(Phospholipase C) from Pseudomonas aeruginosa - Journal of
10
JouRNAL OF BACTERIOLOGY, Oct. 1982, p. 431-440 Vol. 152, No. 1 0021-9193/82/100431-10$02.00/0 Coypright C 1982, American Society for Microbiology Cloning of a Phosphate-Regulated Hemolysin Gene (Phospholipase C) from Pseudomonas aeruginosa MICHAEL L. VASIL,* RANDY M. BERKA, GREGORY L. GRAY,t AND HIROSHI NAKAI Department of Microbiology and Immunology, University of Colorado Medical School, Denver, Colorado 80262 Received 31 March 1982/Accepted 15 June 1982 Phospholipase C (heat-labile hemolysin) of Pseudomonas aeruginosa is a phosphate (Pj-regulated extracellular protein which may be a significant virulence factor of this organism. The gene for this hemolytic enzyme was cloned on a 4.1- megadalton (Mdal) fragment from a BamHI digest of P. aeruginosa PA01 genomic DNA and was inserted into the BamHI sites of the multicopy Escherichia coli(pBR322) and P. aeruginosa(pMW79) vectors. The E. coli and P. aeruginosa recombinant plasmids were designated pGV26 and pVB81, respectively. A restriction map of the 4.1-Mdal fragment from pGV26 was constructed, using double and single digestions with BamHI and EcoRI and several different restriction enzymes. Based on information from this map, a 2.4-Mdal BamHII BglII fragment containing the gene for phospholipase C was subcloned to pBR322. The hybrid plasmids pGV26 and pVB81 direct the synthesis of enzymatically active phospholipase C, which is also hemolytic. The plasmid-directed synthesis of phospholipase C in E. coli or P. aeruginosa is not repressible by Pi as is the chromosomally directed synthesis in P. aeruginosa. Data are presented which suggest that the synthesis of phospholipase C from pGV26 and pVB81 is directed from the tetracycline resistance gene promoter. The level of enzyme activity produced by E. coli(pGV26) is slightly higher than the levels produced by P. aeruginosa(pMW79) under repressed conditions. In contrast, the levels produced by P. aeruginosa(pVB81) are at least 600-fold higher than the levels produced by P. aeruginosa(pMW79) under repressed conditions and approximately 20-fold higher than those produced by P. aeruginosa(pMW79) under derepressed condi- tions. The majority (85%) of the enzyme produced by E. coli(pGV26) remained cell associated, whereas >95% of the enzyme produced by P. aeruginosa(pVB81) was extracellular. Analysis of extracellular proteins from cultures of P. aerugino- sa(pMW79) and P. aeruginosa(pVB81) by high-performance liquid chromoto- graphy and sodium dodecyl sulfate-polyacrylamide gel electrophoresis revealed that the phospholipase C gene was cloned intact, and it is likely that several additional genes were cloned on the 4.1-Mdal fragment of DNA. It was also found that some of these genes encode proteins which are the same molecular weight as some previously described Pi-repressible proteins of P. aeruginosa. The existence of a Pi regulon of P. aeruginosa is proposed. It is likely that one of these genes also regulates the level of pyocyanin production by P. aeruginosa and that one or more play a role in transport or binding of Pi. The availability of the hybrid plasmids described herein will be useful in further studies on the role of this hemolysin in the virulence of P. aeruginosa and in the study of the genetics and physiology of Pi-regulated proteins. There is increasing evidence that the hemoly- infections caused by these organisms. In E. coli, sins of several gram-negative bacteria, including for example, hemolysin appears to play a role in Escherichia coli (7, 22, 33), Pseudomonas aeru- permitting this organism to initiate or sustain ginosa (2, 17, 19, 30), Vibrio parahemolyticus extraintestinal infections, particularly those in- (23), and Aeromonas hydrophila (26, 35), con- volving the urinary tract (7, 22). P. aeruginosa tribute to the pathogenesis of certain kinds of produces two hemolysins (19). One of the hemo- lysins is a heat-labile phospholipase C which t Present address: Department of Molecular Biology, Gen- catalyzes the hydrolysis of phosphatidylcholine entech, Inc., South San Francisco, CA 94080. to phosphorylcholine and diacylglycerol. The 431 Downloaded from https://journals.asm.org/journal/jb on 10 December 2021 by 14.33.109.95.
Transcript of (Phospholipase C) from Pseudomonas aeruginosa - Journal of

JouRNAL OF BACTERIOLOGY, Oct. 1982, p. 431-440 Vol. 152, No. 10021-9193/82/100431-10$02.00/0Coypright C 1982, American Society for Microbiology
Cloning of a Phosphate-Regulated Hemolysin Gene(Phospholipase C) from Pseudomonas aeruginosa
MICHAEL L. VASIL,* RANDY M. BERKA, GREGORY L. GRAY,t AND HIROSHI NAKAIDepartment of Microbiology and Immunology, University of Colorado Medical School, Denver, Colorado
80262
Received 31 March 1982/Accepted 15 June 1982
Phospholipase C (heat-labile hemolysin) of Pseudomonas aeruginosa is aphosphate (Pj-regulated extracellular protein which may be a significant virulencefactor of this organism. The gene for this hemolytic enzyme was cloned on a 4.1-megadalton (Mdal) fragment from a BamHI digest of P. aeruginosa PA01genomic DNA and was inserted into the BamHI sites ofthe multicopy Escherichiacoli(pBR322) and P. aeruginosa(pMW79) vectors. The E. coli and P. aeruginosarecombinant plasmids were designated pGV26 and pVB81, respectively. Arestriction map of the 4.1-Mdal fragment from pGV26 was constructed, usingdouble and single digestions with BamHI and EcoRI and several differentrestriction enzymes. Based on information from this map, a 2.4-Mdal BamHIIBglII fragment containing the gene for phospholipase C was subcloned to pBR322.The hybrid plasmids pGV26 and pVB81 direct the synthesis of enzymaticallyactive phospholipase C, which is also hemolytic. The plasmid-directed synthesisof phospholipase C in E. coli or P. aeruginosa is not repressible by Pi as is thechromosomally directed synthesis in P. aeruginosa. Data are presented whichsuggest that the synthesis of phospholipase C from pGV26 and pVB81 is directedfrom the tetracycline resistance gene promoter. The level of enzyme activityproduced by E. coli(pGV26) is slightly higher than the levels produced by P.aeruginosa(pMW79) under repressed conditions. In contrast, the levels producedby P. aeruginosa(pVB81) are at least 600-fold higher than the levels produced byP. aeruginosa(pMW79) under repressed conditions and approximately 20-foldhigher than those produced by P. aeruginosa(pMW79) under derepressed condi-tions. The majority (85%) of the enzyme produced by E. coli(pGV26) remainedcell associated, whereas >95% of the enzyme produced by P. aeruginosa(pVB81)was extracellular. Analysis of extracellular proteins from cultures of P. aerugino-sa(pMW79) and P. aeruginosa(pVB81) by high-performance liquid chromoto-graphy and sodium dodecyl sulfate-polyacrylamide gel electrophoresis revealedthat the phospholipase C gene was cloned intact, and it is likely that severaladditional genes were cloned on the 4.1-Mdal fragment ofDNA. It was also foundthat some of these genes encode proteins which are the same molecular weight assome previously described Pi-repressible proteins ofP. aeruginosa. The existenceof a Pi regulon of P. aeruginosa is proposed. It is likely that one of these genesalso regulates the level of pyocyanin production by P. aeruginosa and that one ormore play a role in transport or binding of Pi. The availability of the hybridplasmids described herein will be useful in further studies on the role of thishemolysin in the virulence of P. aeruginosa and in the study of the genetics andphysiology of Pi-regulated proteins.
