
LC–NMR–MS in drug discovery
8
1359-6446/03/$ – see front matter ©2003 Elsevier Science Ltd. All rights reserved. PII: S1359-6446(03)02749-1 Complex mixtures constitute a ubiquitous challenge in drug discovery and development regimes. Natural products are mined for novel molecular diversity. Drug metabolites are iden- tified for risk assessment at the drug discov- ery–development interface. In development, regulatory bodies demand the identification of impurities at >0.1% levels. Selective, spe- cific and sensitive analyses that are preferably nondestructive are clearly of paramount im- portance throughout the drug discovery and design process. NMR as a versatile analytical platform Nuclear magnetic resonance spectroscopy (NMR) is the study of molecules by recording the interaction of radiofrequency electromag- netic radiation with the nuclei of molecules placed in a strong magnetic field. Modern NMR provides an enormously powerful toolkit to probe the molecular structure of organic com- pounds. Three distinct sub-disciplines of NMR are solid-state NMR, NMR imaging (MRI) and solution-state NMR, which is the most widely applicable in drug discovery. There are many advantages in using NMR: it is a non-selective, non-destructive detector of low molecular weight molecules in solution; both endogenous and xenobiotic metabolites can be monitored simultaneously in complex biofluids; and qualitative, and in many cases quantitative, data are achieved. NMR provides uniquely rich structural content, such as chemical shifts, multiplicity (the interaction between neigh- bouring nuclei), integrals, intramolecular relationships and a wide range of dynamic processes, including molecular motions in so- lution, chemical exchange and ligand bind- ing. NMR requires minimal sample prepara- tion and requires as little as 5 µl of sample with the latest microcoil probes. As NMR is non- destructive, the sample can be recovered for further analysis. Bioactivity and toxicity testing is then possible by reconstituting the sample in an appropriate solvent. These technical ad- vantages are crucial in natural product and drug metabolism studies in which sample is limited and minor novel metabolites are to be characterized [1]. Complex mixture analysis is further enhanced by the effective coupling of powerful chromatographic techniques with on-line NMR detection [2]. Methodological aspects are reviewed elsewhere [2,3], and this review details recent hyphenated advances for identifying drug-like molecules in complex mixtures. Interfacing LC, NMR and MS It is seldom possible to solve the structure of a novel compound by NMR alone. Common functional groups such as carboxylic acid, phe- nol and amino groups are NMR-silent in many solvents because of proton–deuterium ex- change. Nitro groups and sulfate conjugates do not contain protons and although these functional groups are not directly detectable in proton NMR spectra, they can be readily detected by mass spectrometry (MS). Conversely, MS data might give molecular weight, frag- mentation and molecular formulae that are insufficient to unambiguously assign the mol- ecular structure of an unknown compound. In the most difficult cases closely eluting isobars and isomers are indistinguishable by LC–MS LC–NMR–MS in drug discovery Olivia Corcoran and Manfred Spraul Olivia Corcoran Department of Pharmacy Franklin-Wilkins Building 150 Stamford Street King’s College London UK SE1 9NN e-mail: [email protected] Manfred Spraul Bruker BioSpin GmbH Silberstreifen 76287 Rheinstetten Germany reviews research focus 624 DDT Vol. 8, No. 14 July 2003 Nuclear magnetic resonance spectroscopy (NMR) is arguably the most versatile analytical platform for complex mixture analysis. Specifically, interfacing liquid chromatography with parallel NMR and mass spectrometry (LC–NMR–MS) gives comprehensive structural data on metabolites of novel drugs in development. Applications in natural product, combinatorial chemistry and drug metabolism studies are reviewed. Microcoil probes and capillary separation methods have enormous potential. Recent innovations to improve NMR detection limits include CryoFlowProbes TM and on-line solid-phase extraction (LC–SPE–NMR). These state-of-the-art analytical platforms are widely applicable to identifying novel candidate drugs from diverse complex mixtures within a drug discovery strategy. www.drugdiscoverytoday.com ▼
-
Upload
olivia-corcoran -
Category
Documents
-
view
219 -
download
1
Transcript of LC–NMR–MS in drug discovery

1359-6446/03/$ – see front matter ©2003 Elsevier Science Ltd. All rights reserved. PII: S1359-6446(03)02749-1
Complex mixtures constitute a ubiquitouschallenge in drug discovery and developmentregimes. Natural products are mined for novelmolecular diversity. Drug metabolites are iden-tified for risk assessment at the drug discov-ery–development interface. In development,regulatory bodies demand the identificationof impurities at >0.1% levels. Selective, spe-cific and sensitive analyses that are preferablynondestructive are clearly of paramount im-portance throughout the drug discovery anddesign process.
NMR as a versatile analytical platformNuclear magnetic resonance spectroscopy(NMR) is the study of molecules by recordingthe interaction of radiofrequency electromag-netic radiation with the nuclei of moleculesplaced in a strong magnetic field. Modern NMRprovides an enormously powerful toolkit toprobe the molecular structure of organic com-pounds. Three distinct sub-disciplines of NMRare solid-state NMR, NMR imaging (MRI) andsolution-state NMR, which is the most widelyapplicable in drug discovery. There are manyadvantages in using NMR: it is a non-selective,non-destructive detector of low molecularweight molecules in solution; both endogenousand xenobiotic metabolites can be monitored
simultaneously in complex biofluids; andqualitative, and in many cases quantitative,data are achieved. NMR provides uniquely richstructural content, such as chemical shifts,multiplicity (the interaction between neigh-bouring nuclei), integrals, intramolecular relationships and a wide range of dynamicprocesses, including molecular motions in so-lution, chemical exchange and ligand bind-ing. NMR requires minimal sample prepara-tion and requires as little as 5 µl of sample withthe latest microcoil probes. As NMR is non-destructive, the sample can be recovered forfurther analysis. Bioactivity and toxicity testingis then possible by reconstituting the samplein an appropriate solvent. These technical ad-vantages are crucial in natural product anddrug metabolism studies in which sample islimited and minor novel metabolites are to becharacterized [1]. Complex mixture analysis isfurther enhanced by the effective coupling ofpowerful chromatographic techniques withon-line NMR detection [2]. Methodologicalaspects are reviewed elsewhere [2,3], and thisreview details recent hyphenated advances foridentifying drug-like molecules in complexmixtures.
