
Arsenic inhibits autophagic flux activating the Nrf2-Keap1 pathway
11
Arsenic Inhibits Autophagic Flux, Activating the Nrf2-Keap1 Pathway in a p62-Dependent Manner Alexandria Lau, a Yi Zheng, a Shasha Tao, a Huihui Wang, a Samantha A. Whitman, a Eileen White, b,c Donna D. Zhang a,d Department of Pharmacology and Toxicology, University of Arizona, Tucson, Arizona, USA a ; Cancer Institute of New Jersey, New Brunswick, New Jersey, USA b ; Department of Molecular Biology and Biochemistry, Rutgers University, Piscataway, New Jersey, USA c ; Arizona Cancer Center, University of Arizona, Tucson, Arizona, USA d The Nrf2-Keap1 signaling pathway is a protective mechanism promoting cell survival. Activation of the Nrf2 pathway by natural compounds has been proven to be an effective strategy for chemoprevention. Interestingly, a cancer-promoting function of Nrf2 has recently been observed in many types of tumors due to deregulation of the Nrf2-Keap1 axis, which leads to constitutive acti- vation of Nrf2. Here, we report a novel mechanism of Nrf2 activation by arsenic that is distinct from that of chemopreventive compounds. Arsenic deregulates the autophagic pathway through blockage of autophagic flux, resulting in accumulation of au- tophagosomes and sequestration of p62, Keap1, and LC3. Thus, arsenic activates Nrf2 through a noncanonical mechanism (p62 dependent), leading to a chronic, sustained activation of Nrf2. In contrast, activation of Nrf2 by sulforaphane (SF) and tert-bu- tylhydroquinone (tBHQ) depends upon Keap1-C151 and not p62 (the canonical mechanism). More importantly, SF and tBHQ do not have any effect on autophagy. In fact, SF and tBHQ alleviate arsenic-mediated deregulation of autophagy. Collectively, these findings provide evidence that arsenic causes prolonged activation of Nrf2 through autophagy dysfunction, possibly pro- viding a scenario similar to that of constitutive activation of Nrf2 found in certain human cancers. This may represent a previ- ously unrecognized mechanism underlying arsenic toxicity and carcinogenicity in humans. T he Nrf2-Keap1 pathway is the main cellular defense mecha- nism against oxidative and electrophilic stresses (1–3). Nrf2 is a ubiquitously expressed transcription factor that regulates the expression of genes bearing an antioxidant response element (ARE) in the promoter region (4). These genes include antioxi- dant proteins, phase II detoxification enzymes, transport proteins, proteasome subunits, chaperones, growth factors, receptors, and other transcription factors (4, 5). Under normal, unstressed con- ditions, Keap1 tightly regulates Nrf2. Keap1 is a substrate adaptor protein that associates with cullin 3 (Cul3) and Rbx1 to form an E3 ubiquitin ligase complex to ubiquitinate Nrf2, which is then de- graded by the 26S proteasome to maintain low basal Nrf2 levels (6, 7). Critical cysteine residues in Keap1 act as regulatory sensors and are modified upon exposure to oxidative or electrophilic stress (8–10). It has been hypothesized that modification of critical cys- teine residues causes a conformational change of the Keap1- Cul3-E3 ubiquitin ligase complex and prevents Nrf2 from being ubiquitinated. Structurally diverse small molecules, such as sul- foraphane (SF), tert-butylhydroquinone (tBHQ), cinnamalde- hyde, and resveratrol, as well as environmental toxicants, includ- ing the heavy metals arsenic, cadmium, and chromium, are able to activate the Nrf2-Keap1 pathway (10–12). It is well accepted that activation of the Nrf2 pathway by certain organic compounds, or chemopreventive agents, can reduce the risk of cancer. The observation that Nrf2 / mice are more sus- ceptible to the deleterious effects of chemical toxicants and car- cinogens, coupled with them being refractory to the protective actions of chemopreventive compounds, clearly demonstrates the role of Nrf2 in chemoprevention (1, 13). Therefore, Nrf2 has gen- erally been considered to be a protective mechanism in normal cells. Paradoxically, the “dark side” of Nrf2 has recently been re- vealed. Genetic mutations (14–17) in either Keap1 or Nrf2 that disrupt the Keap1-mediated negative regulation of Nrf2, leading to constitutive activation of Nrf2, have been identified in many human tumors and cell lines. Epigenetic alteration of Keap1 re- sulting in reduced expression of Keap1 is yet another mechanism of Nrf2 overexpression in many cancer types (18–20). Further- more, cancer cells have hijacked the Nrf2 response to protect themselves against cell death, resulting in intrinsic or acquired chemoresistance (21–25). This was experimentally confirmed when knockdown or inhibition of Nrf2 increased the sensitivity of cancer cells to chemotherapeutics (23, 26). Uncovering the dark side of Nrf2 in cancer has led us to question whether Nrf2 activa- tion by arsenic is associated with the cancer-promoting role of Nrf2 and/or arsenic. Chronic exposure to inorganic arsenic from contaminated drinking water has long been associated with high incidences of cancer in various organs, particularly in the skin, lung, bladder, liver, and kidney (27). Arsenic affects a multitude of biological systems; however, the mechanism by which arsenic elicits its toxic and carcinogenic effects remains largely unknown. Numerous studies have been conducted to elucidate the molecular events associated with arsenic exposure, and resulting data suggest mul- tiple mechanisms. For instance, arsenic is able to alter DNA meth- ylation and repair, regulate gene expression through an epigenetic mechanism, affect cell proliferation, generate reactive oxygen spe- cies (ROS), and modulate various intracellular signaling pathways (28–30). Arsenic has also been shown to induce Nrf2 and the expression of Nrf2-dependent downstream genes in a variety of cell lines, including UROtsa cells, HaCaT cells, osteoblasts, and Received 2 January 2013 Returned for modification 28 January 2013 Accepted 22 February 2013 Published ahead of print 15 April 2013 Address correspondence to Donna D. Zhang, [email protected]. A.L. and Y.Z. contributed equally. Copyright © 2013, American Society for Microbiology. All Rights Reserved. doi:10.1128/MCB.01748-12 2436 mcb.asm.org Molecular and Cellular Biology p. 2436 –2446 June 2013 Volume 33 Number 12 Downloaded from https://journals.asm.org/journal/mcb on 17 January 2022 by 125.202.238.18.
Transcript of Arsenic inhibits autophagic flux activating the Nrf2-Keap1 pathway

Arsenic Inhibits Autophagic Flux, Activating the Nrf2-Keap1 Pathwayin a p62-Dependent Manner
Alexandria Lau,a Yi Zheng,a Shasha Tao,a Huihui Wang,a Samantha A. Whitman,a Eileen White,b,c Donna D. Zhanga,d
Department of Pharmacology and Toxicology, University of Arizona, Tucson, Arizona, USAa; Cancer Institute of New Jersey, New Brunswick, New Jersey, USAb;Department of Molecular Biology and Biochemistry, Rutgers University, Piscataway, New Jersey, USAc; Arizona Cancer Center, University of Arizona, Tucson, Arizona, USAd
The Nrf2-Keap1 signaling pathway is a protective mechanism promoting cell survival. Activation of the Nrf2 pathway by naturalcompounds has been proven to be an effective strategy for chemoprevention. Interestingly, a cancer-promoting function of Nrf2has recently been observed in many types of tumors due to deregulation of the Nrf2-Keap1 axis, which leads to constitutive acti-vation of Nrf2. Here, we report a novel mechanism of Nrf2 activation by arsenic that is distinct from that of chemopreventivecompounds. Arsenic deregulates the autophagic pathway through blockage of autophagic flux, resulting in accumulation of au-tophagosomes and sequestration of p62, Keap1, and LC3. Thus, arsenic activates Nrf2 through a noncanonical mechanism (p62dependent), leading to a chronic, sustained activation of Nrf2. In contrast, activation of Nrf2 by sulforaphane (SF) and tert-bu-tylhydroquinone (tBHQ) depends upon Keap1-C151 and not p62 (the canonical mechanism). More importantly, SF and tBHQdo not have any effect on autophagy. In fact, SF and tBHQ alleviate arsenic-mediated deregulation of autophagy. Collectively,these findings provide evidence that arsenic causes prolonged activation of Nrf2 through autophagy dysfunction, possibly pro-viding a scenario similar to that of constitutive activation of Nrf2 found in certain human cancers. This may represent a previ-ously unrecognized mechanism underlying arsenic toxicity and carcinogenicity in humans.