There is increasing evidence that the hemoly- infections caused by these organisms. In E. coli,sins of several gram-negative bacteria, including for example, hemolysin appears to play a role inEscherichia coli (7, 22, 33), Pseudomonas aeru- permitting this organism to initiate or sustainginosa (2, 17, 19, 30), Vibrio parahemolyticus extraintestinal infections, particularly those in-(23), and Aeromonas hydrophila (26, 35), con- volving the urinary tract (7, 22). P. aeruginosatribute to the pathogenesis of certain kinds of produces two hemolysins (19). One of the hemo-
lysins is a heat-labile phospholipase C whicht Present address: Department of Molecular Biology, Gen- catalyzes the hydrolysis of phosphatidylcholine
entech, Inc., South San Francisco, CA 94080. to phosphorylcholine and diacylglycerol. The
431
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

432 VASIL ET AL.
other is a heat-stable glycolipid (15). Both hemo-lysins are produced during stationary phase inlow-phosphate media but are repressed in high-phosphate media (8, 9, 15). The proposedphysiological role of these hemolysins in theorganism is to act cooperatively with alkalinephosphatase in the liberation of Pi from phos-pholipids in the environment. In terms of patho-genesis, it has been proposed that the phospholi-pase C of P. aeruginosa is important in theinitiation of infection, particularly in the lung,where there is ample substrate for this enzymein the form of the phospholipids of surfactant(19). Studies on the substrate specificity of thishemolytic enzyme indicate that it is very activeon the phospholipids found in eucaryotic cellsbut has very little, if any, activity on phospholip-ids found in procaryotic membranes (2a). Thephospholipase C also appears to be a necrotic orcytolytic toxin, like the phospholipase C (alpha-toxin) of Clostridium perfringens (19).
In P. aeruginosa several studies have impli-cated the heat-labile hemolysin as a determinantof virulence in the pathogenesis of lung infec-tions (17, 30). These studies are of particularinterest in terms of patients with cystic fibrosis,in whom P. aeruginosa lung infections are asso-ciated with high mortality rates. Also, this labo-ratory has reported a significant association ofincreased heat-labile hemolysin production withisolates from urinary tract infection (2). Thepathogenesis of P. aeruginosa infections is com-plex and appears to involve the production ofseveral distinct toxins which have been implicat-ed as virulence factors (36). Therefore, geneticmethods including recombinant DNA technolo-gy, offer a rational approach for dissecting thecomplex mechanisms of the virulence of P.aeruginosa. We report here the molecular clon-ing and stable expression of the heat-labile he-molysin gene of P. aeruginosa in E. coli and inP. aeruginosa, a significant step in the study ofthe role of this toxin in disease. (In this paperhemolysin and phospholipase C will be usedinterchangeably.)
MATERIALS AND METHODSChemicals. Propidium iodide and p-nitrophenyl-
phosphorylcholine were obtained from Sigma Chemi-cal Co., St. Louis, Mo.
Bacterial strains and plasmids. The bacterial strainsand plasmids used in these studies are given in Table 1.
Assay of phospholipase C and hemolysin production.Conditions for the production of phospholipase C havebeen previously described and were used in this studyexcept that in some cases, where noted, dialysate ofTrypticase soy broth (BBL Microbiology Systems,Cockeysville, Md.) (10) was used instead of tryptoseminimal media (2, 8, 9) because of the better growthobtained, particularly with auxotrophic E. coli and P.aeruginosa PA02003 Hly+ derivatives. The concen-
TABLE 1. Bacterial strains and plasmidsa
Stock no. Genotype or Source/referencephenotypeBacterial strains
E. coliHB101 leu thr pro lac gal A. L. Taylor
Strr recA EndoIhsdR hsdM
KL320 trp his ilv pro lac R. K. HolmesrspL (4)
H22 htx mutant of R. K. HolmesKL320 (4)
P. aeruginosaPAO1 Prototroph chl-3 B. W. Holloway
(8-10)PA02003 argH3 str-39 cml- V. Krishnapillai
2 rec-2 FP- (34)PlasmidspBR322 Ap Tc Bolivar and
Backman (3)pMW79 Ap/Cb Sm (hybrid D. 0. Wood
of RSF1010 and (34)pBR322)
pGV26 Ap Hly+ This studypVB81 Ap Cb Sm Hly+ This studypRM82 Ap Hly+ This studya Genotype symbols are described by Bachmann
and Low (1). Ap, Tc, Sm, and Cb refer to resistance toampicillin, tetracycline, streptomycin, and carbenicil-fin, respectively. Hly refers to the hemolytic or phos-pholipase C phenotype of the recombinant plasmid.
tration of Pi in dialvsate of Trypticase soy broth (10mM) is sufficient to repress synthesis of phospholipaseC by 20- to 100-fold (9). Phospholipase C activity wasmeasured by p-nitrophenylphosphorylcholine hydro-lysis as previously described (2, 8, 9). Blood agar forscreening the genomic libraries consisted of basalmedium previously described by Johnson and Boese-Marrazzo (15) and was supplemented with 5% defi-brinated sheep erythrocytes.
Isolation and manipulation of DNA. Genomic DNAof P. aeruginosa PA01 was prepared by growing thecells overnight at 37°C and then extracting the DNA aspreviously described (6). Plasmid DNA from E. coliand P. aeruginosa was isolated by a modification ofthe procedure of Hirt (13). Briefly, 300 ml of an 18- to24-h culture of E. coli or P. aeruginosa grown at 37°Cin L-broth was centrifuged at 5,000 rpm for 10 min,and the cells were suspended in 10 ml of 5 mM Tris-hydrochloride (pH 8) in 25% sucrose. Lysozyme wasadded to a final concentration of 1 mg/ml, and the cellswere put on ice for 5 min. Then 5 ml of 0.2 M EDTA(pH 8.0) was added to each tube followed by 2 ml of10% sodium dodecyl sulfate (SDS), and the tubecontents were then blended in a Vortex mixer. Next,2.5 ml of 5 M NaCl was added to each tube and mixedgently by inversion of the tubes. This lysate was left onice overnight and then centrifuged at 12,000 rpm for 1h. The supernatant was precipitated with an equalvolume of isopropyl alcohol for 2 h at -20°C, and itwas then centrifuged at 10,000 rpm for 20 min. Thepellet was redissolved in water at room temperature.
