
STUDENTS MINI PROJECT REPORT SUBMITTED - … 15-16 40 projects/6 - M. Saranya Devi.pdfSTUDENTS MINI...
60
INHIBITIVE BEHAVIOUR OF GLYCINE UREA AND SUCROSE ON THE CORROSION OF TIN PLATE IN ACID MEDIUM STUDENTS MINI PROJECT REPORT SUBMITTED BY M. SARANYA DEVI TAMILNADU STATE COUNCIL FOR HIGHER EDUCATION CHENNAI-600005
Transcript of STUDENTS MINI PROJECT REPORT SUBMITTED - … 15-16 40 projects/6 - M. Saranya Devi.pdfSTUDENTS MINI...

INHIBITIVE BEHAVIOUR OF GLYCINE UREA AND SUCROSE ON THE
CORROSION OF TIN PLATE IN ACID MEDIUM
STUDENTS MINI PROJECT REPORT SUBMITTED
BY
M. SARANYA DEVI
TAMILNADU STATE COUNCIL FOR
HIGHER EDUCATION
CHENNAI-600005

CONTENTS
Chapters page no.
Chapter-I …………………. 1-34
1. Introduction
1.1. Definition of Corrosion
1.2. Example of Corrosion
1.3. Principle of Corrosion
1.4. Theories of Corrosion
1.5. Factors influencing Corrosion
1.6. Common types of Corrosion
1.7. Prevention from Corrosion
1.8. Application of protective coatings
1.9. Corrosion Inhibitor
1.10. Inhibitor classification
1.11. Review of Literature
Chapter-II …………………. 35-36
2. Objectives
Chapter-III …………………. 37-38
3. Experimental methods
Chapter-IV …………………. 39-54
4. Results and Discussions
Chapter-V …………………... 55
5. Conclusions

ACKNOWLEDGEMENT
At first I thank God the Almighty for enabling me to work on such a
wonderful project. I express my sincere gratitude to “Tamil Nadu State Council
for Higher Education” for selecting and providing fund for this project.
My sincere thanks to my college principle Dr. P. Parvathi, M.Sc., M.Phil.,
Ph.D., for giving permission to do this project.
I express my sincere thanks to my guide Dr. C.V. Mythili, M.Sc., M.Phil.,
M.Ed., Ph.D., for her valuable guidance and suggestion to complete the project on
time.
Place: Tirunelveli Signature of student
Date:

Date:
CERTIFICATE
This is to certify that Miss. M. Saranya Devi is a bonafide final year P.G.
student of the Department of Chemistry and that this project work is part of the
curriculum.
Signature of the Guide Signature of the HOD
Signature of the Principal

CHAPTER-I
1. INTRODUCTION
Corrosion is a general term used to describe various interactions between a
material and its environment leading to degradation in the material properties.
Interaction with ambient oxygen can cause the formation of oxide layers via
diffusion controlled growth these may passivity the material against further
oxidation.
1.1. DEFINITION OF CORROSION
The word corrosion comes from the Latin ‘corrodere’ to gnew away. It is
defined as any process of deterioration or destruction and consequent loss of a
solid metallic material through an unwanted or unintentional chemical or
electrochemical attack by its environment. Thus corrosion is a process reverse of
extraction of metals. Corrosion is a natural process.
During corrosion the metal is converted into its compounds. In this regard
corrosion may be considered as the reverse process of extraction of the metal from
its ore. This may be represented as follow.
Metal ↔ Metallic compounds + energy
The above equation indicates that energy is released in the corrosion process and
the energy is required for the extraction of metal.
1.2. EXAMPLE OF CORROSION
(i) Rusting of Iron
When Iron metal is exposed to the atmospheric conditions a layer of reddish
scale and powder of oxide (Fe2O3) is formed on the surface of Iron in presence of
moisture and air.

(ii)Formation of Green film of Copper
When Copper is exposed to moist air containing carbon dioxide, it forms a
green film of basic carbonate [CuCO3+ Cu (OH) 2] on the surface of the Copper.
2. PRICINPLE OF CORROSION
If a piece of iron rod in immersed in a polar solvent like water some of the
metal ions leave the crystal and go into solution. They get hydrated, (i.e.) they
form bonds with the polar water molecules.
Fe + 2H2O →Fe (OH) 2+H2
As the metal continues to dissolve more and more electrons are left back and a net
negative charge builds up into the metal. Similarly the presence of excess positive
ions builds up a positive charge in the adjacent solution. These opposing electrical
layers discourage further dissolution of the metal .Soon a dynamic equilibrium is
reached with no net flow of metal ions into the solution.
The potential developed by an electron in equilibrium is a property of the
metal forming the electrode. This potential is measured under standard conditions
with a pure metal an electrode and an electrolyte containing unit concentration of
ions of the same metal. As only potential differences can be measured in an
experiment, the potential of an electrode is determined against a standard hydrogen
electrode whose potential is taken to zero.
Gold at the top of the most Nobel metal and it will not dissolve easily.
Lithium at the bottom of the list in the most active and base metal. It will go into
solution readily.
The standard potential will be changed .If the metal is not pure or if the
metal ion concentration in the electrolyte is not unity. The potential V, under non
standard conditions is given by,

V = Vo – RT/nFln[M/M+]
Where,VO → The standard potential
M+ → The metal ion concentration in the electrolyte
M → The concentration of the metal in the electrode
n → The valency of the metal ion
F →The Faraday constant equal to 96.49 kc/moles of electrons
If M+<1, (i.e.) the electrolyte is deficient in metal ions, the potential decrease,
If M<1, (i.e.) the electrolyte is rich in metal ions, the potential will increase.
2.1. THEORIES OF CORROSION
Corrosion is the primary means by which metals deteriorate. Cast metals
corrode on contact with water, acids, base salts, oils aggressive metal polishers,
and other solid and liquid chemicals. Metals will also corrode when exposed to
gaseous materials like acid vapors, formaldehyde, gas, ammonias gas and sulphur
containing gases. Corrosion specifically refers to any process involving the
deterioration or degradation of metal components. The best known case is that of
the rusting of steel. Corrosion processes are usually electrochemical in nature
having the essential features of a battery.
Humans have most likely been trying to understand and control corrosion for
as long as they have been using metal.
The most important metals are used namely Iron Age, Bronze Age with a few
exceptions, metals is unstable in ordinary aqueous environments. Metals are
usually extracted from ores through the application of a considerable amount of
energy.

a) ACID THEORY OR CARBONATE FORMATION THEORY
According to this theory traces of an acid are necessary for the corrosion to
occur. Even carbon dioxide dissolved in water is sufficient for corrosion, because
carbonic acid is formed. Initial corrosion takes place as a result of combined
chemical action of water oxygen and carbon dioxide.
CO2 + H2O → H2CO3
2H2 +O2 →2H2O
Fe+ H2CO3 →FeCO3+ H2↑
4FeCO3+10H2O+O2→4Fe (OH)3+ 4H2CO3
Carbonic acid is regenerated and is responsible for further attack and hence
for continuation of the corrosion. Excess of oxygen present in the atmosphere
promotes corrosion and other gases such as H2O,SO2,SO3,Cl2 etc. may also react
with Iron. This theory however fails to explain why corrosion of metals such as
iron continues even when no CO2 gas is present in the atmosphere and why
corrosion takes place even in alkaline medium.
b) PEROXIDE THEORY
This is also known as Golding’s chemicals theory according to which iron
reacts with water to liberate hydrogen. The latter reacts with dissolved oxygen to
form H2O2.
Fe+2H2 O → Fe (OH)2 + H2
H2 + O2 → H2O2
The H2O2 thus formed oxidizes Fe(OH)3 to Fe(OH)2 the H2O2 may also react
with iron metal to form Fe(OH)2.
Fe+H2O2 → Fe (OH)2

This theory also has several limitation for example pure H2O2 has no
perceptible action on iron and the reagents which destroy H2O2 are no capable of
preventing corrosion.
c) OXYGEN THEORY
According to this theory metals form oxides have the same colour as that of the
metals.
4Al+3O2→2Al2O3
2Sn+O2 →2SnO
When copper metal is attacked however it gets converted into black copper
oxide and copper is said to be tarnished. This type of corrosion cannot proceed in
the absence of moisture and this theory is not sufficient for explaining the
corrosion in all the cases.
d) ELECTRO CHEMICAL THEORY
This theory satisfactorily explains the process of corrosion. According to this
theory the chemically non uniform surface metals behave like small electric cells
in presence of water containing dissolved oxygen and carbon dioxide.
e) MODERN ELECTROCHEMICAL THEORY
1. The covered end of the metal surface acts as the anode where Fe forms Fe2+
ions by losing electron.(oxidation)
Fe → Fe2+ + 2e-
A less electropositive metal like Cu which is present in the form of impurity
conducts away the electrons and develops the cathode where OH- ions are formed
according to the reaction.
2H2O + 2e-→ H2+ 2 OH-