The Nrf2-Keap1 pathway is the main cellular defense mecha-nism against oxidative and electrophilic stresses (1–3). Nrf2 is
a ubiquitously expressed transcription factor that regulates theexpression of genes bearing an antioxidant response element(ARE) in the promoter region (4). These genes include antioxi-dant proteins, phase II detoxification enzymes, transport proteins,proteasome subunits, chaperones, growth factors, receptors, andother transcription factors (4, 5). Under normal, unstressed con-ditions, Keap1 tightly regulates Nrf2. Keap1 is a substrate adaptorprotein that associates with cullin 3 (Cul3) and Rbx1 to form an E3ubiquitin ligase complex to ubiquitinate Nrf2, which is then de-graded by the 26S proteasome to maintain low basal Nrf2 levels (6,7). Critical cysteine residues in Keap1 act as regulatory sensors andare modified upon exposure to oxidative or electrophilic stress(8–10). It has been hypothesized that modification of critical cys-teine residues causes a conformational change of the Keap1-Cul3-E3 ubiquitin ligase complex and prevents Nrf2 from beingubiquitinated. Structurally diverse small molecules, such as sul-foraphane (SF), tert-butylhydroquinone (tBHQ), cinnamalde-hyde, and resveratrol, as well as environmental toxicants, includ-ing the heavy metals arsenic, cadmium, and chromium, are able toactivate the Nrf2-Keap1 pathway (10–12).
It is well accepted that activation of the Nrf2 pathway by certainorganic compounds, or chemopreventive agents, can reduce therisk of cancer. The observation that Nrf2�/� mice are more sus-ceptible to the deleterious effects of chemical toxicants and car-cinogens, coupled with them being refractory to the protectiveactions of chemopreventive compounds, clearly demonstrates therole of Nrf2 in chemoprevention (1, 13). Therefore, Nrf2 has gen-erally been considered to be a protective mechanism in normalcells. Paradoxically, the “dark side” of Nrf2 has recently been re-vealed. Genetic mutations (14–17) in either Keap1 or Nrf2 thatdisrupt the Keap1-mediated negative regulation of Nrf2, leadingto constitutive activation of Nrf2, have been identified in manyhuman tumors and cell lines. Epigenetic alteration of Keap1 re-
sulting in reduced expression of Keap1 is yet another mechanismof Nrf2 overexpression in many cancer types (18–20). Further-more, cancer cells have hijacked the Nrf2 response to protectthemselves against cell death, resulting in intrinsic or acquiredchemoresistance (21–25). This was experimentally confirmedwhen knockdown or inhibition of Nrf2 increased the sensitivity ofcancer cells to chemotherapeutics (23, 26). Uncovering the darkside of Nrf2 in cancer has led us to question whether Nrf2 activa-tion by arsenic is associated with the cancer-promoting role ofNrf2 and/or arsenic.
Chronic exposure to inorganic arsenic from contaminateddrinking water has long been associated with high incidences ofcancer in various organs, particularly in the skin, lung, bladder,liver, and kidney (27). Arsenic affects a multitude of biologicalsystems; however, the mechanism by which arsenic elicits its toxicand carcinogenic effects remains largely unknown. Numerousstudies have been conducted to elucidate the molecular eventsassociated with arsenic exposure, and resulting data suggest mul-tiple mechanisms. For instance, arsenic is able to alter DNA meth-ylation and repair, regulate gene expression through an epigeneticmechanism, affect cell proliferation, generate reactive oxygen spe-cies (ROS), and modulate various intracellular signaling pathways(28–30). Arsenic has also been shown to induce Nrf2 and theexpression of Nrf2-dependent downstream genes in a variety ofcell lines, including UROtsa cells, HaCaT cells, osteoblasts, and
Received 2 January 2013 Returned for modification 28 January 2013Accepted 22 February 2013
Published ahead of print 15 April 2013
Address correspondence to Donna D. Zhang, [email protected].
A.L. and Y.Z. contributed equally.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/MCB.01748-12
2436 mcb.asm.org Molecular and Cellular Biology p. 2436–2446 June 2013 Volume 33 Number 12
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

fibroblasts (31–35). Our laboratory has demonstrated that arsenicactivates the Nrf2 pathway and upregulates many of its down-stream genes through a Keap1 cysteine 151 (Keap1-C151)-inde-pendent mechanism (31), which is different from well-character-ized Nrf2 inducers such as tBHQ and SF (3, 7, 8, 36). These dataimply that different modes of Nrf2 activation (Keap1-C151 de-pendent versus independent) may determine whether Nrf2 is ben-eficial or detrimental.
Very recently, arsenic was also shown to induce autophagy, abulk-lysosomal degradation pathway where cytoplasmic compo-nents, misfolded proteins, damaged organelles, and specificallytargeted proteins are sequestered into double-membrane vesiclescalled autophagosomes (37–39). Autophagy was thought to be anonselective degradation pathway; however, selective substrateadaptor proteins, such as p62, have been shown to facilitate deg-radation of specific proteins through autophagy (40). p62 inter-acts directly with microtubule-associated protein 1 light chain 3(LC3), a component of the autophagosomal membrane, that iscleaved (LC3-I) and lipidated (LC3-II) and can be used as amarker for autophagy (41). Not only does p62 self-oligomerizeand bind ubiquitin, it also is incorporated into the autophago-some and degraded via autophagy (40–42). The role of p62 in theNrf2-Keap1 pathway was insufficiently studied until very recently,when our group and others independently demonstrated that anincrease in the level of p62, either by overexpression or deregula-tion of autophagy, sequesters Keap1 into autophagosomesthrough direct interactions (43–47). p62-mediated sequestrationof Keap1 stabilized Nrf2 and increased the transcription levels ofits downstream genes (43). This p62-dependent mode of Nrf2activation was termed the “noncanonical” mechanism of Nrf2activation (43).
In the current study, we demonstrate that arsenic blocks au-tophagic flux, resulting in accumulation of p62 and sequestrationof Keap1 into autophagosomes. This was also confirmed in hu-man lung epithelial cells because the lung is a primary target organof arsenic toxicity. Furthermore, this p62-mediated sequestrationof Keap1 prolonged Nrf2 activation, seemingly mimicking thedark side of Nrf2 in cancer. Consistent with the notion that au-tophagic flux is blocked by arsenic, an overall increase in p62protein levels was observed following low-dose arsenic treatment.More importantly, our results show that arsenic-mediated dereg-ulation of autophagy is attenuated by SF and tBHQ, indicatingthat canonical Nrf2 activators (chemopreventive compounds thatactivate Nrf2 in a Keap1-C151-dependent manner) may be usedto protect against arsenic toxicity and carcinogenicity.
MATERIALS AND METHODSMaterials, antibodies, and cell culture. Sodium arsenite, tBHQ, insulin,transferrin, dexamethasone, trypsin inhibitor, and the antibody againstLC3 were all purchased from Sigma-Aldrich. L-Sulforaphane was used inall experiments and was purchased from LKT Laboratories, Inc. Primaryantibodies specifically against Nrf2, Keap1, p62, glyceraldehyde-3-phos-phate dehydrogenase (GAPDH), and �-actin as well as mouse, rabbit, andgoat horseradish peroxidase (HRP)-conjugated secondary antibodieswere purchased from Santa Cruz Biotechnology. Alexa Fluor 488, 594,and 680 were purchased from Invitrogen.