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

CLONING OF A HEMOLYSIN GENE FROM P. AERUGINOSA 433
Undissolved material was centrifuged from the lysateat 5,000 rpm for 5 min. The lysate was then subjectedto propidium iodide-cesium chloride density gradientsas previously described (34).Construction of P. aeruginosa PAO1 genomic librar-
ies in E. coli HB101. P. aeruginosa PAO1 chromo-somal DNA (200 ,ug/ml; 80 ,ul) was digested to comple-tion with BamHI restriction endonuclease (BethesdaResearch Laboratories, Gaithersburg, Md.), mixedwith pBR322 (20 p.g/ml; 50 ,ul) cleaved with BamHI,and ligated in a total volume of 164 pd (T4 ligase[Bethesda Research Laboratories]; 1 U of DNA per,ug, 12°C, 18 to 24 h). The ligated DNA was trans-formed into E. coli HB101 by standard methods (21).Ampicillin-resistant (Ap), tetracycline-sensitive (Tc')transformants were chosen for further study. Thetransformants were initially selected on L-agar con-
taining 50 ,ug of ampicillin per ml. The frequency oftransformants for this experiment was 4 x 105 Aprtransformants per ,ug of input pBR322. The Apr trans-formants were scored for Tcs on L-agar plates contain-ing 30 p.g of tetracycline per ml. It was necessary toscore for Tc' transformants in this manner since therecently developed procedures for selection of Tc' didnot work for E. coli HB101 (20). However, it was
found that Tcs transformants were reasonably frequent(22%), making it relatively easy to choose large num-
bers of potential clones carrying P. aeruginosa genes.From a single ligation and transformation experiment,approximately 3,000 Apr Tc' clones were saved and,according to the formula of Clarke and Carbon (6),represent at least 95% of the genome of P. aeruginosa.A Sail library of P. aeruginosa PAO DNA was con-
structed in a similar manner.Cloning of the hemolysin gene in P. aeruginosa. The
overall scheme for cloning the phospholipase C gene isshown in Fig. 1. The recombinant pBR322 plasmidcarrying the gene for hemolysin production (pGV26)was digested with BamHI endonuclease and thenligated to BamHI-cleaved pMW79 (34), using theconditions described above. The ligated DNA was
transformed to P. aeruginosa PA02003, using meth-ods previously described (34). Transformants wereselected on brain heart infusion agar (Difco Labora-tories, Detroit, Mich.) containing 500 ,ug of carbenicil-lin per ml and scored for Tc' on brain heart infusionagar with 30 ,ug of tetracycline per ml and for Hly+ on
blood agar containing 500 p.g of carbenicillin per ml.Restriction enzyme procedures and mapping strategy.
All restriction enzymes were obtained from BethesdaResearch Laboratories, and conditions for restrictionenzyme reactions were as specified by the manufac-turer. Restriction mapping was performed with thepGV26 recombinant plasmid. Restriction mappingstrategy was as follows. A series of restriction en-
zymes were chosen which would not digest the vectorpBR322 or which had only one site in that vector.Also, enzymes were chosen which only cleaved theinserted DNA once or twice. Then, using doubledigestion with EcoRI or BamHI and the selectedenzyme, cleavage sites were determined after agarosegel electrophoresis, using a HindIII digest of bacterio-phage lambda as a molecular weight standard. In thismanner, the restriction map was constructed.Restriction enzyme-digested samples were run in
0.8% agarose gels with Tris-acetate buffer (pH 7.5) for
3 h at 100 V. DNA fragments were visualized bystaining with ethidium bromide (1 ,ug/ml) and photo-graphed in short-wavelength UV light.
Nick hnslation of plasmids and Southern blot hy-bridization with PAO1 genomic DNA. Southern blothybridization of the nick-translated 2P-labeled (27)pGV26 plasmid with BamHI-digested P. aeruginosaPAO1 DNA was performed as previously described(29).
Preparation of extraceUlular proteins for high-per-formance liquid chromotography (HPLC) and SDS-polyacrylamide gel electrophoresis (PAGE). Strains tobe examined for extracellular proteins were eachgrown in 500 ml of tryptose minimal medium (2) in 2-liter flasks for 18 h at 32°C on a gyratory shaker (-100rpm). Cultures were centrifuged at 5,000 rpm for 10min to remove the cells. The proteins in the superna-tants were precipitated with ammonium sulfate to 70%osaturation. The precipitated proteins were dissolved in5 ml of 10 mM Tris-hydrochloride (pH 7.2) and dia-lyzed against the same buffer for 12 h at 4°C. Thesamples were adjusted to identical volumes for furtherstudy.HPLC of extracellular proteins. High-performance
size exclusion chromatography of extracellular pro-teins was done with a Beckman model 334 liquidchromatograph equipped with a 60-cm SpherogelTSK-2000 column (Altex Scientific, Berkeley, Calif.).An isocratic buffer system composed of 50 mM Tris-hydrochloride buffer, pH 7.2, in 5% (vol/vol) glycerolwas used. Absorbance at 280 nm was monitored with aHitachi model 100-40 spectrophotometer equippedwith a liquid flow cell. Retention times and peakassignments were made with an Altex model C-R1Arecorder-integrator that was interfaced with the liquidchromatograph. The column was calibrated by chro-matographing protein standards and plotting the logM, of each standard against its retention time. Theprotein standards included phosphorylase b (Mr =94,000), bovine serum albumin (M, = 68,000), ovalbu-min (Mr = 43,000), carbonic anhydrase (Mr = 30,000),soybean trypsin inhibitor (M, = 20,100), and a-lactal-bumin (M, = 14,400). The retention times for theabove proteins were 11.68, 16.05, 18.48, 20.52, 25.25,and 27.92 min, respectively.SDS-PAGE. SDS-PAGE was done as described pre-
viously (9). Protein bands were developed with ammo-niacal silver nitrate as described by Oakley et al. (24).
RESULTSIsolation of an E. coli clone carrying the phos-
pholipase C gene from P. aeruginosa. The BamHIand SalI genomic libraries were first screenedfor phospholipase C production. Each of theclones was inoculated into 0.1 ml of tryptoseminimal medium in microtiter plates and incu-bated for 18 to 24 h at 32°C. Enzyme productionwas assayed by the hydrolysis of the artifi-cial substrate p-nitrophenylphosphorylcholine,which yields a yellow color (2). There was noclone found to produce phospholipase C by thismethod. At this point it was considered that theintact gene was not cloned, that it was notexpressed, and that the phospholipase C was notsecreted in E. coli. E. coli produces few extra-
VOL. 152, 1982
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.
