
Huntington’s Disease Leon S. Dure, MD The University of Alabama at Birmingham.
37
Huntington’s Disease Leon S. Dure, MD The University of Alabama at Birmingham
-
Upload
basil-porter -
Category
Documents
-
view
216 -
download
1
Transcript of Huntington’s Disease Leon S. Dure, MD The University of Alabama at Birmingham.

Huntington’s Disease
Leon S. Dure, MDThe University of Alabama at Birmingham

George Huntington and Huntington’s Disease

Huntington’s Disease
Unique among choreas Hereditary, presenting in adult or mid-life
Escapees - “the thread is broken” Never skips a generation
Tendency to insanity and suicide Manifests only in adult life
Distinct from postinfectious or other choreas

Chorea – Historical Aspects
St. Vitus (Guy) – imprisoned, thrown to lions, later boiled in oil (c. 303 A.D.)
Medieval Germany – 14 Holy Helpers Dancing at the statue
would ensure health Protection against
plague, illness Patron saint of dancers,
choreics, epileptics, also actors, comedians, those who oversleep

Chorea - Definition
Adjective-laden involuntary movement Frequent, brief, sudden Twitch-like Chaotic
Flow of movement from one body part to another
Intrusion of “movement fragments”

Sydenham’s Chorea
Example of a postinfectous chorea
Consequence of Rheumatic Fever

Clinical Features of HD
Prevalence Inheritance
Age of Onset
Duration
4-10/100,000
Dominant (Anticipation)Expansion of part of the htt gene
High penetrance
35-40yrs (2-80)10% present <18yo
15-30yrs

Clinical Presentation of HD
Initial signs and symptoms Chorea, incoordination, personality changes Psychiatric diagnoses
Later signs and symptoms Progressive chorea, dystonia Dysarthria Dementia, ongoing psychiatric disturbance

“Typical” HD
Mid-30’s to 40’s Difficulty with job
Employment Household management
Behavioral changes Impulsivity and altered judgment Mood swings

Adult HD Clinical Features

JHD Clinical Features
Onset before 18y Paternal inheritance the
norm Primarily hypokinetic
(Westphal variant) Dystonia, tremor,
dysarthria Rarely, a late juvenile
choreic variant Seizures Rapid course, early
death

Variability of JHD

HD/JHD Genetics
Expansion of translated CAGn, chromosome 4p Huntingtin protein (htt) Polyglutamate motif (similar to MCD, SCA-1, etc.) CAG > 39 correlated with clinical disease
JHD associated with “large” expansions Inheritance
affected father JHD mother/father
Expansions > 200 have been described
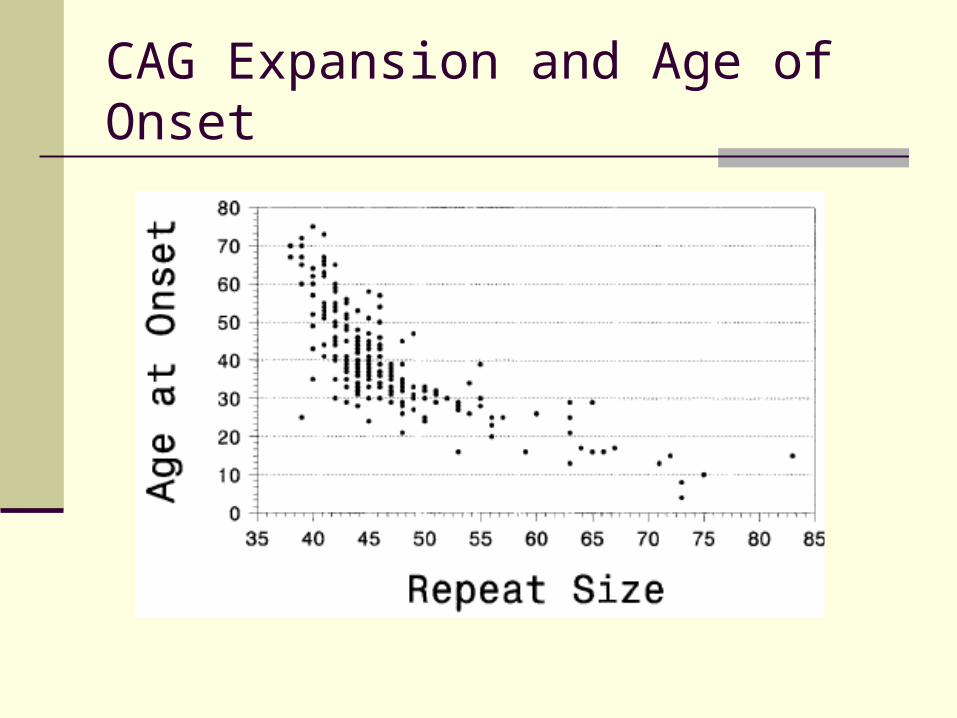
CAG Expansion and Age of Onset

