
HER2 Signaling Drives DNA Anabolism and Proliferation...
14
Molecular and Cellular Pathobiology HER2 Signaling Drives DNA Anabolism and Proliferation through SRC-3 Phosphorylation and E2F1-Regulated Genes Bryan C. Nikolai 1 , Rainer B. Lanz 1 , Brian York 1 , Subhamoy Dasgupta 1 , Nicholas Mitsiades 1,2,3 , Chad J. Creighton 2,4 , Anna Tsimelzon 5 , Susan G. Hilsenbeck 5 , David M. Lonard 1 , Carolyn L. Smith 1 , and Bert W. O'Malley 1 Abstract Approximately 20% of early-stage breast cancers display amplification or overexpression of the ErbB2/HER2 oncogene, conferring poor prognosis and resistance to endocrine therapy. Targeting HER2 þ tumors with trastuzumab or the receptor tyrosine kinase (RTK) inhibitor lapatinib significantly improves survival, yet tumor resistance and progression of metastatic disease still develop over time. Although the mechanisms of cytosolic HER2 signaling are well studied, nuclear signaling components and gene regulatory networks that bestow therapeutic resistance and limitless proliferative potential are incompletely understood. Here, we use biochemical and bio- informatic approaches to identify effectors and targets of HER2 transcriptional signaling in human breast cancer. Phosphory- lation and activity of the Steroid Receptor Coactivator-3 (SRC- 3) is reduced upon HER2 inhibition, and recruitment of SRC-3 to regulatory elements of endogenous genes is impaired. Tran- scripts regulated by HER2 signaling are highly enriched with E2F1 binding sites and define a gene signature associated with proliferative breast tumor subtypes, cell-cycle progression, and DNA replication. We show that HER2 signaling promotes breast cancer cell proliferation through regulation of E2F1- driven DNA metabolism and replication genes together with phosphorylation and activity of the transcriptional coactivator SRC-3. Furthermore, our analyses identified a cyclin-dependent kinase (CDK) signaling node that, when targeted using the CDK4/6 inhibitor palbociclib, defines overlap and divergence of adjuvant pharmacologic targeting. Importantly, lapatinib and palbociclib strictly block de novo synthesis of DNA, mostly through disruption of E2F1 and its target genes. These results have implications for rational discovery of pharmacologic combinations in preclinical models of adjuvant treatment and therapeutic resistance. Cancer Res; 76(6); 1463–75. Ó2016 AACR. Introduction The ErbB/HER receptor tyrosine kinase (RTK) family of mem- brane growth factor receptors activates multiple signaling path- ways and is often associated with cancer. Tyrosine kinase activity in this family of receptors is accomplished through homo- or heterodimerization of ErbB family members (EGFR/ERBB1, HER2/ERBB2, ERBB3, and ERBB4) upon ligand binding and/or membrane juxtaposition, and subsequent auto-and cross-phos- phorylation of intracellular domains (1). Phosphorylated recep- tor tyrosine residues serve as docking platforms for various SH2 domain proteins that function, in part, as signal adaptors and amplifiers for downstream kinase activity. Ras/Raf/MAPK and PI3K/AKT pathways are subsequently activated and the cascading substrates of these phospho-signaling events become effectors of cell division, evasion of apoptosis, and general tumor- igenicity (1, 2). Steroid Receptor Coactivator-3 (SRC-3/AIB1/NCOA3) is a potent transcriptional coregulator for various nuclear receptors and transcription factors, including E2F1, NFkB, and members of the ETS family (3–6). SRC-3 is amplified and/or overexpressed in a wide variety of tumors, including amplification in up to 10% and overexpression in up to 60% of breast tumors (3, 7–10). Importantly, SRC-3 is a coactivating partner for estrogen receptor (ERa), the cognate effector for the mitogenic action of estrogenic steroids in the mammary epithelium (11). ERa expression is an important biomarker for prognosis and treatment and is present in 70% of breast tumors at diagnosis, most of which receive endocrine therapy to block estrogen synthesis or receptor activity (12, 13). The function of SRC-3 in breast cancer has been inten- sively studied because it is a major coactivator for ERa and a prominent, overexpressed oncogene. The coactivation potential of SRC-3 is tightly regulated by posttranslational modifications that alter protein activity, stability, and subcellular localization (14, 15). In the context of ERa and NFkB-regulated transcription, SRC-3 is differentially activated via phosphorylation at multiple residues in response to estradiol or TNFa (14, 16). Collectively, these studies position SRC-3 as a potential "signal integrator" of 1 Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas. 2 Department of Medicine, Baylor College of Medicine, Houston, Texas. 3 Center for Drug Discovery, Baylor College of Medicine, Houston, Texas. 4 Dan L. Duncan Cancer Center Division of Biostatistics, Baylor College of Medicine, Houston, Texas. 5 Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, Texas. Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). C.L. Smith and B.W. O'Malley contributed equally to this article. Corresponding Author: Bert W. O'Malley, Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. Phone: 713-798-6205; Fax: 713-798-5599; E-mail: [email protected] doi: 10.1158/0008-5472.CAN-15-2383 Ó2016 American Association for Cancer Research. Cancer Research www.aacrjournals.org 1463 on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383
Transcript of HER2 Signaling Drives DNA Anabolism and Proliferation...

Molecular and Cellular Pathobiology
HER2 Signaling Drives DNA Anabolism andProliferation through SRC-3 Phosphorylation andE2F1-Regulated GenesBryanC.Nikolai1, RainerB. Lanz1, BrianYork1, SubhamoyDasgupta1, NicholasMitsiades1,2,3,Chad J. Creighton2,4, Anna Tsimelzon5, Susan G. Hilsenbeck5, David M. Lonard1,Carolyn L. Smith1, and Bert W. O'Malley1
Abstract
Approximately 20% of early-stage breast cancers displayamplification or overexpression of the ErbB2/HER2 oncogene,conferring poor prognosis and resistance to endocrine therapy.Targeting HER2þ tumors with trastuzumab or the receptortyrosine kinase (RTK) inhibitor lapatinib significantly improvessurvival, yet tumor resistance and progression of metastaticdisease still develop over time. Although the mechanisms ofcytosolic HER2 signaling are well studied, nuclear signalingcomponents and gene regulatory networks that bestowtherapeutic resistance and limitless proliferative potential areincompletely understood. Here, we use biochemical and bio-informatic approaches to identify effectors and targets of HER2transcriptional signaling in human breast cancer. Phosphory-lation and activity of the Steroid Receptor Coactivator-3 (SRC-3) is reduced upon HER2 inhibition, and recruitment of SRC-3to regulatory elements of endogenous genes is impaired. Tran-
scripts regulated by HER2 signaling are highly enriched withE2F1 binding sites and define a gene signature associated withproliferative breast tumor subtypes, cell-cycle progression, andDNA replication. We show that HER2 signaling promotesbreast cancer cell proliferation through regulation of E2F1-driven DNA metabolism and replication genes together withphosphorylation and activity of the transcriptional coactivatorSRC-3. Furthermore, our analyses identified a cyclin-dependentkinase (CDK) signaling node that, when targeted using theCDK4/6 inhibitor palbociclib, defines overlap and divergenceof adjuvant pharmacologic targeting. Importantly, lapatiniband palbociclib strictly block de novo synthesis of DNA, mostlythrough disruption of E2F1 and its target genes. These resultshave implications for rational discovery of pharmacologiccombinations in preclinical models of adjuvant treatment andtherapeutic resistance. Cancer Res; 76(6); 1463–75. �2016 AACR.
IntroductionThe ErbB/HER receptor tyrosine kinase (RTK) family of mem-
brane growth factor receptors activates multiple signaling path-ways and is often associated with cancer. Tyrosine kinase activityin this family of receptors is accomplished through homo- orheterodimerization of ErbB family members (EGFR/ERBB1,HER2/ERBB2, ERBB3, and ERBB4) upon ligand binding and/ormembrane juxtaposition, and subsequent auto-and cross-phos-phorylation of intracellular domains (1). Phosphorylated recep-tor tyrosine residues serve as docking platforms for various SH2
domain proteins that function, in part, as signal adaptorsand amplifiers for downstream kinase activity. Ras/Raf/MAPKand PI3K/AKT pathways are subsequently activated and thecascading substrates of these phospho-signaling events becomeeffectors of cell division, evasion of apoptosis, and general tumor-igenicity (1, 2).
