Imaging in Spine and Spinal Cord Developmental Malformations · Embryology The development of the...
32
Imaging in Spine and Spinal Cord Developmental Malformations Andrea Rossi Contents Entity ..................................................................................... 3 Definition of Entity ...................................................................... 3 Basic Epidemiology/Demographics/Pathophysiology ................................ 3 Embryology .............................................................................. 3 Pathological Features ................................................................... 4 Clinical Scenario and Indications for Imaging ....................................... 6 Imaging Technique and Recommended Protocol ..................................... 7 Principal Entities ........................................................................ 9 Myelomeningocele ....................................................................... 9 Myelocele and Myeloschisis ............................................................. 9 Hemimyelomeningocele .................................................................. 10 Lipomas with Dural Defect: Lipomyelomeningocele, Lipomyelocele, and Lipomyeloschisis ................................................................. 11 Myelocystocele ........................................................................... 13 Meningocele .............................................................................. 15 Intradural Lipoma ......................................................................... 16 Filar Lipoma .............................................................................. 17 Tight Filum Terminale .................................................................... 17 Persistent Secondary Neural Tube ........................................................ 17 This publication is endorsed by: European Society of Neuroradiology (www.esnr.org) A. Rossi (*) IRCCS Istituto Giannina Gaslini Children’ s Hospital, Genoa, Italy e-mail: [email protected] # Springer Nature Switzerland AG 2019 F. Barkhof et al. (eds.), Clinical Neuroradiology , https://doi.org/10.1007/978-3-319-61423-6_32-1 1
Transcript of Imaging in Spine and Spinal Cord Developmental Malformations · Embryology The development of the...

Imaging in Spine and Spinal CordDevelopmental Malformations
Andrea Rossi
ContentsEntity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Definition of Entity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Basic Epidemiology/Demographics/Pathophysiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Embryology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Pathological Features . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
Clinical Scenario and Indications for Imaging . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Imaging Technique and Recommended Protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
Principal Entities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9Myelomeningocele . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9Myelocele and Myeloschisis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9Hemimyelomeningocele . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Lipomas with Dural Defect: Lipomyelomeningocele, Lipomyelocele,
and Lipomyeloschisis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Myelocystocele . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13Meningocele . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15Intradural Lipoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Filar Lipoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Tight Filum Terminale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Persistent Secondary Neural Tube . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
This publication is endorsed by: European Society ofNeuroradiology (www.esnr.org)
A. Rossi (*)IRCCS Istituto Giannina Gaslini Children’s Hospital,Genoa, Italye-mail: [email protected]
# Springer Nature Switzerland AG 2019F. Barkhof et al. (eds.), Clinical Neuroradiology,https://doi.org/10.1007/978-3-319-61423-6_32-1
1

Persistent Terminal Ventricle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18Split Notochord Syndrome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18Diastematomyelia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20Caudal Regression Syndrome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23Segmental Spinal Dysgenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28Dermal Sinus Tract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
Checklist for Image Reporting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
Clinical Cases and Sample Reports . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30Sample Report 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30Sample Report 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
AbstractSpinal cord development occurs through threeconsecutive periods, gastrulation, primary neu-rulation, and secondary neurulation, compris-ing the second to sixth gestational week.Defects in one of these three embryologicalsteps produce congenital spinal cord abnormal-ities, also called spinal dysraphisms.
Based on their external appearance, spinaldysraphisms may be categorized into two sub-sets. In open spinal dysraphisms (OSD), theplacode (non-neurulated neural tissue) isexposed to the environment through a cutane-ous defect along the child’s back. OSD mainlyinclude myelomeningocele, myelocele/myeloschisis, and hemimyelomeningoceleand are typically associated with the Chiari IImalformation. Myelomeningocele is by far themost common of these forms; the placode pro-trudes through a posterior defect and is ele-vated above the skin surface due toconcurrent dilatation of the subarachnoidspaces. Closed spinal dysraphisms (CSD) arecompletely covered by the skin, often carryingcutaneous stigmata that betray their presence.A sizable subcutaneous mass identifies CSDwith tumefaction, comprising lipomas withdural defect (lipomyelocele/schisis andlipomyelomeningocele), meningocele, andmyelocystocele. CSD without tumefactioncomprise a heterogeneous host of conditions,some characterized by complex associations ofspinal and spinal cord malformation (includingthe split notochord syndrome,
diastematomyelia, caudal regression syn-drome, and segmental spinal dysgenesis) andsome mainly characterized by spinal cord teth-ering (including tight filum terminale, persis-tent secondary neural tube, filar and intradurallipomas).
Clinical neuroradiology plays a fundamen-tal role in the diagnostic workup of patientssuspected of harboring spinal cordmalformations. Among the available radiolog-ical techniques, MRI is paramount to identifyand characterize congenital spinal cord abnor-malities. Along with conventional sequencesincluding triplanar T1- and T2-weightedimages, high-resolution heavily T2-weightedsequences (such as driven equilibrium –DRIVE or equivalent) provide exceptionallydetailed depictions of the abnormality and areextremely useful both for the diagnosis and forthe planning of surgery as well as in the assess-ment of surgical complications.
KeywordsMyelomeningocele · Neural tube defects ·Spina bifida · Spinal dysraphisms · Tetheredcord
AbbreviationsCNS Central nervous systemCRS Caudal regression syndromeCSD Closed spinal dysraphismsCSF Cerebrospinal fluidCT Computerized tomographyDRIVE Driven equilibrium
2 A. Rossi

DWI Diffusion-weighted imagingMRI Magnetic resonance imagingNTCD Neural tube closure defectsOEIS Omphalocele, exstrophy of the
bladder, imperforate anus, spinalanomaly
OSD Open spinal dysraphismsSSD Segmental spinal dysgenesisSTIR Short-tau inversion recoveryT TeslaVACTERL Vertebral abnormality, anal
imperforation, cardiacmalformations, tracheoe-sophageal fistula, renal abnor-malities, limb deformities
Entity
Congenital malformations of the spine and spinalcord (spinal dysraphisms)
Definition of Entity
Congenital malformations of the spine and spinalcord comprise a broad range of anomalies causedby embryologic defects in the process of spinalcord development, which occurs in a time windowthat covers the second to the sixth gestationalweeks. They are also loosely referred to as spinaldysraphisms, a term that implicates a defect ofclosure of the neural tube and should, therefore,strictly be applied only to open defects (such asmyelomeningocele) resulting from defective neu-rulation. However, in this chapter we will adopt abroader definition which includes allmalformations in which the development of thespinal cord and filum terminale is deranged.
Basic Epidemiology/Demographics/Pathophysiology
Spinal dysraphisms are frequently discovered dur-ing prenatal screening for fetal abnormalities.Typically, prenatal ultrasound detects most grossabnormalities, either by direct identification of the
spinal defect or indirectly through the visualiza-tion of the cranial abnormalities that are associ-ated with it (especially the Chiari II sequence inthe case of OSDs). Fetal MRI may then be used toconfirm the diagnosis and assess the globalcraniospinal axis, however, with limitations dueto the frequently difficult assessment of the fetalspine, especially early during the second trimester.In some cases, however, and depending on a num-ber of factors, spinal dysraphisms are onlydetected at birth or even afterwards, when neuro-logical complications (mostly attributable to cordtethering) ensue. In rare cases, they may remainundetected and be discovered in adults duringunrelated imaging studies.
The estimated incidence of spinal dysraphismis about 1–3/1000 live births, whereas the preva-lence has declined in the last few decades due tobetter maternal nutrition, folic acid supplementa-tion, and improved prenatal diagnosis. Recently,fetal surgery has been proposed as a valid treat-ment for OSD.
Embryology
The development of the spine and spinal cord is ahighly coordinated process consisting of severalconsecutive steps occurring during earlygestation.
• Gastrulation: The bilaminar embryonic disc,consisting of the epiblast and hypoblast, isconverted into a trilaminar disc with the for-mation of an intervening third layer, the meso-derm. This process begins by day 14 or15 when the primitive streak, a stripe of thick-ened epiblast composed of totipotential cells, islaid down along the midline of the dorsalaspect of the embryo. Epiblastic cells startmigrating bilaterally toward the primitivestreak and pass inward at Hensen’s node,which is its knob-like cranial termination;there, they enter the interface between the epi-blast and the hypoblast and displace the hypo-blast, to form the endoderm; subsequent wavesof migrating cells course bilaterally above theendoderm to form the mesoderm. Cells that
Imaging in Spine and Spinal Cord Developmental Malformations 3

