
Lecture 25 - Phylogeny Based on Chapter 23 - Molecular Evolution Copyright © 2010 Pearson Education...
25
Lecture 25 - Phylogeny Based on Chapter 23 - Molecular Evolution Copyright © 2010 Pearson Education Inc.
-
Upload
morgan-stevenson -
Category
Documents
-
view
216 -
download
2
Transcript of Lecture 25 - Phylogeny Based on Chapter 23 - Molecular Evolution Copyright © 2010 Pearson Education...

Lecture 25 - Phylogeny Based on Chapter 23 - Molecular Evolution
Copyright © 2010 Pearson Education Inc.
1. Introduction
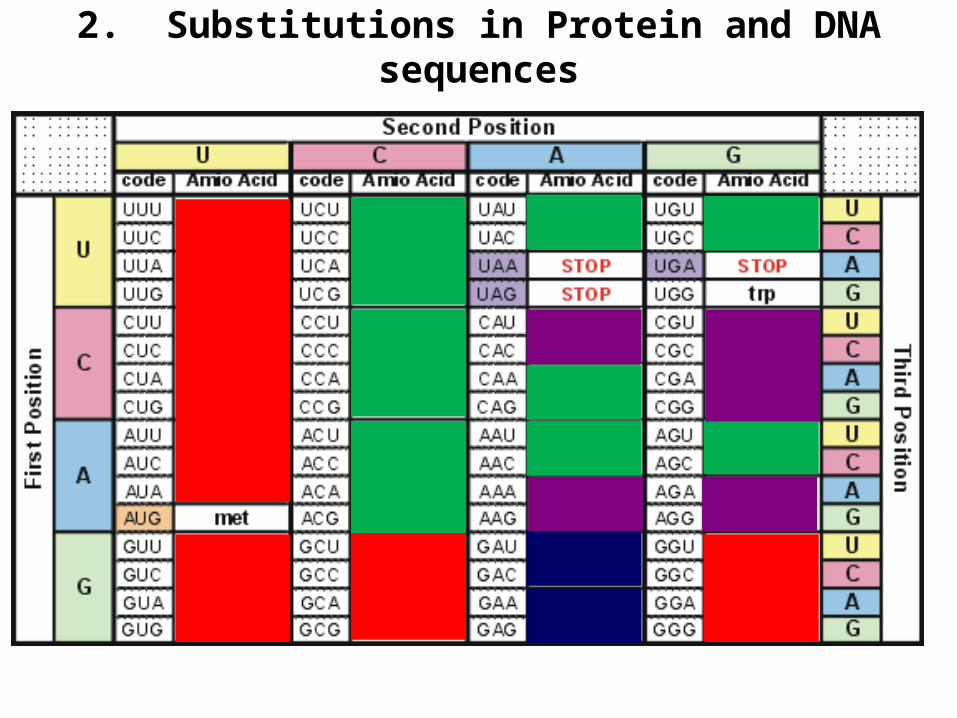
2. Substitutions in Protein and DNA sequences

3. Comparing Sequences Using Sequence Alignments
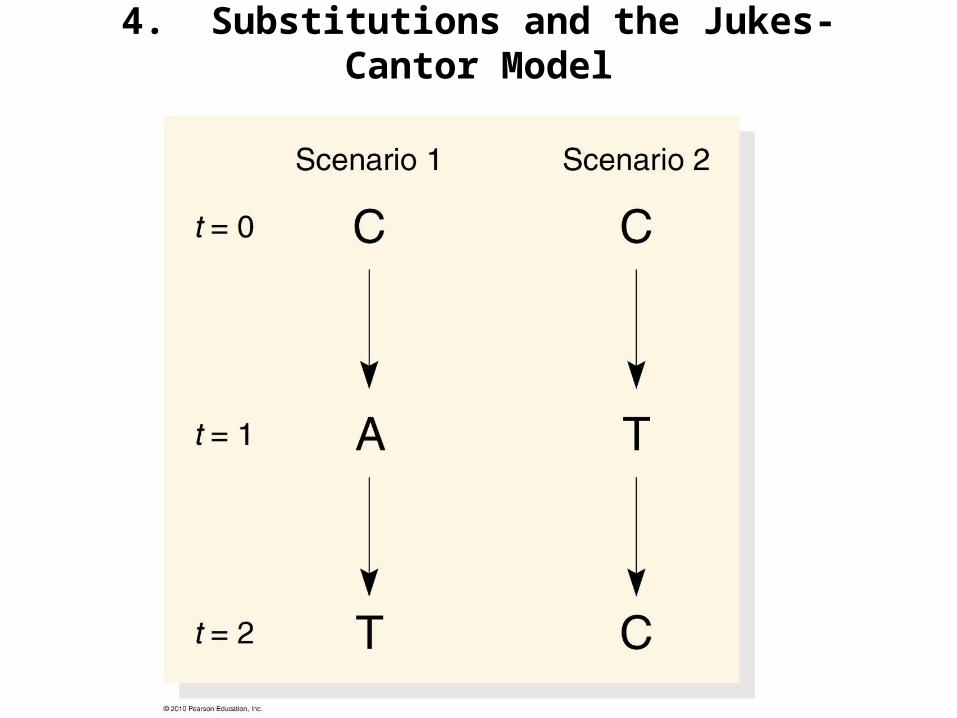
4. Substitutions and the Jukes-Cantor Model

