
Republic of Iraq Ministry of Higher Education and ...scbaghdad.edu.iq/library/Biology/PhD/2013/Role...
125
Republic of Iraq Ministry of Higher Education and Scientific Research University of Baghdad College of Science Role of oprD Gene in Biofilm Formation and Imipenem Resistance in Pseudomoanas aeruginosa A Thesis Submitted to the College of Science/University of Baghdad in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biology/Microbiology By Hadeel Kareem Musafer B.Sc in Microbiology/ Al-Mustansiriya University, College of Science 2005 MSc. in Microbiology/ Al-Mustansiriya University, College of Science 2007 Supervised by Dr. Harith J. Fahad Al-Mathkhury Assistant professor November 2013 Muharrem 1434
Transcript of Republic of Iraq Ministry of Higher Education and ...scbaghdad.edu.iq/library/Biology/PhD/2013/Role...

Republic of Iraq Ministry of Higher Education and Scientific Research University of Baghdad College of Science
Role of oprD Gene in Biofilm Formation and Imipenem Resistance in Pseudomoanas aeruginosa
A Thesis
Submitted to the College of Science/University of Baghdad
in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Biology/Microbiology
By
Hadeel Kareem Musafer
B.Sc in Microbiology/ Al-Mustansiriya University, College of Science 2005
MSc. in Microbiology/ Al-Mustansiriya University, College of Science 2007
Supervised by
Dr. Harith J. Fahad Al-Mathkhury
Assistant professor
November 2013 Muharrem 1434

Committee Certification
We certify that we have read this thesis entitled “Role of oprD Gene in Biofilm Formation and Imipenem Resistance in Pseudomoanas aeruginosa” and as the examination committee examined the student in its content and in our opinion it adequate for award of the doctorate of philosophy in Biology/Microbiology.
Signature Signature Name: Dr. Abdel Kareem Al- Qazaz Name: Dr. Mouruj Abd Alsatar
Scientific Degree: Assistant professor Scientific Degree: Assistant professor Member Member
Date: / / 2013 Date: / / 2013
Signature Signature Name: Dr. Haifa Hadi Hassani Name: Dr. Hanaa Saleem Yossef
Scientific Degree: Professor Scientific Degree: Assistant professor Member Member
Date: / / 2013 Date: / / 2013
Signature Signature Name: Dr. Harith J. F. Al-Mathkhury Name: Dr. Rajwa Hasan Essa
Scientific Degree: Professor Scientific Degree: Professor Member/advisor Chairman
Date: / / 2013 Date: / / 2013 Approved by the deanery of the college of science: Signature: Name: Dr. Saleh Mahdi Ali Scientific Degree: Professor Address: Dean of the College of Science Date: / /2013

Linguistic Certification
I certify that this thesis entitled “Biofilm Formation by Psudomonas aeruginosa in Respect of Imipenem Resistance” was prepared by Hadeel Kareem Musafer, under my linguistic supervision. It was amended to meet the style of the English Language.
Dr. Ayaid Khadem Zgair
Biology Department/ College of Science/ University of Baghdad
/ / 2013
In view of the available recommendations, I forward this thesis for Debate by the examination committee

حمي ن الر� مح �سم هللا الر�
�ن آ�وتوا العمل در�ات (( �ن آمنوا منمك وا�� ا�� �رفع ا�� بما تعملون خبري ))وا��
صدق هللا العظمي
ا�اد� سورة) من ١١آية (

Acknowledgment
I praise and thank Allah for clarifying my way so I could have the aptitude to
accomplish this modest effort.
I gratefully acknowledge dean of college of science and head of department of
Biology/University of Baghdad, to permit me to complete my Ph. D. study.
I would like to express appreciation and deepest thanks to the Iraqi Ministry of
Higher education and scientific research for the financial support to complete my project.
I am grateful to my supervisor Dr. Harith Jabar Fahad Al-Mathkhury for his
guidance, patience and scientific directions.
My deep appreciation to Dr. George A. O’tool, Geisel School of Medicine,
Dartmouth College / USA, for his patience, scientific directions and partially funding my
thesis.
I would like to express my special grateful thanks to Dr. Sherry L. Kuchma,
microbiology and immunology department, Geisel School of Medicine, Dartmouth
College / USA for her kind and scientific assistance during six months.
Special thanks for all members of Microbiology and Immunology department\
Geisel School of Medicine, Dartmouth College/ USA for their assistant.
Also I would like to thank my colleagues in University of Baghdad and Al-
Mustansyria University for their kind help.

Summary Fifty eight Pseudomonas aeruginosa strains evaluated with E test; forty
seven (81.03%), two (3.4٥%), and nine (15.5۲%) strains were susceptible,
intermediate, and resistant to imipenem, respectively. All the resistant and
intermediate susceptible strains, previously isolated from patients with cystic
fibrosis, showed no carbapenemase activity as confirmed by the Hodge test.
The relationship between biofilm formation, imipenem resistance, and
oprD expression were assessed in some imipenem resistant clinical P.
aeruginosa strains (SMC631C, SMC631F, SMC631J, SMC631H,
SMC631K and SMC 4974). All resistant strains showed low oprD
expression and low biofilm values in comparison to the sensitive ones.
The results in this study was revealed that oprD::isphoA/hah resistant
strain developed significantly lower biofilm formation capacity than PAO1
wild type. Moreover, this mutation triggered Imipenem resistance (P < 0.001
by comparison to PAO1 strain).
During this work, an strain with point mutation (SMC631F-ImR) in
oprD gene was obtained. This mutant was resistant to imipenem (MIC> 32
mg/l) and weak biofilm former (OD550= 0.06) compared with the parent
SMC631 biofilm (OD550=0.19).
Expression of poprD::isphoA/hah plasmid in the oprD mutant fully
restores the biofilm defect observed for the oprD mutant. There were no
significant differences (P>0.05) between PAO1 and complemented strain;
nevertheless, significant differences (P<0.05) were noticed between the
complemented strain and the mutant oprD::isphoA/hah with vector.

Furthermore, poprD::isphoA/hah plasmid expression restored
imipenem sensitivity (4.3) to the level of wild type and there were no
significant difference (P>0.05) between them. However, significant
differences were found between the complemented strain and the mutant
oprD::isphoA/hah with empty vector. Similar results were obtained with the
clinical strain SMC631. An SMC631F-ImR/poprD plasmid introduced into
the oprD mutant. Again, expression of SMC631F-ImR / poprD+ in the oprD
mutant fully restores the biofilm defect observed for the oprD mutant
relative to the empty vector as control.
Taking together, these results confirm that the inactivation of oprD is
responsible for the observed defect of biofilm phenotype.
mariner transposon mutagenesis was performed in two clinical strains
comprised the highest biofilm former. Additionally, biofilm deficient strains
were screened by microtiter plate assay, in the mutant derivatives (originated
from SMC576 and SMC214). We mapped four different genes, pilY1, pilW,
pslI and algC. The results revealed that pilY::Mar mutants and pilW::Mar19
mutant showed significantly deficient biofilm formation in comparison to
the parent SMC576, pilY1::Mar represented five independent mutant strains
(five insertion sites in pilY1 gene) and one strain with insertion in pilW gene.
Moreover, mutation of pilY1 and pilW in the SMC576 background lead to
loss of twitching motility.
The swarm phenotypes of SMC576 mutants were shown. The
pilY1::Mar mutants and pilW::Mar19 mutant exhibit a pattern of swarming
motility in comparison with SMC576 (weak swarm, shorter and fewer
tendrils).
The second highest biofilm is formed by SMC214; were mapped two
different genes pilW, pilX I that responsible for biofilm deficient. Those

biofilms formed by the pilX::Mar and pilW::Mar mutants were significantly
lower (P < 0.001) than the biofilm of the SMC214 parent strain.
The pilW::Mar and pilX::Mar mutants exhibit swarming phenotypes
that closely resembled the SMC214 parent strain; while the pilW::Mar and
pilX::Mar showed strong suppressor of twitching motility.
We introduced constructed plasmid, +ppilY1 plasmid into the
pilY1::Mir8 mutant, pilY1:Mir8 expression restored the higher biofilm
formation to the levels comparable to the SMC576. Furthermore, the
expression of +ppilY1 in the pilY1::Mir8 mutant fully restored the swarming
defect observed for the pilY1::Mir8 mutant relative to the vector control.
Regarding twitching motility, the expression of PilY1 was observed to
complement the twitching defect of the pilY1::Mir8 mutant. Collectively,
these results confirmed the inactivation of pilY1 alone which responsible for
the observed suppression of SMC576 mutant phenotypes.
The map of mutants pslI::Mar and algC::Mar showed the differences
in Psl that produced by pslI::Mar and algC::Mar mutants which were highly
deficient biofilm.

List of contents
Page Subject I Summary V List of Contents 1 1. Introduction and literature review 1 1.1. Introduction 3 1.2. Literature review 3 1.2.1. Pseudomonas aeruginosa: general characteristics 4 1.2.2. Pseudomonas aeruginosa and cystic fibrosis 5 1.2.3 Antibiotic resistance in P. aeruginosa 7 1.2.4 Carbapenem resistance in P. aeruginosa 7 1.2.4.1 Carbapenems 8 1.2.4.2 Carbapenemase
10 1.2.4.3 Porin oprD
10 1.2.4.4 Molecular Mechanisms of OprD-Mediated Resistance
11 1.2.5. Biofilm 13 1.2.5.1. Stages of biofilm development
15 1.2.5.2. Roles of extracellular polymeric substances in P. aeruginosa biofilms
19 1.2.5.3. Extracellular DNA 20 1.2.6. Pseudomonas aeruginosa motility on surface 20 1.2.6.1 Swarming motility 22 1.2.6.2 Twitching motility 28 2. Materials and Methods 28 2.1. Materials 28 2.1.1. Apparatuses and Equipment 29 2.1.2. Chemicals and biological materials 31 2.1.3. Culture media 31 2.1.4. Kits

32 2.1.5. Standard strains quality control bacteria 33 2.1.6. Primers
33 2.1.6.1. Primers used in oprD genetic complementation and sequencing
33 2.1.6.2. Quantification real time PCR primers 34 2.1.6.3. Primer used in Arbitrary PCR
34 2.1.6.4. Primers used in pilY1 genetic complementation and sequencing
34 2.1.7. Plasmids and vectors used in this study 35 2.1.8. Buffers and Solutions 35 2.1.8.1. Magnesium sulfate MgSO4 1 M 35 2.1.8.2. Glucose20% 35 2.1.8.3. Lazy Bones Solution 35 2.1.8.4. Sodium Borate (SB) buffer 20X
35 2.1. 8.5. Ethylendiaminetetraacetic Acid (EDTA), 0.5 and 0.05 M
35 2.1. 8.6. Tris buffer (1M) pH 8 35 2.1. 8.7. EDTA (TE) buffer 36 2.1. 8.8. loadeing buffer 6X dye 36 2.1. 8.9. Ethanol (70%) 36 2.1. 8.10. Glycial acetic acid 30% 36 2.1.9.11. Casamino acids 20% (CAA)
2.2. Methods 36 2.2.1. Sterilization 36 2.2.2. Laboratory prepared culture media 36 2.2.2.1. Yeast peptone dextrose (YPD) broth media 37 2.2.2.2. Yeast peptone dextrose (YPD) agar plate 37 2.2.2.3. Lysogeny broth (LB) broth 37 2.2.2.4. Lysogeny broth (LB) agar plates 37 2.2.2.5. Minimal salts medium 5X M63 37 2.2.2.6. Minimal salts medium 5X M8 37 2.2.2.7. Minus uracil medium 38 2.2.2.8. Glycerol for -80ºC 38 2.2.2.9. Stabs 38 2.2.3. McFarland Standard (no. 0.5) Preparation

39 2.2.4. Single Stranded Carrier DNA 39 2.2.5. Preservation of bacterial strains 39 2.2.6. DNA agarose gel electrophoresis 40 2.2.7. Imipenem stock preparation 41 2.2.8. Antibiotic stocks used in this study 41 2.2.9. Biofilm Media 41 2.2.10. Swarming motility media 41 2.2.11. Twitching motility media 41 2.2.12. Standard and clinical strains culture 42 2.2.13. Determination of MIC of Imipenem for strains 42 2.2.13. 1. The E-test method 42 2.2.13. 2. Microdilution method 43 2.2.14. Modified Hodge Test (MHT) 44 2.2.15. Biofilm formation assay 45 2.2.16. Extraction of Genomic DNA 45 2.2.17. Amplification of oprD gene by polymerase chain reaction 46 2.2.18. Sequencing 47 2.2.19. Quantitative reverse transcription-PCR (qRT-PCR) (Kuchma 47 2.2.19.1. Bacterial Harvest 47 2.2.19.2. RNA extraction and cDNA 48 2.2.20. Genetic complementation steps of oprD mutant strain
48 2.2.20.1. Digestion of PMQ72 2.2.20. 2. Yeast transformation
49 2.2.20. 3. Electroporation
50 2.2.21. mariner transposon mutagenesis of the Highest biofilm producers; 576 and 214
50 2.2.21.1. Conjugation and selection for mutants 51 2.2.21.2. Storing/screening the library
2.2.21.3. Screening for Biofilm deficient 2.2.21.4. To store the library
52 2.2.21.5. Mapping Mariner transposons by Arbitrary PCR (ARB PCR)
55 2.2.22. Construction of mutant strains and plasmids 55 2.2.23. Twitching assays 56 2.2.24. Swarming motility

56 2.2.25. Estimation of polysaccharide extracts 56 ۲.3. Statistical analysis 58 3. Results and Discussion 58 3.1. Imipenem susceptibility and carbapenemase detection
59 3.2. Assessing biofilm formation and imipenem resistance in clinical strains
61 3.3. Analysis of Sequencing of oprD gene 65 3.4. Analysis of oprD expression 66 3.5. OprD participates in biofilm formation.
70 3.6. Genes required for biofilm formation in clinical strains are conserved
78 3.7. Biofilm defective mutants of clinical strains SMC576 and SMC214 are sensitive to imipenem.
81 Conclusion 82 Recommendations 83 REFERENCES

chapter one
introduction and literature
review

2. Introduction and literature review 2.1. Introduction
Pseudomonas aeruginosa is an important opportunistic human pathogen
that can cause life-threatening infections, especially in patients with cystic
fibrosis (CF) and individuals with a compromised immune system. This
environmental bacterium is able to survive both in free-swimming planktonic
form and in surface-associated communities known as biofilms. P.
aeruginosa biofilms can be formed on both biotic and abiotic surfaces, thus
likely contributing to this microbe’s ability to cause disease in clinical settings
(Davies et al, 1998; O’Toole et al., 2000).
Although there are several antimicrobial agents that continue to be
effective against P. aeruginosa (i.e., carbapenem, cefepime, ceftazidime,
tobramycin and amikacin), in the last few years this bacterium’s increasing
resistance to antibiotics has been reported (Sanchez-Romero et al, 2007;
Ruiz-Martinez et al, 2011). The emergence and spread of acquired
carbapenem resistance in this species have challenged the success of
therapeutic and control efforts. Therefore, investigation of the molecular
mechanisms leading to resistance is crucial (Riera et al, 2011).
The OprD porin of P. aeruginosa facilitates the uptake across the outer
membrane of basic amino acids, small peptides that contain these amino
acids, and their structural analogue, the antibiotic imipenem. Indeed,
prolonged treatment of patients with P. aeruginosa infections with this
antibiotic leads to imipenem resistant mutants that either lack OprD or
Metallo-β-lactamase (Wolter et al., 2009).

A biofilm is a structured consortium of bacteria embedded in a self-
produced polymer matrix. Bacterial biofilms cause chronic infections
because they show increased tolerance to antibiotics (Høiby et al., 2010).
The goal of the present study was to investigating the link between
imipenem resistance, due to oprD dysfunction, and biofilm formation in
laboratory and clinical isolates of P. aeruginosa, the steps of the study are
listed below:
1. Detecting the Imipenem resistance isolates in P. aeruginosa
associated with cystic fibrosis by E test strip.
2. Investigating the mechanism of imipenem resistance whether it is
related to carbapenemase by Hodge test, or to mutation in oprD gene,
by sequence analysis of oprD gene.
3. Quanatification of Biofilm formation in wild type PAO1 and clinical
isolates.
4. Assessing the expression of oprD gene by quantification reverse
transcription time PCR (qRT-PCR) and compared the expression with
the biofilm values.
5. Genetic complementation of oprD into imipenem resistance isolate
restores imipenem sensitivity and biofilm formation.
6. Explaining why the clinical isolate forms high biofilm by using
transposon mutagenesis with mariner transpose and genetic screening
for the genes responsible for high biofilm formation in clinical isolates
by Arbitrary PCR and sequence analysis for these genes.
7. Perceiving the relationship between biofilm and Twitching test,
polysaccharide synthesis, and swarming motility.

8. Genetic complementation of mutant gene pilY1responsible for high
biofilm.
9. Estimation of polysaccharide production by ELISA test.

1.2 Literature review
1.2.1 Pseudomonas aeruginosa: general characteristics
P. aeruginosa is a gram negative, uniformly stained, straight or slightly
curved rods, measuring 0.5 to 1.0 μm by 1.5 to 5.0 μm in length. They are
aerobic, non-spore forming, motile by one or more polar flagella. They are
either incapable of utilizing carbohydrates as source of energy or degrade
them “oxidatively” rather than fermentative pathway (de Freitas and Luis
Barth, 2002).
P. aeruginosa is a member of the Gamma Proteobacteria class of
bacteria, belonging to the bacterial family Pseudomonadaceae. Since the
revision taxonomy based on conserved macromolecules (e.g. 16S ribosomal
RNA) the family includes only members of the genus Pseudomonas which
are cleaved into eight groups. P. aeruginosa is the type species of its group
(Hall et al., 2004).
Biochemically, they are oxidase and catalase positive, motile, grows
well at 42˚C, P. aeruginosa, utilizes glucose oxidatively, reduces nitrate to
nitrite, Methyl red/Voges Proskauer test is negative, do not decarboxylate
lysine and ornithine, but dihydrolyze agrinine, mannitol not fermented,
produce alkaline slant/alkaline butt with no gas and no H2S in TSI, indole
negative, utilize citrate, do not hydrolyze urea, do not produce phenyl
pyruvic acid, liquefies gelatin, do not hydrolyze aesculin and utilize
acetamide (Trautmann et al., 2008).
The typical Pseudomonas bacterium in nature might be found in a
biofilm, attached to some surface or substrate, or in a planktonic form, as a

unicellular organism, actively swimming by means of its
flagellum. Pseudomonas is one of the most vigorous, fast-swimming
bacteria seen in hay infusions and pond water samples. In its natural
habitat P. aeruginosa is not particularly distinctive as a pseudomonad, but it
does have a combination of physiological traits that are noteworthy and may
relate to its pathogenesis (Bodey et al., 1983).
On nutrient agar, colonies are large and pigmented
pyocyanin/fluorescence and on 5% sheep blood agar, they produce β-
hemolytic, large flat spreading, mucoid, rough, pigmented colonies with
characteristic metallic sheen (high carbon, low nitrogen content). Many
strains may produce a fruity, sweety, musty or grape like odor due to the
presence of 2-aminoacetophenone. On MacConkey agar, they produce non
lactose fermenting colonies with green pigmentation and metallic sheen.
Colonies from respiratory tract infection samples produce large amounts of
alginate, an exopolysaccharide consisting of mannuronic and guluronic acids
which aids in forming mucoid colonies.
Cetrimide agar is a selective and differential medium for the
identification of P. aeruginosa in which Cetrimide acts as detergent which
inhibits most bacteria and enhances the production of two pigments
pyocyanin, pyoverdine. However, about 4% of clinical strains of P.
aeruginosa do not produce pyocyanin (Mackie and MacCartney. 1996).
1.2.2 P. aeruginosa and cystic fibrosis
Cystic fibrosis (CF) is the most common inherited lethal genetic
disorder, Individuals suffering from CF harbor mutations in a gene on the
long arm of chromosome 7 (Riordan et al., 1989). The gene product is the
cystic fibrosis transmembrane conductance regulator (CFTR) which

regulates and facilitates transport of electrolytes across epithelial cell and
other membranes. The mucus in the CF airways is highly viscid, sulphated
and readily forms aggregates (Chace et al., 1985; Welsh and Smith, 1993).
In the CF lung, the viscid mucous cannot be propelled so easily and the
escalator fails, leading to an accumulation of mucus and trapped bacteria
(Bals et al., 1999).
P. aeruginosa is a major pathogen in the CF lung. Chronic
colonization with P. aeruginosa is associated with a more rapid decline in
lung function, especially if the isolate becomes mucoid (Emerson et al.,
2002). Although most patients are initially infected with nonmucoid P.
aeruginosa, it later transitions to a mucoid state (Li et al., 2004). Mucoidy
results from an overproduction of alginate which is thought to play a
protective role in the relatively harsh environment of the CF lung, perhaps
by enhancing the formation of biofilms, The link between CF-derived
mucoid P. aeruginosa isolates and their biofilm lifestyle, has led to the
assumption that alginate is the key secreted polysaccharide in biofilms of
both mucoid and nonmucoid strains (Hentzer et al., 2001).
The studies examining the role of alginate in the initiation or
maturation of biofilms often involved the comparison of mucoid strains
isolated from the CF lung with nonisogenic, nonmucoid strains; these
studies generally compared the ability of these strains to form biofilms and
the antibiotic resistance of established biofilms (Mai et al., 1993).
1.2.3 Antibiotic resistance in P. aeruginosa
P. aeruginosa has become an important and frequent opportunistic
nosocomial pathogen. This organism is characterized by an innate resistance
to multiple classes of antimicrobials, causing difficult-to-treat infections,

which are therefore associated with significant morbidity and mortality
(Obritsch et al., 2004).
Infections by P. aeruginosa are a serious clinical problem, particularly
in immune compromised hosts in hospital settings (Fujitani et al, 2011).
Moreover, the treatment of these infections is often difficult because of the
limited number of effective antimicrobial agents, due to the intrinsic
resistance of P. aeruginosa strains and their different modes of growth
(Khan et al, 2010). This resistance reflects the synergy between the
bacterium’s low outer-membrane permeability, its chromosomally encoded
AmpC β-lactamase, and its broadly specific drug efflux pump (Masuda et al,
2000). Furthermore, P. aeruginosa readily acquires resistance to most
antimicrobials through mutations in its chromosomal genes and through
extrachromosomal elements carrying resistance determinants (Qiu et al,
2009; Livermore and Yang, 1987).
The broad-spectrum resistance of P. aeruginosa is mainly due to a
combination of different factors: (i) low outer membrane permeability
(Nikaido , 1994), (ii) Presence of the inducible AmpC chromosomal ß-
lactamase (Lister et al., 2009), (iii) synergistic action of several multidrug
efflux systems (Poole, 2004), and (iv) prevalence of transferable resistance
determinants, in particular, carbapenemhydrolyzing enzymes (mainly
metallo-ß-lactamases (MBL) (Gutiérrez et al., 2007).
Although there are several antimicrobials (carbapenems, cefepime,
ceftazidime, tobramycin and amikacin) that continue to be effective against
P. aeruginosa, in the last few years the bacterium’s increasing resistance to
many others has been reported (Sanchez-Romero et al, 2007; Ruiz-Martinez
et al, 2011).

