
Introduction to ab initio methods I Kirill Gokhberg.
22
Introduction to ab initio methods I Kirill Gokhberg
-
Upload
randell-hutchinson -
Category
Documents
-
view
259 -
download
0
Transcript of Introduction to ab initio methods I Kirill Gokhberg.

Introduction to ab initio methods I
Kirill Gokhberg

„We‘ve got fascinating results!“
„ ... this is ICD in the Ar dimer ...“

Anatomy of an ICD process
Ar2 GS PEC
Initial vibrational WP
Optical excitation. Dipole TM needed!
Core-Excited state PEC
Satellite Ar+*Ar PEC
Final Ar+Ar+ PEC
Resonant Auger partial rates
ICD rates (R)
Nuclear dynamics in ICD states
ICD electron (and KER) spectra

e NH H T He describes only electronic motion (nuclei fixed).
( , )e NH T T U r R
2 2
,
,2 2
1( , )
| | | |
ie N
i
i i ji i j
p PT T
M
ZU
r Rr R r r
H describes the motion of N electrons and M nuclei.
+ relativistic terms if needed.
Electron interaction term

( ), ( )n nV R R
( , ) ( ) ( , )
( ) ( ) ( ) ( )2
n nn
N n n n nm mm
iT V E
r R R r R
R R R R
( , ) ( ) ( , )e n n nH V r R R r R1. Bound electronic states
PEC or PES Used to obtain the „properties“ :TM, etc
Kirill&Andreas
2. Resonance (bound-in-continuum) states Premysl Electronic widths
3. Nuclear dynamics (in local approximation)
time dependent formulation + coupling to final states when computing ICD spectraNicolas

N-electron SE cannot be solved exactly!
Approximate solutions are found numerically and two questions should be answered before starting the work.
1. What electronic structure method should we use?
2. How do we represent the respective Hilbert space accurately enough?
or
What basis set should we select?

Choosing a method
• Independent particle (mean-field) methods – an electron moves in an average field of (N-1) other electrons.
Typical example - Hartree-Fock (HF) approximation.
• Correlated methods – a motion of an electron is influenced by (correlated with) the motion of (N-1) other electrons at each instant.
Examples – configuration interaction (CI), propagator (ADC), many-body perturbation theory (MBPT), coupled cluster (CC), ... methods.
• Hartree-Fock solution is used as an input for the correlated methods.

Independent-electron wave function
( ) ( ) ( ) x r
1/2 !
0 1 21
1( 1) ( ) ( ) ( )
!n
Np
n NnN
1 2 NP
spin-orbital
spatial orbitalor
molecular orbital (MO)
spin functionSlater determinant
(ground state)
•Electrons with anti-parallel spins are uncorrelated.•Electrons with parallel spins are exchange correlated.

Hartree-Fock approximation
0 00
0 0
,
0
elHE
E
( ) ( ) ( )
( ) ( ) ( ) ( )r r rf
f h
1 1 1
1 1 1 1J K
12
12
* 1
1
* 1
1
( ) ( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) ( )
N
r i i ri
N
r i r ii
d r
d r
2
2
1 1 x 2 2 1
1 1 x 2 2 1
J
K
12
12
1 112 12
1 1 1
1 * 1 *12
1 * 1 *12
1
2
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
N N N
HFi i j
i i j j
i j j i
E i h i ij r ij ij r ji
ij r ij d d r
ij r ji d d r
1 2
1 2
x x 1 1 2 2
x x 1 1 2 2
HF equations
Fock operator
Total electronic energy
Coulomb and Exchange integrals
Coulomb and exchange operators
Orbital energy

