Splice Site Mutations Are a Common Cause of X-Linked Chronic ...
7
Splice Site Mutations Are a Common Cause of X-Linked Chronic Granulomatous Disease By Martin de Boer, Ben G.J.M. Bolscher, Mary C. Dinauer, Stuart H. Orkin, C.I. Edvard Smith, Anders Ahlin, Ron S. Weening, and Dirk Roos Chronic granulomatous disease (CGD) is characterized by the absence of a respiratory burst in activated phagocytes. Defects in at least four different genes lead to CGD. Patients with the X-linked form of CGD have mutations in the gene for the psubunit of cytochrome bm (gp91-phox). We studied the molecular defect in four patients with X-linked CGD. In a fifth family, we studied the mother of a patient with X-linked CGD who had died before our investigations. GpSI-phox messenger RNA (mRNA) was reverse transcribed into cDNA and the coding region was amplified by polymerase chain reaction into three fragments. Sequence analysis showed the absence of the exon 7,5,3, and 2 sequences in patients 1,2,3, and 4, respectively. In carrier 5, we found both normal cDNA and cDNA that lacked 57 3‘-nucleotides of exon 6. We analyzed the splice sites of the flanking introns of the missing exons. In patients 1, 2, and 3, we found single nucleotide HE SUPEROXIDE-producing NADPH:02 oxi- T doreductase of phagocytic leukocytes is an important enzyme for the bactericidal activity of these Cyto- chrome b558 is a membrane-bound component of this enzyme.3 This cytochrome b558 is a heterodimer of a 91-Kd glycoprotein (gp91-phox) and a 22-Kd polypeptide (p22- ~hox).~,~ gp91-phox is encoded by a single gene on the X-chromosome6 and p22-phox by a gene on chromosome 16.7 Chronic granulomatous disease (CGD) is a heteroge- neous disease characterized by the absence of NADPH oxidase activity.8-10 As a result, phagocytes of these patients fail to generate superoxide, and consequently, CGD pa- tients suffer from recurrent life-threatening bacterial and fungal infections. In X-linked CGD, phagocytic cells are defective in gp91-pho4 11,12 and in autosomal cytochrome b558-negative CGD, p22-phox is defe~tive.~,’~ Other autoso- mal recessive forms of the disease are characterized by the absence of the cytosolic proteins p47-phox or p67-phox. 14,15 X-linked CGD patients in whom the molecular defect has been characterized had complete absence of gp91-phox messenger RNA (mRNA) due to large gene deletions,16-18 or had normal amounts of gp91-phox mRNA. In this latter group of patients, single point mutations were found, leading to incorporation of different amino acids19smor a premature stop codon.m Finally, one mRNA-positive pa- tient has been described with a partial mRNA deletion that predicts the synthesis of a truncated protein. This deletion was due to a single-base substitution that leads to abnormal splicing of mRNA.21 The patients reported here have short deletions in the gp91-phox mRNA, caused by point mutations that produce partial or complete exon skipping during mRNA splicing. MATERIALS AND METHODS CGD patients were classified as X-linked when complementation of nitroblue tetrazolium (NBT) reductase activity was found after fusion of their monocytes with monocytes from autosomal cytochrome bs=,s-positive CGD pa- tients,22 or when a mosaic pattern was found in an NBT slide test of Classification of CGDpatients. substitutions within the first five positions of the down- stream 5’ donor splice sites. In patient 4, a similar substitu- tion was found at position -1 of the 3’ acceptor splice site of intron 1. In carrier 5, no mutation was found in the exon 6-intron 6 boundary sequence. Instead, a single substitution was observed in exon 6 (C+A at nucleotide 633) that created a new donor splice site. Apparently, mRNA splicing occurs preferentially at this newly created splice site. We conclude that the absence of the exon sequences in the gp9l-phox mRNA of these patients is due to splicing errors. Of 30 European X-linked CGD patients studied by us so far, five appear to be caused by mutations that affect correct mRNA splicing. Thus, such mutations appear to be a common cause of X-linked CGD. 0 1992 by The American Society of Hematology. the neutrophils from the patients’ mothers. In the fifth family studied, a patient had died before the start of our investigation. His mother and his sister were identified as carriers of X-linked CGD. Cells from the mother were used for this study. Mononuclear leukocytes were purified from 20 to 100 mL of citrated blood by isopycnic centrifugation for 20 minutes at 1,OOOg at 20°C on an isotonic Percoll suspension (Pharmacia Fine Chemicals, Uppsala, Sweden) with a specific gravity of 1.076 g/mL. The cell layer on top of the Percoll suspension was collected and washed twice with phosphate- buffered saline (PBS). The number of monocytes was calculated from a size distribution pattern (Coulter Counter; Coulter, Hi- aleah, FL). RNA was isolated by dissolving the mononuclear cells Preparation of DNA and RNA. From the Central Laboratory of the Netherlands Red Cross Blood Transfusion Service and Laboratory for Experimental and Clinical Immunology of the University ofAmsterdam, Amsterdam, The Nether- lands; the Department of Pediatrics, H.B. Wells Center for Pediatric Research, Riley Children’s Hospital, Indiana University Medical School, Indianapolis; the Division of Hematology-Oncology, Chil- dren ’s Hospital, and Dana-Farber Cancer Institute, Department of Pediatrics, Harvard Medical School and Howard Hughes Medical Instiiute, Boston, MA; the Department of Clinical Immunology and Centerfor Biotechnology, Karolinska Institute at Huddinge Hospital1 NOVUM, Huddinge, Sweden; the Department of Pediatrics, Sachs’ Children’s Hospital, Stockholm, Sweden; and Emma Children’s Hospital,Academic Medical Center, University ofAmsterdam Amster- dam, The Netherlands. Submitted September 3,1991; accepted May 19,1992. Supported by Grant No. 900-503-110 from the Netherlands Organi- zation for Scientijic Research (NWO), and by the Swedish Medical Research Council. Address reprint requests to Dirk Roos, PhD, cIo Publication Secretariat, Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, PO Box 9190, 1006 A D Amsterdam, The Netherlands. The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with I8 U.S.C. section 1734 solely to indicate this fact. 0 1992 by The American Society of Hematology. 0006-4971l92/8OO6-0OO9$3.00l0 Blood, Vol80, No6 (September 15). 1992: pp 1553-1558 1553 For personal use only. on March 17, 2018. by guest www.bloodjournal.org From
Transcript of Splice Site Mutations Are a Common Cause of X-Linked Chronic ...

Splice Site Mutations Are a Common Cause of X-Linked Chronic Granulomatous Disease
By Martin de Boer, Ben G.J.M. Bolscher, Mary C. Dinauer, Stuart H. Orkin, C.I. Edvard Smith, Anders Ahlin, Ron S. Weening, and Dirk Roos
Chronic granulomatous disease (CGD) is characterized by the absence of a respiratory burst in activated phagocytes. Defects in at least four different genes lead to CGD. Patients with the X-linked form of CGD have mutations in the gene for the psubunit of cytochrome b m (gp91-phox). We studied the molecular defect in four patients with X-linked CGD. In a fifth family, we studied the mother of a patient with X-linked CGD who had died before our investigations. GpSI-phox messenger RNA (mRNA) was reverse transcribed into cDNA and the coding region was amplified by polymerase chain reaction into three fragments. Sequence analysis showed the absence of the exon 7,5,3, and 2 sequences in patients 1,2,3, and 4, respectively. In carrier 5, we found both normal cDNA and cDNA that lacked 57 3‘-nucleotides of exon 6. We analyzed the splice sites of the flanking introns of the missing exons. In patients 1, 2, and 3, we found single nucleotide
HE SUPEROXIDE-producing NADPH:02 oxi- T doreductase of phagocytic leukocytes is an important enzyme for the bactericidal activity of these Cyto- chrome b558 is a membrane-bound component of this enzyme.3 This cytochrome b558 is a heterodimer of a 91-Kd glycoprotein (gp91-phox) and a 22-Kd polypeptide (p22- ~ h o x ) . ~ , ~ gp91-phox is encoded by a single gene on the X-chromosome6 and p22-phox by a gene on chromosome 16.7
Chronic granulomatous disease (CGD) is a heteroge- neous disease characterized by the absence of NADPH oxidase activity.8-10 As a result, phagocytes of these patients fail to generate superoxide, and consequently, CGD pa- tients suffer from recurrent life-threatening bacterial and fungal infections. In X-linked CGD, phagocytic cells are defective in gp91-pho4 11,12 and in autosomal cytochrome b558-negative CGD, p22-phox is defe~tive.~,’~ Other autoso- mal recessive forms of the disease are characterized by the absence of the cytosolic proteins p47-phox or p67-phox. 14,15
X-linked CGD patients in whom the molecular defect has been characterized had complete absence of gp91-phox messenger RNA (mRNA) due to large gene deletions,16-18 or had normal amounts of gp91-phox mRNA. In this latter group of patients, single point mutations were found, leading to incorporation of different amino acids19sm or a premature stop codon.m Finally, one mRNA-positive pa- tient has been described with a partial mRNA deletion that predicts the synthesis of a truncated protein. This deletion was due to a single-base substitution that leads to abnormal splicing of mRNA.21
The patients reported here have short deletions in the gp91-phox mRNA, caused by point mutations that produce partial or complete exon skipping during mRNA splicing.
