
Hematopoietic and Leukemic Stem Cells Have Distinct ... · PDF filecal CML model, loss of...
14
Hematopoietic and Leukemic Stem Cells Have Distinct Dependence on Tcf1 and Lef1 Transcription Factors * Received for publication, January 25, 2016, and in revised form, April 3, 2016 Published, JBC Papers in Press, April 4, 2016, DOI 10.1074/jbc.M116.717801 Shuyang Yu ‡1,2 , Fengyin Li §1 , Shaojun Xing § , Tianyan Zhao ‡ , Weiqun Peng ¶3 , and Hai-Hui Xue §4 From the ‡ State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing 100193, China, ¶ Department of Physics, The George Washington University, Washington, D. C. 20052, and § Department of Microbiology, Carver College of Medicine, University of Iowa, Iowa City, Iowa 52242 Hematopoietic and leukemic stem cells (HSCs and LSCs) have self-renewal ability to maintain normal hematopoiesis and leuke- mia propagation, respectively. Tcf1 and Lef1 transcription factors are expressed in HSCs, and targeting both factors modestly expanded the size of the HSC pool due to diminished HSC quies- cence. Functional defects of Tcf1/Lef1-deficient HSCs in multi-lin- eage blood reconstitution was only evident under competitive con- ditions or when subjected to repeated regenerative stress. These are mechanistically due to direct positive regulation of Egr and Tcf3 by Tcf1 and Lef1, and significantly, forced expression of Egr1 in Tcf1/Lef1-deficient HSCs restored HSC quiescence. In a preclini- cal CML model, loss of Tcf1/Lef1 did not show strong impact on leukemia initiation and progression. However, when transplanted into secondary recipients, Tcf1/Lef1-deficient LSCs failed to prop- agate CML. By induced deletion of Tcf1 and Lef1 in pre-established CML, we further demonstrated an intrinsic requirement for these factors in LSC self-renewal. When combined with imatinib ther- apy, genetic targeting of Tcf1 and Lef1 potently diminished LSCs and conferred better protection to the CML recipients. LSCs are therefore more sensitive to loss of Tcf1 and Lef1 than HSCs in their self-renewal capacity. The differential requirements in HSCs and LSCs thus identify Tcf1 and Lef1 transcription factors as novel therapeutic targets in treating hematological malignancies, and inhibition of Tcf1/Lef1-regulated transcriptional programs may thus provide a therapeutic window to eliminate LSCs with minimal side effect on normal HSC functions. Hematopoietic stem cells (HSCs) 5 represent a rare popula- tion that self-renew and continuously replenish all blood cells throughout one’s lifetime (1, 2). HSCs are regulated by both intrinsic factors and extrinsic signals from the microenviron- ment to maintain a homeostatic HSC pool at steady state as well as in response to injuries such as radiotherapy or traumatic blood loss (3–5). Leukemic stem cells (LSCs) share similar properties with HSCs, and the self-renewing LSCs are respon- sible for initiation, maintenance, and propagation of the disease (6, 7). Chronic myelogenous leukemia (CML) is initiated by reciprocal translocation of chromosomes 9 and 22, which gen- erates a constitutively active fusion kinase, BCR-ABL (8). BCR- ABL tyrosine kinase inhibitors (TKIs) including imatinib are effective in inducing remissions and improving survival in CML patients at the chronic phase. Because CML LSCs are not sen- sitive to imatinib, continuous TKI treatment is necessary to prevent relapse. Molecules and/or pathways that are specifi- cally utilized by the LSCs are of great therapeutic value for erad- ication of leukemias. A plethora of data has implicated Wnt signaling pathway in regulation of HSC activities (9, 10). Overexpression of Dkk1 or Wif1, which blocks the interaction between Wnt ligands and their receptors, diminishes HSC quiescence and its repopula- tion capacity (11, 12). Wnt3a deficiency also greatly impaired HSC activity (13). Because Wnt signaling is involved in proper bone formation (14, 15), blocking Wnt/receptor interaction or germline deletion of Wnt proteins may affect the HSC niche, hence indirectly impacting HSCs. Activation of the canonical Wnt signaling pathway leads to stabilization and nuclear trans- location of -catenin. The role of -catenin in HSCs has been a highly contentious issue. Depending on the experimental sys- tems utilized, -catenin activation is reported to have detri- mental (16, 17), beneficial (18), or no effect on adult HSCs (19, 20). A recent study reports that the magnitude of -catenin activation matters, with a narrow window of active -catenin positively regulating HSC repopulation capacity (21). With regard to necessity of -catenin, it is essential for definitive hematopoiesis (by embryonic day 10.5) (22), but its require- ment in adult HSCs is only evident after serial transplantation (23). Despite the reported discrepancies on its roles in normal HSCs, -catenin has been consistently demonstrated to be crit- ical for development and maintenance of LSCs in CML (23–25). * This study is supported in-part by grants from the American Cancer Society (RSG-11-161-01-MPC, to H.-H. X.), the National Institutes of Health (AI105351, AI112579, AI115149, and AI119160, to H.-H. X.), and the U. S. Department of Veteran Affairs (Merit Review Award I01 BX002903 to H.-H. X., Biomedical laboratory Research and Development Program). H.-H. X. is the founder of Cure-it LifeSciences, and this capacity has no roles in experimental design and data interpretation in this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Veteran Affairs. 1 Both authors contributed equally to this work. 2 Supported by the Chinese Universities Scientific Fund (2014RC004) and National Natural Science Foundation of China (31422037 and 31571522). To whom correspondence may be addressed: 2 Yuanmingyuan West Rd., Haid- ian District, Beijing 100193, China. E-mail: [email protected]. 3 Supported by a National Institutes of Health Grant AI113806. 4 To whom correspondence may be addressed: 51 Newton Rd. BSB 3–772, Iowa City, IA 52246. Tel.: 319-335-7937; Fax: 319-335-9006; E-mail: hai-hui- [email protected]. 5 The abbreviations used are: HSC, hematopoietic stem cell; BrdU, bromo-2- deoxyuridine; LSC, leukemic stem cell; CML, chronic myelogenous leuke- mia; TKI, tyrosine kinase inhibitor; BMT, bone marrow transplantation; PBC, peripheral blood mononuclear cell. crossmark THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 291, NO. 21, pp. 11148 –11160, May 20, 2016 © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. 11148 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016 by guest on May 8, 2018 http://www.jbc.org/ Downloaded from
Transcript of Hematopoietic and Leukemic Stem Cells Have Distinct ... · PDF filecal CML model, loss of...

Hematopoietic and Leukemic Stem Cells Have DistinctDependence on Tcf1 and Lef1 Transcription Factors*
Received for publication, January 25, 2016, and in revised form, April 3, 2016 Published, JBC Papers in Press, April 4, 2016, DOI 10.1074/jbc.M116.717801
Shuyang Yu‡1,2, Fengyin Li§1, Shaojun Xing§, Tianyan Zhao‡, Weiqun Peng¶3, and Hai-Hui Xue§4
From the ‡State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing 100193,China, ¶Department of Physics, The George Washington University, Washington, D. C. 20052, and §Department of Microbiology,Carver College of Medicine, University of Iowa, Iowa City, Iowa 52242
Hematopoietic and leukemic stem cells (HSCs and LSCs) haveself-renewal ability to maintain normal hematopoiesis and leuke-mia propagation, respectively. Tcf1 and Lef1 transcription factorsare expressed in HSCs, and targeting both factors modestlyexpanded the size of the HSC pool due to diminished HSC quies-cence. Functional defects of Tcf1/Lef1-deficient HSCs in multi-lin-eage blood reconstitution was only evident under competitive con-ditions or when subjected to repeated regenerative stress. Theseare mechanistically due to direct positive regulation of Egr and Tcf3by Tcf1 and Lef1, and significantly, forced expression of Egr1 inTcf1/Lef1-deficient HSCs restored HSC quiescence. In a preclini-cal CML model, loss of Tcf1/Lef1 did not show strong impact onleukemia initiation and progression. However, when transplantedinto secondary recipients, Tcf1/Lef1-deficient LSCs failed to prop-agate CML. By induced deletion of Tcf1 and Lef1 in pre-establishedCML, we further demonstrated an intrinsic requirement for thesefactors in LSC self-renewal. When combined with imatinib ther-apy, genetic targeting of Tcf1 and Lef1 potently diminished LSCsand conferred better protection to the CML recipients. LSCs aretherefore more sensitive to loss of Tcf1 and Lef1 than HSCs in theirself-renewal capacity. The differential requirements in HSCs andLSCs thus identify Tcf1 and Lef1 transcription factors as noveltherapeutic targets in treating hematological malignancies, andinhibition of Tcf1/Lef1-regulated transcriptional programs maythus provide a therapeutic window to eliminate LSCs with minimalside effect on normal HSC functions.
Hematopoietic stem cells (HSCs)5 represent a rare popula-tion that self-renew and continuously replenish all blood cells
throughout one’s lifetime (1, 2). HSCs are regulated by bothintrinsic factors and extrinsic signals from the microenviron-ment to maintain a homeostatic HSC pool at steady state as wellas in response to injuries such as radiotherapy or traumaticblood loss (3–5). Leukemic stem cells (LSCs) share similarproperties with HSCs, and the self-renewing LSCs are respon-sible for initiation, maintenance, and propagation of the disease(6, 7). Chronic myelogenous leukemia (CML) is initiated byreciprocal translocation of chromosomes 9 and 22, which gen-erates a constitutively active fusion kinase, BCR-ABL (8). BCR-ABL tyrosine kinase inhibitors (TKIs) including imatinib areeffective in inducing remissions and improving survival in CMLpatients at the chronic phase. Because CML LSCs are not sen-sitive to imatinib, continuous TKI treatment is necessary toprevent relapse. Molecules and/or pathways that are specifi-cally utilized by the LSCs are of great therapeutic value for erad-ication of leukemias.