Steroid Receptor Coactivator-3 (SRC-3/AIB1/NCOA3) is apotent transcriptional coregulator for various nuclear receptorsand transcription factors, including E2F1, NFkB, andmembers ofthe ETS family (3–6). SRC-3 is amplified and/or overexpressed ina wide variety of tumors, including amplification in up to 10%and overexpression in up to 60% of breast tumors (3, 7–10).Importantly, SRC-3 is a coactivating partner for estrogen receptor(ERa), the cognate effector for the mitogenic action of estrogenicsteroids in the mammary epithelium (11). ERa expression is animportant biomarker for prognosis and treatment and is presentin �70% of breast tumors at diagnosis, most of which receiveendocrine therapy to block estrogen synthesis or receptor activity(12, 13). The function of SRC-3 in breast cancer has been inten-sively studied because it is a major coactivator for ERa and aprominent, overexpressed oncogene. The coactivation potentialof SRC-3 is tightly regulated by posttranslational modificationsthat alter protein activity, stability, and subcellular localization(14, 15). In the context of ERa and NFkB-regulated transcription,SRC-3 is differentially activated via phosphorylation at multipleresidues in response to estradiol or TNFa (14, 16). Collectively,these studies position SRC-3 as a potential "signal integrator" of
1Department of Molecular and Cellular Biology, Baylor College ofMedicine, Houston, Texas. 2Department of Medicine, Baylor Collegeof Medicine, Houston, Texas. 3Center for Drug Discovery, BaylorCollege of Medicine, Houston, Texas. 4Dan L. Duncan Cancer CenterDivision of Biostatistics, Baylor College of Medicine, Houston, Texas.5Lester and Sue Smith Breast Center, Baylor College of Medicine,Houston, Texas.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
C.L. Smith and B.W. O'Malley contributed equally to this article.
Corresponding Author: BertW. O'Malley, Department of Molecular and CellularBiology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030.Phone: 713-798-6205; Fax: 713-798-5599; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-15-2383
�2016 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 1463
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

various cytokine and hormone elicited cell-signaling events thatspecify distinct patterns of gene expression (17).
Clinical studies have shown that breast cancer patients withtumors expressing high levels of both HER2 and SRC-3 displayresistance to endocrine therapy and have reduced disease-freesurvival (12, 18, 19). Furthermore, SRC-3 knockout mice areresistant to HER2/neu-mediated breast tumorigenesis and dis-play significantly reduced phosphorylation of key signalingcomponents, including HER2, AKT, and JNK (20). In MCF-7breast cancer cells, SRC-3 phosphorylation is influenced byHER2 (21). Together, these studies suggest HER2 and SRC-3are cooperating oncogenes whereby HER2 signaling eventsinfluence SRC-3 activity on discrete gene programs. However,the transcriptional targets of HER2 and SRC-3 are largelyundescribed and it is unknown whether intrinsic SRC-3 activityand specific phosphorylation sites are affected by HER2. Ampli-fication of HER2 occurs in approximately 20% of all breasttumors, and overexpression of HER2 and SRC-3 is associatedwith endocrine resistance and extremely poor prognosis. Giventhe link between HER2 signaling and SRC-3 in breast cancerand the importance of SRC-3 phosphorylation for its coactiva-tion of steroid receptors and other transcription factors, thecombined effects of HER2 and SRC-3 on gene regulation areunderstudied in breast cancer. Additionally, nuclear effectors ofHER2 signaling that regulate transcription of tumorigenic genenetworks are incompletely understood.
Here, we study the effects of HER2-mediated signaling eventson SRC-3 activity in breast cancer cells with amplified HER2and SRC-3 genes. HER2 depletion attenuates site-specific phos-phorylation of SRC-3, leading to a reduction in its intrinsicactivity. Transcriptomic analysis identifies genes that are regu-lated by HER2 signaling, with a massive enrichment for tran-scripts involved in mitotic cell-cycle processes and DNA repli-cation. A striking majority of genes downregulated upon HER2depletion contain canonical and evidence-based binding sitesfor the E2F1 transcription factor. Moreover, bioinformaticsreveals a common HER2/SRC-3/E2F1 target gene (CCND1)that is significantly correlated with sensitivity to RKT inhibitorsand identifies a CDK signaling node amendable to pharmaco-logic targeting. Our results suggest that SRC-3 and E2F1 serve amembrane-to-nuclear conduit function for HER2 signaling,resulting in an anabolic DNA gene signature exploitable bycombinatorial therapies.
Materials and MethodsCell culture, growth assays, and reagents
BT-474 and SKBR-3 cells were cultured in DMEM (Gibco/LifeTechnologies) supplemented with 10% fetal bovine serum(FBS) and 2 mg/L insulin. Cells were obtained from ATCC,and cell identity was verified using short tandem repeat (STR)analysis routinely by the Tissue Culture Core at Baylor Collegeof Medicine, Houston, TX. For growth assays, 5 � 104 cells wereplated per well on 12- or 24-well multiwell plates in completemedia and allowed to settle, and the following day were rinsedonce with phenol red-free DMEM before treatment in identicalconditions supplemented with 5% charcoal-stripped FBS.For Figs. 1A, C, E and 6A and B, crystal violet staining withcolorimetric absorbance was used to measure cell density.For Fig. 6C and D, MTS (Promega) assay was used. Detaileddescriptions of reagents are listed in Supplementary Table S2.
Sample preparation, immunoblotting, and RT-PCRFor protein analysis, cells were lysed (20 mmol/L Tris–HCl pH
8.0, 150 mmol/L NaCL, 1 mmol/L EDTA, and 0.5% NP-40) for 1hour at 4�C, followed by centrifugation at 12,000 � g for 10minutes. The supernatantwas removed to a fresh tube and dilutedfor quantitation using the Bradford method. Samples were sep-arated usingNuPAGE4%to12%Bis–Tris gels (Life Technologies)and transferred to nitrocellulose using methanol transfer buffer.Membranes were blocked in 5% fat-freemilk reconstituted in PBSplus 0.5% Tween-20 (PBST) for 1 hour at room temperature.Primary antibodies were diluted in 1% fat-free milk in PBSTand blots incubated for several hours at room temperature orovernight at 4�C. Species-specific secondary antibodies conju-gated with horseradish peroxidase were used with traditionalenhanced chemiluminescense and X-ray film. Densitometricquantification was performed using ImageJ and numbers infigures represent the area under curve for each band, normal-ized to actin in the identical lane. TRIzol reagent (low pHphenol; Life Technologies) was used to collect and extract totalRNA. RNA concentration was determined using a NanoDropspectrometer and integrity was evaluated using Lab-on-chip(Agilent). Reverse transcription using random hexamer priming(BioRad iScript) was performed before qPCR on ABI OneStepwith Roche Universal Library hydrolysis probes. Data wereanalyzed using the DDCT method (see www.nursa.org/qpcr_tu-torials/ for review). Detailed descriptions of primer sequencesare listed in Supplementary Table S2.
RNAi, transfections, and luciferase assayssiRNA was custom designed and/or ordered as prevalidated
duplexes (Life Technologies; see Supplementary Table S2 fordetails). BT-474 cells were plated on 6-well plates at a density of4 � 105 per well 1 day prior to transfection in reduced serummedia (OPTI-MEM; Life Technologies) at a final concentration of40 nmol/L siRNA. Luciferase reporter and plasmid DNA transfec-tions with siRNA are performed similarly with Lipofectamine2000 (Life Technologies). Cells were harvested in Tris–EDTA–NaCl (TEN) buffer, pelleted at 3500 � g, and suspended inreporter lysis buffer (Promega). Reporter activity was measuredusing a Thermo Luminoskan Ascent. Relative light unit (RLU)was calculated relative to total protein quantity. Detailed descrip-tions of reagents and catalog numbers are listed in SupplementaryTable S2.
Microarray analysis, motif scanning, and bioinformaticsRNA was prepared from BT-474 cells as described above and
hybridized to Affymetrix GeneChip Human Genome U133A2.0 Array chips by the Genomic and RNA Profiling (GARP) corefacility at the Baylor College of Medicine, and analysis wasperformed using BRB Array Tools (http://linus.nci.nih.gov/BRB-ArrayTools.html), dChip (http://www.hsph.harvard.edu/cli/complab/dchip), and ArrayAnalyzer (http://www.solution-metrics.com.au/products/s-plus_arrayanalyzer/default.html).Motif scanning was performed using P-scan Ver. 1.2.2 (http://159.149.160.51/pscan/) with �950 to þ50 criteria of matchedprobesets that were at least 2-fold downregulated upon HER2depletion. Bioinformatics were performed using a collection ofresources in the Dan L. Duncan Cancer Center and Departmentof Molecular and Cellular Biology. Briefly, the Galaxy/Cistromesuite, NCBI Gene ID, and existing data deposits were used incombination with SQL-based database organization to queue
Nikolai et al.
Cancer Res; 76(6) March 15, 2016 Cancer Research1464
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

and display integrated transcriptome and cistrome data basedon expression and protein binding, respectively. Gene ontol-ogy was displayed using DAVID and pathway mapping byKyoto Encyclopedia of Genes and Genomes (KEGG). Genesignature analysis involving the Kessler and colleagues (22)and The Cancer Genome Atlas (TCGA; ref. 23) datasets wascarried out as previously described (24). Microarray data aredeposited in the Gene Expression Omnibus as accessionGSE71347.