migrate along the midline form the notochord.The skull, vertebral column, and meningesoriginate from the mesoderm surrounding theneural tube and notochord; the notochord itselfis required for the ectoderm to further developinto neural ectoderm that will form theneural tube.
• Primary neurulation: The neural ectodermoverlying the notochord along the midline ofthe dorsal aspect of the embryo is initiallyformed by a single layer of columnar cells,constituting the neural plate. On day 18, theneural plate starts bending, forming pairedneural folds that progressively increase in sizeand approximate to each other until they even-tually merge along the midline, forming theneural tube. This process occurs bidirectionallyas a sort of zipper, starting at the rhombenceph-alon and proceeding both cephalad and cau-dad. As the neural tube seals, the cutaneousectoderm on both sides of the neuroectodermdetaches from the latter and seals the overlyingskin (disjunction). The cranial end of the tubecloses on day 30 at the anterior neuropore,while the caudal end closes on day 31 at theposterior neuropore, corresponding to the 32ndsomite (future S3).
• Secondary neurulation: It consists of the for-mation of a neural tube caudally to the primaryneural tube, i.e., below the 32nd somite. Theprocess begins with the formation of the tailbud, a mass of totipotential cells which willthen develop into a secondary neural tube bymeans of internal cavitation (canalization). Thesecondary neural tube merges with the neuraltube formed by primary neurulation to form acontinuous structure. Then, it regresses (retro-gressive differentiation) to eventually form thetip of the conus medullaris (containing theterminal ventricle, a focal expansion of theependymal canal) and the filum terminale, afibroconnectival structure practically devoidof neural elements.
• Development of the vertebral column: Theparaxial trunk mesoderm to both sides of thenotochord matures into the somites, a bilateralcolumn of epithelial spheres that are laid downprogressively in a craniocaudal direction. Each
somite further develops into the dermatome(dorsal), myotome (intermediate), andsclerotome (ventral). The sclerotome will dif-ferentiate into the cartilaginous cells of thevertebrae, intervertebral discs, and ligaments,as well as into the spinal meninges. Thesclerotome spreads from its initially ventrallocation to enwrap the entire neural tube. Indoing so, it forms the dorsal mesoderm thatinsinuates itself between the neural tube andthe surface ectoderm after disjunction. Subse-quently, each sclerotome divides in half hori-zontally; the bottom half of one fuses with thetop half of another to form the vertebral bodies,while notochordal remnants eventually formthe nucleus pulposus within theintervertebral disc.
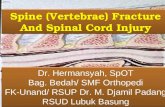
Spinal dysraphisms can be classified accordingto the putative embryological stage at which eachgiven abnormality arises and the presumed mech-anism of formation that generates it (Table 1).
Pathological Features
Spinal dysraphisms can be subdivided into open(OSD) and closed (CSD) depending on whetherthe malformed cord is exposed to the environmentthrough a congenital skin defect or, rather, is cov-ered by the skin. Based on this approach, patientsare further categorized into smaller groups basedon the appearance of the spinal cord (Table 2).
OSD do not usually cause major concern to theradiologist, at least in a postnatal evaluation sincethe diagnosis is usually already made during pre-natal life. The vast majority of OSDs isrepresented by myelomeningoceles, in which theneural placode is elevated and protrudes above thesurrounding cutaneous surface due to expansionof the underlying subarachnoid spaces; muchrarer forms are the myelocele, in which theplacode is flush within the surrounding skin, andthe myeloschisis, in which the placode isdepressed. A much rarer form, the hemimyelome-ningocele, is essentially a diastematomyelia (i.e.,cord splitting) in which one of the two hemicords
4 A. Rossi
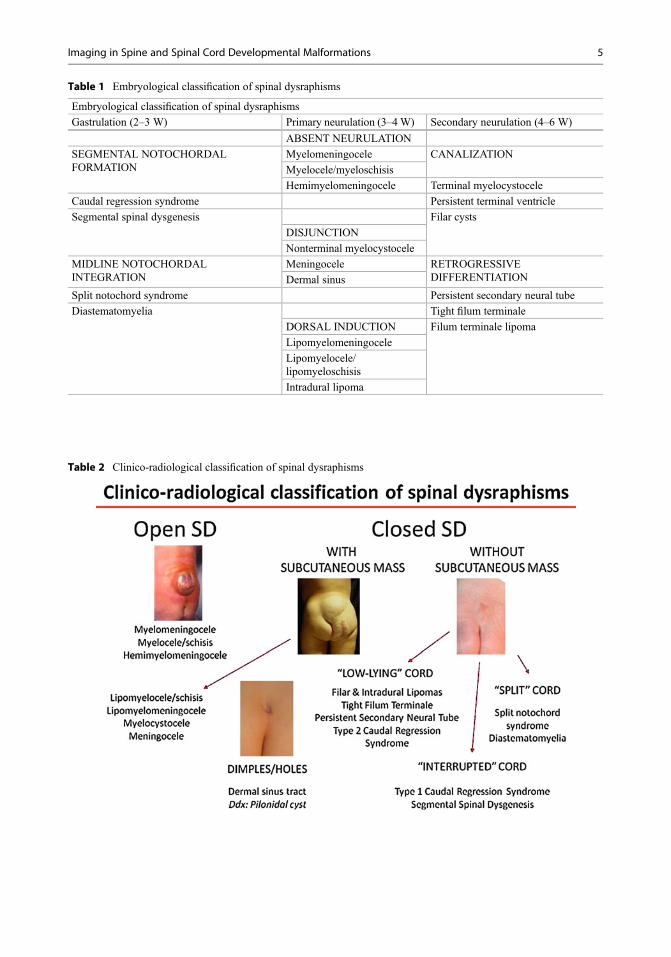
Table 1 Embryological classification of spinal dysraphisms
Embryological classification of spinal dysraphisms
Gastrulation (2–3 W) Primary neurulation (3–4W) Secondary neurulation (4–6 W)
ABSENT NEURULATION
SEGMENTAL NOTOCHORDALFORMATION
Myelomeningocele CANALIZATION
Myelocele/myeloschisis
Hemimyelomeningocele Terminal myelocystocele
Caudal regression syndrome Persistent terminal ventricle
Segmental spinal dysgenesis Filar cysts
DISJUNCTION
Nonterminal myelocystocele
MIDLINE NOTOCHORDALINTEGRATION
Meningocele RETROGRESSIVEDIFFERENTIATIONDermal sinus
Split notochord syndrome Persistent secondary neural tube
Diastematomyelia Tight filum terminale
DORSAL INDUCTION Filum terminale lipoma
Lipomyelomeningocele
Lipomyelocele/lipomyeloschisis
Intradural lipoma
Table 2 Clinico-radiological classification of spinal dysraphisms
Imaging in Spine and Spinal Cord Developmental Malformations 5

fails to neurulate and is therefore exposed to theenvironment.
CSD are much more heterogeneous than OSDsand comprise a larger amount of entities that differsignificantly from one another both on embryo-logical and radiological terms as well as clinically.An initial categorization step is dictated by thepresence of a subcutaneous mass on the patient’sback, which is usually located at the lumbosacrallevel. In this situation, the differential diagnosis isrestricted to very few entities, of which spinallipomas with defective dura (lipomyelocele/lipomyeloschisis and lipomyelomeningocele) areby far more common, and the remainder(meningocele and myelocystocele) isdistinctly rare.
In the absence of a subcutaneous mass alongthe patient’s back, spinal dysraphisms may befurther categorized based on the radiologicalappearance of the spinal cord:
• Low-lying cord: When the position of theconus medullaris is below the normal level(i.e., L2–L3 disc space), a tethering abnormal-ity is usually present; this is often representedby a tight filum terminale, a filar, or anintradural lipoma. In this category, associatedsacrococcygeal agenesis should be activelysought that would incorporate the case intothe caudal regression syndrome.
• Split cord: When the spinal cord is split later-ally into two distinct halves, or hemicords, theterm “diastematomyelia” is used. This termi-nology remains contended but, in our opinion,better describes the fact that a variable degreeof separation of the cord into two distinct struc-tures is found in these patients. Failure toneurulate one of the two hemicords producesa distinctly rare condition known ashemimyelomeningocele, which formallybelongs to the OSD category. More complexforms that involve clefting of the spinal col-umn are grouped within the split notochordsyndrome header.
• Interrupted cord: The absence of a segment ofthe cord most often involves the caudal portion(i.e., conus and lower thoracic cord) and isassociated with major degrees of
sacrococcygeal, and sometimes lumbosacralor even thoracolumbar, agenesis in the contextof the caudal regression syndrome. Much rareinstances involve segmental agenesis of anintermediate, metamerically correspondentsegment of the spine and spinal cord, which isdefined as segmental spinal dysgenesis.
• Dorsal dimples or ostia: Dermal sinuseslocated above the intergluteal crease shouldbe presumed to violate the subarachnoidspace until proven otherwise. They may beassociated with other forms of CSD such as,typically, those with a subcutaneous mass.Notably, skin pits located within theintergluteal crease are related tosacrococcygeal cysts or fistulas and usuallydo not require further diagnostic investigation(Kucera et al. 2015; Sewell et al. 2015).
Clinical Scenario and Indicationsfor Imaging
In OSD, ulceration of the placode and infectioncauses elevated mortality in untreated newborns;additionally, cerebrospinal fluid (CSF) lossthrough the defect causes intracranial hypotensionwhich eventually generates the Chiari II malfor-mation with hindbrain displacement into the cer-vical canal and, ultimately, hydrocephalus(McLone and Knepper 1989). Therefore, thesepatients require early surgical intervention that isaccomplished immediately after birth in themajority of centers. The subsequent clinical pic-ture includes a variable association of sensorimo-tor disturbances, bowel and bladder incontinence,hindbrain dysfunction, and intellectual and psy-chological disabilities. Recently, fetal surgicalrepair has proven effective in reversing hindbraindescent, reducing the incidence of shunting pro-cedures for hydrocephalus, and improving psy-chomotor outcomes; however, an increasedincidence of premature deliveries and uterinedehiscence has been reported (Adzick et al. 2011).
CSD often present with signs of “tetheredcord,”which is a clinical syndrome of progressiveneurologic deterioration (Warder and Oakes1993) composed of variably associated motor
6 A. Rossi