The hydrogen so liberated is accumulated at the cathode and causes
polarization which may stop the corrosion but the atmospheric oxygen acts as a
depolarizer.
H2 + ½ O2 → H2O
2. Now there exists two oppositely charged ions cathodic and anodic on the
surface of the metal and a galvanic flow of ions may take place. As a result two
oppositely charged ions may combine to form a compound.
Fe2+ + 2 OH- → Fe (OH)2
The CO2 and O2 present in atmosphere convert Fe (OH)2to basic carbonate
according to the reaction.
4Fe (OH)2+ 2H2 O + O2 →4Fe (OH)3
Fe (OH)2+CO2→ Fe (OH)(HCO3)
Fe (OH)3+ CO2→ Fe (OH)2 (HCO3)
According to electrochemical theory any corrosion process may be regarded
as an electrochemical process in which cathode and anode are formed on the
surface of metal and an electrolyte which may be water, acid or salt solution must
be present to allow the flow of electron current to form the corrosion products. It
should be, a higher the metal in the electrochemical series the more easily it is
corroded.
2.2. FACTORS INFLUENCING CORROSION
The corrosion depends upon the following factors.
(i) Nature of the metal
(ii) Nature of the corroding environment
(i) NATURE OF THE METAL
(a) Position in emf series.
(b) Relative areas of anode and cathode

(c) Purity of the metal
(d) Over voltage or over potential
a) Position of emf series
Position of a metal in the electrochemical series gives an idea about its
corrodibility metals with very low reduction potential easily the rate and severity
of corrosion depends upon the difference in their positions in galvanic series. If
greater is the difference, the faster is the corrosion rate. The rate of corrosion
decrease as follows: Li > K > Na > Mg > Zn > Cr > Fe > Cu
b) Relative areas of anode and cathode
The rate of the anodic to the cathodic area is a dominant factor in corrosion.
Corrosion is more rapid and severe and highly localized if the anodic area is small
because the current density at a smaller anodic area is much greater. The demand
for electrons of large cathodic area can be met by smaller anodic area by
undergoing corrosion more briskly.
c) Purity of metal
Impurities in a pure metal generally causes heterogeneity and galvanic cell
type of corrosion takes place. Example is zinc metal having iron as impurity.
d) Over voltage or over potential
One of the cathodic reactions possible in corrosion is the evolution of
hydrogen. Evolution of hydrogen gas is called the hydrogen over voltage. It
depends upon the nature of cathode ionic concentration, current density and
reaction temperature.
Corrosion is very slow if hydrogen over voltage of the cathodic metals in a
corrosion cell is very high. For rough cathodes such as planishedpt it is very low

and for soft cathodes such as Hg and Pb it is very high. Thus the reduction in over
voltage of the corroding metal accelerates the corrosion rate.
(ii) NATURE OF CORRODING ENVIRONMENT
(a) Temperature
(b) Humidity
(c) Pressure of corrosive gases
(d) Presence of suspended particles
(e) Effect of pH
(a) Temperature
Corrosion being chemical in nature is expected to be affected by temperature
with the increase of environment the relation as well as rate increase there by
corrosion rate is generally enhanced.
(b) Humidity
Humidity is directly related to the corrosion rate. Critical humidity is the
relative humidity above which the corrosion rate of metal increases sharply. The
enhancement of corrosion in humidity is because of the gases like CO2, O2, etc.
dissolves in water to produce the electrolyte which is essential for electrochemical
corrosion.
(c) Presence of corrosive gases
The gases, NaCl, (NH4)2SO4 in corrosive environment like CO2,O2,H2S and
fumes of HCl, H2SO4etc.Produces the electrochemical corrosion.
(d) Presence of solid suspended particles
Compound like NaCl, (NH4)2SO4 along with moisture are acts as powerful
electrochemical corrosion rate.

(e)Effects of pH
Generally acidic media pH>7 are more corrosive than alkaline and neutral
media. Metals like Pt, Au, Pd, and Ir are not affected by any acid or alkali on the
other hand alkali and alkaline earth metals are affected by acid, bases and even
water. Both dilute acids and alkaline affects the amphoteric metals as Al, Zn, Sn,
Pb and Fe.
2.3. COMMON TYPES OF CORROSION
Based on the mechanism corrosion can be classified into two types.
(i) Dry (or) Chemical corrosion
(ii) Wet (or) Electrochemical corrosion
Dry (or) Chemical corrosion
This type of corrosion occurs mainly through the direct chemical action of
environment atmospheric gases such as oxygen, halogen, hydrogen sulphide,
sulphur dioxide, nitrogen or anhydrous inorganic liquid with metal surface.
Wet (or) Electrochemical corrosion
When a liquid is in contact with a metal corrosion of metal, take place. This
type of corrosion is known as immersed corrosion. This type of corrosion occurs.
(i). Where a conducting liquid is in contact with metal.
(ii). When two dissimilar metals or alloys are either immersed or
dipped partially in a solution.
According to electrochemical theory the corrosion occurs due to the existence of
separate ‘anodic’ and ‘cathodic’ parts between which current flows through the
conducting solution.
At anodic part oxidation reaction take place with the liberation of free electrons. In
cathodic part reduction reaction take place with the absorption of electron.

M → Mn++ne- (oxidation)
Metal Metal ion
Mn+ → dissolves in solution
At cathodic part compound such as oxides are formed
Mn+ → oxide compounds (Metal ion)
Classification of wet or electrochemical corrosion
(i) Chemical corrosion
(ii) Underground corrosion
(iii) Under water corrosion
Chemical corrosion
This type of corrosion is the result of electro chemical or chemical reaction
between a metal and its liquid surroundings.
This chemical corrosion involves the formation of anodic and cathodic areas
of part in contact with each other in contact with each other in presence of a
conducting medium.
Underground (or) soil corrosion
This type of corrosion take place in electric cables and other underground
structures embedded in the soil. The corrosiveness of the soil depends upon various
factors such as
(a) Acidity-The corrosion increases with increase in the concentration of H+
ions.
(b) Degree of aeration
(c) Moisture and soluble matter-These contents are greater, higher is
corrosion.
(d) Electrical conductivity-Greater the electrical conductivity, greater is
corrosion.

(e) Presence of bacteria-Certain types of bacteria in the soil oxidize organic
and other oxidisable matter to produce gases which may cause corrosion.
(f) Texture of soil-The texture of a soil is determined by the percentage of
particles of various sizes.
Under water corrosion
This type of corrosion includes corrosion in water pipes of all types hasting
systems; steam boilers etc. the water is in sufficient amount where as the air in
such type of corrosion in limited.
Corrosion can be categorized in some common types
1. Uniform corrosion
2. Pitting corrosion
3. Galvanic corrosion
4. Atmospheric corrosion
5. Inter-granular corrosion
6. Waterline corrosion
7. Stress corrosion
8. Crevice corrosion
9. Graphite corrosion
10. Fretting corrosion
1. Uniform corrosion
Differences in electrical potential occur on the surface of a piece of
metal due to small differences in chemical composition phase differences
amount of cold work etc. these differences set up small corrosion cells each
with an anode and cathode. Corrosion continues until the metal is consumed
or the film or rust formed on the surface ster up a barrier to the electrolyte.

2. Pitting corrosion
The metal loss is randomly located on the metal surface often combined with
stagnant fluid or in areas, with low fluid velocity.
Pitting corrosion is quite often evident on metal surfaces where no uniform
corrosion is usually proceeds fairly rapidly and is accelerated by the presence of
chlorides and is particularly common at the base of bleaks in coatings.
It often termed “under deposit corrosion”. It is deep penetration of the metal
surface with little general corrosion in the surrounding area. Due to surface
deposited electrical imbalance or corrosion potential attacks a selected number of
individual sites.
Anode reaction : Fe →Fe2+ +2e-
Cathode reaction : H2O+½O2+2e-→2OH-
Overall reaction : Fe2+ +2OH2+→Fe(OH)2 (corrosion product)
The presence of the extraction impurities embedded on the surface of metals
also lead to pitting.
3. Galvanic corrosion (or) bi- metallic corrosion
Galvanic corrosion takes place between two different metals or coatings
which are joined together in the presence of an electrolyte. Each metal has a
potential different from any other metal when placed in an electrolyte. A series can
be built up of all the metals relative to each other.
4. Atmospheric corrosion
This type of corrosion caused the combined effects of oxide film formation
and film break down. An oxide film is formed on the surface of the film because of
oxidation of metal by atmospheric air or oxygen and this oxide film protects the
metal by maintaining the continuity and hence reduces the chances of further
attack. The electrochemical action in presence of moisture or an electrolyte the