A plethora of data has implicated Wnt signaling pathway inregulation of HSC activities (9, 10). Overexpression of Dkk1 orWif1, which blocks the interaction between Wnt ligands andtheir receptors, diminishes HSC quiescence and its repopula-tion capacity (11, 12). Wnt3a deficiency also greatly impairedHSC activity (13). Because Wnt signaling is involved in properbone formation (14, 15), blocking Wnt/receptor interaction orgermline deletion of Wnt proteins may affect the HSC niche,hence indirectly impacting HSCs. Activation of the canonicalWnt signaling pathway leads to stabilization and nuclear trans-location of �-catenin. The role of �-catenin in HSCs has been ahighly contentious issue. Depending on the experimental sys-tems utilized, �-catenin activation is reported to have detri-mental (16, 17), beneficial (18), or no effect on adult HSCs (19,20). A recent study reports that the magnitude of �-cateninactivation matters, with a narrow window of active �-cateninpositively regulating HSC repopulation capacity (21). Withregard to necessity of �-catenin, it is essential for definitivehematopoiesis (by embryonic day 10.5) (22), but its require-ment in adult HSCs is only evident after serial transplantation(23). Despite the reported discrepancies on its roles in normalHSCs, �-catenin has been consistently demonstrated to be crit-ical for development and maintenance of LSCs in CML(23–25).
* This study is supported in-part by grants from the American Cancer Society(RSG-11-161-01-MPC, to H.-H. X.), the National Institutes of Health(AI105351, AI112579, AI115149, and AI119160, to H.-H. X.), and the U. S.Department of Veteran Affairs (Merit Review Award I01 BX002903 toH.-H. X., Biomedical laboratory Research and Development Program).H.-H. X. is the founder of Cure-it LifeSciences, and this capacity has no rolesin experimental design and data interpretation in this study. The content issolely the responsibility of the authors and does not necessarily representthe official views of the National Institutes of Health or the Veteran Affairs.
1 Both authors contributed equally to this work.2 Supported by the Chinese Universities Scientific Fund (2014RC004) and
National Natural Science Foundation of China (31422037 and 31571522). Towhom correspondence may be addressed: 2 Yuanmingyuan West Rd., Haid-ian District, Beijing 100193, China. E-mail: [email protected].
3 Supported by a National Institutes of Health Grant AI113806.4 To whom correspondence may be addressed: 51 Newton Rd. BSB 3–772,
Iowa City, IA 52246. Tel.: 319-335-7937; Fax: 319-335-9006; E-mail: [email protected].
5 The abbreviations used are: HSC, hematopoietic stem cell; BrdU, bromo-2�-
deoxyuridine; LSC, leukemic stem cell; CML, chronic myelogenous leuke-mia; TKI, tyrosine kinase inhibitor; BMT, bone marrow transplantation; PBC,peripheral blood mononuclear cell.
crossmarkTHE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 291, NO. 21, pp. 11148 –11160, May 20, 2016
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
11148 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Although �-catenin itself does not have the ability to bindDNA after translocation into the nucleus, it interacts with Tcf/Lef transcription factors to modulate gene expression (9). Tcf/Lef factors contain 4 members, Tcf1, Tcf3, Tcf4, and Lef1, andall have a highly conserved high mobility group (HMG) DNAbinding domain. Whereas the requirements for Tcf1 and Lef1in blood cells, in particular T lymphocytes, have been well doc-umented (9, 26), none of the Tcf/Lef factors has been studied inHSCs. By focusing on Tcf1 and Lef1 in the study, we demon-strate that these two factors regulate the regenerative fitness ofHSCs and self-renewal of LSCs in a pre-clinical model of CML.
EXPERIMENTAL PROCEDURES
Animals—Tcf7�/�, Tcf7fl/fl, and Lef1fl/fl mice were described(27–29). Vav-Cre, C57BL/6 and B6.SJL mice were from theJackson Laboratory, and Cre-ERT2 mice were kindly providedby Dr. Yingzi Zhang (30). All mouse experiments were per-formed under protocols approved by the Institutional AnimalUse and Care Committee of the University of Iowa.
Flow Cytometry, Analysis of Apoptosis, Cell Cycle, and BrdUIncorporation—Single cell suspension from the bone marrow(BM) was prepared and surface-stained as previously described(31, 32). Apoptotic cells were detected with the PE AnnexinVApoptosis Detection Kit (BD Bioscience), and cell cycle analysiswas performed by intracellular staining for Ki67 and Hoechst33342 as described (31, 32). For BrdU labeling, the mice werei.p. injected with 1 mg of bromo-2�-deoxyuridine (BrdU) andfed in drinking water (1 mg/ml) for 20 h. Cells were surface-stained, fixed, and permeabilized, and intracellularly stainedwith an anti-BrdU antibody using the APC BrdU Flow Kit (BDBiosciences). The stained cells were analyzed on FACSCalibur,FACSVerse, or Becton Dickinson LSR II or LSR Violet flowcytometers (BD Biosciences). The data were processed usingFlowJo software (Version IX or X, TreeStar).
Cell Sorting and Quantitative RT-PCR—For quantification ofTcf7 and Lef1 transcripts in hematopoietic stem and progenitorcells, Flt3�LSK, Flt3�LSK, and Lin�c-Kit� cells were sorted onFACSAria, and the total RNA was extracted from sorted cellsand reverse-transcribed as described (28, 32). Plasmids con-taining Tcf7, Lef1, or Hprt coding sequence were used to gen-erate standard curves for each transcript, and Tcf7, Lef1, orHprt transcript in the sorted cells was determined by quantita-tive PCR. The copy numbers of Tcf7 and Lef1 were then calcu-lated assuming that Hprt is expressed at 10,000 copies. Theprimers for Tcf7 are 5�-cccttcctgcggatatagac and 5�-ggtacacca-gatcccagcat, those for Lef1 are 5�-tgagtgcacgctaaaggaga and5�-ctgaccagcctggataaagc, and those for Hprt are 5�-gcgtcgtgatt-agcgatgatg, and 5�-ctcgagcaagtctttcagtcc.
For comparative analysis of gene expression betweenTcf7�/�Lef1�/� and control Flt3�LSK cells or LSCs, cDNAwas generated as above and used in quantitative PCR. Theexpression of genes of interest was first normalized to Hprt ineach sample, and its relative expression in Tcf7�/�Lef1�/� cellswas then calculated by normalizing to control cells. The prim-ers are: Egr1, 5�-gagcgaacaaccctatgagc and 5�-gagtcgtttggctggg-ataa; Cdkn1b, 5�-ttgggtctcaggcaaactct and 5�-ttctgttctgttggcccttt;Cdkn1c, 5�-ggagcaggacgagaatcaag and 5�-ttctcctgcgcagttctctt;Fbxw7, 5�-tccagcgagacttcatctcc and 5�-ccagtatcgacaagtctgag;
Foxo1, 5�-cccacacagtgtcaagactac and 5�-cctcagttcctgctgtcagac;Foxo3, 5�-accaggctgaaggatcactg and 5�-ctgtccacttgctgagagcag;Gfi1, 5�-caacacaaggcagtgcactc and 5�-gccacagtactgacagggat; Pten,5�-gattacagacccgtggcact and 5�-gggtcctgaattggaggaat; Stat5a, 5�-tcatccagtaccaggagagc and 5�-gacacttgcttctgctggag; Tal1, 5�-gccct-ccccatatgagatgg and 5�-gatcagctttctgagctcag; Tcf3, 5�-gatgt-cccctccagtgctac and 5�-gtctttgggtcgatctctgg.
Isolation of Lin�c-Kit� BM Cells and Chromatin Immuno-precipitation (ChIP)—Total BM cells were isolated from WTC57BL/6 mice and depleted of lineage-positive cells. The Lin�
BM cells were then incubated with mouse CD117 Microbeads(Miltenyi Biotec) for positive selection of c-Kit� cells. TheLin�c-Kit� BM cells were cross-linked for 10 min with 1%formaldehyde in medium, then processed using a truChIPChromatin Shearing Reagent Kit (Covaris) and sonicated for 5min on Covaris S2 ultrasonicator. The sheared chromatin frag-ments, in the range of 200 –500 bp, were immunoprecipitatedwith an anti-Lef1 (C12A5, Cell Signaling Technologies) or con-trol rabbit IgG and was washed as described (29). The immu-noprecipitated DNA segments were used for quantification byPCR. For calculation of enrichment of Lef1 binding, each anti-Lef1 ChIP sample was first normalized to corresponding IgGChIP sample, and the signal at a target region was then normal-ized to that at the Hprt promoter region. The primers fordetecting the Gapdh and Lef1 genomic regions were described(29), and those for Egr1, 5�-cctgctcagttctccctcac and 5�-tgc-cctctgtaggtctctgg; those for Tcf3, 5�-gggctctgggtgttatgtgt and5�-ccatcgtttcctagcctctc; and those for Cdkn1c, 5�-aagagggact-caggtgagca and 5�-agagaactgcgcaggagaac. Note that one Tcf/Lef consensus motif “CAAAG” is found within the PCR ampli-con of the Tcf3 locus. Although no Tcf/Lef motif is within thePCR amplicon of the Egr1 locus, one motif is found at 52 bpupstream, two motifs are found at 55 bp and 216 bp down-stream of the amplicon.
Bone Marrow Transplantation (BMT)—For competitiverepopulation assays, the test BM cells were collected fromCD45.2� gene-targeted mice, and the competitor BM cellswere from CD45.1� B6.SJL mice. Both test and competitor BMcells were measured for LSK frequency, and the whole BM cellswere mixed at 1:1 LSK ratio (each containing 3,000 LSKs) andthen transplanted into lethally irradiated (1,050 rad)CD45.1�CD45.2� recipients. Sixteen weeks after the trans-plantation, contribution of test and competitor BM cells to dif-ferent blood lineages was determined in peripheral nucleatedblood cells (PBCs) by flow cytometry.
For serial transplantation assays, total BM cells (2 � 106)from CD45.2� gene-targeted mice were transplanted intolethally irradiated CD45.1� primary recipients. Eight weekslater, BM cells from the primary recipients were transplantedinto CD45.1� secondary recipients, and this process wasrepeated to obtain tertiary recipients. Sixteen weeks after eachtransplantation, PBCs were analyzed for the contribution fromthe original donors to different blood lineages.
