
Orphan Nuclear Receptors as eLiXiRs and FiXeRs of Sterol ...
22
Orphan Nuclear Receptors as eLiXiRs and FiXeRs of Sterol Metabolism Timothy T. Lu 1 , Joyce J. Repa 1,2 , and David J. Mangelsdorf 1,2 1 Department of Pharmacology, 2 Howard Hughes Medical Institute, University of Texas Southwestern Medical Center 5323 Harry Hines Blvd. Dallas, TX 75390-9050 Running Title: Nuclear receptor control of sterol metabolism Abbreviations: LXR, liver X receptor; FXR, farnesoid X receptor; LRH-1, liver receptor homologue-1; SHP, small heterodimer partner; RXR, retinoid X receptor; CETP, cholesterol ester transfer protein; PLTP, phospholipid transfer protein; ABC, ATP-binding cassette, NTCP, sodium taurocholate cotransporter polypeptide; BSEP, bile salt export pump; IBABP, intestinal bile acid-binding protein; TTNPB, (E)-4-[2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2- naphthylenyl)-1-propenyl] benzoic acid; SR-BI, Scavenger receptor-class B type I; HDL, high density lipoprotein; LDL, low density lipoprotein; VLDL, very low density lipoprotein, HMG- CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; ACAT, acyl-CoA: cholesterol acyltransferase. 1 Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc. JBC Papers in Press. Published on July 17, 2001 as Manuscript R100035200 by guest on March 27, 2018 http://www.jbc.org/ Downloaded from
Transcript of Orphan Nuclear Receptors as eLiXiRs and FiXeRs of Sterol ...

Orphan Nuclear Receptors as eLiXiRs and FiXeRs of Sterol Metabolism
Timothy T. Lu1, Joyce J. Repa1,2, and David J. Mangelsdorf1,2
1Department of Pharmacology, 2Howard Hughes Medical Institute,University of Texas Southwestern Medical Center5323 Harry Hines Blvd.Dallas, TX 75390-9050
Running Title: Nuclear receptor control of sterol metabolism
Abbreviations: LXR, liver X receptor; FXR, farnesoid X receptor; LRH-1, liver receptor homologue-1; SHP, small heterodimer partner; RXR, retinoid X receptor; CETP, cholesterol ester transfer protein; PLTP, phospholipid transfer protein; ABC, ATP-binding cassette, NTCP, sodium taurocholate cotransporter polypeptide; BSEP, bile salt export pump; IBABP, intestinal bile acid-binding protein; TTNPB, (E)-4-[2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthylenyl)-1-propenyl] benzoic acid; SR-BI, Scavenger receptor-class B type I; HDL, high density lipoprotein; LDL, low density lipoprotein; VLDL, very low density lipoprotein, HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; ACAT, acyl-CoA: cholesterol acyltransferase.
1
Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on July 17, 2001 as Manuscript R100035200 by guest on M
arch 27, 2018http://w
ww
.jbc.org/D
ownloaded from

The recent explosion of research on orphan nuclear receptors has provided new insights
into the regulation of lipid metabolism. A recurring theme that continues to emerge is the
evolution of an elaborate, autoregulated system for sensing and metabolizing biologically active
lipids. One branch of this system includes the oxysterol and bile acid receptors, which serve as
sensors for sterol metabolism that regulate the transport and metabolism of cholesterol and bile
acids. This review will focus on the five orphan nuclear receptors that thus far are known to
govern cholesterol and bile acid homeostasis. These receptors include: the liver X receptors
(LXRα and LXRβ); farnesoid X receptor (FXR); liver receptor homologue-1 (LRH-1); and
small heterodimer partner (SHP). Below we will summarize the discovery and molecular biology
of these orphan receptors, their roles in regulating cholesterol and bile acid homeostasis, and
their potential use as therapeutic targets for the treatment of lipid metabolism disorders.
The Nuclear Receptor Players
LXRs, the oxysterol receptors
The first human liver X receptor (LXR) was named because of its initial isolation from a
liver cDNA library and its liver-enriched expression (reviewed in ref. 1). The LXR subfamily
consists of two members, LXRα (NR1H3) and LXRβ (NR1H2). Both subtypes are expressed in
the enterohepatic system, but each has a distinct pattern of expression in other tissues. Whereas
LXRβ is ubiquitously expressed, LXRα expression is restricted to tissues rich in lipid
metabolism (Figure 1). LXRα and LXRβ form obligate heterodimers with the retinoid X
receptor (RXR), which is bound and activated by 9-cis retinoic acid. The RXR-LXR
heterodimer preferentially binds to a DNA hormone response element (termed an LXRE) that
2
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