Chromatin immunoprecipitationChIP was performed as described with minor modifications
(25). Briefly, cells were fixed in 1% formaldehyde before quench-ing with 125 mmol/L glycine. Cells were washed twice with ice-cold TEN buffer and scraped into tubes on ice. Cell pellets wereeither flash frozen and stored at�80�C or resuspended in 500 mLof Tris–EDTA (TE)/1%SDS. All steps beyond this pointwere doneon ice or at 4�C. This suspension was sonicated for 8 cycles of10 pulses at amplitude 90 and duty cycle 3.5 on a BransonSonifier 250. Chromatin extracts were cleared by centrifugationand diluted 1:10 in a buffer containing 20 mmol/L Tris–HCl(pH8.0), 150 NaCl, 2 mmol/L EDTA, and 1% Triton-X-100 toreduce SDS concentration. Four micrograms of normal rabbitIgG antibody (Santa Cruz Biotechnology) was used with 50 mLProtein A/G Agarose plus salmon sperm DNA (Millipore)to preclear the diluted chromatin for 1 hour. The cleared
supernatant was incubated with 4-mg SRC-3 antibody (CellSignaling 5E11) or control IgG overnight. Agarose beads wereused to precipitate immune complexes for 1 hour beforewashing in buffers of increasing stringency as follows: 1X in20 mmol/L Tris–HCl (pH 8.0), 150 mmol/L NaCl, 2 mmol/LEDTA, and 1% Triton X-100; 1� in 20 mmol/L Tris–HCl (pH8.0), 500mmol/L NaCl, 2 mmol/L EDTA, and 1% Triton X-100;1� in 10 mmol/L Tris–HCl (pH 8.0), 1 mmol/L EDTA, 1%deoxycholate, 1% NP-40, and 0.25 M LiCl; and 2� with TE.Beads were centrifuged at 1,000 �g for 1 minute to pellet. DNAwas eluted from beads in 100 mmol/L NaHCO3 and 1% SDS at65�C overnight. Extraction was performed using PCR productpurification columns (Qiagen).
Tritium(3H)-labeled thymidine incorporationCellswere platedon6-well plates at a density of 2�105perwell
and treated with lapatinib, palbociclib, or bufalin for 24 hours innormal growth media. Four mC of 3H-labeled thymidine (PerkinElmer) was added to the media, and cells were grown for twoadditional hours. Media were removed, and cells were fixed for 5minutes with 25% acetic acid in methanol, rinsed once withmethanol, and protein precipitated using 5% trichloric acid for5 minutes. Wells were washed an additional 3 times with meth-anol and allowed to dry. Radioactivity was recovered using 1NNaOH and equilibrated with an equal volume of 1N HCl beforethe addition of biodegradable counting scintillant and liquid
Figure 1.BT-474 cells are estrogen responsiveand display HER2-dependant SERMresistance. A, BT-474 breast cancercell growth curve upon treatment with0.1% ethanol (Veh), 17-b-estradiol(E2), 4-hydroxytamoxifen (4HT), orraloxifene (Ral). B, transcript levels ofcanonical ERa target genes areelevated upon treatment with E2. C,depletion of HER2 using siRNA resultsin loss of estrogen mitogenic activityand SERM resistance. D, induction ofSERM-induced apoptosis in BT-474cells upon knockdown of HER2. E,depletion of either HER2 or SRC-3 inBT-474 cells grown in completemediadecreases cell viability.
HER2 Regulates DNA Replication through E2F1 Target Genes
www.aacrjournals.org Cancer Res; 76(6) March 15, 2016 1465
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

scintillation using a Beckman scintillation counter. For E2F1rescue experiments, cells were plated on 12-well plates at a densityof 5 � 104 per well and infected using 25 virions per cell for 24hours, then treated with lapatinib for 24 hours followed by 2 mCof 3H-thymidine treatment for 2 hours and processing asdescribed.
ResultsBT-474 cells display HER2-dependent estrogen responsivenessand resistance to antiestrogens
Resistance to selective estrogen receptor modulators (SERM) inHER2þ breast cancer is incompletely understood. Specifically,whether HER2 expression is required for sensitivity to estrogenand SERMs inHER2-amplified cells remains unanswered. The BT-474 breast cancer cell line represents the ideal model system tostudy effects of endogenously amplified HER2 and SRC-3 onco-genes on estrogen signaling because it is also positive for ERaexpression (data not shown; ref. 3). We first tested the mitogenicactivity of 17b-estradiol (E2) and effects of the SERMs 4-hydro-xytamoxifen (4HT) and raloxifene (Ral) on cell growth andgene expression. Cell density was significantly increased by E2relative to vehicle treatment, while tamoxifen and raloxifenefailed to decrease cell density relative to the vehicle control,indicating resistance (Fig. 1A). Likewise, transcript levels ofcanonical ERa-target genes were significantly induced upon E2treatment, but not affected by treatment with either SERM(Fig. 1B). Upon HER2 knockdown, E2 is no longer mitogenic inBT-474 cells (Fig. 1C, E2 panel). Moreover, siRNA against HER2significantly sensitized cells to treatment with 4HT or Ral (Fig. 1C,4HT and Ral panels; note the decrease in cell density over time insiHER2 transfected cells). These results confirm that BT-474 cellgrowth requires HER2 expression for estrogen sensitivity andSERM resistance and indicate that HER2 protects cells fromSERM-induced apoptosis. Indeed, when HER2 is depleted usingsiRNA, caspase-3 is cleaved (activated) upon treatment with 4HTor Ral, but not vehicle or estrogen (Fig. 1D, compare T and R to Vor E in siHER2 lanes). The action of SERMs is postulated to occur,at least in part, through disruption of the interaction between ERaand its transcriptional coactivators, such as SRC-3 (26). Depletionof SRC-3 also induced caspase-3 activation (Fig. 1D) anddecreased survival of breast cancer cells grown in steroid repletemedia (Fig. 1E), demonstrating a functional dependency on bothHER2 and SRC-3 survival pathways in this cell model.
HER2 signaling affects SRC-3 phosphorylation and activitySteroid Receptor Coactivator-3 is a potent coactivator for
numerous transcription factors involved in breast cancer and thepreference of SRC-3 for, and activity with, different transcriptionfactors is influenced by combinatorial phosphorylation (14). Todetermine if HER2 signaling affects SRC-3 phosphorylation andfunction, siRNA was used to deplete HER2 in BT-474 cells.Knockdown of HER2 results in decreased SRC-3 phosphorylationat T24, S543, S857, and S860 (Fig. 2A–C). Importantly, this loss ofphosphorylation does not appear to be uniform for SRC-3 proteinbecause S505 phosphorylation remains unchanged upon HER2depletion (Fig. 2A). These data are consistent with a lack of changein total SRC-3 protein levels uponHER2 knockdown, as S505 hasbeen described to function as part of a phospho-degron (27).
Because SRC-3 does not directly bind DNA, a system using aGAL4-DNA binding domain fused to the N-terminus of SRC-3
(pBIND-SRC-3) and a pGL5-Luciferase reporter was used tointerrogate whether HER2 influences intrinsic SRC-3 activity.Upon HER2 knockdown, intrinsic SRC-3 activity is significantlyreduced (Fig. 2D). These results can be pharmacologically reca-pitulated using an inhibitor of HER2, AG-825 (tyrphostin;Fig. 2E). Additionally, transient transfection of HER2 is sufficientto enhance SRC-3 transcriptional activity (Fig. 2F, compare 200vs.300 ng of pBind-SRC-3). These data corroborate previous studiessuggesting a role for SRC-3 in HER2 tumorigenesis and highlightthe ability of this oncogenic signaling pathway to elicit strongeffects on transcriptional processes. Taken together, our resultsshow that HER2 signaling has specific effects on SRC-3 phos-phorylation and is capable of increasing the intrinsic activity ofSRC-3.
HER2 depletion drastically alters the transcriptional landscapeThe transcriptional consequences of HER2 action undoubtedly
play a significant role in its oncogenic activity, but relativelyfew studies have comprehensively investigated the transcriptionaltargets of HER2 signaling. To define the HER2-regulated tran-scriptome, we performed microarray analysis on cells transfectedwith control or HER2 siRNA (Fig. 3A). HER2 knockdowndecreased AKT and Raf phosphorylation, indicating disruptionof the downstream HER2 signaling cascade (Fig. 3B). Substantialtranscriptional changes upon HER2 depletion are evident in theheat map shown in Fig. 3C. Gene Ontology analysis of transcriptsdownregulated upon HER2 depletion indicated an impressivedecrease of transcripts involved in cell cycle andmitotic processes(Fig. 3D), and RT-PCR confirmed loss of transcripts involved in S-phase DNA replication, Ras signaling, and transcription factorgenes (Supplementary Fig. S1A). Moreover, parsing this HER2-regulated transcriptomewith gene signatures associated with Ras/Raf/MAPK or PI3K/AKT signaling revealed significant overlap anddivergence between these signaling pathways (SupplementaryFig. S1B–S1D). Confirming a role for cell-cycle disruption whenHER2 is depleted, a reduction in G1 to S phase transition isobserved using flow cytometry to analyze DNA content (Supple-mentary Fig. S2).