Tamoxifen Treatment—For induced deletion ofTcf7FL/FLLef1FL/FL alleles using CreERT2 in the CML recipients,the mice were treated with tamoxifen at 0.2 mg/g body weightvia oral gavage for 4 consecutive days.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
MAY 20, 2016 • VOLUME 291 • NUMBER 21 JOURNAL OF BIOLOGICAL CHEMISTRY 11149
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from
CML Model and Treatment with Imatinib—The pMIG-p210BCR-ABL retroviral construct was provided by Dr. WarrenPear (34), and the retrovirus was packaged and used for infec-tion of Lin� BM cells as previously described (32). Twenty-fourhours after two rounds of spinofection, the infected Lin� BMcells containing 3,000 – 6,000 GFP� LSK cells along with 2 �105 protector BM cells were transplanted into irradiated B6.SJLrecipients. CML progression in the recipients was monitoredthrough regular evaluation of lethargy, weight loss, splenomeg-aly, and signs of morbidity or detection of GFP�Mac1� leuke-mic cells in the PBCs. For treatment of CML with TKI, imatinib(Novartis) was obtained from the Pharmacy of University ofIowa Healthcare and Clinics and administered to CML recipi-ents at 100 mg/kg body weight by oral gavage twice a day.
For secondary transplantation of LSCs, more than 5 primaryrecipients were sacrificed on day 15 post-BMT to harvest BMcells and sorted for GFP� LSK cells. 10,000 –15,000 sorted LSCswere then transplanted into another cohort of CD45.1� synge-
neic mice along with 2 � 105 protector BM cells for CMLpropagation.
Genetic Complementation with Forced Expression of Egr1—The control and Egr1-pLZRS retroviral vectors were providedby Dr. David Wiest (33), and the retroviruses were packaged toinfect Lin� BM cells as above. The infected cells were trans-planted into irradiated B6.SJL mice at 5 � 105 per recipientwithout additional protector BM cells. Six weeks later,CD45.2�GFP�Lin� BM cells were sorted from the recipientsfor cell cycle analysis as described above. The sorting isolationof GFP� cells was used to avoid quenching of GFP fluorescenceduring intracellular staining for Ki67.
Statistical Analysis—Unpaired comparison among differentgenetic groups was performed using the Student’s t test with atwo-tailed distribution assuming equal sample variance. Forsurvival of CML recipients, statistical significance between dif-ferent genetic and/or treatment conditions was assessed usinglog-rank test using Prism6 software.
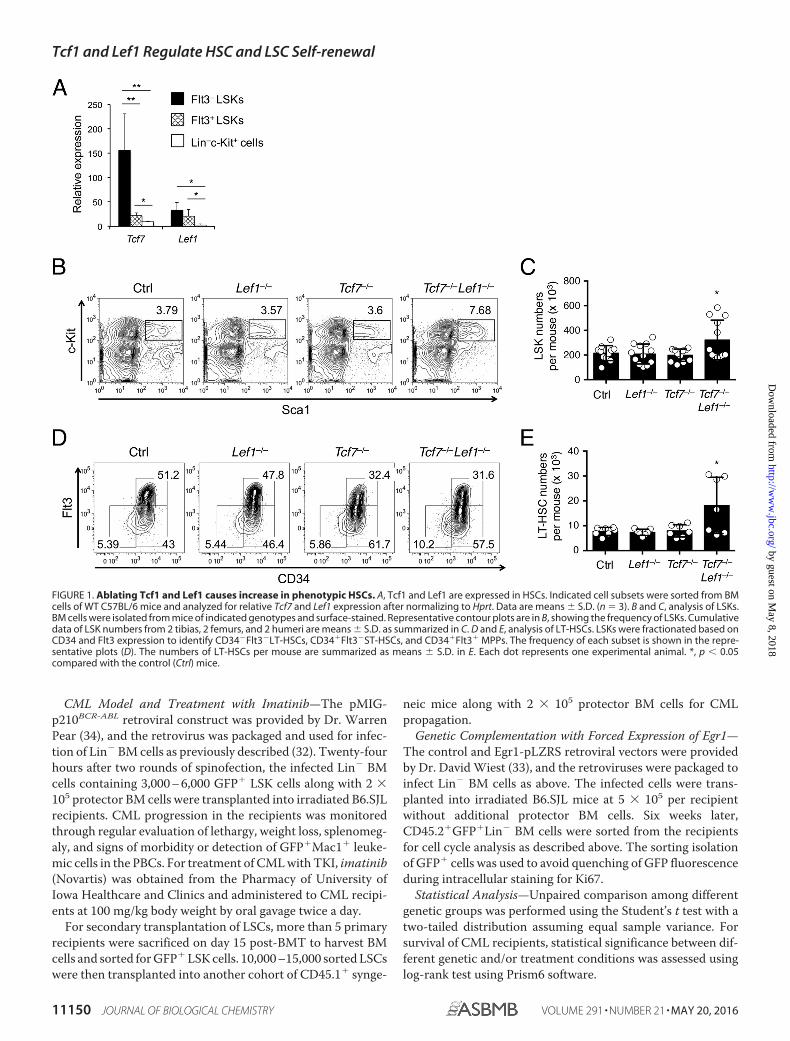
FIGURE 1. Ablating Tcf1 and Lef1 causes increase in phenotypic HSCs. A, Tcf1 and Lef1 are expressed in HSCs. Indicated cell subsets were sorted from BMcells of WT C57BL/6 mice and analyzed for relative Tcf7 and Lef1 expression after normalizing to Hprt. Data are means � S.D. (n � 3). B and C, analysis of LSKs.BM cells were isolated from mice of indicated genotypes and surface-stained. Representative contour plots are in B, showing the frequency of LSKs. Cumulativedata of LSK numbers from 2 tibias, 2 femurs, and 2 humeri are means � S.D. as summarized in C. D and E, analysis of LT-HSCs. LSKs were fractionated based onCD34 and Flt3 expression to identify CD34�Flt3�LT-HSCs, CD34�Flt3�ST-HSCs, and CD34�Flt3� MPPs. The frequency of each subset is shown in the repre-sentative plots (D). The numbers of LT-HSCs per mouse are summarized as means � S.D. in E. Each dot represents one experimental animal. *, p � 0.05compared with the control (Ctrl) mice.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
11150 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Results
Ablation of Tcf1 and Lef1 Causes Moderate Expansion ofthe HSC Pool—Tcf1 and Lef1 are abundantly expressed inT-lineage cells, and are readily detectable in Flt3� lineage-negative Sca1�c-Kit� cells (Flt3� LSKs), which contain bothlong-term (LT) and short-term (ST) HSCs (Fig. 1A). Tcf1expression (encoded by Tcf7) was sharply reduced upon differen-tiation of HSCs to Flt3� LSKs, the multipotent progenitors(MPPs). Both factors were further down-regulated in more differ-entiated Lin�c-Kit� myeloid progenitors (Fig. 1A). These expres-sion changes suggest a potential requirement for Tcf1 and Lef1 inHSCs.
We used VavCre to excise Lef1-floxed alleles (Lef1FL/FL),which resulted in complete ablation of the Lef1 protein inthe BM cells as shown previously (28). We then crossedVavCre-Lef1FL/FL (called Lef1�/�) to germline-targetedTcf7�/� mice to delete both genes. Whereas deleting eitherfactor alone had little effect, Tcf7�/�Lef1�/� mice showed atendency of increased LSK frequency and numbers in theBM cells (Fig. 1, B and C). We next used the CD34 and Flt3markers to identify CD34�Flt3� LT-HSCs within the LSKpopulation. No evident changes were detected in Lef1�/� orTcf7�/� LSKs; however, LT-HSCs exhibited increased fre-quency in Tcf7�/�Lef1�/� LSKs (Fig. 1D). The absolutenumbers of LT-HSCs were elevated in Tcf7�/�Lef1�/� miceaccordingly (Fig. 1E). These data demonstrate that loss ofTcf1 and Lef1 resulted in moderate expansion of the poolsize of phenotypic HSCs.
Loss of Tcf1 and Lef1 Diminishes HSC Function underRepeated Regenerative Stress and Competitive Condition—HSCs have self-renewal capacity to regenerate themselvesunder homeostatic conditions or in response to injuries. Weused serial transplantation assays, the gold standard for mea-suring HSC self-renewal. Utilizing disparate CD45 allele mark-ers, we transplanted the same numbers of CD45.2� BM cellsfrom Lef1�/�, Tcf7�/�, Tcf7�/�Lef1�/� mice or their litter-mate controls into CD45.1� recipients. We then determinedthe contribution of donor BMs to blood reconstitution inperipheral blood mononuclear cells (PBCs) at 16 weeks afterthe BM transplantation (BMT), because this time point allowsexclusive measurement of long-term blood reconstitutioncapacity of donor HSCs. Consistent with previous reports thatTcf1-deficient BM cells fail to generate T cells in the BM chi-meras (35, 36), there were few CD45.2�CD3� T cells producedfrom Tcf7�/� or Tcf7�/�Lef1�/� BM cells (not shown). Toavoid potential bias resulting from loss of T cells in the absenceof Tcf1, we analyzed the non-T compartment in PBCs by focus-ing on CD3� cells. After excluding T cells in the PBCs, we foundthat BM cells from all genotypes were able to restore CD3�
blood lineages similarly in these primary recipients (Fig. 2A).This result also indicates that loss of Tcf1 and/or Lef1 does notaffect HSC homing.