Carbapenems are good antimicrobial activity against P. aeruginosa but
the emergence and spread of acquired carbapenem resistance in this species
have challenged the success of therapeutic and control efforts. Since
carbapenems, especially imipenen, are widely used in the clinical setting
(Riera et al, 2011), investigation of the molecular mechanisms leading to
resistance is crucial.
1.2.4 Carbapenem resistance in P. aeruginosa
1.2.4.1 Carbapenems
Carbapenems are frequently used to treat P. aeruginosa; however,
resistance to the carbapenems is emerging rapidly (Zavascki et al., 2005).
The introduction of carbapenem into clinical practice represented a great
advancement for the treatment of β-lactam resistant bacteria. Due to their
broad spectrum of activity and stability to hydrolysis by most β-lactamase,
the carbapenems have been the drugs of choice for treatment of infections
caused by penicillin or cephalosporin resistant gram negative bacilli
(Jesudason et al., 2005).
Carbapenems exert their action primarily by inhibiting the
peptidoglycan-assembling transpeptidases (penicillin-binding proteins
[PBP]) located on the outer face of the cytoplasmic membrane. In general,
carbapenems can efficiently cross the outer membrane of the bacterium, as
they are small hydrophilic antibiotics. They enter the cell by passing through
the aqueous channels provided by porin proteins (Huang et al., 1995).
Among the several mutation-mediated resistance mechanisms existing
in P. aeruginosa are those conferring decreased susceptibility or resistance
to carbapenems. These antimicrobial agents are commonly used to treat
infections produced by multiresistant strains of P. aeruginosa, as they are

stable against most clinically relevant ß-lactamases (including broad-
spectrum, extendedspectrum, and AmpC-type enzymes). Although
carbapenems remain effective antibiotics for therapy of infections caused by
multidrug resistance (MDR) P. aeruginosa isolates, development of high
carbapenem resistance rates in P. aeruginosa isolates has been reported
worldwide (Davies et al., 2007).
1.2.4.2 Carbapenemase
Metallo-β-lactamase (MBL) was first detected in 1960, in Bacillus
cereus which was chromosomal in location. Then, first plasmid mediated
MBL isolates was found in P. aeruginosa in 1991 in Japan. Since early
1990s, MBL encoding genes have been reported all over the world in
clinically important pathogens, such as Pseudomonas spp., Acinetobacter
spp., and members of the Enterobacteriaceae family (Picao et al., 2008).
MBL in gram negative bacilli is becoming a therapeutic challenge, as these
enzymes usually possess a broad hydrolysis profile that includes all β-lactam
antibiotics including carbapenems (Galani et al., 2008).
The carbapenemases MBLs are the most feared because of their ability
to hydrolyze virtually all drugs in that class, including the carbapenems
(Walsh et al., 2002). In addition to their resistance to all β-lactams, the
MBL producing strains are frequently resistant to aminoglycosides and
fluoroquinolones, Unlike carbapenem resistance due to several other
mechanisms, the resistance due to MBL and other carbapenemase
production has a potential for rapid dissemination, as it is often plasmid
mediated (Walsh et al., 2005). Consequently, the rapid detection of
carbapenemase production is necessary to initiate effective infection control
measures to prevent their dissemination. Various methods like Modified

Hodge test, EDTA disk synergy (EDS) test (Lee et al., 2001), MBL E-test,
EDTA-based microbiological assay are used for the detection of MBLs,
Nevertheless, detection of genes coding for carbapenem resistance by PCR,
usually give reliable and satisfactory results, but this method is of limited
practical use for daily application in clinical laboratories because of the cost
(Marchiaro et al., 2005).
MBLs spread easily on plasmids and cause nosocomial infections and
outbreaks. Such infections mainly concern patients admitted to Intensive
Care Units with several co-morbidities and a history of prolonged
administration of antibiotics (Maltezou, 2009). Moreover, MBL producing
isolates are also associated with higher morbidity and mortality (Walsh et
al., 2005). Early detection of MBL-producing organisms is crucial to
establish appropriate antimicrobial therapy and to prevent their interhospital
and intrahospital dissemination (Picao et al., 2008).
Class 1 integron-containing P. aeruginosa isolates from Australia and
Uruguay were investigated for the genomic locations of these elements.
Several novel class 1 integrons/transposons were found in at least four
distinct locations in the chromosome, including genomic islands (Martinez
et al., 2012). The transmissible MBLs confer high-level resistance to all
carbapenems and are found worldwide (Walsh et al., 2005). AmpC, the
chromosomal β-lactamase, has been found to have very little activity against
carbapenems but can work in synergy with other resistance mechanisms
(Quale et al., 2006).
In the absence of carbapenem-hydrolyzing enzymes, the mechanism
leading to carbapenem resistance is mostly mediated by OprD loss, which
primarily confers resistance to imipenem but also confers low grade
resistance to meropenem (Köhler et al., 1999; Livermore, 1992).

1.2.4.3 Porin oprD
The main porin for uptake of carbapenems in P. aeruginosa is the outer
membrane protein (OMP) OprD, a specialized porin which has a specific
role in the uptake of positively charged amino acids such as lysine and
glutamate (Huang et al., 1995). Other routes for carbapenem uptake have
been proposed (Pérez et al., 1996), but their actual relevance has not been
consistently proved.
Inactivating mutations in OprD have been documented to confer
resistance to imipenem and to a lesser extent to meropenem and doripenem
(Sanbongi et al., 2009). It is also remarkable that mutations leading to the
upregulation of the MexAB-OprM active efflux system may increase the
resistance to meropenem and doripenem but with no effect on the
susceptibility of P. aeruginosa to imipenem, which is not a substrate for this
system (Köhler et al., 1999).
Porin OprD of P. aeruginosa facilitates the uptake across the outer
membrane of basic amino acids, small peptides that contain these amino
acids, and their structural analogue imipenem. Indeed, prolonged imipenem
treatment of patients with P. aeruginosa infections leads to imipenem
resistant mutants that either lack OprD due to an oprD gene mutation (Lynch
et al., 1987) or have strongly reduced OprD levels due to an nfxC-type
mutation (mexT) which suppresses oprD expression at the same time as
upregulation of the mexEF-oprN multidrug efflux operon (Fukuda et al.,
1995; Kohler et al., 1997).

1.2.4.4 Molecular Mechanisms of OprD-Mediated Resistance
The pathway to OprD-mediated resistance can involve mechanisms that
decrease the transcriptional expression of oprD and/or mutations that disrupt
the translational production of a functional porin for the outer membrane. At
the level of oprD transcription, characterized mechanisms include (i)
disruptions of the oprD promoter, (ii) premature termination of oprD
transcription, (iii) coregulation with mechanisms of trace metal resistance,
(iv) salicylate-mediated reduction, and (v) decreased transcriptional
expression through mechanisms of coregulation with the multidrug efflux
pump encoded by mexEF-oprN. oprD promoter disruptions have occurred as
a result of deletions or insertions within the upstream region of oprD.
Yoneyama and Nakae (1993) reported the association of a large deletion
encompassing the putative promoter and initiation codon that prevented
transcription of oprD.
IS1394 and an ISPa16-like insertion (IS) element have been described
upstream of the oprD coding region for imipenem-resistant isolates of P.
aeruginosa exhibiting decreased oprD expression (Wolter et al., 2008;
Wolter et al., 2009).
El Amin et al. (2005) observed that premature termination of
transcription was occurring in clinical isolate, potentially due to mutations
within the structural gene sequence.
The most complex mechanisms are the transcription of oprD that are
linked to the regulation of expression of the mexEF-oprN efflux pump (Ochs
et al., 1999). These mechanisms of coregulation are, showed the complexity
by which P. aeruginosa is able to regulate expression of resistance
mechanisms and why it is sometimes so difficult to definitively link

phenotypes to changes in one specific mechanism (Wolter et al., 2004;
Evans and Segal, 2007).
1.2.5 Biofilm
Biofilms are surface-attached communities of bacteria embedded in an
extracellular matrix of biopolymeric substances and are involved in many
types of chronic infections (Costerton et al., 1995). Biofilm bacteria are
physiologically distinct from free-swimming bacteria of the same species.
Wild-type, nonmucoid P. aeruginosa biofilm formation proceeds through
distinct developmental steps. After initial attachment of single cells to a
surface, the bacteria move on the surface by twitching motility to form
clumps of cells or microcolonies (O’Toole and Kolter, 1998). Figure 1-1
explains the stages of biofilm formation.

Figure 1-1: Essential steps of bacterial biofilm formation inspect with swarming motility
(http://www.pasteur.fr/recherche/RAR/RAR2006/Ggb-en.html).
Common examples of biofilms include dental plaque, endocarditis, and
slime on river stones. Biofilms are increasingly recognized as contributing to
disease pathogenesis in cystic fibrosis and in other bacterial diseases (Parsek
et al., 2003). Bacteria in a biofilm state exhibit increased resistance to
antibiotics (Prince et al., 2002) and host defense factors (Jesaitis et al.,
2003).
Communal bacteria in a biofilm can survive antibiotic concentrations as
much as 1000-fold higher than the same bacteria in an individual, free-
living, planktonic state (Høiby, 2001). Therefore, clinically attainable
antibiotic concentrations may not adequately clear biofilm infections,
allowing the bacterial population to recover, persist, and spread (Singh et al.,
2000).
P. aeruginosa environmental bacterium is capable of living
planktonically or in surface-associated communities known as biofilms. P.
aeruginosa biofilms can form on a variety of surfaces, including in mucus
plugs of the CF lung and abiotic surfaces, such as contact lenses and
catheters (Davies et al., 1998; O’Toole et al., 2000).
1.2.5.1. Stages of biofilm development
· Attachment
Motile (planktonic) bacteria transform to the sessile form prior to
biofilm formation as they adhere to a favourable surface; such as a medical
device or the host tissue. In some cases initial adhesion of biofilm forming
microorganisms is achieved by means of adhesins located on specialised
organelles such as fimbriae (pili) (Sauer et al, 2002; Lasaro et al, 2009).

· Formation of microcolonies
The cells aggregate as they divide on adhesion to a surface but the
daughter cells multiply outward and upward from the point of attachment to
form cell clusters. The dividing cells produce quorum sensing molecules and
extracellular polymeric substances, or polymer matrix. The matrix houses
the aggregating cells in microcolonies and also attaches the biofilm to the
surface on which it is forme, Microcolonies become larger as the number of
organisms increase and the quantity of polymer matrix produced also
increases (Watnick and Kolter, 1999).
More signalling molecules and polymer matrix are produced by the
organisms within the microcolonies at this stage (Tolker - Neilsen et al,
2000; Malic et al, 2009). The fully mature biofilm structure comprises of
bacterial cells, the polymer matrix, and interstitial water channels that
facilitate the exchange of nutrients and wastes in and out of the biofilm into
the surrounding environment, P. aeruginosa displayed multiple phenotypes
during biofilm development and biofilm cells at dispersion were found to be
similar to the planktonic cells in phenotypic expression (Sauer et al, 2007).
· Detachment and dispersal of biofilm organisms
The biofilm environment is innately regulated and studies have shown
that high population density within a mature biofilm induced programmed
detachment of bacteria from biofilm through the secretion of chemical
substances by the organisms (O'Toole et al, 2000). Studies have shown that
detachment occurs when the organisms respond to chemical substances
secreted by them such as signalling molecules (Stoodley et al, 2005),
proteins and degradative enzymes (Barraud et al, 2006) and oxidative or
nitrosative stress-inducing molecules such as nitric oxide (NO) produced as

a result of metabolic processes within a biofilm (Schlag et al, 2007). It has
also been shown that alginate lyase; a degradative enzyme produced by
biofilm organisms cleaves the polymer matrix into short oligosaccharides.
The cleavage antagonises the attachment characteristics of alginate leading
to increased detachment of biofilm organisms (Barraud et al, 2006).
1.2.5.2 Roles of extracellular polymeric substances in P. aeruginosa
biofilms
Exopolysaccharides are an important component of the microbial
biofilm extracellular matrix, since they contribute to overall biofilm
architecture and to the resistance phenotype of bacteria in biofilms. Several
species have been shown to produce a matrix consisting of
exopolysaccharides, proteins and nucleic acids (Pamp et al., 2007). The
major functions ascribed to the matrix are its role as a structural scaffolding
for biofilm cells and as a protective barrier against some antimicrobials.
Matrix production can dictate pattern formation in biofilms in a number of
ways. In some cases where biofilm populations consist of motile and non-
motile subpopulations, matrix production can facilitate the transition from
surface motility to sessility (Merritt et al., 2007; Kuchma et al., 2007).
The common theme in several species, where motility and matrix
production are inversely regulated by intracellular levels of the signalling
molecule cyclic dimeric guanosine monophosphate (c-di-GMP), Although
the matrix in general is considered to spatially fix the cells in a biofilm,
evidence has been presented that matrix components in some cases can
guide migration of the cells (Barken et al., 2008). Matrix production can also
influence average cell-to-cell distances between members of a biofilm
community. The formation of large, tightly packed aggregates is a feature

sometimes observed in a biofilm population overproducing secreted
components of the biofilm matrix (Stapper et al., 2004).
In P. aeruginosa, three major secreted polysaccharides have been
implicated in pattern formation in biofilms; alginate, Psl, and Pel (Colvin et
al., 2012).
· Alginate
AlgC appears to be crucial for general exopolysaccharide biosynthesis,
not just alginate, as it is also required for precursor synthesis of Psl, as well
as LPS and rhamnolipids (Goldberg et al., 1993; Olvera et al., 1999).
Alginate is composed of the uronic acids, mannuronic acid, and its
epimer, guluronic acid (Govan and, Deretic, 1996). In non-mucoid strains,
alginate does not appear to be an important component of pattern
formation/community structure (Wozniak et al., 2003). In clinical biofilms,
it appears to be produced, where its expression is induced under conditions
of low oxygen tension (Worlitzsch et al., 2002).
In the airways of people suffering from Cystic Fibrosis, P. aeruginosa
is seen to undergo a transition to a mucoid phenotype (Govan and, Deretic,
1996). Mucoidy is characterized by alginate overproduction and its impact
on pattern formation in biofilm communities is great (Stapper et al., 2004).
The chemical environment is key in this regard. Extracellular calcium acts as
a cation bridge between the negatively charged alginate polymers (Sarkisova
et al., 2005). In the absence of calcium, alginate overproduction results in
the production of aggregates of tightly packed bacterial cells in the biofilm.
However, in the presence of low calcium (µM–mM), the secreted alginate
forms a gel. This results in individual cells being suspended within the gel,
increasing the average intercellular distance (Hentzer et al., 2001; Sarkisova

et al., 2005). A functional consequence of alginate overproduction in a
biofilm is increased tolerance to antibiotics such as tobramycin (Hentzer et
al., 2001).
· Psl
The psl gene cluster contains 15 cotranscribed genes (pslA to pslO)
encoding proteins predicted to synthesize the Psl EPS, which is important to
initiate and maintain biofilm structure by providing cell-cell and cell-surface
interactions The pslH- and pslI-encoded proteins exhibit homology to
galactosyltransferases and mannosyltransferases, respectively. That means
the Psl EPS is composed mainly of mannose and galactose and that Psl is
indeed a matrix component of the biofilm (Ma et al., 2007), Overhage and
colleagues (2005) mapped the psl operon promoter 41 bp upstream of the
pslA start codon.
Ma et al. (2007) observed the frame deletions of pslH (strain
WFPA818) and pslI (strain WFPA819). The biofilm formation capacities of
these strains were compared with those of wild-type and psl-deficient strains
in a rapid attachment assay. Loss of either PslH or PslI function results in a
profound attachment defect, similar to that observed with the psl null strain
WFPA800. The attachment defect of WFPA818 and WFPA819 was restored
when a plasmid expressing either pslH (pMA10) or pslI (pMA11) was
introduced into the respective strain; these data indicate that PslH and PslI
are key proteins for Psl EPS synthesis. In a prior transposon mutagenesis
screen, pslH and pslI mutants also exhibited reduced biofilm formation
(Friedman, and Kolter, 2004).

Overproduction of the Psl polysaccharide led to enhanced cell-surface
and intercellular adhesion of P. aeruginosa. This translated into significant
changes in the architecture of the biofilm. Consequently, it was proposed
that Psl has an important role in P. aeruginosa adhesion, which is critical for
initiation and maintenance of the biofilm structure. The ability to form
biofilms in the airways of people suffering from cystic fibrosis is a critical
element of P. aeruginosa pathogenesis. The 15-gene psl operon encodes a
putative polysaccharide that plays an important role in biofilm initiation in
nonmucoid P. aeruginosa strains. Biofilm initiation by a P. aeruginosa
PAO1 strain with disruption of pslA and pslB (ΔpslAB) was severely
compromised, indicating that psl has a role in cell-surface interactions. In
previous study showed the adherence properties of this ΔpslAB mutant
using biotic surfaces (epithelial cells and mucin-coated surfaces) and abiotic
surfaces. Accordingly, psl is required for attachment to a variety of surfaces,
independent of the carbon source (Ma et al., 2006).
The actual structure of Psl is not known, although it is rich in
rhamnose, mannose, and glucose monomers (Ma et al., 2007). The primary
function for Psl in non-mucoid strains is attachment; Strains defective for Psl
production are defective in surface attachment on many different surface
types. However, Psl contributes to maintaining biofilm structure at later
stages in development (Matsukawa and Greenberg, 2004).
Ma et al. (2006) demonstrated that a strain that conditionally produces
Psl formed mature biofilms that eroded away once Psl expression was
disrupted. Psl was seen to preferentially localize to the exterior of
aggregates or microcolonies, forming a shell (Wozniak et al., 2003).
The interesting Psl staining pattern suggests that the polysaccharide
plays a key structural role encasing and ultimately holding together cells in

an aggregate. Like alginate/mucoidy, genetic variants that overproduce Psl
have been observed in CF sputum samples (Haussler et al., 2003). Psl
producing strains produce distinctive colony morphology on solid medium,
called rough, small colony variants (RSCVs). Like mucoidy, the CF airways
select for this phenotype (Smith et al., 2006). RSCVs are characterized by
autoaggregation in liquid culture and hyper-attachment to surfaces (Kirisits
et al., 2005).
Overhage et al. (2005) stated that psl expression was localized to the
centers of microcolonies within biofilms. In addition, Kirisits et al. (2005)
showed that the expression of psl and pel was elevated in variants isolated
from aging P. aeruginosa PAO1 biofilms. It has been suggested that the
mechanistic basis for psl and pel overproduction in these variants, as well as
other autoaggregative variants, involves elevated levels of the c-diGMP. P.
aeruginosa has several loci capable of modulating the c-diGMP level,
including the wsp, LadS, and retS signal transduction systems (Kuchma et
al., 2010).
· Pel
The last major exopolysaccharide produced by P. aeruginosa is Pel.
The pel gene cluster consists of seven genes, which encode the enzymatic
activities required for synthesis of the hydrophobic glucose rich Pel
exopolysaccharide (Friedman and Kolter, 2004). Unlike Psl, Pel does not
appear to play a role in attachment, although it is important in maintaining
mature biofilm structures (Vasseur et al., 2005). Its contribution to the
cellular distribution patterns found in biofilms, and where it is produced in a
biofilm is unclear. However, like Psl, Pel is found to be overproduced in
many RSCVs (Kirisits et al., 2005).