Next, we compared genes that were downregulated uponHER2knockdown with publicly available breast cancer gene expressionpatterns in TCGA in relation to HER2 and SRC-3 (NCOA3)expression levels (Supplementary Fig. S3). Strikingly, tumorsamples with the highest expression of HER2 (SupplementaryFig. S3, top) or SRC-3 (Supplementary Fig. S3, bottom) alsodisplayed high expression of HER2-sensitive genes identified bymicroarray upon HER2 depletion. Jointly considered, these dataindicate that loss of HER2 in breast cancer cells results in asubstantial decrease of transcripts involved in cellular prolifera-tion that relates to expressionofHER2 and SRC-3 in a large datasetof breast tumor samples.
Because HER2 is present at extracellular membranes and thusacts as an indirect effector of nuclear transcription, we nextinvestigated transcription factor–binding motifs in the proximalpromoter regions of downregulated genes. Interrogation of 1-kbupstream promoter sequences of putative target genes identifiedan E2F1 consensus sequence as the most significantly enrichedtranscription factor motif (Fig. 3E). Transcript levels for E2F1 andits target genes CCND1 and CDC25A have similar expressionpatterns in our microarray analysis (Fig. 3F) and HER2 knock-down depletes the protein levels of these gene products (Fig. 3G–I). Upon HER2 knockdown, E2F1 protein levels were abolished
Nikolai et al.
Cancer Res; 76(6) March 15, 2016 Cancer Research1466
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

(Fig. 3G), which also was observed upon SRC-3 knockdown,indicating thatHER2 and SRC-3 signaling likely converge onE2F1target genes (Fig. 3H). Quantitative RT-PCR screening of HER2target genes revealed that transcripts for additional mitogenictranscription factors FOXM1 and ETV4, as well as the G1 to Scheckpoint gene CCND1, are significantly decreased in cellstransfected with siRNA against SRC-3 or HER2 (SupplementaryFig. S4C). These results suggest that depletion of HER2 or SRC-3reduces the expression ofmultiple transcription factors, includingE2F1, which are known to regulate the transcription of genesinvolved in mitotic progression and proliferation (28–30).
A majority of genes regulated by HER2 contain E2F1 bindingsites
All E2F family gene transcripts except E2F4 and E2F6 weredecreased upon HER2 knockdown in the microarray, and E2F7and E2F8 were among the most depleted transcripts in the entirearray (Table 1 and Supplementary Fig. S3). Given the largenumber of cell-cycle and mitotic genes downregulated uponHER2 depletion and the enrichment for E2F1 motifs, we inves-tigated E2F1 transcription factor binding to DNA regulatoryelements of HER2-regulated genes. Binding sites for E2F1 from
published chromatin immunoprecipitation-sequencing (ChIP-seq) in breast cancer cells (31) were compared with transcriptsfrom our BT-474 siHER2 microarray. Briefly, chromosomal coor-dinates of significantly downregulated genes were compared withE2F1 binding site intervals within the extended promoter region(EPR, �7.5 to þ2.5 Kb) or anywhere within the gene and includ-ing 10-kb gene-flanking regions (10 kb). A striking majority ofgenes regulated by HER2 (>75%) in our microarray are bound byE2F1 (Fig. 4A). Atypical E2F7 and E2F8 ChIP-seq binding incancer cells also shows significant overlap with the HER2 genesignature, and E2F8 may be a HER2 and SRC-3 target gene(Supplementary Fig. S5A–S5C). Transcript and protein levels ofE2F7 andE2F8were reducedwith siRNA transfectionof siHER2orsiSRC-3 (Supplementary Fig. S5D–S5F). These results are intrigu-ing considering aproposed role of atypical E2Fs inmaintenance ofgenome size and cancer, but E2F7 and E2F8 function was notfurther explored in this study (32, 33). Additionally, HER2-regulated genes with E2F1 promoter binding sites displayedoverlap with PI3K/AKT and Ras/Raf/MAPK gene signatures(Supplementary Fig. S6A).
The CCND1 gene is flanked by E2F1 binding sites including aknown SRC-3 binding element downstream, colloquially termed
Figure 2.HER2 affects phosphorylation andactivity of SRC-3. A and B, BT-474cells were transfected with siRNAtargeting different regions of HER2transcript, and cell lysates wereimmunoblotted using phospho-specific SRC-3 antibodies. SRC-3phosphorylation is decreased uponHER2 depletion at T24, S543, S860,and S857, while phosphorylation ofS505 is unaltered. C, densitometryquantification of S857phosphorylation. D, BT-474 cellstransfected with pBind-SRC-3/GAL4-Luciferase reporter system and siRNAtargeting HER2. Knockdown of HER2using two separate siRNAs decreasesintrinsic SRC-3 activity. E, AG-825(tyrphostin) inhibition of HER2decreases pBind-SRC-3 activity. F,cotransfection of HER2 relieves theactivation plateau of pBind-SRC-3in HeLa cells. � , P < 0.05 for allexperiments.
HER2 Regulates DNA Replication through E2F1 Target Genes
www.aacrjournals.org Cancer Res; 76(6) March 15, 2016 1467
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

enhancer 2 (enh2; Fig. 4B; ref. 34). This enhancer region is boundby E2F1 and SRC-3 in BT-474 cells (Fig. 4C), and upon HER2knockdown, SRC-3 recruitment to CCND1 enh2 is significantlyreduced (Fig. 4D). Moreover, treatment with the HER2 inhibitorlapatinib results in decreased SRC-3 recruitment, and H3K27acetylation (but not H3K14ac) is lost from the enhancer region(Fig. 4E). Taken together, these data show that HER2-mediatedgene regulation involves mechanisms of E2F1 and SRC-3 recruit-ment to gene regulatory elements involved in mitogenic expres-sion of CCND1.
The HER2 transcriptome signature correlates with E2F1 andproliferative breast cancer subtypes
BT-474 cells have previously been characterized as a Luminal Bsubtypemodel for breast cancer owing toHER2 amplification andERa expression (35), and we observe responsiveness to estrogenbut HER2-dependent resistance to antiestrogens (see Fig. 1A). As
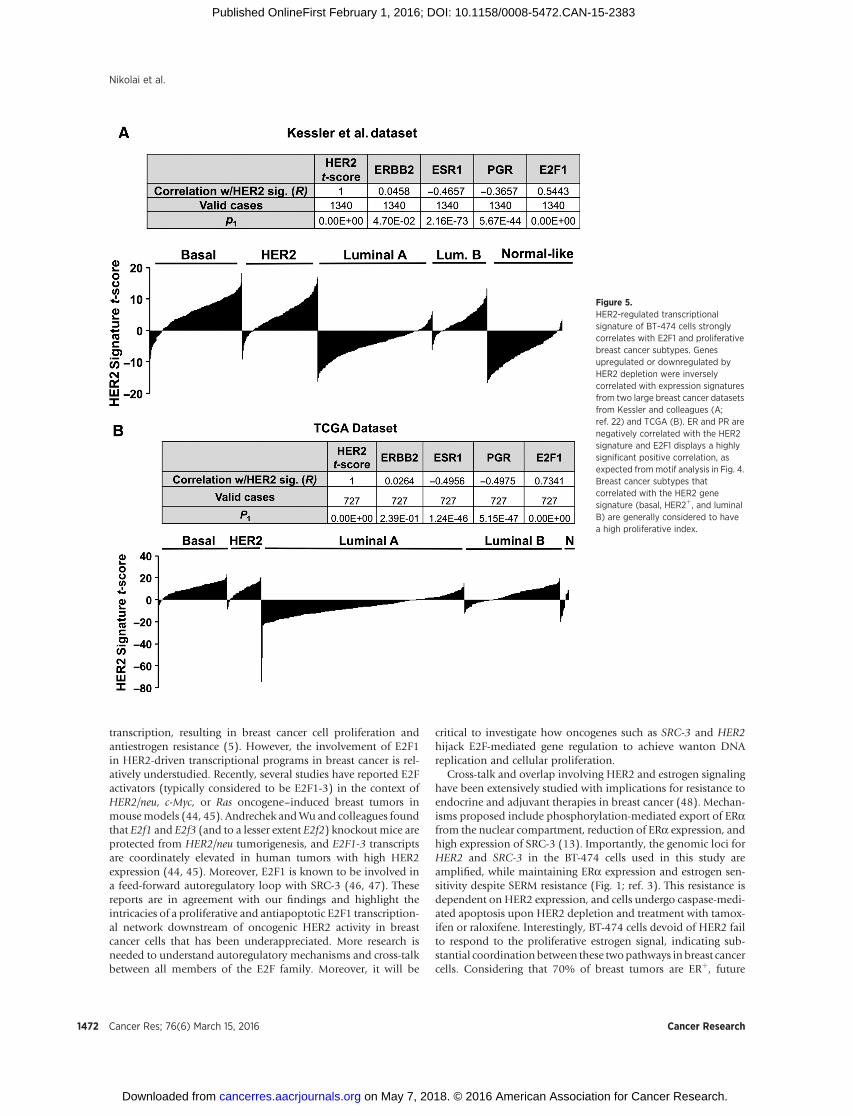
suggested from cistromic and transcriptomic analyses, comparingour HER2 gene signature expression scores with two large breasttumor datasets reveals a strong correlation with E2F1 expression(Table insets, Fig. 5A and B). Additionally, the HER2-regulatedgene signature correlates with the most proliferative breast cancersubtypes: basal, HER2þ, and luminal B (Fig. 5A and B, histo-grams). Tumors considered to be slow growing (luminal A andnormal-like) display a negative correlation with our HER2 genesignature, as do the predictors of luminal A subtype, ERa and PR(Fig. 5). Collectively, gene signatures from patient samples areconsistent with our in vitro findings and indicate that HER2signaling mediates a proliferative regulatory gene network inbreast cancer.