HEK293, NIH 3T3, and BEAS-2B cells were purchased from theAmerican Type Culture Collection (ATCC), and enhanced green fluores-cent protein (EGFP)-LC3-expressing immortalized baby mouse kidney(iBMK) cells were established in the laboratory of Eileen White (CancerInstitute of New Jersey). The 16HBE14o� (HBE) cell line was obtained
from the California Pacific Medical Center, San Francisco, CA. NIH 3T3and iBMK cell lines were maintained in Dulbecco’s modified Eagle’s me-dium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1%L-glutamine, and 0.01% gentamicin. BEAS-2B cells were grown in Ham’sF-12 medium (MediaTech) supplemented with 1 ml bovine hypothala-mus extract (PromoCell), insulin (2 mg/ml) (Sigma), transferrin (2.5 mg/ml) (Sigma-Aldrich), dexamethasone (0.05 mM) (Sigma), cholera toxin(10 �g/ml) (List Biological Laboratories, Inc.), and epidermal growthfactor (10 �g/ml) (Millipore). HBE cells were grown in modified Eagle’smedium supplemented with 10% FBS, 1% L-glutamine, and 0.01% gen-tamicin. HEK293 cells were grown in the same medium as HBE cells andwere supplemented with 0.1 mM nonessential amino acids (Cellgro) and1 mM sodium pyruvate (Gibco). All cell lines were incubated at 37°C in ahumidified incubator containing 5% CO2.
Transfection of siRNA and cDNA and luciferase reporter assay.Transfection of cDNA was performed by using Lipofectamine Plus (In-vitrogen), and HiPerfect was used for transfection of small interferingRNA (siRNA); both were used according to the manufacturer’s instruc-tions. p62 siRNA (SI00057596) and Keap1 siRNA (SI03246439) were pur-chased from Qiagen. Cells were transfected with wild-type Keap1 (Keap1-WT) and Keap1-C151S, along with plasmids encoding mouse glutathioneS-transferase (mGST)–ARE–firefly luciferase and thymidine kinase (TK)-Renilla luciferase (internal control). Luciferase activities were measuredby using the Promega dual-luciferase reporter assay system. The experi-ment was run in triplicate and repeated three times. Results are expressedas means � standard deviations (SD). A P value of �0.05 was consideredto be significant.
Immunoblot analysis, indirect immunofluorescence, and live-cellimaging. Cells were harvested in 1� sample buffer (50 mM Tris-HCl [pH6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 100 mM dithiothre-itol [DTT], 0.1% bromophenol blue) and immediately boiled for 3 to 5min. Samples were sonicated, resolved by SDS-PAGE, and transferredonto a nitrocellulose membrane for immunoblot analysis.
For indirect immunofluorescence, cells were grown on round glasscoverslips (Fisher Scientific) in 35-mm cell culture dishes. Following 20min of fixation with prechilled methanol, coverslips were washed withphosphate-buffered saline (PBS), permeabilized with 0.2% TritonX-100 –PBS for 15 min, and blocked with 2% bovine serum albumin–PBSfor 30 min. Coverslips were then incubated with primary antibody for 1 h,followed by three 10-min washes in PBS. Coverslips were then incubatedwith secondary antibody for 50 min, followed by three 10-min washes inPBS. The coverslips were mounted onto glass slides with antifade mount-ing medium purchased from Invitrogen. NIH 3T3 cells were used forlive-cell imaging due to the cytoplasm-to-nucleus ratio, therefore allow-ing the localization of proteins in the cytoplasm to be more easily distin-guishable. Furthermore, the iBMK stable cell line was used to avoid in-duction of autophagy caused by lipid transfection of LC3 and was the onlystable cell line available to assess formation of autophagosomes. For live-cell imaging, cells were grown on 35-mm glass-bottom dishes from InVivo Scientific. Cells were gently washed once with 1� PBS and imaged inphenol red-free DMEM supplemented with 10% FBS. Images were cap-tured with a Zeiss Observer.Z1 microscope by using the Slidebook4.2.0.11 computer program (Intelligent Imaging Innovations, Inc.).
Electron microscopy. Electron microscopy (EM) was performed atthe SWEHSC cellular imaging facility core at the University of Arizona.Briefly, HEK293 and BEAS-2B cells were treated with the indicated dosesof arsenic, SF, or both for 4 h. Cells were then fixed in 2.5% glutaraldehydein 0.1 M cacodylate buffer, followed by postfixation in 1% osmium tetrox-ide. Cells were then washed, pelleted, stained with 4% aqueous uranylacetate, and dehydrated with ethanol infiltrated with Spurr’s resin. Cellswere incubated at 60°C to allow polymerization. Cut silver-gold sectionswere then mounted onto 150-mesh copper grids and stained with 2% leadacetate for 3 min. Sections were examined by using an FEI CM12 trans-mission electron microscope (TEM) (Philips) operated at 80 kV. Electron
Arsenic Activates Nrf2 in a p62-Dependent Manner
June 2013 Volume 33 Number 12 mcb.asm.org 2437
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

micrograph images were collected on an Optronics Macrofire AMT 542digital camera at the indicated magnifications.
RESULTSArsenic causes accumulation of autophagosomes by inhibitingthe later stage of autophagy. HEK293 cells were treated with lowdoses of arsenic for 4 h. Electron microscopy (EM) showed that250 nM, 500 nM, and 1 �M arsenic induces formation of autopha-gosomes (seen as double-membrane vesicles) in HEK293 cells(Fig. 1). Consistent with the results of EM indicating an upregu-lation of autophagosomes, immortalized baby mouse kidney(iBMK) cells stably expressing EGFP-LC3 also showed an increasein the number of LC3 puncta when cells were exposed to 125 nM,250 nM, 500 nM, or 1 �M arsenic for 4 h, which remained for upto 24 h (Fig. 2). In contrast, 1.25 �M SF did not induce LC3 puncta(Fig. 2), indicating that SF has no effect on autophagy. Although ithas been well established that both arsenic and SF are Nrf2 induc-ers, these results indicate that only arsenic, and not SF, is able toenhance the number of autophagosomes.
An increase in the number of autophagosomes not only is in-dicative of autophagy induction but also could be the result of aninhibition of autophagosome clearance. To differentiate these twophenomena experimentally, a tandem mouse red fluorescent pro-tein (mRFP)-GFP-LC3 construct was transfected into cells to de-termine which stage of the autophagic process is affected by arse-nic. GFP fluorescence is quenched in acidic environments such asthat of the lysosome or when an autophagosome fuses with thelysosome to form an autolysosome, whereas mRFP is more stable.Therefore, colocalization of both GFP and RFP fluorescence (yel-low/orange puncta in the merged image) indicates an autophago-some that has not yet fused with a lysosome or where acidificationof the lysosome is disrupted. In contrast, RFP alone (without GFP)corresponds to an autolysosome. Two positive controls were usedto differentiate increased formation of autophagosomes (starva-tion) from blockage of autophagosome clearance (bafilomycin A1[Baf A1], which has been reported to prevent fusion of autopha-gosomes with lysosomes or inhibit acidification of autolyso-somes). NIH 3T3 cells transfected with the tandem mRFP-GFP-LC3 construct were either starved in Hanks balanced salt solution(HBSS) or treated with 100 nM Baf A1 for 4 h. Starved cells hadboth yellow and red puncta, indicating autophagosomes and au-tolysosomes, respectively (Fig. 3A). Cells treated with Baf A1,however, had mostly yellow/orange puncta (Fig. 3A). Interest-ingly, only yellow/orange puncta were observed in cells treatedwith 500 nM or 1 �M arsenic as early as 2 h and persisted for thelatest time point measured, 12 h (Fig. 3B to E). Cells at every timepoint treated with 1.25 �M SF resembled untreated cells, such thatno or minimal puncta were observed (Fig. 3A to E). These resultssupport that the mechanism by which arsenic affects autophagy issimilar to that of Baf A1, preventing autophagic flux either byinhibiting fusion of autophagosomes with lysosomes or by affect-ing acidification of autolysosomes.