Interfacing LC, NMR and MSIt is seldom possible to solve the structure of anovel compound by NMR alone. Commonfunctional groups such as carboxylic acid, phe-nol and amino groups are NMR-silent in manysolvents because of proton–deuterium ex-change. Nitro groups and sulfate conjugatesdo not contain protons and although thesefunctional groups are not directly detectablein proton NMR spectra, they can be readilydetected by mass spectrometry (MS). Conversely,MS data might give molecular weight, frag-mentation and molecular formulae that areinsufficient to unambiguously assign the mol-ecular structure of an unknown compound.In the most difficult cases closely eluting isobarsand isomers are indistinguishable by LC–MS
LC–NMR–MS in drug discoveryOlivia Corcoran and Manfred Spraul
Olivia CorcoranDepartment of PharmacyFranklin-Wilkins Building
150 Stamford StreetKing’s College London
UK SE1 9NNe-mail:
[email protected] Spraul
Bruker BioSpin GmbHSilberstreifen
76287 RheinstettenGermany
reviews research focus
624
DDT Vol. 8, No. 14 July 2003
Nuclear magnetic resonance spectroscopy (NMR) is arguably the most
versatile analytical platform for complex mixture analysis. Specifically,
interfacing liquid chromatography with parallel NMR and mass
spectrometry (LC–NMR–MS) gives comprehensive structural data on
metabolites of novel drugs in development. Applications in natural product,
combinatorial chemistry and drug metabolism studies are reviewed.
Microcoil probes and capillary separation methods have enormous
potential. Recent innovations to improve NMR detection limits include
CryoFlowProbesTM and on-line solid-phase extraction (LC–SPE–NMR).
These state-of-the-art analytical platforms are widely applicable to
identifying novel candidate drugs from diverse complex mixtures within
a drug discovery strategy.
www.drugdiscoverytoday.com
▼

[4]. It is possible to collect HPLC fractions for off-lineanalysis in NMR tubes. However, parallel on-line NMR andMS detection efficiently provides complementary data andminimizes ambiguities between LC–MS and LC–NMR sys-tems. Indeed, the characterization of certain reactive andvolatile metabolites was impossible by LC–MS and onlyfeasible by LC–NMR [4,5].
Until recent years, interfacing LC with NMR and MS wasdifficult. However, many of the problems in siting the LCand MS physically close to the NMR magnet have nowbeen overcome. With the latest shielded magnets, a bench-top ion trap MS can now be placed <1 m from the center of a 500 MHz magnet without adverse performance. Thisshort distance also limits LC peak broadening, which results in enhanced NMR sensitivity. Practically, a simple hyphenation of LC to NMR and MS is achieved using apost-column splitter. This directs 90–95% of the flow to theNMR via a 1–2 m capillary and the remainder to the MS. Apowerful alternative is the valve-switching interface termedthe BNMI (Bruker NMR-Mass Spectrometry Interface). Thisis a computer-controlled splitter and a double dilutor forproviding an appropriate make-up flow for optimal ioniza-tion in the MS. It also permits proton–deuterium exchangeto simplify MS spectra otherwise obtained in LC–NMR–MS.This BNMI plays an important role in LC–NMR–MS loopstorage mode in which a portion of the loop contents, ontransfer to the NMR, can be stored in a delay loop. DuringNMR acquisition, the dilutor then slowly infuses analytesinto the MS. These integrated LC–NMR–MS systems arehighly versatile, operating as needs dictate. Indeed, the LC–MScan be used routinely during longer NMR experiments.Other researchers have exploited the greater sensitivity ofMS to target minor yet important analytes, which requirelonger NMR acquisition for good quality NMR data [6–8].
The routine production of LC–NMR–MS results are sig-nificantly improved by expert optimization of chromato-graphic separation conditions using a set of pre-definedground rules [2,3]. Typically, analytical scale reversed-phaseHPLC uses columns of 2–4.6 mm i.d. × 50–250 mm lengthand flow rates of 0.2–1.5 ml min−1. Mobile-phase mixturesemploying deuterated water and non-deuterated organicmodifiers are routinely employed (acetonitrile/D2O ormethanol/D2O) and efficient solvent suppression routinesare available [2]. Occasionally, a fully deuterated organicsolvent might be used to minimize solvent artifacts in theNMR spectrum. However, it is seldom possible by one-di-mensional 1H NMR to fully characterize co-eluting analytesin those cases in which NMR signals from two or morecompounds are overlapped, and unless there is a significantdynamic range difference in concentration of the coelutinganalytes. Of course, on-line MS detection can be used to
deconvolute coeluting components, but only if they arenot isobars or isomers. If sufficient material is available,two-dimensional H–H NMR spectroscopy is useful to deter-mine correlations between the resonances within the co-eluting analytes. However, this can be time-consumingand counter-productive, and therefore optimizing thechromatographic resolution is frequently the logical solu-tion. Combining the NMR assignments with the MS dataoften provides unambiguous structural elucidation.
AutomationTo date, by far the majority of LC–NMR–MS applicationsuse stopped-flow mode. In this mode, the sample, as inconventional NMR, is stationary. Once the analyte reachesthe active NMR probe volume, the flow is halted and thesubsequent eluant is simply diverted either to waste or toon-line storage loops. Loop storage mode was a valuableadvance whereby chromatographic peaks are trapped on-line in a 36-loop cassette according to a UV or MS detectionthreshold [9,10]. Automation directs peaks to the loopsand controls the transfer of loop contents, one-at-a-time,to the NMR flow probe for stopped-flow analysis. Loopstorage avoids peak diffusion on-column, which reducesNMR sensitivity when several analytes require long NMRacquisition. Less sample is required because all the analytesin a single injection are stored and acquired later underautomation. If the BNMI is integrated then MS experimentscan be obtained on the loop contents while the NMR acquires data. Loop storage mode is therefore a highly efficient mode of operation.