film undergoes break down on the surface of the metal and thus corrosion take
place at the cracks.
5. Inter-granular corrosion
Corrosion occurs at the grain boundaries due to a difference in potential
between the anodic grain boundaries and the cathodic grains. “Sensitized” stainless
steels, where carbides have been precipitated in the grain boundaries during
improper heat treatment or in the heat affected zone of a weld are particularly
susceptible to inter-granular corrosion.
6. Waterline corrosion
It is otherwise known as differential oxygen concentration corrosion. When
water is stored in a steal tank, it will be found that maximum amount of corrosion
takes place along a line just beneath the level of water meniscus. The area above
the waterline is highly oxygenated and acts as the cathodic section and is
completely unaffected by corrosion however, when the water is relatively free
from acidity little corrosion should take place.
Waterline corrosion is also caused in marine ships and is accelerated by
marine plants which are attacked to the sides of the ships. This type of corrosion is
prevented to a great extent by painting the sides of the ships by special anti fouling
paints.
7. Stress corrosion
Failure is due to the simultaneous influence of static tensile stresses and a
corrosive environment and this is specific to a particular metal. The stresses may
be internal such as those caused by cold work welding heat treatment or external
forces caused by stresses set up by heat treatment or external forces caused by
mechanical stresses set up by assembly practices. A good example of this form of
corrosion is 316 stainless steel was developed to with stand attacks in chloride
environment but if stressed the steel will fail by stress corrosion cracking.

8. Crevice corrosion
It occurs when there is a difference in ion or oxygen concentration between
the metal and its surroundings. Oxygen starvation in an electrolyte at the bottom of
a sharp section will set up an anodic site in the metal that then corrodes rapidly.
9. Graphite corrosion
Cast iron loosing iron in salt water (or) acids, leaves the graphite in place
resulting in a soft weak metal.
10. Fretting corrosion
It occurs as a result of repeated wearing weight and (or) vibration on an
uneven rough surface. Corrosion resulting in pits and grooves occurs on the surface
fretting corrosion is often found in rotation and impact machinery bolted
assemblies and bearings as well as to surfaces exposed to vibration during
transportation.
2.4. PREVENTION FROM CORROSION
Applied coatings
Plating, painting and application of enamel are the most common
anticorrosive treatments. They work by providing a barrier of corrosion – resistant
material between the damaging environment and the structural material.
Reactive coatings
If the environment is controlled (especially in recirculation system),
corrosion inhibitors can often be added to it. These from an electrically insulating
and or chemically impermeable coating on exposed metal surfaces, to suppress
electrochemical reactions such methods obviously make the system less sensitive
to scratches or defects in the coating. Since, extra indicators can be made available
wherever metal becomes exposed.

Anodization
Aluminium alloys often undergo a surface treatment. Electrochemical
conditions in the bath are carefully adjusted so that uniform pores several
nanometers wide appear in the metals oxide film. These pores allow the oxide to
grow much thicker than passivating conditions would allow. At the end of the
treatment, the pores are allowed to seal, forming a harder than usual surface layer.
In this coating is scratched, normal passivating processes take over to protect the
damaged urea. Anodizing is very resilient to weathering and corrosion, so it is
commonly used for building facades and other urea’s that the surface will come
into regular content with the elements.
Controlled permeability form work
Controlled permeability form work (CPF) is a method of preventating the
corrosion of reinforcement by naturally enhancing the durability of the cover
during concrete placement. CPF has been used in environments to combat the
effects of carbonation, chlorides, frost and abrasion.
Cathodic protection
Cathodic protection is a technique to control the corrosion of a metal surface
by making that surface the cathode of an electrochemical cell. It is a method used
to protect metal structures from corrosion. Cathodic protection systems are most
commonly used to protect steel, water and fuel pipelines and tanks, steel piper
pipes, ships and off shore oil platforms.
Anodic protection
Anodic protection impresses anodic current on the structure to be protected
(opposite to the cathodic protection). It is appropriate for metals that exhibit
passivity (e.g. Stainless steel) and suitably small passive current over a wide range
of potentials. It is used in aggressive environments. (e.g.) solutions of sulphuric
acid.

2.5. APPLICATION OF PROTECTIVE COATINGS
The application of protective coating is probably the oldest of the common
procedures for corrosion prevention. A coated surface isolates the underlying
metals from the corroding environment. The limitations of protective coatings:
I. The coating applied must be chemically inert to the environment
under particular conditions of temperature and pressure.
II. Moreover, they must prevent the penetration of the material, which
they project.
The two important process of protective coatings,
Anodic coating
Anodic coating is produced from coating-metals, which are “anodic” to the
metal. E.g. coating of Zn, Al and Cd on steel are anodic, because their electrode
potentials are lower than of the base metal iron.
Cathodic coating
Cathodic coatings are obtained by coating a more noble metal (ie having
higher electrode potential) than the base metal. They protect the base metal,
because they have higher corrosion-resistance than the base metal. Cathodic
coatings provide effective protection to the metal only when they are completely
continuous and free from pores, breaks or discontinuities.
E.g. A tin-coating on a sheet of iron provides protection only as long as the
surface of the metal is completely covered, since tin is lower than iron in
electromotive series. So, the surface coating is punctured, the tin becomes the
cathode. While the iron is above in the electromotive series acts as anodic.

2.6. CORROSION INHIBITOR
A corrosion inhibitor is a chemical compound. It may be defined as “a
substance which when added in small quantities to the aqueous corrosive
environment effectively decreases the corrosion rate of a metal”. In this word is
coming from Old Frenchinibicion and from Latininhibitio. So they were called to
inhibitor.
Corrosion processes are responsible for numerous losses mainly in the
industrial scope. It is clear that the best way to combat it is prevention. Among the
various methods to avoid or prevent destruction or degradation of metal surface,
the corrosion inhibitor is one of the best known methods of corrosion protection
and one of the most useful in the industry. This method is following stand up due
to low cost and practice method. Important researches have being conducted with
government investment mainly in large areas such as development construction of
new pipelines for shale gas and growth in construction. The focus of these
researches has being the inhibitors applications in water and concrete for the
protection of metals. Historically, inhibitors had great acceptance in the industries
due to excellent anti-corrosive proprieties. However, many showed up as a
secondary effect, damage the environment. Thus the scientific community began
searching for friendly environmentally inhibitors, like the organic inhibitors. This
chapter presents a revision of the corrosion inhibitors applications mainly the novel
compositions environmentally friendly. It describes the mechanisms of action of
inhibitors, main characteristics, environmental impact, technical analysis and
calculation of efficiency.

Mechanisms of actions of inhibitors
Inhibitors are substances or mixtures that in low concentration and in
aggressive environment inhibit, prevent or minimize the corrosion. Generally the
mechanism of the inhibitor is one or more of three that are cited below:
The inhibitor is chemically adsorbed (chemisorption) on the
surface of the metal and forms a protective thin film with
inhibitor effect or by combination between inhibitor ions and
metallic surface;
The inhibitor leads a formation of a film by oxide protection of
the base metal.
The inhibitor reacts with a potential corrosive component
present in aqueous media and the product is a complex.
History review
There are many industrial systems and commercial applications that
inhibitors are applicable, such as cooling systems, refinery units, pipelines,
chemicals, oil and gas production units, boilers and water processing, paints,
pigments, lubricants, etc. There are evidences of the use of inhibitor since the early
XIX century. On that time they were already used to protect metals in processes
such as acid picking, protection against aggressive water, acidified oil wells and
cooling systems. Since years 1950's and 1960's, there was significant advances in
the development of technology for corrosion inhibitor as the application of
electrochemistry to evaluate corrosion inhibitors. Recent studies estimate that the
U.S. demand for corrosion inhibitors will raise 4.1% per year to USD$ 2.5 billion
in 2017. In 2012 they estimated that the market demand of inhibitors was divided
on 26.6% to refining petroleum, 16.9% utilities, 16.7% gas and oil production,
15.3% chemical, 9.5% metals, 7.1% pulp and paper and 8.0% other. Now a days,
due to changes occurred on the market of corrosion inhibitors, some industrial

corrosion inhibitors are being unused. Due to high toxicity of chromate, phosphate
and arsenic compounds, related to various environmental and health problems,
strict international laws were imposed. Reducing the use of these and therefore
increasing the need for the development of other inhibitor to supply the lack in this
area. Should, however, present a similar anti corrosive properties similar than a
chromate inhibitor. An important number of papers have been published with the
intention of develop an environmentally friendly corrosion inhibitors and a lot of
research has been doing to development of the called “green” corrosion inhibitors.
Also, has been increasing research in natural products, such as plant extracts,
essential oils and purified compounds to obtain environmentally friendly corrosion
inhibitors. The first evidence of natural product use as corrosion inhibitors is
1930’s. When extracts of Chelidoniummajus(Celadine) and other plants were used
on the first time in H2SO4 pickling baths. Successful developments of researches to
obtain natural corrosion inhibitors are growing as quickly as the environmental
consciousness is gaining ground. Chromates as active inhibitors are being replaced
by other components such as molybdate compounds and rare earth metal salt, like
cerium chloride. Also, drugs have been studied as corrosion inhibitors.
2.7. INHIBITORS CLASSIFICATIONS
The corrosion inhibitors can be chemicals either synthetic or natural
and could be classified by:
The chemical nature as organic or inorganic.
The mechanism of action as anodic, cathodic or a anodic-cathodic mix
and by adsorption action.
As oxidants or not oxidants.
In general, the inorganic inhibitors have cathodic actions or anodic. The organics
inhibitors have both actions, cathodic and anodic and the protective by a film
adsorption.

