
Hematologic Disorders Notes
23
HEMATOLOGIC DISORDERS HEMATOLOGY • The hematologic or hematopoietic system includes the blood, blood vessels, and blood forming organs ( bone marrow, spleen, liver, lymph nodes, and thymus gland). • Major function of blood is to carry necessary materials ( oxygen, nutrients ) to cells and to remove CO2 system and metabolic waste products. • It also plays a role in hormone transport, inflammatory and immune responses, temperature regulation, fluid-electrolyte balance, and acid-base balance. THREE BROAD FUNCTIONS OF BLOOD : Transportation • Respiratory – transport of gases by the RBC • Nutritive – transport of digested nutrients from the GIT to the different cells of the body • Excretory – transport of metabolic wastes to the kidneys and excreted as urine Regulation • Hormones and other molecules that help regulate metabolism are also carried in the blood • Thermoregulation Protection • Blood clotting • Leukocytes COMPONENTS OF THE BLOOD : 1. Plasma 2. Formed Elements • Erythrocytes or RBC • Leukocytes or WBC • Thrombocytes or Platelets BLOOD • Average volume is 5 – 6 liters or approximately 6 quarts • pH is 7.35 – 7.45 • Arterial blood is usually bright red in color compared to venous blood which has a darker color, due primarily to the large concentration of oxyhemoglobin found in arterial blood BLOOD a. ) Plasma - fluid portion of the blood Contains : proteins / albumin fibrinogen clotting factors electrolytes waste products nutrients b. ) Cellular Components 1. Leukocytes (WBC) 2. Erythrocyte (RBC) 3. Thrombocyte (Platelets) Hematopoiesis – occurs in the bone marrow ( pelvis, ribs, vertebrae and sternum. Extramedullary Hematopoiesis - the liver and the spleen produces blood cells PLASMA • The liquid part of the blood • Approximately 90% water • Also contains nutrients, ions (salts, primarily Na), respiratory gases, hormones, plasma proteins, antibodies and various wastes and products of cellular metabolism
-
Upload
mikkagreen -
Category
Documents
-
view
2.458 -
download
2
Transcript of Hematologic Disorders Notes

HEMATOLOGIC DISORDERS
HEMATOLOGY• The hematologic or hematopoietic system includes the blood, blood vessels, and blood forming organs ( bone marrow, spleen,
liver, lymph nodes, and thymus gland). • Major function of blood is to carry necessary materials ( oxygen, nutrients ) to cells and to remove CO2 system and metabolic
waste products. • It also plays a role in hormone transport, inflammatory and immune responses, temperature regulation, fluid-electrolyte
balance, and acid-base balance.
THREE BROAD FUNCTIONS OF BLOOD :Transportation
• Respiratory – transport of gases by the RBC• Nutritive – transport of digested nutrients from the GIT to the different cells of the body• Excretory – transport of metabolic wastes to the kidneys and excreted as urine
Regulation• Hormones and other molecules that help regulate metabolism are also carried in the blood• Thermoregulation
Protection• Blood clotting• Leukocytes
COMPONENTS OF THE BLOOD :1. Plasma2. Formed Elements
• Erythrocytes or RBC• Leukocytes or WBC• Thrombocytes or Platelets
BLOOD • Average volume is 5 – 6 liters or approximately 6 quarts• pH is 7.35 – 7.45• Arterial blood is usually bright red in color compared to venous blood which has a darker color, due primarily to the large
concentration of oxyhemoglobin found in arterial blood
BLOODa. ) Plasma - fluid portion of the bloodContains :proteins / albumin fibrinogenclotting factors electrolyteswaste products nutrients
b. ) Cellular Components1. Leukocytes (WBC)2. Erythrocyte (RBC)3. Thrombocyte (Platelets)
Hematopoiesis – occurs in the bone marrow ( pelvis, ribs, vertebrae and sternum.Extramedullary Hematopoiesis - the liver and the spleen produces blood cells
PLASMA
• The liquid part of the blood• Approximately 90% water• Also contains nutrients, ions (salts, primarily Na), respiratory gases, hormones, plasma proteins, antibodies and various
wastes and products of cellular metabolism• PLASMA PROTEINS – the most abundant solutes in the plasma- Three Types1. Albumin2. Globulin3. Fibrinogen
FORMED ELEMENTS OF THE BLOOD
Erythrocytes• Also called Red Blood Cells or RBC’s• Function primarily to ferry Oxygen in the blood to all cells in the body• Also transports Carbon dioxide out of the body• Lifespan of 120 days only