MATERIALS AND METHODS CGD patients were classified as
X-linked when complementation of nitroblue tetrazolium (NBT) reductase activity was found after fusion of their monocytes with monocytes from autosomal cytochrome bs=,s-positive CGD pa- tients,22 or when a mosaic pattern was found in an NBT slide test of
Classification of CGDpatients.
substitutions within the first five positions of the down- stream 5’ donor splice sites. In patient 4, a similar substitu- tion was found at position -1 of the 3’ acceptor splice site of intron 1. In carrier 5, no mutation was found in the exon 6-intron 6 boundary sequence. Instead, a single substitution was observed in exon 6 (C+A at nucleotide 633) that created a new donor splice site. Apparently, mRNA splicing occurs preferentially at this newly created splice site. We conclude that the absence of the exon sequences in the gp9l-phox mRNA of these patients is due t o splicing errors. Of 30 European X-linked CGD patients studied by us so far, five appear t o be caused by mutations that affect correct mRNA splicing. Thus, such mutations appear to be a common cause of X-linked CGD. 0 1992 by The American Society of Hematology.
the neutrophils from the patients’ mothers. In the fifth family studied, a patient had died before the start of our investigation. His mother and his sister were identified as carriers of X-linked CGD. Cells from the mother were used for this study.
Mononuclear leukocytes were purified from 20 to 100 mL of citrated blood by isopycnic centrifugation for 20 minutes at 1,OOOg at 20°C on an isotonic Percoll suspension (Pharmacia Fine Chemicals, Uppsala, Sweden) with a specific gravity of 1.076 g/mL. The cell layer on top of the Percoll suspension was collected and washed twice with phosphate- buffered saline (PBS). The number of monocytes was calculated from a size distribution pattern (Coulter Counter; Coulter, Hi- aleah, FL). RNA was isolated by dissolving the mononuclear cells
Preparation of DNA and RNA.
From the Central Laboratory of the Netherlands Red Cross Blood Transfusion Service and Laboratory for Experimental and Clinical Immunology of the University ofAmsterdam, Amsterdam, The Nether- lands; the Department of Pediatrics, H.B. Wells Center for Pediatric Research, Riley Children’s Hospital, Indiana University Medical School, Indianapolis; the Division of Hematology-Oncology, Chil- dren ’s Hospital, and Dana-Farber Cancer Institute, Department of Pediatrics, Harvard Medical School and Howard Hughes Medical Instiiute, Boston, MA; the Department of Clinical Immunology and Center for Biotechnology, Karolinska Institute at Huddinge Hospital1 NOVUM, Huddinge, Sweden; the Department of Pediatrics, Sachs’ Children’s Hospital, Stockholm, Sweden; and Emma Children’s Hospital, Academic Medical Center, University ofAmsterdam Amster- dam, The Netherlands.
Submitted September 3,1991; accepted May 19,1992. Supported by Grant No. 900-503-1 10 from the Netherlands Organi-
zation for Scientijic Research (NWO), and by the Swedish Medical Research Council.
Address reprint requests to Dirk Roos, PhD, cIo Publication Secretariat, Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, PO Box 9190, 1006 A D Amsterdam, The Netherlands.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with I8 U.S.C. section 1734 solely to indicate this fact.
0 1992 by The American Society of Hematology. 0006-4971 l92/8OO6-0OO9$3.00l0
Blood, Vol80, No6 (September 15). 1992: pp 1553-1558 1553
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom

1554 de BOER ET AL
Table 1. Pdmers Used for DNA Ampli&.tion Primer Sequences
Family Exon on Flanking Introns
1 7 5'-tactggatcCTAlTACTAAnTGATCTGATCTGG-3' (sense)
CAGG-3' (antisense)
GAACTTG-3 (sense)
TAAGACCTCCGGATG-3' (antisense)
5'-tagtaagctTACATGllTClTAGACA-
2 5 5'-tactggatcCAGTAGCACTCTCT-
5'gaataagctTGTACCAAAAGAClTCAAAG-
3 3 5'-CCTCATGCTAAGAACClTGG-3' (sense) 5'-lTGATGGCCllTGAAAATTAGAG-
GAACTTAG-3' (antisense) 4 2 5'-tactggaTCCAGTCTTGTGTGGAATC-
TAC-3' (sense)
CATG-3' (antisense)
GAGTTGTCATCACGC-3' (sense)
(antisense)
5'gaataagctTGTGACCAGCCAATAlTG-
5 5'-tactggatCCTGlTGGCAGGCATCACTG-
5'-GG AC ATGAAATCClTCACTTCAG-3'
The lower case letters indicate the 8emHl and Hindlll restriction sites for sense and antisense primers, respectively.
