
MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibits interleukin-1-induced matrix...
9
MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibits interleukin-1-induced matrix metalloproteinase-13 gene expression and promoter activity by impairing MAP kinase activation Rasheed Ahmad, Judith Sylvester, Muhammad Zafarullah ⁎ Department of Medicine, University of Montreal and Research Centre of CHUM Notre-Dame Hospital, Montreal, Quebec, Canada H2L 4M1 Received 20 July 2007; accepted 6 August 2007 Available online 25 August 2007 Abstract Interleukin-1 (IL-1) is the major prototypic proinflammatory cytokine that stimulates degradation of cartilage in arthritis by inducing prominent collagen II-degrading matrix metalloproteinase-13 (MMP-13). Nothing is known about the involvement of adaptor proteins, MyD88, IRAK1 and TRAF6 in MMP-13 regulation. Here we investigated for the first time the role of these proteins in IL-1-regulated MMP-13 expression in chondrocytes. MyD88 homodimerization inhibitory peptide diminished the expression of MMP-13 gene, promoter activity, phosphorylation of mitogen-activated protein kinases (MAPKs), c-Jun and activating protein 1 (AP-1) activity. Knockdown of MyD88, IRAK1 and TRAF6 by RNA interference (RNAi) drastically down-regulated the expression of IL-1-induced MMP-13 mRNA and protein levels and MMP-13 promoter-driven luciferase activity. Non-specific control siRNA had no effect. Mechanisms of MMP-13 inhibition involved reduced phosphorylation of ERK, p38, JNK and c-Jun as well as AP-1 transcription factor binding activity. The genetic evidence presented here demonstrates that MyD88, IRAK1 and TRAF6 proteins are crucial early mediators for the IL-1-induced MMP-13 regulation through MAPK pathways and AP-1 activity. These proteins could constitute important therapeutic targets for arthritis-associated cartilage loss by MMP-13. © 2007 Elsevier Inc. All rights reserved. Keywords: Interleukin-1; Signal transduction; MyD88; IRAK1; TRAF6; RNA interference; Transcription factors; Matrix metalloproteinase-13; Gene regulation; Rheumatoid arthritis; Cartilage 1. Introduction Interleukin-1 (IL-1) is a major pleiotropic and proinflamma- tory cytokine implicated in arthritic cartilage and bone destruc- tion. IL-1 levels are increased in the synovial fluid and cartilage of patients with rheumatoid arthritis (RA) and osteoarthritis (OA) [1,2] and after injuries to cartilage [3]. OA chondrocytes are more sensitive to IL-1 due to augmented receptor levels [4]. IL-1 inhibits the cartilage extracellular matrix (ECM) synthe- sis and promotes its degradation by matrix metalloproteinases (MMPs) in human chondrocytes [5,6]. IL-1 treatment of carti- lage explants leads to aggrecan loss within 1 week followed by collagen loss after 3 weeks [7]. Blocking IL-1 actions with specific antibodies and IL-1 receptor antagonist reduce the arthritis-associated cartilage and bone loss and synovial invasion of cartilage in animal models, human cartilage explants and patients [8–12]. MMP-13 is the major collagenase in OA cartilage [13] where it preferentially cleaves type II collagen [14] and aggrecan [15], the principal components of cartilage ECM as well as cartilage fibromodulin [16] and small leucine-rich proteoglycans [17]. MMP-13-cleaved type II collagen fragments can further induce MMP-13 gene expression starting a vicious cycle of collagen and aggrecan degradation in arthritis [18,19]. Elevated expres- sion of MMP-13 is associated with severe and destructive type II synovitis in RA patients [20] and loosened hip replacement implants [21]. Transgenic cartilage-specific or adenovirus-me- diated overexpression of human MMP-13 in mouse joints in- duced OA-like cartilage destruction or inflammatory reactions including cytokine production, synovial hyperplasia and pannus formation, features found in RA patient joints [22,23]. Thus, IL- Available online at www.sciencedirect.com Cellular Signalling 19 (2007) 2549 – 2557 www.elsevier.com/locate/cellsig ⁎ Corresponding author. K-5255 Mailloux, Hôpital Notre-Dame du CHUM, 1560 Sherbrooke E, Montréal, Québec, Canada H2L 4M1. Tel.: +1 514 890 8000 25690; fax: +1 514 412 7612. E-mail address: [email protected] (M. Zafarullah). 0898-6568/$ - see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2007.08.013
-
Upload
rasheed-ahmad -
Category
Documents
-
view
214 -
download
2
Transcript of MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibits interleukin-1-induced matrix...

Available online at www.sciencedirect.com
007) 2549–2557www.elsevier.com/locate/cellsig
Cellular Signalling 19 (2
MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibitsinterleukin-1-induced matrix metalloproteinase-13 gene expression and
promoter activity by impairing MAP kinase activation
Rasheed Ahmad, Judith Sylvester, Muhammad Zafarullah ⁎
Department of Medicine, University of Montreal and Research Centre of CHUM Notre-Dame Hospital, Montreal, Quebec, Canada H2L 4M1
Received 20 July 2007; accepted 6 August 2007Available online 25 August 2007
Abstract
Interleukin-1 (IL-1) is the major prototypic proinflammatory cytokine that stimulates degradation of cartilage in arthritis by inducing prominentcollagen II-degrading matrix metalloproteinase-13 (MMP-13). Nothing is known about the involvement of adaptor proteins, MyD88, IRAK1 andTRAF6 in MMP-13 regulation. Here we investigated for the first time the role of these proteins in IL-1-regulated MMP-13 expression inchondrocytes. MyD88 homodimerization inhibitory peptide diminished the expression of MMP-13 gene, promoter activity, phosphorylation ofmitogen-activated protein kinases (MAPKs), c-Jun and activating protein 1 (AP-1) activity. Knockdown of MyD88, IRAK1 and TRAF6 by RNAinterference (RNAi) drastically down-regulated the expression of IL-1-induced MMP-13 mRNA and protein levels and MMP-13 promoter-drivenluciferase activity. Non-specific control siRNA had no effect. Mechanisms of MMP-13 inhibition involved reduced phosphorylation of ERK, p38,JNK and c-Jun as well as AP-1 transcription factor binding activity. The genetic evidence presented here demonstrates that MyD88, IRAK1 andTRAF6 proteins are crucial early mediators for the IL-1-induced MMP-13 regulation through MAPK pathways and AP-1 activity. These proteinscould constitute important therapeutic targets for arthritis-associated cartilage loss by MMP-13.© 2007 Elsevier Inc. All rights reserved.