1.2.5.3 Extracellular DNA
Extracellular DNA was shown to be present in high concentrations in
the outer part of the microcolonies in young P. aeruginosa biofilms and
between the stalk-forming and cap-forming subpopulations in mature
glucose grown biofilms (Allesen-Holm et al., 2006). Type IV pili bind with
high affinity to DNA (Aas et al., 2002; van Schaik et al., 2005), and
evidence has been presented that the high concentration of extracellular
DNA on the microcolonies in developing P. aeruginosa biofilms may cause
accumulation of the migrating piliated cells and thereby facilitate formation
of the mushroom caps (Barken et al., 2008).
Production of extracellular DNA during P. aeruginosa biofilm
development has been shown to be dependent on the quorum-sensing system
(Allesen-Holm et al., 2006). Expression of the pqs-genes in developing P.
aeruginosa biofilms was shown to occur specifically in the outer layer of the
stalk forming microcolonies, correlating with the location of the
extracellular DNA (Yang et al., 2007).
1.2.6 P. aeruginosa motility on surface 1.2.6.1 Swarming motility
Swarming motility is operationally defined as a rapid multicellular
bacterial surface movement powered by rotating flagella. Although simple,
accurate, and mechanistically meaningful, the definition does not do justice
to the wide array of phenotypes associated with swarming motility, nor does
it emphasize all that remains unknown about this behavior (Copeland et al.,
2010).
A. Factores important for swarming motility
· Flagella

During swarming, P. aeruginosa retains its polar flagella but
synthesizes an alternative motor specifically required to propel movement on
surfaces and through viscous environments, Thus the expression of
alternative motors is at least one other way to facilitate swarming motility
besides use of peritrichous flagella (Toutain et al., 2005).
· Rafting
Whereas bacteria swim as individuals, swarming bacteria move in side-
by-side cell groups called rafts (Copeland et al., 2010). Raft formation is
dynamic: cells recruited a raft move with the group whereas cells lost from a
raft quickly become non-motile. The dynamism in cell recruitment and loss
suggests that no substance or matrix maintains raft stability save perhaps the
flagella themselves. Indeed, scanning electron microscopy of a swarm of
Proteus mirabilis revealed extensive rafting and perhaps intercellular
bundling of flagella (Jones et al., 2004). As with hyperflagellation, the
reason that swarming motility requires raft formation is at present unclear.
· Surfactant synthesis
Initial characterization of P. aeruginosa implicated rhamnolipids as the
swarming surfactant (Köhler et al., 2000). Di-rhamnolipid is composed of
two rhamnose sugars attached to the complex fatty acid β-hydroxydecanoyl-
β-hydroxydecanoate (HAA) (Caiazza et al., 2005). Subsequent investigation
has shown that the di-rhamnolipid precursors HAA and mono-rhamnolipid
also act as surfactants to promote swarm expansion (Tremblay et al., 2007).
Surfactant production is commonly regulated by quorum sensing
(Ochsner and Reiser, 1995). Surfactants are shared secreted resources and
are only effective at high concentration. Therefore, quorum sensing may
have evolved to regulate surfactant production to ensure that the surfactants

are only made when there are sufficient bacteria present to make surfactants
beneficial (Kato et al., 1999).
The flagellum and the chemotaxis system, consisting of
chemoreceptors and a signal relay system similar to that of E. coli, allow the
bacterium to respond to attractants and repellents (Kato et al., 1999).
Swarmer cells, which are usually elongated and hyperflagellated,
differentiate from vegetative cells probably by sensing the viscosity of the
surface or in response to nutritional signals (Harshey, 1994).
The swarming of the normally polar, monotrichously flagellated
bacterium P. aeruginosa is induced on 0.5 to 0.7% agar when certain amino
acids are provided as the sole source of nitrogen. The swarmer cells of P.
aeruginosa are elongated and can possess two polar flagella. Unlike all other
swarming bacteria, P. aeruginosa also requires type IV pili for this type of
motility.
B. Biofilm and swarming motility inversely regulate by c-diGMP
Cyclic-di-GMP is a ubiquitous second messenger in bacteria, the c-di-
GMP antagonistically controls motility and virulence of single, planktonic
cells on one hand and cell adhesion and persistence of multicellular
communities on the other has spurred interest in this regulatory compound
(Aldridge et al., 2003).
C-di-GMP controls cellular processes associated with the sessile-motile
transition in eubacteria, including exopolysaccharide (EPS) production,
attachment, and motility. Low concentrations of c-di-GMP are associated
with cells that move by virtue of flagellar motors or retracting pili. In
contrast, increasing concentration of c-di-GMP promote the expression of
adhesive matrix components and results in multicellular behavior and

biofilm formation that mean coordinate regulation of these two surface
behaviors depends upon common downstream effectors, such as regulation
of flagellar function and production of the Pel-derived EPS (Lim et al.,
2006).
Although Events during early biofilm formation by P. aeruginosa
PA14 require proper control of flagellar function for the reversible to
irreversible attachment phase, as well as robust production of the Pel EPS
(Caiazza et al., 2007; Merritt et al., 2007). C-di-GMP-mediated regulation
can occur through a variety of mechanisms, including stimulation of
exopolysaccharide (EPS) production, cell surface adhesin expression/
localization, and/or repression of various forms of motility (Newell et al.,
2009). Conversely, reduced levels of c-di-GMP generally lead to stimulate
of motile behaviors concomitant with reduced biofilm formation and hence
promote the switch to a motile lifestyle (Hengge, 2009).
Swarming motility is a flagellum-driven process and therefore the
sensitive to changes in flagellar function (Caiazza et al., 2007; Merritt et al.,
2007). Moreover, strains defective for production of the Pel EPS show
enhanced swarming relative to the wild type (WT), indicating that
production of the polysaccharide negatively impacts on swarming motility in
P. aeruginosa PA14 (Caiazza et al., 2007).
Strains with mutations in the bifA gene in PA14 elevate the levels of c-
di-GMP, and this accumulation is largely dependent upon the cyclase
activities of both SadC and RoeA, the resulting excess c-di-GMP produced
in the bifA mutant leads to hyper-biofilm formation and repression of
swarming motility (Kuchma et al., 2007, Merritt et al., 2010).
Moreover, it was shown that enhanced production of the Pel EPS is
required for the hyper-biofilm-forming phenotype but contributes only

marginally to the swarming defect of the bifA mutant (Kuchma et al., 2007).
Always the question was witch factors are required for repression of
swarming motility when c-di-GMP levels are elevated, (Kuchma et al.,
2010) performed a genetic screen to identify suppressors in the bifA
background that restored the ability to swarm. They identified a role for the
pilY1 gene in c-di-GMP-mediated repression of swarming motility. Strains
with mutations in the pilY1 gene show robust suppression of both the
swarming deficiency as well as the hyper-biofilm-forming phenotype
exhibited by the bifA mutant.
1.2.6.2 Twitching motility
Twitching motility is believed to result from the extension and
retraction of the pilus filament, which propels the cells across a surface.
Pilus synthesis and assembly require at least 40 genes which are located in
several unlinked regions on the chromosome (Hobbs et al., 1993).
It is clear that active extension and retraction of type IV pili is involved
in twitching motility (Skerker and Berg, 2001). These pili are about 6 nm in
diameter and up to 4 µm in length; they are typically found at one or both
cell poles (Bradley, 1972). Radial expansion rates of colonies via twitching
motility can approach 0.3 µm/s (Mattick, 2002), involved in biofilm
formation (O’Toole and Kolter, 1998).
Twiching motility like swarmer cells, rafts or spearhead-like clusters of
aggregated cells in P. aeruginosa have been observed during twitching
motility. Within the rafts, cells are highly aligned in close cell-cell contact.
The rafts move radially outward, following the long axis of the cells. Cells
from one group join into another and form a lattice, many like swarming
bacteria. Such cells at first move end forward toward the other cells until

they touch with their poles and then rapidly snap into an aligned position,
which accounts for the characteristic jerky twitching motion observed with
this form of motility (Mattick, 2002). Although twitching motility is
primarily a social activity, individual cells can show limited movement when
in contact with inert substrates or on agar at low concentrations (Mattick,
2002).
In vitro and in vivo studies show that mutants lacking functional type
IV pili have a significant reduction in colonization, biofilm formation, and
ability to spread (O’Toole and Kolter, 1998; Wozniak and Keyser, 2004).
Control of type IV pili expression and twiching motility is complex.
One system that controls twiching motility is a sensor kinase and a response
regulator pair referred to as FimS/AlgR (Whitchurch et al., 1996).
However, type IV pili are polar organelles that are composed of a
single protein subunit, PilA. PilA is exported out of the cell and polymerized
to form the surface fimbrial strand. Prepilin genes are located in one of the
many v gene clusters and appear to play a role in type IV pili assembly,
export, localization, and maturation and the general efficiency of the type IV
pili machinery (Mattick, 2002). A microarray study revealed that the fimU-
pilVWXY1Y2E prepilin cluster is under the control of algR (Lizewski et al.,
2004).
The pilY1 gene from P. aeruginosa was discovered by Richard Alm
and colleagues at the University of Queensland in 1996, it was identified at
the same time as three other previously uncharacterized genes, pilW, pilX,
and pilY2, all of which were initially implicated in pilus biogenesis. PilY1 is
a large (127 kDa) protein with ~1163 amino acids (value varies between
different strains).

However, mutation of the pilA gene, encoding the major subunit of the
type IV pilus, showed only weak suppression of the bifA swarming and
biofilm defects, indicating that it is loss of PilY1 specifically, and not loss of
pili, that fully suppresses the bifA mutant defects (Kuchma et al. 2010).
PilY1 protein plays two distinct cellular roles: one role promoting pilus
assembly and a second role repressing swarming motility. Also the minor
pilins, PilW and PilX, impact both pilus assembly and swarming motility,
and this subset of pilus assembly proteins functions to allow P. aeruginosa
to coordinately regulate two distinct motility behaviors, swarming and
twitching, when associated with a surface (Kuchma et al., 2012).
Finally Surface induction of PilY1 not only facilitates pilus biogenesis
but also stimulates synthesis of the intracellular signaling molecule, c-di-
GMP, via an interaction with the SadC diguanylate cyclase (figure 1-3).
These data are consistent with the minor pilins, PilX and PilW, also
participating in the modulation of c-di-GMP levels, either in conjunction
with PilY1 or possibly via a separate pathway. Increased synthesis of c-di-
GMP then leads to repression of swarming motility, most likely by
influencing flagellar function as previously proposed (Caiazza et al., 2007).
Elevated c-di-GMP levels also stimulate biofilm formation (Caiazza et al.,
2007; Kuchma et al., 2007; Merritt et al., 2007), allowing P. aeruginosa
cells to coordinately regulate all three of these distinct surface behaviors
(Kuchma et al., 2012).

Figure 1-3: Model for coordinate regulation of surface-based swarming and
twitching motility, surface growth induces expression of pilus assembly proteins,
including PilY1, PilX, and PilA, likely to prepare cells for twitching motility (Kuchma et
al., 2012).

chapter two
materials and methods

2. Materials and Methods
2.1. Materials 2.1.1. Equipment
Apparatuses and equipment used in this study were listed in table 2-1.
Table 2-1: Apparatuses and equipment used in this study
Manufacturing Company/ Origin Equipment Id
Coastar/ USA 96 well flat bottom plate (poly styrene) 1
Coastar/ USA 96 well U bottom plate (Vinyl) 2 Fischer / USA ABI 7500 Fast System 3 Hirayama /Japan Autoclave 4 Hettich /Germany Centrifuge 5 Sanyo/Japan Deep-freezer 6 Canon /Japan Digital camera 7 Fischer /USA Electrical balance 8 Fischer /USA Electroporation unit 9 Fischer / USA Eppendorf tubes 10 Fischer / USA Gel electrophoresis unit 11 IKA /Germany Hot plate with magnetic stirrer 12 Memmert /Germany Incubator 13 Fischer / USA Laminar air flow hood 14 Schleicher and Schuel / USA Membrane filters (0.22µm) 15 Hettich /Germany Microcentrifuge 16 Brand /Germany Micropipette 17 Fischer / USA Microwave 18 Siga / USA Microwave oven 19 Fischer / USA Nanodrop 20 SherWood /USA Oven 21 IKA /Germany PCR Unit 22 Fischer / USA pH meter 23 Fischer / USA Power supply 24 Fischer / USA Refrigerator 25 BBL /USA Screw capped test tubes 26

Table 2-1 continued
2.1.2. Chemicals and biological materials
Chemicals and biological material used in this study are listed in table
2.2.
Table 2-2: Chemical and biological materials
Shimadzu /Japan Spectrophotometer 27 Millipore/ USA Stericup filter unit 28
Manufacturing Company/ Origin Equipment Id
Ultraviolet products institute /USA Ultraviolet light 29
Labcoo /Germany Vortex 30 Memmert /Germany Water bath 31 Fischer / USA Water distillatory 32
Id Chemical and biological materials Company (origin) 1 1 KB plus DNA Ladder Invitrogen/USA 2 5X HF Buffer Ivitrogene / USA 3 Agarose gel Promega /USA 4 Ammonium sulfate (NH4)2SO4 Difco /USA 5 Ampicillin (Ap) Siga/ Aldrich /USA 6 Arabinose Himedia /India 7 Barium chloride (BaCl2. 2H2O ) Difco/USA 8 Boric acid Difco /USA 9 Bromothymol blue dye BDH/UK
10 Carbincillin (CA) Siga-Aldrich /USA 11 Cassamino acid Difco /USA 12 Crystal violet Merck /Germany 13 Dipotassium phosphate (K2HPO4) Siga- Aldrich /USA 14 Disodium phosphate (Na2HPO4.7H2O) Difco /USA 15 DMSO Difco /USA 16 EDTA Difco /USA 17 Ethanol Absolute Merck /Germany

Table 2-2 continued
2.1.3 Culture media
All culture media used for the isolation and identification of bacteria
through this study are listed in table 2- 3.
Table 2-3: Ready to use culture media
18 Gentamicin (Gm) Siga- Aldrich /USA 19 Glacial acetic acid Difco / USA 20 Glucose Difco/ USA 21 Glycerol Siga- Aldrich /USA
Id Chemical and biological materials Company (origin) 22 Imipenem powder 25mg Siga-Aldrich / USA 23 Ladder 1 Kb Promega / USA
24 Lithium acetate Difco / USA 25 Magnesium sulfat (MgSO4) Merck / Germany 26 Monopotassium phosphate (KH2PO4) Difco / USA 27 Nalidixic acid (NA) Siga-Aldrich /USA 28 Nucleotide mix dNTP Roch-Germany /USA 29 Pepton Difco / USA 30 Phusion enzyme Invitrogene /USA 31 Polyethylene glycol Siga /USA 32 Sodium chloride (NaCl) Difco / USA 33 Sodium hydroxide (NaOH ) Difco /USA 34 Sodium phosphate (NaHPO4) Difco / USA 35 Sucrose Fluka-Swizerland/ Germany 36 Sulphuric acid (H2SO4) Difco /USA
37 Syper safe stain Invetrogen / USA 38 Taq polymerase BioLabs / USA
39 Taq polymerase 5U/µl Promega / USA
40 Tri-HCl Difco / USA
41 Tris base Difco / USA 42 Tryptone Difco / USA 43 Yeast extract Biomerieux /France 44 Yeast Nitrogen Base Difco / USA

Id Medium Company(Origion) 1 Cetrimide Agar Base
Difco (USA) 2 MacConkey Agar 3 Muller Hinton Agar 4 Muller Hinton Broth
2.1.4. Kits
All kits used in this study are listed in table 2-4.
Table 2-4: Kits used in the present work ID The kit Company (Origin)
۱ E-test strip kit of Imipenem (from 0.002 to 32 μg/ml)
BioMérieux (France)
۲ Gentra Puregen Yeast/Bactacteria Kit Qiagene (USA)
۳ Phusion enzyme kit, High fidelity DNA polymeraseare.
Invitrogen (USA)
4 QIAquick PCR purification kit. Qiagen (USA) 5 Qiagen Kit for yeast plasmid DNA prep Qiagen (USA)
6 Qiagen Kit for plasmid DNA prep from Bacteria
Qiagen (USA)
7 RNeasy Mini kit for purification total RNA from bacteria
Qiagen (USA)
2.1.5. Standard strains quality control bacteria
All standard strain and quality control bacteria used throughout this
study are listed in table 2-5:

Table 2-5: The standard strains and quality control bacteria used in the present study .
Name Relevant Genotype, description, or sequence Source
Saccharomyces cerevisiae (InvSc1)
MATa/MATα leu2/leu2 trp1-289/trp1-289 ura3-52/ura3-52 his3-Δ1/his3-Δ1
Invitrogen/USA
Escherichia coli S17-1(λpir)
thi pro hsdR-hsdM+ ΔrecA RP4-2::TcMu-Km::Tn7
Simon et al., 1983
DH5α lˉ f80dlacZDM15D (lacZYA-argF) U169 recA1 endA hsdR17(rKˉ mKˉ ) supE44 thi-1 gyrA relA1
Life Technologies/ USA
Table 2-5 continued
Name Relevant Genotype, description, or sequence Source
Pseudomonas aeruginosa PAO1 Wild type Jacobs et
al.,2003
oprD::isphoA/hah PAO1 with isphoA/hah insertion in oprD;Tcʳ
Jacobs et al.,2003
Escherichia coli ATCC 25922/ Sensitive for all antibiotic ATCC/USA
Pseudomonas aeruginosa
ATCC 27853/ Sensitive for all antibiotic ATCC/USA
2.1.6 Primers
2.1.6.1. Primers used in oprD genetic complementation and sequencing
All primers used in oprD complementation and sequencing were
designed according to http://www.idtdna.com website listed in table 2-6.

Table 2-6: oprD gene primer used in Genetic complementation and
sequencing
Primers Name Primer sequence (5′ – 3′) Origin
oprD comp 5′ ttctccatacccgtttttttggggaaggagatatacatATGAAAGTGATGAAGTGGAG
Integrated DNA Technology (IDT)/USA
oprD comp 3′ taatctgtatcaggctgaaaatcttctctcatccgccTCACAGGATCGACAGCGGATAG
oprD seq a-f ATGAAAGTGATGAAGTGGAGC
oprD seq a-r AGGGAGGCGCTGAGGTT
oprD seq b-f AACCTCAGCGCCTCCCT
P730 GCAACTCTCTACTGTTTCTCC
Note: In primer sequences, lowercase letters indicate sequence homology to the cloning vector, uppercase letters indicate a Pseudomonas gene-specific sequence, boldface and lower case letters indicates a His tag sequence.
2.1.6.2. Quantification real time PCR primers
All primer used in oprD complementation and sequencing were
designed according to http://www.idtdna.com website are listed in table 2-9.
Table 2-9: oprD primers used in q RT-PCR
Primer name Primer sequence (5′ – 3′) Origin
oprD-RT Forward CCGCAGGTAGCACTCAGTTCG IDT/USA
oprD-RT Reverse GTAGTTGCGGAGCAGCAGGTC

2.1.6.3. Primer used in Arbitrary PCR
All primers used in Arbitrary PCR are listed in table 2-7.
Table 2-7: Primer used in Arbitrary PCR
Primer name Primer sequence (5′ – 3′) Origin
P235 GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT
IDT/USA
P236 GGCCACGCGTCGACTAGTACNNNNNNNNNNACGCC
P237 GGCCACGCGTCGACTAGTACNNNNNNNNNNAGAG
P238 TATAATGTGTGGAATTGTGAGCGG P239 GGCCACGCGTCGACTAGTAC P240 ACAGGAAACAGGACTCTAGAGG P241 CACCCAGCTTTCTTGTACAC
2.1.6.4. Primers used in pilY1 genetic complementation and sequencing
All primer used in pilY1Genetic complementation and sequencing were
designed according to http://www.idtdna.com web site are listed in table 2-
8.
Table 2-8: pilY1 primers used in Genetic complementation and sequencing
Primer name Primer sequence (5′ – 3′) Origin pilY1 comp 5′ tctccatacccgtttttttgggctagcgaattcgaaggagatataca
tATGAAATCGGTACTCCACCAG
IDT/USA
pilY1His comp 3′ tcttctctcatccgccaaaacagccaagcttgcatgcctTCAgtggtgatggtggtggtgGTTCTTTCCGATGGGGC
pilY1 seq Rev 2 TGAACGGACAGGTACAGATCC
pilY1 seq 3 GGATCTGTACCTGTCCGTTC
pilY1 seq 4 GGCGAGTTTCTCAAGAAGACC
pilY1 seq 5 CTTCCAGGACATCCTCAACCG
pilY1 seq 6 AGCCCAGCGGTAACTACTCC
pilY1 seq 7 CAAGGTCAACCAGGACGATC P730 GCAACTCTCTACTGTTTCTCC
Note: In primer sequences, lowercase letters indicate sequence homology to the cloning vector, uppercase letters indicate a Pseudomonas gene-specific sequence, boldface and lower case letters indicates a His tag sequence.
2.1.7. Plasmids and vectors used in this study
All the plasmids and vector were used in this study were summarized in
table 2-10.
Table 2-10: Plasmids and vectors
Plasmids description, or sequence Origin
pMQ72 Shuttle vector for cloning in yeast and for arabinose-inducible gene expression; GmR
Shanks et al., 2006
PMQ70 Shuttle vector for cloning in yeast and for arabinose-inducible gene expression; ApR
Shanks et al., 2006
pBT20 Vector carrying mariner transposon; ApR GmR * (marker on transposon)
Kulasekara et al., 2005
poprD oprD gene cloned in pMQ72; GmR This study
pPilY1His His-tagged pilY1 gene cloned in pMQ70; ApR This study
*GmR: Gentamicin resistance, ApR: Ampicillin resistance
2.1.8. Buffers and solutions
All buffers and solutions were prepared according to British
Pharmacopeia (2012) unless it is mentioned elsewhere.
2.1.8.1. Magnesium sulfate MgSO4 1 M
MgSO4 (24.65) g was dissolve in ~75 ml distilled water, then brought
up to 100 ml volume and then sterilized with autoclave (15 minutes, 121°C).