Classification of inhibitors:
Inorganic inhibitors
Anodic inhibitors
Anodic inhibitors (also called passivation inhibitors) act by a reducing
anodic reaction, that is, blocks the anode reaction and supports the natural reaction
of passivation metal surface, also, due to the forming a film adsorbed on the metal.
In general, the inhibitors react with the corrosion product, initially formed,
resulting in a cohesive and insoluble film on the metal surface. Figure 1 shows a
potentiostatic polarization diagram of a solution with behaviour inhibitor anodic.
The anodic reaction is affected by the corrosion inhibitors and the corrosion
potential of the metal is shifted to more positive values. As well, the value of the
current in the curve decreases with the presence of the corrosion inhibitor.
Figure 1: Potentiostatic polarization diagram: electrochemical behaviour of a
metal in a solution with anodic inhibitor (a) Versus without inhibitor (b).

The anodic inhibitors reacts with metallic ions M+ produced on the anode,
forming generally, insoluble hydroxides which are deposited on the metal surface
as insoluble film and impermeable to metallic ion. From the hydrolysis of
inhibitors results in OH- ions. Figure 2 shows how is the mechanism of the anodic
inhibitory effect.
Figure 2: Illustration of anodic inorganic inhibitors effect and their
mechanism of action.
When the concentrations of inhibitor becomes high enough, the cathodic
current density at the primary passivation potential becomes higher than the critical
anodic current density, that is, shift the potential for a noble sense, and,
consequently, the metal is passivated. For the anodic inhibitors effect, it is very
important that the inhibitor concentrations should be high enough in the solution.
The inappropriate amount of the inhibitors affects the formation of film protection,
because it will not cover the metal completely, leaving sites of the metal exposed,
thus causing a localized corrosion.
Concentrations below to the critical value are worse than without inhibitors
at all. In general can cause pitting, due reduction at the anodic area relative to
cathodic, or can accelerate corrosion, like generalized corrosion, due to full

breakdown the passivity. Some examples of anodic inorganic inhibitors are
nitrates, molybdates, sodium chromates, phosphates, hydroxides and silicates.
Cathodic inhibitors
During the corrosion process, the cathodic corrosion inhibitors prevent the
occurrence of the cathodic reaction of the metal. These inhibitors have metal ions
able to produce a cathodic reaction due to alkalinity, thus producing insoluble
compounds that precipitate selectively on cathodic sites. Deposit over the metal a
compact and adherent film, restricting the diffusion of reducible species in these
areas. Thus, increasing the impedance of the surface and the diffusion restriction of
the reducible species, that is, the oxygen diffusion and electrons conductive in
these areas. These inhibitors cause high cathodic inhibition. The Figure 3 shows an
example of a polarization curve of the metal on the solution with a cathodic
inhibitor. When the cathodic reaction is affected the corrosion potential is shifted
to more negative values.
Figure 3: Potentiostatic polarization diagram electrochemical behavior of the
metal in a cathodic inhibitors solution (a), as compared to the same solution,
without inhibitor (b).
The cathodic inhibitors form a barrier of insoluble precipitates over the
metal, covering it. Thus, restricts the metal contact with the environment, even if it
is completely immersed, preventing the occurrence of the corrosion reaction. Due

to this, the cathodic inhibitor is independent of concentration, thus, they are
considerably more secure than anodic inhibitor. The Figure 4 shows the illustration
of mechanical effect of cathodic inhibitors to restrain the corrosion process.
Figure 4: Illustration has shown the mechanism of actuation of the cathodic
inhibitors.
Some examples of inorganic cathodic inhibitors are the ions of the
magnesium, zinc, and nickel that react with the hydroxyl (OH-) of the water
forming the insoluble hydroxides as (Mg(OH)2, Zn(OH)2, Ni(OH)2) which are
deposited on the cathodic site of the metal surface, protecting it. Also can be cited
polyphosphates, phosphonates, tannins, lignins and calcium salts as examples that
presents the same reaction mechanism. It seen in hard waters a kind of this
mechanism of inhibiting, due to the effect of the magnesium or calcium
bicarbonate on it. When temporary hard water flows over the metal it can assist on
the nucleation of carbonates, allowing the reactions near to the equilibrium and
forming precipitations on the metal surface. These precipitations, like a CaCO3,
cover the cathodic area, protecting the metal. So these cathodic inhibitor depends
only on the chemistry of the water, is not due the metal composition, because of
this they are applicable to all metals. As example, may be mentioned the oxides
and salts of antimony, arsenic and bismuth, which are deposited on the cathode

region in acid solutions. These cathodic inhibitors minimize the release of
hydrogen ions due to a phenomena that can difficult the discharge of the hydrogen,
called overvoltage.
Organic inhibitor
Organic compounds used as inhibitors, occasionally, they act as cathodic,
anodic or together, as cathodic and anodic inhibitors, nevertheless, as a general
rule, act through a process of surface adsorption, designated as a film- forming.
Naturally the occurrence of molecules exhibiting a strong affinity for metal
surfaces compounds showing good inhibition efficiency and low environmental
risk. These inhibitors build up a protective hydrophobic film adsorbed molecules
on the metal surface, which provides a barrier to the dissolution of the metal in the
electrolyte. They must be soluble or dispersible in the medium surrounding the
metal. In the Figure 5, that shows a theory potentiostatic polarization curve, it can
be seen that the effect of the solution containing organic inhibitor on the metal
presents an anodic and cathodic behaviour. After the addition of the inhibitor, the
corrosion potential remains the same, but the current decreases from Icor to I'cor.
Figure 5: Theoretical potentiostatic polarization diagram electrochemical
behavior a metal on a solution containing a cathodic and anodic inhibitor (a)
compared to the same solution without the inhibitor (b).

is showed in Figure 6 the mechanism of actuation of organic inhibitors, when it is
adsorbed to the metal surface and forms a protector film on it.
Figure 6: Illustration of the mechanism of actuation of the organic
inhibitor acting through adsorption of the inhibitor on the metal surface.
where the Inh represents the inhibitor molecules.
The efficiency of an organic inhibitor depends of the:
Chemical structure, like the size of the organic molecule;
Aromaticity and/or conjugated bonding, as the carbon chain length;
Type and number of bonding atoms or groups in the molecule (either
π or σ);
Nature and the charges of the metal surface of adsorption mode like
bonding strength to metal substrate;
Ability for a layer to become compact or cross-linked,
Capability to form a complex with the atom as a solid within the metal
lattice;
Type of the electrolyte solution like adequate solubility in the
environment.

The efficiency of these organic corrosion inhibitors is related to the presence
of polar functional groups with S, O or N atoms in the molecule, heterocyclic
compounds and pi electrons, generally have hydrophilic or hydrophobic parts
ionisable. The polar function is usually regarded as the reaction centre for the
establishment of the adsorption process. The organic acid inhibitor that contains
oxygen, nitrogen and/or sulphur is adsorbed on the metallic surface blocking the
active corrosion sites. Although the most effective and efficient organic inhibitors
are compounds that have π-bonds, it present biological toxicity and environmental
harmful characteristics. Due to the metal surface covered is proportional to the
inhibitor concentrates, the concentrations of the inhibitor in the medium is critical.
Some examples are amines, urea, Mercaptobenzothiazole (MBT), benzotriazole e
toliotriazol, aldehydes, heterocyclic nitrogen compounds, sulfur-containing
compounds and acetylenic compounds and also ascorbic acid, succinic acid,
tryptamine, caffeine and extracts of natural substances. There are still some
inhibitors that act in vapour phase (volatile corrosion inhibitor). Some examples
are: dicicloexilamônio benzoate, diisopropylammonium nitrite or benzoate,
ethanolamine benzoate or carbonate and also the combination of urea and sodium
nitrite.
Techniques for analysis of inhibitors
The most usefully technique to analysis the effectiveness of an inhibitor are
weight loss experiment and electrochemical measurements, like polarization curve
method and the impedance measurement analysing. In addition, microscopy
techniques are used to characterize the corrosion process.
Considerations to employ inhibitors
For all types of inhibitors, we should consider some environmental actions
factors because some elements such as metals, pH, composition, impurities,
agitation, and temperature, geometry of the system, the concentration of inhibitor