Keywords: Interleukin-1; Signal transduction; MyD88; IRAK1; TRAF6; RNA interference; Transcription factors; Matrix metalloproteinase-13; Gene regulation;Rheumatoid arthritis; Cartilage
1. Introduction
Interleukin-1 (IL-1) is a major pleiotropic and proinflamma-tory cytokine implicated in arthritic cartilage and bone destruc-tion. IL-1 levels are increased in the synovial fluid and cartilageof patients with rheumatoid arthritis (RA) and osteoarthritis(OA) [1,2] and after injuries to cartilage [3]. OA chondrocytesare more sensitive to IL-1 due to augmented receptor levels [4].IL-1 inhibits the cartilage extracellular matrix (ECM) synthe-sis and promotes its degradation by matrix metalloproteinases(MMPs) in human chondrocytes [5,6]. IL-1 treatment of carti-lage explants leads to aggrecan loss within 1 week followed bycollagen loss after 3 weeks [7]. Blocking IL-1 actions with
⁎ Corresponding author. K-5255 Mailloux, Hôpital Notre-Dame du CHUM,1560 Sherbrooke E, Montréal, Québec, Canada H2L 4M1. Tel.: +1 514 8908000 25690; fax: +1 514 412 7612.
E-mail address: [email protected] (M. Zafarullah).
0898-6568/$ - see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.cellsig.2007.08.013
specific antibodies and IL-1 receptor antagonist reduce thearthritis-associated cartilage and bone loss and synovial invasionof cartilage in animal models, human cartilage explants andpatients [8–12].
MMP-13 is the major collagenase in OA cartilage [13] whereit preferentially cleaves type II collagen [14] and aggrecan [15],the principal components of cartilage ECM as well as cartilagefibromodulin [16] and small leucine-rich proteoglycans [17].MMP-13-cleaved type II collagen fragments can further induceMMP-13 gene expression starting a vicious cycle of collagenand aggrecan degradation in arthritis [18,19]. Elevated expres-sion of MMP-13 is associated with severe and destructive typeII synovitis in RA patients [20] and loosened hip replacementimplants [21]. Transgenic cartilage-specific or adenovirus-me-diated overexpression of human MMP-13 in mouse joints in-duced OA-like cartilage destruction or inflammatory reactionsincluding cytokine production, synovial hyperplasia and pannusformation, features found in RA patient joints [22,23]. Thus, IL-

2550 R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
1 and MMP-13 are principal therapeutic targets for protectingcartilage integrity.
Transmission of proinflammatory cytokine signals stimu-lates multiple cascades including extracellular signal-regulatedkinase (ERK), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinases (MAPK) pathways [24,25]. Binding ofIL-1 to the receptor (IL-1R1) induces the formation of a receptorcomplex that includes the IL-1R accessory protein (IL-1RAcP).An intracellular adaptor protein, MyD88 is then recruitedthrough its Toll-ILR-1 (TIR) domain to the complex, whichinteracts through its death domain with the correspondingdomain of IL-1R-associated kinase (IRAK). IRAK subsequent-ly undergoes phosphorylation and becomes associated withTRAF6, a downstream transducer required for NF-κB and AP-1activation. TRAF6 has to be ubiquinated for its activity [26]. Wehave previously demonstrated that IL-1β-induced activation ofMAPKs plays an important role in the regulation of MMP-13gene [6]. However, role of adaptor proteins in MMP-13 induc-tion has not been previously investigated. By using cell perme-able inhibitory peptide and RNA interference, we demonstratefor the first time, critical requirement of adaptor proteins in IL-1-induced MMP-13 gene expression.
2. Materials and methods
2.1. Chondrocyte cultures and inhibitory peptide treatments
The normal human knee articular chondrocytes (Cambrex, Walkerville, MD)were grown to confluence as high-density passage 2 primary monolayer culturesin Differentiation Bullekit medium for maintaining their differentiated phenotype(Cambrex). These cells express cartilage-specific type II collagen marker up topassage 3, as examined by Western blot analysis. After trypsinization, the cellswere grown in 6-well plates in Dulbecco's modified Eagle's medium (DMEM)(Invitrogen Life Sciences Inc, Burlington, ON) with 10% fetal calf serum (FCS).Chondrocytes were washed with phosphate buffered saline (PBS) and kept inserum-free DMEM for 24 h and incubated with the 150 μM of MyD88 dimer-ization inhibitory peptide (DRQIKIWFQNRRMKWKKRDVLPGT) or control-scrambled peptide (DRQIKIWFQNRRMKWKK) ([27], IMGENEX, SanDiego,CA) for 12 h. Subsequently, cells were either treated with 0.1% BSA (control) orstimulatedwith IL-1β (10 ng/ml), (R&DSystems,MinneapolisMN) for 24 h andRNA levels measured by RT-PCR and proteins by Western blotting. For MAPKmeasurements, chondrocytes were first treatedwith the peptides for 12 h and thenstimulated with IL-1β for 20 min and protein extracts analyzed by Westernblotting.
2.2. RT-PCR
MMP-13 and GAPDH mRNA levels were measured by RT-PCR withMMP-13 specific primers [28] as described before [29] yielding 491 and 226 bpcDNA bands.
2.3. Western blotting
Cells were harvested and incubated for 30 min with lysis buffer (Tris62.5 mM pH 7.5, 1% Triton X 100, 10% Glycerol). The lysates were thencentrifuged at 14,000 rpm for 10min and the supernatants were collected. Proteinconcentration in the lysates was measured by Bio-Rad Protein Assay kit. Protein(20 μg) samplesweremixedwith sample loading buffer, heated for 5min at 95 °Cand separated by SDS-PAGE. Cellular proteins were transferred to Immobilin-Pmembrane by electroblotting. The membranes were then blocked with 5% non-fat milk in PBS for 1 h, incubated with the desired primary antibody diluted eitherin 5% milk in PBS or 5% BSA overnight at 4 °C. Antibodies against phos-phorylated ERK 1/2, p38, JNK and c-Jun and corresponding total proteins were
from Cell signaling Technology (Beverley, MA) and were used at 1:1000dilutions. The blots were then washed four times with TBS and incubated for 2 hwith HRP-conjugated secondary antibody. Immunoreactive bands were devel-oped using an Enhanced Chemiluminescent substrate (ECL, Amersham) andvisualized by autoradiography. For monitoring the efficiency of knockdown,protein extracts were probed with MyD88, IRAK1 (2 μg/ml, from ProSci Inc.,Poway CA) and TRAF6 (1: 200 dilution, Santa Cruz Biotechnology Inc. SantaCruz, CA) antibodies. The same blots were subsequently used for measuring thelevels of beta-actin (Sigma) loading control.
