
Joint Use SAXS MX EM - EMBL Hamburg · X--ray anray andt ttif lld neutron scattering from...
56
EMBO Global Exchange Lecture Course 2 May 2011 Beijing China Joint use of SAXS with MX and EM Peter Konarev European Molecular Biology Laboratory, Hamburg Outstation BioSAXS group
Transcript of Joint Use SAXS MX EM - EMBL Hamburg · X--ray anray andt ttif lld neutron scattering from...

EMBO Global Exchange Lecture Course 2 May 2011 Beijing China
Joint use of SAXS o owith MX and EM
Peter Konarev
European Molecular Biology Laboratory,Hamburg Outstation
BioSAXS group

Information content in SAXSIn SAXS, the molecule’s rotationally averaged scattering pattern is measured as a function of spatial frequency, typicallyt 1 3 l tito 1–3-nm resolution
Because of rotational averaging, the information content of a SAXS spectrum is dramatically reduced compared to aSAXS spectrum is dramatically reduced compared to a diffraction pattern in X-ray crystallography or even a density map from EM.
S lDetector
Monochromatic beamSample
2θ
Log (Intensity)
0
1
2
Scattering vector s=4π sinθ/λ
2θ
s=4π sinθ/λ, nm-10 1 2 3
-1X-ray generator Synchrotron
Nevertheless, SAXS can provide important shape information about proteins and assemblies in the wide size range, which are not amenable to cryo- EM and NMR spectroscopy
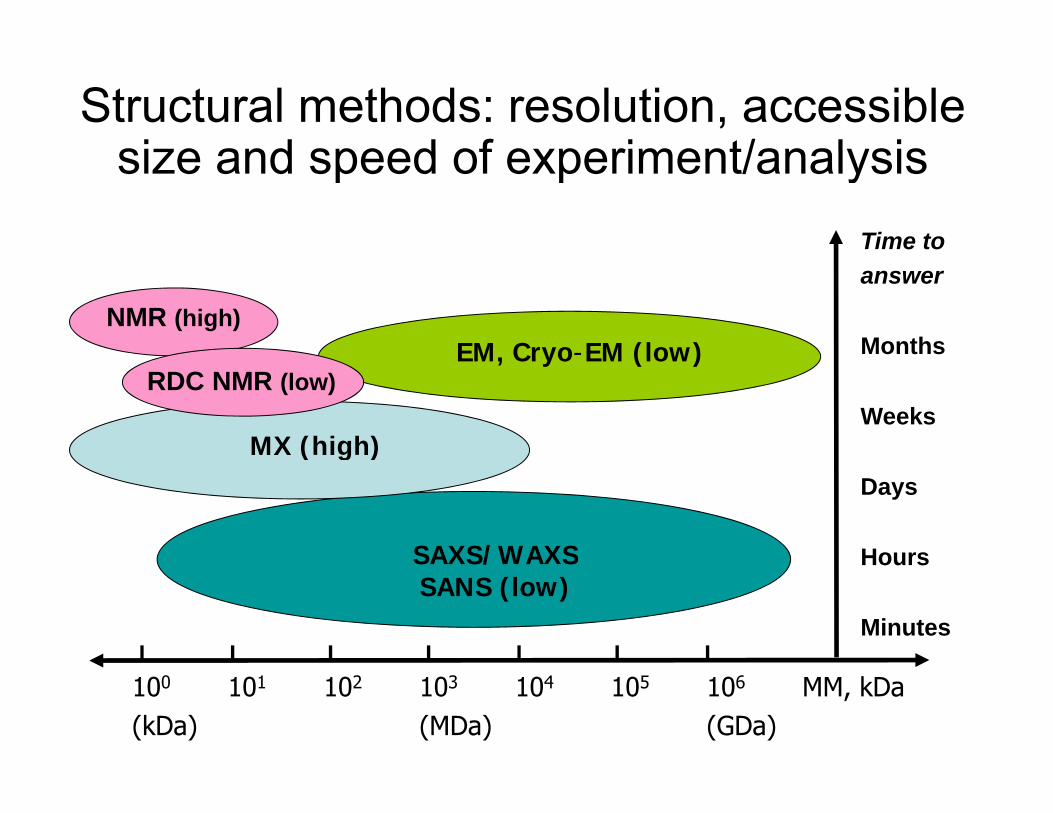
Structural methods: resolution, accessible size and speed of experiment/analysissize and speed of experiment/analysis
Time to answer
MonthsNMR (high)
EM Cryo-EM (low)
Weeks
EM, Cryo EM (low)RDC NMR (low)
MX (high)Days
HoursSAXS/WAXS
( g )
Hours
Minutes
SAXS/WAXSSANS (low)
100 101 102 103 104 105 106 MM, kDa(kDa) (MDa) (GDa)
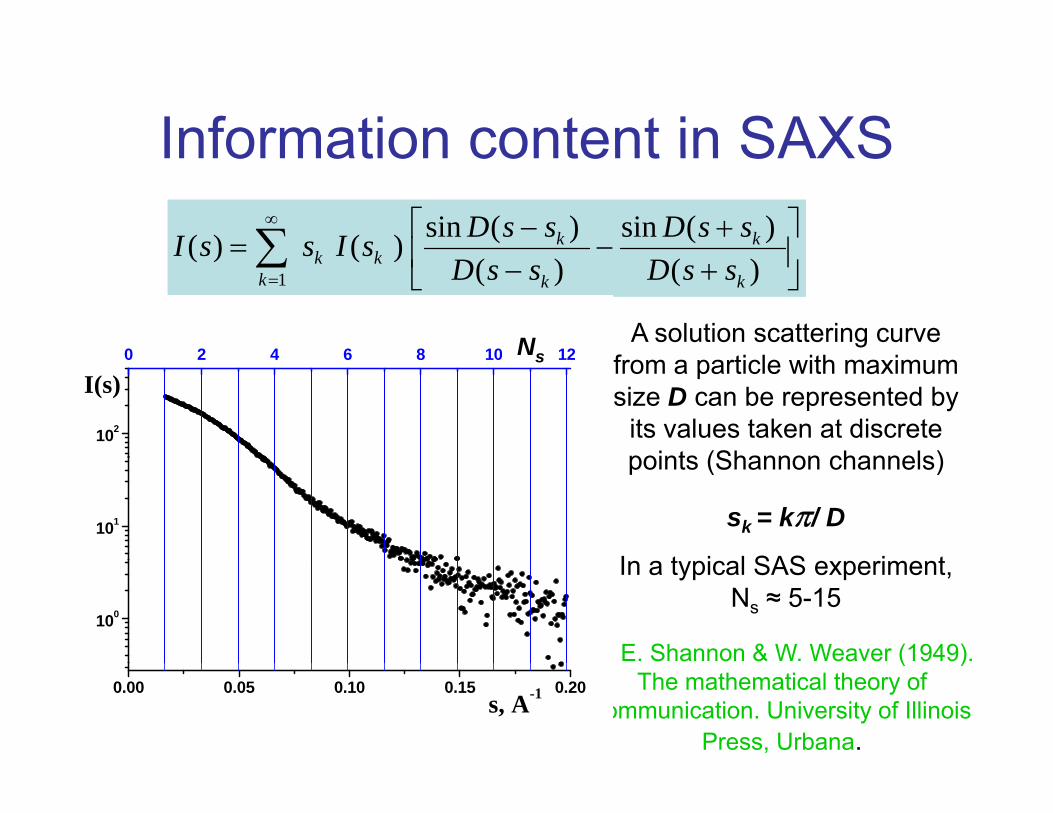
Information content in SAXS
∑∞
⎥⎤
⎢⎡ +
−−
=)(sin)(sin)()( kk
kkssDssDsIssI
Information content in SAXS
A solution scattering curve f ti l ith i
∑=
⎥⎦
⎢⎣ +−1 )()(
)()(k kk
kk ssDssDsIssI
0 2 4 6 8 10 12
Ns0 2 4 6 8 10 12
Ns from a particle with maximum size D can be represented by
its values taken at discrete points (Shannon channels)
102
I(s)0 2 4 6 8 10 12s
102
I(s)0 2 4 6 8 10 12s
points (Shannon channels)
sk = kπ/ D
I t i l SAS i t101 101
In a typical SAS experiment, Ns ≈ 5-15
C. E. Shannon & W. Weaver (1949). 100100
( )The mathematical theory of
communication. University of Illinois Press, Urbana.
0.00 0.05 0.10 0.15 0.20s, A-1
0.00 0.05 0.10 0.15 0.20s, A-1

Information content in SAXSSAXS spectrum can be transformed into a radial distributionfunction which is essentially a histogram of all pairwise
Information content in SAXS
function, which is essentially a histogram of all pairwise distances of the atoms in an assembly weighted by their respective atomic numbers.
drsrrpsID
∫=sin)(4)( π
For structure determination, additional information is needed because the radial distribution function alone is relatively
drsr
rpsI ∫0
)(4)( π
yuninformative about the details of molecular structure.
The recent renaissance of SAXS is to a large extent the result ofThe recent renaissance of SAXS is to a large extent the result of efforts on integrating SAXS with other structural information from additional complementary sources (e.g. MX, EM, NMR, bioinformatics etc.).