434 VASIL ET AL.
EcoR IBamH I
I ~ Tc AA~rApr pBR322
pBR3222.9 M del P. aeruginosa PAOI
Chromosome BSmHlComplete digestion
BsmH 41\
|T4 Ligase
4.1 Mdaiinsertion ofPAOI DNA
N T4 Ligase
BsmHlEcoR ISamHoRi I
asmHl1 pMW7O 9 sPic+MHSy2T4 Ligse .6 Mdc 6
BCmHH/Bglll
EcoRI
pVB-S1Cbr TcO mr 8ur Pic*(HIy+)
12.7 Mdal
Smr
Cbr aBmH IEcoRli
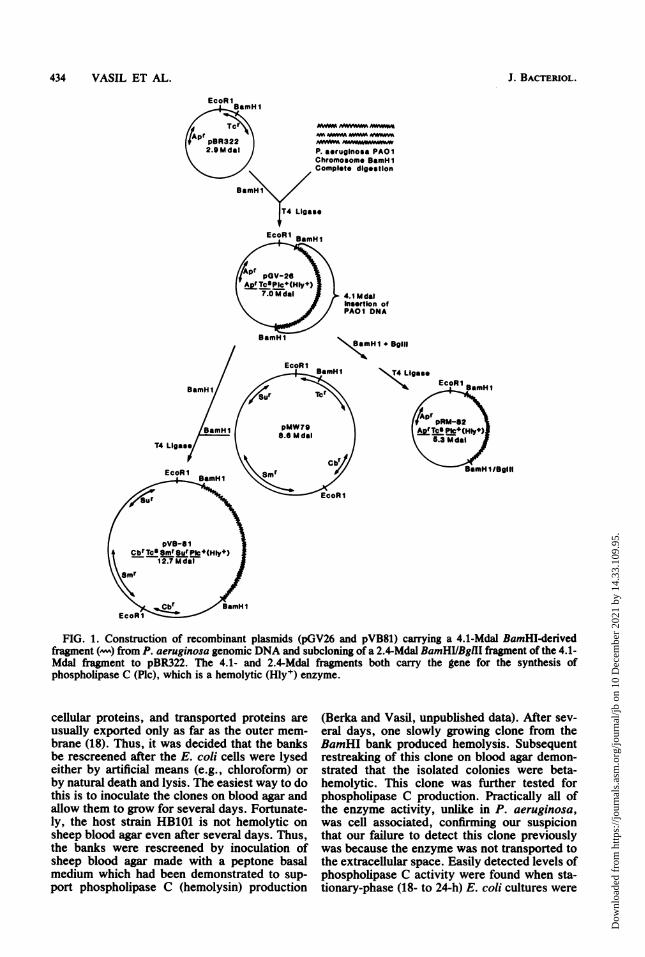
FIG. 1. Construction of recombinant plasmids (pGV26 and pVB81) carrying a 4.1-Mdal BamHI-derivedfragment (-) from P. aeruginosa genomic DNA and subcloning of a 2.4-Mdal BamHVBgllI fragment of the 4.1-Mdal fragnent to pBR322. The 4.1- and 2.4-Mdal fragments both carry the gene for the synthesis ofphospholipase C (Plc), which is a hemolytic (Hly+) enzyme.
cellular proteins, and transported proteins areusually exported only as far as the outer mem-brane (18). Thus, it was decided that the banksbe rescreened after the E. coli cells were lysedeither by artificial means (e.g., chloroform) orby natural death and lysis. The easiest way to dothis is to inoculate the clones on blood agar andallow them to grow for several days. Fortunate-ly, the host strain HB101 is not hemolytic onsheep blood agar even after several days. Thus,the banks were rescreened by inoculation ofsheep blood agar made with a peptone basalmedium which had been demonstrated to sup-port phospholipase C (hemolysin) production
(Berka and Vasil, unpublished data). After sev-eral days, one slowly growing clone from theBamHI bank produced hemolysis. Subsequentrestreaking of this clone on blood agar demon-strated that the isolated colonies were beta-hemolytic. This clone was further tested forphospholipase C production. Practically all ofthe enzyme activity, unlike in P. aeruginosa,was cell associated, confirming our suspicionthat our failure to detect this clone previouslywas because the enzyme was not transported tothe extracellular space. Easily detected levels ofphospholipase C activity were found when sta-tionary-phase (18- to 24-h) E. coli cultures were
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

CLONING OF A HEMOLYSIN GENE FROM P. AERUGINOSA 435
lysed with lysozyme, EDTA, and SDS or withchloroform. The enzyme had all of the proper-ties of the enzyme synthesized by P. aeruginosa(Table 2), and the enzyme activity from the E.coli clone was neutralized by antiserum to P.aeruginosa phospholipase C (Table 2).
Characterization of the DNA carrying the he-molysin gene and transfer to a P. aeruginosa host-vector system. A 7.0-megadalton (Mdal) plasmiddesignated pGV26 was isolated from the HB101Hly+ cells. A BamHI digest ofpGV26 containedtwo fragments: the 2.9-Mdal band correspondingto the molecular weight of the pBR322 vectorand a 4.1-Mdal fragment, presumably of P.aeruginosa origin (Fig. 2A). That the 4.1-Mdalfragment was in fact P. aeruginosa DNA wasconfirmed by Southern blot hybridization (29) ofthe nick-translated 32P-labeled (27) pGV26 plas-mid with BamHI-digested P. aeruginosa PA01genomic DNA and the lack of a detectable signalwith E. coli DNA (Vasil, unpublished data). Thehybridization results were positive with a frag-ment ofPA01 genomic DNA which had a mobil-ity identical to the 4.1-Mdal fragment frompGV26. HB101 was transformed to ampicillinresistance by pGV26, and 100% of the Aprtransformants tested produced the hemolyticenzyme. The 4.1-Mdal fragment is of more thansufficient size to encode for the phospholipase Cwhich has recently been purified in this labora-tory and found to be a 78,000-dalton protein (2a).
TABLE 2. Comparison of the cloned phospholipaseC (heat-labile hemolysin) in E. coli HB101(pGV-26)and the native enzyme from P. aeruginosa PA01"
Characteristic PA01 HB101(pGV26)Heat-labile reactivity with + +NPPCb
Resistance to 2% SDS + +Resistance to 1% 1- + +
mercaptoethanolSensitivity to 2 M urea + +Sensitivity to 1 mM ZnCl2 + +Neutralization of enzyme activ- + +
ity by antiserum against P.aeruginosa phospholipase C
aHB101(pGV26) cells (20 ml) were grown for 18 to24 h, centrifuged, treated with 10 ml of STET buffer(8% sucrose, 5% Triton X-100, 50 mM EDTA, 50 mMTris, pH 8.0), and incubated for 15 min at 370C.Lysozyme (1 ml; 10 mg/ml in water) was added, andthe mixture was incubated for 15 min at 37°C. SDS (1ml; 10%) was then added, and the mixture was incu-bated at 37°C for an additional 15 min. Enzyme fromP. aeruginosa PAO1 was previously purified in thislaboratory from culture supematants, and the aboveproperties have been documented elsewhere (2a).
b Enzyme activity was measured as described previ-ously (2).
