Outline - Unife
52
Outline • Introduction; • Ion Torrent platform – how does it work?; • Library preparation; • NGS data analysis; • Pros and cons. Amplicon sequencing project
Transcript of Outline - Unife

Outline
• Introduction;
• Ion Torrent platform – how does it work?;
• Library preparation;
• NGS data analysis;
• Pros and cons.
Amplicon sequencing project

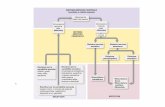
Amplicon sequencing project – NGS data analysis
Ion Torrent report

Amplicon sequencing project – NGS data analysis
“Ion Server” approach: • Alignment; • Duplicate removal; • Variant caller.
Customised approach: • Up to you!
When it is useful: • Standard approaches (such as cancer panel, etc.); • Medical genetics;
When it is useful: • Not straightforward projects/organisms; • Structural variation; • Explore the data; • Customise some steps.
There are no strict/standard rules to analyses NGS data, there are some standard pipelines but it most depends on your case-study.

Amplicon sequencing project – NGS data analysis
What shall I do next?
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
It is not THE way to analyse NGS/Ion Torrent data but it is one possible way to analyse my data (hopefully the best one!).
What is it needed to produce the raw data?
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

Amplicon sequencing project – before the raw data
Basecalling
Per-base quality scoring
Trimming
Raw data
Base recalibration
Before the raw data:
Io
n
To
rr
en
t
su
it
e
(I
on
S
er
ve
r)
Filtering

Amplicon sequencing project – before the raw data
Basecalling Calling the base for each well
Base recalibration
Before the raw data:
Base recalibration is a process to improve base calls by relearning the homopolymer flow signal distribution from the alignment of a
fraction of library reads.
Before recalibration After recalibration

Per-base quality scoring
Trimming
Amplicon sequencing project – before the raw data
Base quality score (BQ): • Phred-scale value; • -10*log_10(error rate);
Phred Quality Score
Probability of incorrect base call
Base call accuracy
10 1 in 10 90%
20 1 in 100 99%
30 1 in 1000 99.9%
40 1 in 10000 99.99%
50 1 in 100000 99.999%
• Removal of adapter sequence; • Removal of lower-quality 3' Ends with Low Quality Scores.
Filtering • Removal of short reads; • Removal of adapter dimers; • Removal of reads lacking sequencing key; • Removal of reads with off-scale signal; • Removal of polyclonal reads.

Amplicon sequencing project – before the raw data
Filtering • Removal of short reads; • Removal of adapter dimers; • Removal of reads lacking sequencing key; • Removal of reads with off-scale signal; • Removal of polyclonal reads.
An Ion Sphere Particle is clonal if all of its DNA fragments are cloned from a single original template. All the fragments on such a bead are identical, and they respond in unison as each nucleotide is flowed in turn across the chip.
A adaptper Barcode
DNA fragment to be sequenced
P1 adapter

Amplicon sequencing project – before the raw data
• Clonal amplification, ~50% of flows are 0-signal flows; • The positive flows cluster around integer values.
• Polyclonal amplification, 19% of flows are 0-signal flows; • The positive flows no longer cluster exclusively around integer values.
• Super-mixed beads, ~0% of flows are 0-signal flows; • The positive flows do not cluster around integer values at all.

Amplicon sequencing project – Raw data
FASTQ file
Raw data

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

Amplicon sequencing project – Alignment
alignment – process of determining the most likely location within the genome for the observed DNA read
raw reads reference genome

Amplicon sequencing project – Alignment
trade-off: speed vs sensitivity – the higher the accuracy the longer the alignment run
two classes of methods:
Burrows-Wheeler
• Fast • less robust at high divergence with
reference genome • e.g. bwa
Hashing
• slow (needs more memory) • robust at high divergence with
reference genome • e.g. stampy
short reads: ranging between 150bp and 200bp, the shorter the read the harder is to find its location in the genome big amount of data: computationally challenging for memory and speed

Amplicon sequencing project – Alignment
TMAP – Ion Torrent suite
Burrows-Wheeler based software Hashing based software

raw reads reference genome
low MQ: the probability of mapping to different locations is high, but no perfect multiple matches
high MQ: a single match
MQ0: a perfect multiple match
What if there are several possible places to align your sequencing read? This may be due to: - Repeated elements in the genome - Low complexity sequences - Reference errors and gaps MQ is a phred-score of the quality of the alignment
Amplicon sequencing project – Alignment, Mapping Quality (MQ)