consists of two hexanucleotide repeats separated by 4 nucleotides (i.e., DR4) (2,3).
The first LXR activators were identified by screening organic tissue extracts and natural
compound libraries. These activators consisted of a select group of oxysterols derived from
tissue-specific cholesterol metabolism in the liver, brain and gonads. The most potent LXR
activators identified were 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 24(S),25-
epoxycholesterol (4,5). A binding assay based on scintillation proximity technology demonstrated
that these compounds bind with Kds of 0.1-0.4 µM (6,7). Since the identification of these natural
ligands, extensive structure-activity relationship studies on LXR ligands have been performed
and have yielded several synthetic LXR agonists, including a potent, high-affinity (Kd = 50
nM), non-steroidal ligand (8,9).
FXR, the bile acid receptor
FXR (NR1H4) was first isolated from a rat liver cDNA library, and structurally is most
closely related to the insect ecdysone receptor and the LXRs (10). Expression of FXR is restricted
to the enterohepatic system, kidneys and adrenals (Figure 1). FXR forms an obligate heterodimer
with RXR and binds to an inverted hexanucleotide repeat spaced by one nucleotide (i.e., IR1) (10).
The name FXR (farnesoid X receptor) derives from early studies that revealed
supraphysiological concentrations of farnesol could activate some species of FXR (10). However,
more recently, three independent groups identified bile acids as the endogenous ligands for FXR
(11-13). Together, these researchers demonstrated that physiologic concentrations of biologically
active bile acids can directly bind and transactivate FXR. The primary bile acid
chenodeoxycholic acid (CDCA) was shown to be the most potent FXR ligand in vitro and in
3
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

cells at an EC50 of 10-50 µM. FXR can also be activated by the secondary bile acids lithocholic
acid and deoxycholic acid (11-13). Importantly, cholic acid and several conjugated bile acids also bind
to FXR in vitro, but because of their membrane impermeability they only transactivate FXR in
cells that express a bile acid transporter, such as the sodium taurocholate cotransporter (NTCP)
or the ileal bile acid transporter (IBAT). Interestingly, this makes FXR the only known nuclear
receptor that requires active transport of its endogenous ligands into its target cell. This
requirement selectively limits the ligand-dependent activity of FXR to tissues (e.g., the ileum)
that co-express a bile acid transporter (12). In addition to bile acids, a number of synthetic
compounds are able to bind and activate FXR. These include the synthetic retinoid, TTNPB (14),
and a novel, high affinity (Kd = 50 nM) non-steroidal agonist (15,16).
LRH-1, a tissue-specific competence factor
LRH-1 (NR5A2) is a monomeric orphan receptor that has been isolated by several
groups and shown to be the mammalian homolog of the Drosophila fushi tarazu F1 receptor gene
(17,18). Expression of LRH-1 is limited to the liver, pancreas, intestine and ovary. LRH-1 binds
DNA as a monomer to an extended nuclear receptor half-site (Figure 1). LRH-1 is the
enterohepatic paralog of mammalian steroidogenic factor-1 (SF-1), which has been shown to be
essential for the development of the hypothalamic-pituitary-adrenal and -gonadal axes. SF-1
serves as a tissue-specific competence factor for transcription of steroidogenic P450 enzymes (19).
In similar fashion, LRH-1 is believed to serve as a tissue-specific competence factor for bile
acid synthesis. To date, no ligands have been reported for LRH-1, but several target genes have
been identified including alpha-fetoprotein (17), SHP (20), CETP (21) and a number of crucial bile
acid-synthesizing enzymes, such as cholesterol 7α-hydroxylase (CYP7A1) (18) and sterol 12α-
4
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

hydroxylase (CYP8B) (22,23).
SHP, a tissue-specific repressor
SHP (NR0B2) was isolated by yeast two-hybrid techniques because of its ability to
heterodimerize with several other nuclear receptors (24). Structurally, SHP is an atypical nuclear
orphan receptor that has a ligand-binding domain, but no DNA-binding domain. SHP is
expressed in the liver, intestine, heart, pancreas and adrenal glands (Figure 1) (24). It shows
greatest similarity to DAX1, another atypical nuclear orphan receptor without a DNA-binding
domain that heterodimerizes with SF-1 and strongly represses SF-1 activity (25,26). In a similar
fashion, SHP has been shown to be a potent repressor of LRH-1 and its cognate target genes
(discussed below). The current understanding of SHP action suggests that it may not have an
endogenous ligand, but rather it functions as a constitutive repressor (16,24,27-32).
Nuclear Receptor Control of Sterol Homeostasis
The cholesterol sensor: LXR
A number of studies over the past three years have elucidated the role of LXRs as the
body’s key sensing apparatus for maintaining cholesterol homeostasis by regulating cholesterol
catabolism, storage, absorption and transport through the transcriptional control of the key target
genes involved in these processes. This work has been facilitated by the use of mouse models to
identify LXR target genes (Table 1) and by characterization of the phenotype of Lxr-null
animals. The LXR regulatory pathways are summarized in Figure 2 and discussed below.
The liver serves as the primary site for the elimination of cholesterol from the body (see
Figure 2). In the hepatocyte excess cholesterol has been shown to generate oxysterols (33), the
signaling molecules that stimulate transcription of cholesterol 7α-hydroxylase (CYP7A1), the
5
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