Manual, semi-automated or fully automated operationis possible in both stopped-flow and loop storage modes.For flexibility, it is also possible to switch between modesduring a run. A state-of-the-art integrated system for LC–NMR–MS and on-line solid phase extraction (LC–SPE–NMR–MS) is described in Box 1, as well as the associatedhardware and software. The goal of LC–NMR–MS is to tar-get important analytes for efficient NMR analysis. In drugdiscovery, in which novel diverse molecules are present incomplex mixtures, UV and MS provide intelligence for fur-ther NMR experiments [6–8]. Mass-directed peak selectionhas been used to rapidly target urinary metabolites of the novel drug candidates GW420867 and GI265080 forstopped-flow NMR analysis [6]. MS–MS has also been usedto selectively discriminate anti-oxidant quercetin andphloretin glycosides from apple peel extract at concen-trations of 0.2–5 mg g−1 apple peel. MS–MS data triggeredeither direct transfer to the magnet or to loops for auto-mated NMR analysis. NMR data unequivocally identifiedboth the position and α/β conformations of the knownquercetin and phloretin glycosides [7].
625
DDT Vol. 8, No. 14 July 2003 reviewsresearch focus
www.drugdiscoverytoday.com
DetectionThe limits of detection at the 1H observation frequency of600 MHz for 500 MW analytes are ~100 ng in stopped-flowand loop storage mode. Although continuous flow or on-flow modes use NMR as the real-time detector, sensitivityand resolution are limited by the short residence time ofanalytes at 0.5–1.5 ml min−1, and typically >10 µg per analyte
are needed for quality results at the 1H observation fre-quency of 500 MHz [2]. Fluorine-containing molecules arerelatively common in agrochemical and drug design so 19F-NMR can be exploited for selective detection of xenobioticmetabolites from complex mixtures [11,12]. Because radio-labelled drug is not usually available until a candidate isselected for development, 14C-detection is less useful in
626
DDT Vol. 8, No. 14 July 2003reviews research focus
www.drugdiscoverytoday.com
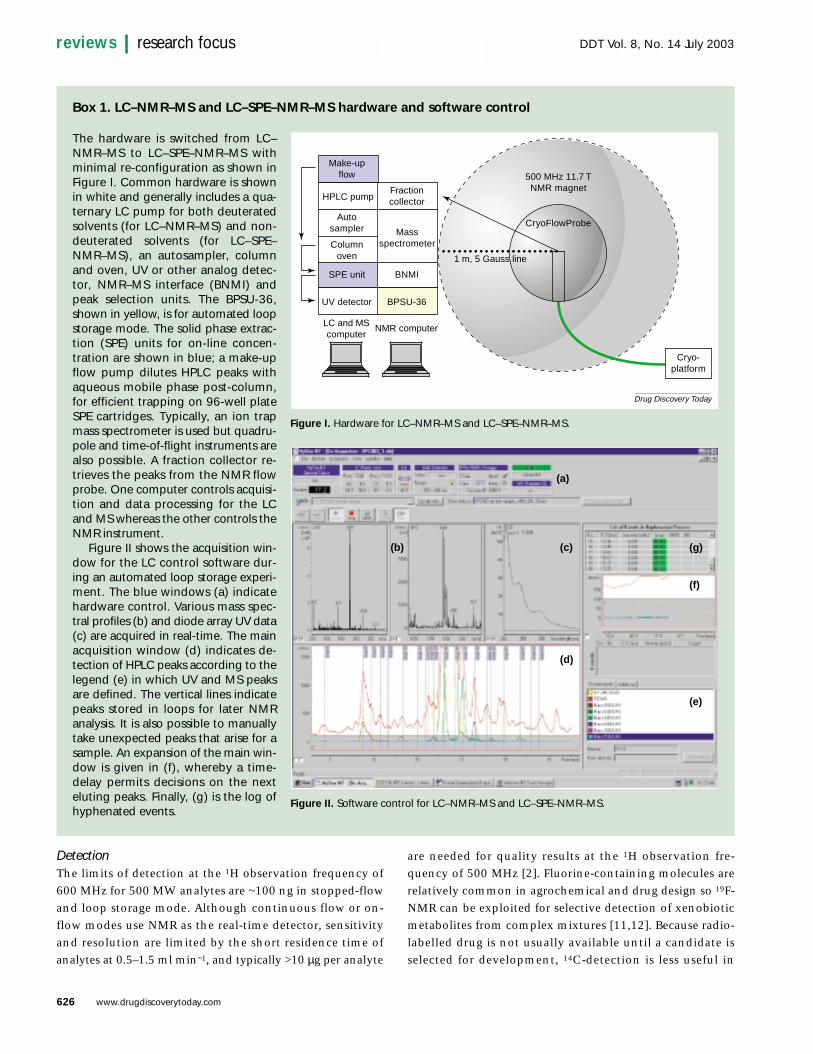
Box 1. LC–NMR–MS and LC–SPE–NMR–MS hardware and software control
The hardware is switched from LC–NMR–MS to LC–SPE–NMR–MS withminimal re-configuration as shown inFigure I. Common hardware is shownin white and generally includes a qua-ternary LC pump for both deuteratedsolvents (for LC–NMR–MS) and non-deuterated solvents (for LC–SPE–NMR–MS), an autosampler, columnand oven, UV or other analog detec-tor, NMR–MS interface (BNMI) andpeak selection units. The BPSU-36,shown in yellow, is for automated loopstorage mode. The solid phase extrac-tion (SPE) units for on-line concen-tration are shown in blue; a make-upflow pump dilutes HPLC peaks withaqueous mobile phase post-column,for efficient trapping on 96-well plateSPE cartridges. Typically, an ion trapmass spectrometer is used but quadru-pole and time-of-flight instruments arealso possible. A fraction collector re-trieves the peaks from the NMR flowprobe. One computer controls acquisi-tion and data processing for the LCand MS whereas the other controls theNMR instrument.