and the mixture of one or more inhibitors may change the anti-corrosive
mechanism. To employment of the inhibitors is quite satisfying that certain factors
should be seen as the real cause of the corrosion, the cost X benefit and possible
interactions of the inhibitor with the environment, such as the influence of a
catalyst, deposition or contamination. Four fundamental aspects must be analyzed
to obtain a satisfactory result from the use of the inhibitor.
Industrial application
Acid pickling: Prevent the attack in the metal due to the acid solution in
which metal gets cleaned of mill scale (bark lamination), and also prevented the
subsequent hydrogen evolution inhibitors are added, typically organic, must be
soluble or dispersed in the solution. Examples: thiourea and amino and its
derivatives, propargyl alcohol.
Oil industry: sodium carbonates or organic amines complex are employed
to reduce the corrosive effect of CO2, H2S and organic acids, enabling the use of
more cheaper materials and less resistant to corrosion in wells extracting crude oil.
Pipes for gasoline and kerosene are employed sulphonated oils, sodium nitrite. Oil
well uses up fatty amines, fatty acids, imidazolines and quaternary ammonium
salts. Internal pipe corrosion occurs in wet gas transportation due to condensation
of water containing dissolved corrosive gases. Corrosion is caused by the
dissolution of the corrosive gases, such as carbon dioxide and hydrogen sulphide as
well as condensation of acid vapors.
Water transmission and distribution systems: is used corrosion inhibitor
in combination with pH adjusters and alkalinity control towards an efficient
protection. The most common inhibitors are phosphates, amines volatiles
(cyclohexylamine, morphine)

Concrete: To improve the durability of reinforced concrete structures,
which are impaired due the high alkalinity, are used corrosion inhibitors, mixed
with cement or concrete paste. An example is phosphate ion.
Boiler: Thermoelectric use, in general, Ammonia, Cyclohexylamine, alkanol
and Morpholine as inhibitors in boilers in various processes. The inhibitors, also,
are added by the hydrochloric acid used for the solubilization of limescaleto
prevent the attack on pipes.
Application of corrosion inhibitors for steels in acidic media for the oil and
gas industry
Human demand for fossil fuels is still growing even though alternatives to
such energy are currently being sought. Oil and natural gas account for 60% of all
global energy demands. It is thus not expected that the conventional method of
extracting fossil fuels will disappear within the next few decades. The extraction of
geothermal water for use as an energy source is also of paramount importance and
its usage is increasing. The methods required to maximize production typically
comprise formation stimulation and subsequent well cleaning, both of which can
induce a corrosive environment for the steel involved, as it is the main construction
material of wells. Corrosion is worth investigating in oilfield applications, because
corrosion problems represent a large portion of the total costs for oil and gas
producing companies every year worldwide. Moreover, appropriate corrosion
control can help avoid many potential disasters that can cause serious issues
including loss of life, negative social impacts, and water resource and
environmental pollution. Corrosion in oilfields occurs at all stages from down hole
to surface equipment and processing facilities. It appears as leaks in tanks, casings,
tubing, pipelines, and other equipment and. Corrosion problems are usually
connected with operating problems and equipment maintenance, leading to
recurrent partial and even total process shutdown, resulting in severe economic

losses. Moreover, Garcia-Arriaga et al. reported that the economic costs linked to
the corrosion of natural gas sweetening (CO2 corrosion) and oil refining plants
range between 10% and 30% of the maintenance budget.
In the petroleum industry, general and localized corrosion are the most
common types of corrosion occurrences. The other large problem in operating pipe
flow lines is internal corrosion, mainly due to stress corrosion cracking. Martinez
et al. claim that the combination of corrosion and erosion is the main problem in
pipe deterioration. Also noted recently is an increase in the occurrence of galvanic
corrosion problems associated with the use of different dissimilar materials, which
has garnered much attention. Wilhelm reported that the most common situation of
coupling dissimilar materials in wells consists of a tubing string made of corrosion-
resistant alloy in contact with lower-grade steel casing. Moreover, the metal
contacts also cause crevice corrosion in the occluded area between tubing and
casing.
The primary focus of this review is to summarize different research relating
to corrosion and its inhibition regarding mild, carbon, and low-alloy steel – lower-
grade steels – in different acidic solutions encountered in the crude oil and natural
gas sector. These materials are used in well construction. In the petroleum industry,
one facet of the development of new oil and gas production is the stimulation
process. Overall, the stimulation process involves many different aspects,
including the acidizing portion utilized to stimulate the carbonate reservoir or for
dissolving fines. Typically, highly concentrated acids, between 5 and 28 wt.%, are
used which make the environment corrosive to mild, carbon, and low-alloy steels.
Hydrochloric, hydrofluoric, acetic, or formic acids are injected into the well during
the acidizing stimulation process and cause serious corrosion issues. In the absence
of corrosion inhibitors (CIs), the general CR (corrosion rate) can be extremely high
(>100 mm/y) and can increase exponentially with increasing temperatures and acid

concentrations. Due to the extreme corrosion conditions of this process, developed
technology can then be translated to other industries. In particular, this can be
relevant for acid pickling, industrial cleaning, and acid descaling, where corrosion
conditions are usually milder. This may be a secondary source of information for
readers of this review. It has to be pointed out that the petroleum industry is the
largest consumer of CIs. This review only addresses individual CIs for application
in HCl mediums with different steels because HCl is the most prevalent acid used
in stimulation.
An effort has been made herein to combine different works by the same
authors in a single paragraph, even though not all authors of different articles or
patents appear together all the time. In this review, when steel materials in general
are written about, lower-grade steels are being referred to. All concentrations in %
are always reported as a mass fraction if not stated otherwise. Moreover, when
concentrations in various articles were reported in parts per million (ppm), herein
they are converted to mg/L.
This work discusses the well acidizing procedure in general so that readers
of this review can gain an impression of the severe corrosion conditions during that
process. Moreover, the steel materials used for well construction and associated
with corrosion problems are discussed. The corrosion of these steel materials and
previously tested CIs for HCl solutions are reviewed. This review also explains
aspects of a corrosion inhibitor formulation design in order to increase the success
of these CIs at elevated temperatures or under other well environmental conditions.
Furthermore, it also presents environmental concerns in corrosion inhibition
processes, environmental friendly methane, sulphonic acid, and some
recommendations for correct test methods regarding acid CIs.

The well acidizing procedure
Limestone formations or carbonate-bearing sandstone carry many
hydrocarbon reservoirs. A very important step in the oil, gas, and geothermal water
drilling industry is the well acidizing procedure, which is a rock reservoir (the
origin of the natural resource or water – a geological subterranean formation)
stimulation technique used to improve productivity. Acids are forced under high
pressure through the borehole into the pore spaces of the rock formation, where
they react chemically with rocks to dissolve them (usually calcite, limestone, and
dolomite), which enlarges the existing flow channels and opens new ones to the
wellbore. Acidizing is used in conjunction with hydraulic fracturing techniques and
matrix acidizing techniques. In fracture acidizing treatments, one or more fractures
are produced in the formation and acidic solution is introduced into the fracture to
etch flow channels in the fracture face. The acid also enlarges the pore spaces in
the fracture face and in the formation. The fractures are then filled with sand or
other material in order to prevent the fractures from closing and allow the
penetration of natural resources or water. Acids are often also employed for scale
removal treatments (pickling of the well tubing) and for the removal of drilling
mud damage in newly drilled wells before being brought into production. For
example, the combination of fluorosilicate with metal ions such as Na+ may cause
the precipitation of gelatinous compounds, which need to be removed. Scale
removal treatments are usually done with 15% HCl at temperatures up to 60 °C in
order to remove iron oxides and carbonated minerals. Acidizing steps are
frequently repeated. All these procedures involve the injection of acids into the
well system made of steel tubes. In deep wells the down hole temperature may
exceed 200 °C. During the acidizing process metallic materials can also come into
contact with acid solution and sometimes with H2S and CO2 at elevated
temperatures. Due to the above listed problems, the acidizing process requires a