in 4 mollL guanidine thiocyanate and centrifuged through 5.7 mollL cesium ch1oride.n Genomic DNA was isolated from circulat- ing blood leukocyte^.^^
Nonhem Mor unulysis. RNA corresponding to IO7 monocytes was electrophoresed in 1.2% (wtlvol) agarose gels in the presence of formaldehyde, and blotted onto GeneScreen Plus membrane filters (NEN Research Products, Boston, MA)." Blots were hybridized with the H64 fragment of gp91-phm cDNA6 or the EcoRIlHincll fragment of p22-phm c D N A . ~ Probes were labeled by random ~riming.2~ Hybridization with a probe for a-actin was used as a control for the quantification and integrity of the RNA.
Amp/ificurion of DNA. The coding region of gp91-phm mRNA was amplified by polymerase chain reaction (PCR) in three overlapping fragments as described previ~usly. '~.~ The mutations were identified in genomic DNA as follows. Exons plus intron boundary sequences were amplified with PCR using primers corresponding to sequences on the flanking introns as indicated in Table 1. Conditions of the PCR were as described.x' The PCR product was purified by the Geneclean I1 kit (B10 101 Inc, La Jolla, CA) to remove the primers and nucleotides. Two hundred nanograms of the purified DNA samples was annealed with 40 ng of one of the primers used for amplification by heating for 3 minutes at IWC, followed by chilling on ice water in the presence of 10% dimethylsulfoxide (DMSO). Direct sequence analysis was performed with the dideoxynucleotide chain termination method using the Sequenase Version 2.0 kit (US Biochemical Corp, Cleveland, OH).
RESULTS
Heme absorbance spectroscopy and Western blot analy- sis of the cytochrome bSm subunits gp91-phox and p22-phar with monoclonal antibodies= showed complete absence of cytochrome h5s8 in the neutrophils of the four X-linked CGD patients studied. In addition, there was n o residual NADPH oxidase activity detectable after stimulation of patients'neutrophils with phorbol myristate acetate (PMA), serum-treated zymosan, or both. Analysis of NADPH oxidase activity in the cell-free s y ~ t e m ~ . ~ ' showed that the
defect was localized exclusively to the particulate fraction of the patients' neutrophils.
We analyzed the mRNA of the four patients' monocytes for the cytochrome b5ss subunits. Both gp91-phar mRNA and p22-phox mRNA of patients 1 through 4 were present on a Northern blot. The amount and mobility of these mRNAs were similar t o normal (not shown).
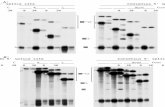
The gp9l-phox mRNAs of the four patients and carrier 5 were amplified in three overlapping fragments as de- scribed.19.2" Figure l shows the analysis of the PCR frag- ments on an agarose gel. The PCR fragment I is apparently smaller in patients 2, 3, and 4. In patient 1 and carrier 5, apparently smaller fragments 11 are observed. In addition, in carrier 5, a normal-sized fragment is present, in agree- ment with the carrier state. All five individuals gave normal-sized fragments 111 (not shown). The additional smaller sized fragment I in lane b was found in three separate RNA samples from patient 1, and was not due to intronexon mutations. Therefore, the origin of this smaller- sized fragment remains obscure.
Sequence analyses of all the PCR fragments were per- formed. As is shown in Fig 2, the sequences of exons 7 and 5 were absent in patients 1 and 2, respectively. In patient 3, the sequence of exon 5 was absent from the cDNA, and in patient 4, the sequence of exon 2 was absent from the cDNA. In carrier 5, two coinciding sequence ladders are observed for fragment I1 (Fig 2.5): panel one is identical t o
Fig 1. Sise ana)vrh of the PCR-amplified fragments of gp91ghox cDNA Ten percent of the PCR amplified DNA products were run on a NuSiwe 2 1 agarow gel and stained with 0.05% (wtIvoI) ethidium bromide. Lane g. molecular weight markers (1.500 to 200 bp, with 100 bp difference); lanes a through 1, PCR fragments I (6 to 633); lanes h through m, PCR fragments II (530 to 1,210). Lanes a and h are of a control and lanes b through f and i through m of patients 1 through 5, respectively.