SAM/BAM format
SAM – sequence alignment map BAM – binary alignment map Standard formats for alignment BAM is the binary version of SAM – reduced size, easier to store and to access but the full information is not readable by human eye
Amplicon sequencing project – Alignment, BAM and SAM file

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

Amplicon sequencing project – Indexing/sorting/RG line
Index (.bai) file: This file acts like an external table of contents, and allows programs to jump directly to specific parts of the bam file without reading through all of the sequences. (samtools). Sort: sort the reads in the bam file by either chromosome position or name (samtools). Add RG line: line containing information about read group identifier, platform name, sample name, library name, etc....
For each bam file we have to:

SAM – sequence alignment map BAM – binary alignment map Tools to visualise bam files such as IGV (http://www.broadinstitute.org/igv/home) and Tablet (http://ics.hutton.ac.uk/tablet/ ).
Amplicon sequencing project – Alignment, BAM and SAM file

Amplicon sequencing project – Alignment, BAM and SAM file

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

Amplicon sequencing project – Local realignment
Short indels in the sample relative to the reference sequence can pose difficulties for alignment programs. Indels occuring towards the ends of the reads are often not aligned correctly, introducing an excess of SNPs.
It uses the full alignment context to determine whether the indel exists. Two-step process: 1. RealignerTargetCreator: it determines the small suspicious intervals which
are likely in need of realignment (GATK software); 2. IndelRealigner: it runs the realignment on those intervals (GATK software).

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

PCR is used during library preparation. This can results in duplicate DNA fragments in the final library prep. PCR-free protocols exist but require a large amount of DNA.
It can result in false SNPs calls. Duplicates may fake a high coverage thus giving high support to some variants (picard software).
Amplicon sequencing project – NGS data analysis
C
C
C
C
A
Possible heterozygote, SNP call
Ref call

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g
We can check the coverage!

Amplicon sequencing project – Coverage
coverage per position GATK/BEDtools
Chromosome name Position Coverage phax5574-500bp_up_down_ref 16994 154 phax5574-500bp_up_down_ref 16995 153 phax5574-500bp_up_down_ref 16996 152 phax5574-500bp_up_down_ref 16997 149 phax5574-500bp_up_down_ref 16998 148 phax5574-500bp_up_down_ref 16999 145 phax5574-500bp_up_down_ref 17000 149 phax5574-500bp_up_down_ref 17001 151 phax5574-500bp_up_down_ref 17002 149

Amplicon sequencing project – Coverage

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

variant calling
SNPs indels SV
samtools GATK: 1. Unified Genotyper 2. Haplotype caller
samtools GATK: 1. Unified Genotyper 2. Haplotype caller Dindel
SVMerge – pipeline combining many
different tools
Amplicon sequencing project – Variant calling and filtering
Haplotype caller: not for non-diploid organisms and pooled samples.
GATK Samtools
SNP true positive rate 0.769 0.851
SNP false positive rate 0.231 0.148
samtools

Amplicon sequencing project – Variant calling and filtering
Factors to consider: - Base call qualities of each supporting base - Proximity to indels and homopolymer run - Mapping qualities of the reads supporting the SNP (increased read length or paired-
end help MQ scores) - Sequencing depth - Individual vs multi-sample calling
Multi-sample calling → better rescue of low frequency SNPs
VCF file: Standardised format for storing DNA polymorphism data - SNPs, indels, SV - Rich annotations Can store variant information over many samples Record meta-data about the site - dbSNP accession, filter status Very flexible - Tags can be introduced to describe new types of variants - Different VCF files may contain different information/annotations

Amplicon sequencing project – Variant calling and filtering
VCF file had two sections: - Header - Data
Header lines starting with ##: arbitrary number of meta-information lines line starting with #: column definition – mandatory columns include: CHROM chromosome POS position of the start of the variant ID unique identifier of the variant (e.g. rs number for SNPs) REF reference allele ALT comma separated list of alternate non-reference alleles QUAL phred-scaled quality score FILTER site filtering information INFO user extensible annotation (e.g. samtools and GATK may differ in this) samples follow
Data one line per site (all columns described above per line); useful information per site and per sample