5. Rates of Nucleotide Substitution

6. Comparative Genomics1. Genome sequencing provides a map to genes but does not reveal their
function. Comparative genome analysis:a. Compares genes with low evolutionary rate and high functional significance.
b. Pseudogenes, which are free to mutate, are used to calculate expected mutation rates.
c. Regions of high sequence similarity in distantly related species are likely to contain functional genes.
2. Between mice and humans, for example, pseudogenes show about five times as many changes as regions that encode proteins or regulate gene expression.
3. Natural selection evaluates the consequences of an enormous number of changes, on an evolutionary time scale.
4. Comparative genome analysis can point the way to meaningful experiments by:
a. Saving the effort of saturation mutagenesis.
b. Allowing use of model organisms (e.g., yeast).
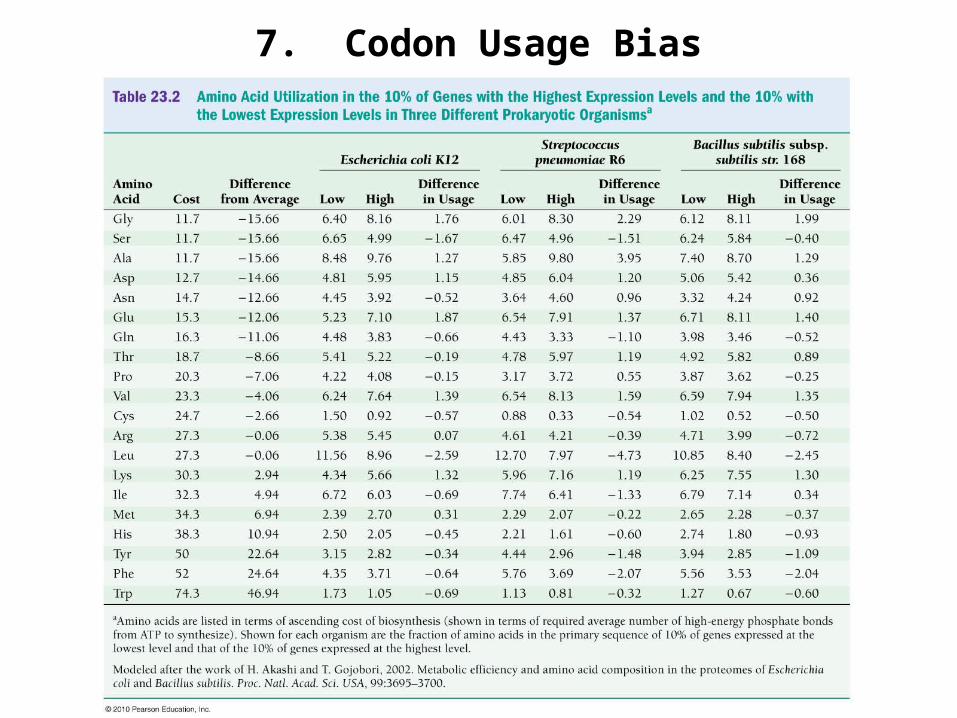
7. Codon Usage Bias

