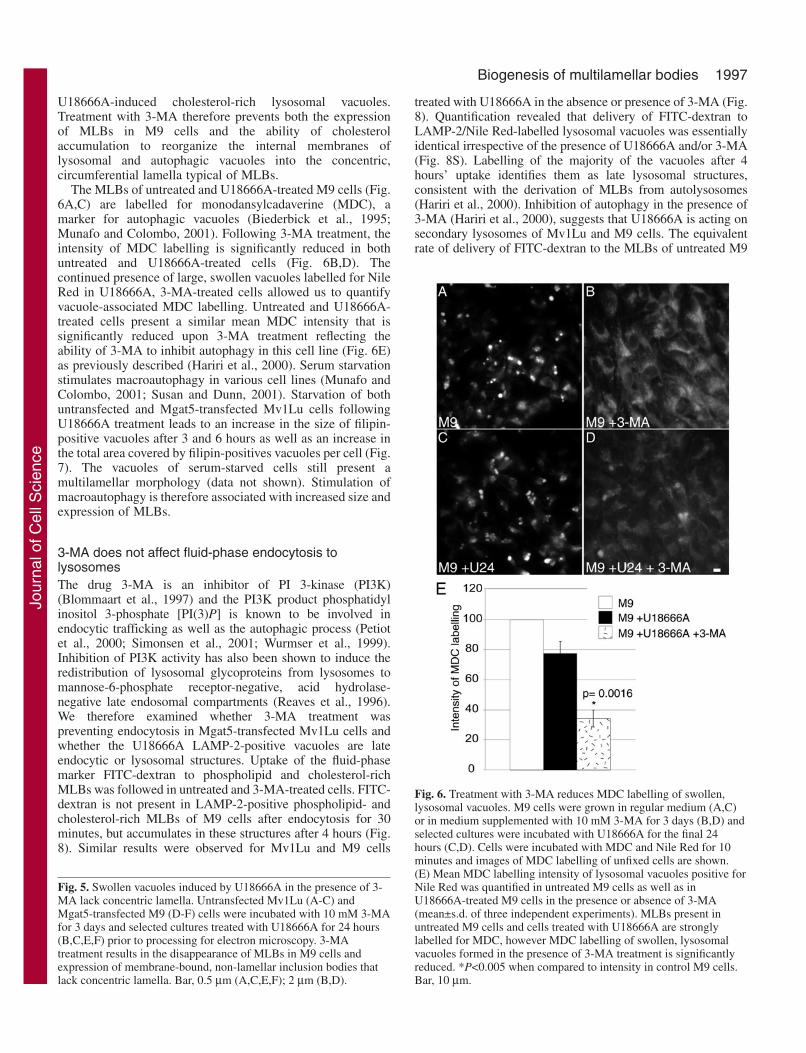
Addition of 3-MA inhibits autophagy atthe sequestration step (Seglen and Gordon,1982) and was previously shown to preventMLB formation in Mgat5-transfectedMv1Lu cells (Hariri et al., 2000). M9 cellsincubated with 3-MA for 3 days no longerexpressed large LAMP-2/Nile Red-positivevacuoles and Nile Red labelling presenteda diffuse cytoplasmic distribution (Fig. 4E-H), similar to that of untreated (Fig. 1) and3-MA-treated Mv1Lu cells (Fig. 4A-D).Addition of U18666A to 3-MA-treatedMv1Lu and M9 cells for 24 hours inducesthe formation of large LAMP-2/NileRed/filipin-positive vacuoles (Fig. 4I-P).As previously reported (Hariri et al., 2000),3-MA treatment of both Mv1Lu- andMgat5-transfected M9 cells results in theappearance of vacuoles that do not presentinternal concentric lamella and that aremorphologically distinct from MLBs (Fig.5A,D). U18666A treatment of 3-MA-treated Mv1Lu and M9 cells resulted inthe expression of non-lamellar vacuolesmorphologically similar to those inducedby 3-MA in the absence of U18666A butlarger in size (Fig. 5B,C,E,F and Table 1).In the presence of U18666A, the non-lamellar vacuoles represent the onlycytoplasmic structures that couldcorrespond to the filipin-labelled LAMP-2-positive vacuoles observed byimmunofluorescence labelling (Table 1).The non-lamellar vacuoles observed in thepresence of 3-MA therefore correspond to
Fig. 2. U18666A induces MLB expression.Mv1Lu (A,C) and Mgat5-transfected M9 (B,D)cells were incubated with U18666A for 24hours (C,D) and processed for electronmicroscopy. Cytoplasmic MLBs are present inM9 cells as well as in both untransfectedMv1Lu and M9 clones following treatmentwith U18666A. Bar, 2.5 µm.
Fig. 3. U18666A induces MLB expressiondespite leupeptin treatment. Mv1Lu (A,C) andMgat5-transfected M9 (B,D) cells wereincubated with leupeptin (2 µg/ml) for 96 hoursand U18666A was added to selected cultures(C,D) for the final 24 hours before processingfor electron microscopy. In the absence ofU18666A, leupeptin induces the accumulationof dense autophagic vacuoles howeverU18666A still induces MLB production inleupeptin-treated cells. Bar, 2.5 µm.
Jour
nal o
f Cel
l Sci
ence

1996 Journal of Cell Science 118 (9)
Fig. 4. U18666A treatmentinduces accumulation ofswollen LAMP-2-positivevacuoles in the presence of theautophagy inhibitor 3-MA.Untransfected Mv1Lu (A-D,I-L) and Mgat5-transfected M9(E-H,M-P) cells were grown inmedium supplemented with 10mM 3-MA for 3 days andselected cultures were incubatedwith U18666A for the final 24hours (I-P). The cells were fixedand then triple labelled withanti-LAMP-2 (A,E,I,M), NileRed (B,F,J,N) and filipin(C,G,K,O). Merged images(D,H,L,P) show LAMP-2 ingreen, Nile Red in red andfilipin in blue. Largephospholipid-rich, LAMP-2-positive vacuoles are notobserved following 3-MAtreatment but U18666A is stillable to induce the formation ofswollen, filipin-positivelysosomal vacuoles. Bar, 5 µm.
Jour
nal o
f Cel
l Sci
ence
1997Biogenesis of multilamellar bodies
U18666A-induced cholesterol-rich lysosomal vacuoles.Treatment with 3-MA therefore prevents both the expressionof MLBs in M9 cells and the ability of cholesterolaccumulation to reorganize the internal membranes oflysosomal and autophagic vacuoles into the concentric,circumferential lamella typical of MLBs.