Integration of SAXS data with other i f iinformation
Similarly to other types of experimental information, SAXS data can be used as a filter for a set of models generated independently by other methodsindependently by other methods.
SAXS data can also be a term in a scoring function that is optimized to obtain a model consistent with the data. (e.g. ab initio modellig, rigid body modelling, addition of missing fragments)
∑+=i
ii PsIsIXE αχ )](),([(})({ exp2
missing fragments)
i

Possible use of solution structure in crystallography
• Determine a solution scattering structure (Dammin/f, Gasbor)
• Place it in unit cell (location & orientation) Place it in unit cell (location & orientation)• Calculate initial phases for phase extension andmolecular replacement
Possible challenges• Resolution and fidelity of initial structure Resolution and fidelity of initial structure• Same solution and crystal structures?• Limitation of using uniform e- densities (flexibleregion hydration layer)region, hydration layer)

Possible use of solution structure in EM
• Use a solution scattering structure (Dammin/f, Gasbor) as a starting reference for EM reconstruction
Bron, T. et.al. (2008) Biol. Cell 100, 413
Hsp90 heat-shock protein
• Superposition of SAXS models and independent EM reconstructions
Tumour suppressor p53 and its complex with DNA
independent EM reconstructions
Tidow, H et. al. (2007) Proc Natl Acad Sci USA, 104, 12324
Tumour suppressor p53 and its complex with DNA

Possible use of SAXS information in EM
Solution Structure of the E coli 70S RibosomeSolution Structure of the E. coli 70S Ribosomeat 11.5 A° Resolution
In the Cryo-EM density map reconstructionIn the Cryo-EM density map reconstruction,Fourier amplitudes at higher spatial frequencies are always underrepresented due to charging, instrument instabilities, specimen drift.
To compensate for these effects, scattering intensities bt i d i X l ti tt iwere obtained using X-ray solution scattering
measurements for E.coli ribosomes in the range up to 1/8 Å-1, and a correction to the Fourier amplitudes up to the 1/11.5 Å-1 was applied.
I.S. Gabashvili, R.K. Agrawal, C.M.T. Spahn, R.A. Grassucci, D.I. Svergun, J. Frank, P. Penczek (2000) Cell, 100, 537–549
pp

SAS experiment Data analysis Additional information
Complementary techniquesRadiation sources:
Shape determination
EMSearch volume
Detector
X-ray tube (λ = 0.1 - 0.2 nm)Synchrotron (λ = 0.05 - 0.5 nm)Thermal neutrons (λ = 0.1 - 1 nm)
2θ
Rigid body modelling
Crystallography
NMR
Atomic models
Incident beam
Sample
Wave 2θ
Missing fragments
NMR
BiochemistryOrientationsSolvent
Resolution nm:
Wave
vector k, k=2π/λ Scattered
beam, k1
I,
rela
tive
2
3
Oligomeric mixtures
FRET
Interface
Resolution, nm:
3.1 1.6 1.0 0.8
lg
1
2 mixturesmapping
BioinformaticsScattering curve I(s)
s, nm -10 2 4 6 8
Flexible systems Secondary
structure prediction
Scattering vector s=k1-k, s=4π sinθ/λ

Ab initio modelling (DAMMIN/F)g ( )Using simulated annealing, finds a compact dummy atomsconfiguration X that fits the scattering data by minimizing
f (X) = χ2[Iexp(s), I(X,s)] + ΣαiPi(X)
Discrepancy from the experimental data
Set of penalties formulating various restraints
where χ is the discrepancy between the experimental andwhere χ is the discrepancy between the experimental andcalculated curves, P(X) is the penalty to ensure compactness andconnectivity, α>0 its weight.
compactcompactcompactcompact
looseloose
disconnecteddisconnected
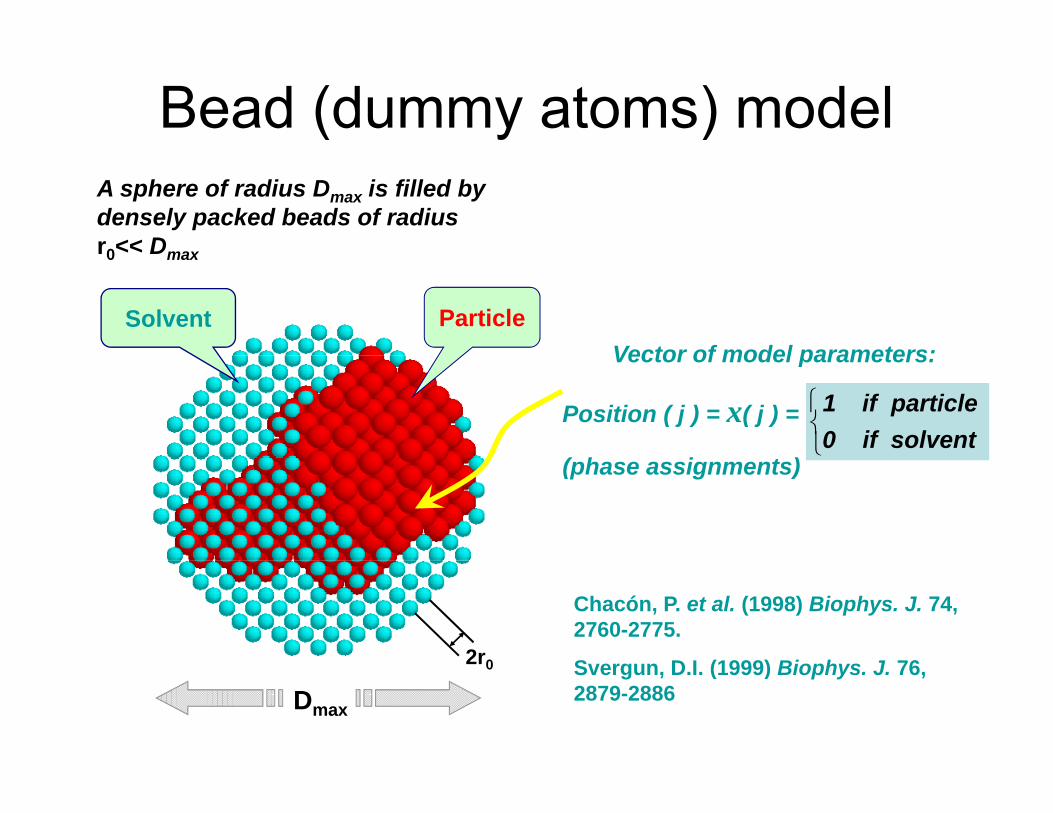
Bead (dummy atoms) modelA sphere of radius Dmax is filled by densely packed beads of radiusr0<< Dmax
Vector of model parameters:Solvent Particle
r0 Dmax
Vector of model parameters:
Position ( j ) = x( j ) =
( h i t )⎩⎨⎧
solventif0particleif1
(phase assignments)
Chacón, P. et al. (1998) Biophys. J. 74, 2760-2775.
S D I (1999) Bi h J 762r0 Svergun, D.I. (1999) Biophys. J. 76, 2879-2886
2r0
Dmax

Validation of Electron Microscopy Modelspy
EM2DAMEM2DAM
Contour levellevel
DENSITY MAP (MRC format) from EMDB
BEAD MODEL
13

Validation of Electron Microscopy Modelspy
CRYSOL
BEAD MODEL EM2DAM: surface layer th h ld+
SAXS EXPERIMENTAL DATAthreshold
14
Refinement with DAMMIN

Ab initio modelling (GASBOR)Using simulated annealing, finds a spatialdistribution of K dummy residues within a
g ( )
ysphere with diameter Dmax to fit thescattering data by minimizing
[ ] })({}){,(),(})({ exp2
iiDRi rPrsIsIrf αχ +=
where χ is the discrepancy between theexperimental and calculated curves, P({ri})
Number of neighbours
4
5
6
experimental and calculated curves, P({ri})is the penalty to ensure a chain-likedistribution of neighbors, α>0 its weight.
0.2 0.4 0.6 0.8 1.00
1
2
3
Shell radius, nm
Neighbors distribution
•Has potential for future development (e.g. phase problem in low resolution crystallography)

The use of high resolution models gin SAXS
• Validation of theoretically predicted models
• Analysis of similarities between macromoleculesin solution and in the crystal
• Modelling of the quaternary structure ofmultisubunit particles/complexes by rigid bodyp / p y g yrefinement
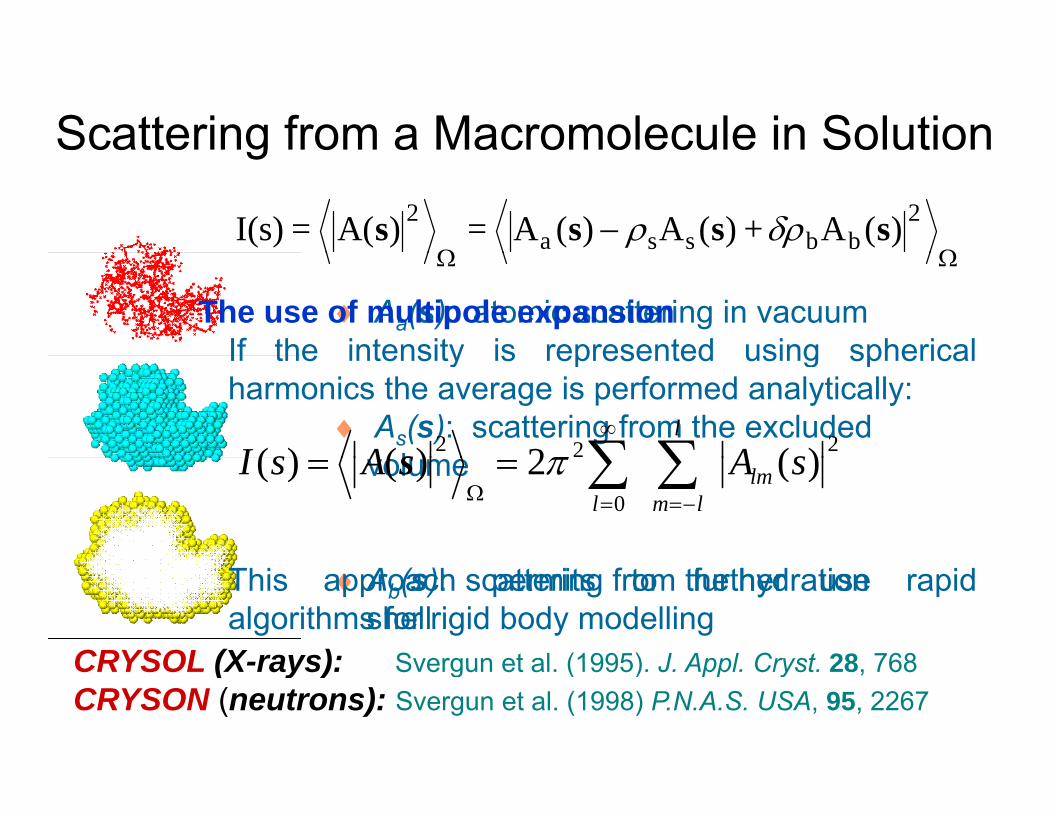
Scattering from a Macromolecule in Solutiong
ΩΩ− 2
bbssa2 )(A+ )(A)(A=)A(=I(s) ssss δρρ
♦ Aa(s): atomic scattering in vacuum
ΩΩ
The use of multipole expansionIf the intensity is represented using spherical
♦ As(s): scattering from the excluded l
If the intensity is represented using sphericalharmonics the average is performed analytically:
222 )(2)()( sAAsIl
∑∑∞
πsvolume0
)(2)()( sAAsI lmlml
∑∑−==Ω
== πs
♦Ab(s): scattering from the hydration shell
CRYSOL (X-rays): Svergun et al (1995) J Appl Cryst 28 768
This approach permits to further use rapidalgorithms for rigid body modelling
CRYSOL (X-rays): Svergun et al. (1995). J. Appl. Cryst. 28, 768 CRYSON (neutrons): Svergun et al. (1998) P.N.A.S. USA, 95, 2267