For MMP-13 Western blots, supernatants were collected from the cellcultured in the 6-well plates and mixed with TCA (10% final) to precipitate theproteins. Samples were kept at −20 °C for 30 min and then centrifuged at14,000 rpm for 20 min at 4 °C. Supernatants were discarded and precipitatedproteins were dissolved in 0.1 M NaOH. Sample loading buffer was added andboiled for 5 min and run on SDS-PAGE for proteins separation. Antibodyagainst MMP-13 (Sigma, Saint Louis, Missouri) was used at 1:500 dilution.
2.4. Plasmid vectors, siRNA, transfections and luciferase assay
Cells were transfected with different vectors or siRNA via calciumphosphate precipitation method for adherent cells in suspension as follows.Cells were removed by trypsinization and trypsin-free suspension incubatedwith respective siRNA (200–250 nM)-calcium phosphate precipitate for 30 minand plated in serum-containing medium without antibiotics for 3 h. MyD88Validated Stealth Duopak siRNA and control siRNA (Invitrogen), SilencerNegative Control #2 siRNA or IRAK1 Silencer Validated siRNA (Ambion,Austin TX). Similarly, control siRNA (sc-44236) and TRAF6 siRNA (sc-36717)(Santa Cruz Biotechnology Inc) was utilized for transfections. Cells werewashed with PBS, allowed to recover in serum-containing medium, maintainedin serum-free medium and then stimulated with IL-1. Equal amount (20 μg) ofprotein was analyzed for measuring respective protein levels. In some experi-ments, 4 μg of MMP-13 promoter–luciferase (−1000 to +71 region; p1000-LUC, [30]), cytomegalovirus (CMV)-Renilla luciferase (4 μg, transfectioncontrol) and respective siRNA were co-transfected by the modified calciumphosphate procedure described above and after recovery for 36 h, treated withIL-1 and luciferase activity measured with Promega Dual-Luciferase Reporterassay System and Turner Designs Luminometer TD-20/20 according to theirrecommended procedures.
2.5. AP-1 binding assay
Chondrocytes were treated with the peptide inhibitor or transfected withsiRNA as above and then stimulated with IL-1β for 40 min. For nuclear proteinisolation, NucBuster Protein Extraction kit (Novagen) was used according tomanufacturer's instructions and nuclear pellets were resuspended in 50 μl ofcomplete lysis buffer provided by Active Motif kit. After 30 min incubation onice, samples were centrifuged and proteins were measured with the Bio-RadProtein Assay kit. AP-1 consensus nucleotide binding activity from nuclearextracts (10 μg) was measured by using TransAM AP-1 family (Active Motif,Carlsbad, CA) colorimetric transcription factor measurement system as recom-mended. Briefly nuclear extract was added to the immobilized oligonucleotidesfollowed by the addition of primary transcription factor antibody and secondaryhorse radish peroxidase (HRP)-conjugated antibody and HRP substrate andcolorimetric values measured at 450 nm were plotted as bar graphs. As a controlof specificity, excess wild type consensus and mutant AP-1 oligonucleotideswere used as competitors for AP-1 binding.
All the experiments were performed at least three times.
3. Results
3.1. Peptide-mediated inhibition of MyD88 dimerizationsuppresses IL-1β-induced MMP-13 gene expression andpromoter activity
MyD88 is a major mediator of IL-1 signal transductiondownstream of IL-1 receptor as it forms homodimers and

2551R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
promotes the recruitment to the plasma membrane and acti-vates IL-1 receptor-associated kinases, IRAK1 and IRAK-4where the latter phosphorylates the former [26]. Despite theavailability of this knowledge, role of MyD88 in up-regulationof MMP-13 by IL-1β is not known and needs to bedeciphered. For this purpose, we incubated human chondro-cytes with cell permeable MyD88 dimerization inhibitorypeptide [27] or scrambled peptide (control peptide) prior tostimulation with IL-1β. Chondrocytes pretreated with thisinhibitor displayed significantly reduced IL-1β-induced ex-pression of MMP-13 mRNA compared to the cells pretreatedwith scrambled peptide without altering the constant levels ofGAPDH mRNA. MMP-13 protein expression was similarly
Fig. 1. Down-regulation of IL-1β inducible MMP-13 gene expression by peptide-medwith the 150 μM of inhibitory peptide or scrambled peptide (cont. peptide) for 12 h.MMP-13 protein were measured by RT-PCR and Western blotting. B) Chondrocytes wluciferase reporter plasmid and after 24 h, either kept untreated or exposed to peptideto the analysis of luciferase activity. Data are normalized with the Renilla luciferase.and control peptide-treated chondrocytes were exposed to IL-1β for 20 min and phosppretreated with control and inhibitory peptides were exposed to IL-1β for 40 min anspecific ELISA. Positive control was tumor promoter agent (TPA)-stimulated K-562competitor for AP-1 binding prevented binding of IL-1β-stimulated nuclear extract wthus demonstrating specificity of the assay.
inhibited by the peptide (Fig. 1A). To examine the impactof MyD88 homodimerization inhibition on MMP-13 pro-moter activity, chondrocytes were first transfected with theMMP-13 promoter (−1000 to +71)–luciferase reporter plas-mid and then treated with the inhibitory or scrambled pep-tide as above. Analysis of luciferase activity revealed thatMyD88 homodimerization inhibitor reduced the IL-1β-induced MMP-13–luciferase activity but scrambled peptidedid not (Fig. 1B).
MyD88 homodimerization stimulates IRAK1 phosphoryla-tion and its interaction with TRAF6 leads to the activation ofIκB kinase and MAPKs, p38, ERK1/2, JNK and c-Jun [26].These kinases activate several transcription factors including
iated inhibition of MyD88 homodimerization. A) Chondrocytes were incubatedCells were treated with IL-1β for 24 h and MMP-13 and GAPDH mRNA andere transfected with MMP-13 promoter–luciferase reporter plasmid and Renilla
inhibitor for 12 h and stimulated with IL-1β for 24 h. Cell lysates were subjectedData are expressed as mean±SD from three separate experiments. C) Inhibitoryhorylated and total proteins were analyzed byWestern blotting. D) Chondrocytesd AP-1 DNA-binding activity was measured from the nuclear extracts by AP-1-nuclear extract. Excessive wild type consensus oligonucleotide that served asith immobilized AP-1 DNA on the plate while mutant oligonucleotide did not,
2552 R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
NF-κB and AP-1 important in the regulation of MMP-13 [6]. Toinvestigate the mechanism of MMP-13 inhibition, we deter-mined the effect of MyD88 homodimerization on the ERK, p38,JNK and c-Jun phosphorylation and observed that the inhibitorypeptide partially reduced the phosphorylation of these proteins(Fig. 1C). Since c-Jun is an essential component of AP-1transcription factor complex, we also examined the effect ofinhibitory peptide on the AP-1 binding activity. Chondrocyteswere preincubated with each peptide for 12 h followed by40 min exposure to IL-1β. Analysis of nuclear extracts for AP-1binding activity by ELISA revealed that the inhibitory peptidesignificantly reduced IL-1β-induced AP-1 DNA-binding activ-ity (Fig. 1D). These results suggest that MyD88 homodimer-ization is critical in the IL-1β-regulated MMP-13 geneexpression in chondrocytes.