high degree of corrosion protection of tubular materials and other equipment
employed.
2.8. REVIEW OF LITERATURE CORROSION OF TIN INFRUIT
JUICES AND IT’S INHIBITION
The corrosion of tin cans is major problem for food industries, which causes
enormous wastage of food material due to metallic contamination. Presence of
metal in juices causes change in colour, taste and texture of food leading to heavy
economic losses to canning industries. The corrosion of tin in contact of acidic
fruit juices is attributed to the reversal of polarity of tin and thus tin became anodic
to iron in acidic medium there by dissolving the latter. Internal corrosion of food
cans mainly includes the properties of tin plate, nature of food process and the
processing as well as storage condition. Surgary et.al used conventional method,
such as weight loss, atomic absorption and colorimetric methods for determining
the amount of dissolved tin or in food Katarina et.al studied the corrosion of iron
and tin in apple, cherry, lemon, orange, grapes and pineapple, juices by
electrochemical and weight loss method and tin present in juices were determined
by using atomic absorption methods. Mahadeviah et.al studied the tin plate
corrosion in canned mango, pineapples and orange juices and observed that
corrosion of the tin plate occurred due to organic acid fraction of fruit juices.
Popova et.al and Zuauya and Chpurza studied in the corrosion behavior of
lacquered tin plate in citric acid and acetic acid. Quaraishi et.al studied the
corrosion behavior of tin plate in citric acid in presence and absence of various
concentration of nitrate and sucrose and influence of glycine on tin in corrosion in
presence of three fruit juices namely orange, mango and pineapple by using
potentiodynamic polarization technique. Mahadevish investigated agar, pectin,
gelatin, phospholipids as corrosion inhibitors for food cans. In view of this there

existed a need for developing non-metallic and effective corrosion inhibitors for
food cans. Here the corrosion behaviour of tin in contact with mango, orange and
pineapple fruit juices by potentiodynamic method inhibitours such as DL Alanine,
D1-aspartic acid, L-Lucine, Dt-2 amoniobutryric acid. L-Glutamic acid, DL-
Terleucine and DL aspartic acid were studied.
Six amino acids studied were excellent corrosion inhibitors of tin in three fruit
juices viz, orange, mango and pineapple. They acted as predominantly Cathodic or
mixed type of corrosion inhibitors.
Most of the tin plate produced is used for the manufacture of food containers. The
nontoxic nature of tin salts makes tin ideal materials for the handling of food and
beverages.
An inspection of the galvanic series will indicates that tin is more noble than steel
and, consequently, the steel would corrode at the base of the pores. On the outside
of tinned containers, the tin is cathodic to the steel. However, on the inside of the
containers, there is a reversal of polarity because of the completing of the stannous
ions by many food products. This greatly reduces the activity of stannous ions,
resulting in a change in the potential of tin in the active direction.
This change in polarity is absolutely necessary because most tin coatings are thin
and therefore porous. To avoid perforation of the can, the tin must act as a
sacrificial coating.
The environment inside a hermetically sealed cans various depending upon
the contents, which could include general foods, beverages, oils, aerosol products,
liquid gases, etc. The interior of a can is subject to general corrosion, localized
corrosion, and discoloring. The coating system for tin plate consists of tin oxide,

metallic tin, and alloy. The dissolution of the tin layer in acidic fruits products is
caused by acids such as citric acids.
In carbonated beverages, the potential reversal does not take place; therefore, the
steel dissolves preferentially at the defects the tin layer. Under such conditions,
pitting corrosion sometimes results in perforation, consequently, except for fruit
cans; almost all tin plate cans are lacquered. When tin plate is to be used for
structural purposes such as roofs, an alloy of 12-25 parts of tin to 88-75 parts of
lead is frequently used. This is called Terneplate. It is less expensive and more
resistant to weather than a pure tin coating. Terneplate is used for fuel tanks of
automobiles and is also used in the manufacture of fuel lines and radiators in
automobiles.

CHAPTER-II
OBJECTIVIES
Tin has high industrial percentage of application. Cans made of tin plate are used
for food products. Since tin was not affected by most foods in the absence of air.
Pine apple comes in real tin cans. Tin plate is used in many ways such as roofing
and as the “tin”(or) telltale in squash court. Tin possesses considerable resistance
against atmospheric corrosion. Moreover, because of non-toxic nature of tin,
tinning is widely used for coating steel, copper and brass sheets, used for
manufacturing containers for storing food stuff, ghee, oils, kerosene and packing
food materials. Tinned-copper sheets are employed for making cooking utensils
and refrigeration equipments.
The surface of the tin metal must be freed from rust or oxide scales. Acid
pickling, the immersion of tin in acid solution has become an important
pretreatment process for removing these rusts and scales. During pickling, soon
after the scales are removed, the acid will attack the metal. To minimize the loss of
metal due to pickling, corrosion inhibitors are added to pickling solutions.
Hydrochloric acid and sulphuric acid is employed in pickling baths and occupies
the center stage due to its economic advantage. The weight of the metal is
minimized.
All these studies are found to be aimed at finding a good inhibitor which should
have the following characteristic.
Should inhibit – the dissolution of metal.
Should limit pickling.
Should be stable and effective at low inhibitor concentration and high
temperature.
Should be a good surfactor.

Should resists the permeation of hydrogen into the metal.
The present work focuses on the study of inhibitive behaviour of Glycine, urea
,and Sucrose on the corrosion of the tin plate in acid medium.

CHAPTER-III
3. EXPERIMENTAL METHODS
In this part of the present work, corrosion of Tin plate in various concentrations of
hydrochloric acid and sulphuric acid the effect of Urea, Glycine and Sucrose as
inhibitor in combination the corrosion have been investigated. For the purpose, the
conventional weight loss method has been employed.
3.1. Chemicals
AR Hydrochloric acid, AR Sulphuric acid, Inhibitors- Urea, Glycine and
Sucrose, double distilled water and acetone.
3.2. Materials
Tin length 4 cm and breadth 1 ½ cm, 100 ml standard measuring flask, 100
ml beaker, hooks and paper.
3.3. Principle
Weight loss method is used here which is the most reliable method. In this method
the loss of metal due to corrosion is measured by exposing the metal specimen of
known urea to the environment for a known period of time and the difference in
weight and after exposure is calculated.
3.4. Procedure
Acid solution: 1N, 0.8N and 0.6N hydrochloric acid and 1N, 0.8N and 0.6N
sulphuric acid was prepared from AR hydrochloric acid and AR sulphuric acid
using double distilled water.
Inhibitor solutions : Commercially available AR Urea, Glycine and Sucrose were
used. Standard inhibitor solutions were prepared by dissolving the inhibitors in
double distilled water, diluted to suitable volumes.
Weight loss Measurement
Tin plate 4 cm x 1.5 cm has been used. It is polished using emery papers and
washed with distilled water and finally degreased with the organic solvent acetone.

The specimens were weighed. After weighing the specimens were immersed in 1N
hydrochloric acid with (different Concentrations) and without inhibitors. After 3
hours, the specimens were washed with distilled water, dried and again weighed.
The weight loss was noted. From this weight loss value inhibitor efficiency were
determined. The same procedure was repeated with 0.8N and 0.6N hydrochloric
acid with0.1N, 0.2N, 0.3N, 0.4N, 0.5N, 0.6N, 0.7N, 0.8N and 0.9N inhibitor
solutions of Urea, Glycine and Sucrose.
The same procedure was repeated with the sulphuric acid.
Inhibitor efficiency
Inhibitor efficiency has been determined by using the following relationship.
I.E % = ௐబିௐ
ௐబ × 100
Where, W0 = Weight loss without inhibitor
Wi = weight loss with inhibitor.
Measurement of surface coverage (ϴ)
The surface coverage (ϴ) is calculated using the formula.
Surface coverage (ϴ) =ௐ್ିௐ
ௐ್
Where Wb and Wi are the weight losses per unit urea per unit time without and
with inhibitor respectively.
Adsorption isotherm
After the determination of surface coverage, the type of isotherm into which
the observed data fit into is determined. The following isotherm studies were
carried
i. Langmuir
ii. Temkin
iii. Frumkin

CHAPTER-IV
4. RESULT AND DISCUSSIONS
Table 1
1N HCl and various concentrations of Urea S.NO CONCEN-
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.844 1.782 0 .079 - 2. 0.1 1.776 1.700 0.076 3.8 3. 0.2 1.865 1.788 0.077 2.53 4. 0.3 1.903 1.832 0.071 10.13 5. 0.4 1.830 1.758 0.072 8.6 6. 0.5 1.917 1.844 0.073 7.59 7. 0.6 1.884 1.805 0.079 0 8. 0.7 1.771 1.699 0.072 8.86 9. 0.8 1.851 1.781 0.070 11.39 10. 0.9 1.831 1.766 0.065 17.72
Table 2
0.8N HCl and various concentrations of urea S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.865 1.786 0 .079 - 2. 0.1 1.803 1.738 0.065 17.72 3. 0.2 1.833 1.766 0.067 15.19 4. 0.3 1.809 1.750 0.059 25.31 5. 0.4 1.884 1.827 0.057 27.85 6. 0.5 1.859 1.800 0.059 25.31 7. 0.6 1.824 1.763 0.061 22.28 8. 0.7 1.832 1.779 0.043 32.91 9. 0.8 1.805 1.755 0.050 36.71 10. 0.9 1.847 1.798 0.049 37.97