A1 2 3 4 mw
Mdal
w-.---.- 1----6.4----
-6.4-~
4.3-
2.9/"'
1.6
- 1.4
Bmw I 2
FIG. 2. Restriction endonuclease cleavage analysisof hybrid plasmids carrying the phospholipase C gene.(A) Restriction analysis of recombinant plasmids de-rived from E. coli: lanes-1, BamHI digestion ofpBR322; 2, BamHI digestion of pGV26; 3, BamHI andBglII digestion of pGV26; 4, BamHI and BglII diges-tion of pRM82; MW, HindIII digestion of X. (B)Restriction analysis of recombinant plasmids derivedfrom P. aeruginosa: lanes-MW, HindIII digestion ofX; 1, BamHI digestion ofpMW79; 2, BamHI and BglIIdigestion ofpVB81. Arrows indicate BamHI and BglIIfragments of pVB81.
A preliminary restriction map of the 4.1-Mdalcloned P. aeruginosa DNA is shown in Fig. 3.This map was constructed with the pGV26 plas-mid from E. coli. While constructing a restric-tion map of the cloned P. aeruginosa DNA (Fig.3), we found that there was a single BglII site at2.4 Mdal in the cloned DNA which offered us anopportunity to reduce its size and somewhatlocalize the phospholipase C gene on the 4.1-Mdal insert. To do this, the pGV26 plasmid wasdigested with BamHI and BglII and then theDNA was religated (Fig. 1). The religated DNAwas then transformed to HB101, and Apr Hly+clones which contained plasmids smaller thanthe pGV26 were chosen for further study. It wasfound that the phospholipase C activity wasassociated with a 5.3-Mdal plasmid designatedpRM82 (Fig. 1 and 2A) and not with a plasmidcarrying the 1.7-Mdal fragment from pGV26,indicating that the phospholipase C gene waslocated on the 2.4-Mdal end of the cloned frag-ment. When the 2.4-Mdal BamHI/BglII frag-ment was ligated to BamHI-digested pBR322,the BglII site was lost since digestion of pRM82with BamHI and BglIl only yielded a linear 5.3-Mdal molecule (Fig. 2A).
VOL. 152, 1982
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

436 VASIL ET AL.
EcoR1BamH 1I I
pBR322 I /1
-0/0
B a m H 1mal/ I
I
0 0.1
Kpni PstI Bgl II
I II
I\ I
1 \ 1.5
Snai XheI \
Pvull Kpnl
p I I-10.7 0.8 0.9 1
I
1.9 2.4
Pvull BamHl
.13.6 4.1 Mdal
FIG. 3. Restriction endonuclease cleavage map of the 4.1-Mdal fragment derived from PAO1 genomic DNAand which carries the phospholipase C gene. The cloned fragment was not cleaved by EcoRI, HindIII, XorII,XbaI, or ClaI.
With the recent availability of a P. aeruginosahost-vector system (34), it was possible to re-clone the 4.1-Mdal DNA fragment carrying thephospholipase C from pGV26 and transfer thenew recombinant plasmid (pVB81) to P. aeru-ginosa. This was do-ne as described in Fig. 1 andin Materials and Methods. The new hybrid plas-mid, designated pVB81, directed the synthesisof substantially increased levels of phospholi-pase C in the rec-2 host strain PA02003. ThepVB81 plasmid appears to carry the same sizeinsert as the pGV26 plasmid (Fig. 1 and 2).However, since PA02003 is not a modification-less strain, it is somewhat difficult to digest thepVB81 plasmid isolated from P. aeruginosa withsome restriction enzymes. Even when the plas-mid is digested with as much as 5 U/Ipg for 24 hwith BglII, it is only partially digested by thisenzyme, but it is fully digested with BamHI (Fig.2B). Similar results are seen when the pVB81 isdigested with SalI. Upon digestion of the pVB81plasmid with Sail, there are only two fragmentsseen by electrophoresis. The pMW79 vectoronly has one Sall restriction site in the Tcr gene,like pBR322. In contrast, digestion of the pGV26plasmid isolated from E. coli HB101, a modifica-tionless strain, yields at least seven fragments.
Expression of the cloned phospholipase C genein E. coli and P. aeruginosa. The availability ofthe phospholipase C gene on a multicopy plas-mid in E. coli and P. aeruginosa offered anopportunity to compare the expression of thishemolytic enzyme in these two organisms. In P1-rich cultures of E. coli, the phospholipase C wasexpressed at detectable levels, but the majority(-90%) of the enzyme activity remained cellassociated (Table 3). This is consistent with theobservation that HB1O1(pGV26) only becomeshemolytic on blood agar after 3 to 4 days ofincubation. When the pGV26 plasmid was trans-formed to other E. coli strains, including astrain which has a chromosomal mutation lead-ing to hyperproduction of plasmid-directed syn-thesis of E. coli enterotoxin (4; kindly suppliedby R. K. Holmes), the levels of phospholipase Cwere not changed compared with HB101. Therealso were no differences in levels of enzyme
activity when HB1Ol(pGV26) was grown in P1-deficient or Pi-rich media (data not shown),indicating that in E. coli synthesis of the phos-pholipase C is not regulated by Pi. Interestingly,when HB1O1(pGV26) was grown in the pres-ence of autoclaved chlortetracycline, there wasa reproducible 25% increase in the total phos-pholipase C activity (Table 3), suggesting that itssynthesis in E. coli is directed from the pBR322Tcr promoter. Inducibility of the pBR322 Tcr
TABLE 3. Quantitation of the expression of thephospholipase C gene in E. coli and P. aeruginosa
PhospholipaseStrain C activity'
0.5 h 18 h
E. coli HB101(pBR322)Extracellular NDb 0Cell associated ND 0
E. coli HB101(pBR322)-CTcExtracellular ND 0Cell associated ND 0
E. coli HB101(pGV26)Extracellular ND 17.8Cell associated ND 121.0
E. coli HB101(pGV26)-CTExtracellular ND 25.8Cell associated ND 151.0
P. aeruginosa PA02003(pMW79)Extracellular 0.6 77.6Cell associated 0 ND
P. aeruginosa PA02003(pMW79)-CTExtracellular 0.5 77.8Cell associated 0 ND
P. aeruginosa PA02003(pVB81)Extracellular 393.5 NDCell associated 2.1 ND
P. aeruginosa PA02003(pVB81)-CTExtracellular 442.8 NDCell associated 0 ND
a Activity is expressed as the absorbance at 405 nmdivided by the optical density of the culture at 590 nm(2).
b ND, Not determined.c CT refers to growth in the presence of 20 ,ug of
autoclaved chlortetracycline per ml as described pre-viously (20).