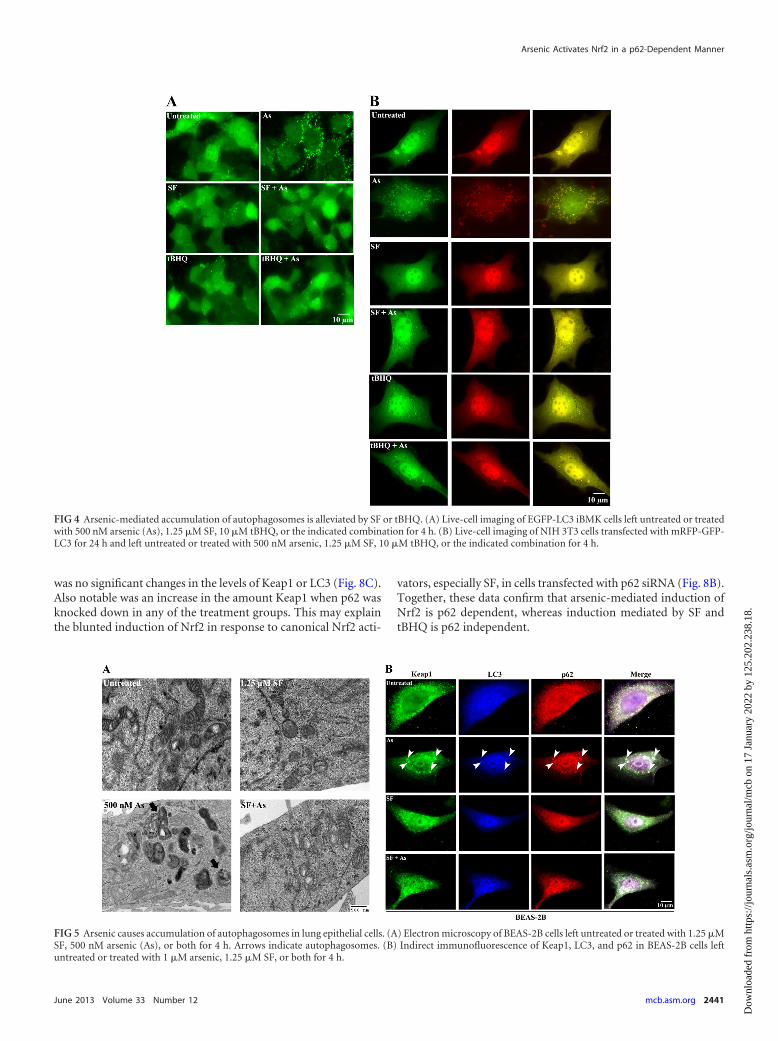
Arsenic-mediated induction of autophagosomes is alleviatedby canonical Nrf2 activators. Nrf2 activators, such as SF andtBHQ, have previously been reported to protect against arsenictoxicity; however, how they impose this protective mechanism hasyet to be elucidated. Therefore, whether these Keap1-C151-de-pendent canonical Nrf2 activators were able to alleviate arsenic-mediated effects on autophagosomes was determined. Live-cellimaging of the EGFP-LC3-expressing stable iBMK cell line treatedwith 500 nM arsenic alone showed an increase in the number ofLC3 puncta compared to untreated or SF- or tBHQ-treated cells(Fig. 4A). Interestingly, SF and tBHQ treatment in combinationwith arsenic showed a reversal or inhibition of arsenic-inducedLC3 puncta (Fig. 4A). Consistent with these results, live-cell im-aging of NIH 3T3 cells transfected with the tandem mRFP-GFP-LC3 construct and treated with either 1 �M arsenic, 1.25 �M SF,25 �M tBHQ, or the indicated combinations also demonstratedthat canonical Nrf2 activators reduced numbers of arsenic-in-duced RFP- and GFP-LC3 puncta (Fig. 4B). Taken together, theseresults indicate that canonical Nrf2 activators are able to suppressarsenic-mediated autophagic deregulation.
Canonical Nrf2 activators protect against arsenic-mediatedautophagic deregulation in lung epithelial cells. One of the pri-mary target organs for arsenic exposure is the lung. In order toconfirm the above-mentioned findings in a relevant in vitro modelfor arsenic toxicity, two types of immortalized but nontrans-formed lung epithelial cell lines, BEAS-2B and HBE, were utilized.EM was conducted on BEAS-2B cells left untreated or treated with
FIG 1 Arsenic increases the number of autophagosomes. Shown are electronmicroscopy images of HEK293 cells left untreated or treated with 250 nM, 500nM, or 1 �M arsenic, as indicated in each panel, for 4 h. Arrows indicatedouble-membrane vesicles (autophagosomes).
Lau et al.
2438 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

500 nM arsenic. Consistent with the results seen with HEK293cells (48), arsenic-treated BEAS-2B cells showed an increase in thenumber of autophagosomes, whereas 1.25 �M SF had no effect.SF in fact reduced the number of autophagosomes when cells weretreated with the combination of SF and arsenic (Fig. 5A). Further-more, indirect immunofluorescence analysis of Keap1, LC3, andp62 in BEAS-2B and HBE cells (HBE cell data not shown) treatedwith 1 �M arsenic, 1.25 �M SF, or the combination was con-ducted. The results further verified that arsenic, and not SF, in-creases the formation of autophagosomes (Fig. 5B). Arsenic in-duced puncta that were positive for Keap1, LC3, and p62 (Fig. 5B),whereas cells treated with 1.25 �M SF were similar to untreatedcells (Fig. 5B). More importantly, SF reversed the effects of arsenicby decreasing the number of Keap1-, LC3-, and p62-positivepuncta (Fig. 5B). These results further suggest that canonical Nrf2activators, such as SF, alleviate arsenic-mediated autophagosomalupregulation and prevent p62-mediated sequestration of Keap1into autophagosomes.
Arsenic-mediated autophagy deregulation is p62 dependentand Nrf2 independent. Thus far, arsenic has been shown to de-regulate autophagy, leading to upregulation of autophagosomesin which p62 and Keap1 accumulate. Previously, it was reportedthat p62 contains an ARE in its promoter region, creating a posi-tive-feedback loop (47). Based on that report, there are two pos-sible mechanisms mediating accumulation of p62 in autophago-somes in response to arsenic: p62 can accumulate either by
activating Nrf2 to increase p62 transcription or by blocking au-tophagic flux, since p62 is itself a substrate of autophagy. There-fore, the p62 or Nrf2 dependence of the observed arsenic-medi-ated autophagosomal upregulation and the alleviation conferredby SF or tBHQ was explored by using indirect immunofluores-cence analysis. BEAS-2B cells transfected with control siRNA, p62siRNA, or Nrf2 siRNA were treated with arsenic, SF, tBHQ, or theindicated combination. Similar to nontransfected cells, arsenic,but not SF or tBHQ, induced puncta in which Keap1, LC3, andp62 colocalized in control-siRNA-transfected cells (Fig. 6A). Incells cotreated with arsenic plus SF or tBHQ, there were minimal(similar to basal) amounts of puncta comparable to those in un-treated cells (Fig. 6A). However, when p62 expression was blockedby p62 siRNA, even arsenic was unable to induce puncta (Fig. 6B),indicating that arsenic-mediated autophagosomal upregulation isp62 dependent. On the other hand, arsenic still induced punctain cells transfected with Nrf2 siRNA (Fig. 6C). More impor-tantly, upon knockdown of Nrf2, SF and tBHQ could no longeralleviate arsenic-induced puncta (Fig. 6C). These data indicatethat arsenic-mediated autophagosomal upregulation and accu-mulation of p62 and Keap1 in the autophagosome are due toblockage of autophagic flux. It is unlikely that arsenic first in-duces Nrf2 that feeds back to further enhance the transcriptionof p62, since upregulation/accumulation of p62 in autophago-somes was still observed when Nrf2 expression was suppressedby Nrf2 siRNA.
