
JRMO Non-CTIMP Protocol Templatelsrg.org.uk/.../uploads/2015/06/PRIME-Protocol-v3.5.docx · Web...
29
LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 1 of 29 CLINICAL TRIAL PROTOCOL Full Title Perioperative Reduction of Inapparent Myocardial Events (PRIME) Pilot Study: a randomised controlled trial Short Title PRIME Pilot Study Sponsor 1 Royal Berkshire NHS Foundation Trust London Road Reading, Berkshire RG1 5AN Tel: 0118 322 8693/7449 Fax: 0118 322 8425 Research Collaborative 2 London Surgical Research Group (LSRG) c/o National Centre for Bowel Research & Surgical Innovation 1 st Floor Abernathy Building 2 Newark St Whitechapel London E1 2AT Tel: 020 7882 8749 REC Reference 13/SC/0306 Clinicaltrials.gov NCT01850927 Chief Investigator Dr Andrew Walden Consultant Intensive Care Royal Berkshire NHS Foundation Trust Principal Investigator Mr Stefan Antonowicz Specialist Registrar General Surgery Oxford Deanery & London Surgical Research Group Steering Committee Mr Stefan Antonowicz 1,2 , Mr Tom Wiggins 2 , Mr James Haddow 2 , Mr Nicholas Symons 2 , Professor Charles Knowles 2 and Dr Andrew Walden 1 Management Group Mr Stefan Antonowicz 1,2 , Mr Dominic Coull 1 , Dr Andrew Walden 1 Reviewers Mr Michael Parker, Medical Statistician, Post- graduate Medical Institute, Anglia Ruskin University
Transcript of JRMO Non-CTIMP Protocol Templatelsrg.org.uk/.../uploads/2015/06/PRIME-Protocol-v3.5.docx · Web...

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 1 of 21
CLINICAL TRIAL PROTOCOL
Full Title Perioperative Reduction of Inapparent Myocardial Events (PRIME) Pilot Study: a randomised controlled trial
Short Title PRIME Pilot Study
Sponsor 1Royal Berkshire NHS Foundation TrustLondon RoadReading, BerkshireRG1 5ANTel: 0118 322 8693/7449Fax: 0118 322 8425
Research Collaborative 2London Surgical Research Group (LSRG) c/o National Centre for Bowel Research & Surgical Innovation1st Floor Abernathy Building2 Newark StWhitechapelLondon E1 2ATTel: 020 7882 8749
REC Reference 13/SC/0306
Clinicaltrials.gov NCT01850927
Chief Investigator Dr Andrew WaldenConsultant Intensive CareRoyal Berkshire NHS Foundation Trust
Principal Investigator Mr Stefan AntonowiczSpecialist Registrar General SurgeryOxford Deanery & London Surgical Research Group
Steering Committee Mr Stefan Antonowicz1,2, Mr Tom Wiggins2, Mr James Haddow2, Mr Nicholas Symons2, Professor Charles Knowles2 and Dr Andrew Walden1
Management Group Mr Stefan Antonowicz1,2, Mr Dominic Coull1, Dr Andrew Walden1
Reviewers Mr Michael Parker, Medical Statistician, Post-graduate Medical Institute, Anglia Ruskin UniversityDr Stuart McKechnie, Consultant Anaesthetist, Oxford University Hospital (Peer reviewer)Dr Robert Parker, Consultant Intensivist, Aintree University Hospitals (Peer reviewer)

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 2 of 21
TABLE OF CONTENTS
1. Glossary of Terms and Abbreviations...................................................................42. Signature Page........................................................................................................53. Summary..................................................................................................................64. Introduction..............................................................................................................75. Study Aims & Endpoints.........................................................................................8
5.1 Aims..................................................................................................................................85.2 Primary endpoint...............................................................................................................85.3 Secondary endpoints.........................................................................................................85.4 Feasibility endpoints..........................................................................................................8
6. Methodology............................................................................................................86.1 Inclusion Criteria................................................................................................................86.2 Exclusion Criteria..............................................................................................................86.3 Study Design and Plan......................................................................................................96.4 Explanatory variables........................................................................................................96.5 Study Scheme Diagram..................................................................................................11
7. Study Procedures..................................................................................................117.1 Study Timetable..............................................................................................................117.2 Recruitment and Informed Consent Procedures.............................................................127.3 Recruitment Rates...........................................................................................................127.4 Randomisation................................................................................................................127.5 Treatment Protocols........................................................................................................127.6 Schedule of Intervention.................................................................................................137.7 Schedule of Assessment.................................................................................................137.8 Criteria for Discontinuation..............................................................................................137.9 Laboratory Assessments.................................................................................................137.10 Subject Withdrawal........................................................................................................147.11 End of Recruitment Definition.......................................................................................14
8. Statistical Considerations....................................................................................148.1 Sample Size....................................................................................................................148.2 Methods of Analysis........................................................................................................14
9. Ethical Considerations..........................................................................................159.1 Consent...........................................................................................................................159.2 Declaration of Helsinski and Good Clinical Practice.......................................................159.3 Ethical Committee Review..............................................................................................15
10. Safety Considerations and Reporting...............................................................1510.1 Adverse Event...............................................................................................................1510.2 Serious Adverse Event (SAE).......................................................................................1510.3 Unexpected Adverse Event...........................................................................................1510.4 Expected Adverse Events (recognised to be caused by the RIPC stimulus)................1510.5 Expected Serious Events Related to Usual Clinical Care.............................................1610.6 Reporting Unexpected Adverse Events........................................................................16
10.6.1 Assessment of Intensity..........................................................................................1610.7 Urgent Safety Measures................................................................................................16
11. Data Handling and Record Keeping..................................................................1612. Laboratories.........................................................................................................1613. Devices and Techniques.....................................................................................17
13.1 Devices..........................................................................................................................17