pBR322a I I
Ir- w x
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

CLONING OF A HEMOLYSIN GENE FROM P. AERUGINOSA
protein has been previously reported (P. W.Rueckert and F. Reusser, Abstr. Annu. Meet.Am. Soc. Microbiol. 1982, H40, p. 119). Similarresults with the pVB81 plasmid in PA02003were found, perhaps not surprisingly, since thepMW79 vector is a hybrid plasmid of pBR322and RSF-1010 (34). Additional support of theidea that phospholipase synthesis is being direct-ed from the Tcr promoter is that the synthesis ofthe enzyme from the chromosomal gene inPA02003(pMW79) is not increased in the pres-ence of autoclaved chlortetracylcline (Table 3).As in E. coli, the plasmid-directed synthesis in
P. aeruginosa PA02003(pVB81) is not P1 regu-lated; however, >95% of the enzyme activityfrom P. aeruginosa is extracellular in contrast toE. coli HB101(pGV26) enzyme. On low-Pi bloodagar plates, after only 24 h, the hemolytic zonesurrounding PA02003(pVB81) is 20 to 50 timesgreater in surface area than that surroundingPA02003(pMW79) (data not shown). The levelsof enzyme activity from PA02003(pVB81) in P,-rich media are 500 to 1,000 times the level ofPA02003(pMW79), whereas in Pi-deficient me-dia the levels of enzyme produced byPA02003(pVB81) are 10 to 20 times that foundin cultures of PA02003(pMW79) (Table 3; Vasiland Berka, unpublished data). These data areconsistent with previously reported resultswhich indicate that phospholipase C productionis 20- to 100-fold repressible by Pi, dependingon the conditions and methods used for assay(8, 9).Comparisons were made of the extracellular
proteins from Ps-deficient cultures ofPA02003(pMW79) and PA02003(pVB81), usingHPLC and SDS-PAGE (Fig. 4A and B). TheHPLC chromatogram reveals that there is asubstantial increase in protein in the 75,000- to80,000-dalton range from the extracellular pro-teins of PA02003(pVB81) compared withPA02003(pMW79) (Fig. 4A). Assay of the frac-tions collected from the HPLC indicated that themajority of the phospholipase C activity fromboth chromatograms eluted between 11 and 13min. When the total enzyme activity from frac-tions eluting between 10 and 16 min were as-sayed, it was found that the activity from thePA02003(pVB81) fractions was 143 U/ml (1 U =1 ,mol of p-nitrophenylphosphorylcholine hy-drolyzed per min), whereas that from thePA02003(pMW79) chromatography fractionswas 7 U/ml. These data taken together indicatethat PA02003(pVB81) is synthesizing approxi-mately 20 times more enzyme thanPA02003(pMW79) and that the gene for phos-pholipase C was cloned intact.Examination of SDS-PAGE profiles (Fig. 4B)
of extracellular proteins of PA02003(pVB81)and PA02003(pMW79) revealed that in addition
to the phospholipase C there are five otherproteins which appear to have been cloned alongwith this enzyme on the 4.1-Mdal piece ofDNA.The molecular weights of these proteins are37,000, 34,000, 31,000, 29,000, and 28,000 andare indicated by arrows on Fig. 4B. The HPLCprofile also indicates that there is an increase inextracellular protein from PA02003(pVB81) inthis molecular weight range (Fig. 4A). Interest-ingly, three of these proteins have the samemolecular weight (37,000, 34,000, and 31,000) asthree Pi-repressible extracellular proteins fromP. aeruginosa PAO1 (8, 9). It is possible that oneor more of these proteins function in binding ortransport or both of Pi. In support of this hypoth-esis is the observation that the rate of P1 trans-port by PA02003(pVB81) is fourfold greaterthan that of PA02003(pMW79) (Vasil andBerka, unpublished data). It should be notedthat the conditions used for growth in theseexperiments were the same as those used for thestudy of extracellular Pi-repressible proteins inP. aeruginosa (8, 9). A protein band with in-creased intensity at 78,000 daltons correspond-ing to the known molecular weight of phospholi-pase C can be seen in the PAO2003(pVB81)track in Fig. 4B, supporting the HPLC data andindicating that the entire phospholipase C genehas been cloned. While performing some of theabove experiments, we noticed thatPA02003(pVB81) appeared to be producing asubstantially greater amount of a blue-greenpigment than did PA02003(pMW79). Furtherinvestigation revealed that this blue-green pig-ment was pyocyanin, which is a phenazine pig-ment produced by P. aeruginosa. Interestingly,it has been previously reported that the produc-tion of pyocyanin is repressed by Pi in themedium (14). We found that the production ofpyocyanin by PA02003(pVB81) was 10-to 14-foldincreased compared with PA02003(pMW79) andthat production ofpyocyanin was only repressed2-fold by >10 mM Pi (Berka and Vasil, unpub-lished data). Because the synthesis of pyocyanininvolves an extensive pathway, it is unlikely thatwe have cloned genes directly involved in thesynthesis of pyocyanin, but rather it seemslikely that we have cloned a gene involved in theregulation of pyocyanin production (2a).Using 135 as an average molecular weight for
an amino acid, the amount ofDNA necessary tocode for all of the proteins identified abovewould be 6.2 kilobases (4.1 Mdal), which isessentially the same size of cloned DNA carriedby pGV26 or pVB81.
DISCUSSION
In this report we have described the construc-tion of hybrid plasmids carrying several chromo-
VOL. 152, 1982 437
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

438 VASIL ET AL.
B 2A14
*S.8 K
,. =.i£.,j
1u-m-.F
.. -~
: K j;
._l.:
Rm
t
! e..X
FIG. 4. HPLC and SDS-PAGE of extracellular protein produced in low-Pi medium (tryptose minimalmedium) by PA02003(pMW79) and PA02003(pVB81). (A) HPLC chromatogram of extraceliular protein.Retention times for major protein peaks are designated by numbers above the peaks, and elution positions for themolecular weight standards are designated by the molecular weights with arrows. Protein samples were preparedas described in the text, and 1.0 ml ofeach of the samples was injected onto the gel permeation column TSK2000.(B) SDS-PAGE of the same samples of extracellular proteins; 10 1l of sample was applied to each lane. Therelative mobility of the molecular weight standards is designated on the left side, and the positions of the proteinswhich have been proposed to be coded for by the 4.1-Mdal BamHI fragment of PA01 genomic DNA areindicated by arrows. (Lane 1) Extracellular proteins of PA02003(pMW79); (lane 2) extracellular proteins ofPAO2003(pVB81).