rate-limiting enzyme in the classic bile acid biosynthesis pathway. Increased CYP7A1 catalyzes
conversion of cholesterol into bile acids. In this pathway, bile acids serve as the direct end-
products of cholesterol catabolism and stimulate the excretion of excess cholesterol into the bile
and feces (34,35). LXRα has been shown to control this regulatory cascade by activating CYP7A1
transcription through an LXRE in the CYP7A1 promoter (5,36). Physiological evidence for this
process has been provided by the analysis of Lxrα-null mice, which fail to upregulate CYP7A1
expression in response to cholesterol, and as a result, rapidly accumulate toxic levels of hepatic
cholesterol. Although LXRβ is also expressed in the liver, its presence does not rescue the loss of
LXRα in these mice (36). This conclusion has been confirmed by the generation of Lxrβ-null
mice, which like wildtype animals are resistant to increased dietary cholesterol (37, J.M. Lobaccaro
and D.J. Mangelsdorf, unpublished observation). Interestingly, the Lxrα/Lxrβ double-knockout
mice have a more severe liver phenotype than the Lxrα-null mice (J.M. Lobaccaro and D.J.
Mangelsdorf, unpublished observation). Together these studies have provided unequivocal
evidence that the LXRs serve as one of the body’s key sensors of dietary cholesterol and thereby
regulate an important feedforward pathway in cholesterol catabolism.
Since the process of cholesterol catabolism is liver-specific, other tissues in the body
must deal with elevated cholesterol by alternative means. There are two primary mechanisms by
which these tissues deal with excess cholesterol: (1) esterification and storage of cholesterol
within the cell and (2) efflux of free cholesterol out of the cell and transport of this cholesterol
back to the liver for further catabolic elimination.
LXRs regulate the esterification and storage of cholesterol by an indirect means that
involves the coordinate regulation of another important lipid metabolic pathway, fatty acid
6
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

synthesis. Under high cholesterol conditions, LXRs upregulate transcription of sterol regulatory
element-binding protein 1c (SREBP-1c) (9,38), the master regulator of genes involved in fatty acid
synthesis (39). Increased SREBP-1c protein results in increased cleavage of this membrane-bound
bHLH-transcription factor and the transcription of a number of fatty acid synthesizing enzymes,
including stearoyl-CoA desaturase-1 (SCD-1). SCD-1 is an enzyme responsible for the ∆9-cis
desaturation of stearoyl-CoA and palmitoyl-CoA, converting them to oleoyl-CoA and
palmitoleoyl-CoA, respectively. Increased oleoyl-CoA, the preferred substrate for acyl-CoA:
cholesterol acyltransferase (ACAT), enables increased esterification of cholesterol for storage
under high cholesterol conditions. Furthermore, increased fatty acid synthesis yields
phospholipids that are required for lipoprotein transport of excess cholesterol and for maintaining
plasma membrane integrity by providing the correct phospholipid/cholesterol ratio. Lipid
metabolism studies in Lxr-null mice have confirmed the essential role of LXRs in this pathway
(36-38).
When the amount of cholesterol exceeds the storage capacity of the cell, the increased
cholesterol load is alleviated by effluxing the excess cholesterol back into the serum, where it is
transported to the liver by a process that is termed reverse cholesterol transport. Cellular efflux of
free cholesterol is achieved through a number of membrane ATP-binding cassette (ABC)
transporters that deliver cholesterol to high density lipoproteins (HDL) that serve as the primary
serum transporter of cholesterol back to the liver. This process is especially important in the
enterocyte and macrophage, both of which can be exposed to large surges in sterols due to
unsaturable uptake of free cholesterol from the diet and serum.
Several recent studies have now shown that LXRs prevent the overaccumulation of
7
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

sterols in the intestine and macrophage by the induction of multiple ABC transporters and
acceptor proteins involved in this pathway (8,40-44). In the macrophage, activation of the RXR/LXR
heterodimer by either naturally occurring oxysterols or RXR/LXR agonists stimulates
transcription of ABCA1 and ABCG1 (8,43). ABCA1 is the protein mutated in Tangier Disease, a rare
autosomal recessive disorder characterized by low circulating levels of HDL and the appearance
of cholesterol-engorged macrophages and reticuloendothelial cells (45). ABCA1 is responsible for
the cellular efflux of free cholesterol and phospholipids to apoliprotein acceptors. In vitro and in
vivo studies have shown that ABCA1 is a direct target of the RXR/LXR heterodimer and that
LXRs are required for cholesterol-induced regulation of the ABCA1 and ABCG1 promoters (8,41,46,47).
ABCG1 is another transporter expressed in macrophages that has been suggested to play a role in
cellular efflux of cholesterol and phospholipids (42). In addition, LXRs also increase the
availability of HDL acceptor particles through transcriptional upregulation of ApoE, which
contributes to the formation of HDL particles (44,48).
After free cholesterol is accepted by the HDL particle, it is esterified by lecithin-
cholesterol acyltransferase (LCAT). The HDL particle then has two fates. Through LXR
upregulation of cholesterol ester transfer protein (CETP), HDL can exchange cholesterol esters
for triglycerides carried by other lipoproteins (49). Alternatively, HDL can deliver cholesterol
esters to the liver for excretion and catabolism (discussed above) (50).
Similar to the macrophage, in the small intestine increased dietary and/or secreted biliary
cholesterol activates LXR and increases transcription of at least three ABC transporters, ABCA1,
ABCG5 and ABCG8. In the enterocyte, these transporters are hypothesized to increase
cholesterol efflux into the intestinal lumen and thereby prevent net sterol absorption. In vivo
8
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