CRYSOLCRYSOL andand CRYSONCRYSON::XX d t tt i f l ld t tt i f l lXX--ray and neutron scattering from macromoleculesray and neutron scattering from macromolecules
∑∑ +L l
sBsEsAsI 22 )()()(2)( δρρπ
• The programs:
∑∑= −=
+−=l lm
lmlmlm sBsEsAsI0
0 )()()(2)( δρρπ
either fit the experimental data by varying the densityof the hydration layer δρ (affects the third term) andy y ρ ( )the total excluded volume (affects the second term)or predict the scattering from the atomic structure(particle is surrounded by an angular envelope and(particle is surrounded by an angular envelope and0.3 nm thick border layer is built around the envelope)provide output files (scattering amplitudes) for rigidb d fi t tibody refinement routinescompute particle envelope function F(ω)

Scattering components (lysozyme)Scattering components (lysozyme)
1) Atomic1) Atomic2) Shape3) B d3) Border4) Difference
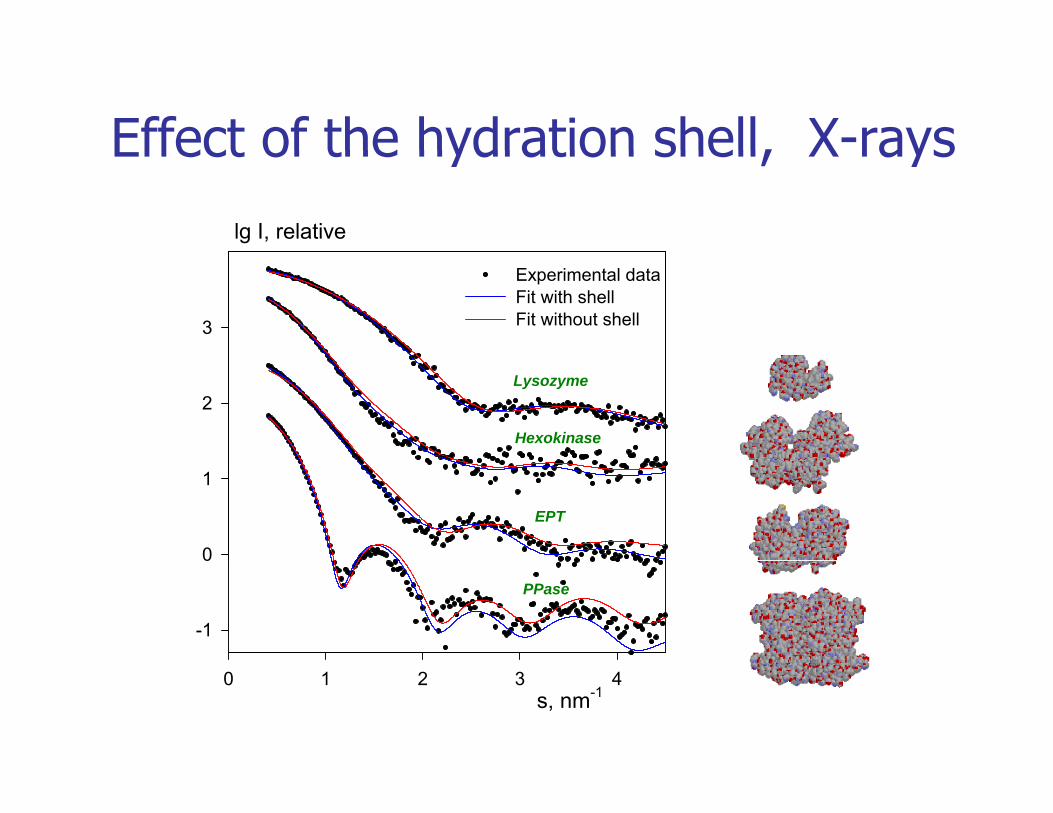
Effect of the hydration shell, X-rayslg I, relative
Effect of the hydration shell, X rays
3
Experimental dataFit with shellFit without shell
2Lysozyme
Hexokinase
0
1
EPT
-1
0
PPase
s, nm-10 1 2 3 4

Other approaches/programs to calculate the scattering Other approaches/programs to calculate the scattering f bi l i l l lf bi l i l l lfrom biological macromolecules:from biological macromolecules:
1) ‘Cube’ method ensures uniform filling of the excluded volume1) Cube method - ensures uniform filling of the excluded volume
2) SoftWaxs (L.Makowski group) – A program to compute WAXS
3) Foxs (A.Sali group) – Debye-like calculations, Web server
4) AXES (A. Bax group) – Explicit water modelling, Web server
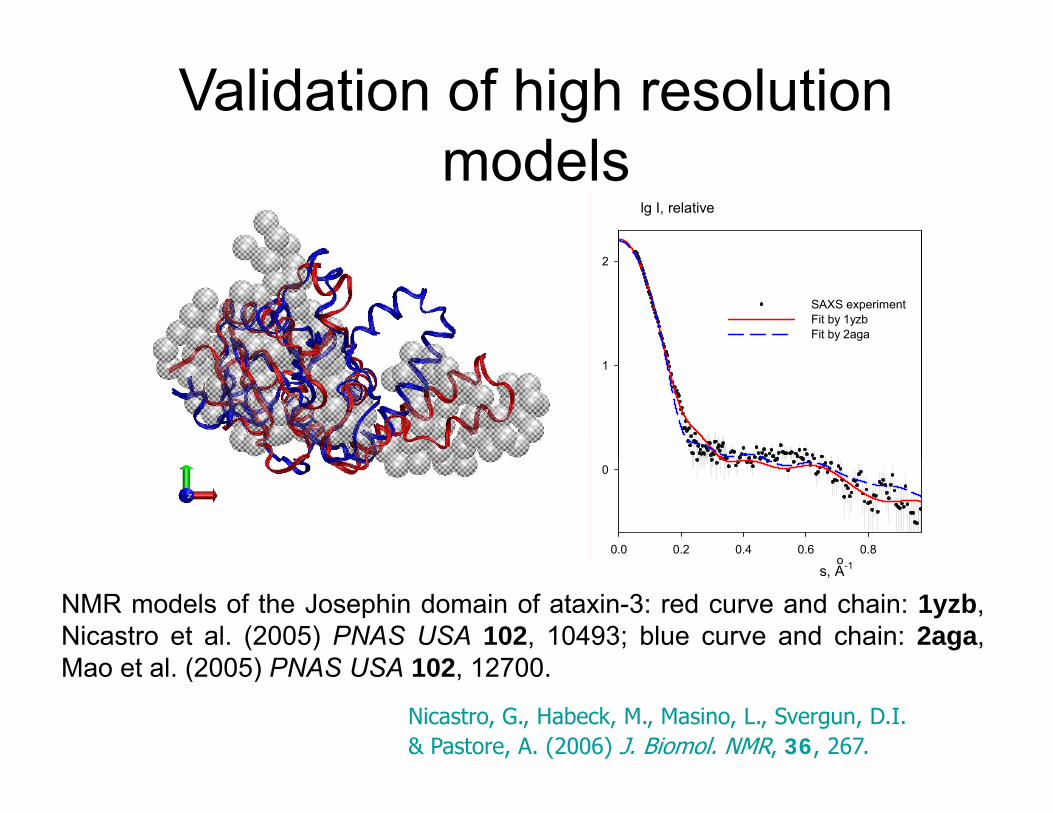
Validation of high resolution modelsmodels
lg I, relative
2
SAXS experimentFit by 1yzbFit by 2aga
1
0.0 0.2 0.4 0.6 0.8
0
o
NMR models of the Josephin domain of ataxin-3: red curve and chain: 1yzb,Nicastro et al. (2005) PNAS USA 102, 10493; blue curve and chain: 2aga,Mao et al (2005) PNAS USA 102 12700
s, A-1o
Nicastro, G., Habeck, M., Masino, L., Svergun, D.I. & Pastore, A. (2006) J. Biomol. NMR, 36, 267.
Mao et al. (2005) PNAS USA 102, 12700.
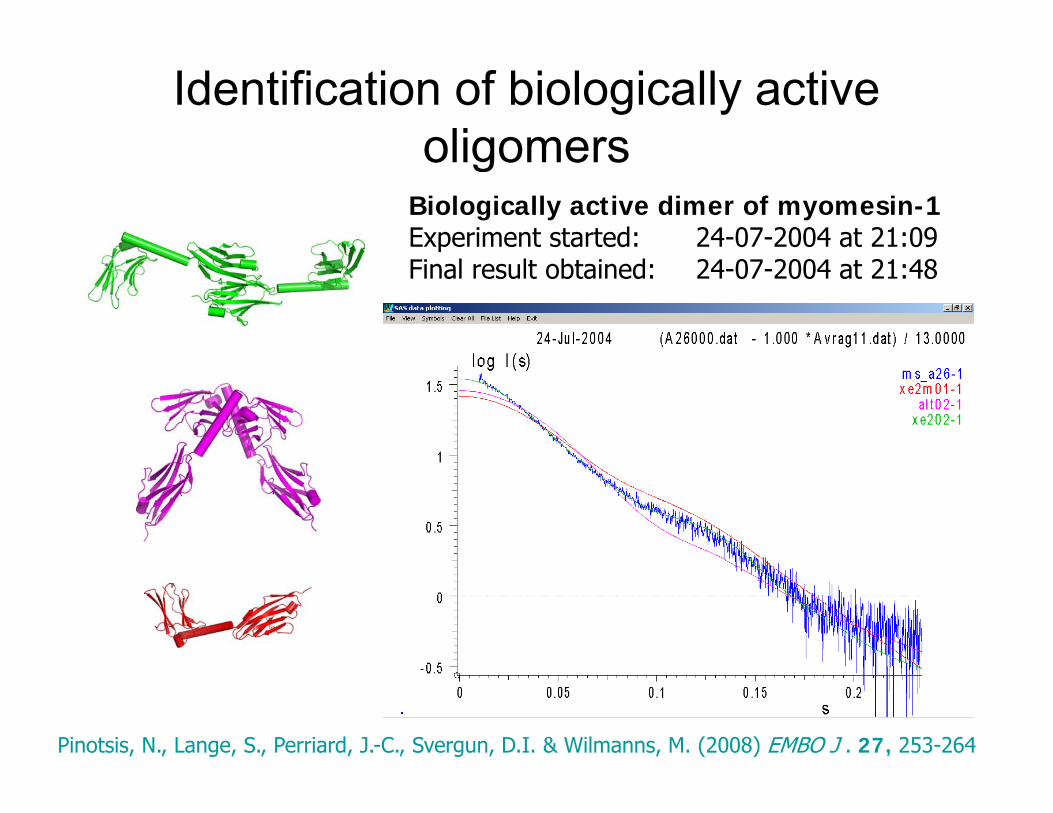
Identification of biologically active oligomersoligomers
Biologically active dimer of myomesin-1Experiment started: 24-07-2004 at 21:09Final result obtained: 24-07-2004 at 21:48
Pinotsis, N., Lange, S., Perriard, J.-C., Svergun, D.I. & Wilmanns, M. (2008) EMBO J . 27, 253-264