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 3 of 21
13.2 Techniques and interventions.......................................................................................17
14. Monitoring and Auditing.....................................................................................1715. Study Committees...............................................................................................17
15.1 Study Steering Committee............................................................................................1715.2 Study Management Group............................................................................................17
16. Finance and Funding..........................................................................................1717. Indemnity.............................................................................................................1718. Dissemination of Research Findings................................................................1819. References...........................................................................................................1820. Appendix I: Clinical endpoint definitions..........................................................19
20.1 MACCE.........................................................................................................................1920.2 Non-fatal Cardiac Arrest................................................................................................1920.3 Acute Myocardial Infarction...........................................................................................1920.4 Angina...........................................................................................................................1920.5 New Cardiac Arrhythmia...............................................................................................1920.6 Congestive Heart Failure...............................................................................................1920.7 Stroke............................................................................................................................1920.8 Cerebrovascular Death.................................................................................................1920.9 Cardiovascular Death....................................................................................................1920.10 Clavien-Dindo Classification:.......................................................................................2020.11 30 day significant surgical complications....................................................................2020.12 Acute Kidney Injury (RIFLE criteria)............................................................................20
21. Appendix II: Components of case reporting formS..........................................2121.1 Consent Form Items (to be recorded by PI)..................................................................2121.2 Baseline CRF Data Items (to be recorded by PI)..........................................................2121.3 Treatment CRF Data Items (to be recorded by treatment officer).................................2121.4 Discharge CRF Data Items (to be recorded by PI).......................................................2121.5 30-day CRF data items (to be recorded by PI).............................................................2121.6 Laboratory CRF Data Items (to be collected by PI)......................................................21

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 4 of 21
1. GLOSSARY OF TERMS AND ABBREVIATIONS
AE Adverse EventAKI Acute Kidney InjuryAR Adverse ReactionAUC Area under curveCA Competent AuthorityCI Chief InvestigatorCRF Case Report FormICF Informed Consent FormJRMO Joint Research Management OfficeMACCE Major adverse cardiac or cerebral event (as defined in Appendix I)MI Myocardial infarction (as defined in Appendix I)NHS REC National Health Service Research Ethics CommitteeNHS R&D National Health Service Research & Development Participant A patient who takes part in this studyPI Principal InvestigatorPIS Participant Information Sheet RCT Randomised Controlled TrialREC Research Ethics CommitteeRIPC Remote Ischaemic PreconditioningSAE Serious Adverse EventSMG Study Management GroupSOP Standard Operating Procedure SSC Study Steering CommitteeSST Serum-separating tubeTNT Troponin T (4th generation assay, given in mcg/L)hs-TNT Highly-sensitive Troponin T (5th generation assay, given in ng/L)VISION Vascular events In noncardiac Surgery patIents cOhort evaluatioN study

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 5 of 21
2. SIGNATURE PAGE
Chief Investigator Agreement
The clinical study as detailed within this research protocol (Version 3.5, dated 17 May 2013), or any subsequent amendments will be conducted in accordance with the Research Governance Framework for Health & Social Care (2005), the World Medical Association Declaration of Helsinki (1996) and the current applicable regulatory requirements and any subsequent amendments of the appropriate regulations.
Chief Investigator Name: Dr Andrew WaldenChief Investigator Site: Royal Berkshire NHS Foundation TrustSignature and Date: 17th May 2013
Principal Investigator Agreement The clinical study as detailed within this research protocol (Version 3.5, dated 17 May 2013), or any subsequent amendments will be conducted in accordance with the Research Governance Framework for Health & Social Care (2005), the World Medical Association Declaration of Helsinki (1996) and the current applicable regulatory requirements and any subsequent amendments of the appropriate regulations.
Principal Investigator Name: Mr Stefan Samad AntonowiczPrincipal Investigator Site: Royal Berkshire NHS Foundation TrustSignature and Date: 17TH May 2013