studies have shown that both LXR agonists and RXR agonists (i.e., rexinoids) strongly
upregulate the ABC transporters in enterocytes and inhibit cholesterol absorption (8). Consistent
with these findings, one of the studies performed in Abca1-null mice showed a significant
elevation in cholesterol absorption (51).
Subtraction cloning from mice treated with an LXR agonist has recently led to the
identification of two new ABC half-transporters, ABCG5 and ABCG8 (40). Mutations in these
transporters, which are expressed exclusively in the liver and intestine, are responsible for
sitosterolemia, a rare genetic disorder characterized by hyperabsorption of cholesterol and toxic
plant sterols (40,52). Experiments in Lxr-null mice have confirmed that these transporters are
transcriptional targets of LXR action (J.J. Repa, H.H. Hobbs and D.J. Mangelsdorf, unpublished
observation). Taken together, these studies have established the role of at least four ABC
transporters as essential downstream mediators of the RXR/LXR signaling cascade.
The bile acid sensor: FXR
The ability of bile acids to both activate and repress transcription of genes involved in
bile acid metabolism has been recognized for many years (see Table 1). Similar to the pathways
that regulate cholesterol homeostasis, bile acids upregulate genes that export bile acids out of
cells and repress genes responsible for bile acid synthesis and uptake. As the bile acid receptor,
FXR is the key factor that maintains bile acid homeostasis by modulating the expression of these
genes (summarized in Figure 2).
The best understood process in bile acid homeostasis is that which governs feedback
repression of synthesis. When the bile acid pool size is increased, transcription of CYP7A1 is
9
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

repressed (34,35). Preliminary studies had identified a conserved “bile acid-response element” in the
CYP7A1 promoter of all species, a region later identified as an LRH-1 binding site (18,53). On the
CYP7A1 promoter, LRH-1 serves as a competence factor required for liver-specific expression
of CYP7A1 (18,30). Bile acid-mediated repression of CYP7A1 and other genes occurs through FXR-
and bile acid-induced expression of SHP, the major FXR target gene in the liver (16,30). Increased
SHP protein forms an inactivating heterodimeric complex with LRH-1, turning off transcription
of CYP7A1 and other LRH-1 target genes, including sterol 12α-hydroxylase (CYP8B) and SHP
itself. The ability of SHP to repress its own transcription provides an elegant mechanism by
which the bile acid sensor turns itself on and off (16,30).
While the Shp- and Lrh-1-null mice have not yet been characterized, in vivo evidence
supporting this mechanism has come from studies in Fxr-null mice. In wildtype mice treated
with rexinoids and/or FXR agonists, transcriptional repression of CYP7A1 is coincident with a
diametric induction of SHP (16,30). In contrast, Fxr-null mice fail to upregulate SHP expression and
consequently fail to repress transcription of CYP7A1 (54). Furthermore, analysis of Cyp7a1- and
Cyp27-null mice, where bile acid pool sizes are significantly diminished, has shown that SHP
levels are severely reduced. Consequently, SHP target genes are derepressed. As would be
predicted, restoration of the bile acid pool by feeding bile acids restores SHP expression and
appropriately downregulates SHP target genes (30,55).
In addition to bile acid synthesis, FXR regulates transport and prevents overaccumulation
of bile acids in the hepatocyte. The two principal transporters governing bile acid levels in the
hepatocyte are the sodium-taurocholate cotransporter polypeptide (NTCP) and the bile salt
export pump (BSEP). NTCP is responsible for bile acid uptake into the hepatocyte and its
10
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

expression is downregulated by FXR (54). Simultaneously, FXR upregulates transcription of
BSEP, which increases bile acid efflux into the bile (54,56). In the intestine, bile acids upregulate
transcription of the ileal bile acid-binding protein (IBABP) (11,57-60). This protein is believed to assist
in decreasing the effective free concentration of bile acids to limit their toxicity and recirculation.
Recent studies have identified IBABP as a direct target gene of FXR (11,59).
Definitive evidence that FXR serves as the bile acid sensor has come from analysis of the
Fxr-null mice (54). In addition to being unable to downregulate bile acid synthesis, Fxr-null mice
fail to regulate appropriately BSEP, NTCP, and IBABP. As a result, Fxr-null mice develop
cholestasis and severe liver damage.
Therapeutic potential of nuclear orphan receptors
eLiXiRs
The model in Figure 2 summarizes the regulation of cholesterol and bile acid metabolism
by nuclear receptors. Given the importance of other nuclear hormone receptors as drug targets,
the biology illustrated in Figure 2 suggests the potential to develop a new series of therapeutic
targets for the treatment of cholesterol and bile acid metabolism disorders. Firstline treatments
for hypercholesterolemia currently entail the use of HMG-CoA reductase inhibitors (or
“statins”). While statins have been very effective, they are not able to prevent full progression of
atherosclerosis and some patients are non-responsive to statin treatment.
The identification of LXR as a cholesterol sensor that governs transport, absorption and
catabolism of sterols provides new possibilities for intervention in the treatment of
hypercholesterolemia. Ideally, an LXR drug (i.e., an eLiXiR) would have three potent anti-
atherogenic effects: (1) increased catabolism of cholesterol through upregulation of CYP7A1; (2)
11
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