FIG 2 Arsenic increases the number of LC3 puncta. Shown is live-cell imaging of iBMK cells stably expressing EGFP-LC3. Cells were either left untreated (left)or treated with 125 nM, 250 nM, 500 nM, or 1 �M arsenic, as indicated (middle), or with 1.25 �M SF (right) for 1, 2, 4, 6, 12, or 24 h (top to bottom).
Arsenic Activates Nrf2 in a p62-Dependent Manner
June 2013 Volume 33 Number 12 mcb.asm.org 2439
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

Arsenic induces Nrf2 in a p62-dependent but Keap1-C151-independent manner. The notion that arsenic treatment in-creases the number of autophagosomes and leads to the accumu-lation of p62 and Keap1 in autophagosomes prompted us to test ifarsenic-mediated induction of Nrf2 is p62 dependent (nonca-nonical mechanism). Therefore, a dual-luciferase assay was con-ducted to determine whether arsenic-mediated Nrf2 activation isindeed p62 and/or Keap1-C151 dependent. HBE cells were firsttransfected with an siRNA specific for the 3= untranslated regionof Keap1 to suppress endogenous Keap1 expression in combina-tion with control siRNA or p62-siRNA. Cells were subsequentlytransfected with either Keap1-WT or Keap1-C151S. After trans-fection, cells were treated with 10 �M arsenic, 5 �M SF, or 25 �MtBHQ for an additional 16 h. Relative firefly and Renilla luciferaseactivities were then measured. As expected, treatment with arse-nic, SF, or tBHQ significantly induced mGST-ARE-mediated lu-ciferase activity in the Keap1-WT/control siRNA group (Fig. 7,top). Furthermore, when Keap1-C151S was cotransfected withcontrol siRNA, luciferase activity was still significantly induced byarsenic but not by SF or tBHQ (Fig. 7, top). Although SF andtBHQ were still able to induce mGST-ARE luciferase activity, arsenicwas no longer able to induce activity in the Keap1-WT/p62 siRNAgroup. None of the compounds were able to induce luciferase activitywhen p62 was knocked down and Keap1-C151S was expressed. Im-munoblot analysis was also conducted in parallel. The results wereconsistent with the results of the dual-luciferase assay (Fig. 7, bot-
tom). These results demonstrate a distinct mechanism of Nrf2 acti-vation: SF or tBHQ activates Nrf2 in a Keap1-C151-dependent, p62-independent manner, whereas arsenic activates the Nrf2 pathway in ap62-dependent, Keap1-C151-independent manner.
Next, the p62 dependence of Nrf2 induction in response toarsenic was further compared to that in response to SF and tBHQ.BEAS-2B and HBE cells transfected with either control siRNA orp62 siRNA were treated with different doses of arsenic or SF for 4h. Immunoblot analysis of Nrf2 and p62 showed a dose-depen-dent increase in the control siRNA group in both cell lines whentreated with arsenic (Fig. 8A). When p62 was knocked down ineither cell line, Nrf2 induction by arsenic was completely abro-gated (Fig. 8A). A slight dose-dependent increase in the amount ofLC3-II was observed in cells transfected with control siRNA andtreated with arsenic, indicating an increase in the number of au-tophagosomes, which was unchanged in the p62 siRNA group(Fig. 8A). In contrast, SF increased the amount of Nrf2 in a dose-dependent manner in cells transfected with either control siRNAor p62 siRNA, although the fold induction of Nrf2 with p62 siRNAwas slightly decreased (Fig. 8B). Furthermore, treatment of cellswith SF elicited no change in p62 or LC3 in either cell lines (Fig.8B). When BEAS-2B and HBE cells were treated with 25 �MtBHQ for 4 h, a dose-dependent increase in the amount Nrf2 wasobserved in both the control siRNA and p62 siRNA groups (Fig.8C). Interestingly, there was also a dose-dependent increase in theamount of p62 in the control-siRNA-transfected cells, but there
FIG 3 Arsenic causes accumulation of autophagosomes by inhibiting the later stage of autophagy. (A) Live-cell imaging of NIH 3T3 cells transfected witha tandem mRFP-GFP-LC3 construct for 24 h and then either left untreated, starved for 4 h in Hanks balanced salt solution, or treated with 100 nMbafilomycin A1 for 4 h. (B to E) Live-cell imaging of NIH 3T3 cells transfected with mRFP-GFP-LC3 for 24 h and treated with 1.25 �M SF, 500 nM arsenic(As), or 1 �M arsenic for 2, 4, 6, or 12 h. Note that arsenic-treated cells resemble those treated with bafilomycin A1, indicating that arsenic interferes withlate-stage autophagy.
Lau et al.
2440 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.
was no significant changes in the levels of Keap1 or LC3 (Fig. 8C).Also notable was an increase in the amount Keap1 when p62 wasknocked down in any of the treatment groups. This may explainthe blunted induction of Nrf2 in response to canonical Nrf2 acti-
vators, especially SF, in cells transfected with p62 siRNA (Fig. 8B).Together, these data confirm that arsenic-mediated induction ofNrf2 is p62 dependent, whereas induction mediated by SF andtBHQ is p62 independent.
FIG 4 Arsenic-mediated accumulation of autophagosomes is alleviated by SF or tBHQ. (A) Live-cell imaging of EGFP-LC3 iBMK cells left untreated or treatedwith 500 nM arsenic (As), 1.25 �M SF, 10 �M tBHQ, or the indicated combination for 4 h. (B) Live-cell imaging of NIH 3T3 cells transfected with mRFP-GFP-LC3 for 24 h and left untreated or treated with 500 nM arsenic, 1.25 �M SF, 10 �M tBHQ, or the indicated combination for 4 h.
FIG 5 Arsenic causes accumulation of autophagosomes in lung epithelial cells. (A) Electron microscopy of BEAS-2B cells left untreated or treated with 1.25 �MSF, 500 nM arsenic (As), or both for 4 h. Arrows indicate autophagosomes. (B) Indirect immunofluorescence of Keap1, LC3, and p62 in BEAS-2B cells leftuntreated or treated with 1 �M arsenic, 1.25 �M SF, or both for 4 h.
Arsenic Activates Nrf2 in a p62-Dependent Manner
June 2013 Volume 33 Number 12 mcb.asm.org 2441
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

Arsenic-mediated activation of Nrf2 is prolonged comparedto activation mediated by SF or tBHQ. We hypothesized that thenoncanonical mechanism of Nrf2 activation by arsenic requires alonger time to resolve p62-mediated sequestration of Keap1 inautophagosomes and thus causes prolonged Nrf2 activation.Therefore, BEAS-2B and HBE cells were treated with arsenic, SF,or tBHQ for the indicated duration, and Nrf2 protein levels wereanalyzed. In both BEAS-2B and HBE cells, arsenic induced Nrf2within 4 h yet persisted for as long as 48 h (Fig. 9A and B). Treat-ment with SF or tBHQ increased Nrf2 protein levels by as early as4 h, and these levels returned to basal levels by approximately 36 hin both cell lines (Fig. 9A and B). Furthermore, a time-dependentincrease in the level of p62 was observed in arsenic- or tBHQ-treated cells, whereas SF did not change the p62 level (Fig. 9A andB). The Keap1 level did not change upon treatment with any com-pound (Fig. 9A and B). To further confirm that Nrf2 activation byarsenic is prolonged compared to activation by SF, the half-life ofNrf2 under different treatment conditions was measured. The re-sults suggest that the half-life of Nrf2 in the presence of arsenic is44.9 min, whereas the half-life in the presence of SF-treated cells isonly 18.6 min. These data support a scenario whereby arsenic-mediated activation of Nrf2 is prolonged, resembling the consti-tutive activation of Nrf2 found in certain types of cancers, whichmay contribute to arsenic carcinogenicity.