8. Variation in Evolutionary Rates between Genes
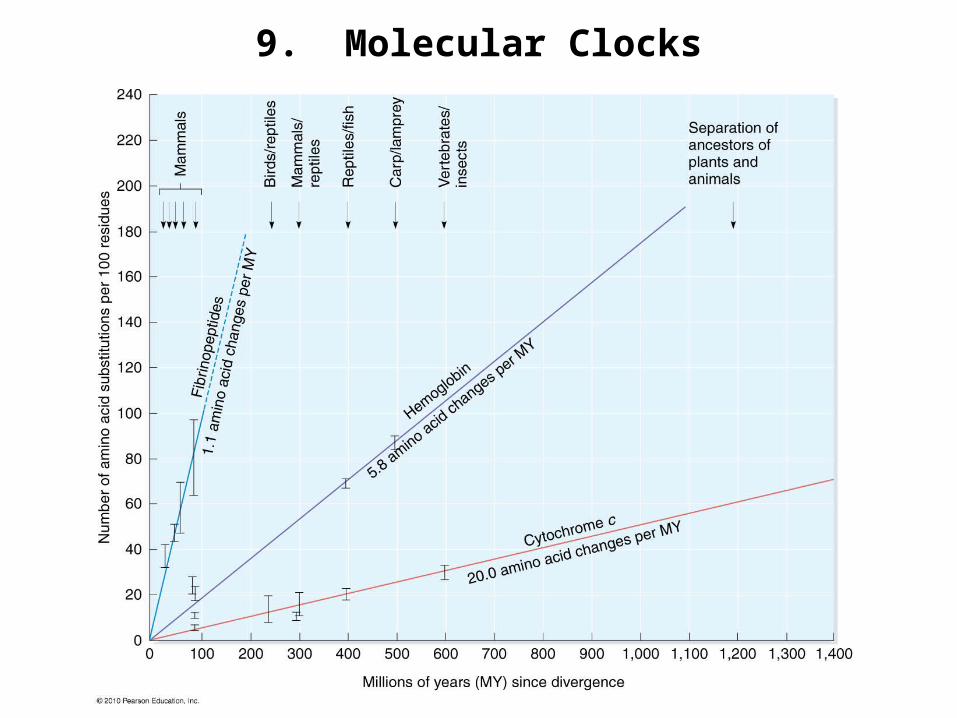
9. Molecular Clocks
10. Molecular Phylogeny
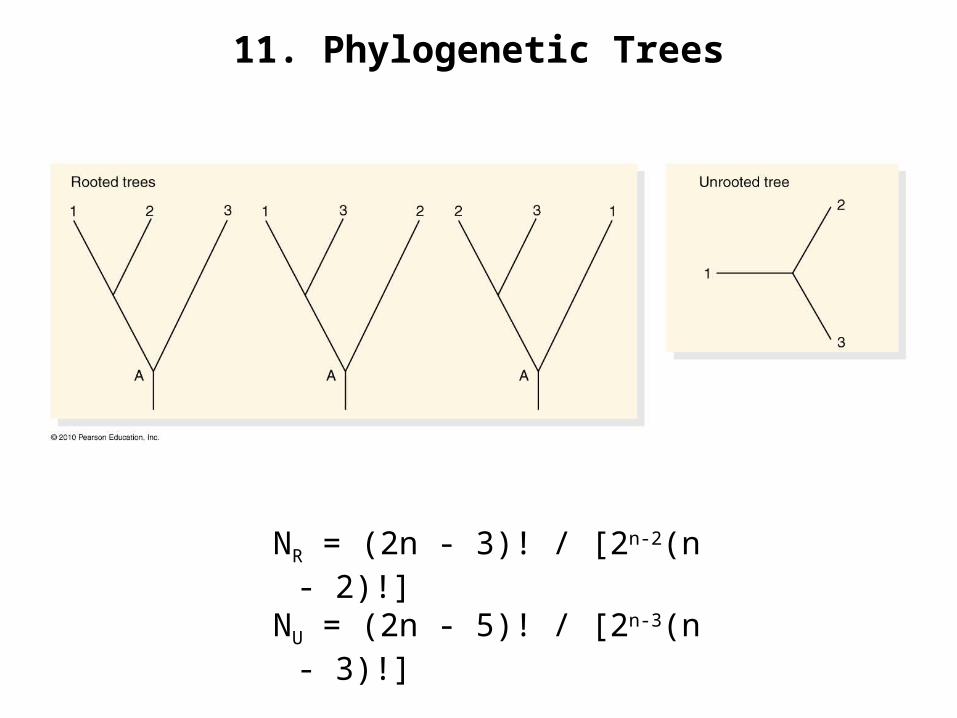
11. Phylogenetic Trees
NR = (2n - 3)! / [2n-2(n - 2)!]NU = (2n - 5)! / [2n-3(n - 3)!]