Solution structure of eucaryotic release factor RF1
• For Release factor 1 (RF1), responsible for terminationof translation in E. coli, a more compact form of the protein was
b d b X t ll h d ith th t b d iobserved by X-ray crystallography compared with that observed incomplex with the ribosome by electron microscopy (EM).
• Small-angle X-ray scattering was able to resolve these differencesS a a g e ay sca e g as ab e o eso e ese d e e cesby demonstrating that the more extended (EM) form was present insolution, and that the compaction was an artefact of crystallization.
Vestergaard, B., Sanyal, S., Roessle, M., Mora, L., Buckingham, R. H., Kastrup, J. S., Gajhede, M., Svergun, D. I. & Ehrenberg, M. (2005) Mol. Cell, 20, 929–938.

Solution structure of eucaryotic release factor RF1
• Cryo-EM: extended; spans the distance between the ribosomal decoding and peptidyldecoding and peptidyl transferase centers •Crystal: compact, does not span this distance
lg I, relative2
p
Red: cryo-EMOrange: Xtal
1
(1)(2)(3)(4)(5)(6)(7)
0A
Vestergaard, B., Sanyal, S., Roessle, M., Mora, L., Buckingham, R. H., Kastrup, J. S., Gajhede, M., Svergun, D. I. & Ehrenberg, M. (2005) Mol. Cell, 20, 929–938.
s0.0 0.1 0.2 0.3

Domain closure of 3-phosphoglycerate kinase b d b SAXSobserved by SAXS
3-Phospho-D-glycerate kinase (PGK) is p g y ( )a typical hinge-bending enzyme with two structural domains of about equal
size (MM=43.5 kDa)
OPEN (Pig, Bs)CLOSED (Tb, Tm)
PGK catalyses the phospho-transfer from 1,3-bisphosphoglycerate (1,3-BPG) to MgADP and produces 3 phospho-glycerate (3-PG) and MgATP during the carbohydrate metabolism.
Closure of the two domains of PGK upon substrate binding is essential for the enzyme function.
A. Varga, B. Flachner, P.V. Konarev, E. Gráczer, J. Szabó, D.I. Svergun, P. Závodszky, & M. Vas (2006) FEBS Lett. 580, 2698-2706.

Domain closure of 3-phosphoglycerate kinase b d b SAXSobserved by SAXS
Numerous crystal structures do not yield
OPEN (Pig, Bs)CLOSED (Tb, Tm)
conclusive answer, which conditions are required for the closure.
The known X ray structures of open and
CRYSOL fits
The known X-ray structures of open and closed conformations were compared to SAXS data.
CRYSOL fits
A. Varga, B. Flachner, P.V. Konarev, E. Gráczer, J. Szabó, D.I. Svergun, P. Závodszky, & M. Vas (2006) FEBS Lett. 580, 2698-2706.
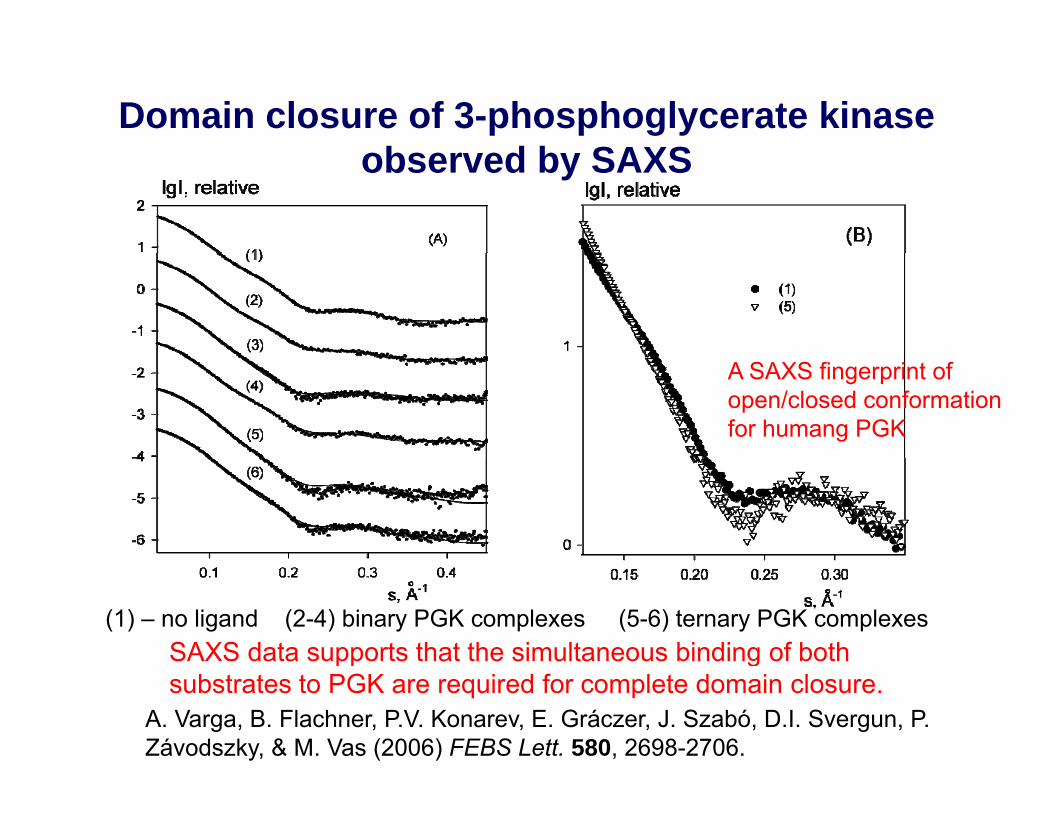
Domain closure of 3-phosphoglycerate kinase b d b SAXSobserved by SAXS
A SAXS fingerprint of open/closed conformation for humang PGK
(1) – no ligand (2-4) binary PGK complexes (5-6) ternary PGK complexesSAXS data supports that the simultaneous binding of both
A. Varga, B. Flachner, P.V. Konarev, E. Gráczer, J. Szabó, D.I. Svergun, P. Závodszky, & M. Vas (2006) FEBS Lett. 580, 2698-2706.
pp gsubstrates to PGK are required for complete domain closure.

Interaction of human 3-phosphoglycerate kinase with L M ADP th i i f D M ADPL-MgADP, the mirror image of D-MgADP.
hPGK can accommodate the mirror image L-enantiomer of MgADP into itsof MgADP into its nucleotide-binding siteand can phosphorylate it, almost as effectively as the natural D-enantiomer.
L-MgADP D-MgADP
A. Varga, J. Szabó, B. Flachner, B. Roy, P.V. Konarev, D.I. Svergun, P. Závodszky, C. Périgaud, T. Barman, C. Lionne, & M. Vas, M. (2008) Biochem. Biophys. Res. Comm. 366, 994-1000.

Idea of Rigid Body Modelling• Large macromolecular complexes are
more difficult to study by high resolution methods
• High resolution models of subunits can be used to model the quaternary structure of complexes based on low resolution methods
• Assuming the tertiary structure is notAssuming the tertiary structure is notchanged by complex formation,arbitrary complex can be constructedby moving and rotating the subunits.
• For each subunit this operationdepends on three orientational anddepends on three orientational andthree translational parameters.