and sensory dysfunction, muscle atrophy,decreased or hyperactive reflexes, urinary incon-tinence, spastic gait, and orthopedic deformitiessuch as scoliosis or foot and hip deformities, thatresults from metabolic derangement caused byconfined motion of, and traction exerted onto,the spinal cord. Such traction (“tethering”) occursin the majority of spinal dysraphisms because thecord is abnormally connected to the thecal sac orsubcutaneous tissues as a consequence of abnor-mal development. After surgical correction, cordtethering often occurs as a consequence of scar-ring phenomena at the level of the surgical site. Inthese cases, the term “retethering” is used.
From a radiological perspective, cord tetheringcan be divided into “typical” and “atypical”forms.
• Typical forms: the conus medullaris terminatesin a lower than normal position (i.e., below theL2–L3 disc space level).
• Atypical forms: the conus position is normal,and tethering occurs at other levels along thelength of the spinal cord.
Not uncommonly, and especially in the contextof split cord malformations (i.e.,diastematomyelia), more than one site of cordtethering exists along the longitudinal length ofthe spinal cord.
Among presenting signs of a tethered cord,scoliosis is especially relevant, due to its signifi-cant prevalence in the general population, espe-cially adolescents (2–3%). Although the vastmajority (>80%) of scoliosis cases are idiopathic,structural causes must be ruled out as surgicalcorrection may stabilize or even reverse the pro-gression of scoliosis. In case of cord tetheringsuch as tight filum terminale or filar lipoma, sco-liosis may develop to counteract cord traction, asthe spinal cord will lodge along the concave sideof the curvature which corresponds to the shorterdistance (Barutçuoğlu et al. 2016). However, inpatients harboring a spinal dysraphism, scoliosiscan also be caused by vertebral malformations(such as hemivertebrae, butterfly vertebrae, and
others) which may involve different spinal seg-ments than the dysraphic abnormality.
Imaging Techniqueand Recommended Protocol
Imaging of the spine and spinal cord in childrensuspected of harboring spinal malformations isbest accomplished with magnetic resonance imag-ing (MRI) in the vast majority of cases, while othermodalities play a complementary role in selectedindications. MRI offers a basis for the classificationof spinal dysraphisms and is greatly helpful to boththe diagnosis and treatment planning, thanks to itsintrinsic multiplanarity, spatial and contrast resolu-tion, and lack of ionizing radiation. CT-basedmodalities have been essentially abandoned becauseof insufficient sensitivity, radiation issues, and inva-siveness (in the case of CT-myelography). In prin-ciple, CT should be reserved to the elucidation ofspecific features and should always be tailored to theminimum possible field of view so as to minimizeunnecessary radiation exposure. Ultrasound is, inthe neonatal period, an extremely useful techniqueto study the incompletely ossified spine and offers agreatly detailed visualization of the spinal cord andcaudal structures but is limited by the degree ofossification of the neural arches of the vertebralcolumns other than by individual operator expertise.
A significant issue in pediatric MRI in generalis the capability of small patients to cooperatelong and well enough to obtain quality imagingstudies. Generally speaking, children may be suf-ficiently cooperative at age 5 years, although spe-cific conditions such as acute illness orpsychomotor delay may change this. Younger orseverely ill children will typically require seda-tion, which is administered differently accordingto individual center protocols. Imaging duringspontaneous sleep with a feed-and-swaddle tech-nique is a viable option in the neonatal period andobviates the need for sedation in this age group.Availability of dedicated rooms for the prepara-tion of the patient and subsequent awakeninggreatly improves the chances of success for imag-ing small infants without sedation.
Imaging in Spine and Spinal Cord Developmental Malformations 7
Recent technical advancements regarding theMRI equipment have made a great impact on thepossibilities of spinal imaging in children as well asin older age groups. The use of multichannel phasedarray coils and applications combining multipleimages into a single full field of view has greatlyimproved the visualization of the entire spine in thesagittal plane, making it realistically possible toacquire whole-spine imaging including thecraniocervical junction and sacrococcyx in a reason-able amount of time. 1.5 Tesla (1.5 T) scannersremain the most widely available for clinical MRimaging of children. However, 3 T units are increas-ingly used in several centers. Advantages of 3 TMRI over lower-field scanners include better imagequality, thanks to a higher spatial and contrast reso-lution, and improved clinical efficiency, thanks to ahigher temporal resolution. However, the scannersare more expensive, and various artifacts caused byfield inhomogeneity, susceptibility, vascular pulsa-tion, and chemical shift are exaggerated. Spinal cordimaging remains especially challenging at 3 T,although technical adjustments may significantlycounteract these setbacks.
In patients suspected of harboring spinaldysraphisms, the MRI protocol (Table 3) shouldalways include high-resolution sagittal T1- and
T2-weighted images covering the whole spine andat least one panoramic coronal sequence; also incase of indications to the study of a specific segmentof the spine, it is useful to include a whole-spinesagittal view to obtain a panoramic appraisal, to ruleout coexisting abnormalities, and to correctly num-ber the vertebral levels. Axial sequences on eitherT1- or T2-weighted imaging are used to study spe-cific regions based on the clinical indications orfindings evidenced on sagittal images. Optimalslice thickness for these sequences should be3 mm or less. High-resolution heavilyT2-weighted images, obtainable with different tech-nical modalities (such as driven equilibrium –DRIVE, among others), provide an exquisite depic-tion of cord/root/CSF interfaces and are particularlyuseful to look for subtle structural abnormalities,such as those often found in spinal dysraphisms.Diffusion-weighted imaging sequences or adminis-tration of gadolinium-based contrast agents are usu-ally not required in patients with spinal dysraphismsbut may be helpful in select indications, such as theidentification and assessment of dysontogeneticmasses.
Table 3 Proposed MRI protocol for spinal dysraphisms
Sequence Orientation Minimum parameters Notes
Required sequences
T1W TSE Sagittal 3 mm slice thickness Entire spine
T2W TSE Sagittal 3 mm slice thickness Entire spine
T2W Coronal Fat suppression (i.e.,STIR or equivalent)
Entire spine (including panoramic paravertebral regionevaluation)
T2 DRIVE(or equivalent)
Sagittal 0.6 mm partitionthickness
3D isotropic voxel centered on suspected abnormalityReformat into axial and coronal planes
T1 TSE Axial 3–4 mm slicethickness
Conus/cauda region
Optional sequences
T2 TSE Axial 3–4 mm slicethickness
Across area of spinal cord abnormality
T1 TSE Coronal 3 mm slice thickness Centered onto, and oriented along major axis of, thesacrum (in case of suspected sacral abnormality)
T1 Sagittal Fat suppression Confirmation of suspected lipoma
DWI Sagittal oraxial
3–4 mm slicethickness
In case of suspected dysontogenetic abnormality (i.e.,epidermoid, etc.)
PostcontrastT1 TSE
Sagittal, axial,and/or coronal
3 mm slice thickness In case of suspected dysontogenetic abnormality (i.e.,epidermoid, etc.) or other mass lesion esp. presacral
8 A. Rossi

Principal Entities
Myelomeningocele
Myelomeningocele is, by far, the most commonform of OSD and is characterized by exposure ofthe neural placode through a midline defect in theback with expansion of the underlying subarach-noid space, which results in extrusion of theplacode above the surrounding cutaneous surface.A myelomeningocele results from absent neuru-lation affecting a segment of the primary neuraltube that typically involves the lumbosacralregion and, more rarely, the thoracic or cervicalregions. An extensive form of open spinal defectin which neurulation is almost completely defec-tive along the entire length of the neural tube iscalled craniorachischisis; it is incompatible withlife. The absent neurulation results in a segment ofthe neural placode that persists in between theunfused edges of the cutaneous ectoderm,explaining the necessary midline skin defect.Therefore, the external surface of the placode,which corresponds to the would-be ependymalsurface of the future spinal cord, is directly visibleon inspection and is surrounded by a translucent,vascularized meningeal membrane called the zonamedullovasculosa. Failed separation between theneural and the cutaneous ectoderms also impairsmesenchymal migration behind the neural tube,causing a defect of the posterior musculoskeletalstructures. The ventral surface of the placode,facing the spinal canal, is in fact the would-beexternal surface of the spinal cord from whichthe nerve roots originate. These nerve roots areusually shorter than normal and course obliquelythrough the subarachnoid space to reach theircorresponding neural foramina.
Fetal MRI studies show a meningocele pro-truding posteriorly into the amniotic sac, withthe spinal cord entering the defect. The surround-ing skin and subcutaneous tissues appearinterrupted; the Chiari II malformation is alsowell depicted on prenatal studies. On postnatalMRI (Fig. 1), the extent and size of the defect,the abnormal course of the spinal cord, and theexposed placode are adequately depicted. High-resolution T2-weighted sequences also identify
the spinal nerve roots that originate from the ven-tral surface of the placode and course anteriorlythrough the meningocele and into the widelydilated subarachnoid spaces of the spinal canal.The subcutaneous fat surrounding the defect isdehiscent. There is a marked individual variabilityas to the size of the posterior defect, which mayinvolve one or several vertebral metameres. Theplacode may be located at the caudal end of thespinal cord (i.e., terminal) or along an intermedi-ate segment (i.e., segmental) below which thecord regains a normal configuration and structure.
After surgery is performed, long-term follow-up is required to assess the various possible com-plications. The evaluation of ventricular size isobviously paramount; in case of hydrocephalus,a ventriculoperitoneal shunt often needs to beplaced. Subsequent deterioration of a previouslystable neurological function may be caused byretethering of the spinal cord to the posteriorwall of the thecal sac due to scarring. Other com-plications that may be recognized with MRIinclude dysontogenetic masses (mostly epider-moids or dermoids) which originate from inadver-tent inclusion of cutaneous ectoderm duringsurgical closure of the defect, hydrosyrin-gomyelia, and scoliosis. Long-term cordretethering often results in a complex picture char-acterized by severe scoliosis, marked thinning ofthe spinal cord, and quasi-holocord syringes.
Myelocele and Myeloschisis
In these rare variants, the subarachnoid spaces arenot expanded, and neural placode is not elevatedabove the skin surface; instead, the placode isflushed with the surrounding skin surface(myelocele) or depressed (myeloschisis). Becauseof the rarity of these conditions and the qualitativenature of their definition, the two terms are oftenused synonymously (Fig. 2). The association withthe Chiari II malformation and the hydrocephalusis equivalent as in myelomeningoceles, and thesurgical and management issues are not dissimi-lar. In fact, the myelomeningocele, myelocele,and myeloschisis should be regarded as a contin-uous spectrum of abnormality in which the sole
Imaging in Spine and Spinal Cord Developmental Malformations 9

discriminant is the degree of expansion (or lackthereof) of the subarachnoid spaces, which in turndepends on the extent and integrity (or lackthereof) of the zone medullovasculosa that sur-rounds the placode.
Hemimyelomeningocele
A hemimyelomeningocele is an exceedingly rarecondition in which splitting of the spinal cord (i.e.,diastematomyelia) is complicated by failed neu-rulation of one hemicord. The degree of
expansion of the subarachnoid spaces generatesa variable picture of extrusion of the hemi-placode, similar to what occurs in themyelomeningocele-myelocele-myeloschisissequence. Clinically, neurological impairment issimilar to that seen in patients withdiastematomyelia but is markedly asymmetric.On visual inspection, a hairy tuft along one sideof an exposed placode is a strong indicator.Embryologically, the abnormality is explainedby failed midline notochordal integration (produc-ing the split cord) with superimposed failure ofprimary neurulation of one hemicord.
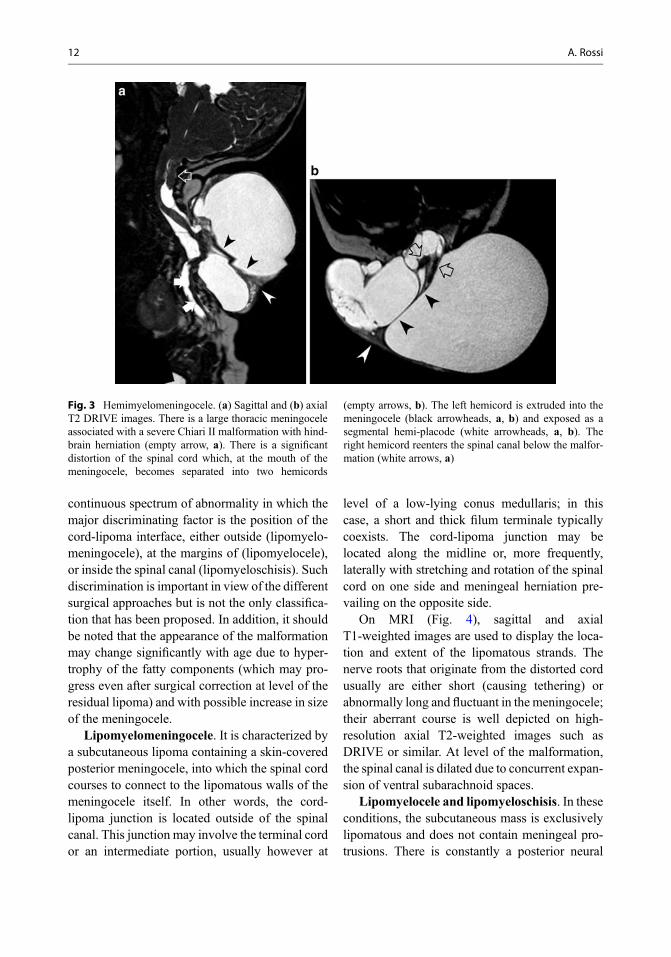
Fig. 1 Myelomeningocele.(a) Sagittal T1-weighedimage and (b) sagittal T2DRIVE image. There is alumbosacral defect withexposure of a segmentalplacode (arrowheads). Theplacode is superelevateddue to expansion of thesubarachnoid spaces ventralto it (asterisks). Notice focalkyphosis at level of thedefect. There is a concurrentsevere Chiari IImalformation withhindbrain herniation(arrow, b)
10 A. Rossi