2.1.8.2. Glucose 20%
Glucose (200) g was added to one litter of water and then autoclaved
for 5 min at 121°C.
2.1.8.3. Lazy bones solution
Polyethylene glycol (40%) and Lithium acetate (0.1 M), 10mM Tris-
HCL (pH 7.5) and 1mM EDTA were mixed
2.1.8.4. Sodium borate (SB) buffer 20X
This buffer was prepared as follows: 8 g NaOH and 40 g boric acid
were added to 1 liter of distilled water. For routine, SB buffer was diluted to
1X concentration.
2.1. 8.5. Ethylendiaminetetraacetic Acid (EDTA), 0.5 and 0.05 M
This buffer was prepared as follows: 18.61 g EDTA in 100 ml distilled
water to achieve 0.5 M concentration followed by pH adjustment to 8.0 and
sterilization was done by filtration. This solution was used for the
preparation of TE buffer. The 0.5 M EDTA solution was diluted by adding
10 ml of this solution to 90 ml distilled water in order to attain the 0.05 M
EDTA solution.
2.1. 8.6. Tris buffer (1M) pH 8
Tris base (122.2) g was added to one liter of distilled water.
2.1. 8.7. Tri- EDTA (TE) buffer
Tris stock (1 M ) 10 ml was add to 2 ml of 0.5 M of EDTA, final
concentration should be 10mM Tris pH8, 1mM EDTA pH8.
2.1. 8.8. loadeing buffer 6X dye

Mixture of 30% sucrose and 0.25% bromophenol blue was made. The
mixture was completed to 100 ml and autoclaved and then kept at 4°C until
used.
2.1. 8.9. Ethanol (70%)
Absolute ethanol (70 ml) was completed to 100 ml with distilled water.
2.1. 8.10. Glycial acetic acid 30%
Absolute glycial acetic acid (70 ml) was completed to 100 ml with
distilled water.
2.1.9.11. Casamino acids 20% (CAA)
200 g of CAA was added to one litter of water and then autoclaved for
15 min at 121°C.
2.2. Methods
2.2.1. Sterilization
Sterilization was achieved by heating; wet heat sterilization by
autoclave at 121˚C/15 psi for 15 min. One more method used is filtration
using 0.22 μm filter unit. All equipment and materials used in this study
were sterilized through these two methods unless mentioned elsewhere.
2.2.2. Laboratory prepared culture media
2.2.2.1. Yeast peptone dextrose (YPD) broth media
The broth was prepared as follows: 10 g yeast extract, 20 g peptone
and
20 g dextrose were dissolve in 1L of distilled water, pH was adjusted to 7.5.
2.2.2. 2. Yeast peptone dextrose (YPD) agar plate

The agar plate was prepared as follows: 10 g yeast extract, 20 g
peptone, 20 g dextrose and 15 g agar were dissolved in one liter of distilled
water, pH was adjusted to 7.5.
2.2.2. 3. Lysogeny broth (LB) broth
The broth was prepared according to Sambrook and Russell (2001) and
was done as follows: 10 g tryptone, 5 g Yeast extract, 5 g NaCl, these
chemicals were dissolved in 1L of distilled water, the pH was adjusted to 7,
autoclaved, then kept at 4°C until used.
2.2.2. 4. Lysogeny broth (LB) agar plates
The agar plate was prepared according to Sambrook and Russell
(2001). In brief, 10 g tryptone, 5 g Yeast extract, 5 g NaCl, and 15 g Agar
these chemicals were dissolved in 1L of distilled water, pH was adjusted to
7, autoclaved, then poured in petri plates.
2.2.2. 5. Minimal salts medium 5X M63
The minimal media was prepared according to Sambrook and Russell
(2001). Briefly, 60g KH2PO4, 140g K2HPO4 and 40g (NH4) 2SO4, the
reagent were mixed well in 4L of distilled water and autoclaved, pH was
adjusted to 7.
2.2.2. 6 Minimal salts medium 5X M8
The minimal media was prepared according to the following method:
64 g of Na2HPO4.7H2O (or 30 g NaHPO4), 15 g KH2PO4 and 2.5 g of
NaCl, were dissolved in four liters of distilled water and autoclaved, pH was
adjusted to 7.
2.2.2. 7 Minus uracil medium

Synthetic medium was prepared by dissolving Yeast Nitrogen Base 6.7
g, 0.76 g supplement mixture minus uracil (CSM-URA), 15g Dextrose, 20 g
Agar and 0.77 g of DoB or DoBA were dissolved in one liter of distilled
water. Afterward, the mixture was autoclave at 121 °C for 15 min.
2.2.2. 8. Glycerol for -80ºC
LB powder no agar ( 20) g was added to 800 ml distilled water and
200 ml glycerol (100%) and mix well.
2.2.2. 9. Stabs
nutrient broth (2.4) g, 3 g agar and 15 g thymine were mixed and
melted in a microwave. Later, 2 ml was added to vial by syringe and
needle, caps were left loosely and autoclaved. Thereafter, caps were
tightened when it became cool.
2.2.3. McFarland standard (no. 0.5) preparation
The 0.5 McFarland standard was prepared in accordance with the
British Society for Antimicrobial Chemotherapy (Vandepitte et al. 2003), by
adding 0.۰٥ ml of 0.048 M barium chloride (1.17% w/v BaCl2. 2H2O) to
99.۹5 ml of 0.18 M sulphuric acid (1% w/v H2SO4) with constant stirring.
The suspension was distributed to five glass tubes of the same size and
volume as those used in growing the broth cultures, the absorbance was
measured in a spectrophotometer at a wavelength of 625 nm and the
acceptable absorbance range for the standard is 0.08-0.13. Afterward, each
tube was thoroughly mixed on a vortex mixer to ensure that it is even. These
turbidity standard tubes were sealed tightly to prevent loss by evaporation.
Stored protected from light at room temperature. Before use, the tubes were

vigorously agitated by hand. It was used for the turbidity standardization for
antibiotic susceptibility test.
2.2.4. Single Stranded Carrier DNA
Single stranded carrier DNA prepared in accordance with (Burke et al.,
2000), high-molecular weight DNA (Deoxyribonucleic acid sodium salt type
III from Salmon testes; sigma), TE buffer (pH 8.0) (10 mM Tris-Hcl pH 8.0,
1mM EDTA).
· DNA (200) mg was weighed and placed into 100 µl TE buffer. This
mixture was mixed vigorously on a magnetic stirrer for 2-3 hours or until
fully dissolve.
· the DNA was aliqouted and stored at -20ºC
· Prior to use, the DNA was boiled an aliquot in water bath for 10 minutes
and quickly placed in ice.
2.2.5. Preservation of bacterial strains
Two methods were used for the preservation of bacterial strains:
A) Glycerol method: A loopful of overnight growth pure culture was added
to LB broth and incubated at 37 ºC. After 18 hr., 0.5 ml of culture was
added to 0.5 ml Glycerol (Vandepitte et al., 2003).
B) The strains of bacteria were subcultured in stabs, kept in cool incubator
4-8 °C and resubcultured every 3 months in a new slant.
2.2.6. DNA agarose gel electrophoresis
Standard method of the (Sambrook and Russell, 2001) was
followed to prepare horizontal agarose gel electrophoresis for genomic
DNA, plasmid and PCR product:

· Agarose at concentrations of 1% was prepared for PCR product for
plasmid and chromosomal DNA electrophoresis. Agarose was dissolve
in 100 ml of 1X SB buffer and solubilized by heating with stirring .The
agarose was left to cool at 60°C before adding Syper safe stain and
poured into the taped plate.
· A comb was placed near one edge of the gel.
· Syper safe was added with the pouring of the agarose
· The gel was left to harden until it became opaque; gently the comb and
tape were removed.
· SB (1X) buffer was poured into gel tank and the slab was placed
horizontally in electrophoresis tank.
· About 2µl of loading buffer was applied to each 5 ml of plasmid, the well
was fill with the mixture.
The power supply was set, 1kp molecular ladder served as marker. Five
microliters of the DNA ladder were mixed with one microliter of blue 6X
loading dye and subjected to electrophoresis in single lane. After
electrophoresis, the gel was exposed to UV using UV transilliuminator and
then photographed.
2.2.7. Imipenem stock preparation
The potency of Imipenem contains is 1000 µg per mg (Wiegand et al.,
2007). The stock solutions were prepared using the formula:
1000 ×V×C W=
P Where P = potency given by the manufacturer (µg/mg), V =volume
required (ml), C = final concentration of solution (multiples of 1000)

(mg/L), and W = weight of antibiotic (mg) to be dissolved in volume V
(mL).
Further stock solutions were prepared from the initial 10 000 mg/L
solution: 1 ml of 10 000 mg/L solution was added to 9 ml diluent = 1000
mg/L, 100 µl of 10 000 mg/L solution was added to 9.9 ml diluent = 100
mg/L.
2.2.8. Antibiotic stocks used in this study
Antibiotic stocks used in this study represent 1000X. Gentamicin
(GM), Nalidixic acid (NA), Ampicillin (Ap) and Carbincillin (CA) were
prepare in concentration of 10, 20, 150, 50 mg/ml respectively, distilled
water represent diluent.
2.2.9. Biofilm Media
M63 (minimal salts medium, 200 ml) 5X was supplemented with 1 M
MgSO4 (1 ml ), 20% glucose ( 10 ml) and 20% CAA ( 25 ml), all the
reagents were mixed and autoclaved at 121 °C for 15 min and then stored at
room temperature 25±2 ºC.
2.2.10. Swarming motility media
M8 (minimal salts medium 200 ml) 5X was supplemented with 1 M
MgSO4 (1 ml), 20% glucose (10 ml) and 20% CAA (25 ml), afterward, they
were mixed and solidified with agar (0.5% final concentration), finally they
were autoclaved at 121 °C for 15 min and stored at room temperature 25±2
ºC.
2.2.11. Twitching motility media
5X M63 (200 ml) was supplemented with 1 M MgSO4 (1 ml), 20%
glucose (10 ml) and 20% CAA (25 ml), thereafter they were mixed and

solidified with agar (1.5% final concentration) , finally they were autoclaved
at 121 °C for 15 min and stored at room temperature 25±2 ºC.
2.2.12. Standard and clinical strains culture
Clinical strains and E. coli DH5α and S17-1 λpir strains carrying the
plasmid pBt20 were routinely cultured on lysogeny broth (LB) medium,
which was solidified with 1.5% agar when necessary. Gentamicin (Gm) was
used at from 25 to 50 µg/ml for P. aeruginosa and at 10 µg/ml for E. coli.
Carbincillin (CA) was used at 50 µg/ml and 10 µg/ml for E. coli. Nalidixic
acid (NA) at 20 µg/ml for E. coli.
Saccharomyces cerevisiae strain InvSc1, used for plasmid construction
via in vivo homologous recombination, was grown with yeast extract-
peptone-dextrose (1% Bacto yeast extract, 2% Bacto peptone, and 2%
dextrose) (Shanks etal., 2006). Selections with InvSc1 were performed using
synthetic defined agar-uracil.
2.2.13. Determination of MIC of Imipenem for strains
2.2.13. 1. The E-test method
All strains of P. aeruginosa were subjected to determine minimum
inhibitory concentrations (MICs) against Imipenem. The E-test method has
been used for MIC determination according to the manufacturer’s
instructions. In brief, bacterial suspensions were prepared from fresh
colonies and the concentration has been adjusted to 1.5 to 2 ×108 cfu/ml
McFarland turbidity. Each strain was inoculated by streaking the bacteria all
over a Mueller Hinton agar (MHA) plate. An E-test strip of Imipenem (from
0.002 to 32 μg/ml) was placed on the surface of cultured media, after
overnight incubation at 37°C, MIC has been determined and the sensitive,
intermediate and resistant phenotypes were tested according to Clinical and

Laboratory Standards Institute (CLSI, 2013). MIC was also determined by
microdilution on microtiter plates to check the similarity of results.
Escherichia coli ATCC 25922 was used as quality control strains for E test.
2.2.13. 2. Microdilution method
The minimum inhibitory concentration (MIC) of P. aeruginosa strains
was determined, using the twofold serial microdilution method (Wiegand et
al., 2007) and using Muller Hinton broth (MHB). A serial dilutions ranging
from 0.5-32 μg/ml for Imipenem were prepared in microtiter plate wells.
Bacterial culture of 1.5 to 2 ×108 cell/ml was prepared using MHB and 1.5
to 2 ×108 cfu/ml McFarland’s standard tube, 10 μl of bacterial culture was
added to each microtiter plate well. The MIC values were taken as the
lowest concentration of the antibiotic in the well that showed no turbidity
after 24 hours of incubation at 37°C. The turbidity of the wells in the
microtiter plate was interpreted as visible growth of the microorganisms, P.
aeruginosa ATCC 27853 was used as quality control strains for
microdilution assay.
Microdilution assay was used to confirm the results of E test in regard
to the resistance strains.
2.2.14. Modified Hodge Test (MHT)
Modified Hodge test is a phenotypic method for detection of
carbapenemases (Anderson et al., 2007; Noyal et al., 2009).
0.5 McFarland dilution of the E. coli ATCC 25922 was prepared in 5
ml of saline. 1:10 dilution was prepared by adding 0.5 ml of overnight
culture to 4.5 ml of saline. Thereafter, a lawn of the 1:10 dilution of E. coli
ATCC 25922 was streaked to a Mueller Hinton agar. Afterward, 10 µg

Imipenem susceptibility disc was placed in the center of the test area. In a
straight line, the test organism was streaked from the edge of the disc to the
edge of the plate. Up to four organisms can be tested on the same plate with
one drug, and then the plate was incubated overnight at 37ºC in ambient air
for 16–24 hours. After incubation period, the plate was examined for a
clover leaf-type indentation at the intersection of the test organism and the E.
coli 25922, within the zone of inhibition of the carbapenem susceptibility
disk. MHT Positive test has a clover leaf-like indentation of the E. coli
25922 growing along the test organism growth streak within the disk
diffusion zone. MHT Negative test has no growth of the E. coli 25922 along
the test organism growth streak within the disc diffusion.
2.2.15. Biofilm formation assay
Biofilm formation in 96-well microtiter plates was assayed and
quantified as previously described by Caiazza and O’Toole (2004). All
biofilm assays were performed using M63 minimal medium supplemented
with glucose, MgSO4, and CAA.
Strains were grown overnight (~16 hours) in LB broth at 37°C, the 96-
well plate(s) were prepared for the assay (label strains). Each strain
suspension was diluted (1:50) into an aliquot of the Biofilm media and
mixed well by swirling and pipetting up and down. The wells were
inoculated (at least 4 wells per strain) of the 96-well plate (100 µl/well) from
the strain mixture using a multi-channel pipet. The 96-well plate was
covered with a lid and placed in the warm room (37°C) for up to 24 hours.
After the incubation period, the wells were shaken out to remove the
unattached bacteria and then were rinsed twice in the water container and
shaken out the excess water by tapping plate on paper towels. Subsequently,

125 µl of Crystal violate (CV) stain (at 0.1% concentration) was added to
each well and the control un-inoculated well, and then the plate was let sit to
10-15 min. The excess stain was shaken out into the waste container and the
plate was rinsed twice. The plate was taped on paper towels to dry.
In order to quantify the biofilms, 125 µl of 30% glacial acetic acid was
added to biofilm wells and to the control well (no bacterial cells, just stained
with crystal violet). This step was done with multichannel pipette. Plates
were allowed to sit at room temp for at least 15 minutes. Later, the
solubilized crystal violet was pipetted up and down gently to evenly mix just
prior to transferring 100 µl from each well to a 96-well flat-bottomed plate
(non-sterile). Finally, the plate was read by a spectrophotometer at an
absorbance of 550 nm.
2.2.16. Extraction of genomic DNA
Extraction of Genomic DNA was accomplished using the Gentra
Puregen Yeast/Bacteria Kit. Phusion enzyme was used to amplify oprD gene
to reduce the chance that errors will be introduced during the PCR reaction
(since we are specifically looking for mutations that have arisen by growth
in the presence of imipenem). The purity was checked by Nanodrop at OD
260/280.
2.2.17. Amplification of oprD gene by polymerase chain reaction
technique (PCR)
The PCR technique was employed for amplifying whole oprD gene in
P. aeruginosa strains. Mastermix reaction and reaction conditions are
summarized in table 2-11 and table 2-12, respectively.

Table 2-11: Mastermix reaction for single reaction
Reagents Volume (µl)
5X HF Buffer* 10
dNTP mix (10 mM) 1
DMSO 1.5
For Primer (25 µM) 1
Rev Primer (25 µM) 1 Template Genomic DNA 1 Phusion enzyme (U/ 50 µl) 0.5
Distilled water 34 *High Fidelity (HF) buffer was used that comes with Phusion enzyme kit, the
primers were listed in table 2-6
Table 2-12: The reaction conditions of PCR
Annealing temperature was determined by adding 3°C to lowest Tm of
primer pair, extension times was determined, (1 kb ) was added in each 30
second (when using complex templates like genomic DNA), the primers
were used in oprD amplification are listed in table 2-6.
Stage Temperature (time) Initial denaturation 98°C (1min)
Denaturation 98°C (10sec) 30 cycles Annealing 69°C (30sec)
Extension 72°C (40sec) Final extension 72°C (10min)
Hold 4°C

2.2.18. Sequencing
After PCR, gel was run if the band of expected size was seen, then
Qiagen PCR clean-up kit was used to clean up the PCR product for the
reactions of sequencing. The mix of reaction summarized in table 2-13.
Table 2-13: The sequence reaction mix
Reagent Volume(µl) PCR product 1 µl
Primer (100 µM) 1 µl Distelled water 18 µl
total volume 20 µl The primers were listed in table 2-6
After reaction, samples were sent to the Core Facility in Dartmouth
College. Sequence analysis was performed according to www.ncbi.org
using, Gene construction kit software and finch TV. The results DNA
sequences were aligned to the PAO1 oprD genomic sequence using the
NCBI BLAST.
2.2.19. Quantitative reverse transcription-PCR (qRT-PCR) (Kuchma et
al., 2005)
The qRT-PCR was used for measuring the oprD expression in clinical
strains
2.2.19.1. Bacterial Harvest
Bacterial suspension were diluted from LB-grown overnight cultures
1:100 into M63 minimal medium (2.2.2.5) and grown for 8 hours to an

optical density of 0.4 at 600 nm (OD600) (Normalized to a volume of 10 ml).
Samples were span down in a centrifuge (37°C at 5000 rpm for 2 minutes).
Thereafter, supernatant was discarded and the tubes were placed in a dry ice
ethanol bath for 10 minutes. The tubes were stored in -80°C freezer, until
use.
2.2.19.2. RNA extraction and cDNA
RNA preparation by Qiagen Rneasy Kit, RNA was run in 1% gel to
assess purity. RNA was quantified by Nanodrop at OD 260/280. 2 to 3 µg of
RNA was taken to make cDNA using the Invitrogen Superscript III cDNA
Kit. Primers were designed according to http://www.idtdna.com. The 96
well plates were set with appropriate number of strains three replica for each
one. 2 µl of 1000 pmol/µl cDNA (read by Nanodrop) was added for each
well and 82.5 µl of Syper green mix to that tube, negative control was
prepared as well. When loading is completed the pressure sensitive sealing
film was added on the plate. The real time plate was sent to core facility
Quantitative reverse transcription-PCR (qRT-PCR) was performed using an
ABI 7500 Fast System and analyzed using ABI Fast System software
version 1.4.
Expression levels were quantified in picograms of input cDNA using a
standard curve method for absolute quantification, and these values were
normalized to rplU expression. Results shown are based on the average from
two independent experiments with three replicates per sample. The primers
used were listed in table 2-9.
2.2.20. Genetic complementation steps of oprD mutant strain
2.2.20. 1. Digestion of PMQ72

PMQ72 was purified from E. coli and 20 µl PMQ72 was digested with
enzyme Sac1 for overnight at 37ºC. Plasmids for complementation and
overexpression were generated using vectors via homologous recombination
in yeast. PMQ72 vector was used for the complementation of oprD mutant
(shanks et al., 2006).
The poprD complementation constructs was generated by PCR
amplification using the high-fidelity DNA polymerase.
2.2.20. 2. Yeast transformation (Burke et al., 2000)
S. cerevisiae (InvSc1) was grown in YPD overnight. After that, couple
of large colonies were picked up with a sterile toothpick and transferred to 5
ml LB broth and incubated for overnight. Then 0.5 ml from overnight
culture was applied in 1.5 ml microfuge tube and spun down pellet at 10000
rpm for 10 second. To the pellet, 0.5 ml of lazy bones solution, 20 µl of
carrier DNA (salmon sperm DNA), 20 µl PCR product and of 5 µl vector
were mixed and then vortexed hard for 1 minute. The mixture was
incubated overnight to 4 days at room temperature (after 1- 4 days the
plasmid efficiency goes down in the yeast). The mixture was exposed to heat
shock for 10-12 minutes at 42ºC. Cell was pelleted, optimally was washed
with T.E., as PEG inhibits growth, and cultured onto selective media. At the
end of the incubation period (5-7 days), the growth was harvested by
spreader from plate and the suspension was taken for plasmid preparation.
Qiagene kit was used for DNA plasmid prep from yeast. Then amount of
plasmid was transferred into E. coli to amplify the plasmid.
2.2.20. 3. Electroporation (Oldenburg et al., 1997)
A. Preparation of E.coli DH5α

A liter of LB was inoculated with 1/100 of fresh overnight E.coli
DH5α, cells grown with vigorous shaking to OD 600 of 0.5 at 1 hour. The
culture was chilled on ice for 15 to 30 min, and then centrifuged in cold
centrifuge at 4000X g for 15 min. Successively, the pellets was resuspended
in a total of 1litter cold water and centrifuged as before, and then the pellet
was resuspended in 0.5 L cold water and centrifuged. The pellet was
resuspended in 10% glycerol and centrifuged. The pellet was resuspended in
a final volume of 2 to 3 ml in 10% glycerol. The cells were aliquoted and
frozen on dry ice. Finally the cell was storaged at -80ºC; the cells are good
for at least six months.
B. Electroporation of E. coli
The cells were thawed at room temperature and placed on ice, 40 µl of
cells was mixed with 1 µl to 2 µl of DNA in low ionic strength buffer and
put in pre-chilled cuvette. The cells were let to sit on ice for a minute.
Thereafter, the gene pulser apparatus was set at 25 µF and 2.5 KV, the
cuvette was put in the slide and the slid was push into the chamber, the
pulsing was once pulsed (time constant should be 4.5 to 5 sec), 500 µl was
added after pulsing immediately of LB and the cells were resuspended.
Subsequently, the cells was transferred to appendroff tube and incubated at
37ºC for 1 hr, the cells were plated on selective media by spreader.
QIAprep Spin Miniprep kit was used for plasmid preparation from E.
coli and run on agarose gel to confirm the presence of the expected band.
Then the sequence analysis was achieved to ensure there is no mutation in
interesting gene that may happen during PCR cycle.
C. Electroporation of P. aeruginosa

From overnight culture, 1 ml of cell were spun to pellet (maximum
speed, 1min), the supernatant was remove and resuspended in 1 ml of 300
mM sucrose, the cell was spun to pellet, the supernatant was removed and
this wash was repeated in sucrose two more times. After the finally spin, the
supernatant was remove carefully and the cells was resuspended in 80 µl of
300mM sucrose, the( DNA no more than 1.5) was pipetted onto the side of
electroporation cuvette to allow cell mixture to go in to the well of the
cuvette (no bubbles), electroporate using the E. coli setting (the putton was
push for the time constant setting so you can see the puls time), immediately,
0.5 ml of LB was added to recover the cells and transfered to fresh
microfuge tube, the cells was recovered at the 37ºC on the wheel for 1.5 – 2
hours, 10 µl of the mixture plus 50 µl was plated on the appropriate selective
media. After that, the phenotypic restoration was screened.
2.2.21. mariner transposon mutagenesis of the highest biofilm producers;
576 and 214 (Kulasekara et al., 2005)
2.2.21.1. Conjugation and selection of mutants
The donor strain E. coli S-17 l pir (strain carrying the plasmid pBT20 –
carries Mariner transposon) and the recipient P. aeruginosa were grown for
overnight with appropriate antibiotic selection (antibiotic markers were
ampicillin marker on plasmid backbone – ampicillin was used for growth of
E. coli; gentamicin marker on Mariner was used for P. aeruginosa). The
transposon carries an outward-directed Ptac promoter so Tn insertions can
lead to overexpression of downstream genes. 1 ml of each fresh strain
overnight culture was pelleted. Enough samples were included to set up
single plating of each strain as negative controls for the conjugations, i.e.

another 1 ml of each P. aeruginosa and E. coli (that was using for the
conjugation) strain was spun.
Each strain was washed twice by resuspending in 1 ml of fresh LB and
pelleted. Then cells were resuspended in a final volume of 100 µl, the
suspensions of each strain were mixed in a tube and 60 µl of this mixture
was spotted onto 2 sterile LB plates (without antibiotics), additional plates
were plated for conjugation were spots to scale this up if necessary. The
conjugation was allowed to proceed for at least 1 hour at 30°C (up to 3
hours). Afterward, the cells were recovered from plate by scraping up the
pellet with a sterile stick and resuspended in a small volume of LB in a 1.5
ml microfuge tube. The cells were vortexed to break up clumps. The
aliquots (10, 20 and 40 µl) of the conjugation mixture were plated to test for
appropriate yield of colonies onto selective LB agar plates (Gentamicin to
select for Mariner; include nalidixic acid to select against E. coli. The
plating was spreaded evenly over entire plate with a sterile glass spreader;
the conjugation plate was stored at 4°C until results of initial plating are
evaluated.
Gentamicin was used to select against P. aeruginosa 25 µg/ml (2.5X),
Nalidixic Acid was used to select against the E. coli strain 20 µg/ml (1X).
The plates were incubated at 37°C overnight, then additional time at room
temperature.
2.2.21.2. Storing/screening the library
The library plates (96-well dishes) were filled with sterile LB plus
antibiotic (Gm), 100 µl per well was added for each well except control
wells for (576 and 214) comparison control wells (e.g. A1, A2) were filled
with LB alone. Colonies were picked into wells using sterile toothpicks, with

taking care to maintain a consistent inoculums, the control strains were pick
into appropriate wells, the plates were allowed to grow at 37°C 12-18 hours.
When substantial uniform growth has been achieved, plates can be screened
right away or stored for later screening.
2.2.21.3. Screening for biofilm deficient
Frog (multipronged device) was used to transfer samples from 96-
well sterile plate into wells of 96-well biofilm plate filled with appropriate
media and incubate for desired time at 37ºC.
2.2.21.4. To store the library
100 µl of sterile 20% glycerol was added to the library (to give final of
10%) to all of the wells, and mixed carefully. Thereafter, the plates were
frozen at -80°C with 2 layers of protection (e.g. parafilm or aluminum foil,
Ziploc bag).
2.2.21.5. Mapping Mariner transposons by Arbitrary PCR (ARB PCR)
Mapping of Mariner transposon was done according to Caetano-
Anolles, (1993) and O’Toole and Kolter (1998), when mutants strains were
isolated and confirmed, the arbitrary PCR was used to identify the insertion
point of the transposon in gene.
In arbitrary PCR there are 2 rounds. Before round1, 5 µl of the
genomic template was extracted by the Gentra kit (2.1.4) and then was
digested with 5 µl of EcoR1 enzyme for overnight at 37ºC. Then use 5µl of
that as template for round 1, the mastermix reaction and ARB PCR program
condition for round 1 are summarized in table 2-14 and table 2-15,
respectively.