Table 3
0.6N HCl and various concentrations of Urea S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.827 1.703 0.124 - 2. 0.1 1.820 1.738 0.082 33.87 3. 0.2 1.847 1.772 0.091 26.61 4. 0.3 1.851 1.778 0.073 41.13 5. 0.4 1.874 1.804 0.070 43.55 6. 0.5 1.807 1.726 0.081 34.68 7. 0.6 1.865 1.786 0.079 36.29 8. 0.7 1.845 1.737 0.108 12.90 9. 0.8 1.843 1.772 0.071 42.74 10. 0.9 1.807 1.754 0.053 57.26
Table 4
1N HCl and various concentrations of Sucrose S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.857 1.748 0.109 -
2. 0.1 1.880 1.805 0.075 31.19
3. 0.2 1.833 1.762 0.071 34.86
4. 0.3 1.814 1.746 0.068 37.61 5. 0.4 1.842 1.767 0.075 31.19
6. 0.5 1.873 1.790 0.083 23.85
7. 0.6 1.750 1.670 0.080 26.61
8. 0.7 1.836 1.736 0.100 8.26
9. 0.8 1.788 1.711 0.077 29.36
10. 0.9 1.814 1.744 0.070 35.78

Table 5
0.8N HCl and various concentrations of Sucrose S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.808 1.723 0.085 - 2. 0.1 1.823 1.757 0.066 22.35 3. 0.2 1.888 1.817 0.071 16.50 4. 0.3 1.896 1.829 0.067 21.18 5. 0.4 1.850 1.787 0.061 25.88 6. 0.5 1.838 1.778 0.060 29.41 7. 0.6 1.895 1.830 0.065 23.53 8. 0.7 1.843 1.758 0.085 0 9. 0.8 1.815 1.745 0.070 17.65 10. 0.9 1.868 1.784 0.084 1.18
Table 6
0.6N HCl and various concentrations of Surose
S.NO CONCENTRATION
INTIAL WEIGHT
W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.875 1.776 0.099 -
2. 0.1 1.830 1.756 0.074 25.25
3. 0.2 1.862 1.777 0.085 14.14
4. 0.3 1.824 1.749 0.075 24.24
5. 0.4 1.842 1.766 0.073 26.26 6. 0.5 1.814 1.753 0.061 22.22
7. 0.6 1.817 1.747 0.077 29.29
8. 0.7 1.829 1.736 0.093 6.06
9. 0.8 1.857 1.769 0.088 11.11
10. 0.9 1.862 1.776 0.086 13.13

Table 7
1N HCl and various concentrations of Glycine S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENC
Y %
1. 0 1.808 1.714 0.094 - 2. 0.1 1.861 1.784 0.077 18.09 3. 0.2 1.897 1.818 0.073 22.34 4. 0.3 1.782 1.657 0.074 21.28 5. 0.4 1.825 1.741 0.084 10.64 6. 0.5 1.850 1.773 0.077 18.09 7. 0.6 1.835 1.763 0.072 23.40 8. 0.7 1.834 1.760 0.074 21.28 9. 0.8 1.862 1.750 0.112 16.68 10. 0.9 1.869 1.784 0.085 9.57
Table 8
0.8N HCl and various concentrations of Glycine
S.NO CONCENTRATION
INTIAL WEIGHT
W1(g)
FINAL WEIGHT
W2(g)
W1-W2 EFFICIENCY %
1. 0 1.844 1.759 0.085 -
2. 0.1 1.826 1.748 0.078 8.24
3. 0.2 1.766 1.668 0.076 10.59
4. 0.3 1.798 1.717 0.081 4.71
5. 0.4 1.828 1.743 0.085 0 6. 0.5 1.795 1.698 0.074 12.94
7. 0.6 1.896 1.764 0.078 8.24
8. 0.7 1.877 1.790 0.079 7.05
9. 0.8 1.856 1.810 0.081 4.71
10. 0.9 1.860 1.760 0.079 7.05

Table 9
0.6 N HCl and various concentrations of Glycine S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.795 1.725 0.07 - 2. 0.1 1.781 1.715 0.066 6.02 3. 0.2 1.774 1.71 0.064 8.57 4. 0.3 1.765 1.699 0.066 6.02 5. 0.4 1.854 1.787 0.067 4.29 6. 0.5 1.744 1.678 0.0665 6.07 7. 0.6 1.817 1.754 0.063 10 8. 0.7 1.867 1.803 0.064 8.57 9. 0.8 1.731 1.665 0.066 6.02 10. 0.9 1.822 1.752 0.07 0
Table 10
1 N H2SO4 and various concentrations of Urea S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2 EFFICIENCY %
1. 0 1.824 1.605 0.219 -
2. 0.1 1.883 1.673 0.210 4.11
3. 0.2 1.838 1.636 0.202 7.76
4. 0.3 1.817 1.622 0.195 10.96
5. 0.4 1.779 1.585 0.194 11.42 6. 0.5 1.830 1.623 0.207 5.48
7. 0.6 1.88 1.666 0.214 2.28
8. 0.7 1.839 1.642 0.197 10.05
9. 0.8 1.842 1.635 0.207 5.48
10. 0.9 1.822 1.628 0.194 11.42

Table 11
0.8 N H2SO4 and various concentrations of Urea S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.814 1.637 0.177 -
2. 0.1 1.850 1.685 0.165 6.78
3. 0.2 1.798 1.655 0.143 19.21
4. 0.3 1.833 1.680 0.153 13.56
5. 0.4 1.876 1.722 0.154 12.99 6. 0.5 1.841 1.687 0.154 12.99
7. 0.6 1.907 1.763 0.144 18.64
8. 0.7 1.868 1.711 0.157 11.31
9. 0.8 1.849 1.692 0.157 11.31
10. 0.9 1.801 1.633 0.168 5.08
Table 12
0.6 N H2SO4 and various concentrations of Urea S.NO CONCENTR
ATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.855 1.698 0.157 -
2. 0.1 1.838 1.708 0.130 15.58
3. 0.2 1.864 1.746 0.118 24.84
4. 0.3 1.808 1.687 0.121 21.43
5. 0.4 1.798 1.675 0.123 20.12 6. 0.5 1.823 1.700 0.123 20.12
7. 0.6 1.820 1.698 0.122 22.29
8. 0.7 1.861 1.727 0.134 14.65
9. 0.8 1.806 1.689 0.117 25.48
10. 0.9 1.880 1.766 0.114 27.39

Table 13
1 N H2SO4 and various concentrations of Sucrose S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 2.125 2.026 0.099 -
2. 0.1 1.895 1.836 0.059 40.40
3. 0.2 1.882 1.802 0.08 19.19
4. 0.3 2.084 2.014 0.07 29.29
5. 0.4 2.107 2.037 0.07 29.29 6. 0.5 2.107 2.037 0.07 29.29
7. 0.6 1.878 1.784 0.094 5.05
8. 0.7 1.792 1.738 0.054 0.445
9. 0.8 1.844 1.782 0.062 37.37
10. 0.9 1.813 1.753 0.06 39.39
Table 14
0.8 N H2SO4 and various concentrations of Sucrose S.NO CONCENTR
ATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.924 1.789 0.135 -
2. 0.1 1.935 1.868 0.067 50.37
3. 0.2 1.924 1.680 0.074 45.19
4. 0.3 1,912 1.830 0.082 39.26
5. 0.4 2.171 2.099 0.072 46.67 6. 0.5 1.997 1.911 0.086 36.29
7. 0.6 1.896 1.790 0.106 21.48
8. 0.7 1.954 1.877 0.077 42.96
9. 0.8 1.943 1.839 0.104 45.93
10. 0.9 1.956 1.883 0.073 0.05

Table 15
0.6 N H2SO4 and various concentrations of Sucrose S.NO CONCENTR
ATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.842 1.719 0.123 -
2. 0.1 2.150 2.084 0.066 46.34
3. 0.2 1.905 1.809 0.096 21.95
4. 0.3 1.878 1.770 0.108 12.19
5. 0.4 1.782 1.730 0.052 57.72 6. 0.5 2.135 2.055 0.08 34.96
7. 0.6 1.954 1.869 0.085 30.89
8. 0.7 1.766 1.694 0.072 41.46
9. 0.8 1.943 1.861 0.082 33.33
10. 0.9 1.875 1.776 0.099 19.51
Table 16
1 N H2SO4 and various concentrations of Glycine S.NO CONCENTR
ATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2
EFFICIENCY
%
1. 0 1.756 1.667 0.089 -
2. 0.1 2.193 2.108 0.085 4.494
3. 0.2 1.895 1.811 0.084 5.618
4. 0.3 1.917 1.834 0.083 6.742
5. 0.4 1.848 1.766 0.082 7.865 6. 0.5 2.186 2.106 0.080 10.112
7. 0.6 2.152 2.068 0.084 5.618
8. 0.7 1.847 1.761 0.086 3.371
9. 0.8 1.820 1.733 0.087 2.247
10. 0.9 1.833 1.751 0.082 7.865