Perturbation analysis (36) of HER2 signature genes with E2F1binding sites using functional annotation with the GenomicRegions Enrichment Annotation Tool (GREAT; ref. 36) revealsa significant enrichment of oncogenic gene networks in diverse
Figure 3.The HER2-regulated transcriptomereveals loss of genes involved in cell-cycle regulation downstream of theHER2/SRC-3/E2F signaling axis. A,schematic of microarray experimentalgroups and HER2 knockdown inbiologic triplicates (right,immunoblot). B, HER2 knockdownresults in disruption of AKT and Ras/Raf signaling cascades, as evidencedby decreased AKT and c-Rafphosphorylation. Decrease of SRC-3phosphorylation was consistentlyobserved upon HER2 knockdown.C, heat map of significantly alteredprobe sets across treatment groups.D, top gene ontology terms returnedfrom HER2-downregulated gene listspecify cell-cycle and mitoticprocesses. E, predicted E2F1 bindingsites are enriched in the promoterregion of HER2-downregulated genes.F, enlarged heat map showingdecreased levels of E2F1, CCND1, andCDC25A upon HER2 knockdown. G,knockdown of HER2 reduces E2F1protein expression. H, knockdown ofSRC-3 reduces E2F1 proteinexpression. I, protein levels of genesfrom F with control and HER2 siRNAtransfection in BT-474 cells.
Nikolai et al.
Cancer Res; 76(6) March 15, 2016 Cancer Research1468
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

Table 1. HER2 regulates DNA anabolism and replication
siHER2 log2FC Gene ID Gene symbol
E2F transcription factors�2.6 144455 E2F7�1.9 79733 E2F8�0.5 1875 E2F5�0.4 1871 E2F3�0.3 1869 E2F1�0.3 1870 E2F2
1874 E2F41876 E2F6
Cyclin D1�0.9 595 CCND1�0.6 896 CCND3
894 CCND2Go, Ichi, Ni, San factors
�1.1 84296 GINS4�1 64785 GINS3�1 9837 GINS1�0.3 51659 GINS2
Replication protein A�0.4 6117 RPA1�0.2 6118 RPA2
6119 RPA329935 RPA4
TK, DHFR, SHMT, and TYMS�1.9 7083 TK1�1.5 1719 DHFR�0.8 6470 SHMT1
6472 SHMT2�0.7 7298 TYMS
IMP synthesis enzymes�1.1 5471 PPAT�0.3 10606 PAICS�0.2 5631 PRPS10.6 2618 GART
DNA ligases and PCNA�1.4 5111 PCNA�1 3978 LIG1�0.4 3980 LIG3
3981 LIG4Topoisomerases
�1.6 7153 TOP2A�0.5 11073 TOPBP1�0.4 7150 TOP10.5 116447 TOP1MT
7155 TOP2B7156 TOP3A8940 TOP3B
Cell division cycle proteins�2.1 8318 CDC45�1.9 990 CDC6�1.9 83879 CDCA7�1.7 8317 CDC7�1.6 113130 CDCA5�1.5 993 CDC25A�1.5 83461 CDCA3�1.4 991 CDC20�1.2 55143 CDCA8�1 157313 CDCA2�1 994 CDC25B�1 995 CDC25C�0.7 55038 CDCA4�0.5 8697 CDC23�0.5 56882 CDC42SE1�0.5 996 CDC27�0.4 998 CDC42�0.4 10435 CDC42EP2�0.3 55561 CDC42BPG
(Continued on the following column)
Table 1. HER2 regulates DNA anabolism and replication (Cont'd )
siHER2 log2FC Gene ID Gene symbol
Cell division cycle proteins�0.3 11140 CDC37�0.3 64866 CDCP10.2 55664 CDC37L10.4 10602 CDC42EP30.4 55536 CDCA7L0.4 8881 CDC160.5 51362 CDC400.8 8555 CDC14B
Ribonucleotide reductases�2.3 6241 RRM2�0.9 6240 RRM1
Origin recognition complex�1.4 23594 ORC6�1.1 4998 ORC1�0.2 4999 ORC2
23595 ORC35000 ORC45001 ORC5
Chromodomain�helicase DNA binding proteins�0.6 9557 CHD1L�0.3 1108 CHD4�0.2 55636 CHD70.3 55349 CHDH0.4 84181 CHD6
1105 CHD11106 CHD21107 CHD3
26038 CHD557680 CHD880205 CHD9
MCM DNA replication factors�1.9 55388 MCM10�1.4 4172 MCM3�1.4 4175 MCM6�1.4 4171 MCM2�1.2 4174 MCM5�0.9 4176 MCM7�0.9 84515 MCM8�0.7 4173 MCM4
254394 MCM9
Replication factor C�1.2 5984 RFC4�1.1 5985 RFC5�0.9 5983 RFC3�0.9 5982 RFC2
5981 RFC1
DNA primases�1.5 145270 PRIMA1�1.2 5557 PRIM1�0.6 5558 PRIM2
DNA polymerases�2 5427 POLE2�1.3 23649 POLA2�1.2 10721 POLQ�1.2 55703 POLR3B�0.9 5425 POLD2�0.8 5424 POLD1�0.8 10714 POLD3�0.7 5426 POLE�0.6 51728 POLR3K�0.5 5422 POLA1�0.4 5441 POLR2L�0.4 661 POLR3D�0.3 56655 POLE4�0.3 5433 POLR2D�0.3 5434 POLR2E
(Continued on the following column)(Continued on the following page)
HER2 Regulates DNA Replication through E2F1 Target Genes
www.aacrjournals.org Cancer Res; 76(6) March 15, 2016 1469
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

cancer types, including resistance to gefitinib (Supplementary Fig.S6B). Importantly, CCND1 is involved in breast cancer sensitivityto the HER/EGFR family inhibitors gefitinib, lapatinib, and afa-tinib (Genomics of Drug Sensitivity in Cancer; ref. 37). Taken incontext, these results suggest that HER2/E2F1 signaling regulatescyclinD1andother genes thatmaypredict sensitivity to therapy inproliferative breast cancer subtypes.
Cyclin D1/CDK cross-talk with HER2 converges ontranscriptional regulation of cell-cycle genes
Proliferation is critical in the etiology and associated pathoge-nicity of cancer. Individual oncogenes and tumor suppressors donot easily define the batteries of genes that regulate proliferationper se, because cell mitosis, DNA replication, and escape fromdifferentiation pathways involve large gene networks working inconcert. Importantly, because the tumor has become dependenton cellular events resulting from amplification or mutation ofsuch genes to maintain a proliferative advantage, these genesrepresent fragility in the network of cancer cell signaling (2, 38).Indeed, this has become the basis for directed therapies targetingoncogenic signaling from tyrosine kinases HER2 and EGFR (2).