Figure II shows the acquisition win-dow for the LC control software dur-ing an automated loop storage experi-ment. The blue windows (a) indicatehardware control. Various mass spec-tral profiles (b) and diode array UV data(c) are acquired in real-time. The mainacquisition window (d) indicates de-tection of HPLC peaks according to thelegend (e) in which UV and MS peaksare defined. The vertical lines indicatepeaks stored in loops for later NMRanalysis. It is also possible to manuallytake unexpected peaks that arise for asample. An expansion of the main win-dow is given in (f), whereby a time-delay permits decisions on the nexteluting peaks. Finally, (g) is the log ofhyphenated events.
Drug Discovery Today
LC and MScomputer
NMR computer
CryoFlowProbeAuto
sampler
Columnoven
Massspectrometer
SPE unit BNMI
UV detector BPSU-36
Fractioncollector
Make-upflow
HPLC pump
500 MHz 11.7 TNMR magnet
Cryo-platform
1 m, 5 Gauss line
(a)
(b) (c) (g)
(f)
(e)
(d)
Figure I. Hardware for LC–NMR–MS and LC–SPE–NMR–MS.
Figure II. Software control for LC–NMR–MS and LC–SPE–NMR–MS.

discovery [13]. Other detection methods of limited use indrug discovery are reviewed elsewhere [2]. Given sufficientmaterial in stopped-flow and loop storage mode, two-dimensional NMR experiments that are routinely used forcharacterization and structural elucidation are feasible[4,7,11,14]. These experiments typically include COSY(H–H correlation spectroscopy, used to identify neighbor-ing protons), TOCSY (H–H total correlation spectroscopy, avariant on COSY that identifies longer range H–H cou-plings in the molecule), and HSQC (heteronuclear singlequantum coherence spectroscopy, one of a family of exper-iments to elucidate the H–C connectivities in a molecule).
Typical stopped-flow detection limits at the 1H observa-tion frequency of 600 MHz using micro coil probes andcapillary systems are in the 5 ng range on overnight acqui-sition, however high concentrations of analyte are requiredin the 1.5 µl NMR-active probe volume. It is possible toachieve quality NMR spectra on routine 500–600 MHzspectrometers equipped with 60–240 µl flow probe cells onovernight accumulation of scans using <100 ng per analytein stopped-flow and loop storage mode [2,3].
Natural products and combinatorial chemistryNovel molecular diversity (NMD) poses a formidable chal-lenge for rational drug design. Bioassay-guided fractiona-tion of natural products is one HTS approach to identifypotent bioactive molecules. Likewise, combinatorial chem-istry potentially provides thousands of novel candidates.Both approaches can provide leads for synthetic strategies.However, neither readily provides optimal NMD, perceivedto be so vital to modern pharmaceutical success. Althoughselectivity and potency are paramount features of a block-buster drug, evaluating safety in man remains a formidablehurdle. Unfortunately, the ultimate measure of drug safetyis clinical use, as illustrated by the many drugs that havebeen withdrawn from the market because of adverse drugreactions [15]. The ability to rapidly identify known or un-desirable molecules at all of the above junctures is thereforecrucial to pharmaceutical companies and LC–NMR–MSwill undoubtedly play an important role in the future ofdrug discovery.
In drug discovery from natural products, it is crucial toavoid duplicate isolation of known or undesirable com-pounds from complex extracts. The term ‘dereplication’describes the commonly used strategy of separating nat-ural product extracts and using UV or MS detection tocheck data against MS and NMR databases or libraries.Although still a field in its infancy, the explosive growth ofLC–NMR and LC–NMR–MS for dereplication in the pastfive years is documented in Refs [16,17]. Even though themajority identify members of known structural classes to
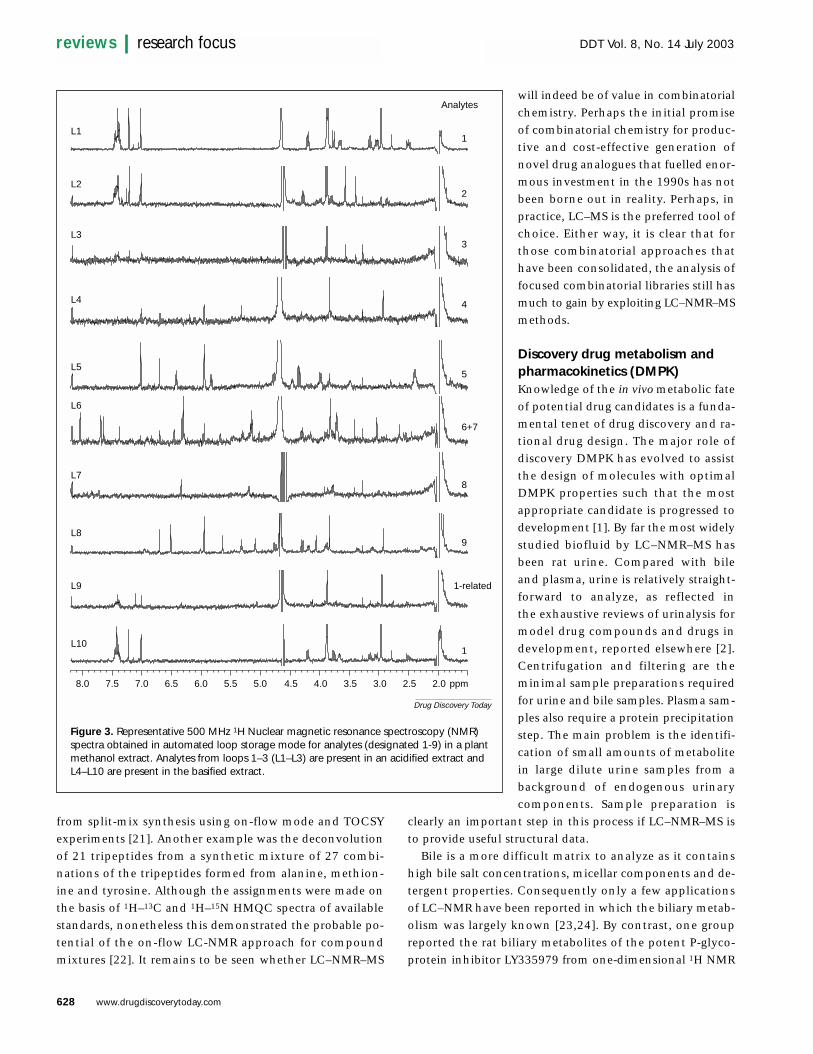
evaluate the use of LC–NMR and LC–MS for dereplication,a few report interesting novel bioactive analogues. One ofthe first studies employed LC–NMR–MS to identify minornovel ecdysteroids from Silene otides that were not detectedby LC–NMR alone, illustrating the complementary natureof NMR and MS data for phytochemical analysis [18].Pharmacognosy and phytochemical laboratories will alsobenefit from faster dereplication using automated loopstorage LC–NMR–MS. This is illustrated by the identifi-cation of nine closely eluting and isomeric aporphine alkaloids in the Taiwanese plant Litsea genus [10] using 50 times less material compared with conventional NMR experiments using 5 mm tubes. Automated loop storageLC–NMR–MS also rapidly identified the main alkaloidclasses from extracts of four medicinal plants from theCape Peninsula region. Figure 3 illustrates the resultingNMR spectra obtained by loop storage of analytes (desig-nated 1–9) from the original methanol extract. Analytesfrom loops 1–3 (L1–L8) are present in an acidified extractand L4–L10 are present in a basified extract. Several differ-ent chemical classes are obvious and co-elution (analytes 6and 7) is observed in the spectrum from loop 6. Traditionaloff-line phytochemical approaches can take several monthscompared with these results which were achieved in 36 husing fully automated loop storage mode on a 500 MHzspectrometer using only 150–300 µg of each extract. It isclear that combining data from such spectra with NMRand MS databases has great potential for efficient derepli-cation strategies in natural product analysis.