Scattering from a Complex ParticleScattering from a Complex ParticleScattering amplitudes from individual subunits in referencepositions/orientations are evaluated using CRYSOL/CRYSON
Shift: x, y, z
positions/orientations are evaluated using CRYSOL/CRYSON
Rotation:α, β, γ
AA0
The partial amplitudes of arbitrarily rotated and displacedsubunit are analytically expressed via the initial amplitudes
, β, γ
and the six positional parameters (three Euler rotation anglesand three Cartesian shifts):
(i) ( ) (i) ( ) { (i) ( ) (i) (i) (i) (i) (i) (i) }A(i)lm(s) = A(i)
lm(s) { A0(i)
lm(s), α (i), β (i), γ (i), x (i), y (i), z (i) }.Svergun, D.I. (1991). J. Appl. Cryst. 24, 485-492
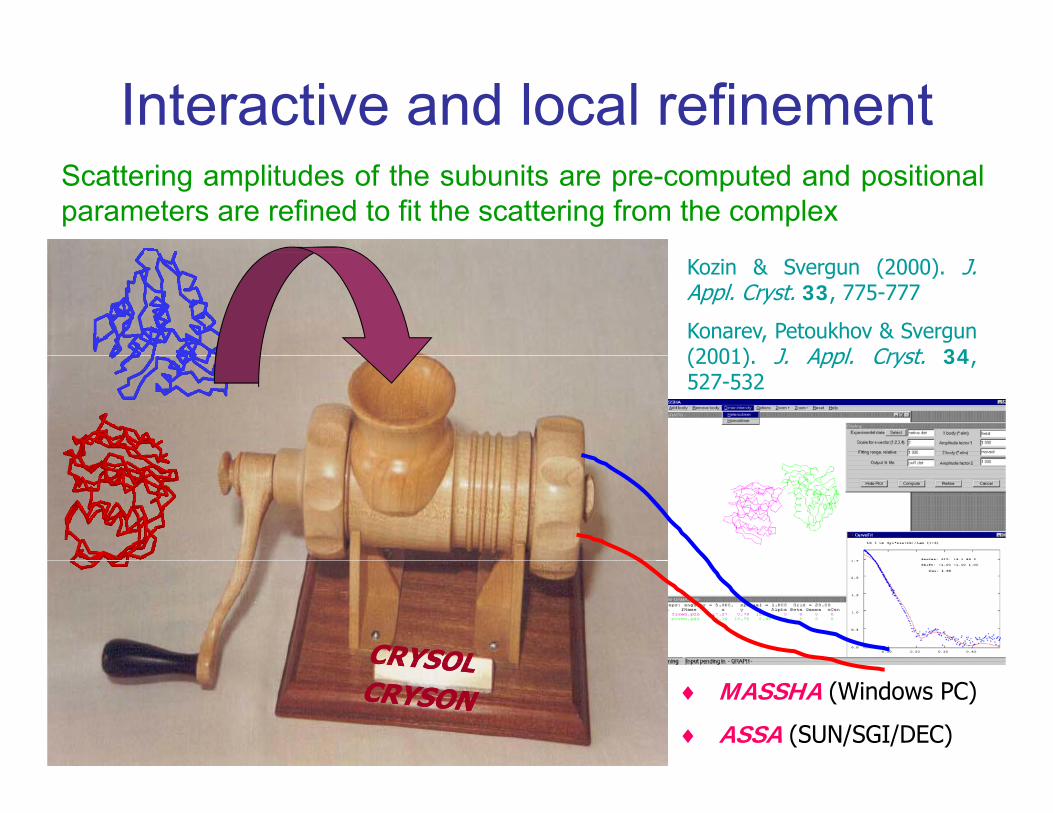
Interactive and local refinementScattering amplitudes of the subunits are pre-computed and positionalparameters are refined to fit the scattering from the complex
Kozin & Svergun (2000). J.Appl. Cryst. 33, 775-777
Konarev, Petoukhov & Svergun(2001) J Appl Cryst 34(2001). J. Appl. Cryst. 34,527-532
♦ MASSHA (Windows PC)
♦ ASSA (SUN/SGI/DEC)

Global rigid body modelling (SASREF)Fit ( lti l X d t ) tt i ( ) f ti lFits (multiple X-ray and neutron) scattering curve(s) from partial constructs or contrast variation using simulated annealing Requires models of subunits, builds interconnected models without steric clashessteric clashes Uses constraints: symmetry, distance (FRET or mutagenesis) relative orientation (RDC from NMR), if applicable
lg I, relative
Petoukhov & Svergun (2005) Biophys J. 89, 1237;(2006) Eur. Biophys. J. 35, 567.
10
11
9
10
s, nm-10.5 1.0 1.5 2.0
8

SASREF Restraints2
Set of penalties formulating various restraints
f (X) = χ2[Iexp(s), I(X,s)] + ΣαiPi(X)
• To ensure the interconnectivity of the entire complexeach subunit should have a contact with at least oneother subunit.The contact distance between C atoms of distinct
Interconnectivity and steric clashes
• The contact distance between Cα atoms of distinctsubunits: 4-7 A.
• Overlap: distance < 4 A.• Not interconnected arrangements of subunits and• Not interconnected arrangements of subunits and
those with steric clashes are penalized.
• From binding affinity studies or from mutagenesisInformation on contactsdata the information on contacting subunits andeven individual residues can be available.
• SASREF allows one to account for thisinformation by specifying the ranges of residuesinformation by specifying the ranges of residuesor nucleotides which can be involved ininteractions between the partners.

Building native-like folds of missing fragments
2Set of penalties formulating various restraints
f (X) = χ2[Iexp(s), I(X,s)] + ΣαiPi(X)
• Using DR-type models and protein-specific penalty functions
Number of neighbours
5
6
0
1
2
3
4
Excluded volume
Shell radius, nm
0.2 0.4 0.6 0.8 1.00
Neighbors distribution
Knowledge-based potentials
Bond angles & dihedrals distributionp
Petoukhov, M.V., Eady, N.A.J., Brown, K.A. & Svergun, D.I. (2002) Biophys. J. 83, 3113

Modelling of multidomain proteins (BUNCH)
• BUNCH combines rigid body and ab initiomodelling to find the optimal positions and orientations of rigid domains and probable conformations of flexible linkers represented as “dummy residues” chains attached to the appropriate termini of domains.pp p
• Multiple experimental scattering data sets from partial constructs (e.g. deletion mutants) can be f f ffitted simultaneously with the data of the full-length protein.
• BUNCH permits to account for the symmetry (the• BUNCH permits to account for the symmetry (the same for all constructs) and offers the possibility to fix some domains.
• Contacts between specific residues can be used as restrains
Petoukhov, M. V. & Svergun, D. I. (2005). Biophys. J. 89, 1237-1250
Contacts between specific residues can be used as restrains.
CORAL – analog of BUNCH for complexes
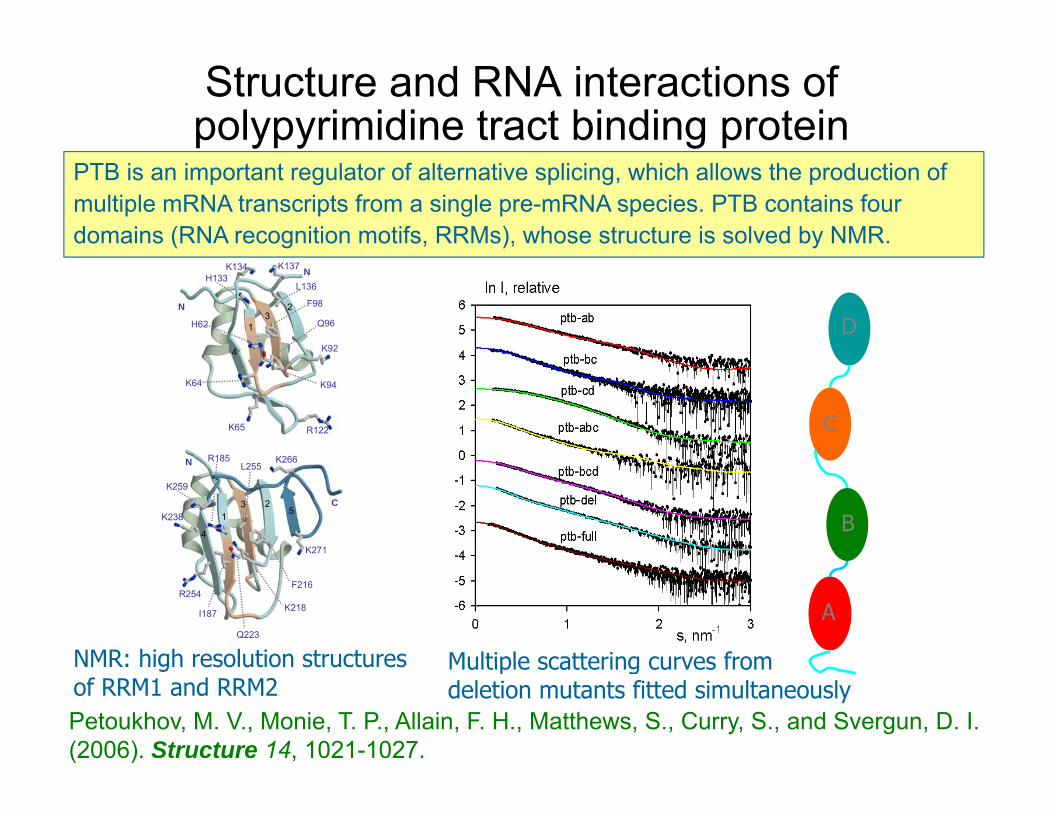
Structure and RNA interactions of polypyrimidine tract binding protein
PTB is an important regulator of alternative splicing, which allows the production of multiple mRNA transcripts from a single pre-mRNA species. PTB contains four domains (RNA recognition motifs, RRMs), whose structure is solved by NMR.
H62
F98
L136
K92
Q96
K137K134H133
4
13
2N
N
D
R185 K266
K94
R122K65
K64
C
L255R185
K238
K271
K266
K259
25
31
4
C
N
B
I187
R254F216
K218
Q223
NMR: high resolution structures
A
Multiple scattering curves fromNMR: high resolution structures of RRM1 and RRM2
Multiple scattering curves from deletion mutants fitted simultaneously
Petoukhov, M. V., Monie, T. P., Allain, F. H., Matthews, S., Curry, S., and Svergun, D. I. (2006). Structure 14, 1021-1027.
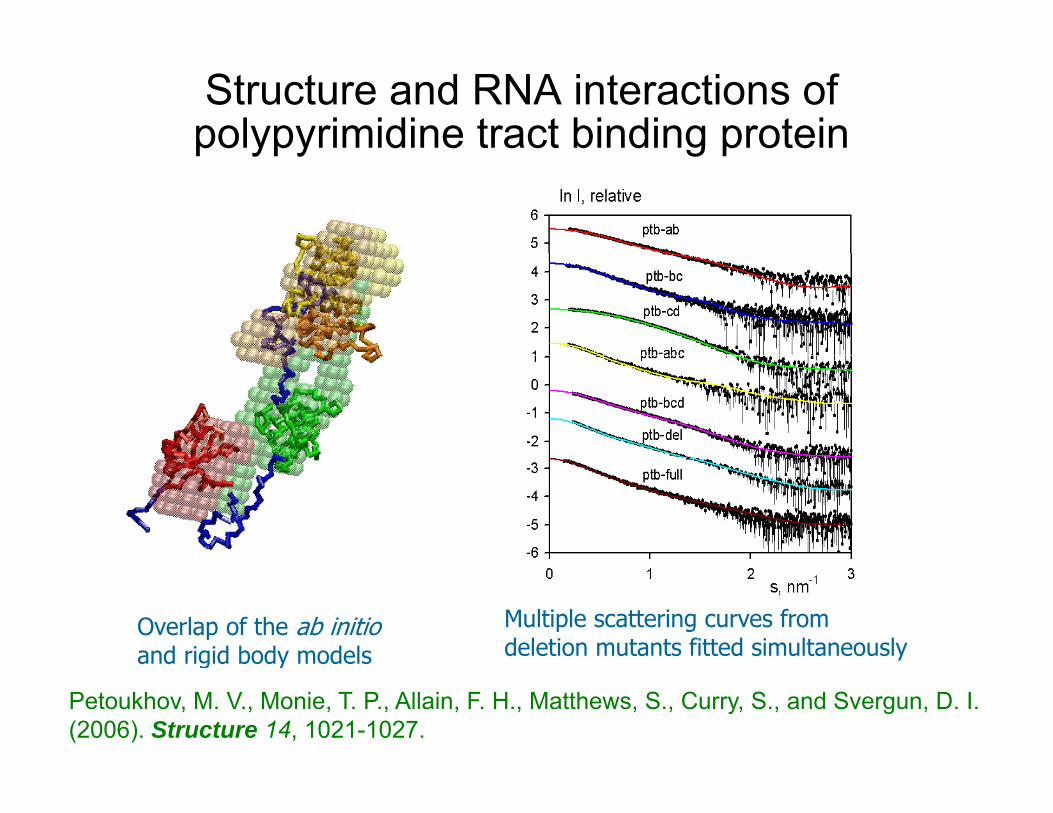
Structure and RNA interactions of polypyrimidine tract binding proteinp ypy g p
Overlap of the ab initio and rigid body models
Multiple scattering curves from deletion mutants fitted simultaneouslyand rigid body models
Petoukhov, M. V., Monie, T. P., Allain, F. H., Matthews, S., Curry, S., and Svergun, D. I. (2006). Structure 14, 1021-1027.