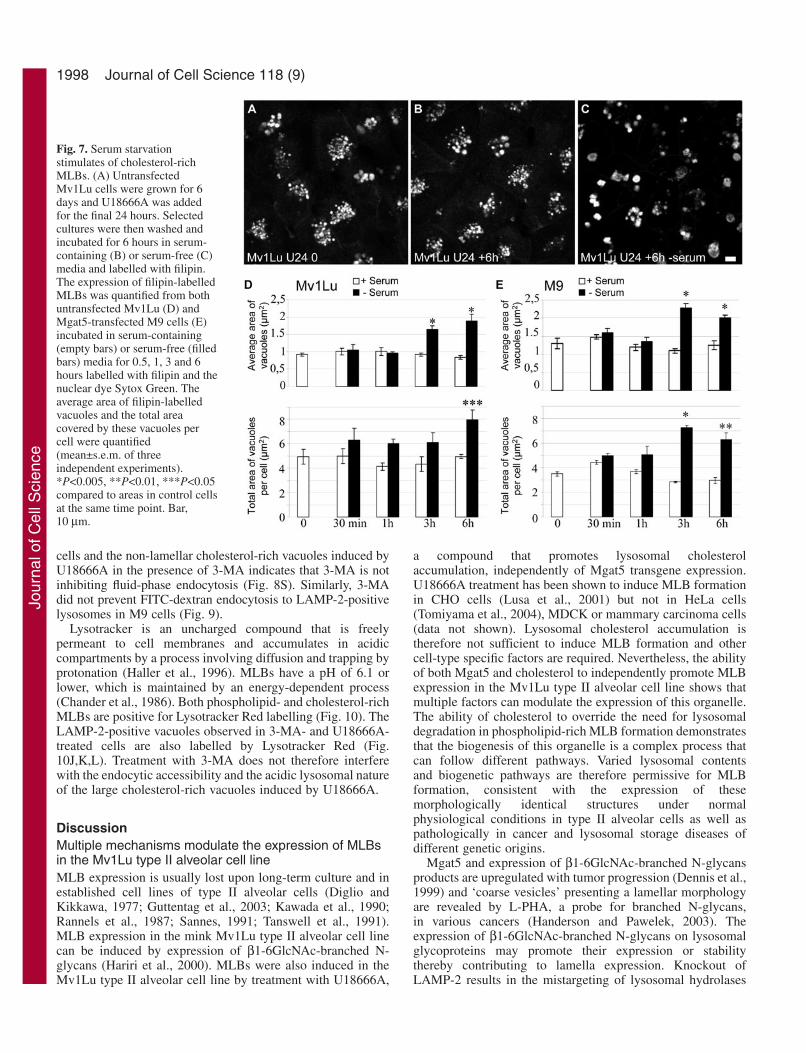
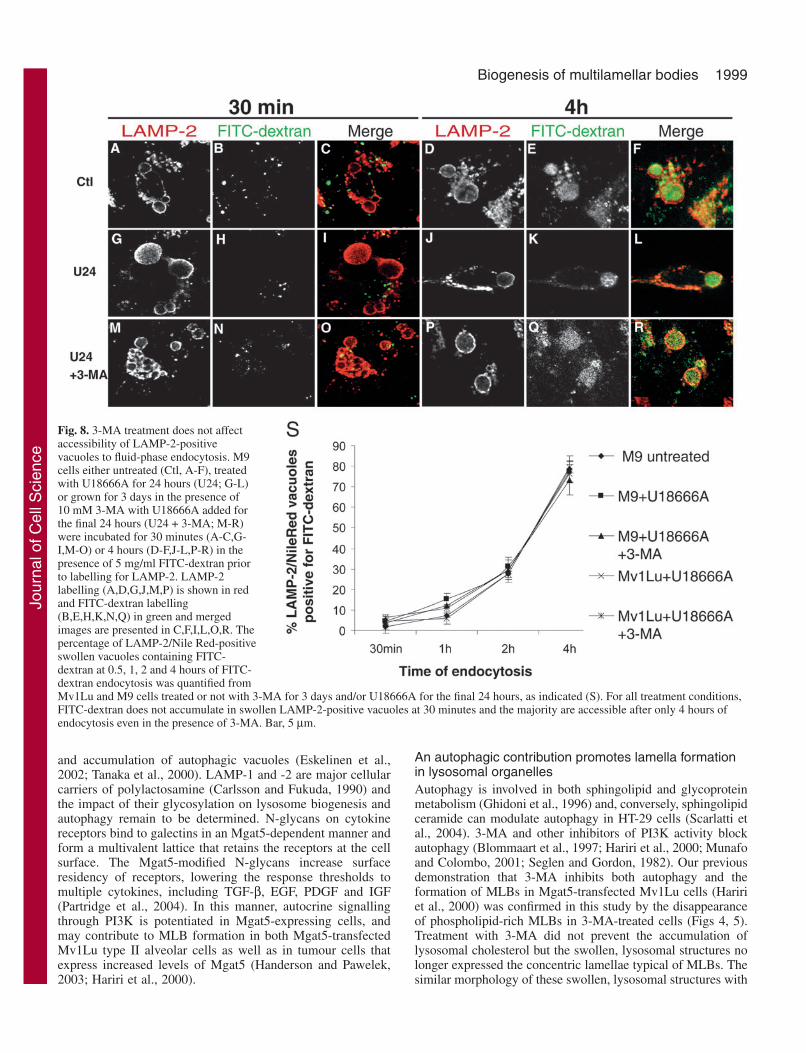
The MLBs of untreated and U18666A-treated M9 cells (Fig.6A,C) are labelled for monodansylcadaverine (MDC), amarker for autophagic vacuoles (Biederbick et al., 1995;Munafo and Colombo, 2001). Following 3-MA treatment, theintensity of MDC labelling is significantly reduced in bothuntreated and U18666A-treated cells (Fig. 6B,D). Thecontinued presence of large, swollen vacuoles labelled for NileRed in U18666A, 3-MA-treated cells allowed us to quantifyvacuole-associated MDC labelling. Untreated and U18666A-treated cells present a similar mean MDC intensity that issignificantly reduced upon 3-MA treatment reflecting theability of 3-MA to inhibit autophagy in this cell line (Fig. 6E)as previously described (Hariri et al., 2000). Serum starvationstimulates macroautophagy in various cell lines (Munafo andColombo, 2001; Susan and Dunn, 2001). Starvation of bothuntransfected and Mgat5-transfected Mv1Lu cells followingU18666A treatment leads to an increase in the size of filipin-positive vacuoles after 3 and 6 hours as well as an increase inthe total area covered by filipin-positives vacuoles per cell (Fig.7). The vacuoles of serum-starved cells still present amultilamellar morphology (data not shown). Stimulation ofmacroautophagy is therefore associated with increased size andexpression of MLBs.