DISCUSSION
Recently, we and other groups independently reported a nonca-nonical mechanism of Nrf2 activation where direct interaction ofp62 inactivates Keap1, causing stabilization of Nrf2 protein andincreasing the transcription of ARE-bearing genes (43–47, 49). Ithas been well established by previous studies conducted in ourlaboratory that arsenic activates Nrf2 in a Keap1-C151-indepen-dent manner, unlike SF and tBHQ, which are common Nrf2 acti-vators that activate the pathway through the canonical Keap1-C151 mechanism (8, 31). Furthermore, arsenic stabilizes Nrf2 bycompromising Keap1-Cul3-dependent ubiquitination (31, 50);
FIG 7 Arsenic induces Nrf2 in a p62-dependent, Keap1-C151-independentmanner. HBE cells were transfected with the indicated siRNA for 24 h. Cellswere then transfected with plasmids encoding mGST-ARE-firefly luciferaseand TK-Renilla luciferase, along with a plasmid for Keap1-WT or Keap1-C151S. Cells were treated with 10 �M arsenic (As), 5 �M SF, or 25 �M tBHQfor 16 h and harvested 96 h after transfection of siRNA. Luciferase activitieswere measured, and the data shown represent the relative ratio of firefly versusRenilla luciferase activity, with the untreated control values normalized to 1(top). The data are expressed as means � SD (n � 3). �, P � 0.05 comparedwith the untreated control in each group. An aliquot of cell lysate was used forimmunoblot analysis (bottom). Con, control.
FIG 6 Arsenic-mediated autophagy deregulation is p62 dependent and Nrf2 independent. BEAS-2B cells transfected with either control siRNA (A), p62 siRNA(B), or Nrf2 siRNA (C) were treated with 1 �M arsenic (As), 1.25 �M SF, 10 �M tBHQ, or the indicated combination for 4 h. Cells were then subjected to indirectimmunofluorescence staining of Keap1, LC3, and p62.
Lau et al.
2442 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

however, the exact mechanism(s) by which arsenic activates theNrf2 pathway has yet to be elucidated. In this investigation, wedemonstrate for the first time that arsenic-mediated activation ofNrf2 is through the noncanonical mechanism (p62 dependent).We report that acute low-dose arsenic blocks autophagic flux andenhances the number of autophagosomes, causing accumulationof p62 and sequestration of Keap1 in autophagosomes. This pro-cess inactivates Keap1, leading to stabilization of Nrf2. Interest-
ingly, arsenic-mediated effects on autophagy were prevented orreversed by cotreatment with SF or tBHQ, indicating that canon-ical Nrf2 activators can prevent the detrimental effects of arsenic.
The mode of action by which arsenic causes cancer is not yetknown. Enhanced ROS production and oxidative stress may playcardinal roles in arsenic toxicity and carcinogenicity, since gener-ation of ROS can lead to genotoxicity, alter signal transductionpathways, inhibit DNA repair, and increase cell proliferation (28–
FIG 8 Arsenic, but not SF or tBHQ, induces Nrf2 in a p62-dependent manner. BEAS-2B and HBE cells transfected with control siRNA or p62 siRNA were treatedwith the indicated doses of arsenic (As) (A), SF (B), or tBHQ (C) for 4 h before cells were harvested 48 h after transfection of siRNA. Note that arsenic increasesNrf2 levels in a dose-dependent manner that is lost with p62 siRNA, which is in contrast to SF and tBHQ. Total cell lysates were subjected to immunoblot analysis.
FIG 9 Arsenic-mediated activation of Nrf2 is prolonged. (A and B) BEAS-2B (A) and HBE (B) cells were treated with arsenic (As), SF, or tBHQ for the indicatedtime points and harvested for immunoblot analysis. Note that arsenic causes a prolonged activation of Nrf2, whereas SF or tBHQ induces Nrf2 transiently. (C)HBE cells were pretreated with arsenic or SF for 4 h prior to the addition of cycloheximide (25 �M) for the indicated time periods. The level of Nrf2 was detectedby immunoblot analysis, the band intensity was quantified and plotted on a semilog graph, and the half-life (t1/2) of Nrf2 was calculated.
Arsenic Activates Nrf2 in a p62-Dependent Manner
June 2013 Volume 33 Number 12 mcb.asm.org 2443
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

30). Acute high-dose arsenic has been shown to increase ROSproduction as well as upregulate a number of oxidative stress-related genes, such as heme-oxygenase 1, NAD(P)H quinone ox-idoreductase, and metallothionein (31, 51). However, low-dosearsenic has been shown to reduce ROS levels. Snow et al. demon-strated that human fibroblast cell lines treated with 500 nM arse-nic for 24 h significantly decreased the amount of ROS present incells by inducing a series of stress response genes compared tountreated cells (48). The 500 nM arsenic dose is considered to bein a subtoxic range and induces changes in overall gene expression(48). In our current study, treatment with 1 �M arsenic for 4 h didnot changes ROS levels in comparison with levels in untreatedcells (data not shown). Therefore, it is unlikely that activation ofNrf2 by low-dose arsenic is through generation of ROS. Further-more, previous studies conducted by Bolt et al. with lymphoblas-toid cell lines showed that arsenic induced autophagy and notapoptosis (38, 39). Also, human urothelial cells exposed to 1 or 4�M arsenic for 48 h exhibited increases in the number of autopha-gosomes as well as protein expression levels of LC3 and Beclin-1,critical regulators for the formation of autophagosomes (37).These studies established an association between arsenic exposureand autophagy; however, the detailed mechanism by which low-dose arsenic causes pathological changes has remained largely un-known until now. Here, we demonstrate that low, environmen-tally relevant doses of arsenic block autophagic flux, causing anincrease in the number of autophagosomes and activating theNrf2 pathway via the noncanonical mechanism, which is indepen-dent of ROS as well.
It has been shown that a lack of essential autophagy genes, suchas Atg5 and Atg7, leads to the formation of p62-positive aggregatesand accumulation of ubiquitin inclusions in the liver (44, 52).Recently, Inami et al. reported that persistent Nrf2 activationthrough p62 in liver-specific-autophagy-deficient mice contrib-utes to the development of hepatocellular carcinoma (53). Anaccumulation of p62 and elevated levels of Nrf2 downstreamgenes were also observed in bronchial epithelial cells of au-tophagy-deficient mice (54). Simultaneous knockout of either p62or Nrf2 in autophagy-deficient mice suppressed ubiquitin accu-mulation and protein aggregation in the liver and brain and alle-viated liver injury (44, 45). Here, we demonstrate that arsenic-mediated activation of Nrf2 is not only p62 dependent but alsoprolonged. Taken together, these studies provide evidence thatderegulation of autophagy resulting in the accumulation of p62and prolonged Nrf2 activation plays a critical role in the patho-genesis of many human diseases, which may include arsenic-in-duced cancers.
Previously, our laboratory demonstrated the protective role ofNrf2 against the cytotoxic effects induced by acute arsenic expo-sure both in cultured cells and in mice (55–57). Specifically,UROtsa cells with reduced Nrf2 expression were more susceptibleto arsenic-induced toxicity than control cells (55). Pretreatmentor cotreatment with an Nrf2 inducer, SF or tBHQ, renderedUROtsa cells more resistant to arsenic and monomethylarsonousacid toxicity (55). We also demonstrated the Nrf2 dependence ofSF- and tBHQ-mediated protection through the use of Nrf2�/�
mouse embryonic fibroblast (MEF) cells (55). Another group alsoshowed that SF suppresses the cellular accumulation of arsenicand decreases toxicity in primary mouse hepatocytes (58). Ourprevious work in mice demonstrated that Nrf2�/� mice are moresensitive than their wild-type counterparts to arsenic-induced
DNA hypomethylation, oxidative DNA damage, and apoptoticcell death in the liver and bladder when exposed to arsenic in theirdrinking water for 6 weeks (56). Our current study demonstratesthat canonical Nrf2 activators prevent or reverse arsenic-mediatedderegulation of autophagy, which may be partly responsible forthe Nrf2-mediated protection against arsenic toxicity observed inprevious studies. Further investigation is necessary to determinethe biochemistry of how SF- or tBHQ-mediated activation of Nrf2in the canonical manner supersedes arsenic-mediated deregula-tion of autophagy. One possible mechanism is that activation ofNrf2 alleviates arsenic-mediated effects through a reduction of theintracellular arsenic concentration, based on previous findings(54). Collectively, these studies clearly demonstrate that activationof the Nrf2 pathway through the canonical Keap1-C151 mecha-nism is important in protecting against the carcinogenic effects ofarsenic and remains an effective strategy for chemoprevention.