On MRI, the cord splitting is variably elon-gated; it may be difficult to recognize withoutthe use of high-resolution axial T2-weightedimages. A septum dividing the dural tube intotwo halves may or may not be present. Theextruded hemicord is often asymmetricallylocated within the meningocele, whose volumeis variable in individual cases (Fig. 3).
Lipomas with Dural Defect:Lipomyelomeningocele,Lipomyelocele, and Lipomyeloschisis
Lipomas with defective dura are characterized bya usually large subcutaneous mass composed offatty tissue and located above the interglutealcrease, often extending asymmetrically into onebuttock. Because the mass is clinically evident atbirth, the diagnosis is usually made early, andsurgical correction may thus be performed beforesignificant neurological deterioration ensues. His-tologically, the mass is composed of clusters ofmature adipocytes separated by collagenousbands, usually intermingled with strands of differ-ent tissues including striated muscle, cartilage,bone, nerve cells, ependyma, and aberrant neuro-glial tissue; thus, these lipomas are indeed betterdescribed as mature teratomas. Sometimes well-defined structures, such as aberrant ribs, aredescribed within the substance of the lipoma.Congenital lipomas may increase in size as anexpression of the normal increase of adipose tis-sue throughout childhood and especially so inearly infancy or near puberty other than in partic-ular conditions such as obesity or pregnancy.
The embryological origin of spinal lipomas isdebated. One leading theory invokes defectiveprimary neurulation with focal premature disjunc-tion of the cutaneous ectoderm from theneuroectoderm, which in turn allows the mesen-chyme to access the interior of the neural tube,contact the ependymal lining, and develop intoadipose tissue. This theory, which requires thepresence of a neural placode (i.e., an area ofincomplete neurulation of the spinal cord), hasnot been conclusively proven experimentally (Liet al. 2001); in addition, spinal lipomas intermin-gle with the outer surface of an otherwise nor-mally neurulated spinal cord, as shown by thepresence of an ependymal canal at level of abnor-mality. This observation suggests that spinal lipo-mas, rather than abnormalities of primaryneurulation, should be considered as defects ofthe induction of the dorsal mesoderm by the roofplate of the neural tube.
It is noteworthy that, quite like in OSDs, lipo-mas with dural defect should be viewed as a
Fig. 2 Myelocele/myeloschisis. Sagittal T2-weightedimage. There is a large thoracolumbar defect with exposureof the placode (arrowheads). Notice that the placode is notelevated above the surrounding skin and is in fact partlydepressed, due to insufficient expansion of the ventralsubarachnoid spaces. Notice severe Chiari II malformationwith hindbrain herniation (arrow)
Imaging in Spine and Spinal Cord Developmental Malformations 11
continuous spectrum of abnormality in which themajor discriminating factor is the position of thecord-lipoma interface, either outside (lipomyelo-meningocele), at the margins of (lipomyelocele),or inside the spinal canal (lipomyeloschisis). Suchdiscrimination is important in view of the differentsurgical approaches but is not the only classifica-tion that has been proposed. In addition, it shouldbe noted that the appearance of the malformationmay change significantly with age due to hyper-trophy of the fatty components (which may pro-gress even after surgical correction at level of theresidual lipoma) and with possible increase in sizeof the meningocele.
Lipomyelomeningocele. It is characterized bya subcutaneous lipoma containing a skin-coveredposterior meningocele, into which the spinal cordcourses to connect to the lipomatous walls of themeningocele itself. In other words, the cord-lipoma junction is located outside of the spinalcanal. This junction may involve the terminal cordor an intermediate portion, usually however at
level of a low-lying conus medullaris; in thiscase, a short and thick filum terminale typicallycoexists. The cord-lipoma junction may belocated along the midline or, more frequently,laterally with stretching and rotation of the spinalcord on one side and meningeal herniation pre-vailing on the opposite side.
On MRI (Fig. 4), sagittal and axialT1-weighted images are used to display the loca-tion and extent of the lipomatous strands. Thenerve roots that originate from the distorted cordusually are either short (causing tethering) orabnormally long and fluctuant in the meningocele;their aberrant course is well depicted on high-resolution axial T2-weighted images such asDRIVE or similar. At level of the malformation,the spinal canal is dilated due to concurrent expan-sion of ventral subarachnoid spaces.
Lipomyelocele and lipomyeloschisis. In theseconditions, the subcutaneous mass is exclusivelylipomatous and does not contain meningeal pro-trusions. There is constantly a posterior neural
Fig. 3 Hemimyelomeningocele. (a) Sagittal and (b) axialT2 DRIVE images. There is a large thoracic meningoceleassociated with a severe Chiari II malformation with hind-brain herniation (empty arrow, a). There is a significantdistortion of the spinal cord which, at the mouth of themeningocele, becomes separated into two hemicords
(empty arrows, b). The left hemicord is extruded into themeningocele (black arrowheads, a, b) and exposed as asegmental hemi-placode (white arrowheads, a, b). Theright hemicord reenters the spinal canal below the malfor-mation (white arrows, a)
12 A. Rossi

arch defect where the lipoma penetrates the spinalcanal; thus, the cord-lipoma interface lies posteri-orly either at level of the adjacent neural arches(i.e., lipomyelocele) or deep within the spinalcanal (i.e., lipomyeloschisis). The latter conditionis more frequent, but owing to the fact that signif-icant stretching of the cord may exist and thelength of the cord-lipoma interface may spanover several vertebral levels, the two conditionsare not clearly separable from one another, and thetwo terms are often used interchangeably (quitelike myelocele and myeloschisis, see above).
On MRI (Fig. 5), T1-weighted images typi-cally show a continuity of adipose tissue thatextends from the subcutaneous mass into the spi-nal canal through a variably sized posterior neuralarch defect. The position of the dural defectthrough which the lipoma communicates withthe spinal canal is often posterior and large, butthe size and location may vary (odd cases being
located laterally), and it may extend over severalvertebral levels. The caliber of the spinal canalmay be increased depending on the size of thelipoma, but the size of the subarachnoid spaceventral to the cord is consistently not expandedand is in fact often reduced due to anterior dis-placement of the spinal cord due to mass effectgenerated by the lipoma. The interface betweencord and lipoma is better resolved by the use ofhigh-resolution axial T2-weighted images; in themajority of cases, this interface is irregular, andthe cord is significantly distorted.
Myelocystocele
A myelocystocele is a rare condition defined asthe herniation of a hydrosyringomyelic portion ofthe spinal cord into a posterior meningocele. Inthe majority of these quite rare cases, the
Fig. 4 Lipomyelomeningocele. (a) Sagittal T1-weightedimage, (b) sagittal, and (c) axial T2 DRIVE images. Thespinal cord is low-lying and enters a meningocele (asterisk,a, b). There is a concurrent subcutaneous lipoma to which
the cord attaches (arrowhead, c). Thus, the cord-lipomainterface is located outside the spinal canal. Notice theenlargement of the subarachnoid spaces at the lumbosacrallevel ventral to the cord
Imaging in Spine and Spinal Cord Developmental Malformations 13

condition involves the terminal portion of the cordat the lumbosacral level and, as such, is defined asterminal myelocystocele. Other rare cases involvean intermediate portion of the spinal cord andhave been called nonterminal myelocystoceles(Rossi et al. 2006). Regardless of location, thecommon feature is a herniated syrinx (the“syringocele”) into a posterior meningocelethrough a posterior neural arch defect, while thesyringocele and meningocele do not communicatewith each other.
Terminal myelocystoceles are frequentlyfound in the context of the OEIS association(Omphalocele, Exstrophy of the cloaca, Imperfo-rate anus, and Spinal anomalies), and affectedpatients typically have a quite severe neurologicalpicture with poor bowel or bladder control andlower extremity dysfunctions. Embryologically,they are classically viewed as abnormalities of
secondary neurulation and, specifically, of theprocess of canalization with persistence of a (usu-ally) cavitated secondary neural tube that balloonsinto a cyst and disrupts the overlyingmesenchyme.
On MRI (Fig. 6), terminal myelocystocelesappear as cystic lesions involving the lumbosacralregion, in which the terminal portion of alow-lying spinal cord displays a trumpet-like flar-ing of the central canal (“syringocele”) whilecoursing within a posterior meningocele to attachto its dome.
Nonterminal myelocystoceles are character-ized by a posterior meningocele located alongthe cervicothoracic spine and present in a rangeof variants that include (Rossi et al. 2006):
• Complete forms: a localized hydrosyrin-gomyelia balloons out into the meningocele
Fig. 5 Lipomyelocele/lipomyeloschisis. (a) SagittalT1-weighted image and (b) axial T2 DRIVE image.There is a large subcutaneous lumbosacral lipoma thatpenetrates the spinal canal through a posterior neural arch
defect, to connect to a low-lying spinal cord. The cord-lipoma interface (arrowheads, b) is located within thespinal canal, and the ventral subarachnoid spaces are notexpanded
14 A. Rossi

• Abortive forms: a fibroneural stalk that origi-nates from the posterior aspect of the spinalcord traverses posteriorly into the meningoceleto connect to its dome
Embryologically, they are believed to resultfrom a phenomenon described as “limited dorsalmyeloschisis” (Pang et al. 2013); this is a limitedarea of incomplete neurulation that leaves behinda connection between the roof plate of the spinalcord and the overlying ectoderm through whichposterior ballooning occurs while not hamperingclosure of the cutaneous ectoderm.
On imaging (Fig. 7), abortive variants present aneural stalk emanating from the dorsal aspect ofthe spinal cord, crossing a narrow posterior bonyspina bifida, and coursing through a posteriormeningocele to attach to the inner aspect of itsdome; complete forms show dissection of ahydromyelic cavity into the stalk, converting itinto a second “cyst” within the meningocele
(Rossi et al. 2006). Concurrent anomalies includefocal hydromyelia immediately cranial to the ori-gin of the posterior stalk and mild forms of ChiariII malformation.
Meningocele
Meningoceles are extraspinal herniations ofCSF-filled sacs lined by dura and arachnoid that,by definition, do not contain any segment of thespinal cord. In the context of a CSD with subcu-taneous mass, the so-called posteriormeningoceles originate from meningeal hernia-tions through a posterior bony spina bifida andare surmounted by a continuous skin coverage;cutaneous dystrophy, hemangiomas, or tail-likeprotrusions are frequently associated. Theirembryogenetic origin is unascertained, but theymight result from failure of detachment of themeninx primitiva from the cutaneous ectoderm,
Fig. 6 Terminal myelocystocele. (a) Sagittal T2-weightedand (b) axial T1-weighted image. The spinal cord islow-lying and enters a meningocele. The terminal portion
of the cord is largely distended by a hydromyelic cavity, thesyringocele (asterisks, a, b); the distended walls of the cord(asterisk) form a trumpet-shaped expansion
Imaging in Spine and Spinal Cord Developmental Malformations 15