12. Number of Possible Trees
# sequences # unrooted # rooted trees trees
2 1 13 1 34 3 155 15 1056 105 9457 945 10,3958 10,395 135,1359 135,135 2,027,02510 2,027,025
34,459,425
NR = (2n - 3)! / [2n-2(n - 2)!]NU = (2n - 5)! / [2n-3(n - 3)!]

13. Gene versus Species Treees

14. Reconstruction Methods
• Many possibilities exist for the REAL phylogenetic trees, and it is generally impossible to know which is the true tree that represents actual events in evolution. Most phylogenetic trees generated with molecular data are considered inferred trees.
• Computer algorithms that generate these inferred trees use three types of approaches:– Distance matrix methods.– Parsimony-based methods.– Maximum likelihood methods.
• We will examine each of these mehtods briefly.

15. Distance Matrix Phylogenetic Reconstruction
Let Ci and Cj be two disjoint clusters:
1di,j = ———————— pq dp,q, where p Ci and q Cj
|Ci| × |Cj|
In words: calculate the average over all pairwise inter-cluster distances for all taxa under consideration.
Ci Cj
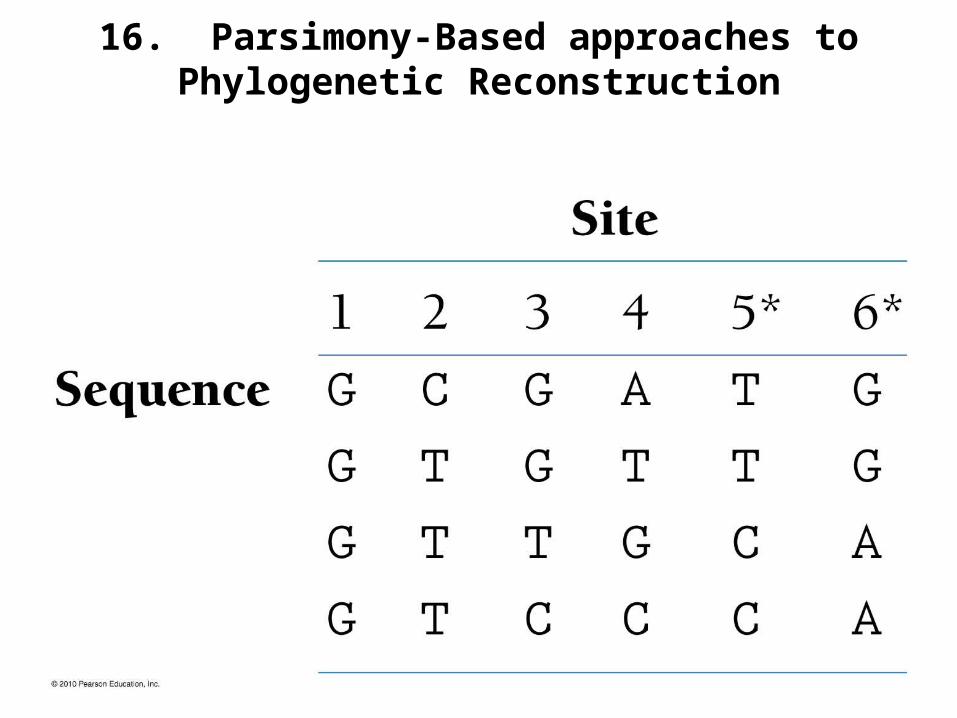
16. Parsimony-Based approaches to Phylogenetic Reconstruction
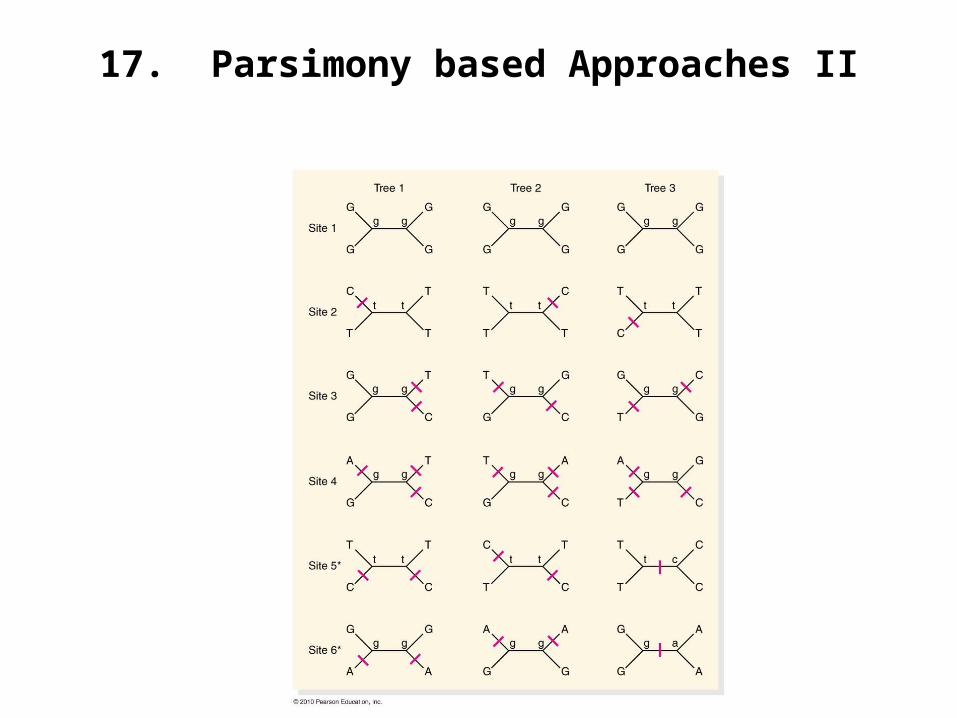
17. Parsimony based Approaches II