Microbial production of secondary metabolites is an-other important and versatile source of novel candidatedrugs, although somewhat limited for NMD purposes.Microbial models of mammalian metabolism also providea biosynthetic source of putative human metabolites forstructural elucidation by NMR at the discovery stage [9,19].LC–NMR provides rapid multiparametric information onmicrobial biotransformations as illustrated by the identifi-cation of novel warfarin metabolites from Streptomyces rimosus [9] and the identification of the antibiotic aris-teromycin from broth supernatants of Streptomyces citri-colour [20]. Similarly, LC–NMR and LC–MS were used toidentify bioactive molecules from marine sources such asthe sponge Aaptos sp. [17] and the starfish Asterias rubens[14]. Clearly the discovery of NMD from such phytochem-ical, microbial and marine sources will be greatly facilitatedby LC–NMR–MS advances.
The likely use of LC–NMR for the analysis of combinato-rial chemistry mixtures was ably shown by the structuredetermination of a four-component mixture of aromaticring positional isomers and a five-component pentapep-tide mixture containing two isomolecular weight peptides
627
DDT Vol. 8, No. 14 July 2003 reviewsresearch focus
www.drugdiscoverytoday.com
from split-mix synthesis using on-flow mode and TOCSYexperiments [21]. Another example was the deconvolutionof 21 tripeptides from a synthetic mixture of 27 combi-nations of the tripeptides formed from alanine, methion-ine and tyrosine. Although the assignments were made onthe basis of 1H–13C and 1H–15N HMQC spectra of availablestandards, nonetheless this demonstrated the probable po-tential of the on-flow LC-NMR approach for compoundmixtures [22]. It remains to be seen whether LC–NMR–MS
will indeed be of value in combinatorialchemistry. Perhaps the initial promiseof combinatorial chemistry for produc-tive and cost-effective generation ofnovel drug analogues that fuelled enor-mous investment in the 1990s has notbeen borne out in reality. Perhaps, inpractice, LC–MS is the preferred tool ofchoice. Either way, it is clear that forthose combinatorial approaches thathave been consolidated, the analysis offocused combinatorial libraries still hasmuch to gain by exploiting LC–NMR–MSmethods.
Discovery drug metabolism andpharmacokinetics (DMPK)Knowledge of the in vivo metabolic fateof potential drug candidates is a funda-mental tenet of drug discovery and ra-tional drug design. The major role ofdiscovery DMPK has evolved to assistthe design of molecules with optimalDMPK properties such that the mostappropriate candidate is progressed todevelopment [1]. By far the most widelystudied biofluid by LC–NMR–MS hasbeen rat urine. Compared with bileand plasma, urine is relatively straight-forward to analyze, as reflected in the exhaustive reviews of urinalysis formodel drug compounds and drugs indevelopment, reported elsewhere [2].Centrifugation and filtering are theminimal sample preparations requiredfor urine and bile samples. Plasma sam-ples also require a protein precipitationstep. The main problem is the identifi-cation of small amounts of metabolitein large dilute urine samples from abackground of endogenous urinarycomponents. Sample preparation is
clearly an important step in this process if LC–NMR–MS isto provide useful structural data.
Bile is a more difficult matrix to analyze as it containshigh bile salt concentrations, micellar components and de-tergent properties. Consequently only a few applicationsof LC–NMR have been reported in which the biliary metab-olism was largely known [23,24]. By contrast, one groupreported the rat biliary metabolites of the potent P-glyco-protein inhibitor LY335979 from one-dimensional 1H NMR
628
DDT Vol. 8, No. 14 July 2003reviews research focus
www.drugdiscoverytoday.com
Figure 3. Representative 500 MHz 1H Nuclear magnetic resonance spectroscopy (NMR)spectra obtained in automated loop storage mode for analytes (designated 1-9) in a plantmethanol extract. Analytes from loops 1–3 (L1–L3) are present in an acidified extract andL4–L10 are present in the basified extract.