We then isolated BM cells from the primary recipients fol-lowed by transplantation into secondary CD45.1� recipients,and repeated this process to generate tertiary recipients.Whereas BM cells from all genotypes remained capable of
FIGURE 2. Ablating Tcf1 and Lef1 moderately compromises HSC functions under regenerative stress or competitive conditions. A, serial transplantationassay. Total BM cells (2 � 106) from test animals (CD45.2�) were transplanted into CD45.1� primary recipients. Eight weeks later, BM cells from the primaryrecipients were transplanted into CD45.1� secondary recipients, and this process was repeated to obtain tertiary recipients. The PBCs were analyzed at � 16weeks after each transplantation to determine the contribution from the original donors. Data on the contribution to CD3� non-T cells are shown to avoid Tlineage-derived bias. Data were pooled from two experiments, with 4 –9 recipients analyzed at each transplantation stage. B and C, competitive repopulationassay. Test (CD45.2�) and competitor (CD45.1�) BM cells, each containing 3,000 LSKs, were mixed and transplanted into CD45.1�CD45.2� recipients. After 16weeks, their contribution to CD3� non-T cells was measured in PBCs. Representative contour plots (B) and cumulative data (C) from three independentexperiments are shown, with 4 –12 recipients analyzed. **, p � 0.01; ***, p � 0.001 compared with control (Ctrl) mice.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
MAY 20, 2016 • VOLUME 291 • NUMBER 21 JOURNAL OF BIOLOGICAL CHEMISTRY 11151
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

reconstituting all non-T blood lineages in the secondary recip-ients (Fig. 2A), in the tertiary recipients, Lef1�/� BM cellsexhibited moderately reduced contribution, and loss of bothTcf1 and Lef1 significantly impaired blood reconstitution (Fig.2A). These results demonstrate that Tcf1 and Lef1 contribute tomaintaining the regenerative fitness of HSCs.
As shown above, HSCs lacking Tcf1 and/or Lef1 were com-petent to reconstitute non-T blood lineages in both the primaryand secondary recipients in the absence of competitors. Wenext investigated their competitive repopulation capacity. Weobtained BM cells from the CD45.2� gene-targeted strains (astest HSCs) and those from CD45.1� WT mice (as competitor
FIGURE 3. Tcf1 and Lef1 contribute to maintaining HSC quiescence. A and B, cell cycle analysis. BM cells from mice of indicated genotypes were surface-stained to identify CD34�Flt3� LT-HSCs and then intracellularly stained for Ki67 and Hoechst 33342. The percentages of HoechstloKi67� G0, HoechstloKi67�
G1, and Hoechstmed-hiKi67� S/G2/M phase cells in LT-HSCs are shown in representative dot plots (A). Cumulative data on LT-HSCs in G0 and G1 phases aresummarized in B. Data are means � S.D. (n � 5–11 from five experiments). C and D, BrdU incorporation in LT-HSCs. Mice were injected i.p. with 1 mg of BrdU,and 24 h later, BM cells were harvested, surface-stained, and intracellularly stained with anti-BrdU antibody. The percentage of BrdU� cells in LT-HSCs is shownin representative contour plots (C). Cumulative data from three experiments are summarized in D. E, detection of apoptotic cells. LT-HSCs were identified bysurface staining and further stained for AnnexinV and 7-AAD. The percentages of AnnexinV�7-AAD� apoptotic cells in LT-HSCs are summarized (n � 4 – 8 from4 experiments). In B, D, and E, the average fold changes of Tcf7�/�Lef1�/� over the control mice are marked in the bars. ns, not statistically significant; **, p �0.01; ***, p � 0.001 compared with control (Ctrl) mice.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
11152 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

HSCs) and determined the LSK frequency. Because the fre-quency of LSK cells was elevated in Tcf7�/�Lef1�/� BM cells(Fig. 1 B), for a fair comparison among the test HSCs, we trans-planted the same numbers of LSKs into lethally irradiatedCD45.1�CD45.2� recipients, instead of using the same num-bers of total BM cells. After 16 weeks, we assessed the relativecontribution of test HSCs to non-T cell blood lineages in PBCs.The control test BM cells moderately outcompeted the com-petitor BM cells, constituting 65% of the non-T blood cells(Fig. 2, B and C). Whereas Tcf7�/� BM cells were similarlycompetent in reconstitution non-T blood cells, the contribu-tion by Lef1�/� BM cells was moderately decreased. On theother hand, the contribution by Tcf7�/�Lef1�/� BM cells wasreduced to 50% of that by control BM cells (Fig. 2, B and C).These observations indicate that Tcf1 and Lef1 have redundantroles in regulating HSC activities, similar to their functions in T
lineage cells (26). These analyses collectively identified arequirement for Tcf1 and Lef1 in maintaining functional HSCs,especially under competitive conditions or repetitive regenera-tive stress.
Tcf1 and Lef1 Contribute to Maintaining HSC Quiescence—To elucidate the mechanisms by which Tcf1 and Lef1 regulatethe HSC biological activities, we first measured its cell cyclestatus. By combination of Ki67 and Hoechst33342, which mea-sures cellular proliferation and DNA content, respectively, wefound that majority of WT CD34�Flt3� LT-HSCs are inHoechstloKi67� G0 phase, with a small portion in Hoechst-loKi67� G1 phase and very few in Hoechstmed-hiKi67� S/G2/Mphase (Fig. 3A). Although loss of Tcf1 or Lef1 alone did not havea discernible impact, Tcf7�/�Lef1�/� LT-HSCs showed mod-est but consistent reduction of the proportion in G0 phase, withcorresponding increase of that in G1 phase (Fig. 3, A and B).
FIGURE 4. Tcf1 and Lef1 act upstream of Egr1 to regulate HSC quiescence. A, gene expression analysis. Flt3� LSK cells were sorted from Tcf7�/�Lef1�/� andlittermate controls and analyzed for expression of indicated genes. Data are means � S.D. (n � 4 from three experiments). *, p � 0.05; **, p � 0.01; ***, p � 0.001by Student’s t test. B, Lef1 is directly associated with 5�-regulatory regions in the Egr1 and Tcf3 genes. ChIP with a Lef1 antibody was performed on Lin�c-Kit�
BM cells, and enriched Lef1 binding was assessed by quantitative PCR at the indicated gene loci. Data are means � S.D. from two independent experimentswith duplicate measurements. C and D, forced expression of Egr1 restores quiescence in Tcf7�/�Lef1�/� HSCs. Lin� BM cells from control or Tcf7�/�Lef1�/�
mice were retrovirally infected with empty pLZRS vector or Egr1-pLZRS and then transplanted into irradiated CD45.1� hosts. Six weeks later,CD45.2�GFP�Lin� BM cells were sorted from the BM chimeras, surfaced stained to identify Flt3� LSK cells, and analyzed for cell cycle status as in Fig. 2A.Representative contour plots (C) and cumulative data (D) are from three experiments (n � 3– 4). ns, not statistically significant. *, p � 0.05 for the indicatedpaired comparison.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
MAY 20, 2016 • VOLUME 291 • NUMBER 21 JOURNAL OF BIOLOGICAL CHEMISTRY 11153
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Consistent with loss of quiescence, Tcf7�/�Lef1�/� LT-HSCsalso exhibited increased incorporation of BrdU into the DNA(Fig. 3, C and D), indicative of more cycling cells. In contrast,AnnexinV� 7-AAD� apoptotic cells in Tcf7�/�Lef1�/� LT-HSCs were not significantly different from those in control LT-HSCs (Fig. 3E). Collectively, Tcf1 and Lef1 contribute to main-taining HSCs at a quiescent state.
Tcf1 and Lef1 Act Upstream of Egr1 and Other HSCRegulators—We next aimed to gain molecular insights into theregulation of HSC quiescence and function by Tcf1 and Lef1. Anumber of factors have been identified to be responsible formaintaining HSC quiescence, and genetic ablation of these fac-tors leads to moderate expansion of HSCs accompanied byfunctional defects due to exhaustion (5). These factors includetranscription factors such as Egr1 (37), Gfi1 (38), Foxo factors(39, 40), Stat5 (41), E2A/E47 (encoded by Tcf3) (42), and Scl(encoded by Tal1) (43); an ubiquitin ligase Fbw7 (44); a signal-
ing molecule Pten (45); and cyclin-dependent kinase inhibitorssuch as p27-Kip1 and p57-Kip2 (encoded by Cdkn1b andCdkn1c, respectively) (46, 47). We sort-purified Flt3� LSK cellsfrom Tcf7�/�Lef1�/� and control mice and scanned for theexpression changes of these known HSC regulators. Theexpression of Egr1, Tcf3, and Cdkn1c was substantially dimin-ished in Tcf1/Lef1-deficient hematopoietic stem/progenitorcells, whereas other genes were not affected or moderatelyreduced in expression (Fig. 4A).
We then investigated if Tcf1 and Lef1 directly regulate Tcf1/Lef1-dependent genes. To this end, we searched the proximalregulatory regions, i.e. �1.5 kb to �0.5 kb sequence flanking thetranscription start site (TSS) of genes of interest for consensusTcf/Lef binding motif “CAAAG.” We found three motifs at�1,378, �1,124, and �963 bp upstream of the Egr1 TSS, one at�180 bp upstream of the Tcf3 TSS, and one at �310 bpupstream of the Cdkn1c TSS. We then performed chromatin
FIGURE 5. Tcf1 and Lef1 are differentially required for CML progression in primary and secondary recipients. A, experimental design for establishing themurine CML model. Lin� BM cells from control or Tcf7�/�Lef1�/� mice were infected with p210BCR-ABL retrovirus. The infected cells (each containing 3,000 to6,000 GFP� LSK cells) along with 2 � 105 protector BM cells were transplanted into irradiated CD45.1� syngeneic mice as the primary recipients. B, leukemiaburden in PBCs of the primary recipients. CD45.2�Mac1�GFP� leukemia cells were detected in PBCs on indicated days post-BMT, and their frequency is shownas means � S.D. (n � 9 –12 from two experiments). C, Kaplan-Meier survival curves of the primary recipients, pooled from two experiments. D, experimentaldesign for testing LSC self-renewal. The CML in primary recipients was established as in A, and CD45.2�GFP� LSK cells were sorted from the primary recipientson day 15 post-BMT. 10,000 –15,000 sorted cells were then transplanted into the secondary CD45.1� recipients along with 2 � 105 protector BM cells. E,leukemia burden in the secondary recipients, as determined in PBCs on day 14 post-BMT. Cumulative data from one of two independent experiments areshown. F, Kaplan-Meier survival curves of the secondary recipients, pooled from two experiments. ns, not statistically significant; *, p � 0.05; ***, p � 0.001compared with control recipient mice.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
11154 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

immunoprecipitation (ChIP) on primary murine Lin�c-Kit�
BM progenitor cells using an anti-Lef1 antibody. The pro-genitor cells were used instead of primary HSCs becauseHSCs constitute only a rare population in mouse bone mar-row but ChIP assay for transcription factor binding usuallyrequires relatively large numbers of purified cells to obtainreliable results. The use of Lef1 antibody is because Lef1protein was more readily detectable than Tcf1 in Lin�c-Kit�
BM cells (not shown). Lef1 itself is a direct target gene of theTcf/Lef factors (28) and used as a positive control. We foundthat Lef1 exhibited enriched binding at the 5�-regulatoryregions of Egr1 and Tcf3 but not that of Cdkn1c (Fig. 4B).These data suggest a direct regulatory role of Lef1, and likelyTcf1 as well, in the expression of key HSC molecules in thehematopoietic progenitor cells.