3-MA does not affect fluid-phase endocytosis tolysosomesThe drug 3-MA is an inhibitor of PI 3-kinase (PI3K)(Blommaart et al., 1997) and the PI3K product phosphatidylinositol 3-phosphate [PI(3)P] is known to be involved inendocytic trafficking as well as the autophagic process (Petiotet al., 2000; Simonsen et al., 2001; Wurmser et al., 1999).Inhibition of PI3K activity has also been shown to induce theredistribution of lysosomal glycoproteins from lysosomes tomannose-6-phosphate receptor-negative, acid hydrolase-negative late endosomal compartments (Reaves et al., 1996).We therefore examined whether 3-MA treatment waspreventing endocytosis in Mgat5-transfected Mv1Lu cells andwhether the U18666A LAMP-2-positive vacuoles are lateendocytic or lysosomal structures. Uptake of the fluid-phasemarker FITC-dextran to phospholipid and cholesterol-richMLBs was followed in untreated and 3-MA-treated cells. FITC-dextran is not present in LAMP-2-positive phospholipid- andcholesterol-rich MLBs of M9 cells after endocytosis for 30minutes, but accumulates in these structures after 4 hours (Fig.8). Similar results were observed for Mv1Lu and M9 cells
treated with U18666A in the absence or presence of 3-MA (Fig.8). Quantification revealed that delivery of FITC-dextran toLAMP-2/Nile Red-labelled lysosomal vacuoles was essentiallyidentical irrespective of the presence of U18666A and/or 3-MA(Fig. 8S). Labelling of the majority of the vacuoles after 4hours’ uptake identifies them as late lysosomal structures,consistent with the derivation of MLBs from autolysosomes(Hariri et al., 2000). Inhibition of autophagy in the presence of3-MA (Hariri et al., 2000), suggests that U18666A is acting onsecondary lysosomes of Mv1Lu and M9 cells. The equivalentrate of delivery of FITC-dextran to the MLBs of untreated M9
Fig. 5. Swollen vacuoles induced by U18666A in the presence of 3-MA lack concentric lamella. Untransfected Mv1Lu (A-C) andMgat5-transfected M9 (D-F) cells were incubated with 10 mM 3-MAfor 3 days and selected cultures treated with U18666A for 24 hours(B,C,E,F) prior to processing for electron microscopy. 3-MAtreatment results in the disappearance of MLBs in M9 cells andexpression of membrane-bound, non-lamellar inclusion bodies thatlack concentric lamella. Bar, 0.5 µm (A,C,E,F); 2 µm (B,D).
Fig. 6. Treatment with 3-MA reduces MDC labelling of swollen,lysosomal vacuoles. M9 cells were grown in regular medium (A,C)or in medium supplemented with 10 mM 3-MA for 3 days (B,D) andselected cultures were incubated with U18666A for the final 24hours (C,D). Cells were incubated with MDC and Nile Red for 10minutes and images of MDC labelling of unfixed cells are shown.(E) Mean MDC labelling intensity of lysosomal vacuoles positive forNile Red was quantified in untreated M9 cells as well as inU18666A-treated M9 cells in the presence or absence of 3-MA(mean±s.d. of three independent experiments). MLBs present inuntreated M9 cells and cells treated with U18666A are stronglylabelled for MDC, however MDC labelling of swollen, lysosomalvacuoles formed in the presence of 3-MA treatment is significantlyreduced. *P<0.005 when compared to intensity in control M9 cells.Bar, 10 µm.
Jour
nal o
f Cel
l Sci
ence
1998
cells and the non-lamellar cholesterol-rich vacuoles induced byU18666A in the presence of 3-MA indicates that 3-MA is notinhibiting fluid-phase endocytosis (Fig. 8S). Similarly, 3-MAdid not prevent FITC-dextran endocytosis to LAMP-2-positivelysosomes in M9 cells (Fig. 9).
Lysotracker is an uncharged compound that is freelypermeant to cell membranes and accumulates in acidiccompartments by a process involving diffusion and trapping byprotonation (Haller et al., 1996). MLBs have a pH of 6.1 orlower, which is maintained by an energy-dependent process(Chander et al., 1986). Both phospholipid- and cholesterol-richMLBs are positive for Lysotracker Red labelling (Fig. 10). TheLAMP-2-positive vacuoles observed in 3-MA- and U18666A-treated cells are also labelled by Lysotracker Red (Fig.10J,K,L). Treatment with 3-MA does not therefore interferewith the endocytic accessibility and the acidic lysosomal natureof the large cholesterol-rich vacuoles induced by U18666A.
DiscussionMultiple mechanisms modulate the expression of MLBsin the Mv1Lu type II alveolar cell lineMLB expression is usually lost upon long-term culture and inestablished cell lines of type II alveolar cells (Diglio andKikkawa, 1977; Guttentag et al., 2003; Kawada et al., 1990;Rannels et al., 1987; Sannes, 1991; Tanswell et al., 1991).MLB expression in the mink Mv1Lu type II alveolar cell linecan be induced by expression of β1-6GlcNAc-branched N-glycans (Hariri et al., 2000). MLBs were also induced in theMv1Lu type II alveolar cell line by treatment with U18666A,
a compound that promotes lysosomal cholesterolaccumulation, independently of Mgat5 transgene expression.U18666A treatment has been shown to induce MLB formationin CHO cells (Lusa et al., 2001) but not in HeLa cells(Tomiyama et al., 2004), MDCK or mammary carcinoma cells(data not shown). Lysosomal cholesterol accumulation istherefore not sufficient to induce MLB formation and othercell-type specific factors are required. Nevertheless, the abilityof both Mgat5 and cholesterol to independently promote MLBexpression in the Mv1Lu type II alveolar cell line shows thatmultiple factors can modulate the expression of this organelle.The ability of cholesterol to override the need for lysosomaldegradation in phospholipid-rich MLB formation demonstratesthat the biogenesis of this organelle is a complex process thatcan follow different pathways. Varied lysosomal contentsand biogenetic pathways are therefore permissive for MLBformation, consistent with the expression of thesemorphologically identical structures under normalphysiological conditions in type II alveolar cells as well aspathologically in cancer and lysosomal storage diseases ofdifferent genetic origins.
Mgat5 and expression of β1-6GlcNAc-branched N-glycansproducts are upregulated with tumor progression (Dennis et al.,1999) and ‘coarse vesicles’ presenting a lamellar morphologyare revealed by L-PHA, a probe for branched N-glycans,in various cancers (Handerson and Pawelek, 2003). Theexpression of β1-6GlcNAc-branched N-glycans on lysosomalglycoproteins may promote their expression or stabilitythereby contributing to lamella expression. Knockout ofLAMP-2 results in the mistargeting of lysosomal hydrolases
Journal of Cell Science 118 (9)
Fig. 7. Serum starvationstimulates of cholesterol-richMLBs. (A) UntransfectedMv1Lu cells were grown for 6days and U18666A was addedfor the final 24 hours. Selectedcultures were then washed andincubated for 6 hours in serum-containing (B) or serum-free (C)media and labelled with filipin.The expression of filipin-labelledMLBs was quantified from bothuntransfected Mv1Lu (D) andMgat5-transfected M9 cells (E)incubated in serum-containing(empty bars) or serum-free (filledbars) media for 0.5, 1, 3 and 6hours labelled with filipin and thenuclear dye Sytox Green. Theaverage area of filipin-labelledvacuoles and the total areacovered by these vacuoles percell were quantified(mean±s.e.m. of threeindependent experiments).*P<0.005, **P<0.01, ***P<0.05compared to areas in control cellsat the same time point. Bar,10 µm.