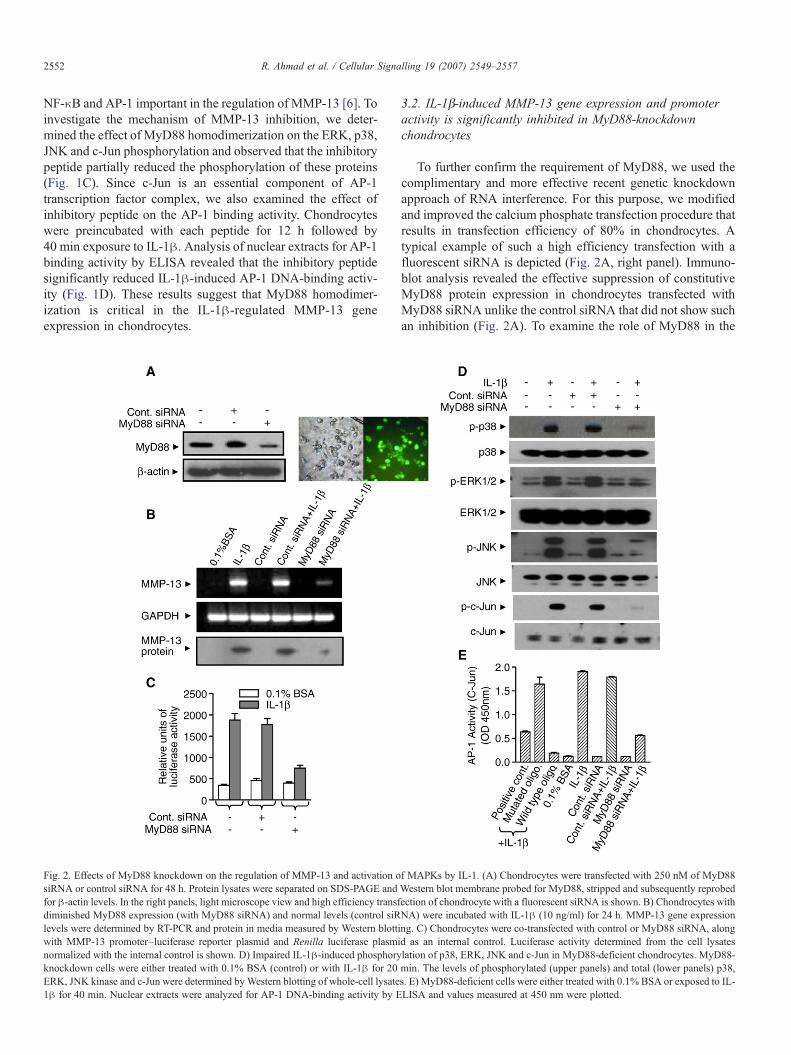
Fig. 2. Effects of MyD88 knockdown on the regulation of MMP-13 and activation osiRNA or control siRNA for 48 h. Protein lysates were separated on SDS-PAGE andfor β-actin levels. In the right panels, light microscope view and high efficiency transfdiminished MyD88 expression (with MyD88 siRNA) and normal levels (control siRlevels were determined by RT-PCR and protein in media measured by Western blottiwith MMP-13 promoter–luciferase reporter plasmid and Renilla luciferase plasminormalized with the internal control is shown. D) Impaired IL-1β-induced phosphoryknockdown cells were either treated with 0.1% BSA (control) or with IL-1β for 20ERK, JNK kinase and c-Jun were determined byWestern blotting of whole-cell lysate1β for 40 min. Nuclear extracts were analyzed for AP-1 DNA-binding activity by E
3.2. IL-1β-induced MMP-13 gene expression and promoteractivity is significantly inhibited in MyD88-knockdownchondrocytes
To further confirm the requirement of MyD88, we used thecomplimentary and more effective recent genetic knockdownapproach of RNA interference. For this purpose, we modifiedand improved the calcium phosphate transfection procedure thatresults in transfection efficiency of 80% in chondrocytes. Atypical example of such a high efficiency transfection with afluorescent siRNA is depicted (Fig. 2A, right panel). Immuno-blot analysis revealed the effective suppression of constitutiveMyD88 protein expression in chondrocytes transfected withMyD88 siRNA unlike the control siRNA that did not show suchan inhibition (Fig. 2A). To examine the role of MyD88 in the
f MAPKs by IL-1. (A) Chondrocytes were transfected with 250 nM of MyD88Western blot membrane probed for MyD88, stripped and subsequently reprobedection of chondrocyte with a fluorescent siRNA is shown. B) Chondrocytes withNA) were incubated with IL-1β (10 ng/ml) for 24 h. MMP-13 gene expressionng. C) Chondrocytes were co-transfected with control or MyD88 siRNA, alongd as an internal control. Luciferase activity determined from the cell lysateslation of p38, ERK, JNK and c-Jun in MyD88-deficient chondrocytes. MyD88-min. The levels of phosphorylated (upper panels) and total (lower panels) p38,s. E) MyD88-deficient cells were either treated with 0.1% BSA or exposed to IL-LISA and values measured at 450 nm were plotted.