The phenotypic and functional HSC impairments are strik-ingly similar between Egr1�/� and Tcf7�/�Lef1�/� mice,including expansion of the HSC pool and loss of HSC quies-cence (37). We next investigated if genetic complementation of
Tcf7�/�Lef1�/� cells with Egr1 restores HSC quiescence (33).We used retroviral transduction to achieve forced expression ofEgr1 in control or Tcf7�/�Lef1�/� Lin� BM cells and thentransplanted into CD45.1� hosts to make BM chimeras. Thebicistronic pLZRS retroviral vector uses GFP as an expressionindicator, and we focused our cell-cycle analysis on sortedCD45.2� GFP�Flt3�LSK cells in the BM chimeras. In theempty pLZRS vector-transduced group, Tcf7�/�Lef1�/� HSCscontinued to exhibit reduced portion in G0 phase with a mod-erate increase in S/G2/M phases (Fig. 4, C and D). Whereasforced expression of Egr1 did not detectably alter the cell cyclestatus of control HSCs, Egr1-complemented Tcf7�/�Lef1�/�
HSCs showed increase in quiescent G0 phase and substantialdecrease in S/G2/M phases (Fig. 4, C and D). In steady state,Tcf7�/�Lef1�/� HSCs showed an increase in G1 phase com-pared with control HSCs (Fig. 3, A and B); however, the differ-ence in G1 phase was not evident between Tcf7�/�Lef1�/� andcontrol HSCs in the BM chimeras, whether transduced with thepLZRS vector or Egr1-expressing retroviruses (Fig. 4, C and D).
FIGURE 6. Tcf1 and Lef1 are intrinsically required for LSC self-renewal. A, experimental design. Lin� BM cells from WT or CreERT2-Tcf7FL/FLLef1FL/FL mice wereused to establish CML in the primary recipients, which were untreated or treated with Tamoxifen during days 12–15 post-BMT. One day later, BM cells from theprimary recipients were lineage-depleted, and the Lin� BM cells containing 12,000 –16,000 of CD45.2�GFP�Sca1� cells were transplanted into CD45.1�
secondary recipients along with 2 � 105 protector BM cells. B, leukemia burden in the secondary recipients, as determined in PBCs on day 25 post-BMT.Cumulative data from two independent experiments are shown. ns, not statistically significant; ***, p � 0.001 compared with WT recipient mice. C, Kaplan-Meier survival curves of the secondary recipients, pooled from two experiments.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
MAY 20, 2016 • VOLUME 291 • NUMBER 21 JOURNAL OF BIOLOGICAL CHEMISTRY 11155
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

A likely explanation is that when the BM chimeras were ana-lyzed at 6 weeks after HSC transplantation, similar amounts ofTcf7�/�Lef1�/� and control HSCs remained in a cycling-readystate during the process of active reconstitution of hematopoi-esis. Collectively, these data support the notion that Tcf1 andLef1 act, at least in part, through transcriptional activation ofEgr1 to maintain HSC quiescence.
Targeting Tcf1 and Lef1 Impairs Self-renewal of LSCs—Wenext investigated a requirement for Tcf1 and Lef1 in LSCs.Because a moderate impact on HSC functionalities was onlyevident in the absence of both factors, we focused onTcf7�/�Lef1�/� cells. We used a preclinical CML model, wherewe infected Tcf7�/�Lef1�/� or control BM cells with a bicis-tronic retrovirus expressing the p210BCR-ABL fusion proteinalong with GFP, followed by transplantation into irradiatedCD45.1� congenic recipients (Fig. 5A) (34). Forced expressionof p210BCR-ABL resulted in malignant transformation of hema-topoietic stem/progenitor cells and accumulation ofGFP�Mac1� myelogenous leukemic cells. The leukemic cellswere readily detectable in the PBCs, and all the recipients suc-cumbed to the disease by day 40 post-BM transplantation(post-BMT) (Fig. 5, B and C). Accumulation of Tcf7�/�Lef1�/�
GFP�Mac1� leukemic cells was not significantly different fromthat of control leukemic cells during the early stages of diseasedevelopment (days 10 and 14 post-BMT), albeit it was lowerthan control on day 18 post-BMT (Fig. 5B). The survival ofTcf7�/�Lef1�/� CML recipients was marginally extendedcompared with that of control recipients (Fig. 5C). These datasuggest that Tcf1/Lef1 deficiency only minimally affected theonset and progression of CML, mirroring their role in HSCs atthe steady state.
We next sort-purified GFP� LSKs from the BM cells of pri-mary recipients as LSCs and transplanted the same numbers ofTcf7�/�Lef1�/� or control LSCs into secondary recipients (Fig.5D), which is the most stringent test for LSC self-renewal. Thecontrol LSCs were able to propagate CML in the secondaryhosts, with leukemic cells readily detected in PBCs on day 14post-BMT; in contrast, Tcf7�/�Lef1�/� LSCs gave rise to sub-stantially lower numbers of leukemic cells (Fig. 5E). Consistentwith the difference in leukemia burden, all the secondary recip-ients of control LSCs succumbed to the disease by day 60 post-BMT, but all those of Tcf7�/�Lef1�/� LSCs survived beyondday 80 post-BMT (Fig. 5F). We demonstrated above that therepopulation capacity of Tcf7�/�Lef1�/� HSCs was only evi-dently diminished in the tertiary recipients. These data in CMLsuggest that LSCs are more dependent on Tcf1 and Lef1 forself-renewal than HSCs.
Tcf1 and Lef1 Are Intrinsically Required for LSCSelf-renewal—Because Tcf7 was targeted in germline in theTcf7�/� model, the Tcf7�/�Lef1�/� BM cells may have beenexposed to Tcf1-deficient niche environment or have otherintrinsic defects, leading to unwanted changes inTcf7�/�Lef1�/� LSCs. To address these potential caveats, wetook advantage of a recently established Tcf7FL/FL strain (29)and generated Tcf7FL/FLLef1FL/FL mice. To achieve inducibleinactivation of both floxed genes, we used a CreERT2 strain inwhich the fusion protein of Cre recombinase and estrogenreceptor (ER) was driven by the ubiquitously expressed Rosa26locus (30). In the resulting CreERT2 Tcf7FL/FLLef1FL/FL mice,treatment with tamoxifen for 4 consecutive days effectivelyablated both Tcf7 and Lef1 genes in hematopoietic cells asdetermined in splenic T cells (not shown). We used BM cells
FIGURE 7. Tcf1 and Lef1 are essential for LSC survival. BM cells from CreERT2-Tcf7FL/�Lef1FL/� or CreERT2-Tcf7FL/FLLef1FL/FL mice were used to establish CML,and the primary recipients were treated with Tamoxifen, and then enriched for LSCs followed by transplantation into secondary recipients as in Fig. 6A. Fifteendays post-BMT, the BM cells were harvested from the secondary recipients for analysis. A and B, cell cycle analysis in LSCs. Lin�GFP� BM cells were sorted andsurface-stained to identify Sca1�c-Kit� cells followed by intracellular staining of Ki67 and Hoechst33342. The percentages of G0, G1, and S/G2/M phase cells inGFP�LSKs are shown in representative contour plots (A). Cumulative data on LSCs in G0 and S/G2/M phases are summarized in B. Data are means � S.D. (n �6 from 2 experiments). C and D, analysis of apoptosis in LSCs. BM cells were surface-stained to identify GFP�LSKs without cell sorting, followed by detection ofAnnexin-V and Hoechst33342. Contour plots (C) and cumulative data (D) are representative of two experiments with similar results. E, detection ofGFP�Lin�cKit� transformed myeloid progenitor cells in the BM of secondary recipients. Data are representative of two experiments with similar results. ns, notstatistically significant; **, p � 0.01 compared with control (Tcf7FL/�Lef1FL/�) mice.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
11156 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

from CreERT2Tcf7FL/FLLef1FL/FL mice to establish CML so thatthe LSCs were generated without pre-existing conditions. Weleft one cohort of primary recipients untreated, and treatedanother cohort with tamoxifen to induce Tcf7 and Lef1 genedeletion, followed by transplantation of Lin� BM cells contain-ing the same numbers of GFP�Lin�Sca1� cells into second-ary recipients (Fig. 6A). This approach of using theGFP�Lin�Sca1� phenotype to enumerate LSCs has beenshown to have similar capacity as using sorted GFP�LSKs topropagate CML in secondary or tertiary recipients (25), withthe advantages of obtaining sufficient LSCs from fewer donormice and avoiding additional stress on LSCs due to cell sorting.In parallel, we used Tcf1/Lef1-sufficient WT BM cells to estab-lish CML coupled with tamoxifen treatment as another controlgroup. LSCs from tamoxifen-treated WT mice and those fromuntreated CreERT2Tcf7FL/FLLef1FL/FL mice had similar capacityto generate leukemic cells and led to death of the secondaryrecipients by day 50 post-BMT (Fig. 6, B and C). This observa-tion indicates that tamoxifen treatment per se had little impacton CML propagation. In contrast, very few leukemic cells weregenerated from LSCs derived from tamoxifen-treatedCreERT2Tcf7FL/FLLef1FL/FL mice, and all the secondary recipi-ents of these LSCs survived beyond day 80 post-BMT. Thesedata demonstrate an intrinsic requirement for Tcf1 and Lef1 inmaintaining self-renewal capacity of LSCs.