One interesting observation is that tBHQ itself, similar to ar-senic, was able to induce p62 at the protein level in a dose-depen-dent and time-dependent manner (Fig. 8C and 9A and B). Never-theless, tBHQ did not result in upregulation of autophagosomes.tBHQ may enhance the transcription of p62 though its reportedARE in the promoter, leading to an increase in the level of p62(47). Furthermore, since autophagic flux is not affected, clearanceof autophagosomes remains intact, and proteins such as LC3 andp62 continue to be degraded. Conversely, SF had no effect on theprotein level of p62. It is interesting to note that different Nrf2activators can upregulate distinct sets of Nrf2 target genes. Duringthis study, we observed that arsenic, SF, and tBHQ had differenteffects on the induction of these well-studied Nrf2 target genes,including heme-oxygenase, �-glutamylcysteine synthetase, aldosereductase, and NAD(P)H dehydrogenase 1 (data not shown). Itwould be interesting to understand how activation of Nrf2 bydifferent compounds results in upregulation of distinct Nrf2downstream genes.
In conclusion, we propose a model where arsenic induces per-sistent or prolonged Nrf2 activation, mimicking constitutive acti-vation of Nrf2 caused by the dysfunctional Keap1-Nrf2 axis seenin many types of tumors. This study supports the notion that theKeap1-C151-dependent mechanism of Nrf2 activation is protec-tive, while p62-mediated activation or somatic mutations leadingto persistent or prolonged Nrf2 activation comprise the “darkside” of Nrf2 that is expected to promote tumor growth and sur-vival. We believe that our study lays the groundwork for futureinvestigations both in vitro and in vivo to explore the interplaybetween Nrf2, Keap1, p62, and autophagy as well as their roles inarsenic carcinogenicity.
ACKNOWLEDGMENTS
We thank A. S. McElhinny for critical reading and editing of the manu-script.
This study was supported by the following grants: grantsR01ES015010 (NIEHS) and R01CA154377 (NCI), which were awarded toD.D.Z.; an SOT-Novartis graduate student fellowship awarded to A.L.;and grant ES006694 (NIH), a center grant.
REFERENCES1. Osburn WO, Kensler TW. 2008. Nrf2 signaling: an adaptive response
pathway for protection against environmental toxic insults. Mutat. Res.659:31–39.
2. Kobayashi M, Yamamoto M. 2006. Nrf2-Keap1 regulation of cellulardefense mechanisms against electrophiles and reactive oxygen species.Adv. Enzyme Regul. 46:113–140.
Lau et al.
2444 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

3. Zhang DD. 2006. Mechanistic studies of the Nrf2-Keap1 signaling path-way. Drug Metab. Rev. 38:769 –789.
4. Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. 2010.Cancer chemoprevention mechanisms mediated through the keap1-nrf2pathway. Antioxid. Redox Signal. 13:1713–1748.
5. Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. 2008. Dual roles ofNrf2 in cancer. Pharmacol. Res. 58:262–270.
6. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T,Igarashi K, Yamamoto M. 2004. Oxidative stress sensor Keap1 functionsas an adaptor for Cul3-based E3 ligase to regulate proteasomal degrada-tion of Nrf2. Mol. Cell. Biol. 24:7130 –7139.
7. Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. 2004. Keap1is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiq-uitin ligase complex. Mol. Cell. Biol. 24:10941–10953.
8. Zhang DD, Hannink M. 2003. Distinct cysteine residues in Keap1 arerequired for Keap1-dependent ubiquitination of Nrf2 and for stabiliza-tion of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell.Biol. 23:8137– 8151.
9. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, WakabayashiN, Katoh Y, Yamamoto M, Talalay P. 2002. Direct evidence that sulfhy-dryl groups of Keap1 are the sensors regulating induction of phase 2 en-zymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci.U. S. A. 99:11908 –11913.
10. McMahon M, Lamont DJ, Beattie KA, Hayes JD. 2010. Keap1 perceivesstress via three sensors for the endogenous signaling molecules nitric ox-ide, zinc, and alkenals. Proc. Natl. Acad. Sci. U. S. A. 107:18838 –18843.
11. Baird L, Dinkova-Kostova AT. 2011. The cytoprotective role of theKeap1-Nrf2 pathway. Arch. Toxicol. 85:241–272.
12. Wondrak GT, Villeneuve NF, Lamore SD, Bause AS, Jiang T, ZhangDD. 2010. The cinnamon-derived dietary factor cinnamic aldehyde acti-vates the Nrf2-dependent antioxidant response in human epithelial coloncells. Molecules 15:3338 –3355.
13. Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, TalalayP, Kensler TW. 2001. Sensitivity to carcinogenesis is increased and che-moprotective efficacy of enzyme inducers is lost in nrf2 transcription fac-tor-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 98:3410 –3415.
14. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO,Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S.2006. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung can-cer. PLoS Med. 3:e420. doi:10.1371/journal.pmed.0030420.
15. Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, OhtsujiM, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. 2006. Structuralbasis for defects of Keap1 activity provoked by its point mutations in lungcancer. Mol. Cell 21:689 –700.
16. Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M,Hirohashi S. 2008. Genetic alteration of Keap1 confers constitutive Nrf2activation and resistance to chemotherapy in gallbladder cancer. Gastro-enterology 135:1358 –1368.
17. Nioi P, Nguyen T. 2007. A mutation of Keap1 found in breast cancerimpairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Com-mun. 362:816 – 821.
18. Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, Zhang DD.2010. High levels of Nrf2 determine chemoresistance in type II endome-trial cancer. Cancer Res. 70:5486 –5496.
19. Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M,Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M,Hirohashi S. 2008. Loss of Keap1 function activates Nrf2 and providesadvantages for lung cancer cell growth. Cancer Res. 68:1303–1309.
20. Muscarella LA, Parrella P, D’Alessandro V, la Torre A, Barbano R,Fontana A, Tancredi A, Guarnieri V, Balsamo T, Coco M, Copetti M,Pellegrini F, De Bonis P, Bisceglia M, Scaramuzzi G, Maiello E, ValoriVM, Merla G, Vendemiale G, Fazio VM. 2011. Frequent epigeneticsinactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics6:710 –719.
21. Slocum SL, Kensler TW. 2011. Nrf2: control of sensitivity to carcinogens.Arch. Toxicol. 85:273–284.
22. Shim GS, Manandhar S, Shin DH, Kim TH, Kwak MK. 2009. Acquisi-tion of doxorubicin resistance in ovarian carcinoma cells accompaniesactivation of the NRF2 pathway. Free Radic. Biol. Med. 47:1619 –1631.
23. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X,Zheng W, Wondrak GT, Wong PK, Zhang DD. 2008. Nrf2 enhancesresistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2.Carcinogenesis 29:1235–1243.
24. Hu L, Miao W, Loignon M, Kandouz M, Batist G. 2010. Putativechemopreventive molecules can increase Nrf2-regulated cell defense insome human cancer cell lines, resulting in resistance to common cytotoxictherapies. Cancer Chemother. Pharmacol. 66:467– 474.
25. Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H,Coulter J, Blackford A, Goodman SN, Bunz F, Watson WH, GabrielsonE, Feinstein E, Biswal S. 2008. RNAi-mediated silencing of nuclear factorerythroid-2-related factor 2 gene expression in non-small cell lung cancerinhibits tumor growth and increases efficacy of chemotherapy. CancerRes. 68:7975–7984.
26. Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, Zhang DD.2011. Brusatol enhances the efficacy of chemotherapy by inhibiting theNrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. U. S. A. 108:1433–1438.
27. Platanias LC. 2009. Biological responses to arsenic compounds. J. Biol.Chem. 284:18583–18587.
28. Flora SJ. 2011. Arsenic-induced oxidative stress and its reversibility. FreeRadic. Biol. Med. 51:257–281.
29. Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. 2011. Arsenicexposure and toxicology: a historical perspective. Toxicol. Sci. 123:305–332.
30. Lau A, Whitman SA, Jaramillo MC, Zhang DD. 2013. Arsenic-mediatedactivation of the Nrf2-Keap1 antioxidant pathway. J. Biochem. Mol. Toxi-col. 27:99 –105.
31. Wang XJ, Sun Z, Chen W, Li Y, Villeneuve NF, Zhang DD. 2008.Activation of Nrf2 by arsenite and monomethylarsonous acid is indepen-dent of Keap1-C151: enhanced Keap1-Cul3 interaction. Toxicol. Appl.Pharmacol. 230:383–389.
32. Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. 2003. Transcriptionfactor Nrf2 activation by inorganic arsenic in cultured keratinocytes: in-volvement of hydrogen peroxide. Exp. Cell Res. 290:234 –245.
33. Endo H, Sugioka Y, Nakagi Y, Saijo Y, Yoshida T. 2008. A novel role ofthe NRF2 transcription factor in the regulation of arsenite-mediated ker-atin 16 gene expression in human keratinocytes. Environ. Health Perspect.116:873– 879.
34. Aono J, Yanagawa T, Itoh K, Li B, Yoshida H, Kumagai Y, YamamotoM, Ishii T. 2003. Activation of Nrf2 and accumulation of ubiquitinatedA170 by arsenic in osteoblasts. Biochem. Biophys. Res. Commun. 305:271–277.
35. Meng D, Wang X, Chang Q, Hitron A, Zhang Z, Xu M, Chen G, LuoJ, Jiang B, Fang J, Shi X. 2010. Arsenic promotes angiogenesis in vitro viaa heme oxygenase-1-dependent mechanism. Toxicol. Appl. Pharmacol.244:291–299.
36. Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, FreemanML. 2008. Covalent modification at Cys151 dissociates the electrophilesensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 21:705–710.
37. Chai CY, Huang YC, Hung WC, Kang WY, Chen WT. 2007. Arsenicsalts induced autophagic cell death and hypermethylation of DAPK pro-moter in SV-40 immortalized human uroepithelial cells. Toxicol. Lett.173:48 –56.
38. Bolt AM, Douglas RM, Klimecki WT. 2010. Arsenite exposure in humanlymphoblastoid cell lines induces autophagy and coordinated induction oflysosomal genes. Toxicol. Lett. 199:153–159.
39. Bolt AM, Byrd RM, Klimecki WT. 2010. Autophagy is the predominantprocess induced by arsenite in human lymphoblastoid cell lines. Toxicol.Appl. Pharmacol. 244:366 –373.
40. Ichimura Y, Kominami E, Tanaka K, Komatsu M. 2008. Selectiveturnover of p62/A170/SQSTM1 by autophagy. Autophagy 4:1063–1066.
41. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H,Overvatn A, Bjorkoy G, Johansen T. 2007. p62/SQSTM1 binds directlyto Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregatesby autophagy. J. Biol. Chem. 282:24131–24145.
42. Itakura E, Mizushima N. 2011. p62 targeting to the autophagosomeformation site requires self-oligomerization but not LC3 binding. J. CellBiol. 192:17–27.
43. Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, WhiteE, Zhang DD. 2010. A noncanonical mechanism of Nrf2 activation byautophagy deficiency: direct interaction between Keap1 and p62. Mol.Cell. Biol. 30:3275–3285.
44. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A,Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y,Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K,
Arsenic Activates Nrf2 in a p62-Dependent Manner
June 2013 Volume 33 Number 12 mcb.asm.org 2445
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.

Yamamoto M. 2010. The selective autophagy substrate p62 activates thestress responsive transcription factor Nrf2 through inactivation of Keap1.Nat. Cell Biol. 12:213–223.
45. Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, Lage K,Xavier RJ, Ryu KY, Taguchi K, Yamamoto M, Tanaka K, Mizushima N,Komatsu M, Kopito RR. 2010. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response path-way: a potential role for protein aggregation in autophagic substrate selec-tion. J. Cell Biol. 191:537–552.
46. Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, LayfieldR, Foster BJ, Goldring CE, Park BK. 2010. Physical and functionalinteraction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 celldefense pathway. J. Biol. Chem. 285:16782–16788.
47. Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A,McMahon M, Hayes JD, Johansen T. 2010. p62/SQSTM1 is a target genefor transcription factor NRF2 and creates a positive feedback loop byinducing antioxidant response element-driven gene transcription. J. Biol.Chem. 285:22576 –22591.
48. Snow ET, Sykora P, Durham TR, Klein CB. 2005. Arsenic, mode ofaction at biologically plausible low doses: what are the implications for lowdose cancer risk? Toxicol. Appl. Pharmacol. 207:557–564.
49. Riley BE, Kaiser SE, Kopito RR. 2011. Autophagy inhibition engagesNrf2-p62 Ub-associated signaling. Autophagy 7:338 –340.
50. He X, Chen MG, Lin GX, Ma Q. 2006. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 � Keap1 � Cul3 com-plex and recruiting Nrf2 � Maf to the antioxidant response element en-hancer. J. Biol. Chem. 281:23620 –23631.
51. Huerta-Olvera SG, Macias-Barragan J, Ramos-Marquez ME, Armen-
dariz-Borunda J, Diaz-Barriga F, Siller-Lopez F. 2010. Alpha-lipoic acidregulates heme oxygenase gene expression and nuclear Nrf2 activation asa mechanism of protection against arsenic exposure in HepG2 cells. En-viron. Toxicol. Pharmacol. 29:144 –149.
52. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S,Eishi Y, Hino O, Tanaka K, Mizushima N. 2011. Autophagy-deficientmice develop multiple liver tumors. Genes Dev. 25:795– 800.
53. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O,Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K,Komatsu M. 2011. Persistent activation of Nrf2 through p62 in hepato-cellular carcinoma cells. J. Cell Biol. 193:275–284.
54. Inoue D, Kubo H, Taguchi K, Suzuki T, Komatsu M, Motohashi H,Yamamoto M. 2011. Inducible disruption of autophagy in the lung causesairway hyper-responsiveness. Biochem. Biophys. Res. Commun. 405:13–18.
55. Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. 2007.Nrf2 protects human bladder urothelial cells from arsenite and monom-ethylarsonous acid toxicity. Toxicol. Appl. Pharmacol. 225:206 –213.
56. Jiang T, Huang Z, Chan JY, Zhang DD. 2009. Nrf2 protects againstAs(III)-induced damage in mouse liver and bladder. Toxicol. Appl. Phar-macol. 240:8 –14.
57. Du Y, Villeneuve NF, Wang XJ, Sun Z, Chen W, Li J, Lou H, Wong PK,Zhang DD. 2008. Oridonin confers protection against arsenic-inducedtoxicity through activation of the Nrf2-mediated defensive response. En-viron. Health Perspect. 116:1154 –1161.
58. Shinkai Y, Sumi D, Fukami I, Ishii T, Kumagai Y. 2006. Sulforaphane,an activator of Nrf2, suppresses cellular accumulation of arsenic and itscytotoxicity in primary mouse hepatocytes. FEBS Lett. 580:1771–1774.
Lau et al.
2446 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 17
Jan
uary
202
2 by
125
.202
.238
.18.


