somal genes from P. aeruginosa, including onewhich directs the synthesis of the heat-labilehemolysin (phospholipase C). Data have beenpresented that indicate that the entire phospholi-pase C gene was cloned intact and that thesynthesis of the enzyme from the recombinantplasmids is not regulated by Pi in E. coli or P.aeruginosa as is the chromosomal gene in P.aeruginosa. At least one likely reason for thelack of P1 control of the cloned gene expressionis that synthesis is directed from the Tcr genepromoter in pBR322 or pMW79. In support ofthis hypothesis is the observation that the plas-mid-directed synthesis of phospholipase C is
partially inducible by autoclaved chlortetracy-cine, a known inducer of the Tcr gene. Despitethe fact that the plc gene was on a multicopyplasmid, the level of activity of the phospholi-pase C from E. coli was only a little more thantwice that of the P. aeruginosa chromosomalgene-directed synthesis under repressed condi-tions. However, it is not possible to tell at thepresent time whether this is a result of reducedexpression of a Pseudomonas gene in E. coli, ashas been previously reported (11, 12), or wheth-er the activity of the synthesized enzyme isreduced, or both. Because the synthesis of plas-mid phospholipase C is most likely directed from
A
J. BACTERIOL.
A:%%. :.:...4 ::4 -,.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

CLONING OF A HEMOLYSIN GENE FROM P. AERUGINOSA 439
the Tcr promoter and not from its own Pi-regulated promoter, it may be that the phospho-lipase C protein in E. coli is a fusion productwith the Tcr protein. Alternatively, becausephospholipase C is an extracellular protein in P.aeruginosa, it is also possible that the enzymesynthesized in E. coli has a leader sequence thatis not cleaved by E. coli proteases, therebyreducing its potential enzyme activity (e.g., pro-enzyme). It is interesting to note at this pointthat the enzyme from E. coli has both hemolyticactivity and phospholipase C activity. If, in fact,the phospholipase C has a leader sequence andis fused with a Tcr protein, it seems remarkablethat P. aeruginosa is capable of handling theincreased synthesis from the multicopy plasmidand is capable of efficiently processing and se-creting relatively large amounts of the protein inits native form.Based on the data presented in Fig. 4B, three
genes of particular interest, in addition to thephospholipase C gene, which code for proteinswith molecular weights of 37,000, 34,000, and31,000 have also been cloned. The interestingaspect of these proteins is that their molecularweights are identical to three previously de-scribed Pi-repressible proteins from P. aerugin-osa (8, 9; R. E. W. Hancock, personal communi-cation). The conditions for production of theseextracellular proteins were identical in all cases.Thus, it is tempting to speculate that phospholi-pase C is a part of a Pi regulon similar to thatreported in E. coli (32) and that the gene for thishemolytic enzyme shares a common promoterwith several other Pi-regulated genes, some ofwhich were cloned on the 4.1-Mdal piece ofDNA. The function for these other Pi-regulatedproteins is not totally clear; however, it appearsthat we have cloned a gene which regulates theproduction of pyocyanin, a secondary metabo-lite which has been shown to be regulated by Pi(14), and that we have cloned a gene whichencodes a Pi transport or Pi-binding protein(Hancock, personal communications; Vasil andBerka, unpublished data). It is clear that thealkaline phosphatase gene of P. aeruginosa wasprobably not cloned on the 4.1-Mdal insert be-cause no alkaline phosphatase activity could bedetected in Pi-rich cultures of PA02003(pVB81)(Vasil and Berka, unpublished data). In thisregard, it has recently been shown that thesynthesis of alkaline phosphatase and phospholi-pase C are coordinately regulated by a singlegenetic locus in P. aeruginosa (8, 9). Therefore, ifboth alkaline phosphatase and phospholipase Care part of the same operon in P. aeruginosa, itmay be that alkaline phosphatase is more proxi-mal to the promoter in relation to phospholipaseC.The availability of recombinant plasmids car-
rying the phospholipase C gene ofP. aeruginosashould facilitate studies on the role of this hemo-lytic enzyme in the pathogenesis of Pseudomo-nas infections and its function in the Pi-scaveng-ing system of P. aeruginosa. Further studieswhich could be of interest, using the recombi-nant plasmid described herein, are as follows. (i)With regard to the study of the role of thehemolysin in the virulence of P. aeruginosa, itshould be possible to: (a) induce, in vitro or invivo, an inactivating mutation in the structuralgene for hemolysin (16, 28); (b) transfer thismutant gene to a variety of P. aeruginosa clini-cal strains from different sources; and (c) isolatestable recombinants which are altered only inhemolysin (phospholipase C) activity (16). Itwould then be possible to more precisely assessthe role of heat-labile hemolysin in Pseudomo-nas pathogenesis by comparing virulence ofsuch recombinant strains with their parentalstrains in several well-characterized experimen-tal animal models (5, 25, 31, 37). (ii) With thecloned hemolysin gene from P. aeruginosa as aprobe, it is now possible to do comparativehomology studies on the various hemolysins ofgram-negative bacteria at the molecular level.(iii) Because of the Pi-repressible nature of phos-pholipase C synthesis, it will be of interest to usethe cloned plc gene as well as the genes for theother Pi-repressible proteins and a series ofrecently well-characterized Pi regulatory mu-tants (2, 8, 9) to examine the genetic and physio-logical mechanisms of the regulation of Pi-re-pressible proteins in P. aeruginosa.The restriction map shown in Fig. 3 should be
useful for subcloning the phospholipase C geneand insertional inactivation of this gene and anyother gene of interest found on this DNA. Thismap also is probably sufficient to sequence atleast the phospholipase C (Hly+) gene of P.aeruginosa.
Finally, because the phospholipase C has sev-eral unusual properties, including a significantnumber of ornithine and hydroxyproline resi-dues, unusually avid hydrophobicity, and astrong preference for phospholipids with substi-tuted ammonium groups (2a), it will be of inter-est to obtain a DNA sequence of the gene forstudies of the structure-function relationships ofthis enzyme as well as of other similar hemoly-sins.
ACKNOWLEDGMENTSThis study was supported by Public Health Service grant
AI15940 from the National Institute of Allergy and InfectiousDiseases to M.L.V.We thank Peter Melby for excellent technical assistance.
LITERATURE CITED1. Bachmann, B. J., and K. B. Low. 1980. Linkage map of
Escherichia coli K-12, edition 6. Microbiol. Rev. 44:1-56.2. Berka, R. M., G. L. Gray, and M. L. Vasil. 1981. Studies
VOL. 152, 1982
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.

440 VASIL ET AL.
of phospholipase C (heat-labile hemolysin) in Pseudomo-nas aeruginosa. Infect. Immun. 34:1071-1074.
2a.Berka, R. M., and M. L. Vasil. 1982. Phospholipase C(heat-labile hemolysin) ofPseudomonas aeruginosa: puri-fication and preliminary characterization. J. Bacteriol.152:239-245.
3. Bollvar, F., and K. Beckman. 1979. Plasmids ofEscherich-ia coli as cloning vectors. Methods Enzymol. 68:245-267.
4. Bramccwl, M. G., E. M. Twiddy, W. B. Balne, and R. K.Holmes. 1981. Isolation and characterization of hypertox-inogenic (htx) mutants of Escherichia coli KL320 (pCG86). Infect. Immun. 32:1034-1044.
5. Cash, H. A., D. E. Woods, B. McCulough, W. G. Johan-son, Jr., and J. A. Ba. 1979. A rat model of chronicrespiratory infection with Pseudomonas aeruginosa. Am.Rev. Respir. Dis. 119:453-359.