Drug Discovery Today
L1
L2
L3
L4
L5
L6
L7
L8
L9
L10
2.02.53.03.54.04.55.05.56.06.57.07.58.0 ppm
Analytes
1
2
3
4
5
6+7
8
9
1-related
1

spectra obtained in 2–3 h on an estimated 100 ng materialusing a 500 MHz instrument [25]. Sample preparation forurine and bile often involves a simple pre-chromatographicconcentration step that passes the sample through a solidphase extraction (SPE) cartridge to retain the analytes ofinterest and elute interferents. Finally, the analyte is elutedusing organic solvent and injected onto the chromato-graphic column. However, when lyophilization and selec-tive solvent extractions are used, many labile and reactivemetabolites might be lost. Consequently, intelligent sam-ple preparation is required if accurate biliary and urinaryprofiles are to be determined by LC–NMR–MS as part ofrisk assessment in drug discovery.
The in vivo use of animals in drug discovery also raisesemotive ethical issues. Unfortunately, often the only wayto generate sufficient quantities of metabolites for struc-tural elucidation and risk assessment is to dose multipleanimals. Multiple compound ‘cassette’ dosing, first reportedby Potts and co-workers [26], is a relatively new approachunder evaluation for increasing throughput of in vivo metab-olism studies while reducing animal numbers [26,27].Although limited to fluorine-containing drugs, we reporteda preliminary evaluation of 19F NMR spectroscopy as ameans of providing metabolic as opposed to pharmacokineticdata following cassette dose of six model drug compoundsto the rat [28].
Innovations in HTS and laboratory robotics during the1990s have enabled a high level of automation for in vitromodels of drug metabolism. Various models are used at anincreasingly early stage in drug discovery and these includeisolated hepatocytes, human liver slices, subcellular matri-ces, microbial systems and genetically engineered humandrug metabolizing enzymes (DMEs). The putative risks forcertain drug metabolites can therefore be evaluated. Thechoice of a suitable model species for man is also of funda-mental importance. Although the subcellular matrix is rel-atively clean by NMR standards, metabolite identificationat the typically low substrate concentration range of 5–100µm poses a substantial challenge for LC–NMR studies usingroutinely available 500 MHz spectrometers [29]. Semi-preparative LC–MS-directed purification was therefore re-quired to isolate metabolites of the candidate anti-schizo-phrenic drug iloperidone in SPE-concentrated extracts ofdog liver microsomes and cytosolic fractions for off-lineLC–NMR analysis at 500 MHz [23]. Using a 600 MHz spec-trometer it was soon possible to identify metabolites of thepotential monoamine oxidase (MAO) inhibitor BW1370U87directly from SPE extracts of human liver microsomes [30].Directly coupled LC–NMR at 750 MHz identified phenoxy-pyridine metabolites down to the 0.6% level in extracts of rat microsomal incubations containing 1 mM substrate
[31]. Stopped-flow LC–NMR spectra were obtained in 15min with ~200 ng of material in the NMR flow probe.
Microbial models of mammalian metabolism are also animportant tool at the discovery stage. These can provide abiosynthetic source of putative human metabolites as apossible alternative to dosing animals to generate suffi-cient metabolite quantities for structural determination byNMR [9,19]. However, the rate-limiting step is screeningfor microbial species that parallel the biochemical machin-ery (such as cytochrome P450 enzymes). Once microbialspecies that produce the required metabolites are found,experiments can be scaled-up to produce milligram quanti-ties of putative human metabolites. A recent study exploitedthe presence of fluorine in drug candidates to evaluate byautomated 19F NMR 48 taxonomically diverse organismsfor biosynthesis of minor rat urinary metabolites [19].These diverse applications of LC–NMR–MS in DMPK amplydemonstrate that recent advances will greatly facilitaterapid metabolic information within the timescale of high-throughput lead optimization exercises in drug discovery.
Capillary separations and microcoil NMR probesMiniaturization of separation techniques is potentially im-portant in hyphenated NMR, particularly for mass-limitedsamples in drug discovery. Microcoil probes have a highfilling factor as the RF coil is wound directly onto the cell.Coupling capillary LC (CHPLC) and capillary electrochro-matography (CEC) to these probes permits less sample andeluant requirements and shorter analysis times [32,33]. Ahigher sample loading than CE-NMR is also achievable,which bodes well for NMR sensitivity. One group reporteda system using a 240 nl NMR cell that readily configuresfor CHPLC, CE and CEC–NMR and this was applied to theanalysis of natural products [33], drugs and their metab-olites [34]. We recently reported a commercial capLC–NMRsystem using a capNMR™ (Waters; http://www.waters.comand Protasis; http://www.protasis.com) probe (1.5 µl active-NMR volume) in automated stopped-flow mode at 600MHz to obtain 1H NMR spectra with detection limits of5–25 ng per drug metabolite in an extract of urine 4 h aftera 1 g dose of acetaminophen [35]. Capillary isotachophoresisand capillary zone electrophoresis have also been success-fully coupled to NMR microcoils for improved sampleconcentration [36] and trace impurity analysis [37] andthis is an area of great potential. As the newer capillary-NMRtechniques become commercialized, they will undoubtedlybe applied at the discovery stage for mass-limited samples.