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom

SPLICE SITE MUTATIONS IN X-LINKED CGD 1555
FIg 2 Sequence analyses of amplified gp9l.phox cDNA. Sequence Iadden of tha mutated regions of tha four patients and tha mother of patient 5 am as indicated. The control sequences am shown t o the left. The horizontal lines point to the splice junctions whew exons 7,5,3, and 2 are skipped In patients 1,2,3, and 4, mspecthrely. In carrier 5, only part of exon 6 Is skipped. Splice sites between exons 6 and 7 fall within codon 225, and between exons 7 and 8 between codons 268 and 269. Splice sites between exons 4 and 5 fall within codon 113, and between exons 5 and 6 between codons 161 and 162. Between exons 2 and 3 the splice site falls between codons 47 and 48. and between exons 3 and 4 between codons 84 and 85. Splice sites between exons 1 and 2 fa11 between codons 15 and 16, and between exons 2 and 3 between codons 47 and 48. The newly created splice site on exon 6 (carrier 5) lies within codon 206. Codon numbering is according to Orkin.= T h e (-1 strand has been sequenced.
the control sequence, while in the other sequence the 3' end of exon 6 (nucleotide 630 through 686) is missing. These results are consistent with the apparently smaller PCR fragments shown in Fig 1.
To investigate whether the absence of the exon se- quences in the patients was due to splicing errors, we amplified the flanking intron sequences from genomic DNA by PCR (see Table 1 for primer sequences). In all five individuals, PCR products of the expected sizes were obtained, indicating that the absence of the exons in the gp91-phox mRNAs was not due to a gene deletion of these
sequences. When the donor-splice regions of the missing exons were sequenced, single-base substitutions were ob- served in the consensus donor splice sequences of introns 7 (t + a at position +2 in patient 1). 5 (a + t at position +3 in patient 2), and 3 (g + a at position +5 in patient 3) (Fig 3). In patient 4. a single-base substitution was observed in the consensus acceptor splice sequence of intron 1 (g + a at position - 1). No other mutations were identified. There- fore, we conclude that the absence of exons 7,5,3, and 2 in the gp91-phar mRNA of patients 1,2,3, and 4, respectively, is due to mRNA splicing defects resulting in exon skipping.
FIg 3. !Sequence analysis of PCR-amplified g e m " DNAcontainlng tha mutated shea. (1) Sequence of exon 7-Intron 7 boundary; (2) seqmnm of exon blntron 5 boundary; (3) sequence of exon 3-intron 3 boundary; (4) sequence of exon 2-Intron 1 boundary; (5) sequence of the newty created splice site on exon 6. The sequence of each patient is as indicated in the figure, with the control sequence at the left. Arrow point to the mutations. 'The (-1 strand has been sequenced.
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom

1556 de BOER ET AL
Genomic PATIENT I
Exon 6 Intron 6 Exon 7 Intron 7 Exon 8
... GGA GCT GAIgtgag ... CcaglA CGA AAT...CCT ATGIgtatg ... acagrACT TGG A... control
patient 1
cDNA
... GGA GCT GAIgtgag ... ccagl~ CGA AAT...CCT ATGIgaatg ... acagjACT TGG A... t
223 . . .267 Gly Ala Glu Arg Ile Pro Met Thr Trp
control ... GGA GCT G M CGA ATT...CCT ATG ACT TGG A... A A
A Gly Ala Glu Leu Gly
,..GOA OCT O M CTT GGA.. . patient 1
G m k PATIENT 2
Exon 4 Intron 4 Exon 5 Intron 5 Exon 6
... CAC TCT Glgtaag ... tcaglffi ATT...ATA AAGIgtaag . . .g caglMC CCT GA...
... CAC TCT Glgtaag ... tcaglffi ATT...ATA AAGlgttag ... gcag AAC CCT GA... control
patient 2
cDNA t
111 . . .160 His Ser Ala Ile Ile Lys Asn Pro
control . . .CAC TCT Gffi ATT.. .ATA AAG AAC CCT GA.. A A
H i 0 Ser Glu Pro Stp ... CAC TCT GAA CCC TGA... patient 2
Genomic PATIENT 3
A
Exon 2 Intron 2 Exon 3 Intron 3 Exon 4
. . .CTT CTT GGG Igtaag.. ..tag[ TCA GCA.. .ACT GCG Igtaag. . .aaag
. ..CTT CTT GGGIgtaag.. .ctaglTCA GCA.. .AGT GCGIgtaaa.. . a a a g l m
control
patient 3
cDNA t
45.. . . . .s3 Leu Leu Gly Ser Ala Ser Ala Cy8 Cys
control ... CTT CTT GGG TCA GCA... AGT GCG TGC TGC... A - Leu Leu Gly Cye Cye
patient 3 ... CTT CTT GGG TGC TGC... A Genomic PATIENT 4
Exon 1 Intron 1 Exon 2 Intron 2 Exon 3
...OTCATT( gtaag. . . gcagl CTG GTT.. .CTT w l g t a a g . . . ctag ~TCA OCA CTG. . . -1 gtaag .. .g caalcx GTT...CTT GGGIgtaag ... ctag(~CA OCA CTG...