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 6 of 21
3. SUMMARY
Short Title PRIME Pilot Study
Methodology Single-blind randomised controlled trial
Research Site Royal Berkshire NHS Foundation Trust
Aims To assess whether remote ischaemic preconditioning (RIPC) can reduce the mean peak post-operative troponin measured within 72 hours of major gastrointestinal surgery
To allow accurate estimation of the incidence of a positive post-operative troponin level (≥20 ng/L) and the effect size of RIPC in major gastrointestinal surgery, to inform an accurate sample size calculation for a clinically-powered parent study
To assess study feasibility, in order to better inform the design of a clinically-powered parent study
Main Inclusion Criteria Any patient who:1. is ≥ 45 years old; and2. is undergoing elective major colorectal or upper GI surgery.
Number of Participants Target 98 (within 9 months recruitment window; minimum 64), allocated 1:1 RIPC to sham-RIPC prior to surgery
Endpoints Primary endpoint Peak post-operative 5th generation hs-TNT level within 72 hours
Secondary endpoints Post-operative 5th generation hs-TNT AUC within 72 hours Positive post-operative 5th generation hs-TNT level (≥20 ng/L)
within 72 hours (binary endpoint) Any MACCE within 30 days Any significant surgical events within 30 days Any mortality within 30 days
Feasibility endpoints Rate of recruitment Crossover events Dropout rate Study completion
Statistical Methodology and Analysis
Descriptive statistics For continuous variables (peak hs-TNT concentration and AUC
hs-TNT concentration), means tested using a permutation two-sample, two-sided, t-test; confidence intervals obtained using a bootstrap approach.
Associations between categorical variables (including MACCE) will be tested using Fisher’s Exact Test.
Binary logistic multiple regression models will be sought to predict and explain myocardial injury in participants (positive troponin and MACCE) in terms of other available variables
Study Duration April 2013 to Aug 2014 – 4 months ethics and R&D approval, 9 months recruitment and follow-up, 3 months analysis, write-up, dissemination.
LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 7 of 21
4. INTRODUCTION
Of more than 65,000 patients admitted for elective major abdominal surgery in England and Wales each year, 3-5% of patients will die within 30 days of their surgery. Major adverse clinical cardiovascular events (MACCE) such as myocardial injury and stroke make a significant contribution to this mortality; the recent Vascular events In noncardiac Surgery patIents cOhort evaluatioN study (VISION) investigators attributed 30% of general surgical mortality to a vascular event. Moreover, subclinical myocardial injury characterised by an elevated serum troponin concentration is proportionally linked to increased morbidity and mortality both in short and long-term follow-up, and may occur in 12-39% of major general surgery. Risk factors for perioperative myocardial injury include length of surgery, intraoperative haemodynamic instability, and the severity of the postoperative inflammatory response. This makes myocardial injury an important complication of major surgery and a potential quality metric for surgical care.
One potential method to reduce intraoperative myocardial injury is the use of ischaemic preconditioning (IPC), first described by Murry et al in 1986. IPC involves applying a period of non-lethal ischaemia followed by reperfusion to a target organ, which generates a metabolic response that protects the tissue against further ischaemia-reperfusion insults. Remote ischaemic preconditioning (RIPC) exploits the same effect, however the preconditioned tissue is away from the area of interest, most commonly a limb. In meta-analyses of patients undergoing coronary artery bypass grafting, limb-RIPC led to a 40% reduction in post-operative troponin release, and up to a 50% reduction in myocardial infarction. Similar effect sizes have been reported in smaller series in aortic surgery. Lastly, there are suggestions that RIPC protects against other ischaemia-reperfusion pathologies such as pre-renal acute kidney injury, although conclusions from meta-analyses are less clear.
No studies have investigated whether RIPC may benefit patients undergoing major gastro-intestinal operations, and only limited data is available regarding post-operative troponin release in these patients as a surrogate for clinical outcomes. Eventually, our aim is to investigate whether RIPC reduces vascular-associated clinical events. In this initial pilot study, we will use peak 5th generation highly-sensitive troponin-T (hs-TNT) concentration for the primary endpoint as a surrogate for clinical outcomes for the following reasons. First, VISION data suggests that myocardial damage as characterised by troponin release is proportionally linked to morbidity and mortality, although no good data exists for the incidence of hs-TNT release (published VISION data used 4th rather than 5th generation troponin-T assays). Second, there is little data estimating the effect size of RIPC in general surgical patients, and therefore designing a multi-centre parent study powered to detect differences in clinical outcomes is difficult. Third, subclinical troponin release is much more common (12-39% of major general surgical patients), and therefore an adequately powered study is more easily achievable. Lastly, as opposed to more accurate measurement such as area under the curve (AUC), peak hs-TNT is more familiar to clinicians, assisting practical application of the results. Secondary endpoints will be AUC 5th
generation hs-TNT concentration and clinical outcomes including MACCE (see Appendix I), non-vascular morbidity and mortality, and length of stay.
Study design will be single-blind randomised controlled trial. We found no good data on which we could build a reliable sample size calculation, so we estimated recruitment targets using VISION supplementary data. With this 4th generation assay data, estimate the mean peak hs-TNT rise in positive patients to be 0.20 mcg/L, with a standard deviation of 0.06 mcg/L. Meta-analysed data reports an effect size of about 40% in patients undergoing CABG. With a two-tailed significance of 0.05, power 0.8, dropout rate of 10%, and a significant (i.e. ≥20 ng/L) hs-TNT rise occurring in 20% of patients this returns sample sizes of 49 in each group. If the proportion is raised to 25%, it is 39 in each group, and if 30%, it is 32 in each group. Therefore we want to recruit as close to 98 participants as possible within the 9-month recruitment window, with a minimum of 64 participants.

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 8 of 21
It should be stressed that although we have attempted to inform our recruitment targets, these values can only be arbitrary given available data. Therefore, the main aim of the PRIME Pilot is to allow accurate estimation of the incidence of a positive post-operative troponin level (≥20 ng/L) and the effect size of RIPC in major gastrointestinal surgery, and to assess feasibility of the methodology. A further aim is to assess whether RIPC can reduce the mean peak post-operative troponin within 72 hours of major gastrointestinal surgery, although we cannot reliably power the study for the reasons given above.
5. STUDY AIMS & ENDPOINTS
5.1 Aims To assess whether remote ischaemic preconditioning (RIPC) can reduce the mean peak
post-operative troponin measured within 72 hours of major gastrointestinal surgery To allow accurate estimation of the incidence of a positive post-operative troponin level
(≥20 ng/L) and the effect size of RIPC in major gastrointestinal surgery, to provide an accurate sample size calculation for a clinically-powered parent study
To assess study feasibility, in order to better inform the design of a clinically-powered parent study
5.2 Primary endpoint Peak post-operative 5th generation hs-TNT level within 72 hours
5.3 Secondary endpoints Post-operative 5th generation hs-TNT AUC within 72 hours Positive post-operative 5th generation hs-TNT level (≥20 ng/L) within 72 hours (binary
endpoint) Any MACCE* within 30 days from surgery Any significant surgical complications** within 30 days from surgery All cause mortality within 30 days from surgery Length of post-operative stay (not inclusive of any readmission)
*MACCE (Major adverse clinical cardiovascular events) is a composite binary endpoint and is any of acute myocardial infarction, angina, arrhythmia, congestive heart failure, stroke cerebrovascular death, cardiovascular death (see Appendix I for definitions).
**Significant surgical complications is a composite binary endpoint and is any post-operative complication of Clavien-Dindo grade III-IV (see Appendix I).
5.4 Feasibility endpoints Rate of recruitment Crossover events Dropout rate Study completion
6. METHODOLOGY
6.1 Inclusion Criteria
Any patient who: is ≥ 45 years old; is undergoing elective major colorectal or upper GI surgery.
6.2 Exclusion Criteria
Diabetic patients that are taking glibenclamide medication*