resulting in a posterior meningeal extrusion thatballoons out through a posterior spina bifida withrelentless CSF pulsations. Most posteriormeningoceles are lumbar or sacral in location,but cervicothoracic meningoceles may be found.By definition, the spinal cord is not extruded,although it may be tethered to the neck of themeningocele.
On imaging (Fig. 8), the contents of the sac areisointense with CSF; high-resolution T2-weightedimages display the possible presence of redundantnerve roots within the sac. The presence of teth-ering bands that connect the cord to the innersurface of the meningocele dome, also welldepicted on high-resolution T2-weighted images,defines the abnormality as an abortivemyelocystocele (see below). It has been specu-lated that true meningoceles, lacking whateverconnection with the spinal cord, are exceedinglyrare (Pang et al. 2013).
Intradural Lipoma
Intradural lipomas differ from lipomas with duraldefects in that they are contained within an intactdural sac in a subpial location. They lie along themidline in contact with the dorsal surface of thespinal cord and may bulge posteriorly in the sub-arachnoid spaces elevating the pia mater. Largelipomas may displace the cord laterally, resultingin an off-midline cord-lipoma interface. In rareinstances, lipomas infiltrate the cord extensively,so that an interface with the spinal cord is noteasily identified. Intradural lipomas are com-monly located at lumbosacral level and usuallypresent with signs of cord tethering, whereascervicothoracic lipomas generally produce insidi-ous signs of spinal cord compression.
OnMRI (Fig. 9), lipomas appear as masses thatare isointense to fat in all sequences, includingthose acquired with fat suppression techniques.There is a clear separation between the intraspinal
Fig. 7 Nonterminal myelocystocele. (a) Sagittal T2DRIVE sequence. In this complete form, a neural stalkemanating from the dorsal aspect of the spinal cord (thinwhite arrow) penetrates a large posterior meningocele andis dissected by hydromyelia (arrowhead); the conusmedullaris is low (thick white arrow) and not involved in
the extrusion. (b) Sagittal T2 DRIVE sequence. In thisabortive form, the neural stalk originates from the posterioraspect of the spinal cord (white arrow) and courses within ameningocele (black thin arrows) to connect to its dome.Again, the terminal portion of the cord, albeit low (thickarrow), is not directly involved
16 A. Rossi

lipoma and the adipose tissue in the subcutaneouscompartment.
Filar Lipoma
The occurrence of incidental fat within the filumterminale in the normal adult population is esti-mated to be 1.5–5% in unselected MRI studies. Afilar lipoma is properly described when a short-ened, thickened, and lipomatous filum terminaleis observed, frequently in association with alow-lying conus medullaris (i.e., below themid-L2 level). Embryologically, abnormal retro-gressive differentiation with persistence of cellscapable of maturing into adipocytes is a likelymechanism.
MRI detects fatty tissue within a thickenedfilum terminale as a stripe of increased signal
intensity on T1-weighted images (Fig. 10). Insome cases, the lipoma may extend to the poste-rior aspect of the conus medullaris along the entirelength of the embryological secondary neuraltube.
Tight Filum Terminale
The tight filum terminale is characterized by ashort, hypertrophic filum terminale that producescord tethering and impaired ascent of the conusmedullaris, resulting in a low position of the conusmedullaris, i.e., below the mid-L2 level. Isolatedcases are uncommon, whereas the abnormality ismore frequent in patients with othermalformations, such as diastematomyelia, spinallipomas, or dermal sinus tracts. Embryologically,the tight filum terminale is due to abnormal retro-gressive differentiation of the secondary neuraltube, producing a thicker and shorter filum.
On MRI (Fig. 11), the principal abnormalityconsists in a low position of the conus medullaris,which may also appear stretched and adherent tothe posterior aspect of the thecal sac. The filumterminale should not exceed 2 mm in diameter innormal individuals, but the exact thickness of thefilum terminale may be difficult to measure evenwhen heavily T2-weighted sequences such asDRIVE are used. In patients with neurologicalsigns compatible with a tethered cord syndromebut without a significantly low position of theconus, MRI sequences acquired in the prone posi-tion have been proposed as a useful adjunct,showing lack of anterior gravity displacement ofthe filum in case of occult tethering (Nakanishiet al. 2013); however, prone positioning can bedifficult to achieve in uncooperative or sedatedpatients, and its real usefulness in the routineclinical practice has been questioned (Singhet al. 2012).
Persistent Secondary Neural Tube
This abnormality, also denominated the “abnor-mally elongated spinal cord” or “retained medul-lary cord,” is embryologically related to a
Fig. 8 Meningocele. Sagittal T2-weighted image. There isa posterior thoracolumbar meningeal outpouching filledwith CSF. The spinal cord is normal and is not connectedto or attracted toward the neck of the meningocele
Imaging in Spine and Spinal Cord Developmental Malformations 17

complete lack of retrogressive differentiation ofthe secondary neural tube. It may occur in isola-tion or in association to other CSDs, such asdermal sinuses or intradural lipomas, and is clin-ically associated with signs of cord tethering.
On imaging (Fig. 12), there is an absence of thenormal taper of the conus medullaris, and thespinal cord does not show significant changes incaliber down to the sacrum, forming a robustelongated structure that connects to the bottomof the thecal sac.
Persistent Terminal Ventricle
The persistent terminal ventricle appears as anovoid cyst-like cavity located within the conusmedullaris, occupying a central position and fre-quently bulging either anteriorly or posteriorly.The fluid filling the cyst is isointense with CSFin all MR sequences and does not enhance withgadolinium chelate administration. High-
resolution T2-weighted sequences (i.e., DRIVEor similar) are especially suited to depictit. Differentiation from hydrosyringomyelia isbased on location at the conus, whereasintramedullary tumors are ruled out by theabsence of gadolinium enhancement. The size ofthe “cyst” usually remains unchanged on follow-up scans but may rarely increase when a ball valvemechanism allows for entrance, but not exit, ofCSF from the ependymal canal above theventricle (Fig. 13).
Split Notochord Syndrome
The split notochord syndrome is a severe, albeitrare, condition caused by failed midline noto-chordal integration that occurs during gastrula-tion. Separation of the notochord into twohalves, perhaps caused by persistence of theneurenteric canal, causes a segmental cleft of thespine through which intestinal segments extend
Fig. 9 Intradural lipoma. (a) Sagittal and (b) axialT1-weighted images. There is a large cervicomedullarylipomatous mass in an intradural location, displacing and
compressing the spinal cord (arrowheads, b). The lipoma isentirely intraspinal, with no connection with thesubcutaneous fat
18 A. Rossi