control
patient 4
CDNA t
13 ... 46 Phe Val Ile Leu Val...Leu Gly Ser Ala Leu
A A Phe Val Ile set Ala Leu
A
control ... TTT GTC ATT CTG GTT...CTT GGG TCA OCA CTG...
patient 4 ...TlY GTC ATT TCA GCA CTG...
G d PATIENT 5
Intron 5 Exon 6 Intron 6 Exon 7
control
patient 5
cDNA
...g caglMC CCT...TpT TGG TAC ACA...GGA GCT GAlgtgag ... CCaglA CGA ATT...
... g c a g l ~ ~ ~ CCT...TTT TGG TAA ACA...GGA GCT GAIgtgag ... ccag(A CGA ATT... t
162 ... 205 . . .223 Asn Pro Phe Trp Tyr Thr
control ... AAC CCT...TTT TGG TAC ACA...GGA GCT GAR CGA ATT... Gly Ala Glu Arg Ile
A A m Pro Phe Stp
A patient 5 ... AAC CCT...TTT TGA CGA ATT...
Fig 4. Mutations in the gp91-phox gene caus- ing X910 CGD. Short stretches are shown of the nucleotide sequences and predicted amino acid sequences that contain the mutations. Upper case letters indicate coding sequences, and lower case letters indicate intron sequences. Exon se- quences in genomic DNA are shown within boxes. Arrowheads in the cDNA sequences show the splice sites. Arrows indicate the mutations in the patients. The amino acid numbering on top of the sequences is according to 0rkin.a In contrast to the published gene structure,a we found a CCAG sequence at the acceptor site of intron 6.
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom

SPLICE SITE MUTATIONS IN X-LINKED CGD 1557
Figure 4 summarizes the results of the mutation analyses of these four patients.
In the female carrier of family 5, no mutations in the exon 5-intron 5 boundary sequence were observed. Instead, a single-base substitution was found within exon 6 (C +. A at position 633) in addition to the normal sequence, consistent with the X-linked carrier state (Fig 3). No other mutations were identified. Because part of the mRNA of this individ- ual lacked the 3' part of exon 6, starting at nucleotide 631, it appears that this mutation has created a new donor splice site at nucleotide 630 to 635 (GTAAA). This is shown in Fig 4.
To confirm the mutations found, fragments containing the mutated sites were PCR amplified from genomic DNA isolated from all five individuals, some available family members, and 72 control alleles. For patients 1 to 4, these PCR products were hybridized with allele-specific oligonu- cleotides (data not shown). Only the PCR fragments amplified from the DNA of the four patients hybridized to the oligonucleotides containing the corresponding mutant sequence. Because the mutation in family 5 predicts the disappearance of a recognition site for the restriction enzyme Rsa I, the PCR fragments (205 bp) amplified from the genomic DNA derived from the mother and sister of patient 5 and 36 unrelated female donors were digested with this enzyme. The PCR products of the 72 control alleles were completely digested by Rsa I into two frag- ments of 112 and 93 bp, respectively, whereas only half of the PCR product of the alleles of the mother and sister of patient 5 were digested (not shown).
DISCUSSION
In the inherited disease CGD, phagocytes fail to produce superoxide upon stimulation due to a defect in the NAD- PH:02 oxidoredu~tase.~.~ CGD is a very heterogeneous disorder, and defects in at least four different gene products can cause CGD.15 The most common form of CGD is the X-linked form, in which gp91-phox (the @-subunit of cyto- chrome b558) is affected."J2
The molecular defect has been characterized in a number of X-linked CGD patients. Interestingly, all patients were found to have a different genetic defect, ranging from gene deletions6J6-18 to point mutations in the coding r e g i o n ~ . l ~ , ~ ~ In the four patients we describe in this report, deleted gp91-phox mRNAs were found. Sequence analysis of the
gp91-phox mRNA showed the absence of exon 7,5,3, and 2 sequences, respectively. Analyses of the patients' genomic DNA showed that these exons were not deleted in the gene. Point mutations in the consensus donor splice regions were identified in patients 1,2, and 3, which must cause aberrant mRNA splicing with exon skipping. Because the abnormal splicing alters the reading frame in patients 1 and 2, these mutations predict the occurrence of frameshifts and prema- ture termination of the synthesized protein in these pa- tients at positions 230 and 133, respectively. The absence of exon 3 in the mRNA of patient 3 leads to an in-frame deletion and therefore to a shortened polypeptide. In patient 4, a point mutation was found at position -1 of the consensus acceptor splice region at the intron 1-exon 2 boundary, which apparently causes aberrant mRNA splic- ing with skipping of exon 2. This also leads to an in-frame deletion and therefore to a shortened polypeptide.