Amplicon sequencing project – Variant calling and filtering

Amplicon sequencing project – Variant calling and filtering
GT: genotype 0=ref, 1=alt; PL: phred-scaled genotype likelihoods (For a phred-scaled likelihood of P, the raw likelihood of that genotype L = 10-P/10 , so the higher the number, the less likely it is that your sample has that genotype); DP: depth of coverage; SP: phred-scaled strand bias P-value, it tests if variant bases tend to come from one strand; GQ: genotype quality, encoded as a phred quality -10log_10p(genotype call is wrong).
GT:PL:DP:SP:GQ 0/0;0,255,255;138;0;99

240 samples, 3 PHAX regions spanning 49070bp:
• Variant calling with BQ13 and MQ0 (standard parameters): 512 sites;
• Variant calling with BQ20 and MQ50: 419 sites;
• Filtering for DP10 (DP>=10) and excluding heterozygotes positions (converted in
missing data);
• Sites with >= 5% of missing data were removed (113):306 sites;
Amplicon sequencing project – Variant calling and filtering

Amplicon sequencing project – Variant calling and filtering
Missing data per site (filtered per 5% but several threshold were tested):
Threshold missing data # removed sites
2.5% 121
5% 113
10% 99
20% 90
30% 70
50% 60
80% 46
100% 13
0
20
40
60
80
100
120
140
2.5% 5% 10% 20% 30% 50% 80% 100%
# si
tes
Threshold for missing data filtering
Variant_sites_240_samples
missing_data

Amplicon sequencing project – Variant calling and filtering
• Annotate the variants using dbSNP 138;
• SnpGap 10 (each snp within a 10bp around a gap will be filtered) and GapWin 3 (window size for filtering adjacent gaps);
Threshold QUAL (0-999) # kept sites
QUAL > 0 306
QUAL > 10 302
QUAL > 50 300
QUAL > 100 297
QUAL > 200 283
QUAL > 300 283
QUAL > 500 283
QUAL >= 999 283
270
275
280
285
290
295
300
305
310
> 0 > 10 > 50 > 100 > 200 > 300 > 500 >= 999#
kep
t si
tes
QUAL threshold
Variant_sites_240samples
QUAL
QUAL >= 200 would be the best value, 23 sites would be discarded, 3 out of 23 are dbSNP annotated...I did not filter for it.
QUAL phred-scaled quality score for the assertion made in ALT. i.e. -10log_10 prob(call in ALT is wrong). If ALT is ”.” (no variant) then this is -10log_10 p(variant), and if ALT is not ”.” this is -10log_10 p(no variant).

• Samples with >= 5% of missing data were removed (2);
Amplicon sequencing project – Variant calling and filtering
Final dataset: • 238 samples (96_NTH005 and SP15); • 297 variant sites;
PHAX 3115 PHAX 5574 PHAX 8913 Total
# variant sites 33 214 50 297
PHAX region 3115 5574 8913
Average coverage 169.3x 208.3x 164.6x
# variants (SNPs) 33 214 50
SNP density (SNP/kb) 6.6 5.6 8.3

PAR
1
PAR
2
PH
AX
31
15
P
HA
X 5
57
4
PH
AX
89
13
X c
hro
mo
som
e
Average coverage across 238 samples Amplicon sequencing project – Variant calling and filtering

Amplicon sequencing project – Variant calling and filtering
42% 68%
32%
58%
Singletons and non-singleton sites
non-singletonsites
singletons indbSNP
newsingletons
0%
20%
40%
60%
80%
100%
vari
ant
site
s
singletons - EU
singletons - ME
singletons - YRI
non-singleton sites

Amplicon sequencing project – NGS data analysis
Data processing
Raw data
Alignment TMAP
Indexing/sorting/RG line samtools + picard
Local realignment GATK
Duplicate removal Picard
Fo
r
al
l
sa
mp
le
s
Multi-sample variant calling samtools
Validation
False Positive (FP%) and False Negative percentages (FN%)
Chromosome position and reference allele concordance
V a l i d a t i o n
Filtering (vcf file) Validation
(SNP chip, Complete Genomics)
Filtering variant sites
Sets of filters for several parameters
(BQ, MQ, DP, missing data per site/sample)
F i l t e r i n g

Amplicon sequencing project – Validation
• Any NGS dataset needs a validation step to check the quality of the data and to estimate the error rate in our experiment;
• Validation can be performed in different ways:
• Sanger sequencing of a subset of new
SNPs; • Custom SNP chip approach; • NGS with a different platform; • Comparison with already sequenced
samples in publicly available dataset (i.e. 1000 Genome Project, Complete Genomics, etc….).