Advancing sensitivityThe highest magnetic field strengths with cryogenicallycooled NMR probes and preamplifiers provide unsurpassed
629
DDT Vol. 8, No. 14 July 2003 reviewsresearch focus
www.drugdiscoverytoday.com

NMR sensitivity. Currently, the highest magnetic field usedfor LC–NMR–MS is 800 MHz [38] and LC–NMR has yet to be reported at 900 MHz. An alternative for increasingsensitivity on existing LC–NMR systems is an on-line SPEadd-on, particularly for routine field strengths of 400–600MHz [35]. In the integrated LC–SPE–NMR system the ana-lytes are detected post-column by UV or MS and automati-cally trapped on SPE cartridges in 96-well plate format usingthe modified Spark-Prospekt SPE instrument (http://www.spark.nl). Multiple trapping of a given peak on a given SPEcartridge is possible, the cartridge is then dried under nitrogen and the contents are finally eluted from the car-tridge into the NMR flow probe. This is highly economicalas nondeuterated solvents and a wider choice of HPLCbuffers are now permissible for the chromatography andonly the final transfer volume of 200–500 µl is deuterated.Exchangeable protons are observed in the NMR spectrumwhich can aid assignment. As the probe only ever receivesthe same deuterated solvent, shimming is trivial and solvent suppression is mostly not required. The elutingvolume of 30–50 µl is best matched to a 30 µl flow probe,and because of an automated wash and dry procedure theresulting spectra show no cross-contamination. The signal-to-noise ratio is improved up to 6.8-fold on triple trappingcompared with conventional loop collection mode for a60 µl probe at 500 MHz and this procedure is fully auto-mated [35].
Finally, the cryogenic cooling of the NMR RF coils andelectronics to give greatly enhanced sensitivity is arguablythe most significant recent advance in NMR spectroscopy[8,39]. The four-fold increase in sensitivity enables a four-fold lower detection limit which reduces the experimenttime 16-fold for a given sample. This is of critical impor-tance in drug discovery with mass-limited analyte, reactivemetabolites and necessary short experimental time becauseof a high-throughput analytical regime. We recently applied the first CryoFlowProbe™ (Bruker; http://www.bruker-biospin.com) to the identification of acetaminophenmetabolites in human urine using LC–NMR–MS [8]. Besidesthe known glucuronide and sulfate metabolites, threemetabolites previously undetected at 500 MHz were di-rectly observable in a 15 min on-flow experiment. MS datacombined with stopped-flow spectra for greater signal-to-noise ratios enabled unambiguous assignment of thesecompounds.
ConclusionsLC–NMR–MS in automated loop storage mode clearly pro-vides a versatile analytical platform for complex mixtureanalysis. The most demanding problems require two-di-mensional NMR and 13C NMR data and the continuing
pace of SPE and CryoFlowProbe™ developments will nodoubt drive these applications forward. The diversity of ap-plications for LC–NMR–MS is a powerful testament to thecontributions LC–NMR–MS can make to rational drug design strategies. In future, automated MS-directed LC–SPE–NMR–MS systems equipped with CryoFlowProbes™ shouldprovide the ultimate in NMR sensitivity for drug discovery.
AcknowledgementsOlivia Corcoran holds a Maplethorpe Fellowship of theUniversity of London.
References1 Watt, A.T. et al. (2003) Metabolite identification in drug discovery.
Curr. Opin. Drug Disc. Dev. 6, 57–652 Alberts, K., ed. (2002) On-line LC-NMR and Related Techniques, John
Wiley & Sons3 Holzgrabe, U. et al. (1998) NMR Spectroscopy in Drug Development and
Analysis, Wiley-VCH4 Corcoran, O. et al. (2001) HPLC/1H NMR spectroscopic studies of the
reactive α-1-O-acyl isomer formed during acyl migration of S-naproxenβ-1-O-acyl glucuronide. Chem. Res. Toxicol. 14, 1363–1370
5 Ruhl, R. et al. (2001) Synthesis, high-performance liquid-chromatography-nuclear magnetic resonance characterization andpharmacokinetics in mice of CD271 glucuronide. J. Chromatogr. BBiomed. Appl. 757, 101–109
6 Dear, G.J. et al. (2000) Mass directed peak selection, an efficient methodof drug metabolite identification using directly coupled liquid-chromatography-mass spectrometry-nuclear magnetic resonancespectroscopy. J. Chromatogr. B Biomed. Appl. 748, 281–293
7 Lommen, A. et al. (2000) Application of directly coupled HPLC-NMR-MS to the identification and confirmation of quercetin glycosides andphloretin glycosides in apple peel. Anal. Chem. 72, 1793–1797
8 Spraul, M. et al. (2003) Advancing sensitivity for LC-NMR-MS using acryogenic probe: application to the metabolites of acetaminophen.Anal. Chem. 75, 1546–1551
9 Cannell, R.J.P. et al. (1997) Novel metabolites of warfarin produced byBeauveria bassiana and Streptomyces rimosus: a novel application ofHPLC-NMR. Xenobiotica 27, 147–157
10 Tseng, L.H. et al. (2000) Structure identification of aporphine alkaloidsby on-line coupling of HPLC-NMR with loop storage. J. Chin. Chem.Soc. 47, 1231–1236
11 Shockcor, J.P. et al. (2000) Application of directly coupled LC-NMR-MSto the structural elucidation of metabolites of the HIV-1 reversetranscriptase inhibitor BW935U83. J. Chromatogr. B Biomed. Appl. 784,269–279
12 Tugnait, M. et al. (2002) The metabolism of 4-trifluoromethoxyanilineand [13C]-4-trifluoromethoxyacetanilide in the rat: detection andidentification of metabolites excreted in the urine by NMR and HPLC-NMR. J. Pharm. Biomed. Anal. 28, 875–885
13 Dear, G.J. et al. (2002) The potential of serially coupled alkyl-bondedsilica monolithic columns for high resolution separations ofpharmaceutical compounds in biological fluids. Chromatographia 55,177–184
14 Sandvoss, M. et al. (2000) Isolation and structural elucidation of steroidoligoglycosides from the starfish Asterias rubens by means of directonline LC-NMR-MS hyphenation and 1 and 2 dimensional NMRinvestigations. Eur. J. Org. Chem. 7, 1253–1262