into the spinal canal. Complete (dorsal entericfistula) and localized (neurenteric cysts) variantsexist.
Dorsal enteric fistula. In this exceedingly rarecondition, a fistulous tract comprising gastrointes-tinal or respiratory epithelium forms a structurethat traverses the prevertebral soft tissue, the ver-tebral bodies, and the spinal canal with its con-tents; sometimes it may become exteriorizedposteriorly through an open spinal defect, produc-ing a complete gastrointestinal evisceration.
On MRI (Fig. 14), a dorsal enteric fistula givesa complex picture that basically involves the asso-ciation of a spinal cleft with the presence of atubular structure that extends from the intraspinalcompartment (often in association with a cordsplitting) into the chest or abdomen. Splitting ofthe spinal cord (i.e., a diastematomyelia) can bevariably associated.
Neurenteric cysts. These are related to endo-dermal differentiation of primitive streak rem-nants that remain trapped between a splitnotochord; as such, they may be viewed as alocalized form of dorsal enteric fistula, in whichany portion of the tract involutes or becomesfibrous, leading to a fistula or a cyst. They arelined with a mucin-secreting, cuboidal, or colum-nar epithelium that resembles the alimentary tract.
OnMRI (Fig. 15), neurenteric cysts are usuallyfound in an intradural cervicothoracic locationanterior to the cord, usually in close connectionwith vertebral abnormalities; however, they mayalso be found in the lumbar spine and even in theposterior fossa. They appear as cystic structuresthat are isointense to hyperintense relative to CSFon both T1- and T2-weighted images, dependingon their protein content.
Fig. 10 Filar lipoma. (a) Sagittal and (b) axialT1-weighted images. There is a thickened and short filumterminale that is hyperintense consistent with a fatty
infiltration (arrowheads). The conus medullaris is stretchedand terminates in a low position at L4
Imaging in Spine and Spinal Cord Developmental Malformations 19
Diastematomyelia
The term diastematomyelia refers to a variablyelongated separation of the spinal cord in twousually, but not necessarily, symmetric halves.Diastematomyelia may be best considered as acontinuous spectrum of abnormality, rangingfrom a completely duplicated cord with four col-umns and segmental roots to forme fruste with apartially cleft cord contained within a single duraltube. The term “split cord malformations” hasbeen suggested to describe this continuum. Weprefer the traditional denomination, which hasthe advantage of being widely recognized andaccepted.
Embryologically, failed midline integration ofthe notochord results into two paired notochordalprocesses separated by intervening primitivestreak cells. Each heminotochord induces a
separate hemineural plate and, as such, the forma-tion of two “hemicords.” The latter term is some-what misleading, in that each hemicord presents arelatively better preserved lateral aspect fromwhich a single pair of anterior and posteriornerve roots emerge, and a more dysplastic medialaspect which may be devoid of roots or originateaberrant roots that course haphazardly and mayproduce cord tethering. As such,diastematomyelia as a whole may be viewed as“incompletely duplicated cord,” rather than “splitcord,” malformations. The resulting abnormalityessentially depends on the developmental fate ofthe intervening primitive streak tissue, which maypersist and develop into the bone or instead bereabsorbed and disappear. This represents thefoundation of the classification into two groups.
Fig. 11 Tight filum terminale. Sagittal T2 DRIVE image.The filum terminale (arrowheads) is short and tethers thecord into a low position at L3–L4. There is a concurrentsyrinx, which disappeared after surgical detethering (notshown)
Fig. 12 Persistent secondary neural tube. Sagittal T2DRIVE image. The spinal cord extends downward withouta significant taper to connect to the bottom of the thecal sac
20 A. Rossi
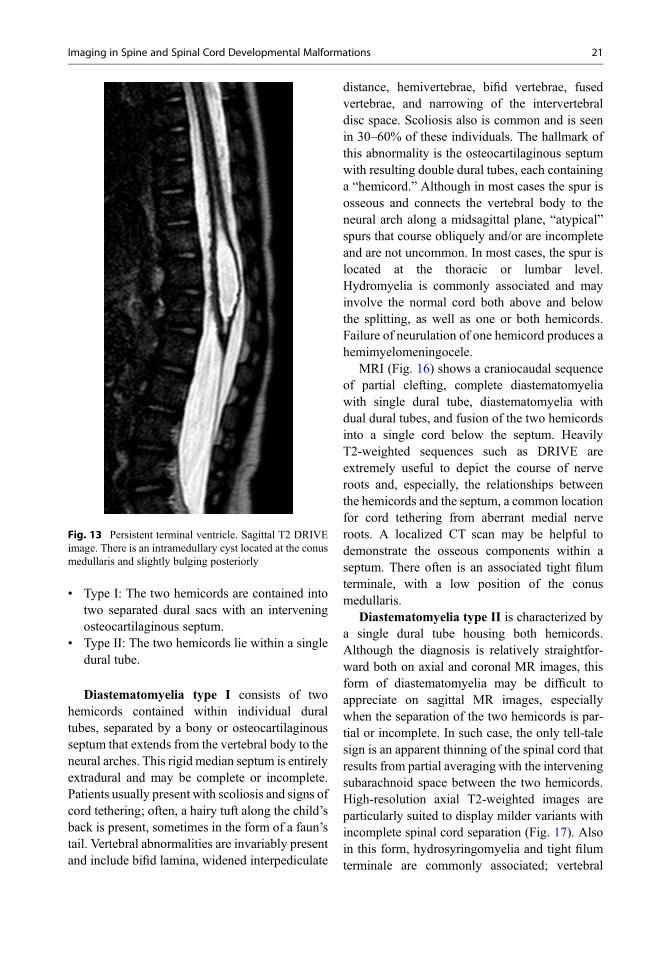
• Type I: The two hemicords are contained intotwo separated dural sacs with an interveningosteocartilaginous septum.
• Type II: The two hemicords lie within a singledural tube.
Diastematomyelia type I consists of twohemicords contained within individual duraltubes, separated by a bony or osteocartilaginousseptum that extends from the vertebral body to theneural arches. This rigid median septum is entirelyextradural and may be complete or incomplete.Patients usually present with scoliosis and signs ofcord tethering; often, a hairy tuft along the child’sback is present, sometimes in the form of a faun’stail. Vertebral abnormalities are invariably presentand include bifid lamina, widened interpediculate
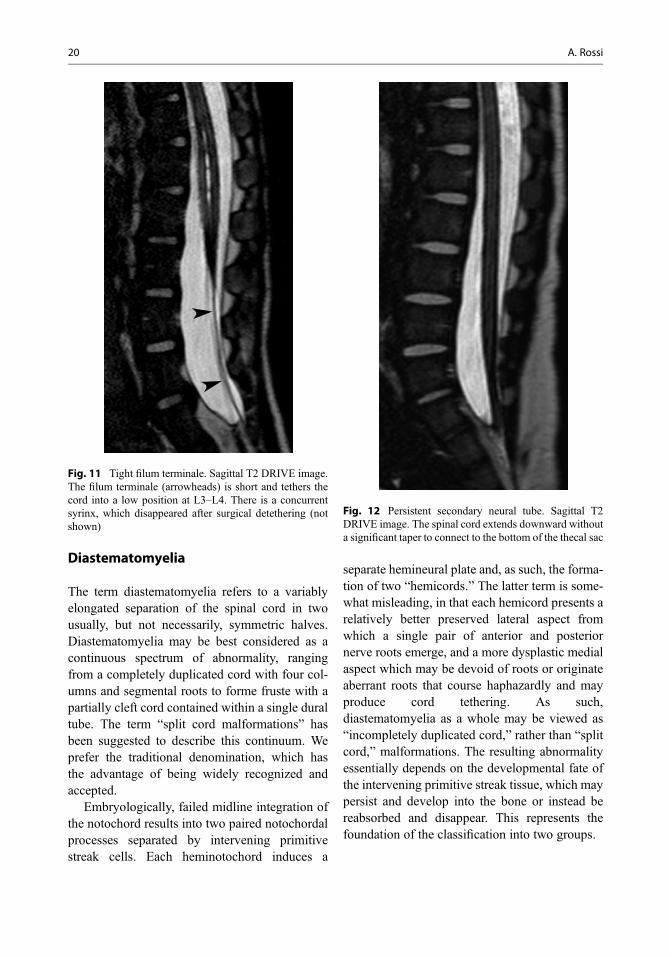
distance, hemivertebrae, bifid vertebrae, fusedvertebrae, and narrowing of the intervertebraldisc space. Scoliosis also is common and is seenin 30–60% of these individuals. The hallmark ofthis abnormality is the osteocartilaginous septumwith resulting double dural tubes, each containinga “hemicord.” Although in most cases the spur isosseous and connects the vertebral body to theneural arch along a midsagittal plane, “atypical”spurs that course obliquely and/or are incompleteand are not uncommon. In most cases, the spur islocated at the thoracic or lumbar level.Hydromyelia is commonly associated and mayinvolve the normal cord both above and belowthe splitting, as well as one or both hemicords.Failure of neurulation of one hemicord produces ahemimyelomeningocele.
MRI (Fig. 16) shows a craniocaudal sequenceof partial clefting, complete diastematomyeliawith single dural tube, diastematomyelia withdual dural tubes, and fusion of the two hemicordsinto a single cord below the septum. HeavilyT2-weighted sequences such as DRIVE areextremely useful to depict the course of nerveroots and, especially, the relationships betweenthe hemicords and the septum, a common locationfor cord tethering from aberrant medial nerveroots. A localized CT scan may be helpful todemonstrate the osseous components within aseptum. There often is an associated tight filumterminale, with a low position of the conusmedullaris.
Diastematomyelia type II is characterized bya single dural tube housing both hemicords.Although the diagnosis is relatively straightfor-ward both on axial and coronal MR images, thisform of diastematomyelia may be difficult toappreciate on sagittal MR images, especiallywhen the separation of the two hemicords is par-tial or incomplete. In such case, the only tell-talesign is an apparent thinning of the spinal cord thatresults from partial averaging with the interveningsubarachnoid space between the two hemicords.High-resolution axial T2-weighted images areparticularly suited to display milder variants withincomplete spinal cord separation (Fig. 17). Alsoin this form, hydrosyringomyelia and tight filumterminale are commonly associated; vertebral
Fig. 13 Persistent terminal ventricle. Sagittal T2 DRIVEimage. There is an intramedullary cyst located at the conusmedullaris and slightly bulging posteriorly
Imaging in Spine and Spinal Cord Developmental Malformations 21

Fig. 14 Dorsal enteric fistula. (a) Sagittal, (b) coronal,and (c) axial T2 DRIVE images; (d) contrast-enhancedaxial T1-weighted image; (e) coronal reformatted; and (f)3D CT images. There is a cystic formation (asterisk, a–c)that communicates through an anterior spinal cleft (whitearrow, c) with a large pluriconcamerated mass (empty
arrows, a, b, d) whose enhancing walls were composedof gastrointestinal epithelium consistent with gut duplica-tion (surgical-pathological report). There is a concurrentdiastematomyelia (arrowheads, b, c). CTscans show spinalduplication with multiple vertebral malformations
22 A. Rossi

anomalies are usually milder than in the type I andsometimes limited to a bifid spinous process.
Caudal Regression Syndrome
The caudal regression syndrome (CRS) comprisesa constellation of abnormalities that include totalor partial agenesis of the spinal column, analimperforation, genital anomalies, bilateral renaldysplasia or aplasia, pulmonary hypoplasia, andlower limb abnormalities. The most severe variantis sirenomelia, characterized by extreme hypopla-sia of the lower limbs with variable degrees ofnonseparation. Etymologically, the term CRSimplies a concept of excessive regression of theembryonic tail that cannot be adequately appliedin tail-less animals, such as humans (M. Catala,personal communication 2004); therefore, it is arelatively inappropriate term that could bereplaced by “caudal dysgenesis syndrome”; how-ever, its use is so widespread that it is probablybest retained. There is a definite association of
CRS with maternal diabetes mellitus (1% of off-spring of diabetic mothers). CRS may be part ofsyndromic complexes such as:
• OEIS (Omphalocele, Exstrophy of the cloaca,Imperforate anus, and Spinal deformities)
• VACTERL (Vertebral abnormality, Analimperforation, Cardiac malformations,Tracheoesophageal fistula, Renal abnormali-ties, Limb deformities)
• The Currarino syndrome is a particular formcaused, in some cases, by heterozygous muta-tion in the HLXB9 homeobox gene on chro-mosome 7q36 and consists of a triad of:• Sacral agenesis with asymmetric
hemisacrum and intact first sacral vertebra(“sickle-shaped sacrum”)
• A presacral mass – anterior meningoceleand/or teratoma
• Anorectal malformation
Fig. 15 Neurenteric cyst. (a) Sagittal T2-weighted imageand (b) post-contrast sagittal T1-weighted image. There isan intradural cyst (asterisk) causing spinal cord
compression at the C2–C4 level; the cyst is unenhancingand isointense to CSF in both sequences
Imaging in Spine and Spinal Cord Developmental Malformations 23