Dimer model for Filamin C (domains 23-24) obtained by SAXS
Filamins are dimeric actin-binding proteins that contribute to organization of the actin based cytoskeleton and to its remodelling by integrating differentthe actin-based cytoskeleton and to its remodelling by integrating different signalling pathways.
The crystal structure of domain 23 of filamin C e c ys a s uc u e o do a 3 o a Cshowed that the protein adopts the expected immunoglobulin (Ig)-like fold.
Filamin C domain 24 forms an antiparallel dimer exploiting strands C and D, and it was proposed that these two strands create a dimerization interface in all vertebrate filamins.
In order to investigate if the domain 23 influences dimerization of filamins the tandem domains 23 and 24 of filamin C were used for structural studies.
L.Sjekloca, R. Pudas, B. Sjoblom, P. Konarev, O. Carugo, V. Rybin, T.R. Kiema,D. Svergun, J. Ylanne, & K.D. Carugo, (2007) J Mol Biol. 368, 1011-1023.

Dimer model for Filamin C (domains 23-24) bt i d b SAXSobtained by SAXS
DAMMIN and BUNCH models
No symmetry5 nm
o sy e y
P2 symmetry
The results of the SAXS study on construct 23–24 clearly indicate that domain 23 is not involved in dimerization but protrudes away from the dimer core
L.Sjekloca, R. Pudas, B. Sjoblom, P. Konarev, O. Carugo, V. Rybin, T.R. Kiema,D. Svergun, J. Ylanne, & K.D. Carugo, (2007) J Mol Biol. 368, 1011-1023.
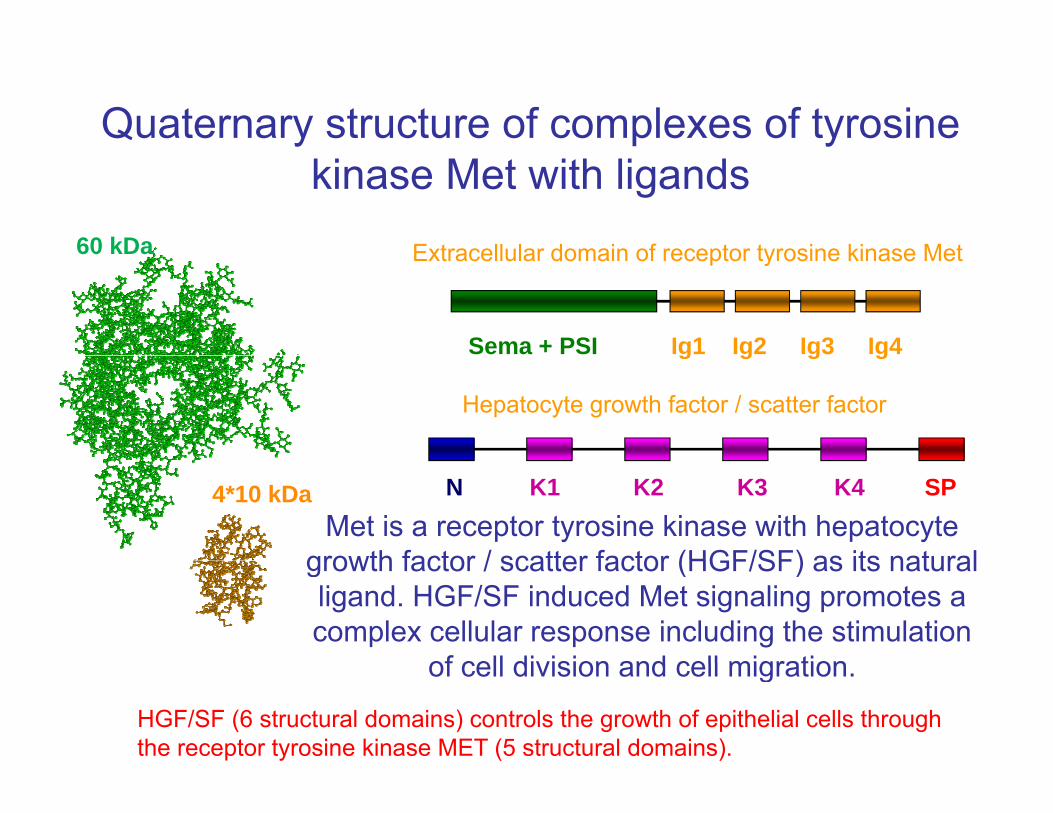
Quaternary structure of complexes of tyrosine kinase Met with ligands
Extracellular domain of receptor tyrosine kinase Met60 kDa
Sema + PSI Ig1 Ig2 Ig3 Ig4
Extracellular domain of receptor tyrosine kinase Met 60 kDa
g g g g
Hepatocyte growth factor / scatter factor
Met is a receptor tyrosine kinase with hepatocyte growth factor / scatter factor (HGF/SF) as its natural
4*10 kDa N K1 K2 K3 K4 SP
growth factor / scatter factor (HGF/SF) as its natural ligand. HGF/SF induced Met signaling promotes a complex cellular response including the stimulation
of cell division and cell migrationof cell division and cell migration.
HGF/SF (6 structural domains) controls the growth of epithelial cells through the receptor tyrosine kinase MET (5 structural domains).
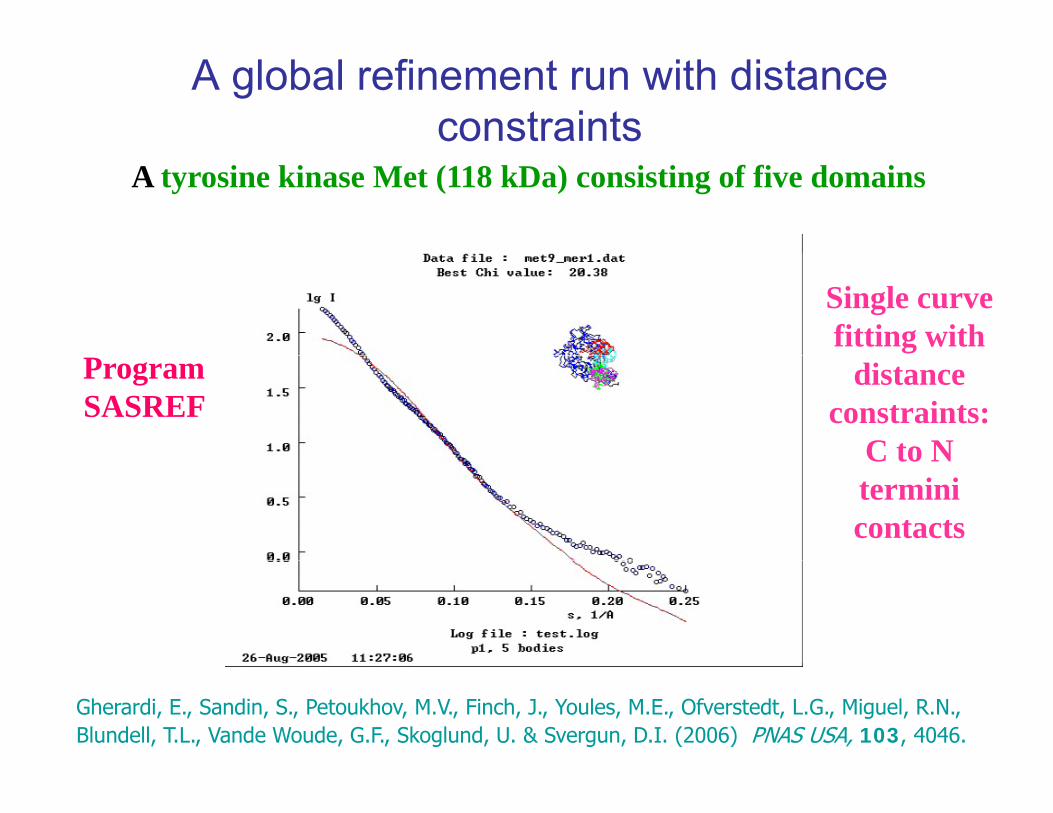
A global refinement run with distance constraints
A tyrosine kinase Met (118 kDa) consisting of five domains
Single curve fitting with
ProgramSASREF
distance constraints:
C to NC to N termini contacts
Gherardi, E., Sandin, S., Petoukhov, M.V., Finch, J., Youles, M.E., Ofverstedt, L.G., Miguel, R.N., Blundell, T.L., Vande Woude, G.F., Skoglund, U. & Svergun, D.I. (2006) PNAS USA, 103, 4046.
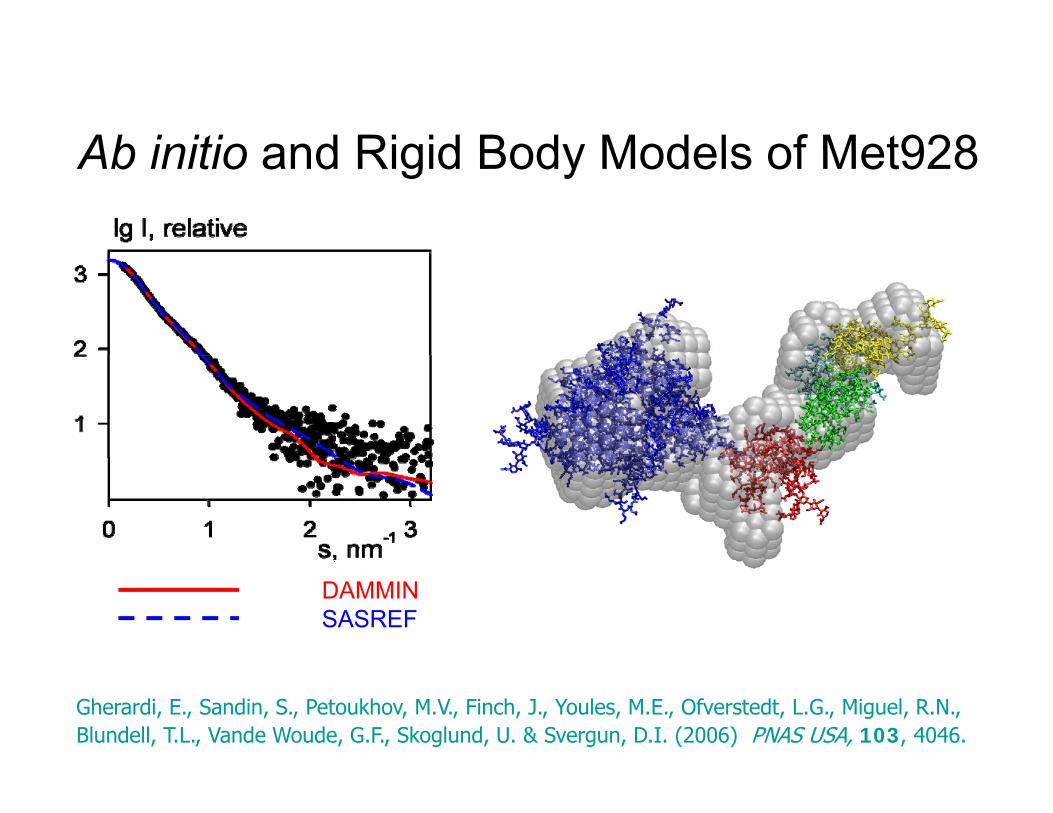
Ab initio and Rigid Body Models of Met928Ab initio and Rigid Body Models of Met928
DAMMINSASREF
Gherardi, E., Sandin, S., Petoukhov, M.V., Finch, J., Youles, M.E., Ofverstedt, L.G., Miguel, R.N., Blundell, T.L., Vande Woude, G.F., Skoglund, U. & Svergun, D.I. (2006) PNAS USA, 103, 4046.