Table 17
0.8 N H2SO4 and various concentrations of Glycine S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2 EFFICIENCY %
1. 0 1.974 1.812 0.162 -
2. 0.1 1.775 1.692 0.083 48.77
3. 0.2 1.965 1.834 0.131 19.14
4. 0.3 2.164 2.079 0.085 47.53
5. 0.4 1.894 1.771 0.123 24.07 6. 0.5 2.238 2.152 0.086 46.91
7. 0.6 1.774 1.716 0.058 64.2
8. 0.7 1.958 1.843 0.115 29.01
9. 0.8 2.123 2.048 0.075 53.7
10. 0.9 2.143 2.066 0.077 52.47
Table 18
0.6 N H2SO4 and various concentrations of Glycine S.NO CONCEN
TRATION INTIAL
WEIGHT W1(g)
FINAL WEIGHT
W2(g)
W1-W2 EFFICIENCY %
1. 0 2.189 2.115 0.074 -
2. 0.1 1.960 1.842 0.118 37.29
3. 0.2 1.944 1.836 0.108 8.47
4. 0.3 1.815 1.724 0.091 22.88
5. 0.4 2.231 2.151 0.08 32.20 6. 0.5 1.960 1.847 0.113 4.24
7. 0.6 1.818 1.759 0.059 50
8. 0.7 1.963 1.862 0.101 14.41
9. 0.8 1.840 1.736 0.104 11.86
10. 0.9 1.836 1.733 0.103 12.71

Temkin isotherm, Frumkin isotherm and Langmuir isotherm Graph for 1N,
0.8N, 0.6N HCl versus concentration of Urea
Temkin isotherm Frumkin isotherm
Langmuir isotherm

Temkin isotherm, Frumkin isotherm and Langmuir isotherm Graph for 1N,
0.8N, 0.6N HCl versus concentration of Sucrose
Temkin isotherm Frumkin isotherm
Langmuir isotherm

Temkin isotherm, Frumkin isotherm and Langmuir isotherm Graph for 1N,
0.8N, 0.6N H2SO4 versus concentration of Urea
Temkin isotherm Frumkin isotherm
Langmuir isotherm
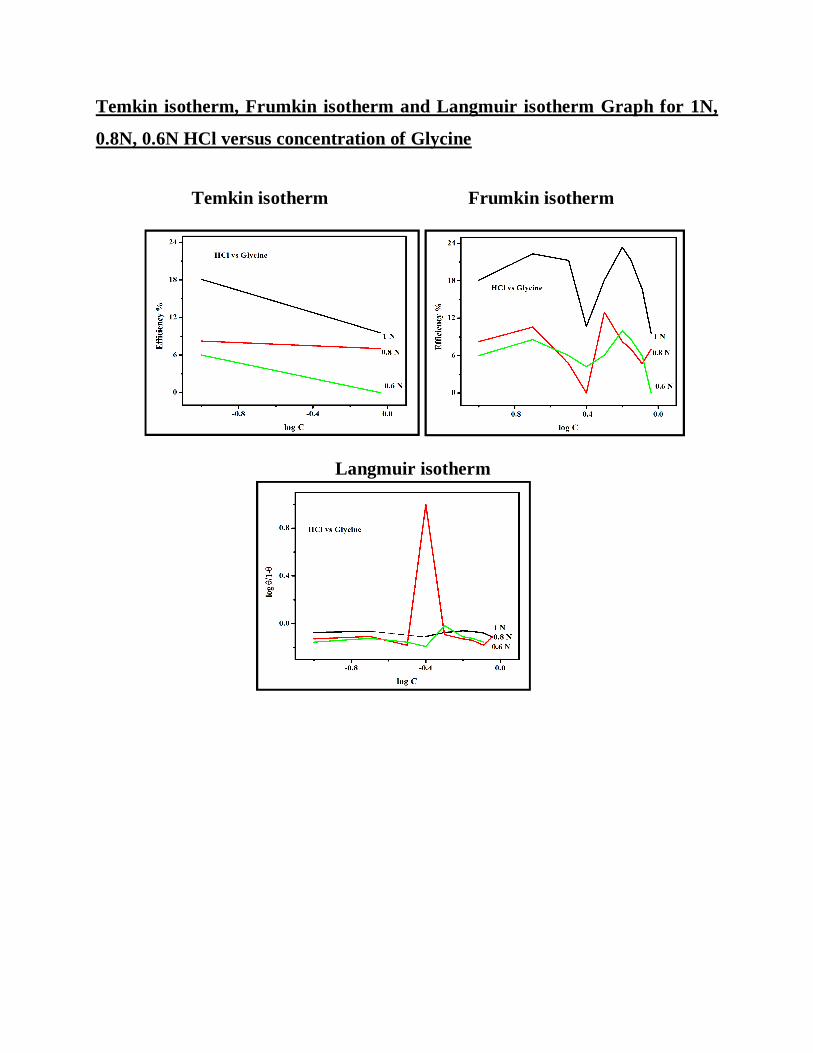
Temkin isotherm, Frumkin isotherm and Langmuir isotherm Graph for 1N,
0.8N, 0.6N HCl versus concentration of Glycine
Temkin isotherm Frumkin isotherm
Langmuir isotherm

Temkin isotherm, Frumkin isotherm and Langmuir isotherm Graph for 1N,
0.8N, 0.6N H2SO4 versus concentration of Glycine
Temkin isotherm Frumkin isotherm
Langmuir isotherm

Temkin isotherm, Frumkin isotherm and Langmuir isotherm Graph for 1N,
0.8N, 0.6N H2SO4 versus concentration of Sucrose
Temkin isotherm Frumkin isotherm
Langmuir isotherm

From the table it is observed that,
HCl medium:
The inhibitor efficiency of urea in 0.6 N HCl is better when compared to 0.8
and 1N HCl, Temkin, Frumkin and Langmuir isotherm also reveals the same
result. The order of inhibitor efficiency 0.6 N > 0.8N > 1N.
In the case of Sucrose, the inhibitor efficiency is high in 1N HCl. The order
of inhibitor efficiency 1N > 0.6N >0.8N.
In the case of glycine, the inhibitor efficiency is high in 1N HCl. The order
of inhibitor efficiency 1N > 0.8N > 0.6N.
Sulphuric acid medium:
In the case of urea, the inhibitor efficiency is high in 0.6N. The order of
inhibitor efficiency 0.6N > 0.8N > 1N. The inhibitor efficiency of Sucrose in
0.8N is better when compared to 1N and 0.6N The order of inhibitor
efficiency 0.8N > 1N > 0.6N. Frumkin and Temkin follow the above order
whereas Langmuir isotherm reveals 1N > 0.8N > 0.6N. In the case of
glycerine, the inhibitor efficiency follows the order
0.8N > 0.6N > I N

CHAPTER 5
5. CONCLUSION
From the above result the inhibitor efficiency is better in Hydrochloric acid
than sulphuric acid
Inhibitor efficiency is high in 1N HCl in case of Sucrose and glycine.
The inhibitor efficiency is high in 0.8N Sulphuric acid in the case of Sucrose
and glycine
The inhibitor efficiency of urea in both hydrochloric acid and sulphuric acid
follows the same order 0.6N > 0.8N > 1N

BIBLIOGRAPHY
Materials Science and Engineering by V. Raghavan , 3rd edition (1990).
Fundamentals of inorganic glasses by A.K. Varshneya, Society of glass
technology, Sheffied, pp. 628 (2006).
New development in glassy nuclear waste forms by M.I.Ojovan, W.E.Lee,
Nova Science publishers, New York, pp.136 (2007).
Corrosion of glass, Ceramics and Superconductors, by D.E.Clark,
B.K.Zoitos, Willam Andrew Publishing/Noyes, pp.672 (1992).
Management of accelerated low water corrosion in steel maritime structures
by J. E. Breakell, M.Siegwart, K. Foster, D. Marshall, M.Hodgson, R.
Cottis, S. Lyon.
Corrosion-causes and prevention by F. N. Speller, 3rd edition (London: The
English Universities Press 1971).
Corrosion by L.L.Shrier, Geroge Newness Ltd, London, 2 (1994).
Bulleting of Electrochemistry volume 19(5) May 2003.


