inhibition of dietary cholesterol absorption by upregulating intestinal transporters (ABCA1,
ABCG5 and ABCG8); and (3) increased cholesterol transport from peripheral tissues through
upregulation of efflux transporters (ABCA1 and ABCG1) and apolipoproteins (ApoE). One
potential problem with an LXR agonist is its ability to also upregulate fatty acid synthesis,
resulting in hypertriglyceridemia. Future design of LXR agonists would have to dissociate this
process from the beneficial effects on cholesterol metabolism. One possibility might be to design
specific agonists for LXRα and LXRβ. Studies of LXRα and LXRβ single- and double-
knockout mice have provided evidence that the LXRs have overlapping, but distinct roles. LXR-
subtype specific agonists would be useful in identifying these pathways and would aid in the
design of drugs that have selective activities.
FiXeRs
While it is clear that FXR mediates repression of bile acid synthesis, it is less clear
whether it would be therapeutically useful to inhibit this process with an FXR drug (i.e., a
FiXeR). For example, an FXR antagonist would increase conversion of cholesterol into bile acids
and lower hepatic cholesterol levels, which would be therapeutically desirable. Indeed,
transgenic studies overexpressing CYP7A1 have been shown to lower serum cholesterol levels
(61). On the other hand, when the FXR-RXR heterodimer is activated by an agonist, bile acid
synthesis and dietary cholesterol absorption are repressed (8). Furthermore, Fxr-null mice on a
low fat diet have elevated serum triglycerides and cholesterol levels, in particular elevated VLDL
and LDL. In addition, phospholipid transfer protein (PLTP), a serum protein that is believed to
help form HDL, has recently been demonstrated to be a direct target gene of FXR (62,63).
Collectively, these data suggest that activation rather than antagonism of the FXR-RXR
12
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