Table 2-14: The mastermix reaction for ARB PCR round 1
Table 2-15: PCR1 program for round 1 – The annealing temperature was very low.
*-1 °C for each cycle
The PCR product was cleaned up using Qiagen kit and eluted with 50
µl distilled water.
In regard to round 2, the mastermix reaction and ARB PCR program
conditions are summarized in table 2-16 and table 2-17, respectively.
Reagent Volume P235 (1µg/µl) 1µl P236 (1µg/µl) 1 µl P237 (1µg/µl) 1 µl P238 (1µg/µl) 1 µl Taq Buf. 10X 5 µl dNTP mix (10mM each) 2 µl MgCl2 (50mM) 0.5 µl DMSO 2.5 µl H2O 30 µl Taq DNA polymerase (U/ 50 µl) 1 µl Digested gDNA 5 µl Total Volume 50µl
Stage Temperature (time) Temperature (time) Initial denaturation 94°C (2min) 94°C (2min)
Denaturation 94°C (30sec) 5 cycles
94°C (30sec) 25 cycles Annealing 42°C (30sec)* 65°C (30sec)
Extension 72°C (3min) 72°C (3min) Hold None 4°C (∞) 1cycle
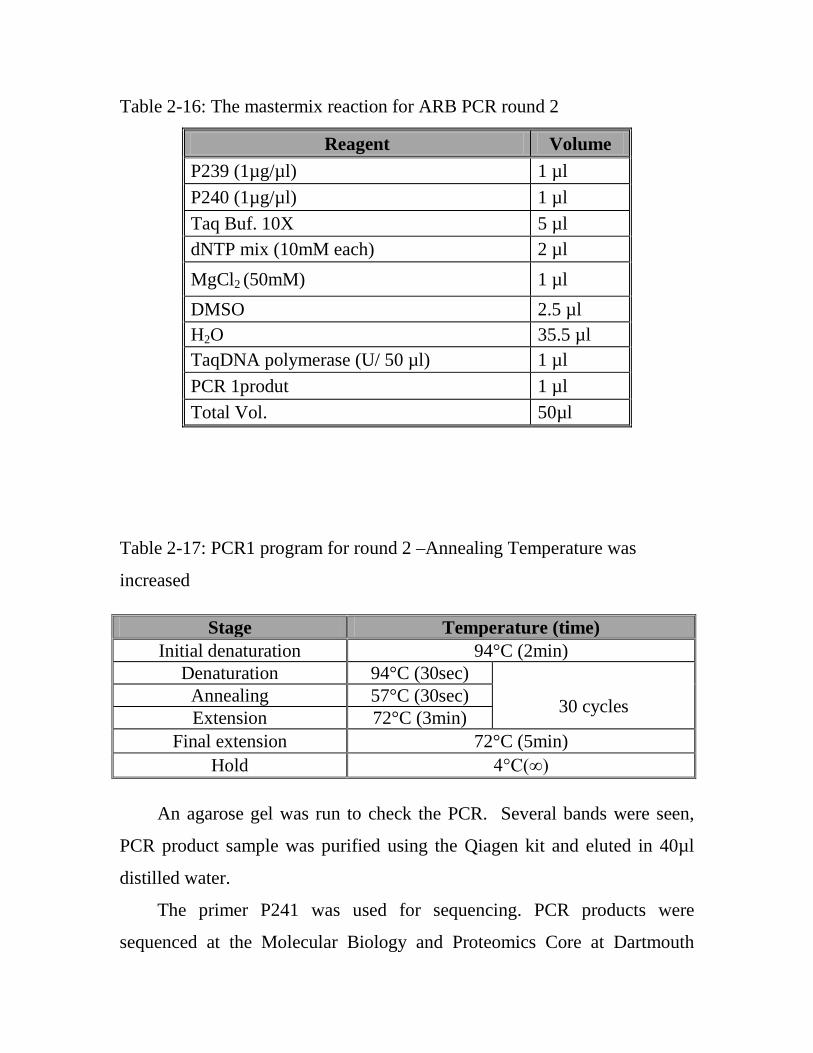
Table 2-16: The mastermix reaction for ARB PCR round 2
Table 2-17: PCR1 program for round 2 –Annealing Temperature was
increased
An agarose gel was run to check the PCR. Several bands were seen,
PCR product sample was purified using the Qiagen kit and eluted in 40µl
distilled water.
The primer P241 was used for sequencing. PCR products were
sequenced at the Molecular Biology and Proteomics Core at Dartmouth
Reagent Volume P239 (1µg/µl) 1 µl P240 (1µg/µl) 1 µl Taq Buf. 10X 5 µl dNTP mix (10mM each) 2 µl MgCl2 (50mM) 1 µl DMSO 2.5 µl H2O 35.5 µl TaqDNA polymerase (U/ 50 µl) 1 µl PCR 1produt 1 µl Total Vol. 50µl
Stage Temperature (time) Initial denaturation 94°C (2min)
Denaturation 94°C (30sec) 30 cycles Annealing 57°C (30sec)
Extension 72°C (3min) Final extension 72°C (5min)
Hold 4°C(∞)

College. The resulting DNA sequences were aligned to genomic sequence
using the NCBI BLAST program, the primers was used in ARB PCR are
listed in Table (2-7)
2.2.22. Construction of mutant strains and plasmids
PMQ70 was purified from E. coli and digest 20 µl with 4 µl of enzyme
Sac1 for overnight in 37 ºC. Then use 5 µl of was used as template. PMQ70
vector was used for complementation of PilY1 mutant (shanks et al, 2006),
pPilY1 complementation construct was generated by PCR amplification of
pilY1 gene using the high-fidelity Phusion DNA polymerase. The reaction
conditions for the amplification pilY1 gene are sumerized in table 2-18.
Table 2-18: The condition reaction for amplification pilY1 gene
The primers that used for the pilY1 amplification are listed in table 2-
18.
2.2.23. Twitching assays
Twitch motility plates consisted of M63 medium supplemented with
MgSO4, 20% glucose and 20% CAA and solidified with 1.5% agar. Twitch
Stage Temperature (time) Initial denaturation 98°C (30min)
Denaturation 98°C (10sec) 30 cycles Annealing 72°C (20sec)
Extension 72°C (2min) Final extension 72°C (10min)
Hold 4°C(∞)

assays were performed as previously reported (Whitchurch, 1990; O’Toole
& Kolter 1998). Briefly, cells were stab inoculated with a toothpick through
a thin (approximately 3 mm) LB agar layer to the bottom of the Petri dish.
After overnight growth at 37ºC, the zone of twitching motility between the
agar and Petri dish interface was visualized by staining with crystal violet.
2.2.24. Swarming motility
Swarm motility plates were comprised of M8 medium supplemented
with MgSO4, 20% glucose, and 20% CAA and solidified with 0.5% agar.
Swarming assays were performed as previously reported (Köhler et al.,
2000). Agar was allowed to cool slightly and then was poured thick plate
approximately 25 ml/ plate; these plates were set at room temperature for
few hours. For testing individual strains, 2 µl of grown overnight cultures
was inoculated onto the surface of the swarm plates and incubated for 16 h
at 37°C. Wild type was included as apositive control.
2.2.25. Estimation of polysaccharide extracts
We quantified Psl production via ELISA with some modifications from
an existing protocol (Honko et al., 2006). Flat-bottom 96-well MaxiSorp
plates (Nunc) were coated in triplicate overnight at 4°C with 100 µl /well.
Plate was washed thrice with PBS plus 0.1% Tween-20 for 3 minutes each
and tapped to mix block plate with 300µL/well of PBS plus 1% Bovine
serum albumin (BSA), 60 min at 4°C.
Primary antibody diluted in PBS plus 0.1% BSA was added for 1-2 hr
at room temp (or 4°C). Thereafter, Horseradish peroxidase (HRP)-conjugate
secondary antibody diluted in PBS plus 0.1% BSA was added. 1:5,000
dilutions from reconstituted stock were made and finally the plate was

incubated for 1 hr at room temp or 4°C. Subsequently, plate was washed and
100 µl/well TMB SureBlue development reagent to half of the plate at a
time was added as well. 100 µl /well 0.2 N H2SO4 was added to terminate
development in the hood. Measurement was achieved at 450 nm.
3.3. Statistical analysis
The data were analyzed by GraphPad Prism software was originally
designed for experimental biologists in medical schools and drug companies,
which was applied for the comparison among different values when the
enumerative data are qualitative using ANOVA One way Analysis
(http://www.graphpad.com). P<0.05 was considered as significant
difference.

chapter three
results and discussion

3. Results and Discussion
3.1. Imipenem susceptibility and carbapenemase detection
Among fifty eight P. aeruginosa strains evaluated with E test, forty
seven (81.03%) strains were susceptible, two (3.4٥%) strains were
intermediate, and nine (15.5۲%) strains were resistant to imipenem.
All these 9 strains were previously isolated from patients with cystic
fibrosis. No carbapenemase activity was found among the intermediate and
resistant strains, as it is confirmed by the Hodge test. A positive strain
develop a ‘cloverleaf shaped’ zone of inhibition due to carbapenemase
production. The strains showed negative results (undistorted zone of
inhibition) as it is depicted in figure 3-1. These results suggested that the
imipenem resistance is due to oprD malfunction rather than carbapenemase.
Carbapenems are not prone to inactivation by extended spectrum ß-
lactamases and penetrate across the outer membrane of P. aeruginosa
through a porin OprD, which allows selective penetration of basic amino
acids, small peptides containing these amino acids, and carbapenems, their
structural analogs. Prolonged treatment of P. aeruginosa-infected patients
with imipenem has often allowed for the emergence of imipenem-resistant
mutants. These resistant strains have either lost OprD or have strongly
reduced OprD levels due to a nfxC type of quinolone-resistant mutation
(mexT) which represses oprD expression and activates the mexEF-oprN
multidrug efflux operon (Yoneyama and Nakae, 1993). Ochs et al. (1999)
reported the possible mechanisms by which resistance to imipenem emerged
in 17 imipenem-resistant P. aeruginosa clinical strains, related to the loss of
OprD was the predominant reason of imipenem resistance, OprD loss was
caused by a chromosomal oprD mutation.

In the present work, five imipenem resistant variants (SMC631C, F, H,
J, K) were originated from SMC631strain that grew as discrete colonies in
the zone of inhibition at 32 µg/ml while performing an MIC assay with an
imipenem E Test strip, upon retesting in a microdilution assay. These
mutants were confirmed to be resistant to imipenem (MIC > 8 µg/l).
Figure 3-1: Modified Hodge test, the negative strain shows an undistorted zone of
inhibition by p. aeruginosa.
3.2. Assessing biofilm formation and imipenem resistance in clinical
strains
To explore the relationship between biofilm formation and imipenem
resistance, these phenotypes were assessed for an imipenem sensitive
clinical strain SMC631, a number of imipenem resistant variants of
SMC631, and several additional P. aeruginosa clinical strains. As shown in
table 3-1, all these variants were tested for their ability to form a biofilm and
compared to the parent SMC631.

Table 3-1: Biofilm and imipenem resistance phenotypes of clinical
strains
Strains Imipenem MIC (µg/ml)a Biofilm (A550 ± SDb)
SMC4973 0.5 0.96 ± 0.0509
SMC576 1 0.84 ± 0. 2305
SMC4972 1 0.77 ± 0.0257
SMC631 1.5 0.19 ± 0.0226
PAO1 4 0.88 ± 0.0134
SMC214 4 0.70 ± 0.1634
SMC4979 4 0.53 ± 0.0626
SMC631C 16 0.12 ± 0.0153
SMC631F 16 0.06 ± 0.0161
SMC631H 16 0.10 ± 0.0188
SMC631J 16 0.11 ± 0.0151
SMC5810 23 0.02 ± 0.0121
SMC631K 24 0.12 ± 0.0344
SMC5811 24 0.01 ± 0.0325
SMC4974 32 0.04 ± 0.0051 aSensitive (≤2 µg/ml), intermediate (4 =µg/ml) or resistant (≥8 µg/ml). bLSD= standard deviation. LSD= 0.13, P= 1.755 *10-18.
Markedly, an inverse relationship (r= -0.83) was found between biofilm
formation and imipenem resistance in P. aeruginosa. The imipenem resistant
strains SMC4974, SMC5810, and SMC5811 showed significantly low (P <
0.01) biofilm formation compared to the imipenem sensitive or intermediate
P. aeruginosa clinical strains SMC576, SMC214, SMC4972, SMC4973, and
SMC4979, which all formed relatively robust biofilms (Table 3-1).

Noticeably, table 3-1 demonstrates nonuniformity in clinical strains
biofilm results. Even though the imipenem sensitive SMC631 has low
biofilm value (0.19 ± 0.0226) in comparison to its spouses of MIC category
≤ 4 µg/ml (SMC576, SMC214, SMC4972, SMC4973, and SMC4979), all its
imipenem resistance variants (SMC631C, SMC631H, SMC631F and
SMC631J) formed low biofilm by comparison to the parent SMC631. These
data confirmed the possibility of an inverse relationship between biofilm
formation and P. aeruginosa imipenem resistance.
3.3. Analysis of Sequencing of oprD gene
Loss of oprD is one of the most important mechanisms of resistance to
imipenem in P. aeruginosa. Multiple studies have evaluated the importance
of oprD mutation in clinical strains of P. aeruginosa resistant to
carbapenems.
Always authors demonstrated a correlation between the levels of
expression of oprD and the degrees of susceptibility to imipenem (Dib et al.,
1995; Ocampo-Sosa et al., 2012; Lee et al., 2012).
The relationship between oprD mutation type and imipenem
susceptibility profiles in imipenem-susceptible ,intermediate, and resistance
clinical strains of P. aeruginosa was investigated. The impact of oprD
mutations in 19 clinical strains, 11 were metallo-ß-lactamase- negative P.
aeruginosa and 8 sensitive for imipenem was analyzed (Table 3-2).
Selection of the strains was based on their imipenem susceptibility
profiles, including organisms with a broad range of susceptibility:
susceptible (MICs ≤ 2 µg/ml), intermediately susceptible (MIC = 4 µg/ml)
or resistance (MICs ≥ 8 µg/ml) to imipenem. The evaluation of the

mutations found in oprD of clinical strains was compared with the analysis
of the oprD of PAO1.
Sequencing of the entire oprD gene regions (Appendix A) permitted
analysis of the mutation in oprD gene in such strains. Most of the strains
presented point mutations consisting of single nucleotide insertion or
nucleotide deletion in positions −167 and −177, respectively (relative to the
ATG start codon). Such mutations were observed in both susceptible and
resistant strains; therefore, it considered the involvement in decreased
imipenem susceptibility not relevant. It is likely there is another passage for
the imipenem or this mutation does not affect the OprD function as a porin.
Wolter et al. (2009) suggested that carbapenems can enter through an
alternative route. These data highlight the complex interactions of resistance
mechanisms in P. aeruginosa and their roles in drug susceptibility.
Patterns of mutations found in oprD (Tables 3-2) were assembled into
three groups: group I showed full length comprising PAO1, SMC 4963,
SMC 4972, SMC 4986 and clinical strains (SMC 631, SMC 631 K, SMC
4974). Witch were included several oprD allelic variants, due to amino acid
substitutions. Group II substitution of a nucleotide in the oprD gene resulted
in a premature termination of translation whereas the last pattern group III
comprises those oprD genes harboring a frameshift mutation due to
nucleotide insertions or deletions. Group II and group III called deficient
types, as their mutations resulted in loss of oprD porin (Ocampo-Sosa et al.,
2012).
Following the usual pattern, most of the oprD full-length type strains
were susceptible strains. Several oprD allelic variants compared to PAO1
oprD were found (Table 3- 2). The most frequent amino acid substitutions
were T103S, K115T, and F170L, found in the parent SMC631 susceptible

strains and the five resistance derivatives of 631 (SMC631C, F, H, J, K) and
SMC4974 resistance strains, oprD variants showing these amino acid
substitutions, were described before in clinical strains of P. aeruginosa (El
Amin et al., 2005; Gutiérrez et al., 2007; Rodríguez-Martínez et al., 2009;
Ocampo-Sosa et al., 2012). Other frequently found amino acid substitutions
are shown in (Table 3-2); some of them were previously described in strains
with different susceptibility profiles to imipenem (El Amin et al., 2005).
The OprD “deficient types” included one strain, SMC631F, showing an
earlier termination of translation due to premature stop codons.
Most strains have frameshift mutations caused by nucleotide insertions
or deletions in the oprD structural gene. The nt 152Gnt 153 mutation in SMC631
H, SMC 631J and SMC631C in variants strains from clinical strain
SMC631.
The remaining mutations (D 43 E, nt 167Gnt 168, nt 177ΔC) were found in
susceptible, intermediately susceptible and resistance (Table 3-2).
The majority of mutations found within this oprD type consisted of
single nucleotide insertions at different positions in the oprD gene. The
insertion of a G at 167 the most frequent mutation, observed in susceptible
and resistance strains and intermediately susceptible. Most of the strains
harboring these mutations remained susceptible to imipenem (Table 3-2).
3.4. Analysis of oprD expression
The relationship between biofilm formation, imipenem resistance, and
oprD expression (Appendix B) was assessed in some imipenem resistant
clinical strains (SMC631C, SMC631F, SMC631J, SMC631H, SMC631K
and SMC 4974). As it is illustrated in table 3-3, all resistant strains showed
low oprD expression and low biofilm values as comparison with the

sensitive strains. Remarkably, the correlation coefficient (r=0.5) appreciates
the possibility of a direct relationship between oprD expression and biofilm
formation.
Table 3-2: Different oprD mutation types found in clinical strains of P.
aeruginosa shown different Imipenem susceptibility.
Name of strain Imipenem MIC (µg/ml)*
mutation(s)
Type of mutation
PAO1 2 None Full-length Wild type SMC 631 1.5 T 103 S, K 115 T, F 170 L
Amino acid substitutions SMC 631 K 24 T103 S, K 115 T, F 170 L
SMC 4974 32 T 103 S, K 115 T, F 170 L
SMC 631 F 16 T103 S, K 115 T, F 170 L, W 417 opa Amino acid substitutions Premature stop codon
SMC 631 H 16 T103 S, K 115 T, nt 152Gnt 153 Amino acid substitutions, Frame shift mutations due to
insertions SMC 631 J 16 T 103 S, K 115 T, nt 152Gnt 53
SMC 631 C 16 T 103 S, K 115 T, nt 152Gnt 153
SMC 5810
23
nt 167Gnt 168 , S 56 K, G 57 V, nt 177ΔC, I 210 V, E 230 K, S 240 T,
N 262 T, A 267 S, A 280 G, K 295 Q , Q 300 E, R 310 G, V 359 L Amino acid substitutions,
Frame shift mutations due to insertions and deletions
SMC 5811 24
D 43 N, nt 167Gnt 168, S 56 K, G 57 V, nt 177ΔC, nt 567ΔC, E 102 Q,
I 210 V, E 230 K, S 240 T, N 262 T, A 267 S, A 280 G, K 295 Q, Q 300 E, R
310 G , V 359 L
SMC 4980 32 nt 167Gnt 168, nt 177ΔC, nt861ΔG Frame shift mutations due to insertions and deletions
SMC 4963 1 None Full-length type SMC 4972 1 None
SMC 4986 1 None SMC 4970 0.5 D 43 E, nt 167Gnt 168, nt 177ΔC
Amino acid substitutions, Frame shift mutations due to
insertions and deletions,
SMC 4973 0.5 D 43 E, nt 167Gnt 168, nt 177ΔC SMC 4966 0.5 D 43 E, nt 167Gnt 168, nt 177ΔC SMC 4964 0.75 D 43 E, nt 167Gnt 168, nt 177ΔC SMC 4979 4 D 43 E, nt 167Gnt 168, nt 177ΔC