It has been proposed that self-renewal of stem cells is deter-mined by a balance among three major cellular events, prolif-
eration, survival, and differentiation (48). To further investigatethe impact of Tcf1/Lef1 deficiency on self-renewing LSCs, weused Lin� BM cells from CreERT2 Tcf7FL/FLLef1FL/FL or Cre-ERT2 Tcf7FL/�Lef1FL/� mice to establish CML, treated the pri-mary recipients with tamoxifen and transplanted enrichedLSCs into secondary recipients as in Fig. 6A. Fifteen days post-BMT, we harvested BM cells from the secondary recipients andfound that the total BM cells and GFP�LSKs from Tcf1/Lef1-deficient LSC recipients were 29 � 5.4% and 19.2 � 3.8% ofthose from control LSC recipients, respectively. Cell cycle anal-ysis in GFP�LSK cells in the secondary recipients revealed thatboth Tcf1/Lef1-sufficient and -deficient LSCs showed similarfrequency in the quiescent G0 state or in the actively cyclingS/G2/M state (Fig. 7, A and B). On the other hand, Tcf1/Lef1-deficient LSCs in the secondary recipients were severely com-promised in survival compared with control LSCs, showing3– 4-fold increase in Annexin-V�Hoechest33342– apoptoticpopulation (Fig. 7, C and D). Because of the extreme paucity ofTcf1/Lef1-deficient LSCs in the secondary recipients, it was notfeasible to perform colony formation assays which requiredsorting separation. Nonetheless, the GFP�Lin�c-Kit� trans-formed myeloid progenitor cells, which are derived from GFP�
LSKs, exhibited similar frequency in the BM of Tcf1/Lef1-defi-cient or control LSC recipient mice (Fig. 7E). We infer that lossof Tcf1 and Lef1 did not potently affect LSC differentiation.These data collectively indicate that the impaired self-renewal
FIGURE 8. Targeting Tcf1 and Lef1 synergizes with imatinib therapy in controlling CML. A, experimental design. The primary CML recipients wereestablished and treated with imatinib from day 8 post-BMT. B and C, characterization of LSCs. The primary recipients were treated with imatinib for 3 weeks. Onday 29 post-BMT, the BM cells were harvested, and LSKs were detected in CD45.2�GFP� BM cells with the frequency shown in representative contour plots (B).The frequency and numbers of CD45.2�GFP� LSKs in BM cells in each recipient were in C. Data are means � S.D. (n � 8 –9 from two experiments). **, p � 0.01;***, p � 0.001 compared with control (Ctrl) mice. D, Kaplan-Meier survival curves of the CML recipients, untreated or treated with imatinib during days 8 – 80post-BMT. Data are from two experiments.
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
MAY 20, 2016 • VOLUME 291 • NUMBER 21 JOURNAL OF BIOLOGICAL CHEMISTRY 11157
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

of Tcf1/Lef1-deficient LSCs can be largely attributed to a strongdependence of CML LSCs on Tcf1 and Lef1 for survival.
Targeting Tcf1 and Lef1 Synergizes with TKI in CMLTherapy—The TKI therapy is effective in controlling thegrowth of CML leukemic blasts but lacks strong effect on qui-escent LSCs and thus cannot prevent the disease relapse (49).Given the potent impact of targeting Tcf1 and Lef1 on LSCself-renewal, we investigated if the combination of Tcf1/Lef1deficiency with imatinib could better control CML in the pri-mary recipients (Fig. 8A). After establishing CML usingTcf7�/�Lef1�/� or control BM cells, we treated the recipientswith imatinib for 3 weeks. By the end of treatment, all the CMLrecipients were protected from the lethal disease and survivedbeyond day 50 post-BMT. Both control and Tcf7�/�Lef1�/�-derived LSCs engrafted recipients similarly during the earlystage of CML onset, as measured by leukemia burdens in therecipient PBCs (Fig. 5B). To determine if imatinib treatmentaffects LSC persistence, we detected GFP� LSKs as LSCs inthe BM from imatinib-treated recipients and found thatTcf7�/�Lef1�/�-derived LSCs were greatly diminished infrequency and numbers (Fig. 8, B and C). Continuous ima-tinib treatment further prolonged survival of recipients ofp210BCR-ABL-infected control BM cells, albeit majority ofthem eventually succumbed to CML (Fig. 8, A and D). Incontrast, recipients of p210BCR-ABL-infected Tcf7�/�Lef1�/� BMcells were better protected by imatinib treatment, with morethan 80% of the recipients survived at the end of observationperiod (Fig. 8D). These findings suggest that targeting Tcf1 andLef1 has synergistic effect with TKI treatment to better controlCML progression and relapse.
Discussion
In this study, we identified differential requirements for theTcf1 and Lef1 transcription factors for the normal HSCs andtransformed LSCs. For the HSCs, there is a modest requirementfor Tcf1 and Lef1. Loss of both factors resulted in moderatelydiminished quiescence of HSCs without discernible effect onHSC survival, which leads to an expansion of the HSC pool.Despite the increase in phenotypic HSCs in Tcf1/Lef1-deficientmice, they are functionally inferior, especially under competi-tive conditions or regenerative stress. HSCs lacking Lef1 alonealso showed a moderate reduction in reconstitution of multipleblood lineages under competitive condition. This is line with arecent report that shRNA-mediated knockdown of Lef1 inmobilized BM progenitor cells diminishes spleen-colony for-mation capacity on day 12 post-BMT (50). Nonetheless, theHSC impairments were more evident in the absence of Tcf1and Lef1, indicating a functional redundancy. HSCs lackingboth Tcf1 and Lef1 were able to successfully repopulate allnon-T blood lineages in primary and secondary hosts, but theircapacity for blood regeneration was compromised in the terti-ary hosts. This functional deficiency remarkably resembles thatobserved in transgenic expression of Wnt inhibitors such asDkk1 and Wif1 and in Wnt3a-deficient fetal liver cells (11–13).In those studies where Wnt activity is manipulated, a generalconcern is that such intervention has a side effect on bonestructure and/or stromal cells and thus may have altered HSCniches. Our data on Tcf1 and Lef1, the nuclear effector proteins
downstream of the canonical Wnt pathway, provide supportingexperimental evidence to the conclusion that Wnt ligands havedirect impact on HSCs per se to modulate their regenerativecapacity.
On the mechanistic side, the expression of Egr1, Tcf3 andCdkn1c depended on Tcf1 and Lef1 in hematopoietic stem/progenitor cells. In addition, the 5�-regulatory regions of Egr1and Tcf3 were bound by Lef1 in BM progenitor cells, albeit wecannot exclude the possibility that Cdkn1c may be regulated byTcf1/Lef1 factors through distal elements. Egr proteins are theimmediate early response transcription factors and areinvolved in stress responses in many tissues. The molecularconnection between Tcf1/Lef1 and Egr proteins has beenobserved in immature thymocytes as well (51). By genetic com-plementation, we showed that forced expression of Egr1 wassufficient to rectify the diminished quiescence in Tcf1/Lef1-deficient HSCs. These data suggest that Egr1 is one of the keytarget genes regulated by Tcf1 and Lef1 in preventing excessiveHSC cycling. Cell cycle regulators are essential for maintainingHSC quiescence (52). Among the Cip/Kip family of cyclin-de-pendent kinase inhibitors, only genetic targeting of Cdkn1c (thep57-Kip2) caused evident loss of HSC quiescence, albeit it doeshave cooperative roles with other family members, p21-Cip(encoded by Cdkn1a) and p27-Kip1, in the maintenance ofHSCs (46, 47). The role of Tcf1 and Lef1 in HSC quiescence maybe also mediated by regulation of the p57-Kip2, suggesting amultifaceted regulatory circuit under the control of Tcf/Lef fac-tors. Future studies should address a possible redundancy ofTcf1/Lef1 with Tcf3 and Tcf4 factors in the same family, whichwill further elucidate their downstream genes that contribute toregulation of HSC homeostasis.
�-Catenin stabilization and nuclear translocation are keyevents upon activation of the canonical Wnt pathway. The roleof �-catenin in HSCs has been a highly contentious issue.Induced deletion of �-catenin alone or in combination with itshomologue �-catenin did not have a detectable impact on HSCsand blood lineages under homeostatic conditions (19, 20).Under competitive conditions, the �-catenin-deficient BMcells showed diminished contribution to all blood lineages, andtheir contribution was further reduced when transplanted intosecondary recipients (23). Deletion of both Tcf1 and Lef1showed similar effect, with no obvious impact on HSCs at thesteady state but diminishing their competitive repopulationcapacity. Our new observations on Tcf1/Lef1 deficiency reiter-ate the intrinsic requirements of Wnt-�-catenin pathway inHSCs. Our data are also in line with a recent finding that�-catenin is required for HSC regeneration and BM recoveryafter radiation or chemotherapy (53).
Unlike the moderate impact on normal HSCs, Tcf1 and Lef1deficiency more potently affected LSCs in the preclinical CMLmodel. Imatinib treatment prolonged the survival of primaryCML recipients and hence facilitated detection of LSCs at alater time point after BM transplantation. In this context, tar-geting Tcf1 and Lef1 greatly diminished numbers of LSCs in theBM. Furthermore, Tcf1/Lef1-deficient LSCs failed to propagatethe disease after transplantation into the secondary recipients.Mechanistically, the self-renewing CML LSCs critically dependon Tcf1 and Lef1 for survival, whereas their proliferation and
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
11158 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

differentiation were not significantly altered due to Tcf1/Lef1deficiency. Therefore, Tcf1 and Lef1 are essential for maintain-ing the pool size and self-renewal capacity of LSCs. In supportof this notion, it was previously reported that forced expressionof Lef1 in BM progenitors causes B lymphoblastic and acutemyeloid leukemia, which are likely propagated by LSCs withlymphoid characteristics (54). An important goal in chemo-therapy is to eliminate malignant cells and minimize damage tonormal cells. Our studies show that HSCs and LSCs have dif-ferential requirements for Tcf1/Lef1 in their self-renewalcapacity. Tcf1 and Lef1 can therefore be exploited as novel ther-apeutic targets in eradicating LSCs in hematological malignan-cies with less adverse effect on normal hematopoiesis.