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 9 of 21
Patients with upper limb peripheral vascular disease, including those with arteriovenous fistula for dialysis
Untreated hypertension (defined as two or more readings >180mmHg systolic on admission for surgery)
Current participation in any study investigating troponin levels or ischaemic preconditioning
Unable or lacks capacity to give informed consent to participation
*There is limited evidence in animal models that glibenclamide abrogates the RIPC effect.
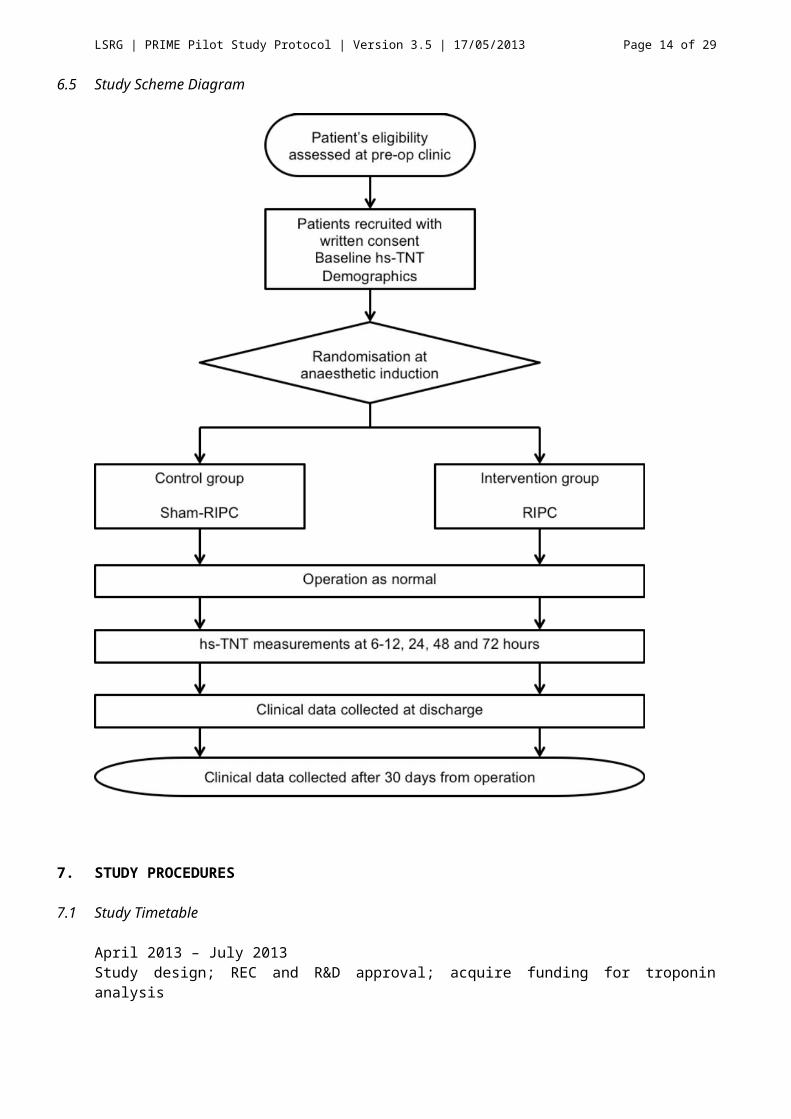
6.3 Study Design and Plan
This is a pilot single-blind randomised controlled trial with participant and observer blinded and will be carried out at a single site. This cannot strictly be double-blind, as the anaesthetic team who carry out the treatment will know the allocation, although additional bias arising from this design is expected to be minimal. Eligible participants will be identified at the pre-operative outpatient assessment, will have the research discussed, and will be provided with the Participant Information Sheet. Informed written consent will be taken after time to consider the research, normally at least 24 hours, together with baseline clinical and demographic data (the Baseline CRF) and a baseline troponin-T.
On the day of surgery, the participant will be randomised into either the treatment or control group. Based on this allocation, the anaesthetic team or research nurse will carry out either RIPC or sham-RIPC after induction of anaesthesia, and will be trained in both techniques well in advance. The anaesthetics team will be asked to complete a Treatment CRF detailing treatment, and this will be returned to the investigator in an opaque envelope. In this way, both the participant and the study investigators will be blinded to the treatment given.
Post-operatively, participants will have at least three blood samples taken at either 6-12, 24, 48 and 72 hours for hs TNT. At discharge further participant characteristics and clinical events will be recorded, including MACCE events (see Appendix I), significant surgical events, mortality, and length of stay. After 30 days from surgery, the participant will be assessed either in clinic or by telephone interview to determine if any of these events had occurred following discharge, and in this pilot study they will also be asked to reflect on their experience in this study with a view to adapting this protocol for a clinically powered parent study.
6.4 Explanatory variables
The following variables will be recorded to demonstrate and compare the characteristics and potential confounders of the participants in each group.Participant variables:
Age at operation Gender BMI Angina (previous or current) Arrhythmia (previous or current) Congestive Heart Failure Diabetes Hypercholesterolaemia Hypertension Pacemaker Previous myocardial infarction Previous stroke Peripheral vascular disease Smoking history (current / ex / never) Family history of IHD