Patients with the Currarino triad are usuallyolder children or adults complaining withlow-back pain, urinary incontinence, or constipa-tion, in whom an MRI study reveals a presacral
CSF-filled mass that is associated with partialsacrococcygeal agenesis. Solid, nodularteratomatous components are often located alongthe walls of the meningocele.
Fig. 16 Diastematomyelia type I. (a) SagittalT1-weighted image; (b) sagittal and (c) axial T2 DRIVEimages; (d) axial CT scan. The spinal cord has a lowtermination with persistence of the secondary neural tube(arrow, a, b). There is an incomplete septum (arrowheads,
a–c) projecting anteriorly from an abnormal neural archand separating the dural tube into two portions, eachcontaining a hemicord (arrows, c). The septum has abony composition (arrowhead, d). Notice concurrenthydrosyringomyelia cranially (a, b)
24 A. Rossi
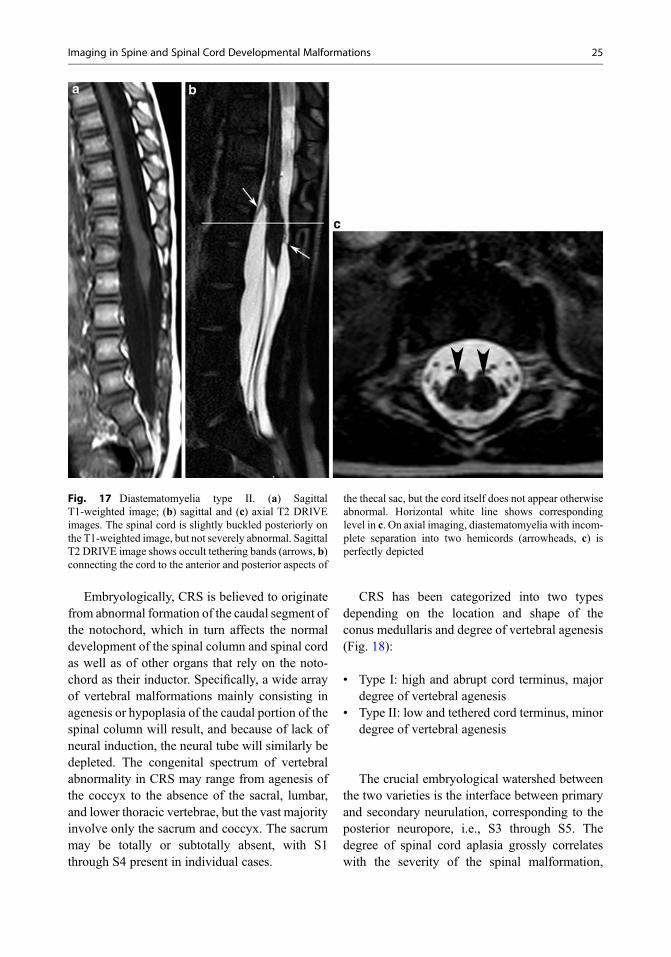
Embryologically, CRS is believed to originatefrom abnormal formation of the caudal segment ofthe notochord, which in turn affects the normaldevelopment of the spinal column and spinal cordas well as of other organs that rely on the noto-chord as their inductor. Specifically, a wide arrayof vertebral malformations mainly consisting inagenesis or hypoplasia of the caudal portion of thespinal column will result, and because of lack ofneural induction, the neural tube will similarly bedepleted. The congenital spectrum of vertebralabnormality in CRS may range from agenesis ofthe coccyx to the absence of the sacral, lumbar,and lower thoracic vertebrae, but the vast majorityinvolve only the sacrum and coccyx. The sacrummay be totally or subtotally absent, with S1through S4 present in individual cases.
CRS has been categorized into two typesdepending on the location and shape of theconus medullaris and degree of vertebral agenesis(Fig. 18):
• Type I: high and abrupt cord terminus, majordegree of vertebral agenesis
• Type II: low and tethered cord terminus, minordegree of vertebral agenesis
The crucial embryological watershed betweenthe two varieties is the interface between primaryand secondary neurulation, corresponding to theposterior neuropore, i.e., S3 through S5. Thedegree of spinal cord aplasia grossly correlateswith the severity of the spinal malformation,
Fig. 17 Diastematomyelia type II. (a) SagittalT1-weighted image; (b) sagittal and (c) axial T2 DRIVEimages. The spinal cord is slightly buckled posteriorly onthe T1-weighted image, but not severely abnormal. SagittalT2 DRIVE image shows occult tethering bands (arrows, b)connecting the cord to the anterior and posterior aspects of
the thecal sac, but the cord itself does not appear otherwiseabnormal. Horizontal white line shows correspondinglevel in c. On axial imaging, diastematomyelia with incom-plete separation into two hemicords (arrowheads, c) isperfectly depicted
Imaging in Spine and Spinal Cord Developmental Malformations 25
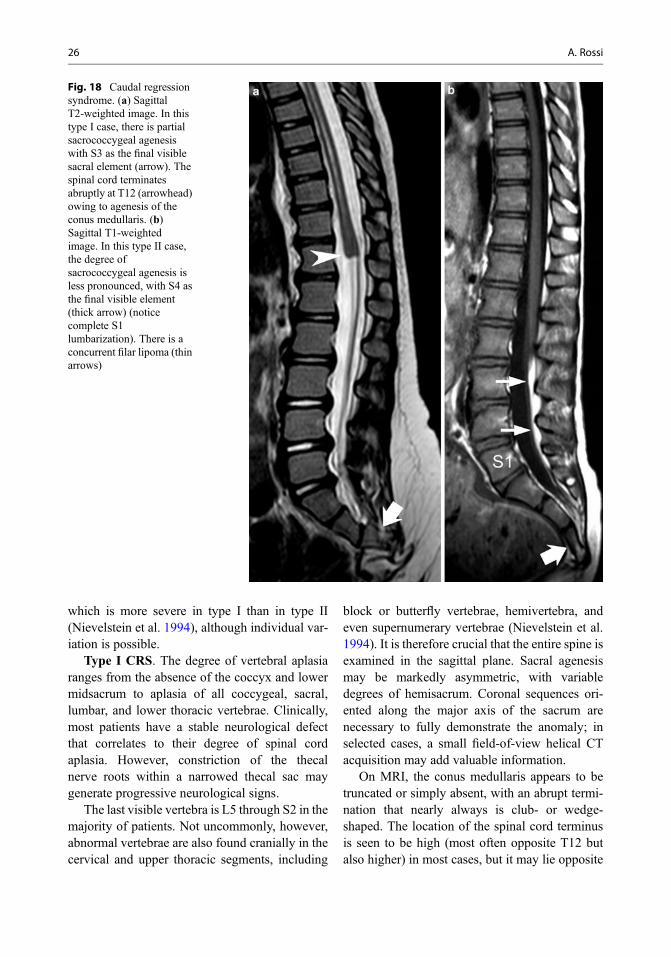
which is more severe in type I than in type II(Nievelstein et al. 1994), although individual var-iation is possible.
Type I CRS. The degree of vertebral aplasiaranges from the absence of the coccyx and lowermidsacrum to aplasia of all coccygeal, sacral,lumbar, and lower thoracic vertebrae. Clinically,most patients have a stable neurological defectthat correlates to their degree of spinal cordaplasia. However, constriction of the thecalnerve roots within a narrowed thecal sac maygenerate progressive neurological signs.
The last visible vertebra is L5 through S2 in themajority of patients. Not uncommonly, however,abnormal vertebrae are also found cranially in thecervical and upper thoracic segments, including
block or butterfly vertebrae, hemivertebra, andeven supernumerary vertebrae (Nievelstein et al.1994). It is therefore crucial that the entire spine isexamined in the sagittal plane. Sacral agenesismay be markedly asymmetric, with variabledegrees of hemisacrum. Coronal sequences ori-ented along the major axis of the sacrum arenecessary to fully demonstrate the anomaly; inselected cases, a small field-of-view helical CTacquisition may add valuable information.
On MRI, the conus medullaris appears to betruncated or simply absent, with an abrupt termi-nation that nearly always is club- or wedge-shaped. The location of the spinal cord terminusis seen to be high (most often opposite T12 butalso higher) in most cases, but it may lie opposite
Fig. 18 Caudal regressionsyndrome. (a) SagittalT2-weighted image. In thistype I case, there is partialsacrococcygeal agenesiswith S3 as the final visiblesacral element (arrow). Thespinal cord terminatesabruptly at T12 (arrowhead)owing to agenesis of theconus medullaris. (b)Sagittal T1-weightedimage. In this type II case,the degree ofsacrococcygeal agenesis isless pronounced, with S4 asthe final visible element(thick arrow) (noticecomplete S1lumbarization). There is aconcurrent filar lipoma (thinarrows)
26 A. Rossi

to L1 in a minority. On sagittal images, the nerveroots in the thecal sac appear to be grouped intoanterior and posterior clumps (“double bundle”shape), whereas axial planes below the cord ter-minus show the so-called ghost conus sign gener-ated by the absence of the conus medullaris inbetween the caudal nerve roots. The thecal sactapers below the cord terminus and ends at anunusually high level.
Type II CRS. The degree of vertebral dysgen-esis is less severe than in the type I, with up to S4present as the last vertebra; most cases showagenesis of the last sacral vertebra and of thecoccyx. MRI shows that only the tip of theconus medullaris (corresponding to the meta-meres formed by secondary neurulation) is absent.However, in most cases, partial agenesis of the
conus is difficult to recognize because the conusitself is stretched caudally and tethered to anabnormality such as a tight filum terminale or afilar lipoma, which justifies a clinical presentationwith a tethered cord syndrome. Not uncommonly,sacrococcygeal agenesis is found in patients withsubcutaneous masses including terminalmyelocystocele, spinal lipoma, or anterior sacralmeningocele. Patients with the Currarino syn-drome present with a presacral CSF-filled massthat is associated with partial sacrococcygealagenesis. Solid, nodular teratomatous compo-nents are often located along the walls of themeningocele. These may produce restricted diffu-sion (epidermoid components) or enhance withgadolinium administration; enhancement is betterappreciated with fat-suppression techniques.
Fig. 19 Segmental spinal dysgenesis. (a and (b) SagittalT2-weighted images. There is a complete separation of thespinal column into two independent segments. The spinal
cord is also separated into two segments (arrows), locatedabove and below the kyphosis apex. Notice concurrenthorseshoe kidney lodged in the concavity of the kyphosis
Imaging in Spine and Spinal Cord Developmental Malformations 27

Segmental Spinal Dysgenesis
Segmental spinal dysgenesis (SSD) is a conditionthat shares embryological similarities with CRS inthat abnormal development of an intermediate(rather than caudal) segment of the notochordgenerates a complex picture with associated seg-mental hypoplasia of the spine and spinal cord(Tortori-Donati et al. 1999). Affected patients typ-ically have a palpable bony gibbus along theirback, are paraplegic or at least severelyparaparetic at birth, and invariably show hypo-trophic and deformed lower limbs withequinocavovarus feet.
The clinico-radiological definition includes:
• Segmental agenesis or dysgenesis of the lum-bar or thoracolumbar spine
• Segmental abnormality of the underlying spi-nal cord and nerve roots
• Congenital paraplegia or paraparesis• Congenital lower limb deformities
MRI (Fig. 19) shows segmental vertebralanomalies of the thoracolumbar, lumbar, or lum-bosacral spine which, in the most severe cases,determine a complete disconnection of the spinalcolumn into two segments that collide with oneanother generating short-angle kyphosis. At thislevel, the spinal canal is severely narrowed, andthe spinal cord is focally aplastic or hypoplastic.Thus, both the spine and the spinal cord appear tobe separated into two portions, with the upperspinal cord segment terminating bluntly and alower spinal cord segment appearing bulky andlow-lying. A horseshoe kidney is typically lodgedin the concavity of the kyphosis (Tortori-Donatiet al. 1999).
Dermal Sinus Tract
The dermal sinus is a fistula lined by epitheliumthat extends from a cutaneous orifice for a variabledepth into the underlying structures, often termi-nating within the thecal sac to connect with the
spinal cord. It is located more frequently in thelumbosacral region, although thoracic, cervical,and occipital locations are possible. On clinicalexamination, a midline dimple or pinpoint ostiumis found, often in association with hairy nevus,capillary hemangioma, or hyperpigmentedpatches. The location of the ostium is invariablyabove the intergluteal cleft, which differentiatesthem from simple coccygeal dimples. Complica-tions of dermal sinuses include local infection,meningitis, and abscesses that may result frombacteria invading the CNS along the tract. Embry-ologically, dermal sinus tracts result from focalincomplete disjunction of the neuroectodermfrom the cutaneous ectoderm; as such, they canbe viewed as a variant of the limited dorsalmyeloschisis continuum (Pang et al. 2013),which also includes nonterminal myelocystocelesand, possibly, meningoceles.
On imaging, dermal sinus tracts are easily rec-ognized on midsagittal T1-weighted images as athin hypointense stripe crossing the subcutaneousfat. The intrathecal portion of the tract is welldepicted on high-resolution heavily T2-weightedimages such as DRIVE (Fig. 20), whereas it maynot be picked up by other conventional sequences.Dermal sinus tracts may become encysted andform epidermoid cysts, appearing as hyperintenseon DWI and exhibiting contrast enhancementwhen superimposed infection occurs.
Conclusions
MRI features of spinal dysraphisms may appearcomplicated and puzzling even to the experiencedobserver. However, a rational approach foundedon a correlation of clinical, embryological, andneuroimaging information greatly facilitates thediagnosis in the vast majority of cases. MRI withhigh-resolution sequences is paramount for adetailed representation of the findings, whichallows a correct classification and adds crucialinformation for surgical planning.
28 A. Rossi