In the mother of patient 5, a point mutation was found in the coding region of exon 6. Apparently, this mutation has created a new donor splice site that is preferred over the original, nonmutated donor splice site of intron 6. The abnormal mRNA splicing results in the inframe deletion of the 3' 57 nucleotides of exon 6, and creates a premature stop codon at amino acid position 206. An alternative consequence of the C-633 +. A substitution found in family 5 is the creation of a TAA stop codon at amino acid position 207 in a correctly spliced product, which predicts premature termination of protein synthesis. However, this transcript was not detected by DNA sequencing of cDNAs (Fig 2), which showed a truncated exon 6 sequence in apparently the same amount as the wild-type sequence (Fig 2). We presume, therefore, that patient 5 suffered from CGD due to alternative splicing of exon 6 in the gp91-phox mRNA.
Mutations in or around splice junctions are a well- recognized cause of inherited human disease, with P-thalas- semia being the first and perhaps best studied example.30 Mutations around the 5' splice site can lead to either alternative splicing with use of cryptic splice s i t e ~ ~ l . 3 ~ or exon skipping, as is reported here. Other reports of exon skipping due to donor splice site mutations include Ehlers- Danlos syndrome type IV32,33 and a~ata lasemia .~~ Of 30 European X-linked CGD patients studied by us so far, 5 appear to be caused by mutations that affect correct mRNA splicing. Thus, such mutations are a common cause of X-linked CGD.
REFERENCES 1. Sbarra AJ, Kamovsky ML The biochemical basis of phagocy-
tosis. 1. Metabolic changes during the ingestion of particles by polymorphonuclear leucocytes. J Bioi Chem 234:1355,1959
2. Babior BM: Oxygen-dependent microbial killing by phago- cytes. N Engl J Med 298:659,1978
3. Segal AW, Jones OTG, Webster D, Allison A C Absence of a newly described cytochrome b from neutrophils of patients with chronic granulomatous disease. Lancet 2:446,1978
4. Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ: Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest 80732,1987
5. Segal A W Absence of both cytochrome b-245 subunits from neutrophils in X-linked chronic granulomatous disease. Nature 326:88,1987
6. Royer-Pokora B, Kunkel LM, Monaco AP, Goff SC, New- burger PE, Baehner RL, Cole FS, Cumutte JT, Orkin SH: Cloning of the gene for an inherited human disorder-chronic granuloma- tous d i s e a s e 4 n the basis of its chromosomal location. Nature 322:32,1986
7. Dinauer MC, Pierce EP, Bruns GAP, Curnutte JT, Orkin S H Human neutrophil cytochrome b light chain (p22-phox). J Clin Invest 86:1729,1990
8. Tauber AI, Borregaard N, Simons E, Wright J: Chronic
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom

1558 de BOER ET AL
granulomatous disease: A syndrome of phagocyte oxidase deficien- cies. Medicine (Baltimore) 62286,1983
9. Baehner RL, Nathan DG: Leukocyte oxidase: Defective activity in chronic granulomatous disease. Science 155835,1967
10. Segal A W The molecular and cellular pathology of chronic granulomatous disease. Eur J Clin Invest 18:433,1988
11. Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos C A The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature 327717,1987
12. Teahan C, Rowe P, Parker P, Totty N, Segal AW: The X-linked chronic granulomatous disease gene codes for the p-chain of cytochrome b-245. Nature 327720,1987
13. Weening RS, Corbeel L, de Boer M, Lutter R, Van Zwieten R, Hamers MN, Roos D: Cytochrome b deficiency in an autosomal form of chronic granulomatous disease. A third form of chronic granulomatous disease recognized by monocyte hybridization. J Clin Invest 75:915,1985
14. Clark RA, Malech HL, Gallin JI, Nunoi H, Volpp BD, Pearson DW, Nauseef WM, Cumutte J T Genetic variants of chronic granulomatous disease: Prevalence of two cytosolic compo- nents of the NADPH oxidase system. N Engl J Med 321547, 1989
15. Smith RM, Curnutte J T Molecular basis of chronic granulo- matous disease. Blood 77:673,1991