CAG Repeats in Other Disorders
Correlation with age of onset
Suggests explanation of anticipation

CAG Expansions
General rule – lower number relates inversely to rate of decline and severity Younger people with symptoms have higher
repeat lengths Older people present with milder symptoms
Large expansions a phenomenon of meiosis Unexplained mechanism CAGn quite unstable in spermatocytes

Genetic Testing - Types
Confirmatory Symptomatic adults and children
Predictive Unaffected adults at risk
Prenatal Testing Unborn children
Unaffected children are not tested (usually)

Testing for HD
Presymptomatic testing available since 1980’s Simple test since 1994 ~16% of at-risk adults opt for testing
HSG (among others) does not recommend testing of at-risk children Exceptions
Symptomatic children Competent children

HD Testing in Children
Should have some of the cardinal features of the disorder (rigidity, dystonia, etc.)
Psychiatric/behavioral manifestations alone are insufficient to warrant testing Psychiatric disorders common in HD families Not always in presymptomatic individuals

Treatment of Chorea
Benign neglect if at all possible Neuroleptics can temporarily help chorea Not a permanent solution, and may do more
harm than good OT/PT evaluations Speech therapy for swallowing issues Other medications
Atypical antipsychotics Amantidine Tetrabenazine

Tetrabenazine in HD

Behavioral and Psychiatric Care
Emotional lability/dyscontrol Cause or consequence of the disease? Education of family and patient
Other more serious psychopathology Treat as you would any other patient!

Management of JHD
No cure No recommendations regarding agents used in adults
Symptomatic therapy Anticonvulsants Psychopharmacology therapy Little data on treating movement disorder
Supportive care OT/PT Speech/swallowing issues Psychosocial support

Neurobiology – What have we learned? Trinucleotide repeat diseases
Basis for anticipation Translated proteins
Gain in function New or impaired?
JHD vs. HD neuropathology HD a disease of whole brain
Most extensive neuronal loss in caudate/putamen Earliest cell loss – MSN projecting to LGP
JHD – less discrete, more profound
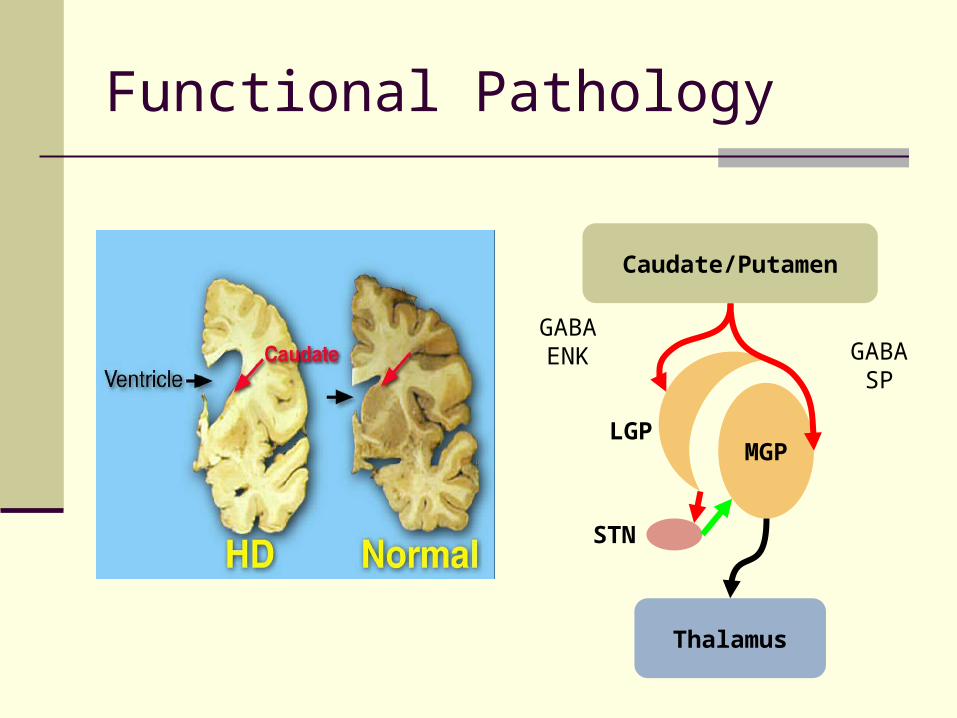
Functional Pathology
Caudate/Putamen
MGP
Thalamus
LGP
STN
GABAENK GABA
SP
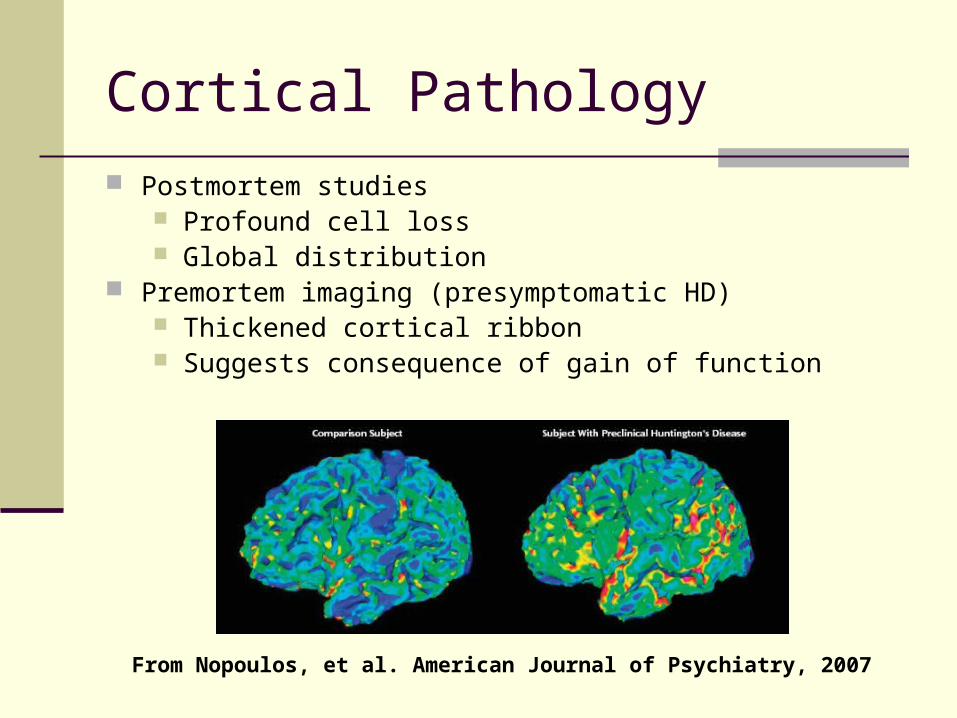
Cortical Pathology
Postmortem studies Profound cell loss Global distribution
Premortem imaging (presymptomatic HD) Thickened cortical ribbon Suggests consequence of gain of function
From Nopoulos, et al. American Journal of Psychiatry, 2007