heterodimer may be more therapeutically useful. As demonstrated by the Fxr-null mice, FXR
defends the liver against cholestasis. Therefore, designing more potent and efficacious FXR
agonists (or partial agonists) may provide better therapeutic regimens for the treatment of
cholestasis. While to date no ligands have been found for SHP and LRH-1, each of these orphan
receptors contains a conserved hydrophobic ligand binding pocket characteristic of all nuclear
receptors. Whether specific pharmacophores designed for binding to these pockets will have
therapeutic utility remains to be seen.
Summary
With the recent characterizations of several nuclear orphan receptors, an emerging theme has
developed placing these receptors as master regulators of lipid metabolism. The identification of
the oxysterol receptors (LXRs) and a bile acid receptor (FXR) has opened up new and exciting
fields of research in cholesterol and bile acid metabolism. Understanding the physiological
pathways that these receptors regulate and designing pharmaceutical drugs for these receptors
may provide alternative treatments for cholesterol and bile acid disorders.
Acknowledgements
We thank our colleagues in the Mango lab for their input and help in preparing this manuscript.
This work was supported by the Howard Hughes Medical Institute and grants from the Human
Frontiers Science Program and the Robert A. Welch Foundation.
13
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES
1. Willy, P.J. and Mangelsdorf, D.J. (1998) Nuclear orphan receptors: the search for novel ligands and signaling pathways in Hormones and Signaling, Volume 1, O’Malley, B.W., editor, Academic Press, San Diego
2. Willy, P.J. and Mangelsdorf, D.J. (1997) Genes Dev. 11, 289-298
3. Wiebel, F.F. and Gustafsson, J.-Å. (1997) Mol. Cell. Biol. 17, 3977-3986
4. Janowski, B.A., Willy, P.J., Rama-Devi, T., Falck, J.R., and Mangelsdorf, D.J. (1996) Nature 383, 728-731
5. Lehmann, J.M., Kliewer, S.A., Moore, L.B., Smith-Oliver, T.A., Oliver, B.B., Su, J.-L., Sundseth, S.S., Winegar, D.A., Blanchard, D.E., Spencer, T.A., and Willson, T.M. (1997) J. Biol. Chem. 272, 3137-3140
6. Janowski, B.A., Grogan, M.J., Jones, S.A., Wisely, G.B., Kliewer, S.A., Corey, E.J., and Mangelsdorf, D.J. (1999) Proc. Natl. Acad. Sci. USA 96, 266-271
7. Spencer, T.A., Li, D., Russel, J.S., Collins, J.L., Bledsoe, R.K., Consler, T.G., Moore, L.B., Galardi, C.M., McKee, D.D., Moore, J.T., Watson, M.A., Parks, D.J., Lambert, M.H., and Willson, T.M. (2001) J. Med. Chem. 44, 886-97
8. Repa, J.J., Turley, S.D., Lobaccaro, J.-M.A., Medina, J., Li, L., Lustig, K., Shan, B., Heyman, R.A., Dietschy, J.M., and Mangelsdorf, D.J. (2000) Science 289, 1524-1529
9. Schultz, J.R., Tu, H., Luk, A., Repa, J.J., Medina, J.C., Li, L., Schwendner, S., Wang, S., Thoolen, M., Mangelsdorf, D.J., Lustig, K.D., and Shan, B. (2000) Genes Dev. 14, 2831-2838
10. Forman, B.M., Goode, E., Chen, J., Oro, A.E., Bradley, D.J., Perlmann, T., Noonan, D.J., Burka, L.T., McMorris, T., Lamph, W.W., Evans, R.M., and Weinberger, C. (1995) Cell 81, 687-693
11. Makishima, M., Okamoto, A.Y., Repa, J.J., Tu, H., Learned, R.M., Luk, A., Hull, M.V., Lustig, K.D., Mangelsdorf, D.J., and Shan, B. (1999) Science 284, 1362-1365
12. Parks, D.J., Blanchard, S.G., Bledsoe, R.K., Chandra, G., Consler, T.G., Kliewer, S.A., Stimmel, J.B., Willson, T.M., Zavacki, A.M., Moore, D.D., and Lehmann, J.M. (1999) Science 284, 1365-1368
13. Wang, H., Chen, J., Hollister, K., Sowers, L.C., and Forman, B.M. (1999) Mol. Cell 3, 543-553
14. Zavacki, A.M., Lehmann, J.M., Seol, W., Willson, T.M., Kliewer, S.A., and Moore, D.D. (1997) Proc. Natl. Acad. Sci. USA 94, 7909-7914
14
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15. Maloney, P.R., Parks, D.J., Haffner, C.D., Fivush, A.M., Chandra, G., Plunket, K.D., Creech, K.L., Moore, L.B., Wilson, J.G., Lewis, M.C., Jones, S.A., and Willson, T.M. (2000) J. Med. Chem. 43, 2971-2974
16. Goodwin, B., Jones, S.A., Price, R.R., Watson, M.A., McKee, D.D., Moore, L.B., Galardi, C., Wilson, J.G., Lewis, M.C., Roth, M.E., Maloney, P.R., Willson, T.M., and Kliewer, S.A. (2000) Mol. Cell 6, 517-26
17. Galarneau, L., Paré, J.F., Allard, D., Hamel, D., Lévesque, L., Tugwood, J.D., Green, S., and Bélanger, L. (1996) Mol. Cell. Biol. 16, 3853-3865
18. Nitta, M., Ku, S., Brown, C., Okamoto, A.Y., and Shan, B. (1999) Proc. Natl. Acad. Sci. USA 96, 6660-6665
19. Parker, K.L. (1998) Mol. Cell. Endocrinol. 145, 15-20
20. Lee, Y.K., Parker, K.L., Choi, H.-S., and Moore, D.D. (1999) J. Biol. Chem. 274, 20869-20873
21. Luo, Y., Liang, Cp.C., and Tall, A.R. (2001) J. Biol. Chem. epub ahead of print
22. Del Castillo-Olivares, A. and Gil, G. (2000) Nucleic Acids Res. 28, 3587-3593
23. Del Castillo-Olivares, A. and Gil, G. (2000) J. Biol. Chem. 275, 17793-17799
24. Seol, W., Choi, H.S., and Moore, D.D. (1996) Science 272, 1336-1339
25. Crawford, P.A., Dorn, C., Sadovsky, Y., and Milbrandt, J. (1998) Mol. Cell. Biol. 18, 2949-2956
26. Ito, M., Yu, R., and Jameson, J.L. (1997) Mol. Cell. Biol. 17, 1476-1483
27. Johansson, L., Båvner, A., Thomsen, J.S., Färnegårdh, M., Gustafsson, J.-Å., and Treuter, E. (2000) Mol. Cell. Biol. 20, 1124-1133
28. Johansson, L., Thomsen, J.S., Damdimopoulos, A.E., Spyrou, G., Gustafsson, J.-Å., and Treuter, E. (1999) J. Biol. Chem. 274, 345-353
29. Lee, Y.-K., Dell, H., Dowhan, D.H., Hadzopoulou-Cladaras, M., and Moore, D.D. (2000) Mol. Cell. Biol. 20, 187-195
30. Lu, T.T., Makishima, M., Repa, J.J., Schoonjans, K., Kerr, T.A., Auwerx, J., and Mangelsdorf, D.J. (2000) Mol. Cell. 6, 507-15
31. Seol, W., Hanstein, B., Brown, M., and Moore, D.D. (1998) Mol. Endocrinol. 12, 1551-1557
15
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