We used network analysis of CCND1 and another HER2/E2F1-regulated gene from the microarray, CDC6, to identify a fragilecomponent of this signalingnetwork amendable topharmacologictargeting. Network analysis of CCND1 and CDC6 revealed strongoverlapwith theHER2 transcriptome.Remarkably, 50%ofgenes inthe CCND1-associated network, and 59% of CDC6 network genesare regulated by HER2 in our microarray (Supplementary TableS1). Not surprisingly, members of the cyclin-dependent kinase(CDK) family and their regulators are enriched in the CCND1 andCDC6 networks (Supplementary Fig. S7; ref. 39), which is consis-tent with KEGG pathway analysis. Because CDK4 and CDK6
function with CCND1 is well described and recent clinical trialshave reported optimistic results and recent FDA approval of theCDK4/6-specific inhibitor palbociclib (PD 0332991, Pfizer;ref. 40), we questioned whether this drug could similarly altercell-cycle gene transcription and sensitize breast cancer cells toother pharmacologic agents. As shown in Fig. 6A, palbociclibprevented the mitogenic effects of estradiol similar to knockdownof HER2 (see Fig. 1C). Additionally, combination treatment ofpalbociclib with 4HT significantly reduced cell survival (Fig. 6A).
Integrators of cellular signaling, such as transcriptional coacti-vators, may represent an additional "weak link" (and therefore atherapeutic target) in cancer gene regulation networks owing totheir pleiotropic effects onmultiple proliferative pathways.Whenpalbociclib treatment is used in combination with bufalin, arecently described inhibitor of SRC-3 (41), we observe a signif-icant combinatorial effect on the decrease in cell survival (Fig. 6B).Similarly, cellular sensitivity to lapatinib is augmented by palbo-ciclib or bufalin in a dose-dependent manner (Fig. 6C and D).Notably, bufalin is active in the nanomolar range and sensitizescells to lower concentrations of lapatinib (Fig. 6D). Similar resultsare observed in the HER2þ/ERa� breast cancer cell line SKBR-3,suggesting that these pathways signal cooperatively in other cellmodels (Supplementary Fig. S8). Palbociclib treatment reducedtranscript levels of cell-cycle genes E2F1, CDC6, and CDC45, butnot CCND1 (Fig. 6E). Transcript levels of E2F1, CDC6, andCCND1, but not CDC45, are depleted upon lapatinib treatment,suggesting that the HER2 pathway converges on similar transcrip-tional targets as CDK signaling (Fig. 6F). Additional genesinvolved in cell-cycle regulation were decreased upon palbociclib(i.e., E2F7, E2F8, andMCM10) or lapatinib (i.e., E2F7, E2F8, andETV4) treatment, indicating that these compounds influencebroad cell-cycle transcriptional networks (Supplementary Fig.S9A and S9B). Treatment with lapatinib also decreases proteinlevels of CCND1, CDC6, CDC45, MCM10, and ribonucleotidereductase (RRM2), a rate-limiting enzyme in DNA nucleosidemetabolism (Supplementary Fig. S9C).
Acute lapatinib or palbociclib treatment restrictsDNA synthesisin HER2þ breast cancer cells
Given the broad effect ofHER2 andCDK signaling pathways ontranscriptional regulation of genes involved in cell-cycle andDNAreplication (Fig. 6G), we reasoned that pharmacologically block-ing HER2 or CDK should also block replicative synthesis of DNAin HER2þ breast cancer cells. Indeed, lapatinib potently restrictsDNA synthesis at low nanomolar concentrations after 24 hours oftreatment, before any major changes occur to cellular health(Supplementary Fig. S9D–S9G) but concomitant with decreasedphosphorylation of HER2 and cell-cycle protein expression (Sup-plementary Fig. S9H). Tritiated (3H)-thymidine incorporation todirectly measure DNA synthesis revealed that acute (24-hour)treatment with lapatinib or palbociclib blocks de novo DNAreplication, without affecting cellular size, shape, or viability (Fig.6H). Bufalin treatment blocks DNA synthesis to a lesser extent(Fig. 6H), suggesting the combined effect of bufalinwith lapatinibor palbociclib on cellular viability (see Fig. 6B andD) is consistentwith its mechanistic role of inducing apoptosis (41). Consistentwith the effect of lapatinib and palbociclib on transcription,protein levels of E2F1, CDC6, and CCND1 are reduced in BT-474 and SKBR-3 breast cancer cells after acute treatment (Fig. 6I).Moreover, the lapatinib-induced reduction of replication forkproteins and block of DNA replication can be rescued by ectopic
Table 1. HER2 regulates DNA anabolism and replication (Cont'd )
siHER2 log2FC Gene ID Gene symbol
DNA polymerases�0.3 5436 POLR2G�0.3 5423 POLB�0.3 11128 POLR3A�0.2 55718 POLR3E�0.2 171568 POLR3H�0.2 26073 POLDIP20.3 246721 POLR2J20.4 5430 POLR2A0.4 57804 POLD40.5 84172 POLR1B0.5 51426 POLK0.5 51082 POLR1D0.6 84265 POLR3GL0.7 11232 POLG20.9 11201 POLI
391811 POLD2P184271 POLDIP354107 POLE35428 POLG5429 POLH27343 POLL27434 POLM
353497 POLN
NOTE: Selection of genes from enriched gene ontology terms involved innucleotide synthesis, DNA replication fork formation, and replication proces-sivity. Numbers represent log2 fold change, with negative values indicatingdecrease upon HER2 knockdown and positive numbers indicating increasedexpression. Boxed gene symbols are validated targets representative of a largernetwork of transcriptional targets downstream of HER2 signaling.
Nikolai et al.
Cancer Res; 76(6) March 15, 2016 Cancer Research1470
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

expression of E2F1 (Fig. 6J). Collectively, these results demon-strate that HER2 and CCND1/CDK signaling pathways regulateDNA replication through E2F1 and its associated transcriptionaltargets, many of which are directly involved in nucleotide bio-synthesis and replicative processivity. Moreover, our findingsdemonstrate the potential for combined targeting of HER2 andCDK signaling pathways or DNA replication may be a rationalmodel that warrants further investigation in preclinical studies.
DiscussionWe describe an E2F1 transcriptional network downstream of
HER2 signaling that is influenced by SRC-3 phosphorylation inbreast cancer cells. Overexpression of SRC-3 contributes to theetiology and progression of breast cancer, andmice lacking SRC-3are resistant to oncogene and carcinogen-induced breast tumor-igenesis (20, 42, 43). Moreover, SRC-3 is a coactivator for E2F1
Figure 4.Cooperative regulation of cyclin D1 byHER2 and SRC-3. A, E2F1 ChIP-seqpeaks were called from publishedresources and aligned to hg19 forparsing with HER2-regulated genesfrom the microarray. The pie graphindicates that a majority of HER2-regulated genes contained at least oneE2F1 binding site in the extendedpromoter region (EPR). B, E2F1 andSRC-3 binding from published ChIP-seq at the enhancer 2 (enh2) region ofCCND1 (stripedbar). C, E2F1 andSRC-3binding by the ChIP assay to theCCND1 enhancer region. D, HER2depletion decreases SRC-3 binding tothe CCND1 enhancer. E, lapatinibtreatment reduces CCND1 transcriptlevels and dismisses SRC-3 andH3K27ac from the CCND1 enhancer.
HER2 Regulates DNA Replication through E2F1 Target Genes
www.aacrjournals.org Cancer Res; 76(6) March 15, 2016 1471
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383
transcription, resulting in breast cancer cell proliferation andantiestrogen resistance (5). However, the involvement of E2F1in HER2-driven transcriptional programs in breast cancer is rel-atively understudied. Recently, several studies have reported E2Factivators (typically considered to be E2F1-3) in the context ofHER2/neu, c-Myc, or Ras oncogene–induced breast tumors inmousemodels (44, 45). Andrechek andWu and colleagues foundthat E2f1 and E2f3 (and to a lesser extent E2f2) knockout mice areprotected from HER2/neu tumorigenesis, and E2F1-3 transcriptsare coordinately elevated in human tumors with high HER2expression (44, 45). Moreover, E2F1 is known to be involved ina feed-forward autoregulatory loop with SRC-3 (46, 47). Thesereports are in agreement with our findings and highlight theintricacies of a proliferative and antiapoptotic E2F1 transcription-al network downstream of oncogenic HER2 activity in breastcancer cells that has been underappreciated. More research isneeded to understand autoregulatory mechanisms and cross-talkbetween all members of the E2F family. Moreover, it will be
critical to investigate how oncogenes such as SRC-3 and HER2hijack E2F-mediated gene regulation to achieve wanton DNAreplication and cellular proliferation.