Investigative Models of HD
R6/2 mice Knock in construct Stereotypic phenotype
Other mouse variations Drosophila macular degeneration Yeast expression Cell culture

Neuronal Intranuclear Inclusions (NII)
Ubiquitinated aggregates Initial observations of Kowall
and Ferrante Studies from HD knock-in
mice Subsequent identification in
human HD brain
NII colocalize with htt protein NII do not correspond to
regions of primary cell loss in cortex
Question of how/why htt enters nuclear compartment
2.7 mN-Q18
1.7 mN-Q82
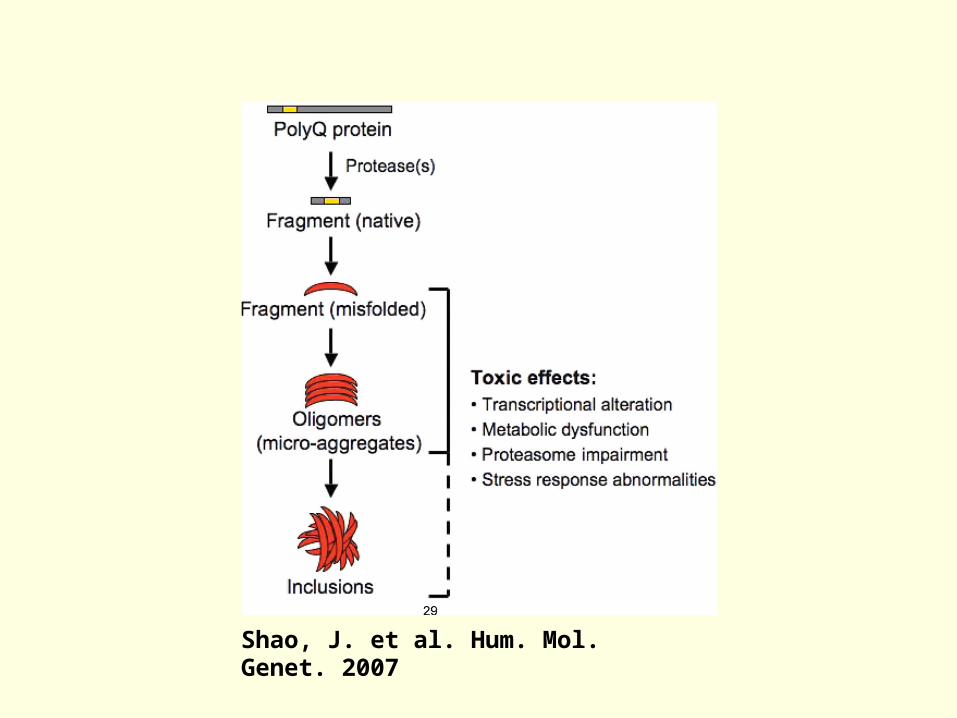
Shao, J. et al. Hum. Mol. Genet. 2007
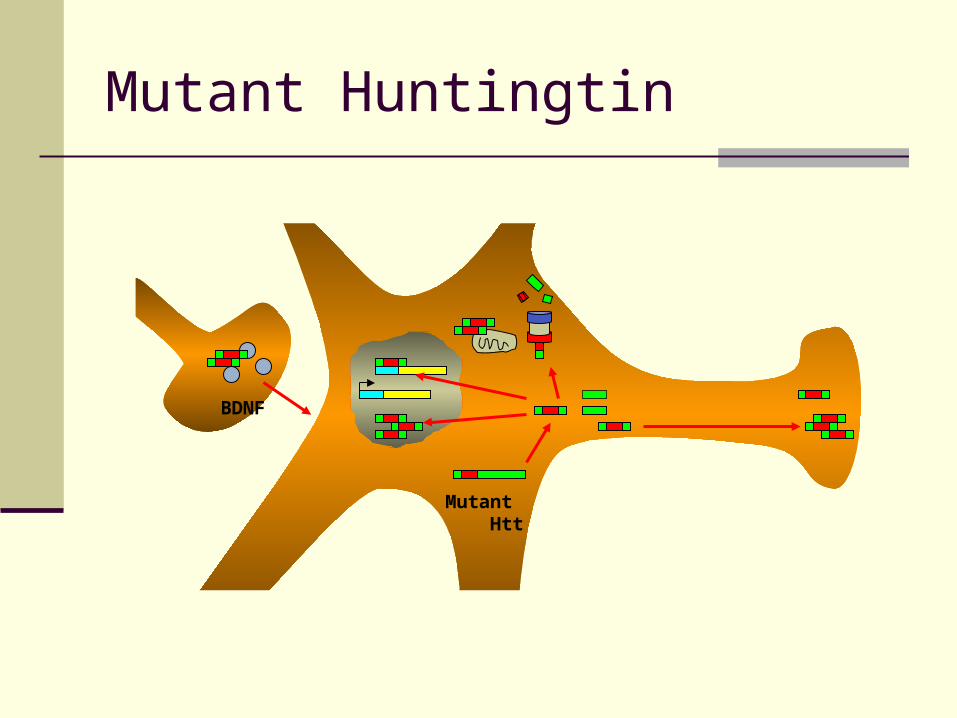
BDNF
Mutant Htt
Mutant Huntingtin

Mutated Huntingtin – Possible Roles
Transcriptional alteration Poly Q domains affect CREB, Sp1, other
transcriptional regulators Protein expression altered
Variety of potential markers identified No “leading” candidates
Metabolic dysfunction Pathology greatest in regions prone to
oxidative stress Clinical observation of HD muscle

Possible Therapeutic Approaches
Excitotoxicity Mitochondria Oxidative damage Caspases Aggregates Mutant htt
NMDA/glu inhibition
Enhance function
Free radical scavengers
Inhibitors
Prevention
Disrupt expression

Ambiguity vs. Progress
How to proceed?
Employment of accepted preclinical assays In vitro and in vivo models Hypothesis-driven testing of compounds
Creatine, minocycline, etc. High-throughput screening of compound
libraries Move forward into human clinical trials

Clinical Research
HSG – dedicated to development and administration of clinical trials in HD with promising compounds Multicenter Phase 1-4 trials
Results of studies have been disappointing – no effective agents to date
New evidence indicating early clinical features Cognitive and behavioral impairments Need to study younger subjects

Recent/New Studies
HART ARC-16 – a dopaminergic modulator Studies in HD and AD with promise 2 year treatment trial
CREST-HD Creatine of possible benefit in prior studies High dose creatine (45-50mg/kg/day) 2 year treatment trial

Research in Younger Subjects
Scientific rationale Early cognitive findings HD is probably a lifelong disease
Problems with inclusion Family dynamics Knowledge of disease risk Consent/assent

Summary
HD is hereditary and progressive Genetic research has been productive
Basis for anticipation Development of screening/diagnostic testing Has helped drive neuroscience investigations
Neurobiology of HD is less definitive Clinical research will remain a challenge


