32. Seol, W., Chung, M., and Moore, D.D. (1997) Mol. Cell. Biol. 17, 7126-7131
33. Zhang, Z., Li, D., Blanchard, D.E., Lear, S.R., Erickson, S.K., and Spencer, T.A. (2001) J. Lipid. Res. 42, 649-58
34. Russell, D.W. and Setchell, K.D.R. (1992) Biochemistry 31, 4737-4749
35. Princen, H.M.G., Post, S.M., and Twisk, J. (1997) Curr. Pharmaceut. Des. 3, 59-84
36. Peet, D.J., Turley, S.D., Ma, W.Z., Janowski, B.A., Lobaccaro, J.-M.A., Hammer, R.E., and Mangelsdorf, D.J. (1998) Cell 93, 693-704
37. Alberti, S., Schuster, G., Parini, P., Feltkamp, D., Diczfalusy, U., Rudling, M., Angelin, B., Björkhem, I., Pettersson, S., and Gustafsson, J.-Å. (2001) J. Clin. Invest. 107, 565-573
38. Repa, J.J., Liang, G., Ou, J., Bashmakov, Y., Lobaccaro, J.-M.A., Shimomura, I., Shan, B., Brown, M.S., Goldstein, J.L., and Mangelsdorf, D.J. (2000) Genes Dev. 14, 2819-2830
39. Horton, J.D., Shimomura, I., Brown, M.S., Hammer, R.E., Goldstein, J.L., and Shimano, H. (1998) J. Clin. Invest. 101, 2331-2339
40. Berge, K.E., Tian, H., Graf, G.A., Yu, L., Grishin, N.V., Schultz, J., Kwiterovich, P., Shan, B., Barnes, R., and Hobbs, H.H. (2000) Science 290, 1771-1775
41. Costet, P., Luo, Y., Wang, N., and Tall, A.R. (2000) J. Biol. Chem. 275, 28240-28245
42. Klucken, J., Büchler, C., Orsó, E., Kaminski, W.E., Porsch-Özcürümez, M., Liebisch, C., Kapinsky, M., Diederich, W., Drobnik, W., Dean, M., Allikmets, R., and Schmitz, G. (2000) Proc. Natl. Acad. Sci. USA 97, 817-822
43. Venkateswaran, A., Repa, J.J., Lobaccaro, J.-M.A., Bronson, A., Mangelsdorf, D.J., and Edwards, P.A. (2000) J. Biol.Chem. 275, 14700-14707
44. Laffitte, B.A., Repa, J.J., Joseph, S.B., Wilpiltz, D.C., Kast, H.R., Mangelsdorf, D.J., and Tontonoz, P. (2001) Proc. Natl. Acad. Sci. USA 98, 507-512
45. Young, S.G. and Fielding, C.J. (1999) Nature Genet. 22, 316-318
46. Wade, D.P. and Owen, J.S. (2001) Lancet 357, 161-163
47. Venkateswaran, A., Laffitte, B.A., Joseph, S.B., Mak, P.A., Wilpitz, D.C., Edwards, P.A., and Tontonoz, P. (2000) Proc. Natl. Acad. Sci. USA 97, 12097-12102
48. Linton, M.F., Atkinson, J.B., and Fazio, S. (1995) Science 267, 1034-7
49. Luo, Y. and Tall, A.R. (2000) J. Clin. Invest. 105, 513-520
50. Glass, C.K. and Witztum, J.L. (2001) Cell 104, 503-16
16
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