3D Modelling of Sc and Tc HGF/SF3D Modelling of Sc and Tc HGF/SF
• TC HGF/SF - MetTC HGF/SF Met
X
N K1 K2 K3 K4 SP
Conversion of pro(single-chain)-HGF/SF into the active two-chain form is associated with a major structural transition from a compact, closed
X
j p ,conformation to an elongated, open one.
Gherardi, E., Sandin, S., Petoukhov, M.V., Finch, J., Youles, M.E., Ofverstedt, L.G., Miguel, R.N., Blundell, T.L., Vande Woude, G.F., Skoglund, U. & Svergun, D.I. (2006) PNAS USA, 103, 4046.
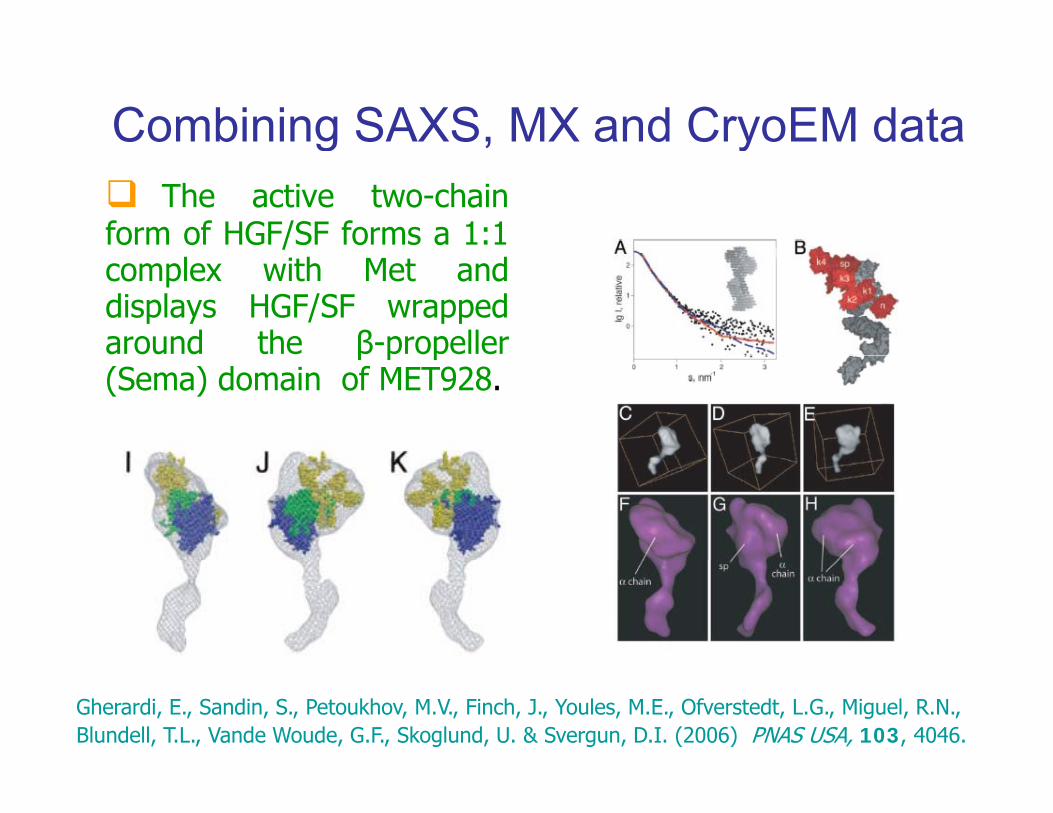
Combining SAXS, MX and CryoEM datag yThe active two-chain
form of HGF/SF forms a 1:1complex with Met anddisplays HGF/SF wrappedaround the β-propellerβ p p(Sema) domain of MET928.
Gherardi, E., Sandin, S., Petoukhov, M.V., Finch, J., Youles, M.E., Ofverstedt, L.G., Miguel, R.N., Blundell, T.L., Vande Woude, G.F., Skoglund, U. & Svergun, D.I. (2006) PNAS USA, 103, 4046.

Hepatocyte growth factor/scatter factor and MET signalling
A truncated Metectodomain ( Met5 = Sema+PSI ) builds a 2:2 complexwith two-chain HGF/SFwith two chain HGF/SFassembled around thedimerization interface seenin the crystal structure ofin the crystal structure ofthe NK1 fragment ofHGF/SF, which displays thefeatures of a functionalfeatures of a functional,signaling unit.
Gherardi, E., Sandin, S., Petoukhov, M.V., Finch, J., Youles, M.E., Ofverstedt, L.G., Miguel, R.N., Blundell, T.L., Vande Woude, G.F., Skoglund, U. & Svergun, D.I. (2006) PNAS USA, 103, 4046.