15 Li, A.P. (2002) A review of the common properties of drugs withidiosyncratic hepatotoxicity and the ‘multiple determinant hypothesis’for the manifestation of idiosyncratic drug toxicity. Chem. Biol. Interact.142, 7–23
630
DDT Vol. 8, No. 14 July 2003reviews research focus
www.drugdiscoverytoday.com

16 Wolfender, J.L. et al. (1998) Liquid chromatography coupled to massspectrometry and nuclear magnetic resonance spectroscopy for thescreening of plant constituents. J. Chromatogr. A 794, 299–316
17 Bobzin, S.C. et al. (2000) Application of liquid chromatography-nuclearmagnetic resonance spectroscopy to the identification of naturalproducts. J. Chromatogr. B Biomed. Appl. 748, 259–267
18 Wilson, I.D. et al. (1999) High performance liquid chromatographycoupled to nuclear magnetic resonance spectroscopy and massspectrometry applied to plant products: identification of ecdysteroidsfrom Silene Otides. Chromatographia 49, 374–378
19 Corcoran, O. et al. (2001) The potential of 19F NMR spectroscopy forrapid screening of cell cultures for models of mammalian drugmetabolism. Analyst 126, 2103–2106
20 Abel, C.B.L. et al. (1999) Characterization of metabolites in intactStreptomyces citricolor culture supernatants using high resolution nuclearmagnetic resonance spectroscopy and directly coupled high-pressureliquid chromatography-nuclear magnetic resonance spectroscopy. Anal. Biochem. 270, 220–230
21 Chin, J. et al. (1998) HPLC-NMR in combinatorial chemistry. J. Org.Chem. 63, 386–390
22 Lindon, J.C. et al. (1995) Separation and characterization ofcomponents of peptide libraries using on-flow coupled HPLC-NMRspectroscopy. Magn. Reson. Chem. 33, 863–875
23 Mutlib, A.E. et al. (1995) Application of hyphenated LC/NMR andLC/MS techniques in rapid identification of in vitro and in vivometabolites of iloperidone. Drug Metab. Dispos. 23, 951–964
24 Spraul, M. et al. (1995) Evaluation of liquid chromatography coupledwith high field 1H NMR spectroscopy for drug metabolism anddetection and characterization: the identification of paracetamolmetabolites in urine and bile. NMR Biomed. 7, 295–303
25 Ehlhardt, W.J. et al. (1998) Liquid chromatography/nuclear magneticresonance spectroscopy and liquid chromatography/mass spectrometryidentification of novel metabolites of the multidrug resistancemodulator LY335979 in rat bile and human liver microsomalincubations. Drug Metab. Dispos. 26, 42–51
26 Potts, W. et al. (1995) Pharmacokinetic assessment of a mixture ofcompounds in the rat using simultaneous dosing and simultaneousLC/MS/MS quantitation. ISSX Proc. 8, 404
27 Tarbit, M.H. and Berman, J. (1998) High-throughput approaches for
evaluating absorption, distribution, metabolism and excretion of leadcompounds. Curr. Opin. Chem. Biol. 2, 411–416
28 Corcoran, O. et al. (1999) Rapid multi-component detection offluorinated drug metabolites in whole urine from a cassette dose studyusing high resolution 19F NMR spectroscopy. Anal. Commun. 36, 259–261
29 Zhang, K.E. et al. (2001) Liquid-chromatography mass spectrometryand liquid chromatography-NMR characterization of in vitrometabolites of a potent and irreversible peptidomimetic inhibitor ofrhinovirus 3C protease. Drug Metab. Dispos. 29, 729–734
30 Shockcor, J.P. et al. (1996) Characterization of in vitro metabolites fromhuman liver microsomes using directly coupled HPLC-NMR:application to a phenoxathiin monoamine oxidase-A inhibitor.Xenobiotica 26, 41–48
31 Corcoran, O. et al. (1997) 750 MHz HPLC-NMR spectroscopicidentification of rat microsomal metabolites of phenoxypyridines. J. Pharm. Biomed. Anal. 16, 481–489
32 Gfrorer, P. et al. (1999) On-line coupling of capillary separationtechniques with 1H NMR. Anal. Chem. 71, 315A–321A
33 Pusecker, K. et al. (1998) On-line coupling of capillary electro-chromatography, capillary electrophoresis, and capillary HPLC withnuclear magnetic resonance spectroscopy. Anal. Chem. 70, 3280–3285
34 Gfrorer, P. et al. (1999) Gradient elution capillaryelectrochromatography and hyphenation with nuclear magneticresonance. Electrophoresis 20, 3–8
35 Corcoran, O. et al. (2002) Advancing sensitivity for flow NMRspectroscopy: LC-SPE-NMR and capillary-scale LC-NMR. Am. Lab. 34,18–21
36 Kautz, R.A. et al. (2001) Sample concentration and separation fornanoliter-volume NMR spectroscopy using capillary isotachophoresis.J. Am. Chem. Soc. 123, 3159–3160
37 Wolters, A.M. et al. (2002) Capillary isotachophoresis/NMR: extensionto trace impurity analysis and improved instrumental coupling. Anal.Chem. 74, 2306–2313
38 Sidelmann, U.G. et al. (1997) Directly coupled 800 MHz HPLC-NMRspectroscopy of urine and its application to the identification of themajor phase II metabolites of tolfenamic acid. Anal. Chem. 69, 607–612
39 Styles, P. et al. (1984) A high-resolution NMR probe in which the coiland preamplifier are cooled with liquid helium. J. Magn. Reson. 60,397–404
631
DDT Vol. 8, No. 14 July 2003 reviewsresearch focus
www.drugdiscoverytoday.com
Do you want to reproduce material from Drug Discovery Today?
This publication and the individual contributions contained in it are protected by the copyright of Elsevier Science.
Except as outlined in the terms and conditions (see p. X), no part of Drug Discovery Today can be reproduced,
either in print or in electronic form, without written permission from Elsevier
Please send any permission requests to:
Elsevier, PO Box 800, Oxford, UK OX5 1DX





![“NMR in drug discovery. The power of resolution...I will discuss (i) how we used NMR spectroscopy to elucidate the mechanism of T-cell receptor signaling [1], (ii) the re-discovery](https://static.fdocuments.us/doc/165x107/60179f40921ed839e528047a/aoenmr-in-drug-discovery-the-power-of-resolution-i-will-discuss-i-how-we.jpg)