2553R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
induction of MMP-13 by IL-1β, the MyD88-deficient cellswere treated with IL-1β for 24 h and impact on MMP-13 geneexpression was studied by RT-PCR. Control siRNA did notinhibit MMP-13 induction while specific siRNA-drivenMyD88-deficient chondrocytes showed a diminished responsefor IL-1β induction of MMP-13 (Fig. 2B). None of the treat-ments affected constitutively expressed GAPDH control mRNAlevels. Results of MMP-13 protein analysis were comparable tothose of RT-PCR (Fig. 2B, lower panel). To assess whetherMyD88 deficiency influenced the promoter of MMP-13, cellswere transfected with MyD88 siRNA together with humanMMP-13 promoter-driven luciferase reporter plasmid. IL-1β-induced MMP-13 promoter activity was suppressed in MyD88-
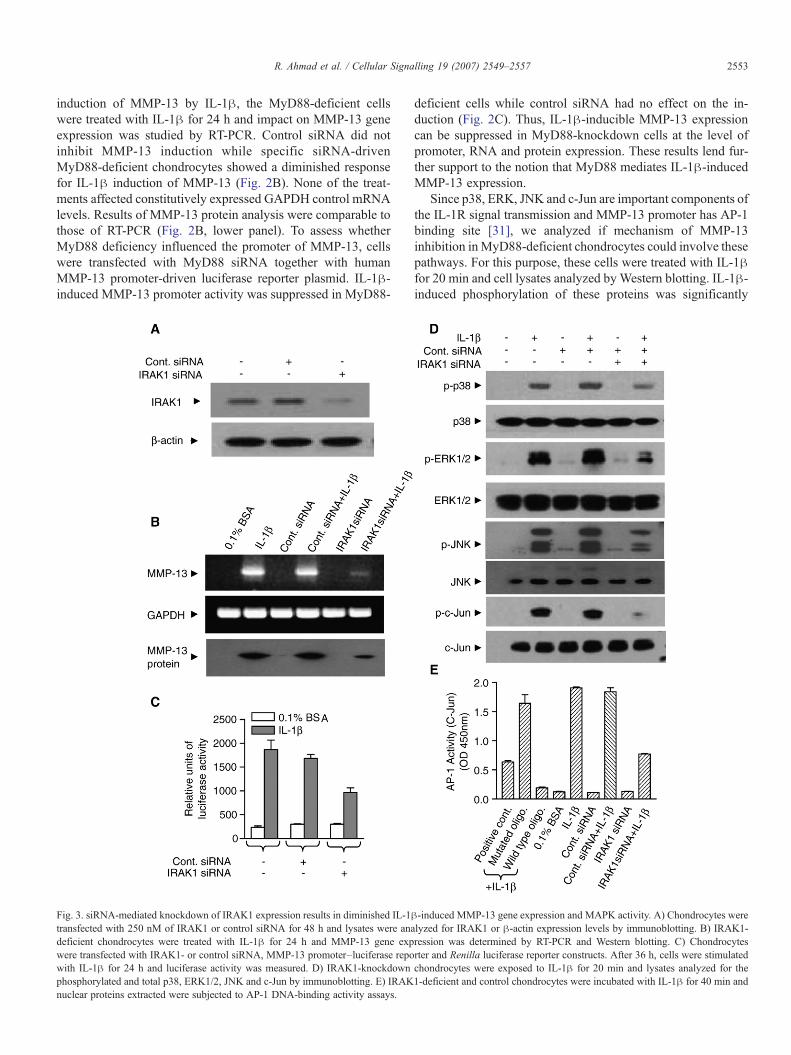
Fig. 3. siRNA-mediated knockdown of IRAK1 expression results in diminished IL-1βtransfected with 250 nM of IRAK1 or control siRNA for 48 h and lysates were anadeficient chondrocytes were treated with IL-1β for 24 h and MMP-13 gene expwere transfected with IRAK1- or control siRNA, MMP-13 promoter–luciferase repowith IL-1β for 24 h and luciferase activity was measured. D) IRAK1-knockdownphosphorylated and total p38, ERK1/2, JNK and c-Jun by immunoblotting. E) IRAKnuclear proteins extracted were subjected to AP-1 DNA-binding activity assays.
deficient cells while control siRNA had no effect on the in-duction (Fig. 2C). Thus, IL-1β-inducible MMP-13 expressioncan be suppressed in MyD88-knockdown cells at the level ofpromoter, RNA and protein expression. These results lend fur-ther support to the notion that MyD88 mediates IL-1β-inducedMMP-13 expression.
Since p38, ERK, JNK and c-Jun are important components ofthe IL-1R signal transmission and MMP-13 promoter has AP-1binding site [31], we analyzed if mechanism of MMP-13inhibition inMyD88-deficient chondrocytes could involve thesepathways. For this purpose, these cells were treated with IL-1βfor 20 min and cell lysates analyzed byWestern blotting. IL-1β-induced phosphorylation of these proteins was significantly
-induced MMP-13 gene expression and MAPK activity. A) Chondrocytes werelyzed for IRAK1 or β-actin expression levels by immunoblotting. B) IRAK1-ression was determined by RT-PCR and Western blotting. C) Chondrocytesrter and Renilla luciferase reporter constructs. After 36 h, cells were stimulatedchondrocytes were exposed to IL-1β for 20 min and lysates analyzed for the1-deficient and control chondrocytes were incubated with IL-1β for 40 min and

2554 R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
reduced in MyD88-deficient cells as compared to non-specificcontrol siRNA-transfected cells (Fig. 2D). These results indicatethat MyD88 may be a key upstream regulator of all three majorMAPK pathways and IL-1β-mediated MMP-13 expression. Iftrue, c-Jun activation by IL-1β in MyD88-deficient cells couldbe impaired. In agreement with this hypothesis, IL-1β-mediatedc-Jun activation was blocked inMyD88-deficient cells but not incontrol siRNA-transfected chondrocytes (Fig. 2D). Since c-Junis a major component of AP-1 transcription factor complex, weexamined AP-1 DNA-binding activity in these cells. Consistentwith the results of c-Jun and JNK activation, IL-1β inducedstrong AP-1 DNA-binding activity in control siRNA-transfected
Fig. 4. Impaired induction of MMP-13 and activation of MAPKs by IL-1β in TRAFcontrol siRNA and cell extracts analyzed after 48 h for inhibition of TRAF6 exprchondrocytes were treated with IL-1β for 24 h andMMP-13 mRNA and protein levelssiRNA, MMP-13 promoter–luciferase and Renilla luciferase vectors. After 36 h, cellin Materials and methods. Results are expressed in mean±SD of three independent ex1β or 0.1% BSA and cell lysates analyzed by Western immunoblotting. Specificphospho-c-Jun were used to determine MAPK activation. E) TRAF6-deficient chondand AP-1 DNA-binding activities were determined by ELISA.
cells that was partially blocked in MyD88-deficient cells(Fig. 2E). These results strongly suggest that inhibition ofMMP-13 induction in MyD88-knockdown chondrocytes ispartly due to down-regulation of AP-1 DNA-binding activity.