Aberrant activation of �-catenin is a hallmark event in tumorinitiation, growth and metastasis, and has been a well sought-after drug target in cancer therapy (55). It has been demon-strated that genetic ablation or pharmacological inhibition of�-catenin expression synergizes with imatinib in improvingsurvival of CML mice; however, this effect is only observed afterthe LSCs were serially transplanted into tertiary recipients (25).In contrast, the synergy of Tcf1/Lef1 deletion and imatinib wasevident in the primary recipients where the CML was firstestablished. These observations imply that the role of Tcf1 andLef1 in LSCs may not be mediated exclusively by �-catenin. Infact, our recent studies showed that Tcf1 and Lef1 directlyinteract with Runx3 transcription factor and this interactioncontributes to Cd4 gene silencing in CD8� T cells (29). Anotherstudy mapped genome-wide binding locations of Tcf1 andRunx1 transcription factors in a mouse EML (erythroid, mye-loid, and lymphocytic) multipotent hematopoietic precursorcells (56). Significantly, the Tcf1 and Runx1 binding peaks over-lap at more than 40% of all their binding locations. The differ-ential use of cofactors (�-catenin versus Runx proteins) by Tcf1and Lef1 merits further investigation. It is currently recognizedthat transcriptional programs can be druggable targets (57).Detailed delineation of Tcf1/Lef1-dependent transcriptionalprogram in HSCs and LSCs will facilitate identification of smallmolecules to antagonize Tcf1/Lef1 activities in LSCs and fur-ther in vivo assessment of their therapeutic potentials in treat-ing leukemia.
Author Contributions—S. Y. and H. H. X. designed the experimentsand wrote the manuscript. S. Y., F. Y., and S. X. performed the exper-iments and analyzed the data. W. P. directed the ChIP studies. T. Z.assisted the overall experiments.
Acknowledgments—We thank Dr. H. Clevers (Hubrecht Institute, theNetherlands) for the permission of using Tcf7�/� mice, Dr. DavidWiest (Fox Chase Cancer Center) for providing the Egr1-pLZRS andcontrol retroviral constructs, the University of Iowa Flow CytometryCore facility (J. Fishbaugh, H. Vignes, and G. Rasmussen) for cell sort-ing, and Radiation Core facility (A. Kalen) for mouse irradiation.
References1. Orkin, S. H., and Zon, L. I. (2008) Hematopoiesis: an evolving paradigm for
stem cell biology. Cell 132, 631– 6442. Doulatov, S., Notta, F., Laurenti, E., and Dick, J. E. (2012) Hematopoiesis:
a human perspective. Cell Stem Cell 10, 120 –1363. Zon, L. I. (2008) Intrinsic and extrinsic control of haematopoietic stem-
cell self-renewal. Nature 453, 306 –3134. Wilson, A., and Trumpp, A. (2006) Bone-marrow haematopoietic-stem-
cell niches. Nat. Rev. Immunol. 6, 93–1065. Rossi, L., Lin, K. K., Boles, N. C., Yang, L., King, K. Y., Jeong, M., Mayle, A.,
and Goodell, M. A. (2012) Less is more: unveiling the functional core ofhematopoietic stem cells through knockout mice. Cell Stem Cell 11,302–317
6. Huntly, B. J., and Gilliland, D. G. (2005) Leukaemia stem cells and theevolution of cancer-stem-cell research. Nat. Rev. Cancer 5, 311–321
7. Visvader, J. E., and Lindeman, G. J. (2012) Cancer stem cells: current statusand evolving complexities. Cell Stem Cell 10, 717–728
8. Sawyers, C. L. (1999) Chronic myeloid leukemia. NEJM 340, 1330 –13409. Staal, F. J., Luis, T. C., and Tiemessen, M. M. (2008) WNT signalling in the
immune system: WNT is spreading its wings. Nat. Rev. Immunol. 8,581–593
10. Malhotra, S., and Kincade, P. W. (2009) Wnt-related molecules and sig-naling pathway equilibrium in hematopoiesis. Cell Stem Cell 4, 27–36
11. Fleming, H. E., Janzen, V., Lo Celso, C., Guo, J., Leahy, K. M., Kronenberg,H. M., and Scadden, D. T. (2008) Wnt signaling in the niche enforceshematopoietic stem cell quiescence and is necessary to preserve self-re-newal in vivo. Cell Stem Cell 2, 274 –283
12. Schaniel, C., Sirabella, D., Qiu, J., Niu, X., Lemischka, I. R., and Moore,K. A. (2011) Wnt-inhibitory factor 1 dysregulation of the bone marrowniche exhausts hematopoietic stem cells. Blood 118, 2420 –2429
13. Luis, T. C., Naber, B. A., Fibbe, W. E., van Dongen, J. J., and Staal, F. J.(2010) Wnt3a nonredundantly controls hematopoietic stem cell functionand its deficiency results in complete absence of canonical Wnt signaling.Blood 116, 496 – 497
14. Guo, J., Liu, M., Yang, D., Bouxsein, M. L., Saito, H., Galvin, R. J., Kuhstoss,S. A., Thomas, C. C., Schipani, E., Baron, R., Bringhurst, F. R., and Kro-nenberg, H. M. (2010) Suppression of Wnt signaling by Dkk1 attenuatesPTH-mediated stromal cell response and new bone formation. CellMetab. 11, 161–171
15. Nemeth, M. J., Mak, K. K., Yang, Y., and Bodine, D. M. (2009) �-Cateninexpression in the bone marrow microenvironment is required for long-term maintenance of primitive hematopoietic cells. Stem Cells 27,1109 –1119
16. Scheller, M., Huelsken, J., Rosenbauer, F., Taketo, M. M., Birchmeier, W.,Tenen, D. G., and Leutz, A. (2006) Hematopoietic stem cell and multilin-eage defects generated by constitutive �-catenin activation. Nat. Immu-nol. 7, 1037–1047
17. Kirstetter, P., Anderson, K., Porse, B. T., Jacobsen, S. E., and Nerlov, C.(2006) Activation of the canonical Wnt pathway leads to loss of hemato-poietic stem cell repopulation and multilineage differentiation block. Nat.Immunol. 7, 1048 –1056
18. Reya, T., Duncan, A. W., Ailles, L., Domen, J., Scherer, D. C., Willert, K.,Hintz, L., Nusse, R., and Weissman, I. L. (2003) A role for Wnt signalling inself-renewal of haematopoietic stem cells. Nature 423, 409 – 414
19. Jeannet, G., Scheller, M., Scarpellino, L., Duboux, S., Gardiol, N., Back, J.,Kuttler, F., Malanchi, I., Birchmeier, W., Leutz, A., Huelsken, J., and Held,W. (2008) Long-term, multilineage hematopoiesis occurs in the combinedabsence of �-catenin and �-catenin. Blood 111, 142–149
20. Cobas, M., Wilson, A., Ernst, B., Mancini, S. J., MacDonald, H. R., Kemler,R., and Radtke, F. (2004) �-Catenin is dispensable for hematopoiesis andlymphopoiesis. J. Exp. Med. 199, 221–229
21. Luis, T. C., Naber, B. A., Roozen, P. P., Brugman, M. H., de Haas, E. F.,Ghazvini, M., Fibbe, W. E., van Dongen, J. J., Fodde, R., and Staal, F. J.(2011) Canonical wnt signaling regulates hematopoiesis in a dosage-de-pendent fashion. Cell Stem Cell 9, 345–356
22. Ruiz-Herguido, C., Guiu, J., D’Altri, T., Inglés-Esteve, J., Dzierzak, E., Es-pinosa, L., and Bigas, A. (2012) Hematopoietic stem cell developmentrequires transient Wnt/�-catenin activity. J. Exp. Med. 209, 1457–1468
23. Zhao, C., Blum, J., Chen, A., Kwon, H. Y., Jung, S. H., Cook, J. M., Lagoo, A.,and Reya, T. (2007) Loss of �-catenin impairs the renewal of normal andCML stem cells in vivo. Cancer Cell 12, 528 –541
24. Hu, Y., Chen, Y., Douglas, L., and Li, S. (2009) �-Catenin is essential forsurvival of leukemic stem cells insensitive to kinase inhibition in mice withBCR-ABL-induced chronic myeloid leukemia. Leukemia 23, 109 –116
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
MAY 20, 2016 • VOLUME 291 • NUMBER 21 JOURNAL OF BIOLOGICAL CHEMISTRY 11159
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

25. Heidel, F. H., Bullinger, L., Feng, Z., Wang, Z., Neff, T. A., Stein, L., Kalait-zidis, D., Lane, S. W., and Armstrong, S. A. (2012) Genetic and pharma-cologic inhibition of �-catenin targets imatinib-resistant leukemia stemcells in CML. Cell Stem Cell 10, 412– 424
26. Steinke, F. C., and Xue, H. H. (2014) From inception to output, Tcf1 andLef1 safeguard development of T cells and innate immune cells. Immunol.Res. 59, 45–55
27. Verbeek, S., Izon, D., Hofhuis, F., Robanus-Maandag, E., te Riele, H., vande Wetering, M., Oosterwegel, M., Wilson, A., MacDonald, H. R., andClevers, H. (1995) An HMG-box-containing T-cell factor required forthymocyte differentiation. Nature 374, 70 –74
28. Yu, S., Zhou, X., Steinke, F. C., Liu, C., Chen, S. C., Zagorodna, O., Jing, X.,Yokota, Y., Meyerholz, D. K., Mullighan, C. G., Knudson, C. M., Zhao,D. M., and Xue, H. H. (2012) The TCF-1 and LEF-1 Transcription FactorsHave Cooperative and Opposing Roles in T Cell Development and Malig-nancy. Immunity 37, 813– 826