18 . Maximum Likelihood Approaches to Phylogenetic Tree Reconstruction
• Maximum likelihood approaches offer a purely statistical alternative method to reconstruct phylogenies.– In a set of sequence alignments, probability is considered for
every possible nucleotide substitution.– Transitions, for example, are three times more likely than
transversions, and so individuals with transition divergences may be considered more closely related than those with transversions.
• Complications of the maximum likelihood method include:– Lack of knowledge about the ancestral sequence.– The probability that multiple substitutions have occurred.– The fact that sites are not necessarily independent or
equivalent.
• A vast number of trees are possible with this computation-intensive method. The one with the highest aggregate probability is most likely to reflect the true phylogenetic tree.

19. Bootstrapping and Tree Reliability
• Large numbers (e.g., >30 species) of long sequences are difficult to analyze, even with fast computers and streamlined algorithms.
• Neither distance matrix nor maximum parsimony methods can guarantee the correct tree; but generally, if a similar tree results from both of these fundamentally different methods, it is considered fairly reliable.
• The confidence level for portions of inferred trees can be determined by bootstrap tests, in which a subset of the original data is drawn with replacement and a new tree inferred.
• Caution is needed in interpreting bootstrap results.
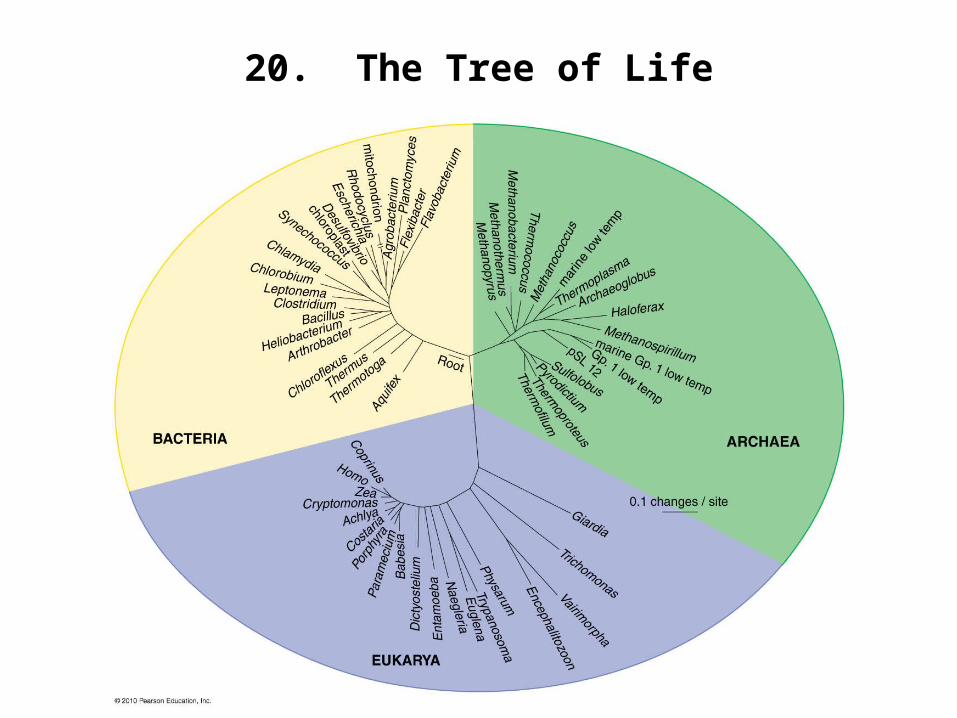
20. The Tree of Life
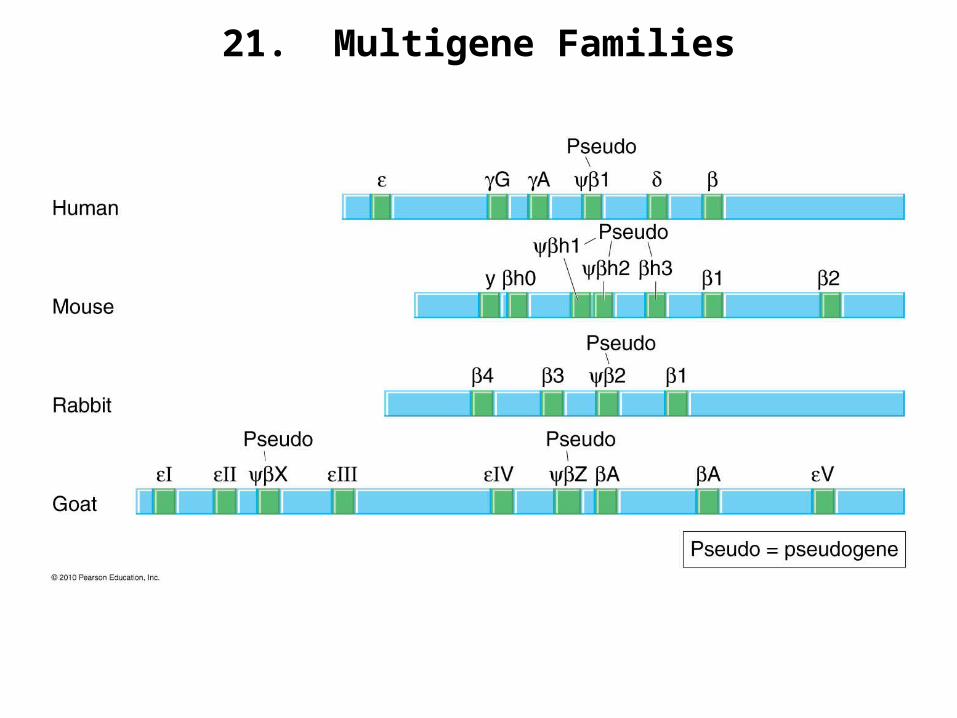
21. Multigene Families