3.3. IRAK1 knockdown by RNA interference inhibits IL-1-inducedMMP-13 expression and promoter activity by impairingdownstream MAPK signaling
Signaling between MyD88 and TRAF6 is mediated byIRAK family members such as IRAK1 [26]. IL-1β stimulationtriggers rapid recruitment of IRAK1 to an activated receptor
6-deficient chondrocytes. A) Cells were transfected with 200 nM of TRAF6- oression by immunoblotting. β-actin levels are also shown. B) TRAF6-deficientare shown. C) Chondrocytes were co-transfected with TRAF6 siRNA or controls were treated with IL-1β and lysates assayed for luciferase activity as describedperiments. D) TRAF6-deficient chondrocytes were incubated for 20 min with IL-antibodies directed against phospho-p38, phospho-ERK1/2, phospho-JNK androcytes were incubated for 40 min with IL-1β and nuclear extracts were prepared

2555R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
resulting in IRAK1 hyperphosphorylation that is necessary forsignal transduction. To assess the role of IRAK1 in the activa-tion of MMP-13 gene by IL-1β, we used IRAK1 siRNA togenerate IRAK1-deficient chondrocytes. The endogenousIRAK1 protein levels were significantly down-regulated inIRAK1 siRNA-transfected cells compared to those transfectedwith control siRNA (Fig. 3A). IL-1β-induced MMP-13 mRNAand protein expression was reduced in IRAK1-deficient cellscompared with control siRNA-transfected chondrocytes(Fig. 3B). The levels of constitutively expressed GAPDHmRNA remained constant and were not affected by the treat-ments. Furthermore, involvement of IRAK1 in IL-1-inducedMMP-13 promoter activation was tested by co-transfectionexperiments. IRAK1 siRNA and MMP-13 promoter–luciferasereporter plasmid transfection down-regulated IL-1-inducedMMP-13 promoter activity most likely at the transcriptionlevel. In contrast, control siRNA did not show any effect(Fig. 3C). These data demonstrate that IRAK1 knockdown bysiRNA suppresses IL-1β-induced MMP-13 expression at thepromoter, mRNA and protein levels.
To gain further insight into the mechanism of IRAK1siRNA-mediated inhibition of MMP-13 induction, we analyzedwhether IRAK1 deficiency affected phosphorylation of MAPkinases. Indeed, IL-1-stimulated phosphorylation of p38, ERKand JNK was reduced in IRAK1-deficient but not in controlsiRNA-transfected chondrocytes. The total levels of the threeMAPKs were not affected (Fig. 2D). Since c-Jun phosphory-lation is important in the activation of AP-1 transcription factorcomplex and MMP-13 promoter is regulated by AP-1 bindingsite [31], we also tested if IL-1β-induced c-Jun phosphorylationwas affected by IRAK1 deficiency. IL-1-evoked c-Jun phos-phorylation was significantly suppressed in IRAK1-deficientbut not in control siRNA-transfected chondrocytes (Fig. 3D).IL-1β-enhanced AP-1 DNA-binding activity was also signifi-cantly reduced in the IRAK1-deficient cells. Control siRNA didnot decrease this activity, demonstrating specificity of theIRAK1 siRNA actions (Fig. 3E).
3.4. siRNA-driven TRAF6 deficiency decreases IL-1β-inducedMMP-13 gene expression and promoter activity by inhibitingMAPK signaling
Following the activation of IRAK1, TRAF6 plays a criticalrole in IL-1R signaling [26]. Therefore, we investigated theimportance of TRAF6 in the induction of MMP-13 gene ex-pression by RNA interference (RNAi) approach. The endoge-nous TRAF6 protein expression levels were significantlyknockdown by transfection of TRAF6 siRNA and not bycontrol siRNA. The constitutive beta-actin levels were not af-fected by the transfections (Fig. 4A). The ability of IL-1β to up-regulate MMP-13 mRNA and protein expression was severelyimpaired in TRAF6 siRNA-transfected chondrocytes comparedto control siRNA-transfected cells (Fig. 4B). In accordance withthe gene expression results, TRAF6 deficiency also decreasedIL-1-induced MMP-13 promoter-driven luciferase activity.Control siRNA did not have any effect on the induction(Fig. 4C). Mechanistic experiments revealed that similar to
previous mediators, TRAF6 knockdown by siRNA inhibitedIL-1-induced phosphorylation of three MAPKs (Fig. 4D), c-Junphosphorylation and AP-1 DNA-binding activity (Fig. 4E) inhuman chondrocytes. The control siRNA had no such effects.Thus, all the components of signal transmission leading toMMP-13 gene induction were impaired by TRAF6 knockdown.
4. Discussion
MMP-13 is the principal enzyme that efficiently cleavescollagen II and aggrecan the major structural proteins of carti-lage in response to IL-1 exposure during the course of arthritis.By using state-of-the-art inhibitory peptide and RNA interfer-ence approaches, we have elucidated the signaling cascadeleading from IL-1RI to the activation of MAPKs that result inup-regulation of MMP-13 in chondrocytes. By analyzing theeffects of adaptor proteins inhibition, we have demonstrated forthe first time that MyD88, IRAK1 and TRAF6 are absolutelyrequired for IL-1 induction of MMP-13 in human articularchondrocytes.
MyD88 is the first adaptor molecule recruited to the IL-1receptor complex after IL-1 treatment of cell. The MyD88 formshomodimerize prior to IL-1 signal transduction. Partial inhibi-tion of MMP-13 expression and promoter activity by a specificMyD88 homodimerization inhibitory peptide and not by itsscrambled homologue suggests specificity of the peptide for itseffects. Partial effectiveness of the peptide to impair downstreamsignaling events (MAPK activation) may be due to factors suchas lower efficiency of penetration, concentration and stability ofthe peptide. To further confirm the validity of the observed data,we used complementary and more powerful RNAi approaches,which strongly supported the critical role of MyD88 in IL-1-induced activation of MMP-13. Impaired MMP-13 induction inMyD88-deficient cells and strong correlation between decreasedpromoter activity, mRNA and protein expression suggest down-regulation at the transcription level. Inhibition of IL-1-activatedMAPKs suggests that blockade of MyD88 inhibits all down-stream signaling events and their target gene, MMP-13. Inparticular, decrease in ERK and JNK pathways could impair theability of essential c-Fos and c-Jun components to form AP-1transcription factor complex. JNK is an important therapeutictarget for reducing cartilage destruction in arthritis [32]. JNKinhibition by immunosuppressant, FK506 suppresses MMP-13[33]. Indeed, c-Jun phosphorylation and AP-1 binding activitywas significantly decreased by MyD88 inhibition. This mech-anism is specific as these effects were observed only in MyD88-deficient chondrocytes and not in control siRNA-transfectedcounterparts. Thus, comparable results from two alternativeapproaches of inhibitory peptide and siRNA strongly support thekey role for MyD88 in regulation of MMP-13 through MAPKsand AP-1 activation. It is interesting to note that MyD88-deficient mice were protected against streptococcal cell wall-induced and IL-1-mediated arthritic inflammation and cartilagedegradation [34]. This protection may be due to MyD88-dependent blockade of proteases (MMP-13) expression demon-strated here. Indeed, we have observed that MyD88 knockdownalso suppresses the induction of principal aggrecan-degrading

2556 R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
enzyme, aggrecanase-1 expression (our unpublished results).MyD88-deficient mice did not display Escherichia coli cellwall- and lipopolysaccharide (a TLR4 ligand)-induced pawswelling, an-arthritis-like inflammation [35,36]. MyD88-defi-cient mice also had reduced gout-like inflammatory responses tomonosodium urate crystals suggesting its important role indifferent forms of arthritis [37,38].