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 10 of 21
ASA Grade Pre-operative creatinine, Black ethnicity (binary) to calculate eGFR (function of
creatinine, age, gender and black ethnicity)Operative variables:
Operation Anaesthetic agents of induction, maintenance and paralysis Laparoscopic/Lap-assisted/Lap converted to open/Open Estimated blood loss Lowest intraoperative MAP Lowest intraoperative heart rate Intraoperative pathological bradyarrhythmia
Postoperative variables: All infections (defined in Appendix II) Acute kidney injury (defined by RIFLE criteria; Appendix II) Other post-operative complications by Clavien-Dindo Grade (defined in Appendix II) Any cause mortality within 30 days Length of post-operative stay (not inclusive of any readmission) Any cause mortality within 6 months

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 11 of 21
6.5 Study Scheme Diagram
7. STUDY PROCEDURES
7.1 Study Timetable
April 2013 – July 2013 Study design; REC and R&D approval; acquire funding for troponin analysis
Aug 2013 – May 2014Participant recruitment

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 12 of 21
June – Aug 2014Data input, cleaning, analysis and write-up
Aug 2014Dissemination of findings by submission for publication
7.2 Recruitment and Informed Consent Procedures
Eligible patients will be identified at the outpatient clinic during their referral visit. A member of the research team (i.e. PI or a research nurse) will introduce the study and provide written and verbal information detailing why it is important, what is involved and what they can expect. The patient will be given a period of at least 24 hours to consider the research proposed. Written consent will be taken at a follow-up visit, which will normally be the pre-operative assessment visit. If this is not planned, a mutually convenient second visit will be offered. Only patients that give written consent will be included in the study. If fully informed consent is not possible the patient will not be recruited into the study. The consent form will stipulate that they have read the information sheet, that they have had any questions answered fully, that their participation is voluntary, that they agree to additional blood tests, that they agree to give their serum to future research, that their anonymised data will be used in analysis, and that they agree to participation in the study. Written consent will be recorded in the site file, together will baseline data.
Inpatients awaiting surgery at the recruiting hospital will be identified by the research team and provided with an information sheet to read. The patient will be given sufficient time to consider the study, recommended to be 24 hours, after which informed written consent will be taken.
7.3 Recruitment Rates
Recruitment will be at a single NHS Trust. This trust carries out an average of 12 colorectal and upper gastrointestinal operations every week that fit the inclusion criteria. Our recruitment target is 2-4 per week (16-33% of eligible patients) until the target of 98 is met. If the 9-month recruitment window closes, a minimum of 64 participants will be needed to permit analysis of the primary outcome (see 8.1)
7.4 Randomisation
Randomisation will be in ratio of 1:1 by way of pre-prepared sealed opaque envelopes, which will contain instructions to the anaesthetics team as to how to proceed. This will be undertaken by the study statistician who will also number the envelopes before recruitment begins. This number will be recorded in the participant’s baseline data. An online random number generator based on atmospheric noise will be used to assign numbers (http://www.random.org/).
7.5 Treatment Protocols
Members of the anaesthetic team or a research nurse will carry out either RIPC or sham-RIPC treatments after the induction and stabilisation of anaesthesia. Preparation prior to recruitment will be to train the relevant anaesthetic teams in RIPC/sham-RIPC treatment delivery. This will involve a face-to-face training session with an investigator to cover aspects of the study protocol, the RIPC procedure, safety precautions and procedures for adverse events. Training will be augmented with a written standard operating procedure and this will be reinforced on the day of surgery.
The RIPC treatment is started after induction and stabilisation of anaesthesia. It involves inflating a standard, CE-marked, manual blood pressure cuff to 200mmHG around either arm, for 5 minutes, followed by a deflated rest phase of a further 5 minutes. This is repeated three times. It can be carried out concurrently with other pre-operative anaesthetic activities such as