22a. Gene Duplication and Gene Conversion
1. Duplication frees a copy of the sequence to undergo changes, since a functional copy will still exist.
a. Most changes would produce less functional products, or even nonfunctional pseudogenes.
b. A few changes, however, might alter function and/or pattern of expression to something more advantageous for the organism. Selection would allow these genes to become widespread in the population.
Gene X
Gene X Gene X
Gene X Gene x’

22b. Gene Duplication and Gene Conversion2. Misalignment between a pseudogene and a functional copy
can result in gene conversion through recombination events.
a. The allele on one homolog is copied and replaces the DNA sequence of the allele on the other homolog; it is not reciprocal exchange.
b. Gene conversion gives organisms even more opportunities to create a gene with a new function.
3. Gene conversion continues to operate in modern humans. An example is two genes for red-green color vision on the X chromosome that undergo gene conversion in most of the known cases of spontaneous deficiencies in green color vision.

23. Domain (Exon) Shuffling
1. Often, less than an entire gene is duplicated, resulting in copies of protein domains.
a. Example - human serum albumin.
b. Internal duplication not a rapid method of producing proteins with new functions.
2. Most complex proteins arise from assemblages of several protein domains performing different functions
a. The beginnings and ends of exons and protein domains often correspond.
b. Gilbert (1978) - most gene families arose through domain shuffling involving duplication and rearrangement.
c. Domain shuffling theory proposes that introns were a feature of early life on Earth, even though they are now missing from prokaryotes.
d. Numerous examples of complex genes made from segments of other genes are known, and clearly some novel functions have been created in this way.
![Molecular Phylogeny and Evolution. Introduction to evolution and phylogeny Nomenclature of trees Five stages of molecular phylogeny: [1] selecting sequences.](https://static.fdocuments.us/doc/165x107/56649e265503460f94b155ae/molecular-phylogeny-and-evolution-introduction-to-evolution-and-phylogeny.jpg)

