* Sensitive (≤ 2 µg/ml), intermediate (4 µg/ml) or resistant (≥ 8 µg/ml) The imipenem sensitive or intermediate P. aeruginosa clinical strains
(SMC576, SMC4972, SMC4973, and SMC4979) formed relatively robust
biofilms; however, the oprD expression in SMC576 and SMC4972 was
high. Such finding is compatible with our theory (there is an inverse
correlation between oprD expression in respect to imipenem resistance and
biofilm formation). Decrease in oprD expression that appeared to be relevant
was also detected in imipenem-susceptible and intermediate susceptible in
two strains SMC4973, SMC4979 respectively, indicated these low
expressions of oprD related to other mechanism. Less commonly there is a
mutation or deletions within mexT convert inactive MexT into an active
form. Somehow mutations occur in mexS located upstream of mexT, lead to
accumulate various metabolites that serve as effector molecules for MexT,
which, in turn or in both cases, the expression of mexEF-oprN occurs at high
level, alongside with a decline in the expression of oprD which is inadequate
to elaborate quantities of OprD in the outer membrane sufficient for normal
cellular function (Fukuda et al., 1995; Köhler et al., 1997).
Wolter et al. (2009) also showed down-regulation in the production of
the carbapenem channel OprD despite carbapenem hypersusceptibility.
These strains had decreased expression of the mexAB–oprM pump involved
with intrinsic antibiotic resistance but overexpressed the mexCD–oprJ and
mexEF–oprN efflux systems normally associated with acquired resistance.
Once again this might means there are other routes for carbapenems
entrance.
It can be concluded that oprD inactivating mutations in clinical strains
of P. aeruginosa are not restricted only to imipenem-resistant strains but are

also found in strains with imipenem MICs ≤ 2 µg/ml during sequence
analysis, and from oprD expression experiment we appreciated may be there
is a link between oprD gene and biofilm formation.
Table 3-3: correlation between oprD expression and biofilm
formation of P. aeruginosa strains.
Imipenem susceptibility Name of strains oprD expression values
± SDb Biofilm values (A550 ±
SD)
Resistance
SMC631 C 0.060 ± 0.034 0.12 ± 0.0153
SMC 631F 0.136 ± 0.007 0.06 ± 0.0161 SMC 631J 0.066 ± 0.003 0.10 ± 0.0188 SMC 631H 0.063 ± 0.028 0.11 ± 0.0151 SMC 631K 0.101 ± 0.033 0.12 ± 0.0344 SMC 4974 0.015 ± 0.004 0.04 ± 0.0051
Sensitive SMC 4972 0.202 ± 0.094 0.77 ± 0.0257
SMC 4973 0.072 ± 0.007 0.96 ± 0.0509 SMC 576 1.231 ± 0.227 0.84 ± 0.2305
Intermediate SMC 4979 0.079 ± 0.014 0.53 ± 0.0626
aSensitive (≤ 2 µg/ml), intermediate (4 µg/ml) or resistant (≥ 8 µg/ml). bSD= standard deviation.
There is little literature available that correlates mechanisms of
planktonic antimicrobial resistance and biofilm production. Dheepa et al.
(2011) indicated the resistance to antibiotics such as ceftazidime, cefepime
and pipercillin in Acinetobacter baumannii was comparatively higher among
biofilm producers than non-biofilm producers. Similarly, Rao et al. (2008)
and Ibrahim et al. (2012) investigated clinical strains of A. baumannii, that
had a significant of biofilm associated with multiple drug resistance. These

data indicate the possibility that there may be an under-appreciated link
between planktonic resistance mechanisms and biofilm formation.
3.5. OprD participates in biofilm formation.
In the present study the critical role of OprD in imipenem resistance
well-established, the defected in the oprD gene that may display an altered
biofilm phenotype using the 96 well microtiter assay. To test this hypothesis,
the ability of an oprD transposon mutant was investigated for biofilm and
compared to the PAO1 strain. Results revealed that oprD mutant showed a
significant reduction in biofilm formation compared to P. aeruginosa PAO1
(Figure 3-2A). The biofilm formation defect of the oprD mutant was
complemented by introducing a wild-type copy of the oprD gene on
plasmid, but the vector control (pMQ72) was not able to rescue this defect
(Figure 3-2A).
To confirm the role of oprD mutant with imipenem resistance, we
performed an E-test assay on the strains described in figure 3-2A. The oprD
transposon mutant and the mutant carrying the vector control (pMQ72)
showed significantly higher resistance to imipenem compared to the parental
P. aeruginosa PAO1 and the oprD mutant complemented with a plasmid
carrying the wild-type copy of this gene (Figure 3-2B).
To evaluate whether these phenotypes might be observed in a clinical
strain as well as the PAO1 laboratory strain, we investigated the biofilm
formation and imipenem resistance phenotypes of a clinical strain of P.
aeruginosa isolated from the sputum of a cystic fibrosis patient. Non-mucoid
strain, designated SMC631, could form a biofilm in the 96 well microtiter
plate and was sensitive to imipenem (Table 3-1).
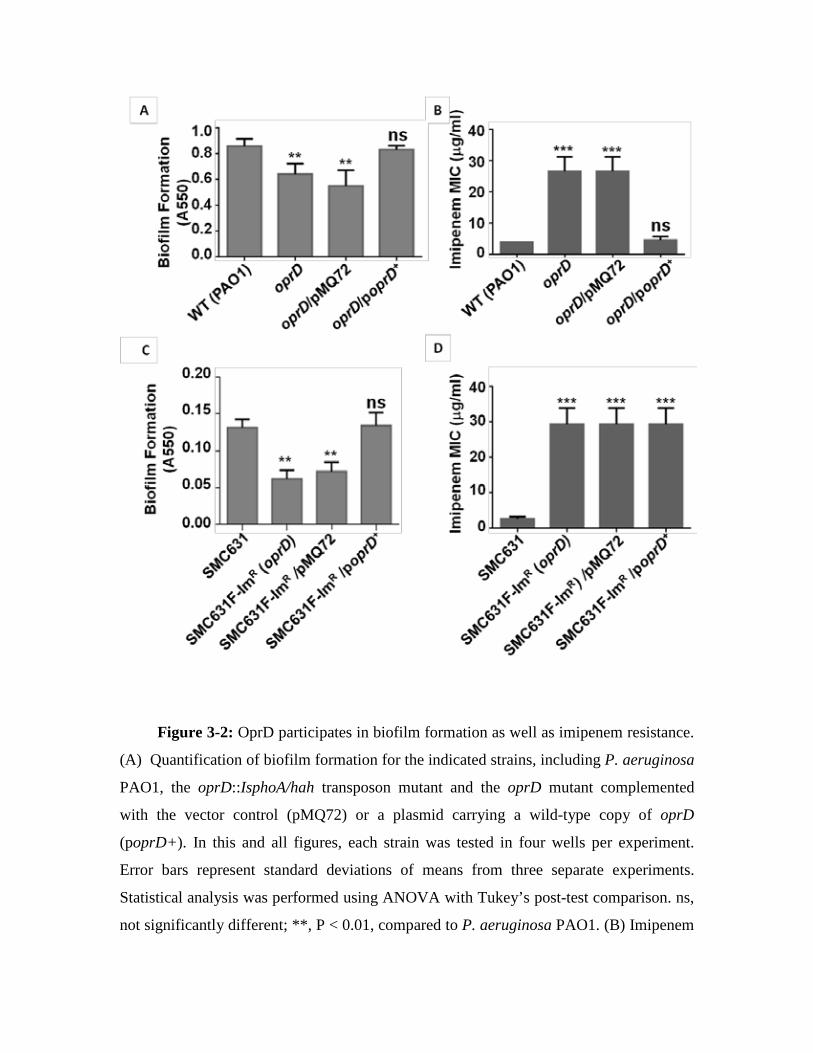
Figure 3-2: OprD participates in biofilm formation as well as imipenem resistance.
(A) Quantification of biofilm formation for the indicated strains, including P. aeruginosa
PAO1, the oprD::IsphoA/hah transposon mutant and the oprD mutant complemented
with the vector control (pMQ72) or a plasmid carrying a wild-type copy of oprD
(poprD+). In this and all figures, each strain was tested in four wells per experiment.
Error bars represent standard deviations of means from three separate experiments.
Statistical analysis was performed using ANOVA with Tukey’s post-test comparison. ns,
not significantly different; **, P < 0.01, compared to P. aeruginosa PAO1. (B) Imipenem

susceptibility was investigated by Etest strips for the same strains described in panel A.
Error bars represent standard deviations of averages from three independent experiments.
Statistical analysis was performed using ANOVA with Tukey’s post-test comparison. ns,
not significantly different; ***, P<0.001, compared to P. aeruginosa PAO1. (C)
Quantification of biofilm formation for the indicated strains, as described in panel A. The
strains tested are the P. aeruginosa imipenem-sensitive clinical strain SMC631, its
imipenem-resistant derivative SMC631F-ImR (oprD), which carries a premature stop
codon mutation in the oprD gene, SMC631F-ImR carrying the vector control (pMQ72)
and SMC631F-ImR carrying a wild-type copy of oprD (poprD+ ). Error bars represent
520 standard deviations of the averages of three experiments with four replicates per
experiment. Data were analyzed by ANOVA with Tukey’s post-test comparison. ns, not
significantly different; **, P < 0.01, compared to SMC631. (D) Imipenem susceptibility
was investigated by Etest strips for the same strains described in panel C, as outlined for
the studies performed in panel B. Error bars represent standard deviations of averages
from three independent experiments with four wells per experiment. Data were analyzed
by ANOVA with Tukey’s post-test comparison. ***,P<0.001, compared to SMC631.
Several imipenem resistant (ImR) variants of the SMC631 strain were
isolated, sequencing of the oprD gene in one of these resistant strains (631F-
ImR), mutation in the oprD gene of the clinical strain SMC631 reduced
biofilm formation (Table 3-1), and this phenotype could be complemented
by introducing a plasmid carrying the wild-type oprD gene of strain P.
aeruginosa PAO1, but not the corresponding vector control (Figure 3-2C).
Interestingly, the imipenem-resistant variant of SMC631 could not be
complemented for its antibiotic resistance phenotype by introduction of a
plasmid carrying the wild-type oprD gene (Figure 3-2D), indicating either
the possibility of a second mutation in this strain, or alternatively, that
providing the oprD gene in multicopy in this strain cannot rescue imipenem
sensitivity.

Taken together, these data indicate that loss of OprD function in a lab
strain and clinical strain results in a reduction in biofilm formation compared
to the parental strain.
3.6. Genes required for biofilm formation in clinical strains are
conserved
To find out the reciprocal relationship between biofilm formation and
imipenem resistance, and to understand whether there might be a
mechanistic link between these processes beyond the role described for
OprD above, the factors that required for biofilm formation in two clinical
strains were identified. These clinical strains of P. aeruginosa (SMC576 and
SMC214) were isolated from the sputum of CF patients, displayed robust
biofilm formation, were imipenem sensitive (MIC ≤ 2 µg/ml) and
intermediate (MIC = 4 µg/ml), respectively, and could be effectively
mutagenized via Mariner transposon mutagenesis. A number of the other
clinical strains were also tested, but they either did not form reproducible
biofilms in the 96 well plate assay in the large-scale screening conditions (4
strains) or the strains showed high level gentamicin resistance, which is the
selectable marker on the Mariner transposon (3 strains). This study had
focused on strains SMC576 and SMC214.
We screened in approximately 1,500 Mariner transposon mutants in the
SMC576 strain for reduction biofilm formation using the microtiter plate
assay. Accordingly, several biofilm formation-defective strains were
isolated and these mutations were mapped (Appendix C) to seven different
genes, with several of these genes being hit more than once. Mutations
mapped to the pilY1, pilW, algC, pslI, cupA2, pilA and pilO genes, all of
which have documented roles in biofilm formation (Friedman and Kolter,

2004a; Li et al., 2007; Kuchma et al., 2012; Zegans et al., 2012 ). We further
characterized the pilY1, pilW, algC and pslI mutants in the studies below
(Figure 3-3A,B).
As expected based on the previous studies (Kuchma et al., 2012), the
single pilW::Mar19 and five independent pilY::Mar mutants isolated
showed increase swarming motility and loss of twitching motility, in
addition to a significant reduction in biofilm formation compared with the
parental strain, SMC576 (Figure 3-3B,C). Additionally, we identified as
biofilm-defective strain with a mutation in algC, this gene contributes in the
biosynthesis of LPS (Goldberg et al., 1993; Olvera et al., 1999) and alginate,
as well as pslI – both of them required for the synthesis of the Psl
polysaccharide (Davies et al., 1993; Davies & Geesey, 1995; Friedman and
Kolter, 2004b; Zegans et al., 2012).
To confirm a role of the pilY1 gene in biofilm formation by strain
SMC576, we performed complementation studies. Mutating pilY1 gene in
the SMC576 background resulted increase swarming phenotype, which
could be complemented by the introduction of a plasmid carrying a wild-
type copy of the pilY1 gene, but not by the vector control (pMQ70, Figure 3-
3D). Similarly, the twitching (Figure 3-3D) and biofilm formation (Figure
3-3D,E) defect of the pilY1 mutant could be complemented by the
introduction of a plasmid carrying a wild-type copy of the pilY1 gene, but
not by the vector control (pMQ70).

Figure 3-3: Identification of biofilm-deficient mutants in the clinical strain
SMC576. (A) Schematic diagram of the pilY1, pilX and pilW genomic loci of P.
aeruginosa PAO1. (www.pseudomonas.com). The transposon insertion sites are
indicated by inverted triangles above the genes. The dark gray triangles indicate

insertions in strain SMC576 and the light gray triangles indicate insertions in strain
SMC214. The numbers above each inverted triangle indicate the nucleotide after which
the transposon is inserted. (B) Representative swarming (top), twitching (middle) and
biofilm (bottom) phenotypes of SMC576 and selected transposon mutants. (C)
Quantification of crystal violet stained biofilms for the indicated strains. Error bars
represent standard deviations of the averages of four experiments with four replicates per
experiment. Data were analyzed by ANOVA with Tukey’s post-test comparison. ns, not
significantly different; ***, P < 0.001 compared to the WT. (D) Representative swarming
(top), twitching (middle) and biofilm (bottom) assays for SMC576, the pilY1::Mar8
mutant of SMC576 and the pilY1::Mar8 mutant carrying either the vector alone (pMQ70)
or the vector encoding a wild-type, His-tagged variant of PilY1 (ppilY1), as indicated at
the bottom of panel E. (E) Quantification of crystal violet-stained biofilms for the
indicated strains, as described in detail . Error bars represent standard deviations of the
averages of four independent experiments with four replicates per experiment. Data were
analyzed by ANOVA with Tukey’s post-test comparison. ns, not significantly different;
***, P < 0.001 compared to the SMC576.
All previous reports showed product of the algC gene is involved in
LPS synthesis, rhamnolipid and alginate production (Goldberg et al., 1993;
Olvera et al., 1999). EPS matrix may play an important role in establishing a
sustainable biofilm. It was postulated that the expression of alginate genes
critical to biofilm formation by P. aeruginosa because expression of algC is
high in the mucoid strain (Davies et al., 1993; Davies & Geesey, 1995). Also
LPS has been demonstrated to affect attachment of bacteria to a surface
(Razatos et al., 1998).
Furthermore, rhamnolipids involved in swarming motility, swarming
occurs when cells move across a hydrated, viscous semisolid surface
(Toutain et al., 2004). According to result, inactivation of the algC gene
could reflect inactivation of many pathways.

Moreover, as expected based on a recent report (Zegans et al., 2012),
the algC mutant shows a reduced production of Psl polysaccharide as judged
of ELISA after using an antibody specific for this polysaccharide (Figure 3-
4). The strain carrying a mutation in pslI shows no obvious swarming or
twitching defect (Figure 3-3B,C).
Figure 3-4: Quantification of Psl production by ELISA for the indicated strains. The
A450 value, a relative measure of Psl production as described in the materials and
methods, is plotted on the Y-axis, and the strains tested are indicated on the x axis.
Clinical strain SMC576 and three transposon mutants are shown. P. aeruginosa PAO1
(PAO1) serves as a Psl-producing positive control, and P. aeruginosa PA14 (PA14)
serves as a control for a strain lacking Psl. Data were analyzed by ANOVA with Tukey’s
post-test comparison. ns, not significantly different; ***, P < 0.001 compared to the
SMC576.

We were able to quantify Psl production by pslI::Mar and algC::Mar
mutants using an ELISA assay, in which Psl-specific antiserum was used to
detect Psl in polysaccharide production on Nunc MaxiSorp plates. The
mutant’s measurement compared with 576 and PAO1 as positive control and
PA14 as negative control and pilY:Mar8 to check whether is there a role of
pilY1gene in Psl production (Figure 3-4).
Similar to quantification of biofilm formation the ELISA revealed that
pslI genes and algC gene are essential for Psl production. Mapped of
Mutants pslI::Mar and algC::Mar showed the differences in Psl produced
by these two pslI::Mar and algC::Mar mutants and showed highly deficient
biofilm.
The pslI production in mutants were highly significant un compared
with 576, PAO1.This provides genetic evidence indicate the pslI and algC
genes are required for Psl synthesis and biofilm phenotype. Finally, as
expected, the strain carrying a mutation in the pilY1:Mar8 gene showed no
difference in Psl production compared to the parent strain SMC576. That
indicates this gene not participate in Psl polysaccharide production.
To extend our findings of the second clinical strain, we screened
~1,500 Mariner transposon mutants of SMC214, another a mucoid, CF strain
of P. aeruginosa. Similar to SMC576, we identified biofilm-defective
mutants that mapped to the pilX and pilW genes (Figure 3-5). These
mutations, as expected, also rendered the strains unable to twitch (Figure 3-
5).
Thus SMC214 and SMC576 require conserved known genes to play a
role in biofilm formation in laboratory strains.
The clinical strain (214) included the pilW::Mar and pilX::Mar
mutants, shown biofilm deficient, quantification of Biofilm formation

indicates that biofilms formed by the pilX::Mar and pilW::Mar mutants
were significantly different from the biofilm of the 214 parent strain (Figure
3-3D fourth row).
Figure 3-5: Identification of biofilm-deficient mutants in the clinical strain SMC214.
(A) Representative twitching (top) and biofilm (bottom) assay phenotypes of clinical
strain SMC214 and selected transposon mutants. All assays are performed as described in
the materials and methods. The location of the insertion site is indicated in panel B. (B)
Quantification of crystal violet-stained biofilms for the indicated strains. Error bars
represent standard deviations of the averages of four experiments with four replicates per

experiment. ANOVA following Tukey’s post-test comparison. *, P < 0.05; **, P < 0.01,
compared to SMC214.
The pilW::Mar and pilX::Mar mutants exhibit swarming phenotypes
that closely resemble the 214 parent strain, it is (just a small circular zone)
due to the correlation of high poly saccharide of the strain 214 with high c-
di-GMP that suppress of swarming motility (kuchma et al, 2007)
Nevertheless, this polysaccharides remained at high level even after pilW
and pilX mutation (Figure 3-3 D first row), while the pilW::Mar and
pilX::Mar showed strong suppressor of twitching motility (Figure 3-3 D
second row).
3.7. Clinical strains SMC576 and SMC214 with biofilm defective
mutants and sensitive to imipenem.
Based on the finding loss of oprD resulted loss of biofilm formation
and increased resistance to imipenem. We hypothesized that there a general
link between loss of biofilm formation and increased resistance to this
antibiotic might be there. Therefore, we used the E-test method to determine
imipenem resistance profiles for the biofilm-defective mutants isolated in
both the SMC576 and SMC214 strains. In all cases, none of biofilm-
deficient mutants isolated showed imipenem resistance (MIC≥4µg/ml),
suggesting that there is no general link between loss of biofilm formation
and increase resistance to Imipenem.
Here we show that OprD. Plays a major role in acquired resistance to
imipenem also participates in biofilm formation. Loss of OprD function
blocks entry of imipenem into the cell and resulted in resistance to this drug
(Wolter et al., 2004). It appears that one trade-off for acquiring resistance to

this antibiotic via loss of OprD may be a compromised ability to form a
biofilm, at least on one model abiotic substratum.
In the current study, we utilized an oprD transposon mutant of P.
aeruginosa PAO1 and a clinical strain (SMC631) with a mutation premature
stop mutation in oprD, to establish this link between increased imipenem
resistance and loss of biofilm formation. This finding prompted us to look
more broadly at the relationship between imipenem resistance/sensitivity and
biofilm formation. Thus, we examined both in vitro-selected imipenem
resistant variants of the sensitive clinical strain SMC631, as well as additional
clinical strains of P. aeruginosa isolated from CF patients. As shown in
Table 3-2, the trend of low biofilm formation correlating with high imipenem
resistance appeared to be maintained (and vice versa), raising the question of
whether loss of biofilm formation in general might confer imipenem
resistance.
To test this idea, we selected two imipenem sensitive clinical strains
which formed robust biofilms (SMC576 and SMC214). Mariner mutagenesis
was used to identified multiple biofilm-deficient strains. Moreover,
combination of complementation studies, phenotypic tests and biochemical
were used to confirm the apparent defects in these strains (Figures 3-2 to 3-4).
Biofilm-deficient strains with mutations in genes known were isolated to
study biofilm formation based on studies of the P. aeruginosa PAO1 and
PA14 lab strains, including mutations in factors required for pili biogenesis
and function (pilA, pilO, pilY1, pilW, pilX) (Alm et al., 1996; Alm et al.,
1996; Bohn et al., 2009; Giltner et al., 2010; Kuchma et al.,2012; ) and
exopolysaccharide production (pslI, algC) (Friedman and Kolter, 2004;
Davies et al., 1993; Davies & Geesey, 1995; Zegans et al., 2012). While,
these findings may not be surprised. But they are useful as they extend

findings from laboratory strains to clinically relevant strains. Importantly,
none of the biofilm-deficient mutants showed imipenem resistance (MIC ≥16
µg/ml), thus arguing against a general link between these two phenotypes.

conclusions and
recommendations

Conclusion
1. OprD plays a major role in acquired resistance to imipenem also participates in biofilm formation. Loss of OprD function, which blocks entry of imipenem into the cell and that ultimately, creates resistance to this drug. It appears that one trade-off for acquiring resistance to this antibiotic via loss of OprD may be a compromised ability to form a biofilm, at least on one model abiotic substratum.
2. An inverse relationship observed between biofilm formation in P. aeruginosa and imipenem resistance, likely due to the participation of OprD in both of these processes showed by oprD expression.
3. Loss of oprD function in a lab strains and clinical strains results in a reduction in biofilm formation as compared to the parental strain.
4. Both imipenem sensitive clinical strains formed robust biofilm (SMC576 and SMC214). Multiple biofilm deficient strains were identifying by using Mariner mutagenesis. Combination of complementation studies, phenotypic tests and biochemical studies were applied to confirm the apparent defects in these strains.
5. The biofilm-deficient strains with mutations in genes known to be involved in biofilm formation based on studies of the P. aeruginosa PAO1 and PA14 lab strains, including mutations in factors required for pili biogenesis and function (pilA, pilO, pilY1, pilW, pilX) exopolysaccharide production (pslI, algC). While a role for these genes in biofilm formation may not be surprised. But they are useful in clearing their extend findings from laboratory strains to clinically relevant isolates.
6. Importantly, none of the biofilm-deficient mutants showed imipenem resistance (MIC ≥8 µg/ml), thus arguing against a general link between these two phenotypes.

Recommendations
1. Identifying the other mechanisms participate in low expression of oprD and their relationship with biofilm formation.
2. Performing the clean deletion of oprD gene in wild type PAO1; thereafter, assessing the biofilm formation.
3. Detecting the role of other outer membrane protein (Efflux pump) in biofilm formation.
4. Studying the role of oprD gene in the biofilm formation by electron
microscopy and confocal laser scanning. 5. Assessing the role of oprD gene in the motility of P. aeruginosa.
6. Screening the role of oprD and pilY1in ecological isolates.
7. Signifying the complete role of pilX and pilW gene in high biofilm
formation in clinical isolate

Appendix A
Agarose gel electrophoresis of amplified oprD gene of P. aeruginosa.