Jour
nal o
f Cel
l Sci
ence
1999Biogenesis of multilamellar bodies
and accumulation of autophagic vacuoles (Eskelinen et al.,2002; Tanaka et al., 2000). LAMP-1 and -2 are major cellularcarriers of polylactosamine (Carlsson and Fukuda, 1990) andthe impact of their glycosylation on lysosome biogenesis andautophagy remain to be determined. N-glycans on cytokinereceptors bind to galectins in an Mgat5-dependent manner andform a multivalent lattice that retains the receptors at the cellsurface. The Mgat5-modified N-glycans increase surfaceresidency of receptors, lowering the response thresholds tomultiple cytokines, including TGF-β, EGF, PDGF and IGF(Partridge et al., 2004). In this manner, autocrine signallingthrough PI3K is potentiated in Mgat5-expressing cells, andmay contribute to MLB formation in both Mgat5-transfectedMv1Lu type II alveolar cells as well as in tumour cells thatexpress increased levels of Mgat5 (Handerson and Pawelek,2003; Hariri et al., 2000).
An autophagic contribution promotes lamella formationin lysosomal organellesAutophagy is involved in both sphingolipid and glycoproteinmetabolism (Ghidoni et al., 1996) and, conversely, sphingolipidceramide can modulate autophagy in HT-29 cells (Scarlatti etal., 2004). 3-MA and other inhibitors of PI3K activity blockautophagy (Blommaart et al., 1997; Hariri et al., 2000; Munafoand Colombo, 2001; Seglen and Gordon, 1982). Our previousdemonstration that 3-MA inhibits both autophagy and theformation of MLBs in Mgat5-transfected Mv1Lu cells (Haririet al., 2000) was confirmed in this study by the disappearanceof phospholipid-rich MLBs in 3-MA-treated cells (Figs 4, 5).Treatment with 3-MA did not prevent the accumulation oflysosomal cholesterol but the swollen, lysosomal structures nolonger expressed the concentric lamellae typical of MLBs. Thesimilar morphology of these swollen, lysosomal structures with
Fig. 8. 3-MA treatment does not affectaccessibility of LAMP-2-positivevacuoles to fluid-phase endocytosis. M9cells either untreated (Ctl, A-F), treatedwith U18666A for 24 hours (U24; G-L)or grown for 3 days in the presence of10 mM 3-MA with U18666A added forthe final 24 hours (U24 + 3-MA; M-R)were incubated for 30 minutes (A-C,G-I,M-O) or 4 hours (D-F,J-L,P-R) in thepresence of 5 mg/ml FITC-dextran priorto labelling for LAMP-2. LAMP-2labelling (A,D,G,J,M,P) is shown in redand FITC-dextran labelling(B,E,H,K,N,Q) in green and mergedimages are presented in C,F,I,L,O,R. Thepercentage of LAMP-2/Nile Red-positiveswollen vacuoles containing FITC-dextran at 0.5, 1, 2 and 4 hours of FITC-dextran endocytosis was quantified fromMv1Lu and M9 cells treated or not with 3-MA for 3 days and/or U18666A for the final 24 hours, as indicated (S). For all treatment conditions,FITC-dextran does not accumulate in swollen LAMP-2-positive vacuoles at 30 minutes and the majority are accessible after only 4 hours ofendocytosis even in the presence of 3-MA. Bar, 5 µm.
Jour
nal o
f Cel
l Sci
ence

2000
the smaller inclusion bodies seen upon 3-MA treatment in theabsence of U18666A suggests that these two structures aresimilar lysosomal organelles that vary in size owing toaccumulation of cholesterol. As seen in Table 1, 3-MAtreatment of U18666A-treated cells resulted in essentially thecomplete transition from expression of MLBs to non-lamellarlysosomal vacuoles. The large size of U18666A-inducedvacuoles enabled us to identify and compare the multilamellarand non-lamellar lysosomal structures in untreated and 3-MA-treated cells by light microscopy.
Inhibitors of PI3K also block vesicular transport from lateendosomes to lysosomes (Punnonen et al., 1994) and impairboth early endosome fusion and maturation of lysosomes(Mousavi et al., 2003; Reaves et al., 1996; Simonsen et al.,1998). More recently, PI3K signalling has been shown to morespecifically regulate endosomal sorting (Petiot et al., 2003). Thelarge non-lamellar LAMP-2-positive, cholesterol-rich vacuolesformed upon addition of U18666A to 3-MA are labelled byfluid-phase uptake of FITC-dextran after 4 hours but not 30minutes and are positive for Lysotracker Red indicating thatthey are acidic, late lysosomal organelles. The non-lamellarvacuoles that accumulate in the presence of 3-MA andU18666A are therefore not pre-lysosomal compartments butcorrespond to late or secondary lysosomes. Furthermore, 3-MAdoes not affect endocytic accessibility of fluid-phase material tothese late, lysosomal organelles. Biosynthetic trafficking ofLAMP to lysosomes can include transit via early endosomesand/or the plasma membrane (Nabi et al., 1991; Peden et al.,2004) and recycling of LAMPs permits uptake of anti-LAMPantibodies to lysosomes (Lippincott-Schwartz and Fambrough,1987; Nabi et al., 1991; Williams and Fukuda, 1990). Asobserved for fluid-phase uptake of FITC-dextran, anti-LAMP-2 antibodies internalized by recycling LAMP-2 access MLBsand non-lamellar vacuoles after 2-4 hours (data not shown).
Multivesicular bodies or endosomes have been observed tofuse with MLBs in type II alveolar cells (Chevalier and Collet,1972; Voorhout et al., 1993; Vorbroker et al., 1995) and sortingof endosomal content to the internal vesicles of multivesicularbodies may represent a requisite aspect of MLB formation(Piper and Luzio, 2001; Stahl and Barbieri, 2002; Weaver et
Journal of Cell Science 118 (9)
Fig. 9. 3-MA treatment does notaffect fluid-phase endocytosis tolysosomes. M9 cells were grown inthe presence of 3-MA for 3 daysand then incubated with FITC-dextran for 30 minutes (A-C) or 4hours (D-F) prior to labelling forLAMP-2 (A,D). FITC-dextranlabelling (B,E) is shown in greenand LAMP-2 in red in the mergedimages (C,F). FITC-dextran doesnot accumulate in lysosomalstructures at 30 minutes but reachesthem after 4 hours of endocytosis.Bar, 5 µm.
Fig. 10. Non-lamellar, cholesterol-rich LAMP-2-positive vacuolesare acidic, lysosomal organelles. M9 cells were grown in regularmedium (A-C,G-I) or in media supplemented with 10 mM 3-MAfor 3 days (D-F,J-L) and selected cultures were incubated withU18666A for the final 24 hours (G-L). Viable cells were labelledwith Lysotracker Red (A,D,G,J) and after fixation, with filipin(B,E,H,K). Merged images (C,F,I,L) show Lysotracker Red in redand filipin in blue. The large cholesterol-rich swollen lysosomalvacuoles induced by U18666A are still acidic in the presence of 3-MA. Bar, 5 µm.