• Hemoglobin in the RBC binds with the Oxygen as it is transported in the blood• Female : 12 – 16 g/100ml• Male : 13 – 18 g/100ml
• Normal RBC count: about 4 – 6 million/mm³• Hematocrit (HCT) – percentage of RBC per given volume of blood and is an important indicator of the Oxygen-carrying
capacity of the blood• Female : 37 – 48%• Male : 45 – 52%
Leukocytes• Also called White Blood Cells or WBC’s• Average value : 4,000 – 11,000 / mm³• Protects the body against any damage• Are able to slip in and out of the blood vessels by ameboid fashion – in a process called diapedesis• When mobilized, the body speeds up production which usually indicates the presence of infection in the body• Leukocytosis – total WBC count above 11,000 / mm³• Leukopenia – an abnormally low WBC count• Types:
o Granulocytes Neutrophils Eosinophils Basophils
o Agranulocytes Lymphocytes Monocytes
Cells of the Immune System• Lymphocytes
• Lymphocytes are created in the bone marrow and migrate to the Thymus where they mature • After becoming immunocompetent, the B & T cells transfer to the lymph nodes & spleen• Types
1. B lymphocytes or B cells – produces antibodies to incapacitate the antigen2. T lymphocytes or T Cells – attacks antigens directly
• Macrophages • Literally means “Big Eaters”• Arise from monocytes formed in the bone marrow• Major role : to engulf foreign particles•
Cellular (Cell-Mediated) Immune Response
• T – Cells• Responds directly to antigens• Will destroy target cells thru secretions of Lymphokines and Perforin ( “Kiss of Death”) which is inserted to the cell
membrane, shortly after that, the target cell ruptures• They have a:
• License to KILL• License to HELP• License to Suppress
• Three types :• Killer T Cells – binds to the surface of invading cells, disrupt the cell membrane & destroy it by altering it’s
environment• Helper T cells – helps to stimulate the B Cells to mature into Plasma Cells which synthetize & secrete
immunoglobulins (Antibodies)• Suppressor T Cells – Reduces the Humoral response•
Humoral (Antibody-Mediated) Immune Response
B – Cells• Matures into Plasma Cells responsible for Antibody production
5 Classes of Immunoglobulins (MADGE) :1. Immunoglobulin M (IgM)
1st immunoglobulin produced in an immune responsepresent in plasma, too big to cross membrane barriers2. Immunoglobulin A (IgA)
Sound in body secretions like saliva, tears, mucus, bile, milk & colostrum3. Immunoglobulin D (IgD)
Present only in the plasma & is always attached to the B Cell4. Immunoglobilin G (IgG)
80% of circulating antibodiesCan cross the placenta and provide passive immunityPresent in all body fluids
5. Immunoglobulin E (IgE) Responsible for Allergic & hypersensitivity reactions