Appendix B
The expression of oprD gene of P. aeruginosa isolates
Name R1 R2 R3 Average Sdev
631 C 0.023247 0.090408 0.06682 0.060158 0.034073
631F 0.140358 0.140728 0.128333 0.136473 0.007052
631H 0.068792 0.061939 0.068712 0.066481 0.003934
631J 0.096998 0.047719 0.04612 0.063612 0.028924
631K 0.069529 0.136932 0.099053 0.101838 0.033788
4974 0.020989 0.013981 0.012189 0.01572 0.00465
4972 0.311376 0.14058 0.155613 0.202523 0.094569
4973 0.080846 0.065342 0.070353 0.072181 0.007912
4979 0.092996 0.064274 0.082453 0.079908 0.014529
576 1.328168 1.394309 0.97086 9.897779 0.227799

Appendix C Agarose gel electrophoresis of un known genes of P.
aeruginosa by Arbitrary PCR Run 1.
Agarose gel electrophoresis of un known genes of P. aeruginosa by Arbitrary PCR Run 2

references

· Aas, F. E., Wolfgang, M., Frye, S., Dunham, S., Lovold, C. and Koomey,
M. (2002). Competence for natural transformation in Neisseria gonorrhoeae: components of DNA binding and uptake linked to type IV pilus expression. Mol. Microbiol. 46:749-760.
· Aldridge, P., Paul, R., Goymer, P., Rainey, P. and Jenal, U. (2003). Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol. Microbiol. 47:1695– 708
· Allesen-Holm, M., Barken, K. B., Yang, L., Klausen, M., Webb, J. S., Kjelleberg, S., Molin, S., Givskov, M. and Tolker-Nielsen, T. (2006). A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 59:1114-1128.
· Alm, R. A., Hallinan, J. P., Watson, A. A. and Mattick, J. S. (1996). Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol. Microbiol. 22:161–173.
· Alm, R. A., Mattick, J. S. (1996). Identification of two genes with prepilin-like leader sequences involved in type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J. Bacteriol. 178:3809 –3817.
· Anderson, K., Lonsway, D. R., Rasheed, J. K., Biddle, J., Jensen, B., McDougal, L. K., Carey, R. B. , Thompson , A. , Stocker, S. , Limbago, B. and Patel , J. B. (2007). Evaluation of Methods to Identify the Klebsiella pneumoniae Carbapenemase in Enterobacteriaceae. J. Clin. Microbiol.45:2723.
· Bals, R., Weiner, D. J. and Wilson, J. M. (1999). The innate immune system in cystic fibrosis lung disease. J. Clin. Invest. 103: 303–7
· Barken, K. B., Pamp, S. J., Yang, L., Gjermansen, M., Bertrand, J. J., Klausen, M., Givskov, M., Whitchurch, C. B., Engel, J. N. and Tolker- Nielsen, T. (2008). Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 10:2331-2343.
· Barken, K.B., Pamp, S.J., Yang, L., Gjermansen, M., Bertrand, J.J., Klausen, M., Givskov, M., Whitchurch, C.B., Engel, J.N. and Tolker-

Nielsen, T. (2008). Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 10:2331-2343.
· Barraud, N., Hassett, D. J., Hwang, S., Rice, R. A., Kjelleberg, S. and Webb, J. S. (2006). Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 188(21): 7344 - 7353.
· Bodey, G. P., Bolivar, R., Fainstein V. and Ladeja L. (1983). Infections caused by Pseudomonas aeruginosa. Rev. Infect. Dis. 5:279–313.
· Bohn, Y. S., Brandes, G., Rakhimova, E., Horatzek, S., Salunkhe, P., Munder, A., van Barneveld, A., Jordan, D., Bredenbruch, F., Haussler, S., Riedel, K., Eberl, L., Jensen, P. O., Bjarnsholt, T., Moser, C., Hoiby, N., Tummler, B. and Wiehlmann, L. (2009). Multiple roles of Pseudomonas aeruginosa TBCF10839 PilY1 in motility, transport and infection. Mol Microbiol. 71: 730- 747. doi: 10.1111/j.1365-2958.2008.06559.x 50.
· Bradley, D. E. (1972). Shortening of Pseudomonas aeruginosa pili after RNAphageadsorption. J. Gen. Microbiol. 72: 303–19.
· British Pharmacopoeia (BP). (2012). Medicin and Healthcare products Regulatory Agency (MHRA). London SW1W 9SZ.
· Burke, D., Dawson, D., and Stearns, T. (2000). Methods in Yeast Genetics. A Cold Spring Harbor laboratory course manual. In, ed., Cold Spring Harbor laboratory Press, New York.
· Caetano-Anolles, G. (1993). Amplifying DNA with arbitrary oligonucleotide primers. PCR Methods Appl. 3:85–92.
· Caiazza, N. C. and O’Toole, G. A. (2004). SadB is required for the transition from reversible to irreversible attachment during biofilm formation by Pseudomonas aeruginosa PA14. J. Bacteriol. 186:4476–4485.
· Caiazza, N. C., Merritt, J. H., Brothers, K. M. and O’Toole, G. A. (2007). Inverse regulation of biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J. Bacteriol. 189:3603–3612.
· Caiazza, N. C., Shanks, R. M. and O'Toole, G. A. (2005). Rhamnolipids modulate swarming patterns of Pseudomonas aeruginosa. J. Bacteriol. 187:7351–7361.
· Chace, K. V., Flux, M. and Sachdev, G. P. (1985) Comparison of physicochemical properties of purified mucus glycoproteins isolated from

respiratory secretions of cystic fibrosis and asthmatic patients. Biochem. 24: 7334–41
· CLSI (2013) Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Third Informational Supplement. CLSI document M100-S23. Clinical and Laboratory Standards Institute, Wayne, PA
· Colvin, K. M., Irie, Y., Tart, C. S., Urbano, R., Whitney, J. C., Ryder, C., Howell, P. L., Wozniak, D. J., Parsek, M. R. (2012). The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 14(8):1913-28.
· Copeland, M. F., Flickinger, S. T., Tuson, H. H. and Weibel, D. B. (2010). Studying the dynamics of flagella in multicellular communities of Escherichia coli by using biarsenical dyes. Appl. Environ. Microbiol. 76:1241–1250.
· Costerton, J. W., Lewandowski, Z., Caldwell, D. E., Korber, D. R. and Lappin-Scott, H. M. (1995). Microbial biofilms. Annual Review of Microbiology; 49:711 - 45.
· Davies, D. G., Chakrabarty, A. M. and Geesey, G. G. (1993). Exopolysaccharide production in biofilms: substratum activation of alginate gene expression by Pseudomonas aeruginosa. Appl. Environ. Microbiol. 59:1181–1186.
· Davies, D. G., Geesey and G. G. (1995). Regulation of the alginate biosynthesis gene algC in Pseudomonas aeruginosa during biofilm development in continuous culture. Appl. Environ. Microbiol. 61: 860–867.
· Davies, D. J., Parsek, M. R., Pearson, J. P., Iglewski, B. H., Costerton, J. W. and Greenberg, E. P. (1998). The involvement of cell to cell signals in the development of a bacterial biofilm. Science 280:295–298.
· Davies, T. A., Marie Queenan, A., Morrow, B. J., Shang, W., Amsler K, He W., Lynch, A. S., Pillar, C. and Flamm, R. K. (2011). Longitudinal survey of carbapenem resistance and resistance mechanisms in Enterobacteriaceae and non-fermenters from the USA in 2007–09. J Antimicrob. Chemother. 66:2298–307.
· de Freitas, A. L. P. and Barth, A. L.(2002). Antibiotic resistance and molecular typing of Pseudomonas aeruginosa: Focus on Imipenem. J. Braz. Chem. Soc.6(1):1-7.

· Dheepa, M., Rashme, V. L. and Appalaraju, B. (2011). Comparision of biofilm production and multiple drug resistance in clinical isolates of Acinetobacter baumanii from a tertiary care hospital in South India. Int J Pharm Biomed Sci. 2: 103-107.
· Dib, C., Trias, D. C. and Jarlier, V. (1995). Lack of additive effect between mechanisms of resistance to carbapenems and other beta-lactam agents in Pseudomonas aeruginosa. Eur. J. Clin. Microbiol. Infect. Dis. 14:979 –986.
· El Amin, N., Giske, C. G., Jalal, S., Keijser, B., Kronvall, G. and Wretlind, B. (2005). Carbapenem resistance mechanisms in Pseudomonas aeruginosa: alterations of porin OprD and efflux proteins do not fully explain resistance patterns observe in clinical isolates. APMIS. 113:187–196.
· Emerson, J., Rosenfeld, M., McNamara, S., Ramsey, B. and Gibson, R. L. (2002). Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 34:91-100.
· Evans, J. C., and Segal, H. (2007). A novel insertion sequence, ISPA26, in oprD of Pseudomonas aeruginosa is associated with carbapenem resistance. Antimicrob. Agents Chemother. 51:3776–3777.
· Friedman, L. and Kolter, R. (2004). Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aerguinosa biofilm matrix. J. Bacteriol. 186:4457-4465.
· Friedman, L. and Kolter, R. (2004). Genes involved in matrix formation of Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675–690.
· Friedman, L. and Kolter, R. (2004). Two genetic loci produce distinct carbohydrate- rich structural components of the Pseudomonas aeruginosa biofilm matrix. J. Bacteriol. 186:4457–4465.
· Fujitani, S., Sun, H.Y., Yu, V.L. and Weingarten, J.A. (2011). Pneumonia due to Pseudomonas aeruginosa. Part I: epidemiology, clinical diagnosis, and source. Chest. 139:909-919.
· Fukuda, H., Hosaka, M., Iyobe, S., Gotoh, N., Nishino, T., Hirai, K. (1995). nfxC-type quinolone resistance in a clinical isolate of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39:790–792.

· Galani, I., Rekatsina, P. D., Hatzaki, D., Plachouras, D., Souli, M. and Giamarellou, H. (2008). Evaluation of different laboratory tests for the detection of metallo-ßlactamase production in Enterobacteriaceae. J. Antimicrob. Chemother. 61:548-53.
· Giltner, C. L., Habash, M., Burrows, L. L. (2010). Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J. Mol. Biol 398: 444-461. doi: 10.1016/j.jmb.2010.03.028.
· Goldberg, J. B., Hatano, K. and Pier, G. B. (1993). Synthesis of lipopolysaccharide O side chains by Pseudomonas aeruginosa PAO1 requires the enzyme phosphomannomutase. J. Bacteriol. 175:1605–1611.
· Goldberg, J. B., Hatano, K. and Pier, G. B. (1993). Synthesis of lipopolysaccharide O side chains by Pseudomonas aeruginosa PAO1 requires the enzyme phosphomannomutase. J. Bacteriol. 175:1605–1611.
· Govan, J. R. and V. Deretic. (1996). Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60:539–574.
· Gutiérrez, O., Juan, C., Cercenado, E., Navarro, F., Bouza, E., Coll, P., Pérez, J. L. and Oliver, A. (2007). Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa isolates from Spanish hospitals. Antimicrob. Agents Chemother. 51:4329–4335.
· Gutiérrez, O., Juan, C., Cercenado, E., Navarro, F., Bouza, E., Coll, P., Pérez, J. L. and Oliver, A. (2007). Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa isolates from Spanish hospitals. Antimicrob. Agents Chemother. 51:4329–4335.
· Hall, J., Hodgson, G. and Kerr, K.G.(2004). Provision of safe potable water for immunocompromised patients in hospital. J. Hosp. Infect. 58:155-158.
· Harshey, R. M. (1994). Bees aren’t the only ones: swarming in gram-negative bacteria. Mol. Microbiol. 13:389–394.
· Haussler, S., Ziegler, I., Lottel, A., Gotz, F. v., Rohde, M., Wehmhohner, D., Saravanamuthu, S., Tummler, B. and Steinmetz, I. (2003). Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J. Med. Microbiol. 52:295-301.

· Hengge, R. (2009). Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7:263–273.
· Hentzer, M., Teitzel, G. M., Balzer, G. J., Heydorn, A., Molin, S., Givskov, M. and Parsek, M. R. (2000). Alginate over-production affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 183:5395-5401.
· Hentzer, M., Teitzel, G. M., Balzer, G. J., Heydorn, A., Molin, S., Givskov, M., Parsek, M. R. (2001). Alginate over-production affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 183:5395-5401.
· Hobbs, J. R., Stickel, M., Martin P, and Edwards, D. (1993). Interpretation as Abduction. Artificial Intelligence Center SRI International. 63: 69-142.
· Høiby, N. (2000). Prospects for the prevention and control of pseudomonal infection in children with cystic fibrosis. Paediatr. Drugs. 2:451-63.
· Høiby, N., Bjarnsholt, T., Givskov, M., Molin, S. and Ciofu, O. (2010). Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob gents. 35(4):322-32. doi: 10.1016.
· Honko, A. N., Sriranganathan, N., Lees, C. J. and Mizel, S. B. (2006). Flagellin Is an Effective Adjuvant for Immunization against Lethal Respiratory Challenge with Yersinia pestis. Infect Immun.74:1113–1120.
· Huang, H., Jeanteur, D., Pattus, F. and Hancock R. E. (1995). Membrane topology and site-specific mutagenesis of Pseudomonas aeruginosa porin OprD. Mol. Microbiol. 16:931–941.
· Huang, H., Jeanteur, D., Pattus, F. and Hancock, R. E. (1995). Membrane topology and site-specific mutagenesis of Pseudomonas aeruginosa porin OprD. Mol. Microbiol. 16:931–941.
· Ibrahim, N. H., Somily, A. M., Bassyouni, R. H. and El-Aabedien, A. Z. (2012). Comparative study 484 assessing the effect of Tigecycline and Moxifloxacin in prevention of Acinetobacter baumannii biofilm. Life Sci. 9: 1016-1024.
· Jacobs, M. A, Alwood, A., Thaipisuttikul, I., Spencer, D., Haugen, E., Ernst, S., Will, O., Kaul, R., Raymond, C., Levy, R., Chun-Rong, L., Guenthner, D., Bovee, D., Olson, M. V. and Manoil, C. (2003)

Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. 100: 14339–14344.
· Jacobs, M. A., Alwood, A., Thaipisuttikul, I., Spencer, D., Haugen, E., Ernst, S., Will, O., Kaul, R., Raymond, C., Levy, R., Chun-Rong, L., Guenthner, D., Bovee, D., Olson, M. V. and Manoil, C. (2003). Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U S A 100: 14339-14344.
· Jesaitis, A. J., Franklin, M. J., Berglund, D., Sasaki, M., Lord, C. I., Bleazard, J. B., Duffy, J. E., Beyenal, H. and Lewandowski, Z. (2003). Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J. Immunol. 171:4329-39.
· Jesudason, M. V., Kandathil, A. J. and Balaji, V. (2005). Comparison of two methods to detect carbapenemase and metallo-β-lactamase production in clinical isolates. Indian J. Med. Res.121:780-3.
· Jones, B. V., Young, R., Mahenthiralingam, E. and Stickler, D. J. (2004). Ultrastructure of Proteus mirabilis swarmer cell rafts and role of swarming in catheter-associated urinary tract infection. Infect. Immun. 72:3941–3950.
· Kato, J., Nakamura, T., Kuroda, A. and Ohtake, H. (1999). Cloning and characterization of chemotaxis genes in Pseudomonas aeruginosa. Biosci. Biotechnol. Biochem. 63:155–161.
· Khan, W., Bernier, S. P., Kuchma, S. L., Hammond, J. H., Hasan, F. and O’Toole, G. A. (2010). Aminoglycoside resistance of Pseudomonas aeruginosa biofilms modulated by extracellular polysaccharide. Int. Microbiol. 13:207-212.
· Kirisits, M. J., Prost, L., Starkey, M., Parsek, M. R. (2005). Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 71:4809-4821.
· Köhler, T., Curty, L. K., Barja, F., Van Delden, C. and Pechère, J. C. (2000). Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J. Bacteriol. 182:5990– 5996.
· Köhler, T., Michéa-Hamzehpour, M., Epp, S. F. and Pechère, J. C. (1999). Carbapenem activities against Pseudomonas aeruginosa: respective

contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 43:424–427.
· Köhler, T., Michéa-Hamzehpour, M., Epp, S. F. and Pechère, J. C. (1999). Carbapenem activities against Pseudomonas aeruginosa: respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 43:424–427.
· Köhler, T., Michéa-Hamzehpour, M., Henze, U., Gotoh, N., Curty, L. K. Pechère, J. C. (1997). Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 23: 345-354.
· Kuchma, S. L., Ballok, A. E., Merritt, J. H., Hammond, J. H., Lu, W., Rabinowitz, J. D. and O'Toole, G. A. (2010). Cyclic-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J. Bacteriol. 192:2950 –2964
· Kuchma, S. L., Brothers, K. M. and Merritt, J. H., Liberati, N. T., Ausubel, F. M. and O’Toole, G. A. (2007). BifA,a cyclic-Di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J. Bacteriol. 189:8165-8178.
· Kuchma, S. L., Connolly, J. P. & O’Toole, G. A. (2005). A threecomponent regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J. Bacteriol. 187: 1441–1454.
· Kuchma, S. L., Griffin, E. F. and O'Toole, G. A. (2012). Swarming Motility Participate in the Negative Regulation of Minor Pilins of the Type IV Pilus System. J. Bacteriol. 194: 5388-5403.
· Kuchma, S. L., Griffin, E. F. and O'Toole, G. A. (2012). Swarming Motility Participate in the Negative Regulation of Minor Pilins of the Type IV Pilus System. J. Bacteriol. 194: 5388-5403.
· Kulasekara, H. D., Ventre, I., Kulasekara, B. R., Lazdunski, A., Filloux, A. and Lory, S. (2005). A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol. Microbiol. 55:368–380.
· Lasaro, M. A., Salinger, N., Zhang, J., Wang Y., Zhong, Z., Goulian, M, Zhu, J. (2009). F1C fimbriae play an important role in biofilm formation and intestinal colonization by the Escherichia coli commensal strain Nissle 1917. Appl. Environ. Microbiol. 75(1): 246-251.

· Lee, J. and Soo, K. K. (2012). OprD mutations and inactivation, expression of efflux pumps and AmpC, and metallo-_-lactamases in carbapenem-resistant Pseudomonas aeruginosa isolates from South Korea. Int. J. Antimicrob. Agents. 40:168– 172.
· Lee, K., Chong, Y., Shin, H. B., Kim, Y. A., Yong, D. and Yum, J. H. (2001). Modifi ed Hodge and EDTA - disk synergy tests to screen metallo-β-lactamase-producing strains of Pseudomonas and Acinetobacter species. Clin Microbiol Infect. 7: 88-91
· Li, Y., Hao, G., Galvani, C. D., Meng, Y., De La Fuente, L., Hoch, H. C, and Burr, T. J. (2007). Type I and type IV pili of Xylella fastidiosa affect twitching motility, biofilm formation and cell–cell aggregation. Microbiol. 153: 719–726.
· Li, Z., Kosorok, M. R., Farrell, P. M., Laxova, A., West, S. E, Green, C. G., Collins, J., Rock, M. J. and Splaingard, M. L. (2005). Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 293:581-8.
· Lim, B., Beyhan, S., Meir, J. and Yildiz, F. H. (2006). Cyclic-diGMP signal transduction systems in Vibrio cholerae: modulation of rugosity and biofilm formation. Mol. Microbiol. 60:331–48.
· Lister, P. D, Wolter, D. J. and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22:582– 616.
· Livermore, D. M. (1992). Interplay of impermeability and chromosomal β- lactamase activity in imipenem-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36:2046-2048.
· Livermore, D. M. and Yang, Y. J. (1987). β-Lactamase lability and inducer power of newer β-lactam antibiotics in relation to their activity against β-lactamase- inducibility mutants of Pseudomonas aeruginosa. J. Infect. Dis. 155:775-782.
· Lizewski, S. E., Schurr, J. R., Jackson, D. W., Frisk, A., Carterson, A. J., and Schurr, M. J. (2004). Identification of AlgR-regulated genes in Pseudomonas aeruginosa by use of microarray analysis. J. Bacteriol. 186:5672–5684.

· Lynch, M. J., Drusano, G. L. and Mobley, H. L. (1987). Emergence of resistance to imipenem in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 31:1892–1896.
· Ma, L., JacksonKD, Landry, R. M., Parsek, M. R. and Wozniak, D. J. (2006). Analysis of Pseudomonas aeruginosa conditional psl variants reveals roles for the psl polysaccharide in adhesion and maintaining biofilm structure postattachment. J. Bacteriol. 188:8213-8221.
· Ma, L., Lu, H., Sprinkle, A., Parsek, M. R., Wozniak, D. J. (2007). Pseudomonas aeruginosa Psl is a galactose- and mannose-rich exopolysaccharide. J. Bacteriol. 189:8353-8356.
· Mah, T. F. and O’Toole, G. A. (2001). Mechanisms of biofilm resistance to antimicrobial agents. Trends. Microbiol. 9:34-39.
· Mai, G. T., Seow, W. K., Pier, G. B., McCormack, J. G. and Thong, Y. H. (1993). Infect. Immun. 61: 559–564.
· Malic, S., Hill, K. E., Hayes, A., Percival, S. L., Thomas, D. W. and Williams, D. W. (2009). Detection and identification of specific bacteria in wound biofilms using peptide nucleic acid fluorescent in situ hybridization (PNA FISH). Microbiol. 155: 2603 – 2611.
· Maltezou, H. C. (2009). Metallo-beta-lactamases in Gram-negative bacteria: introducing the era of pan-resistance?. Int. J. Antimicrob. Agents. 33(5): 1-7.
· Marchiaro, P., Mussi, M. A., Ballerini, V., Pasteran, F., Viale, A. M., Vila A. J. and Limansky A. S. (2005). Sensitive EDTA-based microbiological assays for detection of metallo-β-lactamases in nonfermentative gram-negative bacteria. J. Clin. Microbiol. 43: 5648-52.
· Martinez, E., Marquez, C., Ingold, A., Merlino, J., Djordjevic, S. P., Stokes, H. b. and Chowdhurya, P. R. (2012). Diverse Mobilized Class 1 Integrons Are Common in the Chromosomes of Pathogenic Pseudomonas aeruginosa Clinical Isolates. Antimicrob. Agents Chemother. 56(4):2169-2170.
· Masuda, N., Sakagawa, E., Ohya, S., Gotoh, N., Tsujimoto, H., Nishino, T. (2000). Contribution of the MexX-MexY-OprM Efflux System to Intrinsic Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44(9): 2242–2246.