Jour
nal o
f Cel
l Sci
ence

2001Biogenesis of multilamellar bodies
al., 2002). Similarly, fusion between various elements of theendolysosomal and autophagic pathways is well documented(Fig. 11). Formation of nascent autophagic vacuoles isfollowed rapidly by acquisition of hydrolytic enzymes byfusion with pre-existing lysosomes (Lawrence and Brown,1992; Lawrence and Brown, 1993; Lee et al., 1989; Liou et al.,1997) and early autophagic vacuoles fuse with endosomesbefore fusing with lysosomes (Gordon and Seglen, 1988).Interaction between autophagic vacuoles and both endosomaland lysosomal compartments therefore necessarily contributesto the composition of autolysosomes (Fig. 11).
Autophagic vacuoles have long been noted for the presenceof whorl-like membrane structures (Schmitz and Muller,1991). MDC is a fluorescent probe whose specificity forautophagic vacuoles (Biederbick et al., 1995; Munafo andColombo, 2001) is based, unlike other lysomotropic agents, onits ability to act as a solvent polarity probe exhibiting enhancedfluorescence upon interaction with membrane lipids (Niemannet al., 2000). MDC should therefore selectively label densephospholipid-containing organelles such as autophagicvacuoles and MLBs. LC3 is a specific marker for the isolationmembrane of autophagosomes and autophagosomes (Kabeyaet al., 2000; Mizushima, 2004; Mizushima et al., 2001). MDC-positive structures negative for LC3 are induced by nutrientstarvation and MDC would therefore appear to be a marker formature autophagic vacuoles or autolysosomes (Munafo andColombo, 2001; Gutierrez et al., 2004), consistent with thisreport. Reduced labelling of cholesterol-rich U18666A-induced vacuoles with MDC in the presence of 3-MA (Fig. 6)is therefore indicative of the reduced delivery of phospholipidsto these organelles via autophagy, thereby altering the lipidcomposition of these organelles and preventing theirreorganization into concentric lamella, as observed by EM. Theability of serum starvation to enhance the size of MLBs (Fig.7) further demonstrates that macroautophagy regulates theexpression of these organelles.
This study shows that autophagic delivery of phospholipids
to late lysosomal organelles promotes MLB expression in theMv1Lu type II alveolar cell line and, indeed, argues that MLBsare autolysosomes. However, autoradiographic studies haveshown that phospholipids are delivered to MLBs of type IIalveolar cells in intact lung directly from the Golgi apparatus(Chevalier and Collet, 1972). Furthermore, MLBs are absentfrom Mv1Lu cells, as in most type II alveolar-derived cells inculture (Diglio and Kikkawa, 1977; Guttentag et al., 2003;Kawada et al., 1990; Rannels et al., 1987; Sannes, 1991;Tanswell et al., 1991), and the ubiquitous expression of MLBsin various other cell types argues that multiple mechanismsmay contribute to MLB expression. Indeed, we were able toinduce MLB expression in Mv1Lu cells by modulating bothprotein glycosylation and lysosomal cholesterol content.Whether autophagy plays a critical or regulatory role in MLBformation and surfactant expression in lung type II alveolarcells remains to be determined. Nevertheless, regulation of thelipid composition of late lysosomal organelles by autophagypromotes the expression of MLBs and may modulateexpression of these organelles under both physiological andpathological conditions.
This study was supported by a grant from the Canadian Institutesof Health Research (CIHR). I.R.N. is the recipient of a CIHRInvestigator award.
ReferencesAllegranza, A., Tredici, G., Marmiroli, P., di Donato, S., Franceschetti, S.
and Mariani, C. (1989). Sialidosis type I: pathological study in an adult.Clin. Neuropathol. 8, 266-271.
Amano, N., Yokoi, S., Akagi, M., Sakai, M., Yagishita, S. and Nakata, K.(1983). Neuropathological findings of an autopsy case of adult beta-galactosidase and neuraminidase deficiency. Acta Neuropathol. 61, 283-290.
Berra, B., de Gasperi, R., Rapelli, S., Okada, S., Li, S. C. and Li, Y. T.(1986). Presence of glycoproteins containing the polylactosamine structurein brain and liver of GM1 gangliosidosis patients. Comparative studybetween clinical types I and II, using endo-beta-galactosidase enzyme.Neurochem. Pathol. 4, 107-117.
Biederbick, A., Kern, H. F. and Elsasser, H. P. (1995).
+U
B
phospholipid-rich MLB
cholesterol-rich MLB
plasma membrane
endosome
lysosome
+U
Non-lamellar lysosomal vacuole
endosome
lysosome
+3-MA
+U
+U
A isolationmembrane
plasma membrane
isolationmembrane
autophagosome
autolysosome
leupeptin
Fig. 11. An autophagic contribution isrequired for the formation ofphospholipid and cholesterol-richMLBs. (A) Extensive interaction occursbetween the endocytic (black) andautophagic (red) pathways. Maturationof autolysosomes into MLBs requireslysosomal degradation and is inhibitedby leupeptin. U18666A-mediatedlysosomal cholesterol accumulation(+U) induces the formation ofcholesterol-rich MLBs fromautolysosomes or pre-existingphospholipid-rich MLBs independentlyof lysosomal degradation (blue). (B)Upon inhibition of autophagy with 3-MA, the formation of phospholipid-richMLBs is inhibited and U18666A-mediated lysosomal cholesterolaccumulation results in the formation,not of MLBs, but of non-lamellarlysosomal vacuoles.
Jour
nal o
f Cel
l Sci
ence

2002
Monodansylcadaverine (MDC) is a specific in vivo marker for autophagicvacuoles. Eur. J. Cell Biol. 66, 3-14.
Blanchette-Mackie, E. J. (2000). Intracellular cholesterol trafficking: role ofthe NPC1 protein. Biochim. Biophys. Acta. 1486, 171-183.
Blommaart, E. F., Krause, U., Schellens, J. P., Vreeling-Sindelarova, H.and Meijer, A. J. (1997). The phosphatidylinositol 3-kinase inhibitorswortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes.Eur. J. Biochem. 243, 240-246.
Butler, J. D., Blanchette-Mackie, J., Goldin, E., O’Neill, R. R., Carstea,G., Roff, C. F., Patterson, M. C., Patel, S., Comly, M. E., Cooney, A. etal. (1992). Progesterone blocks cholesterol translocation from lysosomes. J.Biol. Chem. 267, 23797-23805.
Campiche, M. A., Gautier, A., Hernandez, E. I. and Reymond, A. (1963).An electron microscope study of the fetal development of human lung.Pediatrics 32, 976-994.
Carlsson, S. R. and Fukuda, M. (1990). The polylactosaminoglycans ofhuman lysosomal membrane glycoproteins lamp-1 and lamp-2. Localizationon the peptide backbones. J. Biol. Chem. 265, 20488-20495.
Chander, A., Johnson, R. G., Reicherter, J. and Fisher, A. B. (1986). Lunglamellar bodies maintain an acidic internal pH. J. Biol. Chem. 261, 6126-6131.