Checklist for Image Reporting
• Visually inspect the child’s back before startingthe MRI study; identify and make a note ofpossible birthmarks.
• Always include the entire longitudinal lengthof the spine in the sagittal field of view of theMRI study. Count the vertebrae starting fromboth the craniocervical junction and thesacrococcyx and identify possible variations(such as transitional vertebrae, block vertebrae,etc.). Try to number the ribs in order to identifythoracic vertebrae. Check whether an X-ray ofthe spine was already obtained, and use it toassist you in vertebral level numbering.
• Check the position of the tip of the conusmedullaris. Any conus tip at or below theL2–L3 disc space level should be considered
abnormal and should initiate a search for apossible tethering abnormality.
• Always include a heavily T2-weightedsequence (DRIVE or similar) oriented on thesagittal plane. Use an isotropic voxel(0.6 � 0.6 � 0.6 mm) to obtain high-qualityorthogonal reformats. In doubtful cases, add anadditional axial sequence across the suspectedabnormality, using the same parameters.
• Follow an orderly approach to the descriptionof the radiological findings. Is the malforma-tion open or closed? Is there a subcutaneousmass along the child’s back? Is the spinal cordlow-lying, split, or interrupted? Is there a dim-ple or ostium along the back of the child? Andso on.
• Do not inject contrast material unless a masslesion (i.e., suspected epidermoid, presacralmass) is identified on baseline scans, or infec-tion (i.e., abscessation) is suspected.
• Do not use CT scanning routinely. CT can beused as a second-step modality to confirm spe-cific osseous findings, in particular to study aseptum in cases of diastematomyelia. Reducethe field-of-view to the minimum not to unnec-essarily irradiate.
• For reporting, use the following scheme:– Indications: summarize the clinical picture
and results of visual inspection.– Technique: list the obtained sequences
according to spatial orientation andweighting; in case contrast material wasadministered, report the type of material,the dose, and whether adverse reactionsoccurred.
– Results: describe the imaging features, not-ing the conus level and any abnormality(see above for a description of the principalentities).
– Conclusion: draw the final diagnosis or, atleast, a range of differentials. Reportwhether further tests, including other spe-cialist consultations, are indicated. Statewhether follow-up MRI is indicated and atwhat interval.
Fig. 20 Dermal sinus tract. Sagittal T2 DRIVE image.There is a cutaneous orifice (thin arrow) where the tractoriginates. The intradural portion of the tract (arrowheads)is also seen, as well as its insertion onto a slightly buckledconus medullaris (thick arrow)
Imaging in Spine and Spinal Cord Developmental Malformations 29

Clinical Cases and Sample Reports
Sample Report 1
Patient HistoryA 1-year-old boy with a prior history of anorectalatresia which was corrected surgically in the neo-natal period, now presenting with impaired lowerlimb motility and equinocavovarus feet (Fig. 21).
Clinical DiagnosisSuspected tethered cord.
Purpose of MR StudyAssess the spinal cord and sacral region for pos-sible cord tethering.
Imaging Technique(A) Sagittal T1-weighted image, (B) sagittal T2DRIVE image, (C) and (D) axial T1-weightedimages, (E) axial diffusion-weighted image.
Contrast Agent and DoseNo contrast agent was administered.
Full FindingsThere is subtotal sacral agenesis with S1 visible,compatible with a sickle-shaped sacrum. The spi-nal cord is low-lying and tethered by a tight filumterminale (black arrows, B) that attaches to alipomatous component (arrowheads, A–C) at thebottom of the thecal sac. There is a concurrentsyrinx (open arrow, B). There also is an associatedpresacral mass of prevailing adipose composition(thin black arrows, A) which contains a nodularstructure (thick arrows, A, B, D) giving restricteddiffusion (thick arrow, E) consistent with anepidermoid cyst.
InterpretationThe association of anorectal atresia, partial sacralagenesis (with preserved S1), and presacral massis consistent with a diagnosis of Currarinosyndrome.
Fig. 21 see Sample Report 1
30 A. Rossi

CommentsNeonates with anorectal malformations should beinvestigated with MRI for a possible associationwith other features of the Currarino syndrome,which may otherwise remain undetected for vari-able amounts of time (sometimes into adulthood)and to identify causes of cord tethering. Once thediagnosis is established, patients should be inves-tigated for associated renal, ureteral, and uterineanomalies, which have also been described in thissyndrome. Surgery should be performed to releasethe cord tethering and to remove the presacralmass, in order to relieve constipation, but also toremove the dysontogenetic components, whichhave been described, in some cases, to transforminto more aggressive histologies. Genetic investi-gations should also be ordered to look for causalmutations in the MNX1 gene and to identifyfamilial cases.
Sample Report 2
Patient HistoryMale newborn with a nonulcerated open spinaldysraphism composed of a translucent meningealmembrane and a small, nodular placode (arrow, A).
Clinical DiagnosisSuspected myelomeningocele.
Purpose of MR StudyAssess the spinal cord malformation for surgicalplanning and rule out associated Chiari IImalformation.
Imaging Technique(A) picture of the patient’s low back, (B) SagittalT1-weighted image, (C) sagittal T2 DRIVEimage, (D) axial T1-weighted images, (E) axialT2 DRIVE image.
Contrast Agent and DoseNo contrast agent was administered.
Full FindingsSagittal images show a large lumbosacralmeningocele, mainly filled with CSF (Fig. 22).The spinal cord is elongated and reaches the bot-tom of the thecal sac (open arrow, C), with amulticavitary syrinx involving the lumbar seg-ment (asterisks, C). There is a concurrent mildChiari II (arrow, B). Axial images display adiastematomyelia (arrowheads, D,E); the lefthemicord is stretched (arrowhead, C) and entersthe meningocele (thin arrows, D,E) to terminatewith an exposed terminal placode (arrow, D,E)that corresponds to findings in A.
InterpretationA hemimyelomeningocele is defined by the asso-ciation of a split cord malformation (i.e.,
Fig. 22 see Sample Report 2
Imaging in Spine and Spinal Cord Developmental Malformations 31

diastematomyelia) with a failure of neurulation ofone of the two hemicords. A Chiari II malforma-tion is associated, as in other forms of open spinaldysraphism.
CommentsSurgical repair of an open spinal dysraphismshould be performed urgently after birth in orderto prevent subsequent neurological deteriorationand/or infection. MR imaging of the wholeneuraxis should be performed before surgerywhenever possible, so as to assist in surgical plan-ning as well as to identify associatedmalformations, such as diastematomyelia.
References
Adzick NS, Thom EA, Spong CY, Brock JW 3rd, BurrowsPK, Johnson MP, Howell LJ, Farrell JA, DabrowiakME, Sutton LN, Gupta N, Tulipan NB, D’Alton ME,Farmer DL, MOMS Investigators. A randomized trialof prenatal versus postnatal repair ofmyelomeningocele. N Engl J Med.2011;364:993–1004.
Barutçuoğlu M, Selçuki M, Umur AS, Mete M, GurgenSG, Selcuki D. Scoliosis may be the first symptom ofthe tethered spinal cord. Indian J Orthop.2016;50:80–6.
Kucera JN, Coley I, O’Hara S, Kosnik EJ, Coley BD. Thesimple sacral dimple: diagnostic yield of ultrasound inneonates. Pediatr Radiol. 2015;45:211–6.
Li YC, Shin SH, Cho BK, Lee MS, Lee YJ, Hong SK,Wang KC. Pathogenesis of lumbosacral lipoma: a testof the “premature dysjunction” theory. PediatrNeurosurg. 2001;34:124–30.
McLone DG, Knepper PA. The cause of Chiari II malfor-mation: a unified theory. Pediatr Neurosci.1989;15:1–12.
Nakanishi K, Tanaka N, Kamei N, Nakamae T, Izumi B,Ohta R, Fujioka Y, Ochi M. Use of prone positionmagnetic resonance imaging for detecting the terminalfilum in patients with occult tethered cord syndrome. JNeurosurg Spine. 2013;18:76–84.
Nievelstein RAJ, Valk J, Smit LME. Vermeji-Keers C. MRof the caudal regression syndrome: embryologic impli-cations. AJNR Am J Neuroradiol. 1994;15:1021–9.
Pang D, Zovickian J,Wong ST, Hou YJ,Moes GS. Limiteddorsal myeloschisis: a not-so-rare form of primary neu-rulation defect. Childs Nerv Syst. 2013;29:1459–84.
Rossi A, Piatelli G, Gandolfo C, Pavanello M,Hoffmann C, Van Goethem JW, Cama A, Tortori-Donati P. Spectrum of nonterminal myelocystoceles.Neurosurgery. 2006;58:509–15.
Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism:review of cutaneous markers and imaging. PediatrDermatol. 2015;32:161–70.
Singh S, Kline-Fath B, Bierbrauer K, Racadio JM,Salisbury S, Macaluso M, Jackson EC, EgelhoffJC. Comparison of standard, prone and cine MRI inthe evaluation of tethered cord. Pediatr Radiol.2012;42:685–91.
Tortori-Donati P, Fondelli MP, Rossi A, Raybaud CA,Cama A, Capra V. Segmental spinal dysgenesis.Neuroradiologic findings with clinical andembryologic correlation. AJNR Am J Neuroradiol.1999;20:445–56.
Warder DE, Oakes WJ. Tethered cord syndrome and theconus in a normal position. Neurosurgery.1993;33:374–8.
Suggested Readings
Huisman TA, Rossi A, Tortori-Donati P. MR imaging ofneonatal spinal dysraphia: what to consider? MagnReson Imaging Clin N Am. 2012;20:45–61.
Rossi A, Biancheri R, Cama A, Piatelli G, Ravegnani M,Tortori-Donati P. Imaging in spine and spinal cordmalformations. Eur J Radiol. 2004;50:177–200.
Schwartz ES, Rossi A. Congenital spine anomalies: theclosed spinal dysraphisms. Pediatr Radiol. 2015;45(Suppl 3):S413–9.
Tortori-Donati P, Rossi A, Cama A. Spinal dysraphism: areview of neuroradiological features with embryologi-cal correlations and proposal for a new classification.Neuroradiology. 2000;42:471–91.
Tortori-Donati P, Rossi A, Bianchieri R, CamaA. Congenital malformations of the spine and spinalcord. In: Tortori-Donati P, Rossi A, editors. Pediatricneuroradiology. Berlin: Springer; 2005. p. 1551–608.
32 A. Rossi