16. Francke U, Ochs HD, deMartinville B, Giacalone J, Lindren V, Disteche C, Pagon RA, Hofker MH, Van Ommen GJ, Pearson PL, Wedgewood RJ: Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular distrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syn- drome. Am J Hum Genet 37250,1985
17. Baehner RL, Kunkel LM, Monaco AP, Haines JL, Courne- ally PM, Palmer C, Heerema N, Orkin S H DNA linkage analysis of X-chromosome-linked chronic granulomatous disease. Proc Natl Acad Sci USA 83:3398,1986
18. Frey D, Machner M, Seger RA, Schmid W, Orkin S H Gene deletion in a patient with chronic granulomatous disease and McLeod syndrome: Fine mapping of the Xk gene locus. Blood 71952,1988
19. Dinauer MC, Curnutte JT, Rosen H, Orkin SH: A missense mutation in the neutrophil cytochrome b heavy chain in cytochrome- positive X-linked chronic granulomatous disease. J Clin Invest 842012,1989
20. Bolscher BGJM, De Boer M, De Klein A, Weening RS, Roos D: Point mutations in the p-subunit of cytochrome bsss leading to X-linked chronic granulomatous disease. Blood 77:2482, 1991
21. Schapiro BL, Newburger PE, Klempner MS, Dinauer MC
Chronic granulomatous disease presenting in a 69-year-old man. N Engl J Med 325:1786,1991
22. Hamers MN, De Boer M, Meerhof U, Weening RS, Roos D: Complementation in monocyte hybrids revealing genetic heter- ogeneity in chronic granulomatous disease. Nature 307553,1984
23. Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ: Isolation of biological active ribonucleic acid from sources en- riched in ribonuclease. Biochemistry 185294,1979
24. Sambrook J, Fritsch E, Maniatis T Molecular Cloning: A Laboratory Manual (ed 2). Cold Spring Harbor, NY, Cold Spring Harbor, LaboratoIy, 1989
25. Verhoeven AJ, Bolscher BGJM, Meerhof U, Van Zwieten R, Keyer J, Weening RS, Roos D Characterization of two monoclonal antibodies against cytochrome bssg of human neutro- phils. Blood 73:1686,1989
26. Bromberg Y, Pick E Activation of NADPH-dependent superoxide production in a cell-free system by sodium dodecyl sulfate. J Biol Chem 26013539,1985
27. Bolscher BGJM, Van Zwieten R, Kramer IJM, Weening RS, Verhoeven AJ, Roos D: A phosphoprotein of M, 47,000, defective in autosomal chronic granulomatous disease, copuriiies with one of hvo cytosolic components required for NADPH02 oxidoreductase activity in human neutrophils. J Clin Invest 83:757,1989
28. Orkin S H Molecular genetics of chronic granulomatous disease. Ann Rev Immunol7277,1989
29. Skalnik DG, Strauss EC, Orkin SH: CCAAT displacement protein as a repressor of the myelomonocytic-specific gp91phar gene promotor. J Biol Chem 266:16736,1991
30. Orkin SH, Kazazian HH Jr: The mutation and polymor- phism of the human P-globin gene and its surrounding DNA. Ann Rev Genet 18131,1984
31. Treisman R, Proudfoot NJ, Shander M, Maniatis T A single-base change at a splice site in a Bo-thalassemic gene causes abnormal RNA splicing. Cell 2993,1982
32. Cole WG, Chiodo AA, Lamande SR, Janeczko R, Ramirez F, Dah1 HH, Chan D, Bateman J F A base substitution at a splice site in the COL3A1 gene causes exon skipping and generates abnormal type 111 procollagen in a patient with Ehlers-Danlos syndrome type IV. J Biol Chem 265:17070,1990
33. Lee B, Vitale E, Superti-Furga A, Steinmann B, Ramirez F G to T transversion at position +5 of a splice donor site causes skipping of the preceding exon in type 111 procollagen transcripts of a patient with Ehlers-Danlos syndrome type IV. J Biol Chem 288:5256,1991
34. Wen JK, Osumi T, Hashimoto T. Molecular analysis of human acatalasemia, identification of a splicing mutation. J Mol Biol211:383,1990
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom

1992 80: 1553-1558
M de Boer, BG Bolscher, MC Dinauer, SH Orkin, CI Smith, A Ahlin, RS Weening and D Roos granulomatous diseaseSplice site mutations are a common cause of X-linked chronic
http://www.bloodjournal.org/content/80/6/1553.full.htmlUpdated information and services can be found at:
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American
For personal use only.on March 17, 2018. by guest www.bloodjournal.orgFrom