Chen, C. S., Patterson, M. C., Wheatley, C. L., O’Brien, J. F. and Pagano,R. E. (1999). Broad screening test for sphingolipid-storage diseases. Lancet354, 901-905.
Chevalier, G. and Collet, A. J. (1972). In vivo incorporation of choline- 3 H,leucine- 3 H and galactose- 3 H in alveolar type II pneumocytes in relationto surfactant synthesis. A quantitative radoautographic study in mouse byelectron microscopy. Anat. Rec. 174, 289-310.
Choudhury, A., Dominguez, M., Puri, V., Sharma, D. K., Narita, K.,Wheatley, C. L., Marks, D. L. and Pagano, R. E. (2002). Rab proteinsmediate Golgi transport of caveola-internalized glycosphingolipids andcorrect lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 109, 1541-1550.
de Vries, A. C., Schram, A. W., Tager, J. M., Batenburg, J. J. and vanGolde, L. M. (1985). A specific acid alpha-glucosidase in lamellar bodiesof the human lung. Biochim. Biophys. Acta. 837, 230-238.
DeGasperi, R., Alroy, J., Richard, R., Goyal, V., Orgad, U., Lee, R. E. andWarren, C. D. (1990). Glycoprotein storage in Gaucher disease: lectinhistochemistry and biochemical studies. Lab. Invest. 63, 385-392.
Demetriou, M., Nabi, I. R., Coppolino, M., Dedhar, S. and Dennis, J. W.(1995). Reduced contact-inhibition and substratum adhesion in epithelialcells expressing GlcNAc-transferase V. J. Cell Biol. 130, 383-392.
Dennis, J. W., Granovsky, M. and Warren, C. E. (1999). Glycoproteinglycosylation and cancer progression. Biochim. Biophys. Acta. 1473, 21-34.
DiAugustine, R. P. (1974). Lung concentric laminar organelle. Hydrolaseactivity and compositional analysis. J. Biol. Chem. 249, 584-593.
Diglio, C. A. and Kikkawa, Y. (1977). The type II epithelial cells of the lung.IV. Adaption and behavior of isolated type II cells in culture. Lab. Invest.37, 622-631.
Doyle, I. R., Jones, M. E., Barr, H. A., Orgeig, S., Crockett, A. J.,McDonald, C. F. and Nicholas, T. E. (1994). Composition of humanpulmonary surfactant varies with exercise and level of fitness. Am. J. Respir.Crit. Care Med. 149, 1619-1627.
Eskelinen, E. L., Illert, A. L., Tanaka, Y., Schwarzmann, G., Blanz, J., vonFigura, K. and Saftig, P. (2002). Role of LAMP-2 in lysosome biogenesisand autophagy. Mol. Biol. Cell 13, 3355-3368.
Foster, C. D., Zhang, P. X., Gonzales, L. W. and Guttentag, S. H. (2003).In vitro surfactant protein B (SP-B) deficiency inhibits lamellar bodyformation. Am. J. Respir. Cell. Mol. Biol. 29, 259-266.
Ghidoni, R., Houri, J. J., Giuliani, A., Ogier-Denis, E., Parolari, E., Botti,S., Bauvy, C. and Codogno, P. (1996). The metabolism ofsphingo(glyco)lipids is correlated with the differentiation-dependentautophagic pathway in HT-29 cells. Eur. J. Biochem. 237, 454-459.
Gieselmann, V. (1995). Lysosomal storage diseases. Biochim. Biophys. Acta.1270, 103-136.
Gonzales, L. W., Angampalli, S., Guttentag, S. H., Beers, M. F.,Feinstein, S. I., Matlapudi, A. and Ballard, P. L. (2001). Maintenanceof differentiated function of the surfactant system in human fetal lung typeII epithelial cells cultured on plastic. Pediatr. Pathol. Mol. Med. 20, 387-412.
Gordon, P. B. and Seglen, P. O. (1988). Prelysosomal convergence ofautophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 151,40-47.
Gutierrez, M. G., Munafo, D. B., Beron, W. and Colombo, M. I. (2004).
Rab7 is required for the normal progression of the autophagic pathway inmammalian cells. J. Cell Sci. 117, 2687-2697.
Guttentag, S., Robinson, L., Zhang, P., Brasch, F., Buhling, F. and Beers,M. (2003). Cysteine protease activity is required for surfactant protein Bprocessing and lamellar body genesis. Am. J. Respir. Cell Mol. Biol. 28, 69-79.
Haller, T., Dietl, P., Deetjen, P. and Volkl, H. (1996). The lysosomalcompartment as intracellular calcium store in MDCK cells: a possibleinvolvement in InsP3-mediated Ca2+ release. Cell Calcium 19, 157-165.
Handerson, T. and Pawelek, J. M. (2003). Beta1,6-branchedoligosaccharides and coarse vesicles: a common, pervasive phenotype inmelanoma and other human cancers. Cancer Res. 63, 5363-5369.
Hariri, M., Millane, G., Guimond, M. P., Guay, G., Dennis, J. W. and Nabi,I. R. (2000). Biogenesis of multilamellar bodies via autophagy. Mol. Biol.Cell 11, 255-268.
Hass, M. A. and Longmore, W. J. (1979). Surfactant cholesterol metabolismof the isolated perfused rat lung. Biochim. Biophys. Acta. 573, 166-174.
Hatasa, K. and Nakamura, T. (1965). Electron microscopic observations oflung alveolar epithelial cells of normal young mice, with special referenceto formation and secretion of osmiophilic lamellar bodies. Z. Zellforsch.Mikrosk. Anat. 68, 266-277.
Hook, G. E. and Gilmore, L. B. (1982). Hydrolases of pulmonary lysosomesand lamellar bodies. J. Biol. Chem. 257, 9211-9220.
Kawada, H., Shannon, J. M. and Mason, R. J. (1990). Improvedmaintenance of adult rat alveolar type II cell differentiation in vitro: effectof serum-free, hormonally defined medium and a reconstituted basementmembrane. Am. J. Respir. Cell Mol. Biol. 3, 33-43.
Kobayashi, T., Beuchat, M. H., Lindsay, M., Frias, S., Palmiter, R. D.,Sakuraba, H., Parton, R. G. and Gruenberg, J. (1999). Late endosomalmembranes rich in lysobisphosphatidic acid regulate cholesterol transport.Nat. Cell. Biol. 1, 113-118.
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda,T., Kominami, E., Ohsumi, Y. and Yoshimori, T. (2000). LC3, amammalian homologue of yeast Apg8p, is localized in autophagosomemembranes after processing. EMBO J. 19, 5720-5728.
Kolleck, I., Guthmann, F., Ladhoff, A. M., Tandon, N. N., Schlame, M.and Rustow, B. (2002). Cellular cholesterol stimulates acute uptake ofpalmitate by redistribution of fatty acid translocase in type II pneumocytes.Biochemistry 41, 6369-6375.