Using the similar RNA interference approaches, specificinhibition of IRAK1 and TRAF6 expression by the respectivespecific siRNAs and not by the control siRNA was demon-strated. Inhibition of MMP-13 expression at promoter, mRNAand protein levels in IRAK1- and TRAF6-deficient chondro-cytes further reinforced the importance of these adaptor proteinsin IL-1 signal transduction leading to MMP-13 increase. Similarto the results of MyD88, transcriptional inhibition of MMP-13induction may be largely due to impaired ERK, p38 and JNKactivation and subsequent activation of transcription factorssuch as c-Jun and AP-1 complex as demonstrated by Westernblotting and AP-1 binding activities. Once again, these inhibi-tory effects were specific, as control siRNA-transfected chon-drocytes did not impair MMP-13 induction, IL-1-inducedMAPK and c-Jun phosphorylation and AP-1 binding activity.Our in vitro knockdown results are analogous to IRAK knock-out murine embryonic and skin fibroblasts where p38 and JNKactivation was shown to be severely defective due to IRAKdeficiency [39]. Further, IRAK is recruited to focal adhesioncomplex during IL-1 activation of ERK [40]. IRAK has beenshown to mediate IL-1-induced JNK activation without beingphosphorylated [41]. Similarly, another member of the IRAKfamily, IRAK4 knockout mice have been shown to impair IL-1and TLR signaling [42]. Taken together, these results clearlydemonstrate IRAK1 and TRAF6 as critical signaling compo-nents downstream of MyD88 but upstream of ERK, JNK, p38MAPKs, which are required for MMP-13 induction. PartialMMP-13 inhibition indicates that besides these three adaptorproteins, other molecular mediators may also be required for themaximal induction. Thus we have established early receptor-proximal sequence of events responsible for the induction ofthis important IL-1 target gene. Since these adaptor proteins arecommon denominators in IL-1 signal transduction through typeI receptor and microbial products-activated toll-like receptors,these results may have implications for microbial pathogens-induced arthritis and innate immunity [43,44]. IL-1 is alsoknown to up-regulate TLR-2 expression in chondrocytes andsynovial fibroblasts [45–47]. Act1 adaptor protein is an essen-tial component of IL-17 signaling whose receptor has TIR-likeand SEFIR domains needed for recruitment of TAK1 andTRAF6. Act1 deficiency abolishes inflammation [48,49].
5. Conclusions
IL-1 is the principal signal responsible for tissue damage byproteases in arthritis. The findings described here shed new lighton the key function of MyD88, IRAK and TRAF6 in IL-β-induced enhancement of MMP-13 gene expression. Therefore,these adaptor proteins could constitute important therapeutictargets for arthritis-associated cartilage loss by MMP-13.
Acknowledgements
This research was supported by the Canadian Institutes ofHealth Research (CIHR) and The Arthritis Society (TAS)grants. MZ is a member of the Canadian arthritis Network(CAN). Rasheed Ahmad acknowledges CIHR post-doctoralfellowship. We are grateful to Dr Carlos Lopez-Otin (Oviedo,Spain) for the MMP-13 promoter plasmid and Hamid YaqoobQureshi for help with transfections.
References
[1] J.M. Dayer, B. Bresnihan, Arthritis Rheum. 46 (2002) 574.[2] L.C. Tetlow, D.J. Adlam, D.E. Woolley, Arthritis Rheum. 44 (2001) 585.[3] J. Gruber, T.L. Vincent, M. Hermansson, M. Bolton, R. Wait, J. Saklatvala,
Arthritis Rheum. 50 (2004) 2539.[4] J. Martel-Pelletier, R. McCollum, J. DiBattista, M.P. Faure, J.A. Chin, S.
Fournier, M. Sarfati, J.P. Pelletier, Arthritis Rheum. 35 (1992) 530.[5] M.B. Goldring, J.R. Birkhead, L.F. Suen, R. Yamin, S. Mizuno, J.
Glowacki, J.L. Arbiser, J.F. Apperley, J. Clin. Invest. 94 (1994) 2307.[6] A. Liacini, J. Sylvester, W.Q. Li, M. Zafarullah, Matrix Biol. 21 (2002) 251.[7] M.M. Temple, Y. Xue, M.Q. Chen, R.L. Sah, Arthritis Rheum. 54 (2006)
3267.[8] L.A. Joosten, M.M. Helsen, T. Saxne, F.A. van De Loo, D. Heinegard,
W.B. van Den Berg, J. Immunol. 163 (1999) 5049.[9] C.H. Evans, J.N. Gouze, E. Gouze, P.D. Robbins, S.C. Ghivizzani, Gene
Ther. 11 (2004) 379.[10] M. Neidhart, R.E. Gay, S. Gay, Arthritis Rheum. 43 (2000) 1719.[11] S.B. Abramson, A. Amin, Rheumatology (Oxford) 41 (2002) 972.[12] M. Kobayashi, G.R. Squires, A. Mousa, M. Tanzer, D.J. Zukor, J.
Antoniou, U. Feige, A.R. Poole, Arthritis Rheum. 52 (2005) 128.[13] B. Bau, P.M. Gebhard, J. Haag, T. Knorr, E. Bartnik, T. Aigner, Arthritis
Rheum. 46 (2002) 2648.[14] P.G. Mitchell, H.A. Magna, L.M. Reeves, L.L. Lopresti-Morrow, S.A.
Yocum, P.J. Rosner, K.F. Geoghegan, J.E. Hambor, J. Clin. Invest. 97(1996) 761.
[15] A.J. Fosang, K. Last, V. Knauper, G. Murphy, P.J. Neame, FEBS Lett. 380(1996) 17.
[16] T.F. Heathfield, P. Onnerfjord, L. Dahlberg, D. Heinegard, J. Biol. Chem.279 (2004) 6286.
[17] J. Monfort, G. Tardif, P. Reboul, F. Mineau, P. Roughley, J.P. Pelletier, J.Martel-Pelletier, Arthritis Res. Ther. 8 (2006) R26.