29. Steinke, F. C., Yu, S., Zhou, X., He, B., Yang, W., Zhou, B., Kawamoto, H.,Zhu, J., Tan, K., and Xue, H. H. (2014) TCF-1 and LEF-1 act upstream ofTh-POK to promote the CD4� T cell fate and interact with Runx3 tosilence Cd4 in CD8� T cells. Nat. Immunol. 15, 646 – 656
30. Guo, K., McMinn, J. E., Ludwig, T., Yu, Y. H., Yang, G., Chen, L., Loh, D.,Li, C., Chua, S., Jr., and Zhang, Y. (2007) Disruption of peripheral leptinsignaling in mice results in hyperleptinemia without associated metabolicabnormalities. Endocrinology 148, 3987–3997
31. Yu, S., Cui, K., Jothi, R., Zhao, D. M., Jing, X., Zhao, K., and Xue, H. H.(2011) GABP controls a critical transcription regulatory module that isessential for maintenance and differentiation of hematopoietic stem/pro-genitor cells. Blood 117, 2166 –2178
32. Yu, S., Jing, X., Colgan, J. D., Zhao, D. M., and Xue, H. H. (2012) Targetingtetramer-forming GABP� isoforms impairs self-renewal of hematopoieticand leukemic stem cells. Cell Stem Cell 11, 207–219
33. Carleton, M., Haks, M. C., Smeele, S. A., Jones, A., Belkowski, S. M.,Berger, M. A., Linsley, P., Kruisbeek, A. M., and Wiest, D. L. (2002) Earlygrowth response transcription factors are required for development ofCD4(-)CD8(-) thymocytes to the CD4�CD8� stage. J. Immunol. 168,1649 –1658
34. Pear, W. S., Miller, J. P., Xu, L., Pui, J. C., Soffer, B., Quackenbush, R. C.,Pendergast, A. M., Bronson, R., Aster, J. C., Scott, M. L., and Baltimore, D.(1998) Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transducedbone marrow. Blood 92, 3780 –3792
35. Germar, K., Dose, M., Konstantinou, T., Zhang, J., Wang, H., Lobry, C.,Arnett, K. L., Blacklow, S. C., Aifantis, I., Aster, J. C., and Gounari, F. (2011)T-cell factor 1 is a gatekeeper for T-cell specification in response to Notchsignaling. Proc. Natl. Acad. Sci. U.S.A. 108, 20060 –20065
36. Weber, B. N., Chi, A. W., Chavez, A., Yashiro-Ohtani, Y., Yang, Q.,Shestova, O., and Bhandoola, A. (2011) A critical role for TCF-1 in T-lin-eage specification and differentiation. Nature 476, 63– 68
37. Min, I. M., Pietramaggiori, G., Kim, F. S., Passegué, E., Stevenson, K. E., andWagers, A. J. (2008) The transcription factor EGR1 controls both theproliferation and localization of hematopoietic stem cells. Cell Stem Cell 2,380 –391
38. Hock, H., Hamblen, M. J., Rooke, H. M., Schindler, J. W., Saleque, S.,Fujiwara, Y., and Orkin, S. H. (2004) Gfi-1 restricts proliferation and pre-serves functional integrity of haematopoietic stem cells. Nature 431,1002–1007
39. Tothova, Z., Kollipara, R., Huntly, B. J., Lee, B. H., Castrillon, D. H., Cullen,D. E., McDowell, E. P., Lazo-Kallanian, S., Williams, I. R., Sears, C., Arm-strong, S. A., Passegué, E., DePinho, R. A., and Gilliland, D. G. (2007)FoxOs are critical mediators of hematopoietic stem cell resistance to phys-iologic oxidative stress. Cell 128, 325–339
40. Miyamoto, K., Araki, K. Y., Naka, K., Arai, F., Takubo, K., Yamazaki, S.,Matsuoka, S., Miyamoto, T., Ito, K., Ohmura, M., Chen, C., Hosokawa, K.,Nakauchi, H., Nakayama, K., Nakayama, K. I., et al. (2007) Foxo3a is es-sential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell1, 101–112
41. Bunting, K. D., Bradley, H. L., Hawley, T. S., Moriggl, R., Sorrentino, B. P.,and Ihle, J. N. (2002) Reduced lymphomyeloid repopulating activity fromadult bone marrow and fetal liver of mice lacking expression of STAT5.Blood 99, 479 – 487
42. Yang, Q., Esplin, B., and Borghesi, L. (2011) E47 regulates hematopoieticstem cell proliferation and energetics but not myeloid lineage restriction.Blood 117, 3529 –3538
43. Souroullas, G. P., Salmon, J. M., Sablitzky, F., Curtis, D. J., and Goodell,M. A. (2009) Adult hematopoietic stem and progenitor cells require eitherLyl1 or Scl for survival. Cell Stem Cell 4, 180 –186
44. Thompson, B. J., Jankovic, V., Gao, J., Buonamici, S., Vest, A., Lee, J. M.,Zavadil, J., Nimer, S. D., and Aifantis, I. (2008) Control of hematopoieticstem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 205,1395–1408
45. Zhang, J., Grindley, J. C., Yin, T., Jayasinghe, S., He, X. C., Ross, J. T., Haug,J. S., Rupp, D., Porter-Westpfahl, K. S., Wiedemann, L. M., Wu, H., and Li,L. (2006) PTEN maintains haematopoietic stem cells and acts in lineagechoice and leukaemia prevention. Nature 441, 518 –522
46. Matsumoto, A., Takeishi, S., Kanie, T., Susaki, E., Onoyama, I., Tateishi, Y.,Nakayama, K., and Nakayama, K. I. (2011) p57 is required for quiescenceand maintenance of adult hematopoietic stem cells. Cell Stem Cell 9,262–271
47. Zou, P., Yoshihara, H., Hosokawa, K., Tai, I., Shinmyozu, K., Tsukahara, F.,Maru, Y., Nakayama, K., Nakayama, K. I., and Suda, T. (2011) p57Kip2 andp27Kip1 cooperate to maintain hematopoietic stem cell quiescencethrough interactions with Hsc70. Cell Stem Cell 9, 247–261
48. Perry, J. M., He, X. C., Sugimura, R., Grindley, J. C., Haug, J. S., Ding, S., andLi, L. (2011) Cooperation between both Wnt/�-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewaland expansion. Genes Dev. 25, 1928 –1942
49. Ross, D. M., Branford, S., Seymour, J. F., Schwarer, A. P., Arthur, C., Bar-tley, P. A., Slader, C., Field, C., Dang, P., Filshie, R. J., Mills, A. K., Grigg,A. P., Melo, J. V., and Hughes, T. P. (2010) Patients with chronic myeloidleukemia who maintain a complete molecular response after stoppingimatinib treatment have evidence of persistent leukemia by DNA PCR.Leukemia 24, 1719 –1724
50. Edmaier, K. E., Stahnke, K., Vegi, N., Mulaw, M., Ihme, S., Scheffold, A.,Rudolph, K. L., and Buske, C. (2014) Expression of the lymphoid enhancerfactor 1 is required for normal hematopoietic stem and progenitor cellfunction. Leukemia 28, 227–230
51. Xu, M., Sharma, A., Wiest, D. L., and Sen, J. M. (2009) Pre-TCR-induced�-catenin facilitates traversal through �-selection. J. Immunol. 182,751–758
52. Tesio, M., and Trumpp, A. (2011) Breaking the cell cycle of HSCs by p57and friends. Cell Stem Cell 9, 187–192
53. Lento, W., Ito, T., Zhao, C., Harris, J. R., Huang, W., Jiang, C., Owzar, K.,Piryani, S., Racioppi, L., Chao, N., and Reya, T. (2014) Loss of �-catenintriggers oxidative stress and impairs hematopoietic regeneration. GenesDev. 28, 995–1004
54. Petropoulos, K., Arseni, N., Schessl, C., Stadler, C. R., Rawat, V. P., Desh-pande, A. J., Heilmeier, B., Hiddemann, W., Quintanilla-Martinez, L.,Bohlander, S. K., Feuring-Buske, M., and Buske, C. (2008) A novel role forLef-1, a central transcription mediator of Wnt signaling, in leukemogen-esis. J. Exp. Med. 205, 515–522
55. Anastas, J. N., and Moon, R. T. (2013) WNT signalling pathways as ther-apeutic targets in cancer. Nat. Rev. Cancer 13, 11–26
56. Wu, J. Q., Seay, M., Schulz, V. P., Hariharan, M., Tuck, D., Lian, J., Du, J.,Shi, M., Ye, Z., Gerstein, M., Snyder, M. P., and Weissman, S. (2012) Tcf7is an important regulator of the switch of self-renewal and differentiationin a multipotential hematopoietic cell line. PLoS genetics 8, e1002565
57. Ashton, J. M., Balys, M., Neering, S. J., Hassane, D. C., Cowley, G., Root,D. E., Miller, P. G., Ebert, B. L., McMurray, H. R., Land, H., and Jordan,C. T. (2012) Gene sets identified with oncogene cooperativity analysisregulate in vivo growth and survival of leukemia stem cells. Cell Stem Cell11, 359 –372
Tcf1 and Lef1 Regulate HSC and LSC Self-renewal
11160 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 21 • MAY 20, 2016
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Shuyang Yu, Fengyin Li, Shaojun Xing, Tianyan Zhao, Weiqun Peng and Hai-Hui XueLef1 Transcription Factors
Hematopoietic and Leukemic Stem Cells Have Distinct Dependence on Tcf1 and
doi: 10.1074/jbc.M116.717801 originally published online April 4, 20162016, 291:11148-11160.J. Biol. Chem.
10.1074/jbc.M116.717801Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/291/21/11148.full.html#ref-list-1
This article cites 57 references, 16 of which can be accessed free at
by guest on May 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