Kolodny, E. H. (2000). Niemann-Pick disease. Curr. Opin. Hematol. 7, 48-52.
Lange, Y., Ye, J. and Steck, T. L. (1998). Circulation of cholesterolbetween lysosomes and the plasma membrane. J. Biol. Chem. 273, 18915-18922.
Lawrence, B. P. and Brown, W. J. (1992). Autophagic vacuoles rapidly fusewith pre-existing lysosomes in cultured hepatocytes. J. Cell Sci. 102, 515-526.
Lawrence, B. P. and Brown, W. J. (1993). Inhibition of protein synthesisseparates autophagic sequestration from the delivery of lysosomal enzymes.J. Cell Sci. 105, 473-480.
Lee, H. K., Myers, R. A. and Marzella, L. (1989). Stimulation of autophagicprotein degradation by nutrient deprivation in a differentiated murineteratocarcinoma (F9 12-1a) cell line. Exp. Mol. Pathol. 50, 139-146.
Liou, W., Geuze, H. J., Geelen, M. J. and Slot, J. W. (1997). The autophagicand endocytic pathways converge at the nascent autophagic vacuoles. J. CellBiol. 136, 61-70.
Lippincott-Schwartz, J. and Fambrough, D. M. (1987). Cycling of theintegral membrane glycoprotein, LEP100, between plasma membrane andlysosomes: kinetic and morphological analysis. Cell 49, 669-677.
Liscum, L., Ruggiero, R. M. and Faust, J. R. (1989). The intracellulartransport of low density lipoprotein-derived cholesterol is defective inNiemann-Pick type C fibroblasts. J. Cell. Biol. 108, 1625-1636.
Lusa, S., Blom, T. S., Eskelinen, E. L., Kuismanen, E., Mansson, J. E.,Simons, K. and Ikonen, E. (2001). Depletion of rafts in late endocyticmembranes is controlled by NPC1-dependent recycling of cholesterol to theplasma membrane. J. Cell Sci. 114, 1893-1900.
Mizushima, N. (2004). Methods for monitoring autophagy. Int. J. Biochem.Cell. Biol. 36, 2491-2502.
Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y.,Suzuki, K., Tokuhisa, T., Ohsumi, Y. and Yoshimori, T. (2001).Dissection of autophagosome formation using Apg5-deficient mouseembryonic stem cells. J. Cell Biol. 152, 657-668.
Mousavi, S. A., Brech, A., Berg, T. and Kjeken, R. (2003). Phosphoinositide
Journal of Cell Science 118 (9)
Jour
nal o
f Cel
l Sci
ence

2003Biogenesis of multilamellar bodies
3-kinase regulates maturation of lysosomes in rat hepatocytes. Biochem. J.372, 861-869.
Mulugeta, S., Gray, J. M., Notarfrancesco, K. L., Gonzales, L. W., Koval,M., Feinstein, S. I., Ballard, P. L., Fisher, A. B. and Shuman, H. (2002).Identification of LBM180, a lamellar body limiting membrane protein ofalveolar type II cells, as the ABC transporter protein ABCA3. J. Biol. Chem.277, 22147-22155.
Munafo, D. B. and Colombo, M. I. (2001). A novel assay to study autophagy:regulation of autophagosome vacuole size by amino acid deprivation. J. CellSci. 114, 3619-3629.
Nabi, I. R. and Rodriguez-Boulan, E. (1993). Increased LAMP-2polylactosamine glycosylation is associated with its slower Golgi transitduring establishment of a polarized MDCK epithelial monolayer. Mol. Biol.Cell 4, 627-635.
Nabi, I. R., le Bivic, A., Fambrough, D. and Rodriguez-Boulan, E. (1991).An endogenous MDCK lysosomal membrane glycoprotein is targetedbasolaterally before delivery to lysosomes. J. Cell Biol. 115, 1573-1584.
Niemann, A., Takatsuki, A. and Elsasser, H. P. (2000). The lysosomotropicagent monodansylcadaverine also acts as a solvent polarity probe. J.Histochem. Cytochem. 48, 251-258.
O’Hare, K. H. and Sheridan, M. N. (1970). Electron microscopicobservations on the morphogenesis of the albino rat lung, with specialreference to pulmonary epithelial cells. Am. J. Anat. 127, 181-205.
Orgeig, S. and Daniels, C. B. (2001). The roles of cholesterol in pulmonarysurfactant: insights from comparative and evolutionary studies. Comp.Biochem. Physiol. A. Mol. Integr. Physiol. 129, 75-89.
Orgeig, S., Barr, H. A. and Nicholas, T. E. (1995). Effect of hyperpnea onthe cholesterol to disaturated phospholipid ratio in alveolar surfactant of rats.Exp. Lung Res. 21, 157-174.
Orgeig, S., Daniels, C. B., Johnston, S. D. and Sullivan, L. C. (2003). Thepattern of surfactant cholesterol during vertebrate evolution anddevelopment: does ontogeny recapitulate phylogeny? Reprod. Fertil. Dev.15, 55-73.
Ory, D. S. (2000). Niemann-Pick type C: a disorder of cellular cholesteroltrafficking. Biochim. Biophys. Acta. 1529, 331-339.
Pagano, R. E., Puri, V., Dominguez, M. and Marks, D. L. (2000). Membranetraffic in sphingolipid storage diseases. Traffic 1, 807-815.
Partridge, E. A., le Roy, C., di Guglielmo, G. M., Pawling, J., Cheung, P.,Granovsky, M., Nabi, I. R., Wrana, J. L. and Dennis, J. W. (2004).Regulation of cytokine receptors by Golgi N-glycan processing andendocytosis. Science 306, 120-124.
Peden, A. A., Oorschot, V., Hesser, B. A., Austin, C. D., Scheller, R. H. andKlumperman, J. (2004). Localization of the AP-3 adaptor complex definesa novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol.164, 1065-1076.
Pentchev, P. G., Comly, M. E., Kruth, H. S., Tokoro, T., Butler, J., Sokol,J., Filling-Katz, M., Quirk, J. M., Marshall, D. C., Patel, S. et al. (1987).Group C Niemann-Pick disease: faulty regulation of low-densitylipoprotein uptake and cholesterol storage in cultured fibroblasts. FASEB J.1, 40-45.
Petiot, A., Faure, J., Stenmark, H. and Gruenberg, J. (2003). PI3P signalingregulates receptor sorting but not transport in the endosomal pathway. J. CellBiol. 162, 971-999.
Petiot, A., Ogier-Denis, E., Blommaart, E. F., Meijer, A. J. and Codogno,P. (2000). Distinct classes of phosphatidylinositol 3′-kinases are involved insignaling pathways that control macroautophagy in HT-29 cells. J. Biol.Chem. 275, 992-998.
Piper, R. C. and Luzio, J. P. (2001). Late endosomes: sorting and partitioningin multivesicular bodies. Traffic 2, 612-621.