SAXS and EM study of Lymazine synthaseThis enzyme catalyzes the formation of 6,7-dimethyl-8-ribityllumazine in the penultimate step of riboflavinbiosynthesisbiosynthesis.
The enzyme forms icosahedral capsids with a total molecularweight of about 960 kDa.
SAXS measurements were made f
pentamer unit
for native and mutant enzyme species in different solvents and at different pH.
pentamer unitThe formation of mutliple assembly states was observed. They are interconvertable via equilibrium which is sensitive to solvent type and pHwhich is sensitive to solvent type and pH.
X. Zhang, P.V.Konarev, M.V.Petoukhov, D.I.Svergun, L.Xing, R.H.Cheng, I.Haase, M.Fischer, A.Bacher, R. Ladenstein & W. Meining (2006) JMB 362, 753-770

WT
SAXS and EM study of Lymazine synthaseMutant
WT, phosphate buffer
WT, Tris buffer
MIXTURE fitspH 7 WT, Borate bufferpH 7
pH 10
X. Zhang, P.V.Konarev, M.V.Petoukhov, D.I.Svergun, L.Xing, R.H.Cheng, I.Haase, M.Fischer, A.Bacher, R. Ladenstein & W. Meining (2006) JMB 362, 753-770
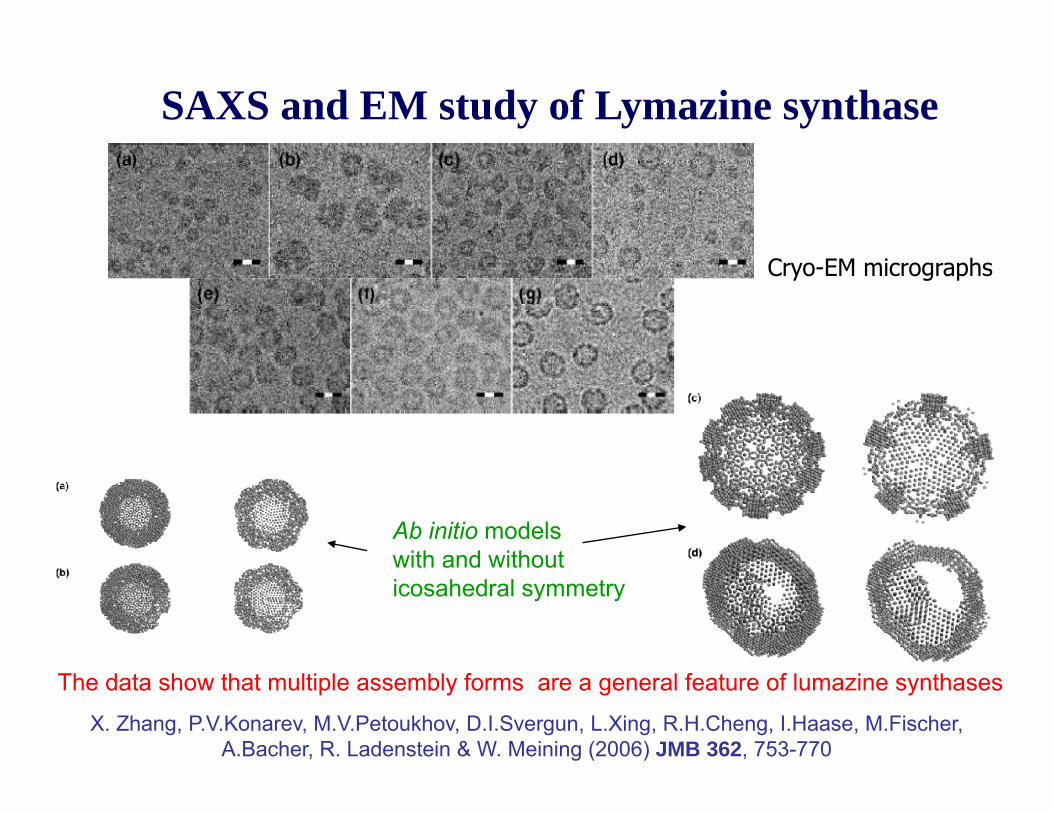
SAXS and EM study of Lymazine synthase
Cryo-EM micrographs
Ab initio models with and withoutwith and without icosahedral symmetry
The data show that multiple assembly forms are a general feature of lumazine synthases.
X. Zhang, P.V.Konarev, M.V.Petoukhov, D.I.Svergun, L.Xing, R.H.Cheng, I.Haase, M.Fischer, A.Bacher, R. Ladenstein & W. Meining (2006) JMB 362, 753-770

pH induced virus maturationNudaurelia capensis Omega Virus (NwV)
Cryo-EM Crystallography
Maturation is an important event associated with establishing virus infectivity
It occurs in many complex viruses in order to accommodate the need for weak interactions between subunits t hi lf bl dto achieve proper self-assembly and the requirement for a robust particleto survive the extra cellular environment.
Maturation results from a program encoded in the initial, often fragile, immature particle that directs largeimmature particle that directs large conformational changes (LCC)resulting in a robust infectious virion.
Immature particle mature particle Matsui T, Tsuruta H, Johnson JE.Biophys J. (2010) 98, 1337

pH induced virus maturationNudaurelia capensis Omega Virus (NwV)
Maturation is often triggered by changes
SAXS
Maturation is often triggered by changes in pH or other electrostatic events within the cell allowing in vitro maturation to be controlled by careful adjustment of the pH.
Time resolved SAXS showed that there were three kinetic stages initiated with an
Time-resolved
gincremental drop in pH; (1) a rapid (<10 ms) collapse to an
incrementally smaller particle, (2) a continuous size reduction over SAXS(2) a continuous size reduction over
the next 5 seconds, (3) a smaller final transition
occurring in 2-3 minutes.
Matsui T, Tsuruta H, Johnson JE.Biophys J. (2010) 98, 1337

Encapsulated Magnetic Iron Oxide Nanoparticles (EM and SAXS)Nanoparticles (EM and SAXS)
Highly monodisperse NPs are prepared by thermal decomposition of i d i l diiron compounds including oxygen-containing ligands in boiling surfactants. The NPs are coated by phospholipids with PEG Tailsphospholipids with PEG Tailsto become soluble.
lg I, relative
5shoulder
lg I, relative
5Ab initio analysis: peculiarities of
TEM image, scale bar 100 nm
1
2
3
4
5123
1st minimum
p(R), relative1
2
3
4
5123
shoulder
1st minimum
p(R), relative
Ab initio analysis: peculiarities of organization of different NPs
s nm-10.0 0.5 1.0 1.5 2.0 2.5 3.0
-2
-1
0
1
R, nm0 5 10 15 2002468
1012
-10.0 0.5 1.0 1.5 2.0 2.5 3.0
-2
-1
0
1
R, nm0 2 4 6 8 100.0
0.5
1.0
1.5
Shtykova, E.V, Huang, X., Remmes, N., Baxter, D., Dixit, S., Stein, B., Dragnea, B., Svergun, D. I. & Bronstein, L. M. (2007) J. Phys. Chem. C, 111, 18078-18086
s, nm s, nm 1
DAMMIN fits

Encapsulated Magnetic Iron Oxide Nanoparticles (EM and SAXS)Nanoparticles (EM and SAXS)
Rigid body analysis reveals equilibrium clusters
lg I, relative lg I, relative
Rigid body analysis reveals equilibrium clusters of the NPs stabilized by magnetic interactions
3
4
512
3
4
512
1
2
3
1
2
3
s, nm-10.0 0.5 1.0 1.5 2.0 2.5 3.0
-1
0
s, nm-10.0 0.5 1.0 1.5 2.0 2.5 3.0
-1
0
Shtykova, E.V, Huang, X., Remmes, N., Baxter, D., Dixit, S., Stein, B., Dragnea, B., Svergun, D. I. & Bronstein, L. M. (2007) J. Phys. Chem. C, 111, 18078-18086
SASREF fits

Data analysisDetector
SAXS in structural biology (biased)
R l ti
Sh2θ
SampleIncident beam
Wave vector k k=2π/λ g
I,
rela
tive
2
3
Scattering I( )
Resolution, nm:
3.1 1.6 1.0 0.8
Shape determination
Rigid body
Solvent
k, k=2π/λ
Scatteredbeam, k1
l
1
curve I(s)
Missing
Rigid body modellingRadiation sources:
X-ray tube (λ = 0.1 - 0.2 nm)Synchrotron (λ = 0.05 - 0.5 nm)Thermal neutrons (λ = 0.1 - 1 nm)
s, nm -10 2 4 6 8
EM
Complementary Complementary techniquestechniques
Oligomeric
gfragments
Homologymodels
Atomicmodels
MS Distances
Crystallography
NMR
i h i
Bioinformatics
mixturesOrientationsInterfaces
AdditionalAdditionalinformationinformation
Biochemistry
FRET
AUCFlexible systems
EPR

Joint use of SAXS,MX and EM for biological macromolecules:biological macromolecules:
conclusionsN hi k b i i i l l i• Nothing known: ab initio low resolution structure (SAXS and EM)
• Complete high resolution structure known: validation in• Complete high resolution structure known: validation in solution and biologically active oligomers (SAXS and MX)
• Incomplete high resolution structure known: probable p g pconfiguration of missing portions (SAXS, MX and EM)
• High resolution structure of domains/subunits known: t t t b i id b d fi tquaternary structure by rigid body refinement
(SAXS, MX and EM)

Acknowledgements:Collaborative projects
Release factor RF1: B.Vestergaard (University of Copenhagen, Denmark)Tyrosine Kinase: E. Gherardi (Medical Research Council Centre, UK)Myomesin-1: M.Wilmanns (EMBL Hamburg Outstation, Germany)Lymazine synthase: R. Ladenstein (Karolinska Institute, Sweden)PTB S C (I i l C ll UK)PTB: S. Curry (Imperial College, UK)3PGK enzyme: M. Vas (Institute of Enzymology, Hungary)Iron Nanoparticles: L.M. Bronstein (Indiana University, USA)Filamin C: K Djinovic-Carugo (University of Vienna Austria)
EMBL H b
Filamin C: K.Djinovic-Carugo (University of Vienna, Austria)Ataxin-3: A. Pastore (National Institute for Medical Research, UK)
EMBL-HamburgD.I. Svergun, M.W. Roessle, M.V. Petoukhov,D. Franke, A.G. Kikhney, W. Shang, H. Mertens
BioSAXS groupBioSAXS group
EMBO Global Exchange Lecture Course


