Stimulates Mast cells & Basophils to release Histamine which mediates inflammation & the allergic response
Thrombocytes• Also called Platelets• Average value : 250,000 – 450,000 / mm³• Lives for about 5 – 10 days• Important in blood clotting
Hematopoiesis (Blood Cell Formation)
• Occurs in the Red Bone Marrow, chiefly in flat bones like Skull, ribs, pelvis, sternum and proximal epiphyses of the humerus and femur
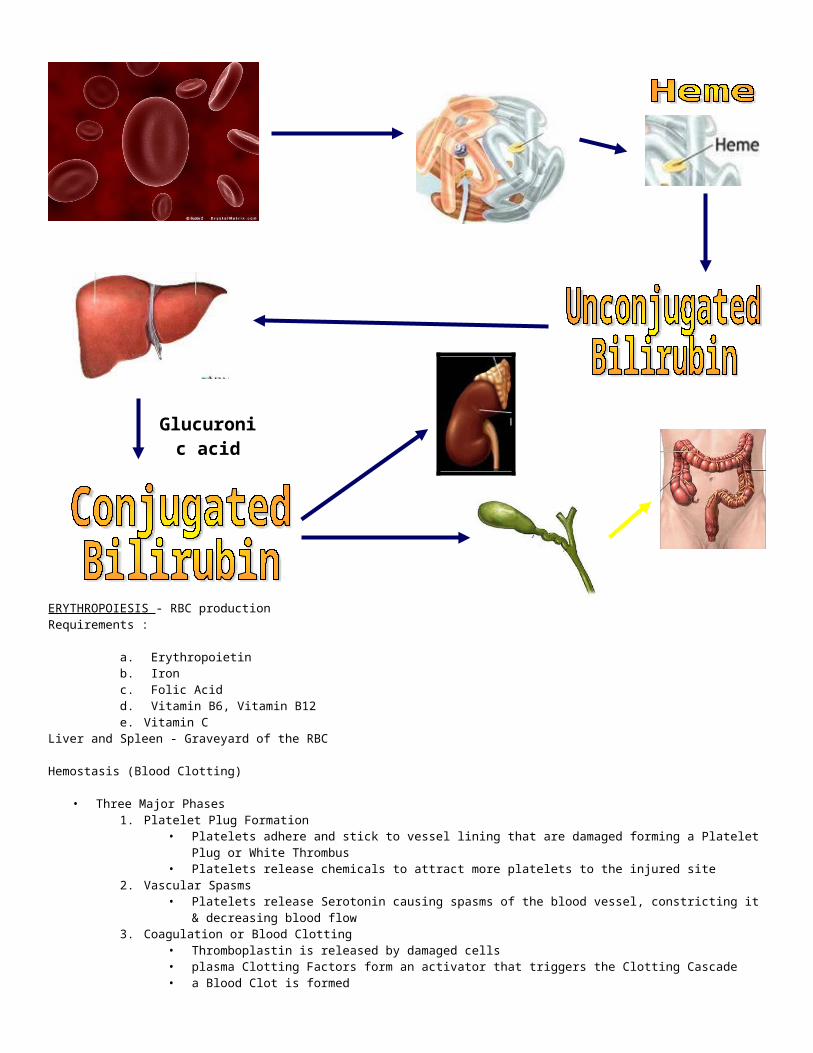
• Erythropoiesis – RBC production, is a very active process• RBC are continuously being destroyed by the liver & spleen• RBC’s have a lifespan of 120 days• As RBC’s are destroyed, iron is recycled to the bone marrow for use in the formation of new RBC’s• Erythropoietin – secreted by the kidneys & released when blood levels of Oxygen begins to decline for any reason; which
stimulates the Red Bone Marrow to produce more RBC’s
ERYTHROCYTES> destruction
- mature cells removed chiefly by spleen & liver
* BILIRUBIN = byproduct of Hgb released when RBC’s destroyed
* IRON = freed from Hgb during bilirubin formation = transported to bone marrow via
TRANSFERIN & reclaimed for new Hgb production
Cell TypeCell Type Normal Value / mm³Normal Value / mm³ FunctionFunctionRBC 4 – 6 million Transport Oxygen
WBC 4,000 – 11,000
Neutrophils 3,000 – 7,00040 – 70% of WBC
Active Phagocytes, increases during acute infections
Eosinophils 100 – 4001 – 4% of WBC
Kills parasitic worms, increases in allergy attacks, helps detoxify foreign substances
Basophils 20 – 500 – 1% of WBC
Contains histamineReleases anticoagulant heparin
Lymphocytes 1,500 – 3,00020 – 45% of WBC
Part of Immune SystemB & T Lymphocytes
Monocytes 100 – 7004 – 8% of WBC
Phagocytes that become macrophages – “clean-up team” & increases in chronic infections
Platelets 250,000 – 450,000 Needed for Blood ClottingInitiates Clotting Cascade

ERYTHROPOIESIS - RBC productionRequirements :
a. Erythropoietinb. Ironc. Folic Acidd. Vitamin B6, Vitamin B12e. Vitamin C
Liver and Spleen - Graveyard of the RBC
Hemostasis (Blood Clotting)
• Three Major Phases1. Platelet Plug Formation
• Platelets adhere and stick to vessel lining that are damaged forming a Platelet Plug or White Thrombus• Platelets release chemicals to attract more platelets to the injured site
2. Vascular Spasms• Platelets release Serotonin causing spasms of the blood vessel, constricting it & decreasing blood flow
3. Coagulation or Blood Clotting• Thromboplastin is released by damaged cells• plasma Clotting Factors form an activator that triggers the Clotting Cascade• a Blood Clot is formed• Serum is squeezed out within the hour pulling the ruptured edges together
Glucuronic acid
Plasma Clotting FactorsI FibrinogenII ProthrombinIII Tissue ThromboplastinIV CalciumV ProacelerinVII ProconvertinVIII Antihemophilic FactorIX Christmas FactorX Stuart – Prower FactorXI Plasma Thromboplastin AntecedentXII Hageman FactorXIII Fibrin Stabilizing Factor