Platt, F. M., Neises, G. R., Reinkensmeier, G., Townsend, M. J., Perry, V.H., Proia, R. L., Winchester, B., Dwek, R. A. and Butters, T. D. (1997).Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science 276, 428-431.
Punnonen, E. L., Reunanen, H., Hirsimaki, P. and Lounatmaa, K. (1988).Filipin labelling and intramembrane particles on the membranes of early andlater autophagic vacuoles in Ehrlich ascites cells. Virchows Arch. B CellPathol. Incl. Mol. Pathol. 54, 317-326.
Punnonen, E. L., Marjomaki, V. S. and Reunanen, H. (1994). 3-Methyladenine inhibits transport from late endosomes to lysosomes incultured rat and mouse fibroblasts. Eur. J. Cell Biol. 65, 14-25.
Puri, V., Watanabe, R., Dominguez, M., Sun, X., Wheatley, C. L., Marks,D. L. and Pagano, R. E. (1999). Cholesterol modulates membrane trafficalong the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol.1, 386-388.
Rannels, S. R., Fisher, C. S., Heuser, L. J. and Rannels, D. E. (1987).Culture of type II pneumocytes on a type II cell-derived fibronectin-richmatrix. Am. J. Physiol. 253, C759-C765.
Reaves, B. J., Bright, N. A., Mullock, B. M. and Luzio, J. P. (1996). Theeffect of wortmannin on the localisation of lysosomal type I integralmembrane glycoproteins suggests a role for phosphoinositide 3-kinaseactivity in regulating membrane traffic late in the endocytic pathway. J. CellSci. 109, 749-762.
Sannes, P. L. (1991). Structural and functional relationships between type IIpneumocytes and components of extracellular matrices. Exp. Lung Res. 17,639-659.
Scarlatti, F., Bauvy, C., Ventruti, A., Sala, G., Cluzeaud, F., Vandewalle,A., Ghidoni, R. and Codogno, P. (2004). Ceramide-mediatedmacroautophagy involves inhibition of protein kinase B and up-regulationof beclin 1. J. Biol. Chem. 279, 18384-18391.
Schmitz, G. and Muller, G. (1991). Structure and function of lamellar bodies,lipid-protein complexes involved in storage and secretion of cellular lipids.J. Lipid Res. 32, 1539-1570.
Seglen, P. O. and Gordon, P. B. (1982). 3-Methyladenine: specific inhibitorof autophagic/lysosomal protein degradation in isolated rat hepatocytes.Proc. Natl. Acad. Sci. USA 79, 1889-1892.
Shulenin, S., Nogee, L. M., Annilo, T., Wert, S. E., Whitsett, J. A. andDean, M. (2004). ABCA3 gene mutations in newborns with fatal surfactantdeficiency. New Engl. J. Med. 350, 1296-1303.
Simons, K. and Gruenberg, J. (2000). Jamming the endosomal system: lipidrafts and lysosomal storage diseases. Trends Cell Biol. 10, 459-462.
Simonsen, A., Lippe, R., Christoforidis, S., Gaullier, J. M., Brech, A.,Callaghan, J., Toh, B. H., Murphy, C., Zerial, M. and Stenmark, H.(1998). EEA1 links PI(3)K function to Rab5 regulation of endosome fusion.Nature 394, 494-498.
Simonsen, A., Wurmser, A. E., Emr, S. D. and Stenmark, H. (2001). Therole of phosphoinositides in membrane transport. Curr. Opin. Cell Biol. 13,485-492.
Stahl, P. D. and Barbieri, M. A. (2002). Multivesicular bodies andmultivesicular endosomes: the “ins and outs” of endosomal traffic. Sci.STKE 2002, PE32.
Stahlman, M. T., Gray, M. P., Falconieri, M. W., Whitsett, J. A. andWeaver, T. E. (2000). Lamellar body formation in normal and surfactantprotein B-deficient fetal mice. Lab Invest. 80, 395-403.
Susan, P. P. and Dunn, W. A., Jr (2001). Starvation-induced lysosomaldegradation of aldolase B requires glutamine 111 in a signal sequence forchaperone-mediated transport. J. Cell Physiol. 187, 48-58.
Tanaka, Y., Guhde, G., Suter, A., Eskelinen, E. L., Hartmann, D.,Lullmann-Rauch, R., Janssen, P. M., Blanz, J., von Figura, K. andSaftig, P. (2000). Accumulation of autophagic vacuoles andcardiomyopathy in LAMP-2-deficient mice. Nature 406, 902-906.
Tanswell, A. K., Byrne, P. J., Han, R. N., Edelson, J. D. and Han, V. K.(1991). Limited division of low-density adult rat type II pneumocytes inserum-free culture. Am. J. Physiol. 260, L395-L402.
Thomas, A. Q., Lane, K., Phillips, J., 3rd, Prince, M., Markin, C., Speer,M., Schwartz, D. A., Gaddipati, R., Marney, A., Johnson, J. et al.(2002). Heterozygosity for a surfactant protein C gene mutation associatedwith usual interstitial pneumonitis and cellular nonspecific interstitialpneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 165, 1322-1328.
Tomiyama, Y., Waguri, S., Kanamori, S., Kametaka, S., Wakasugi, M.,Shibata, M., Ebisu, S. and Uchiyama, Y. (2004). Early-phaseredistribution of the cation-independent mannose 6-phosphate receptor byU18666A treatment in HeLa cells. Cell Tissue Res. 317, 253-264.
Voorhout, W. F., Weaver, T. E., Haagsman, H. P., Geuze, H. J. and vanGolde, L. M. (1993). Biosynthetic routing of pulmonary surfactant proteinsin alveolar type II cells. Microsc. Res. Tech. 26, 366-373.
Vorbroker, D. K., Profitt, S. A., Nogee, L. M. and Whitsett, J. A. (1995).Aberrant processing of surfactant protein C in hereditary SP-B deficiency.Am. J. Physiol. 268, L647-L656.
Weaver, T. E., Na, C. L. and Stahlman, M. (2002). Biogenesis of lamellarbodies, lysosome-related organelles involved in storage and secretion ofpulmonary surfactant. Semin. Cell. Dev. Biol. 13, 263-270.
Williams, M. A. and Fukuda, M. (1990). Accumulation of membraneglycoproteins in lysosomes requires a tyrosine residue at a particularposition in the cytoplasmic tail. J. Cell Biol. 111, 955-966.
Wurmser, A. E., Gary, J. D. and Emr, S. D. (1999). Phosphoinositide 3-kinases and their FYVE domain-containing effectors as regulators ofvacuolar/lysosomal membrane trafficking pathways. J. Biol. Chem. 274,9129-9132.
Jour
nal o
f Cel
l Sci
ence





![Essential Fatty Acid And Cell Culture: Where We Stand · Supplementation with essential fatty acids restored the lipid profile of the cell membrane and vacuoles [1]. Since the work](https://static.fdocuments.us/doc/165x107/5f6f855662ade1060a7b3cca/essential-fatty-acid-and-cell-culture-where-we-stand-supplementation-with-essential.jpg)