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 13 of 21
vascular cannulation and urinary catheterisation. At the end of the third inflated phase, the cuff can be removed and the participant allowed to enter the operating theatre (i.e. after 25 minutes – the third deflated, reperfusion period can occur concurrently with operative positioning and skin preparation). An appropriate cuff size will be selected in accordance with standard operating procedure.
In the sham-RIPC control group, participants will undergo the same process as the RIPC treatment group, however there will be no cuff inflation phases. Therefore, after the induction of anaesthesia, a blood pressure cuff will be attached to their arm and kept on for 25 minutes. The participant will not be allowed to enter the operating theatre until the last cycle of inflation would otherwise have been complete, i.e. 25 minutes. During this time, anaesthetic activities including vascular cannulation and urinary catheterisation can occur as in the treatment group.
7.6 Schedule of Intervention
Initial clinic appointment Identify eligible patients Introduce research and provide participant information sheet if interested in
participating
Pre-assessment clinic Discuss research again, provide opportunity to answer questions If ready to participate, complete the consent form, baseline data form, and
obtain blood sample for pre-operative hs-TNT
Day of operation
Allocation of participant to a pre-numbered, randomised treatment envelope at induction of anaesthesia.
Anaesthetic team or research nurse to carry out treatment following induction and stabilisation of anaesthesia, as set out in the treatment-protocol envelope and in keeping with prior training.
Anaesthetic team to complete and return the Treatment CRFPost-operative course Blood sampling for hs-TNT at 6-12, 24, 48 and 72 hoursDischarge Discharge data form to be completed
Follow-up 30-day data form to be completed at follow-up clinic visit or telephone consultation, after 30 days post-operatively and before 8 weeks.
7.7 Schedule of Assessment
Pre-assessment
clinic
Day of operatio
n
Post-op day 1
Post-op day 2
Post-op day 3
Day of discharge
4 to 8 weeks
Baseline CRF x x
Treatment CRF x
Discharge CRF x
30 day CRF x
hs-TNT x(Baseline)
x(6-12h)
x(24h)
x(48h)
x(72h)
7.8 Criteria for Discontinuation
Patients will be discontinued from the study if they suffer any serious adverse events directly relating to the RIPC or sham RIPC. These events will be reported in accordance with the safety reporting protocols outlined in section 10.
7.9 Laboratory Assessments
Blood samples are to be taken into a 5ml SST tube, and transferred immediately to the pathology department where they will allowed to clot, and then spun at 1300 rpm for 15 minutes. The serum supernatant will be aliquoted into cryotubes and stored in the onsite
LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 14 of 21
laboratory at -80C in accordance with NHS regulatory standards set out for diagnostic services in human subjects. Samples will be analysed in batches on a fortnightly basis, using the Roche Elecsys® STAT™ hs-TNT analyser. This method of storing serum for hs-TNT analysis will not result in any appreciable degradation of the analyte. Once analysed, the serum will be kept for 5 years at -80C within the pathology department. Only members of the research team and the pathology department will have access to this freezer. Measurements will be in absolute values to 1 ng/L increments, rather than “>” or “<” values. Analysing samples fortnightly ensures that these results will not influence participant care.
7.10 Subject Withdrawal
Participants are free to withdraw at any time during the active recruitment and follow-up stages of the research, while the data remains identifiable. This withdrawal policy will form part of the written consent form. Participants will be excluded from analysis if their operation is cancelled after induction of anaesthesia, if they fail to complete follow-up, if they miss the pre-operative hs-TNT sample or if more than one hs-TNT concentration is missing from the post-operative samples. We have allowed for a drop-out rate of 10% within our sample estimation (see 8.1).
7.11 End of Recruitment Definition
This will be when the 98th 30-day proforma has been completed, or when the 9-month recruitment window has closed.
8. STATISTICAL CONSIDERATIONS
8.1 Sample Size
As there is very limited data on troponin release after major elective general surgery using 5 th
generation assays, and no data on the effect size of RIPC in general surgery, we were unable to build a reliable sample size calculation. Therefore, we used surrogate data using 4 th
generation assays, and RIPC in cardiac surgery, to estimate recruitment targets. We conservatively estimated mean peak hs-TNT rise in positive patients to be 0.20 mcg/L, with a standard deviation of 0.06 mcg/L. Meta-analysed data reports an RIPC effect size of approximately 40% in patients undergoing CABG. With a two-tailed significance of 0.05, power 0.8, dropout rate of 10%, and a significant (i.e. ≥20 ng/L) hs-TNT rise occurring in 20% of patients this returns sample sizes of 49 in each group. If the proportion is raised to 25%, it is 39 in each group, and if 30%, it is 32 in each group. Therefore we want to recruit as close to 98 participants as possible within the 9-month recruitment window, with a minimum of 64 participants.
8.2 Methods of Analysis
Following the conclusion of recruitment and follow-up, the data form and hs-TNT values will be digitized on an NHS trust computer, using a password-protected account, in a locked office. The envelope numbers will be matched to a master list held by the study statistician revealing which cases had which treatment. The digitized data will then be anonymised and analysed using R (R Foundation for Statistical Computing, Vienna, Austria). Methods of statistical analysis will include:
Descriptive statistics including mean, median, standard deviation, interquartile range, and range for continuous measures, and counts and percentages for categorical measures
For continuous variables (peak hs-TNT concentration and AUC hs-TNT concentration) the means will be compared using a permutation two-sample, two-sided, t-test, with a 5% significance level, 95% confidence limits for the difference between the means will be obtained using a bootstrap approach.

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 15 of 21
Associations between categorical variables (including MACCE) will be tested using Fisher’s Exact Test.
Binary logistic multiple regression models will be sought to predict and explain myocardial injury in participants (positive troponin and MACCE) in terms of other available variables.
9. ETHICAL CONSIDERATIONS
9.1 Consent
See section 7.2.
9.2 Declaration of Helsinki and Good Clinical Practice
The study will conform to the spirit and letter of the declaration of Helsinki and in accordance with the Good Clinical Practice Guidelines
9.3 Ethical Committee Review
Application to Ethical Committee in progress, under reference 13/SC/0306.
10. SAFETY CONSIDERATIONS AND REPORTING
This is not a study of an investigational medicinal product and therefore all untoward occurrences will be adverse events rather than adverse reactions.
10.1 Adverse Event
An adverse event is defined as any untoward medical occurrence affecting a participant, which does not necessarily have a causal relationship with the RIC stimulus. The terms ‘mild, moderate or severe’ are used to describe the intensity of a specific event or reaction. An event would be considered ‘serious’ based upon participant/event outcome or action criteria as defined below.
10.2 Serious Adverse Event (SAE)
A serious adverse event (SAE) will be considered any untoward medical occurrence/effect that: Results in death Is life-threatening Requires hospitalisation or prolongation of existing inpatient’s hospitalisation. Results in persistent or significant disability or incapacity. Is otherwise considered medically significant by the investigator
‘Life threatening’ refers to an event in which the subject was at risk of death at the time of the event; it does not refer to an event which hypothetically mat have caused death if it had been more severe.
10.3 Unexpected Adverse Event
An unexpected adverse event is considered an adverse event of which the nature and severity is not consistent with the expected consequences of RIPC. Given the ischaemic nature of the treatment, this theoretically could include skin bruising, petechiae, neuropraxia, muscle damage or exacerbation of underlying peripheral vascular disease, although it should be noted that such a diagnosis is a principle exclusion criteria for recruitment.
10.4 Expected Adverse Events (recognised to be caused by the RIPC stimulus)