Restricted vs. Unrestricted HF

• HF recovers up to 99% of the total electronic energy in the
ground state
• However, the energy differences of interest in chemical and spectroscopic processes are a fraction of one percent of the total energy.
• For example HF cannot describe binding between two rare-gas atoms
corr exact HFE E
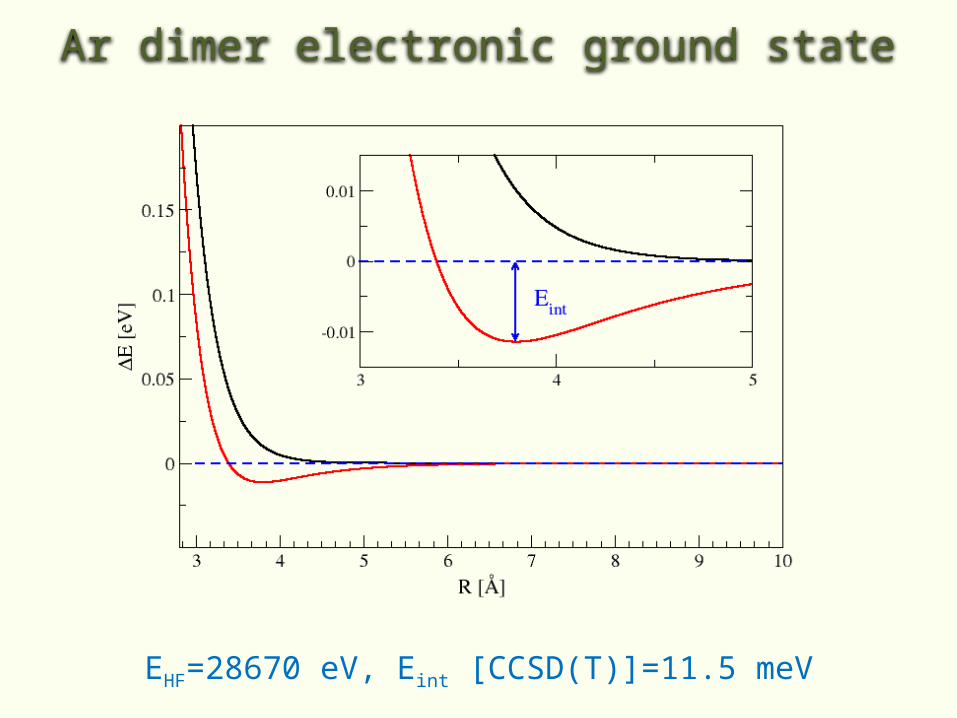
Ar dimer electronic ground state
EHF=28670 eV, Eint [CCSD(T)]=11.5 meV
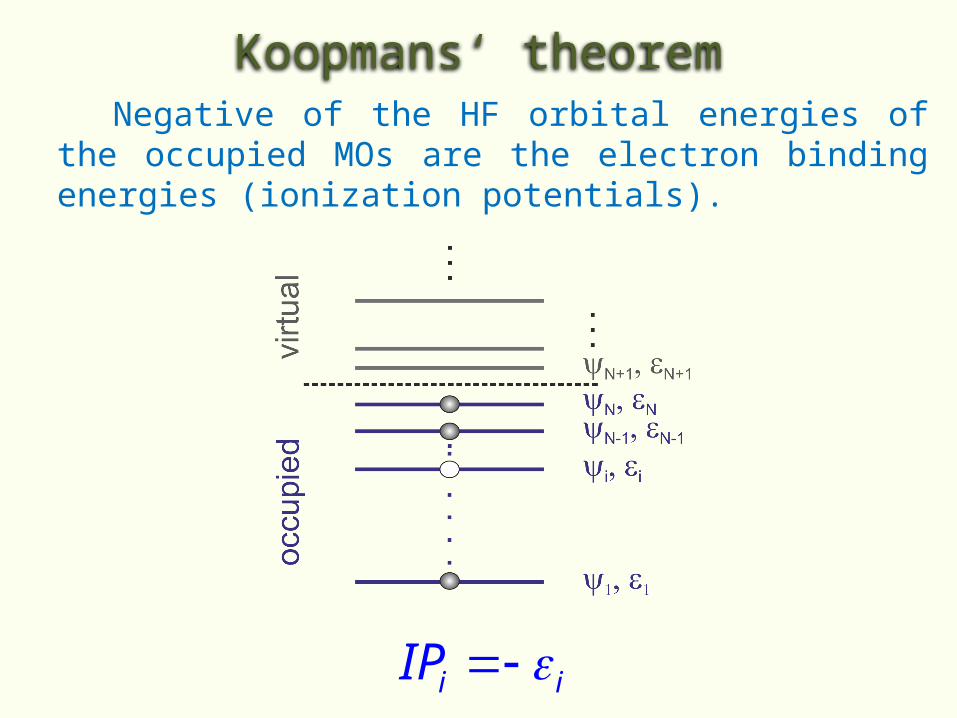
Koopmans‘ theorem Negative of the HF orbital energies of the
occupied MOs are the electron binding energies (ionization potentials).
i iIP

Photoelectron spectrum of H2O
„Breakdown of the MO picture“due to the intra-molecular correlation

Double ionisation threshold
Breakdown of monomer lines due to ICDdriven by inter-molecular correlation

1. MOs and orbital energies serve as input for the correlated methods.
2. HF approximation furnishes us with a vivid picture of many electron system with electrons stacked on shelves called molecular orbitals.
3. HF solutions are of some, albeit limited, use for computing electronic decay rates.
4. The HF approximation usually does not deliver ground state PES in acceptable quality. It generally fails to produce excited state PES at all.
5. It fails to reproduce correlation driven phenomena.

Choosing the basis set,
,
( )M
Ai i
A
c
Ar R
•Proper behaviour at r→0 and r→∞.•Small number of STO basis functions are sufficient to represent a MO.
•Allow for very efficient computation of four-centre two-electron integals:
12
1 * 1 *12 ( ) ( ) ( ) ( )A B C D A C B Dr d d r 1 2x x 1 1 2 2
2exp( )l m nGTO Nx y z r 1 exp( ) ( , )n
STO lmNr r Y

cc-pVDZ basis set for Ne
( ) ( , )i i ii
c r r
Contracted Gaussian function
Primitive Gaussian function
Contraction coefficient
Contraction exponent
Polarisation basis function
Adding diffuse functions
( ) ( )max
diffuse l nn
Valence basis function
Core basis function

RHF calculation of Ne

Augmenting a basis set

Introducing electron correlation

Configuration Interaction scheme
ˆac− creation operator, ic − annihilation operator
0ˆ
I IC
IJH I el JH
ˆ ˆ ˆ ˆ ˆ ˆ ˆ ˆ; , , ; , , , , ;...I a i a b i jC c c a i c c c c a b i j 1
ˆ ˆ ˆ ˆ ˆ, ; , , , ;...I i a i jC c i c c c a i j
ˆ ˆ ˆ ˆ ˆ ˆ ˆ, , ; , , , , ;...I i j a i j kC c c i j c c c c a i j k
Excitation (N electrons)
Ionisation (N-1 electrons)
Double ionisation (N-2 electrons)
E0, E1, ... and corresponding wavefunctions
Diagonalisation More on correlation!
Andreas‘s lecture tomorrow.


