6. Clarke, L., and J. Carbon. 1976. A colony bank containingsynthetic Col E 1 hybrid plasmids representative of theentire E. coli genome. Cell 9:91-99.
7. Evans, D. J., D. G. Evans, C. Hohne, M. A. Noble, E. V.Holdane, H. Liar, and L. S. Young. 1981. Hemolysin andK antigens in relation to serotype and hemagglutinationtype of Escherichia coli isolated from extraintestinal in-fection. J. Clin. Microbiol. 13:171-178.
8. Gray, G. L., R. M. Berka, and M. L. Vadl. 1981. APseudomonas aeruginosa mutant nonderepressible fororthophosphate-regulated proteins. J. Bacteriol. 147:675-678.
9. Gray, G. L., R. M. Berka, and M. L. Vadl. 1982.Phospholipase C regulatory mutation of Pseudomonasaeruginosa that results in constitutive synthesis of severalphosphate-repressible proteins. J. Bacteriol. 150:1221-1226.
10. Gray, G. L., and M. L. Vadl. 1981. Isolation and geneticcharacterization of toxin-deficient mutants of Pseudomo-nas aeruginosa. J. Bacteriol. 147:275-281.
11. Hedgs, R. W., and A. E. Jacob. 1977. In vivo translationof genes ofPseudomonas aeruginosa onto a promiscouslytransmissible plasmid. FEMS Microbiol. Lett. 2:15-19.
12. Hodges, R. W., A. E. Jacob, and I. P. Crawford. 1977.Wide ranging plasmid bearing the Pseudomonas aerugi-nosa tryptophan synthase genes. Nature (London)267:283-284.
13. Hirt, B. 1967. Selective extraction of polyoma DNA frominfected mouse cell cultures. J. Mol. Biol. 26:365-369.
14. Ingledw, M. W., and J. J. R. Campbell. 1969. A newresuspension medium for pyocyanin production. Can. J.Microbiol. 15:595-598.
15. Johnson, M. K., and D. Boese-Marrazzo. 1980. Productionand properties of heat-stable extracellular hemolysin fromPseudomonas aeruginosa. Infect. Immun. 28:1028-1033.
16. rsapa, V. 1979. DNA insertion mutagenesis in aPseudomonas aeruginosa R plasmid. Plasmid 2:237-246.
17. Kurloka, S., and P. V. Liu. 1967. Effect of the hemolysinof Pseudomonas aeruginosa on phosphatides and onphospholipase C activity. J. Bacteriol. 93:670-674.
18. La1aronl, J. C., and R. C. PortaHer. 1981. Genetic andbiochemical characterization of periplasmic-leaky mu-tants of Escherichia coli K-12. J. Bacteriol. 145:1351-1358.
19. Un, P. V. 1979. Toxin of Pseudomonas aeruginosa, p. 63-68. In R. G. Doggett (ed.), Pseudomonas aeruginosa.Academic Press, Inc., New York.
20. Maloy, S. B., and W. D. Nunn. 1981. Selection for loss oftetracycline resistance by Escherichia coli. J. Bacteriol.145:1351-1358.
21. Maendel, M., and A. Hip. 1970. Calcium-dependent bacte-riophage DNA infection. J. Mol. Biol. 53:159-162.
22. Mlusew, B. H., J. jorgemson_, G. W. Counts, and S.Falkow. 1978. Association of hemolysin production, he-magglutination of human erythrocytes, and virulence forchicken embryos of extraintestinal Escherichia coli iso-lates. Infect. Immun. 20:50-54.
23. Mlyamato, Y., Y. Obara, T. N. Nlkkawa, S. Yamal, T.Kato, Y. Yamada, and M. Ohash. 1980. Simplified purifi-cation and biophysicochemical characteristics of Kan-agawa phenomenon-associated hemolysin of Vibrio para-haemolyticus. Infect. Immun. 28:567-576.
24. Oakley, B. R., 0. R. Kirsch, and N. R. Norris. 1980. Asimplified ultrasensitive silver stain for detecting proteinsin polyacrylamide gels. Anal. Biochem. 105:361-363.
25. Petit, J. C., D. Sicord, S. Bago, and G. L. Daguet. 1981.Effect of active and passive immunization on the develop-ment of experimental Pseudomonas aeruginosa pyelone-phritis in mice. Can. J. Microbiol. 27:93-97.
26. RIddle, L. M., T. E. Graham, and R. L. Amborski. 1981.Medium for the accumulation of extracellular hemolysinand protease by Aeromonas hydrophila. Infect. Immun.33:728-738.
27. Rlgby, P. W., J. Dleckman, C. Rhodes, and P. Berg. 1977.Labeling deoxyribonucleic acid to high specific activity invitro by nick translation with DNA polymerase I. J. Mol.Biol. 113:237-251.
28. Ruvkun, G. B., and F. M. Ausubel. 1981. A generalmethod for site-directed mutagenesis in prokaryotes. Na-ture (London) 289:85-88.
29. Soutern, E. M. 1975. Detection of specific sequencesamong DNA fragments separated by gel electrophoresis.J. Mol. Biol. 98:503-517.
30. Southrn, P. M., B. B. Mays, A. D. Perce, and J. P.Sanford. 1970. Pulmonary clearance of Pseudomonasaeruginosa. J. Lab. Clin. Med. 76:548-559.
31. Stlerltz, D. D., and I. A. Holder. 1975. Experimentalstudies of the pathogenesis of infections due to Pseudo-monas aeruginosa: description of a burned mouse model.J. Infect. Dis. 131:688-691.
32. Wanner, B. L., and P. Lattrl. 1980. Mutants affected inallcaine phosphatase expression: evidence for multiplepositive regulators for the phosphate regulon in Esche-richia coli. Genetics 96:353-366.
33. Welch, R. A., E. P. Deger, B. Mlnshew, and S. Falkow.1981. Haemolysin contributes to virulence of extra-intes-tinal E. coli infections. Nature (London) 294:665-667.
34. Wood, D. 0., M. F. Hoflnger, and M. B. Tlndof. 1981.Versatile cloning vector for Pseudomonas aeruginosa. J.Bacteriol. 145:1448-1451.
35. Wretlnd, B., R. MolIby, and T. Wadstrom. 1971. Separa-tion of two hemolysins from Aeromonas hydrophila byisoelectric focusing. Infect. Immun. 4:503-505.
36. Young, L. S. 1980. The role of exotoxins in the pathogene-sis of Pseudomonas aeruginosa. J. Infect. Dis. 142:626-630.
37. Ziegler, E. J., and B. H. Douglas. 1979. Pseudomonasaeruginosa vasculitis and bacteremia following conjuncti-vitis: a simple model of fatal Pseudomonas infection inneutropenia. J. Infect. Dis. 139:288-296.
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
10
Dec
embe
r 20
21 b
y 14
.33.
109.
95.