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 16 of 21
There are not expected to be any serious adverse events caused by the RIPC stimulus.
10.5 Expected Serious Events Related to Usual Clinical Care
These events are well-recognised complications of major General Surgical procedures, and are defined as any complication requiring endoscopic, radiological or operative management, or those resulting in death (Clavien-Dindo III-V). They will be documented in the case reporting forms. Examples include (but are not limited to):
Death, perioperative myocardial injury or infarction. Anastomotic leakage/breakdown potentially requiring revision surgery. Intra-abdominal collection. Wound infection or dehiscence. Bleeding requiring reoperation. Visceral injury, such as chyle leak.
Other minor complications requiring pharmacological intervention (Clavien-Dindo I/II) will not require documentation.
10.6 Reporting Unexpected Adverse Events
Given the nature of the treatment, it is unlikely that any will occur. Nonetheless, should an unexpected adverse events occur, the investigators will make an assessment of severity and report the event to the Research Ethics Committee and the Sponsor as appropriate. Usually, this will mean within 24 hours in the case of severe or moderate events, and within 14 days for mild events.
10.6.1 Assessment of Intensity
Mild: The subject is aware of the event or symptom, but the event or symptom is easily tolerated.Moderate: The subject experiences sufficient discomfort to interfere with or reduce their usual level of activity.Severe: Significant impairment of functioning; the subject is unable to carry out usual activities and/or the subject’s life is at risk from the event.
10.7 Urgent Safety Measures
Should it be necessary to undertake urgent safety measures to ensure the safety and protection of the participants the CI will take these measures immediately. The CI would inform the sponsor and Main research Ethics Committee of this event immediately via telephone and in writing within 3 days in the form of a substantial amendment.
11. DATA HANDLING AND RECORD KEEPING
Information related to study participants will remain confidential and be managed in accordance with the Data Protection Act, NHS Caldecott Principles, The Research Governance Framework for Health and Social Care, and the conditions of Research Ethics Committee Approval.
All case record forms will be held in the site file, which will be kept in a locked drawer in a locked office at the study site. They will be destroyed after 10 years. All digital data will be stored on a password-protected shared drive within the trust inside the N3 network, under the control of the Chief Investigator, and will be erased securely after 10 years.
12. LABORATORIES
LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 17 of 21
See section 7.9.
13. DEVICES AND TECHNIQUES
13.1 Devices
A standard CE approved blood pressure cuff sized appropriately using standard operating procedures will be used to provide the RIPC treatment or sham-RIPC treatment. The device (Lyall Wallis & Co Palm Aneroid Sphygmanometer) is normal hospital issue, complying with a calibration and maintenance schedule necessary for use of clinical equipment on patients. The device will be used as described in section 7.5.
13.2 Techniques and interventions
See section 7
14. MONITORING AND AUDITING
The study will be monitored and audited by the Sponsor, in accordance with usual protocols.
15. STUDY COMMITTEES
15.1 Study Steering Committee
This will comprise Mr Stefan Antonowicz, Mr Tom Wiggins, Mr James Haddow, Mr Nicholas Symons, Professor Charles Knowles and Dr Andrew Walden. The Study Steering Committee will be responsible for drafting the final report and submission for publication, and preparation for the parent multi-centre study.
15.2 Study Management Group
This group will include Mr Stefan Antonowicz, Mr Dominic Coull, and Dr Andrew Walden. This group will be responsible for participant recruitment, data collection and follow-up of the PRIME Pilot.
16. FINANCE AND FUNDING
The only directly incurred costs will be for phlebotomy and hs-TNT assay measurement. By arrangement with the Department of Biochemistry at the host institution, this will total £3.50 per assay including phlebotomy equipment. At 5 assays per participant, 100 participants, this totals £1750, which will be partially met by a local fund to support early stage research (funding agreed), and partially by external funding (application pending). The Principal Investigator will meet indirect costs such as stationary. Unexpected costs from adverse events will be met by the sponsor via NHS Indemnity.
17. INDEMNITY
The nominated sponsor of our research study is Research and Development, Royal Berkshire NHS Foundation Trust, Royal Berkshire Hospital, London Road, Reading, RG2 5AN. The centre will be covered by NHS indemnity for negligent harm providing researchers hold a contract of employment with the NHS, including honorary contracts held by academic staff. Medical co-investigators will also be covered by their own medical defence insurance for non-negligent harm.

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 18 of 21
18. DISSEMINATION OF RESEARCH FINDINGS
Results from this study will be presented in a peer-reviewed journal. Authorship will follow international guidelines.
19. REFERENCES
Hospital Episode Statistics (HES) 2010-11, Department of Health, UK Pearse R M, Moreno R P, Bauer P et al, ‘Mortality after surgery in Europe: a 7 day
cohort study’, Lancet, 2012; 380: 1059-65. Devereaux P J, Chan M T V, Alonso-Coello P et al, ‘Association between postoperative
troponin levels and 30-day mortality among patients undergoing noncardiac surgery’, JAMA, 2012; 307 (21): 2295-2304.
Ali ZA, Callaghan CJ, Lim E, et al. Remote ischemic preconditioning reduces myocardial and renal injury after elective abdominal aortic aneurysm repair: a randomized controlled trial. Circulation 2007;116:I98-105.
Devereaux P J, Xavier D, Pogue J et al, ‘Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study’, Ann Intern Med, 2011; 154(8): 523-528.
Dindo D, Demartines N, Clavien PA. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann Surg. 2004 Aug;240(2):205-13.
Murry CE, Jennings RB, Reimer KA, ‘Preconditioning with ischaemia: a delay of lethal cell injury in ischemic myocardium’, Circulation, 1986; 74: 1124-36.
Kharbanda RK, Mortensen UM, White PA et al, ‘Transient limb ischaemia induces remote ischemic preconditioning in vivo’, Circulation, 2002; 106: 2881-3.
Hausenloy DJ, Mwamure PK, Venugopal V et al, ‘Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial’, Lancet, 2007; 370:575-79.
Brevoord D, Kranke P, Kuijpers M, et al ‘Remote ischemic conditioning to protect against ischemia-reperfusion injury: a systematic review and meta-analysis’, PloS ONE, 2012; 7(7): e42179
D’Ascenzo F, Cavallero E, MOretti C, et al ‘Remote ischaemic preconditioning in coronary artery bypass surgery: a meta-analysis’ Heart 2012;98:1267-1271
ERRICA protocol Bellomo R, Ronco C, Kellum JA, et al, Acute Dialysis Quality Initiative workgroup. Acute
renal failure – definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004; 8:R204 – R212.
Sabaté S, Mases A, Guilera N, Canet J, Castillo J, Orrego C, Sabaté A, Fita G, Parramón F, Paniagua P, Rodríguez A, Sabaté M; ANESCARDIOCAT Group. Incidence and predictors of major perioperative adverse cardiac and cerebrovascular events in non-cardiac surgery. Br J Anaesth. 2011 Dec;107(6):879-90. doi: 10.1093/bja/aer268. Epub 2011 Sep 2.