Amplicon sequencing project – Validation
Drmanac, et al., Science 2010
69 full genomes data: • A Yoruban trio; • A Puerto Rican trio; • A 17-member CEPH pedigree across three generations; • A diversity panel representing unrelated individuals from
nine different populations; Some African (YRI) and European (CEU) samples are included; 9 samples included in my dataset;

Amplicon sequencing project – Validation
Specificity vs Sensitivity = False Positive vs False Negative
our sequenced sample
external source of variation [same sample] – good quality data (i.e. Complete genomics)
TP true positive
FP false positive
TN true negative
FN false negative
high specificity
high sensitivity
low FP
low FN

Amplicon sequencing project – Validation
9 samples included in my dataset; Variant calling all sites (including reference sites and not only SNPS); 427 kb compared across 9 samples;
True calls (%) FP (%) FN (%)
99.9995 0.0005 0
Overall both FP and FN are low confirming the high quality of the data.
True negative
+ True
positive

Outline
• Introduction;
• Ion Torrent platform – how does it work?;
• Library preparation;
• NGS data analysis;
• Pros and cons.
Amplicon sequencing project

Amplicon sequencing project – Pros and cons
Ion PGM Pros: • Ion Torrent platform performs well for small NGS project;
• Fast run time and cost-effective for small scale project;
• Very high data quality;
• Torrent suite software for standard approaches (for not geeky people!);
Cons: • No standard pipeline in analysis NGS data, some general rules though; • Not extremely precise for either small or big indels;
• Not suitable for whole-genome sequencing (see Ion Pronton);
• Remember homopolymer issues (i.e. telomeres).

• Be smart in designing your experiment (i.e. coverage, barcodes, etc…) ;
• Be practical and “creative” in customising the best pipeline for your project ;
• Be critical regarding your data!
• Consider the information you loose at each filtering step;
• Check the error rate in your experiment;
• NGS technology has many advantages but Sanger sequencing was easier!
Amplicon sequencing project – Conclusions

software website
bwa http://bio-bwa.sourceforge.net/
picard http://picard.sourceforge.net/
samtools http://samtools.sourceforge.net/
GATK http://www.broadinstitute.org/gatk/
tablet http://bioinf.scri.ac.uk/tablet/
vcftools http://vcftools.sourceforge.net/
Useful resources:
Jia P et al, Plos One , 2012 – Variant calling. FreeBayes, https://wiki.gacrc.uga.edu/wiki/Freebayes , variant calling software.

Useful resources:
Jia P et al, Plos One , 2012 – Variant calling. FreeBayes, https://wiki.gacrc.uga.edu/wiki/Freebayes , variant calling software.

Acknowledgments
• Mark Jobling
• Alec Jeffreys
• Rita Neumann
• Pille Hallast
• Chiara Batini

Conclusions and future work
• RepeatSeq performs really well for m5753 and m5751…even though this dataset is pretty small, I would expect a low error rate on a bigger scale project;
• LobSTR performs quite well for m2036 (70% accuracy) but probably it is still worth going on with the ABI typing;
• m9053 does not have enough reads to be called by these tools…possibly it depends on the genomic context related to sequencing issues…ABI typing still needed;
• LobSTR error rate looks higher towards long allele compared to the reference sequence and among the wrong calls it prefers the reference allele;
• A validation dataset /subset typed with the ABI seems to be still needed.

Quality Score Predictors Torrent software uses the following six predictors that are correlated with empirical base call quality: P1 Penalty Residual: A penalty based on the difference between predicted and actual flow values. Computed by the base caller. P2 Local noise: Noise (defined as the maximum absolute difference between the flow value and the nearest integer) in the immediate neighborhood (plus/minus 1 base) of the given base. P3 Beverly Events: Number of high-residual flows in the 20-flow window around the flow containing the base. A flow has high residual when the normalized difference between the observed and model-predicted signal exceeds 0.4 or falls below –0.4. The more high-residual flows in the window, the lower quality the base call. P4 Multiple incorporations: Number of incorporated bases in this flow. Length of the homopolymer. For multiple incorporations of the same nucleotide in one flow, the last base in the incorporation order is assigned a value equivalent to the total number of incorporations. All other bases in the sequence of the multiple incorporations are assigned the value 1. P5 Environment noise: The average signal noise (defined as the absolute difference between the flow value and the nearest integer) in the neighborhood (plus/minus 5 bases) of the given base. P6 State Inphase: Live polymerase in phase.