51. McNeish, J., Aiello, R.J., Guyot, D., Turi, T., Gabel, C., Aldinger, C., Hoppe, K.L., Roach, M.L., Royer, L.J., de Wet, J., Broccardo, C., Chimini, G., and Francone, O.L. (2000) Proc. Natl. Acad. Sci. USA 97, 4245-50
52. Lee, M.H., Lu, K., Hazard, S., Yu, H., Shulenin, S., Hidaka, H., Kojima, H., Allikmets, R., Sakuma, N., Pegoraro, R., Srivastava, A.K., Salen, G., Dean, M., and Patel, S.B. (2001) Nat. Genet. 27, 79-83
53. Chiang, J.Y.L. and Stroup, D. (1994) J. Biol. Chem. 269, 17502-17507
54. Sinal, C.J., Tohkin, M., Miyata, M., Ward, J.M., Lambert, G., and Gonzalez, F.J. (2000) Cell 102, 731-744
55. Repa, J.J., Lund, E.G., Horton, J.D., Leitersdorf, E., Russell, D.W., Dietschy, J.M., and Turley, S.D. (2000) J. Biol. Chem. 275, 39685-39692
56. Ananthanarayanan, M., Balasubramanian, N.V., Makishima, M., Mangelsdorf, D.J., and Suchy, F.J. (2001) J. Biol. Chem., epub ahead of print
57. Crossman, M.W., Hauft, S.M., and Gordon, J.I. (1994) J. Cell Biol. 126, 1547-1564
58. Gong, Y.-Z., Everett, E.T., Schwartz, D.A., Norris, J.S., and Wilson, F.A. (1994) Proc. Natl. Acad. Sci.USA 91, 4741-4745
59. Grober, J., Zaghini, I., Fujii, H., Jones, S.A., Kliewer, S.A., Willson, T.M., Ono, T., and Besnard, P. (1999) J. Biol. Chem. 274, 29749-29754
60. Kanda, T., Foucand, L., Nakamura, Y., Niot, I., Besnard, P., Fujita, M., Sakai, Y., Hatakeyama, K., Ono, T., and Fujii, H. (1998) Biochem. J. 330, 261-265
61. Spady, D.K., Cuthbert, J.A., Willard, M.N., and Meidell, R.S. (1995) J. Clin. Invest. 96, 700-709
62. Laffitte, B.A., Kast, H.R., Nguyen, C.M., Zavacki, A.M., Moore, D.D., and Edwards, P.A. (2000) J. Biol. Chem. 275, 10638-10647
63. Urizar, N.L., Dowhan, D.H., and Moore, D.D. (2000) J. Biol. Chem. 275, 39313-39317
17
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Figure 1. Characterization of enterohepatic nuclear orphan receptors involved in sterol
metabolism. Tissue expression of each receptor in the adult mouse is shown using northern blot
analysis (cyclophilin shown as a comparative control). Mode of DNA binding, response element
and ligands are also listed.
Figure 2. Model of nuclear receptor control of sterol metabolism. In the macrophage, intestine,
and hepatocyte, LXRs increase storage, efflux, and transport of cholesterol to decrease
intracellular cholesterol levels. In the hepatocyte, LXRs govern feedforward regulation of
cholesterol catabolism into bile acids. Feedback repression, efflux, and enterohepatic
recirculation of bile acids are regulated by FXR. See text for details. Yellow boxes represent
LXR target genes and green boxes represent FXR target genes. C, cholesterol; HDL, high density
lipoprotein; FA, fatty acid, CE, cholesterol ester; BA, bile acid.
18
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
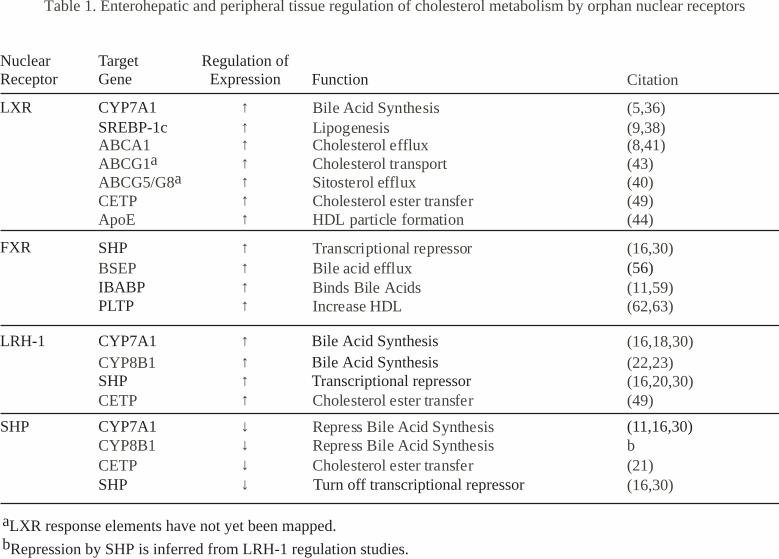
Table 1. Enterohepatic and peripheral tissue regulation of cholesterol metabolism by orphan nuclear receptors
Citation
↑ Bile Acid SynthesisLipogenesisCholesterol efflux Cholesterol transport Sitosterol effluxCholesterol ester transferHDL particle formation
Transcriptional repressorBile acid effluxBinds Bile AcidsIncrease HDL
Bile Acid SynthesisBile Acid SynthesisTranscriptional repressor
(5,36)↑ (9,38)
ABCA1 ↑ (8,41)ABCG1a ↑ (43)ABCG5/G8a ↑ (40)CETP ↑ (49)ApoE ↑ (44)
↑ (16,30)BSEP ↑
↑ (11,59)↑ (62,63)
NuclearReceptor
TargetGene
Regulation ofExpression Function
CYP7A1SREBP-1c
SHP
IBABPPLTP
FXR
LRH-1 CYP7A1 ↑↑↑
CYP8B1SHP
LXR
(16,18,30)(22,23)(16,20,30)
Repress Bile Acid Synthesis↓CYP8B1 ↓ bRepress Bile Acid SynthesisCYP7A1SHP
aLXR response elements have not yet been mapped.bRepression by SHP is inferred from LRH-1 regulation studies.
(56)
Cholesterol ester transferCETP ↑ (49)
Cholesterol ester transferCETP (21)Turn off transcriptional repressorSHP (16,30)
(11,16,30)
↓↓
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

BA
TW
AT
Adr
enal
Bra
inC
olon
Eye
Hea
rt
Kid
ney
Live
rLu
ngM
uscl
eO
vary
Pla
cent
aS
kin
Spl
een
Tes
tisU
teru
s
cyclo
Inte
istin
e
Monomer ?LRH-1 extendedhalf-site
none ?SHP -
Heterodimer OxysterolsLXRα DR4
Heterodimer OxysterolsLXRβ DR4
Heterodimer Bile AcidsFXR IR1
DNA-bindingmode
ResponseElement LigandTissue DistributionReceptor
Figure 1Lu et al.
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Diet
BSEP
SHP
IBABP
Excretion
ABCA1ABCG5/G8
ApoE
ABCA1ABCG1
SREBP-1c
CYP7A1
ABCG8ABCG5
Lu et al.Figure 2
(serum)
(to liver) by guest on M
arch 27, 2018http://w
ww
.jbc.org/D
ownloaded from

Timothy T. Lu, Joyce J. Repa and David J. MangelsdorfOrphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism
published online July 17, 2001J. Biol. Chem.
10.1074/jbc.R100035200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