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 19 of 21
20. APPENDIX I: CLINICAL ENDPOINT DEFINITIONS
20.1 MACCE
Any of (adapted from Sabate et al):a) Non-fatal cardiac arrestb) Acute myocardial infarctionc) Anginad) New cardiac arrhythmiae) Congestive heart failuref) Strokeg) Cerebrovascular deathh) Cardiovascular death
20.2 Non-fatal Cardiac Arrest
An absence of cardiac rhythm or presence of chaotic rhythm requiring any component of basic or advanced cardiac life support.
20.3 Acute Myocardial Infarction
Increase and gradual decrease in troponin level in the company of at least one of the following: ischaemic symptoms, abnormal Q waves on the ECG, ST-segment elevation or depression; or coronary artery intervention (e.g. coronary angioplasty) or a typical decrease in an elevated troponin level detected at its peak after surgery in a patient without a documented alternative explanation for the troponin elevation
20.4 Angina
Dull diffuse substernal chest discomfort precipitated by exertion or emotion and relieved by rest or glyceryl trinitrate.
20.5 New Cardiac Arrhythmia
ECG evidence of atrial flutter, atrial fibrillation, or second- or third-degree atrioventricular conduction block
20.6 Congestive Heart Failure
New in-hospital signs or symptoms of dyspnoea or fatigue, orthopnoea, paroxysmal nocturnal dyspnoea, increased jugular venous pressure, pulmonary rales on physical examination, cardiomegaly, or pulmonary vascular engorgement.
20.7 Stroke
Embolic, thrombotic or haemorrhagic event lasting at least 30 min with or without persistent residual motor, sensory, or cognitive dysfunction; if the neurological symptoms continue for more than 24 h, a person is diagnosed with stroke, and if lasting less than 24 h the event is defined as a transient ischaemic attack.
20.8 Cerebrovascular Death
A death caused by cerebrovascular disease
20.9 Cardiovascular Death

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 20 of 21
Any death, unless an unequivocal non-cardiovascular cause could be established.
20.10 Clavien-Dindo Classification: I. Any physiological deviation from normal post-operative care (allows use of antipyretics,
analgesics, antiemetics, diuretics, fluids, electrolytes, physiotherapy)II. Complication requiring other pharmacological management (includes blood transfusions
and total peripheral nutrition)III. Complications requiring interventional, non-operative managementIV. Complication requiring intensive careV. Death
20.1130 day significant surgical complications
Are any post-operative complications of Clavien-Dindo grade III-IV, i.e. those requiring radiological, endoscopic or operative intervention. Examples would include (but are not limited to):
Anastomotic leak/dehiscence requiring surgical or radiological intervention Surgical space infection Wound infection or dehiscence Bleeding requiring revision surgery
20.12Acute Kidney Injury (RIFLE criteria)
A >2x rise serum creatinine from pre-operative value

LSRG | PRIME Pilot Study Protocol | Version 3.5 | 17/05/2013 Page 21 of 21
21. APPENDIX II: COMPONENTS OF CASE REPORTING FORMS
21.1 Consent Form Items
Identifiers Having read participant information sheet Understood what is involved, including RIPC/sham-RIPC, 5 blood tests of which some
may require additional phlebotomy to usual care, access to clinical records, and follow-up which may include a phone call at one month
Questions answered satisfactorily Understanding of withdrawal policy
21.2 Baseline CRF Data Items (form to be completed by member of research team who takes consent)
Identifiers Participant variables as listed in Section 6.4
21.3 Treatment CRF Data Items (form to be completed by anaesthetic team or research nurse carrying out allocated treatment)
Identifiers Envelope Number Treatment assigned Notes recording any problems, deviations from protocol, insights into treatment.
21.4 Discharge CRF Data Items (form to be completed by PI)
Identifiers Operation variables as listed in Section 6.4 Date of operation Discharge date (of primary admission only) MACCE Significant Surgical Complications Death
21.5 30-day CRF data items (form to be completed by PI)
Identifiers Any further MACCE Any further Significant Surgical Complications Patient involvement (PPI) questionnaire
21.6 Laboratory CRF Data Items (to be completed by PI) Pre-operative 5th generation hs-TNT concentration 6-12, 24, 48 and 72h 5th generation hs-TNT concentration











![Dysrhythmias (002) [Read-Only] - Aventri · Atrial AV node Ventricular Classification of Rhythm Abnormalities Supraventricular Atrial origin Atrial fibrillation Atrial flutter Atrial](https://static.fdocuments.us/doc/165x107/5f024baa7e708231d4038f22/dysrhythmias-002-read-only-aventri-atrial-av-node-ventricular-classification.jpg)