· Matsukawa, M. and Greenberg, E. P. (2004). Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J. Bacteriol. 186:4449-4456.
· Mattick, J. S. (2002). Type IV pili and twitching motility. Annu. Rev. Microbiol. 56:289–314.
· Merritt, J. H., Ha, D., Cowlesb, K. N., Luc, W., Moralesa, D. K., Rabinowitzc, J., Gitaib, Z. and O’Toole, G. A. (2010). Specific control of Pseudomonas aeruginosa surface- associated behaviors by two c-di-GMP diguanylate cyclases. mBio. 1:4 00183-10.
· Merritt, J. H., Brothers, K. M., Kuchma, S. L. and O’Toole, G. A. (2007). SadC reciprocally influences biofilm formation and swarming motility via modulation of exopolysaccharide production and flagellar function. J. Bacteriol. 189:8154-8164.
· Newell, P. D., Monds, R. D. and O’Toole, G. A. (2009). LapD is a bis-(3,5)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc. Natl. Acad. Sci. 106:3461–3466
· Nikaido, H. (1994). Prevention of drug access to bacterial targets: permeability barriers and active efflux. Sci. 264:382–388.
· Noyal ,M.J.C., Menezes, G.A., Harish, B.N., Sujatha, S. and Parija, S.C.. (2009). Simple screening tests for detection of carbapenemases in clinical isolates of nonfermentative Gram-negative bacteria. Indian J. Med. Res. 129: 707-712.
· O’Toole, G. A. and Kolter, R. (1998). Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304.
· O’Toole, G. A., and Kolter, R. (1998). Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304.
· O’Toole, G., Kaplan, H. B. and Kolter, R. (2000). Biofilm formation as microbial development. Annu. Rev. Microbiol. 54:49–79.
· Obritsch, M.D., Fish, D.N., MacLaren, R. and Jung, R. (2004). National surveillance of antimicrobial resistance in Pseudomonas aeruginosa isolates obtained from Intensive Care Unit Patients from 1993 to 2002. Antimicrob. Agents Chemother. 48: 4606-4610.

· Ocampo-Sosa, A. A., Cabot, G., Rodríguez, C., Roman, E, Tubau, F., Macia M. D., Moya, B., Zamorano, L., Suárez, C, Peña, C. and Domínguez, M. A. (2012). Alterations of OprD in Carbapenem-Intermediate and –Susceptible Strains of Pseudomonas aeruginosa Isolated from Patients with Bacteremia in a Spanish Multicenter Study. Antimicrob. Agents Chemother. 56(4):1703.
· Ochs, M. M., McCusker, M. P., Bains, M. and Hancock, R. E. (1999). Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrob. Agents Chemother. 43:1085–1090.
· Ochsner, U.A. and Reiser, J., Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. 92:6424–6428.
· Oldenburg, K. R., Vo, K. T., Michaelis, S. and Paddon, C. (1997). Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 25(2): 451–452.
· Olvera, C., Goldberg, J. B., Sanchez, R. and Soberon-Chavez, G. (1999). The Pseudomonas aeruginosa algC gene product participates in rhamnolipid biosynthesis. FEMS. Microbiol. Lett. 179:85–90.
· Olvera, C., Goldberg, J. B., Sanchez, R. and Soberon-Chavez, G. (1999). The Pseudomonas aeruginosa algC gene product participates in rhamnolipid biosynthesis. FEMS. Microbiol. Lett. 179:85–90.
· Overhage, J., Schemionek, M., Webb, J. S., and Rehm, B. H. A. (2005). Expression of the psl operon in Pseudomonas aeruginosa PAO1 biofilms: PslA performs an essential function in biofilm formation. Appl. Environ. Microbiol. 71:4407–4413.
· Pamp, S. J., Gjermansen, M. and Tolker-Nielsen, T. (2007). The biofilm matrix—a sticky framework. In Bacterial Biofilm Formation and Adaptation. BioSci. 37-69.
· Parsek, M. R. and Singh, P. K. (2003). Bacterial biofilms: an emerging link to disease pathogenesis. Annu. Rev. Microbiol. 57:677-701.
· Pérez, F. J., Gimeno, C., Navarro, D. and García-de-Lomas, J. (1996). Meropenem permeation through the outer membrane of Pseudomonas aeruginosa can involve pathways other than the OprD porin channel. Chemother. 42: 210–214.

· Picao, R. C., Andrade, S. S., Nicoletti, A. G., Campana, E. H., Moraes, G. C., Mendes, R. E. and Gales, A. C. (2008). Metallo-ß-Lactamase Detection: Comparative Evaluation of Double Disk Synergy versus Combined Disk Tests for IMP-, GIM-, SIM-, SPM-, or VIM Producing Isolates. J. Clin. Microbiol. 46(6):2028-37.
· Poole, K. (2004). Efflux-mediated multiresistance in Gram-negative bacteria. Clin. Microbiol. Infect. 10:12–26.
· Prince, A. S. (2002). Biofilms, antimicrobial resistance, and airway infection. N Engl. J. Med. 347:1110-1.
· Qiu, G., Liu, B., Bu, J. and Chen, C. (2009). Expanding Domain Sentiment Lexicon through Double Propagation. In Proceedings of IJCAI. 1199-1204.
· Qiu, X., Kulasekara, B. R. and Lory, S. (2009). Role of horizontal gene transfer in the evolution of Pseudomonas aeruginosa virulence. Genome. Dyn. 6:126-139.
· Quale, J., Bratu, S., Gupta, J. and Landman, D. (2006). Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 50:1633–41.
· Rao, R. S., Karthika, R. U., Singh, S. P., Shashikala, P., Kanungo, R., Jayachandran, S. and Prashanth, K. (2008). Correlation between biofilm production and multiple drug resistance in imipenem resistant clinical isolates of Acinetobacter baumannii. Indian J. Med. Microbiol. 26: 333-337.
· Razatos, A., Ong, Y.-L., Sharma, M. M., Georgiou, G. (1998). Molecular determinants of bacterial adhesion monitored by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 95: 11059–11064.
· Riera, E., Cabot, G., Mulet, X., Garcia-Castillo, M., Del Campo, R., Juan, C., Canton, R. and Oliver, A. (2011). Pseudomonas aeruginosa carbapenem resistance mechanisms in Spain: impact on the activity of imipenem, meropenem and doripenem. J. Antimicrob. Chemother. 66(9):2022-7.
· Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N. and Chou, J.L. (1989). Identification of the cystic fibrosis gene and characterization of complementary DNA. Sci. 245: 1066–73.

· Rodríguez-Martínez, J. M., Poirel, L. and Nordmann, P. (2009). Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53:4783– 4788.
· Ruiz-Martinez, L., Lopez-Jimenez, L., Fuste, E., Vinuesa, T., Martinez, J. P. and Vinas, M. (2011). Class I integrons in environmental and clinical Pseudomonas aeruginosa. Int. J. Antimicrob. Agents. 38(5):398-402.
· Sambrook and Russell, (2001). Molecular Cloning: A Laboratory Manual (3rd ed.). Cold Spring Harbor Laboratory Press. ISBN 978-0-87969-577-4.
· Sanbongi, Y., Shimizu, A., Suzuki, T., Nagaso, H., Ida, T., Maebashi, K., Gotoh, N. (2009). Classification of OprD sequence and correlation with antimicrobial activity of carbapenem agents in Pseudomonas aeruginosa clinical isolates collected in Japan. Microbiol. Immunol. 53:361–367.
· Sanchez-Romero, I., Cercenado, E., Cuevas, O., Garcia-Escribano, N., Garcia-Martinez, J. and Bouza, E. (2007). Evolution of the antimicrobial resistance of Pseudomonas aeruginosa in Spain: Second National Study (2003). Rev. Esp. Quimioter. 20:222-229.
· Sarkisova, S., Patrauchan, M. A., Berglund, D., Nivens, D. E. and Franklin, M. J. (2005). Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J. Bacteriol. 187:4327-4337.
· Sauer, K., Camper, A. K., Ehrlich, G. D., Costerton, J. W. and Davies, D. G. (2002). Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. of Bacteriol. 184(4): 1140 - 1154.
· Sauer, K., Rickard, A. H. and Davies, D. G. (2007). Biofilms and biocomplexity. Microbe. 2(7): 347-353.
· Schlag, S., Nerz, C., Birkenstock, T.A., Altenberend F. and Götz F. (2007). Inhibition of Staphylococcal biofilm formation by nitrite. J. Bacteriol. 189(21): 7911-7919.
· Shanks, R. M., Caiazza, N. C., Hinsa, S. M., Toutain, C. M. and O’Toole, G. A. (2006). Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 72:5027– 5036.

· Shanks, R. M., Caiazza, N. C., Hinsa, S. M., Toutain, C. M. and O'Toole, G. A. (2006). Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. App. Environ. Microbial. 72: 5027-5036.
· Simon, R., Priefer, U. and Pühler, A. (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1:784 –791.
· Singh, P. K., Schaefer, A. L., Parsek, M. R., Moninger, T. O., Welsh, M. J. and Greenberg, E. P. (2000). Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407, 762–764.
· Skerker, J. M., Berg, H. C. (2001). Direct observation of extension and retraction of type IV pili. Proc. Natl. Acad. Sci. 98:6901–4.
· Smith, E. E., Buckley, D. G., Wu, Z., Saenphimmachak, C., Hoffman, L. R., D’Argenio, D. A., Miller, S. I., Ramsey, B. W, Speert, D. P., Moskowitz, S. M., Burns, J. L., Kaul, R. and Olson M. V. (2006). Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. 103:8487-8492.
· Stapper, A. P., Narasimhan, G., Ohman, D. E., Barakat, J., Hentzer, M., Molin, S., Kharazmi, A., Høiby, N. and Mathee, K. (2004). Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J. Med. Microbiol. 53:679-690.
· Stoodley, P., Purevdorj-Gage, B. and Costerton, J. W. (2005). Clinical significance of seeding dispersal in biofilms: a response. Microbiol. 151(11): 3453.
· Tolker-Nielsen, T., Brinch, U. C., Ragas, P. C., Andersen, J. B., Jacobsen, C. S. and Molin, S. (2000). Development and Dynamics of Pseudomonas sp. Biofilms. J. Bacteriol. 182(22): 6482 - 6489.
· Toutain, C. M., Zegans, M. E. and O'Toole, G. A. (2005). Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J. Bacteriol.187:771–777.
· Trautmann, M., Halder, S., Hoegel, J., Royer, H. and Haller, M.(2008). Point-of-use filtration reduces endemic Pseudomonas aeruginosa infections on a surgical intensive care unit. Am. J. Inf. Cont. 36:421-429.

· Tremblay, J., Richardson, A.P., Lepine, F. and Déziel, E. (2007). Self-produced extracellular stimuli modulate the Pseudomonas aeruginosa swarming motility behavior. Environ. Microbiol. 9:2622–2630.
· Trias, J. and Nikaido, H. (1990). Outer membrane protein D2 catalyzes facilitated diffusion of carbapenems and penems through the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 34:52–57.
· Trias, J. and Nikaido, H. (1990). Protein D2 channel of the Pseudomonas aeruginosa outer membrane has a binding site for basic amino acids and peptides. J. Biol. Chem. 265: 15680–15684.
· Van Schaik, E. J., Giltner, C. L., Audette, G. F., Keizer, D. W., Bautista, D. L., Slupsky, C. M., Sykes, B. D. and Irvin, R. T. (2005). DNA binding: a novel function of Pseudomonas aeruginosa type IV pili. J. Bacteriol. 187:1455-1464.
· Vandepitte, J. Verhaegen, J. Engbaek, K. Rohner, P. Piot, P. Heuck, C. C. Second edition (2003). Basic laboratory procedures in clinical bacteriology World Health Organization Geneva.
· Vandepitte, J., Verhaegen, J., Engbaek, K. Rohner, P. Piot, P. and Heuck, C. C. Second edition (2003). Basic laboratory procedures in clinical bacteriology World Health Organization Geneva.
· Vasseur, P., Vallet-Gely, I., Soscia, C., Genin, S., Filloux, A. (2005). The pel genes of the Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation. Microbiol. 151:985-997.
· Walsh, T. R., Bolmstrom, A., Qwarnstrom, A. and Gales, A. (2002). Evaluation of a new Etest for detecting metallo-β-lactamases in routine clinical testing. J. Clin. Microbiol. 40: 2755-9.
· Walsh, T. R., Toleman, M. A., Poirel, L. and Nordmann, P. (2005). Metallo-β-lactamases, the quiet before the storm?. Clin Microbiol Rev. 18:306-25.
· Watnick, P. I. and Kolter, R. (1999). Steps in the development of a Vibrio cholerae El Tor biofilm. Mol.Microbiol. 34(3): 586 - 595.
· Welsh, M. J. and Smith, A. E. (1993). Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 73: 71251-1254.
· Whitchurch, C. B., Engel, J. N. and Tolker- Nielsen, T. (2008). Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the

formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol.10:2331-2343.
· Whitchurch, C. B., Hobbs, M., Livingston, S. P., Krishnapillai, V. and Mattick, J.S. (1990). Characterization of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialized protein export system widespread in eubacteria. Gene. 101: 33-44.
· Wiegand, I., Hilpert, K. and Hancock, R. E. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nature protocol. 3:163-175.
· Wiegand, I., Hilpert, K., Hancock, R.E. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 3: 163-175. doi: 10.1038/nprot.2007.521.
· Wolter, D. J., Acquazzino, D., Goering, R. V., Sammut, P., Khalaf, N. and Hanson, N. D. (2008). Emergence of carbapenem resistance in Pseudomonas aeruginosa isolates from a patient with cystic fibrosis in the absence of carbapenem therapy. Clin. Infect. Dis. 46:137–141.
· Wolter, D. J., Hanson, N. D. and Lister, P. D. (2004). Insertional inactivation of oprD in clinical isolates of Pseudomonas aeruginosa leading to carbapenem resistance. FEMS. Microbiol. Lett. 236:137–143.
· Wolter, D. J., Khalaf, N., Robledo, I. E., Vazquez,G. J., Sante, M. I., Aquino , E. E., Goering, R. V. and Hanson, N. D. (2009). Surveillance of carbapenem- resistant Pseudomonas aeruginosa isolates from Puerto Rican medical center hospitals: dissemination of KPC and IMP-18 beta-lactamases. Antimicrob. Agents Chemother. 53:1660–1664.
· Worlitzsch, D., Tarran, R., Ulrich, M., Schwab, U., Cekici, A., Meyer, K., Birrer, P., Bellon, G., Berger,J., and Weiss, T. (2000). Effectsof reducedmucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109:317-325.
· Wozniak, D. J., Wyckoff, T. J. O., Starkey, M., Keyser, R., Azadi, P. O’Toole, G. A. and Parsek, M. R. (2003). Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. 100:7907-7912.
· Yang, L., Barken, K. B., Skindersoe, M. E., Christensen, A. B., Givskov, M. and Tolker-Nielsen, T. (2007). Effects of iron on DNA release and

biofilm development by Pseudomonas aeruginosa. Microbiol. 153:1318-1328.
· Yoneyama, H. and Nakae, T. (1993). Mechanism of efficient elimination of protein D2 in outer membrane of imipenem-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 37:2385–2390.
· Zavascki, A. P., Gaspareto, P. B., Martins, A. F., Gonçalves, A. L. and Barth A. L. (2005). Outbreak of carbapenem-resistant Pseudomonas aeruginosa producing SPM-1 metallo-B-lactamase in a teaching hospital in southern Brazil. J. Antimicrob. Chemother. 56:1148–1151.
· Zegans, M. E., Wozniak, D., Griffin. E., Toutain-Kidd, C. M., Hammond, J. H., Garfoot, A. and Lamd, J. S. (2012). Pseudomonas aeruginosa Exopolysaccharide Psl Promotes Resistance to the Biofilm Inhibitor Polysorbate 80. Antimicrob. Agents Chemother. 56: 4112–4122.

الخالصة
من عزلة وخمسون ثمانية Pseudomonas aeruginosa بوساطة االميبينيم لمضاد مقاومتها وتقييم عزلها تم حساسة، كانت العزالت هذه من٪) ۱٥.٥۲( وتسعة ،٪)۳.٤٥( اثنان ،٪)۸۱.۰۳( وأربعون سبعة ؛ E test اختبار
والتي ، جميعها المقاومة و الحساسية متوسطة العزالت تبد ولم. التتابع على لإلميبينيم، ومقاومة الحساسية، متوسطةهودج اختبار أكده ما نحو على الكاربابينيميز النزيم فعالية اية الكيسي، التليف من يعانون مرضى من سابقا عزلت
.
جين عن والتعبير اإلميبينيم، ومقاومة الحياتي، الغشاء تكوين بين العالقة قيمت oprD سالالت بعض في P. aeruginosa لإلميبينيم المقاومة السريرية (SMC631C و SMC631F و SMC631J و SMC631H و SMC631K وSMC 4974 ) . لجين منخفضا تعبيرا جميعها المقاومة العزالت أظهرت oprD منخفضة وقيما
الحساسة العزالت مع بالموازنه الحياتي للغشاء .
المقاومة العزلة أن الحالية الدراسة نتائج كشفت oprD :: isphoA /hah الغشاء تكوين على قدرة اظهرت قد الطبيعي النمط ساللة من معنويا اقل الحياتي PAO1. لإلميبينيم مقاومة الطفرة هذه اثارت ذلك من اكثر (P<0.001
ساللة مع بالمقارنة ( PAO1.
نقطية بطفرة طافرة عزلة على الحصول تم العمل هذا أثناء (SMC631F-IMr) جين في oprD الى الطفرة هذه ادت االميبينيم مقاومة (MIC=32 mg/l) ضعيف حياتي غشاء وتكوين (OD550=0.06) االصل العزلة مع مقارنة
SMC631 (OD550=0.19) .
البالزميدي التعبير poprD::isphoA/hah يلحظ لم اذ الحياتي، للغشاء كامل استرجاع الى ادى الطافرة العزلة فيمعنوي فرق أي P>0.05)) العزلة بين PAO1 معنوي فرق لوحظ بينما، جينيا المكملة والعزلة (P<0.05) بين
الطافرة والعزلة جينيا المكملة العزلة oprD::isphoA/hah فارغ بالزميد على الحاوية .
البالزميد تعبير تقدم، ما الى اضافة poprD::isphoA/hah االميبينيم حساسية استرجاع الى ادى (MIC=4 ) الى لدى الحساسية مستوى PAO1 معنوية فروق اية بدون ، (P>0.05) العزلة بين معنوي فرق هنالك بينما. بينهم
فارغ بالزميد على الحاوية الطافرة والعزلة جينيا المكملة oprD::isphoA/hah . النتائج نفس على الحصول تمالعزلة مع SMC631 البالزميد اضفنا حيث SMC631F-Imr/poprD الى البالزميد تعبير وادى الطافرة للعزلة
فارغ بالزميد على الحاوية العزلة ومع االصل العزلة مع موازنة الحياتي للغشاء كامل استرجاع .
جين فعالية عدم ان تثبت سوية النتائج هذه oprD الحياتي الغشاء تكوي نقصان عن المسؤول هو .
السريريتان العزلتان عرضت SMC576, SMC214)) الى حياتي غشاء العلى المكونتان mariner transposon mutagenesis بوساطة عنها التحري بعد الحياتي الغشاء لتكوين فاقدة عزالت على الحصول تم و
مختلفة جينات اربعة على الحصول تم الجينات فحص بعد و الدقيقة المعايرة طبق طريقة pilY1, pilW, pslI and algC الطافرتين العزلتين من كل اظهرت. الحياتي الغشاء نقصان عن مسؤولة pilY::Mar و pilW::Mar19
االصل مع مقارنة الحياتي الغشاء بتكوين معنويا انخفاضا SMC576. مثل pilY1::Mar طافرة عزالت خمسة انحشار نتيجة مستقلة transposon جين في مختلفة مواقع خمسة في pilY1 للجين فقط واحدة عزلة هناك حين في
pilW. الجينين في الطفرات ان عن فضال pilY1 وpilW االرتعاشية الحركة فقدان الى ادت .

لطافرات العج ظاهرة اما SMC576 من كل اظهرت فقد pilY1::Mar و pilW::Mar19 عن مختلفا نمطا SMC576 وقليلة قصيرة اذرع ذو و ضعيفا كان فقد .
حياتي غشاء ألعلى المكونة الثانية للعزلة بالنسبة اما ((SMC214 هما الجينات من اثنين فحص تم فقد ؛ pilW, pilX في كان والذي الحياتي الغشاء قيمة نقصان عن المسؤولة pilX::Mar و pilW::Mar معنويا اقل (P < االصل العزلة من (0.001 SMC214.
الطافرتان العزلتان أظهرت pilW::Mar و pilX::Mar لالصل تماما مشابهة عج ظاهرة SMC214 , بينما االرتعاشية للحركة عاليا تثبيطا العزلتين كلتا اظهرت .
البالزميد وضع تم +ppilY1 الطافرة العزلة في pilY1::Mir8 تكوين الى الطافرة العزلة هذه داخل تعبيره ادى ، منها الطافرة االصل العزلة بمستوى عاليا حياتيا غشاء SMC576 . كلي استرجاع الى ادى البالزميد تعبير ان كما
السيطرة الناقل مع بالموازنة للعج .
جين تعبير ان لوحظ االرتعاشية، الحركة pilY1 الطافرة في النقص تمم قد pilY1::Mir8. النتائج هذه توضح جين فعالية عدم بأجمعها pilY1 الحصول تم التي الطافرة العزالت في السابقة الظواهر تثبيط عن المسؤولة هي
الساللة من عليها SMC576 .
جين ان االليزا فحص بين الحياتي، الغشاء لتكوين الكمي القياس مع تماهيا pslI جين و algC في اساسيان جينان انتاج فحص عند .psl انتاج psl الطافرتين العزلتين في pslI::Mar و algC::Mar ، في تباينا هناك كانت psl
الحياتي الغشاء بتكوين عال نقصان الى ادى التباين وهذا االصل الساللة ومن منهما المنتج .

جمهورية العراق التعليم العالي والبحث العلميوزارة
جامعة بغداد
كلية العلوم
Pseudomonas aeruginosa في مقاومة االميبينيم و تكوين الغشاء الحياتي في بكتريا oprDدور جين
مقدمة الى
كلية العلوم/جامعة بغداد
وهي جزء من متطلبات نيل درجة دكتوراه فلسفة علوم
في علوم الحياة/احياء مجهرية
من
هديل كريم مسافر
۲۰۰٥بكالوريوس احياء مجهرية/كلية العلوم/الجامعة المستنصرية
۲۰۰۷ماجستير احياء مجهرية/كلية العلوم/ الجامعة المستنصرية
بأشراف
أ.م.د. حارث جبار فهد المذخوري
۱٤۳٤ محرم ۲۰۱۳ الثاني تشرين


