Cross-talk and overlap involving HER2 and estrogen signalinghave been extensively studied with implications for resistance toendocrine and adjuvant therapies in breast cancer (48). Mechan-isms proposed include phosphorylation-mediated export of ERafrom the nuclear compartment, reduction of ERa expression, andhigh expression of SRC-3 (13). Importantly, the genomic loci forHER2 and SRC-3 in the BT-474 cells used in this study areamplified, while maintaining ERa expression and estrogen sen-sitivity despite SERM resistance (Fig. 1; ref. 3). This resistance isdependent on HER2 expression, and cells undergo caspase-medi-ated apoptosis upon HER2 depletion and treatment with tamox-ifen or raloxifene. Interestingly, BT-474 cells devoid of HER2 failto respond to the proliferative estrogen signal, indicating sub-stantial coordination between these twopathways inbreast cancercells. Considering that 70% of breast tumors are ERþ, future
Figure 5.HER2-regulated transcriptionalsignature of BT-474 cells stronglycorrelates with E2F1 and proliferativebreast cancer subtypes. Genesupregulated or downregulated byHER2 depletion were inverselycorrelated with expression signaturesfrom two large breast cancer datasetsfrom Kessler and colleagues (A;ref. 22) and TCGA (B). ER and PR arenegatively correlated with the HER2signature and E2F1 displays a highlysignificant positive correlation, asexpected frommotif analysis in Fig. 4.Breast cancer subtypes thatcorrelated with the HER2 genesignature (basal, HER2þ, and luminalB) are generally considered to havea high proliferative index.
Nikolai et al.
Cancer Res; 76(6) March 15, 2016 Cancer Research1472
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

studies should focus on determining the factors regulated byestrogen in the context of HER2 signaling, and what gene pro-grams are responsible for proliferation and cellular immortalityafforded by HER2 and ERa expression. Indeed, it is likely that theHER2/E2F1 gene signature described here may relate to estrogenactivity (49).
The transcriptional consequences of oncogenic signalingpathways have increasingly become a target of study because
downstream gene regulation appears to define tumor subtypesand pathogenicity. We present a model whereby HER2 and E2F1cooperate, in part through SRC-3, to regulateDNA replication andcellular proliferation (Supplementary Fig. S10). HER2 signalingaffects SRC-3 phosphorylation and activity, as well as a vast E2F1transcriptional program that grants promiscuousDNA replicationand induces the hallmarks of cellular transformation. An indi-vidual oncogene (or drug target) responsible for affecting tumor
Figure 6.Combinatorial effects of lapatinib and palbociclib on cell survival and E2F1-driven DNA synthesis. A, BT-474 cells treated with SERMs in the presence of vehicle orpalbociclib. Palbociclib reduced the mitogenic effects of estrogen and sensitized the cells to tamoxifen. B, cells were treated with palbociclib and the SRC-3inhibitor bufalin. Palbociclib and bufalin have additive effects. Asterisks denote significance tested using two-way ANOVA with Bonferroni posttest: � P < 0.05;�� , P < 0.01; ��� , P < 0.001. C, palbociclib cooperatively sensitizes cells to lapatinib treatment. D, bufalin cooperatively sensitizes cells to lapatinib treatment.E, E2F1 and cell-cycle transcripts are reduced upon treatment with palbociclib. F, lapatinib reduces E2F1 and cell-cycle transcripts analogous to HER2 knockdown.G, schematic representation of the HER2/E2F1 signaling axis effects on cell cycle, nucleotide metabolism, and DNA replication gene transcription. DNAreplication fork genes significantly downregulated upon HER2 knockdown are depicted. H, tritiated thymidine (3H) incorporation (DNA replication) inBT-474 and SKBR-3 cells after 24-hour treatment with lapatinib, palbociclib, or bufalin. Note the sharp decrease in DNA synthesis in both cell lines despiteany significant effect on cell size, shape, or viability at this time point. I, cell-cycle proteins E2F1, CCND1, and CDC6 are affected by 24-hour lapatinib orpalbociclib treatment. Phosphorylation of RB and HER2 serve as markers of drug efficacy. J, ectopic expression of E2F1 rescues MCM10, CDC6, and CCND1 proteinexpression and DNA synthesis in the presence of lapatinib in HER2þ breast cancer cells.
HER2 Regulates DNA Replication through E2F1 Target Genes
www.aacrjournals.org Cancer Res; 76(6) March 15, 2016 1473
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

cell proliferation, evasion of apoptosis, and resistance to anti-estrogens or therapeutic drugs is unlikely. In contrast, coregulatorsand associated transcription factors serve as nodal integrators ofcellular signaling pathways and drive transcription of gene net-works required for oncogenesis and tumorigenicity. It is likely thatdiverse oncogenic signaling pathways use discrete coactivators ascritical signaling integrators, perhaps in a tissue-specific manner.Importantly, these coactivators may represent a unique class oftargets to sensitize tumors to existing therapies.
Therapy options for breast and other cancers are ever evolvingand preclinical investigations in tractable models will be infor-mative for hypothesis-driven clinical trials. The data presented inthe current study unveils previously unappreciated interplaybetween known oncogenic and proliferative genes and an ana-bolic DNA replication pathway used to drive cell growth. Impor-tantly, we show that combinations of treatments (siRNA, SERMs,and small-molecule inhibitors) can increase sensitivity to inhibi-tors of alternate pathways of gene regulation that normally lead totreatment-resistant growth and cell survival. Moreover, we iden-tify anabolic metabolism of DNA as an important downstreameffect of theHER2 oncogene. Future studies should be directed atdefining transcriptional and cistromic mechanisms involved intherapy resistance and benefit (or lack thereof) in targeting mul-tiple pathways.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: B.C. Nikolai, B. York, S. Dasgupta, C.L. Smith, B.W.O'MalleyDevelopment of methodology: B.C. Nikolai
Acquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): B.C. NikolaiAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): B.C. Nikolai, R.B. Lanz, N. Mitsiades, C.J. Creighton,A. Tsimelzon, S.G. HilsenbeckWriting, review, and/or revision of the manuscript: B.C. Nikolai, R.B. Lanz,B. York, N. Mitsiades, D.M. Lonard, C.L. Smith, B.W. O'MalleyAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): B.C. NikolaiStudy supervision: C.L. Smith, B.W. O'Malley
AcknowledgmentsThe authors thank Jung-Sun Kim, Ed Bingman, Nancy Weigel, Charles
Foulds, Ping Yi, Qin Feng, Sophia Tsai, andMing-Jer Tsai for technical assistanceand helpful discussion. The authors also thank Cheryl Parker and the CellCulture Core facility at BCM for technical assistance and Travis Willis of SivartGraphics (Houston, TX) for graphic design.
Grant SupportThis project was supported in part by theGenomic and RNAProfiling Core at
Baylor College of Medicine with funding from the NIH NCI grant(P30CA125123) and the expert assistance of Dr. Lisa D. White, Ph.D. and theCytometry and Cell Sorting Core at Baylor College of Medicine with fundingfrom the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and theexpert assistance of Joel M. Sederstrom. B.C. Nikolai was supported by predoc-toral training grants through the Endocrine Society andWelch Foundation. Thiswork was supported by NIH grant (HD8818) and Cancer Prevention andResearch Institute of Texas grants (RP100348 and RP101251) to B.W.O'Malley.
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received August 26, 2015; revised November 19, 2015; accepted November22, 2015; published OnlineFirst February 1, 2016.
References1. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L.
Treatment of HER2-positive breast cancer: current status and future per-spectives. Nat Rev Clin Oncol 2012;9:16–32.
2. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat RevMol Cell Biol 2006;7:505–16.
3. Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y, et al.AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer.Science 1997;277:965–8.
4. Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160coactivators, the acetyltransferase p300 and the receptor tyrosine kinaseHER2/Neu. J Biol Chem 2004;279:14909–16.
5. Louie MC, Zou JX, Rabinovich A, Chen H-W. ACTR/AIB1 functions as anE2F1 coactivator to promote breast cancer cell proliferation and anties-trogen resistance. Mol Cell Biol 2004;24:5157–71.
6. WuR-C,Qin J, Hashimoto Y,Wong J, Xu J, Tsai SY, et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappaB kinase. Mol Cell Biol 2002;22:3549–61.
7. Xie D, Sham JST, ZengW-F, LinH-L, Bi J, Che L-H, et al. Correlation of AIB1overexpression with advanced clinical stage of human colorectal carcino-ma. Hum Pathol 2005;36:777–83.
8. He L-R, Zhao H-Y, Li B-K, Zhang L-J, Liu M-Z, Kung H-F, et al. Over-expression of AIB1 negatively affects survival of surgically resected non-small-cell lung cancer patients. Ann Oncol 2010;21:1675–81.
9. Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN.Expression of RAC 3, a steroid hormone receptor co-activator in prostatecancer. Br J Cancer 2001;85:1928–36.
10. Henke RT, Haddad BR, Kim SE, Rone JD, Mani A, Jessup JM, et al. Over-expression of the nuclear receptor coactivator AIB1 (SRC-3) duringprogression of pancreatic adenocarcinoma. Clin Cancer Res 2004;10:6134–42.