[18] T. Yasuda, E. Tchetina, K. Ohsawa, P.J. Roughley, W. Wu, A. Mousa, M.Ionescu, I. Pidoux, A.R. Poole, Matrix Biol. 25 (2006) 419.
[19] M. Fichter, U. Korner, J. Schomburg, L. Jennings, A.A. Cole, J.Mollenhauer, J. Orthop. Res. 24 (2006) 63.
[20] D. Wernicke, C. Seyfert, E. Gromnica-Ihle, P. Stiehl, Autoimmunity 39(2006) 307.
[21] G.F. Ma, A. Ali, N. Verzijl, R. Hanemaaijer, J. TeKoppele, Y.T. Konttinen,J. Salo, Arthritis Rheum. 54 (2006) 2928.
[22] K. Joronen, R. Ala-aho, M.L. Majuri, H. Alenius, V.M. Kahari, E. Vuorio,Ann. Rheum. Dis. 63 (2004) 656.
[23] L.A. Neuhold, L. Killar, W. Zhao, M.L. Sung, L. Warner, J. Kulik, J.Turner, W. Wu, C. Billinghurst, T. Meijers, A.R. Poole, P. Babij, L.J.DeGennaro, J. Clin. Invest. 107 (2001) 35.
[24] Y. Geng, J. Valbracht, M. Lotz, J. Clin. Invest. 98 (1996) 2425.[25] G.S. Firestein, A.M. Manning, Arthritis Rheum. 42 (1999) 609.[26] L.A. O'Neill, C. Greene, J. Leukoc. Biol. 63 (1998) 650.[27] M. Loiarro, C. Sette, G. Gallo, A. Ciacci, N. Fanto, D. Mastroianni, P.
Carminati, V. Ruggiero, J. Biol. Chem. 280 (2005) 15809.[28] B.V. Shlopov, W.R. Lie, C.L. Mainardi, A.A. Cole, S. Chubinskaya, K.A.
Hasty, Arthritis Rheum. 40 (1997) 2065.[29] A. Liacini, J. Sylvester, W.Q. Li, W. Huang, F. Dehnade, M. Ahmad, M.
Zafarullah, Exp. Cell Res. 288 (2003) 208.[30] A.M. Pendas, M. Balbin, E. Llano, M.G. Jimenez, C. Lopez-Otin,
Genomics 40 (1997) 222.

2557R. Ahmad et al. / Cellular Signalling 19 (2007) 2549–2557
[31] J.A. Mengshol, M.P. Vincenti, C.E. Brinckerhoff, Nucleic Acids Res. 29(2001) 4361.
[32] Z. Han, D.L. Boyle, L. Chang, B. Bennett, M. Karin, L. Yang, A.M.Manning, G.S. Firestein, J. Clin. Invest. 108 (2001) 73.
[33] K. Migita, T. Miyashita, Y. Maeda, T. Aoyagi, Y. Kawabe, M. Nakamura,H. Yatsuhashi, H. Ishibashi, K. Eguchi, Immunol. Lett. 98 (2005) 194.
[34] L.A. Joosten, M.I. Koenders, R.L. Smeets, M. Heuvelmans-Jacobs, M.M.Helsen, K. Takeda, S. Akira, E. Lubberts, F.A. van De Loo, W.B. van DenBerg, J. Immunol. 171 (2003) 6145.
[35] J.Y. Choe, B. Crain, S.R. Wu, M. Corr, J. Exp. Med. 197 (2003) 537.[36] F. Kyo, H. Futani, K. Matsui, M. Terada, K. Adachi, K. Nagata, H. Sano,
H. Tateishi, H. Tsutsui, K. Nakanishi, Arthritis Rheum. 52 (2005) 2530.[37] C.J. Chen, Y. Shi, A. Hearn, K. Fitzgerald, D. Golenbock, G. Reed, S.
Akira, K.L. Rock, J. Clin. Invest. 116 (2006) 2262.[38] R. Liu-Bryan, P. Scott, A. Sydlaske, D.M. Rose, R. Terkeltaub, Arthritis
Rheum. 52 (2005) 2936.[39] P. Kanakaraj, P.H. Schafer, D.E. Cavender, Y. Wu, K. Ngo, P.F. Grealish,
S.A. Wadsworth, P.A. Peterson, J.J. Siekierka, C.A. Harris, W.P. Fung-Leung, J. Exp. Med. 187 (1998) 2073.
[40] M.K. MacGillivray, T.F. Cruz, C.A. McCulloch, J. Biol. Chem. 275 (2000)23509.
[41] X. Li, M. Commane, Z. Jiang, G.R. Stark, Proc. Natl. Acad. Sci. U. S. A.98 (2001) 4461.
[42] N. Suzuki, S. Suzuki, G.S. Duncan, D.G. Millar, T. Wada, C. Mirtsos, H.Takada, A. Wakeham, A. Itie, S. Li, J.M. Penninger, H. Wesche, P.S.Ohashi, T.W. Mak, W.C. Yeh, Nature 416 (2002) 750.
[43] T. Kawai, S. Akira, Arthritis Res. Ther. 7 (2005) 12.[44] C. Ospelt, D. Kyburz, M. Pierer, R. Seibl, M. Kurowska, O. Distler, M.
Neidhart, U. Muller-Ladner, T. Pap, R.E. Gay, S. Gay, Ann. Rheum. Dis.63 (Suppl 2) (2004) ii90.
[45] S.L. Su, C.D. Tsai, C.H. Lee, D.M. Salter, H.S. Lee, Osteoarthr. Cartil. 13(2005) 879.
[46] H.A. Kim, M.L. Cho, H.Y. Choi, C.S. Yoon, J.Y. Jhun, H.J. Oh, H.Y. Kim,Arthritis Rheum. 54 (2006) 2152.
[47] R. Seibl, T. Birchler, S. Loeliger, J.P. Hossle, R.E. Gay, T. Saurenmann,B.A. Michel, R.A. Seger, S. Gay, R.P. Lauener, Am. J. Pathol. 162 (2003)1221.
[48] S.H. Chang, H. Park, C. Dong, J. Biol. Chem. 281 (2006) 35603.[49] Y. Qian, C. Liu, J. Hartupee, C.Z. Altuntas, M.F. Gulen, D. Jane-Wit, J.
Xiao, Y. Lu, N. Giltiay, J. Liu, T. Kordula, Q.W. Zhang, B. Vallance, S.Swaidani, M. Aronica, V.K. Tuohy, T. Hamilton, X. Li, Nat. Immunol.8 (2007) 247.


