11. Xu J, Liao L, Ning G, Yoshida-KomiyaH,DengC,O'Malley BW. The steroidreceptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is requiredfor normal growth, puberty, female reproductive function, and mammarygland development. Proc Natl Acad Sci U S A 2000;97:6379–84.
12. Kirkegaard T, McGlynn LM, Campbell FM, Muller S, Tovey SM, Dunne B,et al. Amplified in breast cancer 1 in human epidermal growth factorreceptor–positive tumors of tamoxifen-treated breast cancer patients. ClinCancer Res 2007;13:1405–11.
13. OsborneCK, Schiff R.Mechanisms of endocrine resistance in breast cancer.Annu Rev Med 2011;62:233–47.
14. Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, et al. Selective phosphoryla-tions of the SRC-3/AIB1 coactivator integrate genomic responses to mul-tiple cellular signaling pathways. Mol Cell 2004;15:937–49.
15. Amazit L, Pasini L, Szafran AT, Berno V,WuR-C,MielkeM, et al. Regulationof SRC-3 intercompartmental dynamics by estrogen receptor and phos-phorylation. Mol Cell Biol 2007;27:6913–32.
16. Zheng FF, Wu R-C, Smith CL, O'Malley BW. Rapid estrogen-inducedphosphorylation of the SRC-3 coactivator occurs in an extranuclear com-plex containing estrogen receptor. Mol Cell Biol 2005;25:8273–84.
17. Wu R-C, Smith CL, O'Malley BW. Transcriptional regulation by steroidreceptor coactivator phosphorylation. Endocr Rev 2005;26:393–9.
18. Su Q, Hu S, Gao H, Ma R, Yang Q, Pan Z, et al. Role of AIB1 for tamoxifenresistance in estrogen receptor-positive breast cancer cells. Oncology2008;75:159–68.
19. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, FuquaSAW, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) andHER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst2003;95:353–61.
20. Fereshteh MP, Tilli MT, Kim SE, Xu J, O'Malley BW, Wellstein A, et al. Thenuclear receptor coactivator amplified inbreast cancer-1 is required forNeu
Nikolai et al.
Cancer Res; 76(6) March 15, 2016 Cancer Research1474
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

(ErbB2/HER2) activation, signaling, andmammary tumorigenesis inmice.Cancer Res 2008;68:3697–706.
21. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al.Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2–positive breast cancer. J Natl Cancer Inst2004;96:926–35.
22. Kessler JD, Kahle KT, Sun T, Meerbrey KL, SchlabachMR, Schmitt EM, et al.A SUMOylation-dependent transcriptional subprogram is required forMyc-driven tumorigenesis. Science 2012;335:348–53.
23. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMi-chael JF, et al. Comprehensive molecular portraits of human breasttumours. Nature 2012;490:61–70.
24. FengQ, Zhang Z, SheaMJ, Creighton CJ, Coarfa C, Hilsenbeck SG, et al. Anepigenomic approach to therapy for tamoxifen-resistant breast cancer. CellRes 2014;24:809–19.
25. York B, Yu C, Sagen JV, Liu Z, Nikolai BC,WuRC, et al. Reprogramming theposttranslational code of SRC-3 confers a switch in mammalian systemsbiology. Proc Natl Acad Sci U S A 2010;107:11122–7.
26. Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. Thestructural basis of estrogen receptor/coactivator recognition and the antag-onism of this interaction by tamoxifen. Cell 1998;95:927–37.
27. Wu R-C, Feng Q, Lonard DM, O'Malley BW. SRC-3 coactivator functionallifetime is regulated by a phospho-dependent ubiquitin time clock. Cell2007;129:1125–40.
28. Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1btranscription factor is essential for hepatocyte DNA replication andmitosisduring mouse liver regeneration. Proc Natl Acad Sci U S A 2002;99:16881–6.
29. Hollenhorst PC, Paul L, Ferris MW, Graves BJ. The ETS gene ETV4 isrequired for anchorage-independent growth and a cell proliferation geneexpression program in PC3 prostate cells. Genes Cancer 2011;1:1044–52.
30. Mudryj M, Hiebert SW, Nevins JR. A role for the adenovirus inducible E2Ftranscription factor in a proliferation dependent signal transduction path-way. EMBO J 1990;9:2179–84.
31. Khatun J, Epstein CB, Shoresh N, Ernst J, Kheradpour P, Ward LD, et al. Anintegrated encyclopedia of DNA elements in the human genome. Nature2012;489:57–74.
32. Chen H-Z, Ouseph MM, Li J, Pecot T, Chokshi V, Kent L, et al. Canonicaland atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol2012;14:1192–202.
33. Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PCJ, Corne-lissen PWA, et al. E2F8 is essential for polyploidization in mammaliancells. Nat Cell Biol 2012;14:1181–91.
34. Eeckhoute J, Carroll JS, Geistlinger TR, Torres-ArzayusMI, BrownM. A cell-type-specific transcriptional network required for estrogen regulation ofcyclin D1 and cell cycle progression in breast cancer. Genes Dev2006;20:2513–26.
35. Neve RM,Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection ofbreast cancer cell lines for the study of functionally distinct cancer subtypes.Cancer Cell 2006;10:515–27.
36. McLeanCY, BristorD,HillerM,Clarke SL, Schaar BT, LoweCB, et al. GREATimproves functional interpretation of cis-regulatory regions. Nat Biotech-nol 2010;28:495–501.
37. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al.Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeuticbiomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–61.
38. Rambaldi D, Giorgi FM, Capuani F, Ciliberto A, Ciccarelli FD. Lowduplicability and network fragility of cancer genes. Trends Genet 2008;24:427–30.
39. An O, Pendino V, D'Antonio M, Ratti E, Gentilini M, Ciccarelli FD. NCG4.0: the network of cancer genes in the era ofmassivemutational screeningsof cancer genomes. Database 2014;2014:bau015–5.
40. Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib inhormone-receptor-positive advanced breast cancer. N Engl J Med2015;373:209–19.
41. Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O'Malley BW. Smallmolecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1.Mol Endocrinol 2011;25:2041–53.
42. Kuang S-Q, Liao L, Zhang H, Lee AV, O'Malley BW, Xu J. AIB1/SRC-3deficiency affects insulin-like growth factor i signaling pathway and sup-presses v-Ha-ras-induced breast cancer initiation and progression in mice.Cancer Res 2004;64:1875–85.
43. Kuang S-Q, Liao L,Wang S, Medina D, O'Malley BW, Xu J. Mice lacking theamplified in breast cancer 1/steroid receptor coactivator-3 are resistant tochemical carcinogen–induced mammary tumorigenesis. Cancer Res2005;65:7993–8002.
44. Andrechek ER. HER2/Neu tumorigenesis and metastasis is regulated byE2F activator transcription factors. Oncogene 2015;34:217–25.
45. Wu L, de Bruin A, Wang H, Simmons T, Cleghorn W, Goldenberg LE, et al.Selective roles of E2Fs for ErbB2- and Myc-mediated mammary tumori-genesis. Oncogene 2015;34:119–28.
46. Mussi P, Yu C, O'Malley BW, Xu J. Stimulation of steroid receptor coacti-vator-3 (SRC-3) gene overexpression by a positive regulatory loop of E2F1and SRC-3. Mol Endocrinol 2006;20:3105–19.
47. LouieMC, RevenkoAS, Zou JX, Yao J, ChenH-W.Direct control of cell cyclegene expression by proto-oncogene product ACTR, and its autoregulationunderlies its transforming activity. Mol Cell Biol 2006;26:3810–23.
48. Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, et al.Advanced concepts in estrogen receptor biology and breast cancer endo-crine resistance: implicated role of growth factor signaling and estrogenreceptor coregulators. Cancer Chemother Pharmacol 2005;56:10–20.
49. Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, et al.ERalpha-dependent E2F transcription can mediate resistance to estrogendeprivation in human breast cancer. Cancer Discov 2011;1:338–51.
www.aacrjournals.org Cancer Res; 76(6) March 15, 2016 1475
HER2 Regulates DNA Replication through E2F1 Target Genes
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383

2016;76:1463-1475. Published OnlineFirst February 1, 2016.Cancer Res Bryan C. Nikolai, Rainer B. Lanz, Brian York, et al. SRC-3 Phosphorylation and E2F1-Regulated GenesHER2 Signaling Drives DNA Anabolism and Proliferation through
Updated version
10.1158/0008-5472.CAN-15-2383doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2017/02/09/0008-5472.CAN-15-2383.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/76/6/1463.full#ref-list-1
This article cites 48 articles, 18 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/76/6/1463.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/76/6/1463To request permission to re-use all or part of this article, use this link
on May 7, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2016; DOI: 10.1158/0008-5472.CAN-15-2383


