Anemia• Conditions in which the number of RBC’s or amount of hemoglobin is lower than normal• leads to hypoxia and ischemia•
Classifications of Anemia According to EtiologyBleeding Hypoproliferative Hemolytic
Resulting from RBC Loss low RBC production increased RBC destruction
accidents / trauma Iron deficiency enlarged spleen
surgery Vit. B12 deficiency sickle cell
childbirth Folic acid deficiency thalassemia
ruptured blood vessel Vit. C deficiency G6PD
menorrhagia chronic disease drug-induced
epistaxis low erythropoietin
hemorrhoids cancer
GI bleeding / ulcers
cancer
Iron Deficiency Anemia• most common type of anemia• Iron stores are depleted, resulting in a decreased supply of iron for the manufacture of hemoglobin in RBC’s
Commonly results from blood loss, increased metabolic demands, syndromes of gastrointestinal malabsorption, and dietary inadequacy
• cause : inadequate absorption or excessive loss of iron• Bleeding – principal cause in adults• Vegetarian diets• Vitamin C – increases iron absorption
Most definitive way to diagnose anemia : Bone marrow aspiration
PathophysiologyPathophysiology
Stage 1Iron loss exceeds intake, depleting iron reserves
Stage 2
Fewer RBC are produced
Stage 4
Bone marrow compensates :Speeds up production – Microlytic /
Small red blood cellsStage 5
Symptoms of anemia worsens
Stage 3Anemia develops
RBC – normal but few in numberHgb & Hct – normal levels

Assessment FindingsMild cases - asymptomaticfatiguedyspneaPalpitations & dizzinessPallorBrittle hair and nailsPicaGlossitisCheilosisIrritabilityKoilonychia
Laboratory findings : RBC’s are small / microcytic and pale
• decreased hemoglobin decreased hematocrit• decreased serum iron decreased ferritin
Nursing Interventions1. Identify the cause2. Monitor S/Sx of bleeding – stool, urine and GI contents3. Provide rest4. Give iron preparations ( 6 – 12 months )
Ferrous Sulfate Ferrous Gluconate Ferrous Fumarate a. always give after meals or snacks b. dilute liquid preps and give thru straw c. give with orange juice (Vitamin C enhances absorption) d. warn clients the stool will become black and can cause constipation5. For clients with poor absorption or continuous blood loss
IM or IV of Iron Dextrana. Use 1 needle to withdraw and another for injectionb. Use z-track methodc. don’t massage but encourage ambulationd. usually, deep IM at buttocks
6. Give dietary teaching – liver, meats, nuts, egg yolk, shellfish, legumes, etc.7. Increase intake of roughage and fluids to prevent constipation.
Pernicious Anemia• Vitamin B12 Deficiency Anemia• caused by inadequate Vit. B12 intake or deficiency in intrinsic factor• Vit. B12 combines with intrinsic factor so it can be absorbed in the ileum into the bloodstream`• the result is abnormally large erythrocytes and hypochlorhydria ( a deficiency of hydrochloric acid in gastric secretions).• Lack of intrinsic factor is caused by gastric mucosal atrophy (possibly due to heredity, prolonged iron deficiency, or an
autoimmune disorder), can also result in client who have had a total gastrectomy • Usually occurs in men and women over age 50, with an increase in blue eyed persons.
PATHOPHYSIOLOGY
Assessment :Anemia - symptoms are :Fatigue, weaknessdyspneaPalpitations & dizzinessPallorconfusion
Intrinsic factor + Vit. B12 for absorption
Result : decreased or no Vit. B12Result : decreased or no Vit. B12
Lead to : decreased RBC productionLead to : decreased RBC production

Decreased intellectual fxnSore tongue : Beefy red tongueParesthesiasWeight loss
Lab ResultsDecrease RBCDecreased free Hydrochloric acidLarge RBC / MegaloblastPositive Schilling Test – definitive test for Pernicious anemia
- used to detect lack of intrinsic factorPositive schilling test
• Measures absorption of radioactive vitamin B12 both before and after parenteral administration of intrinsic factor.• Definitive test for pernicious anemia.• Used to detect lack of intrinsic factor.• Fasting client is given radioactive vitamin B12 by mouth and nonradioactive vitamin B12 IM to saturate tissue binding sites
and to permit some excretion of radioactive vitamin B12 in the urine if it is absorbed.• 24-48 hour urine collection is obtained; client is encouraged to drink fluids. • If indicated, a second stage Schilling test will be performed 1 week after first stage. • Fasting client is given radioactive vitamin B12 combined with human intrinsic factor and the test will be repeated.
Nursing Interventions / Treatment1. Drug Therapy
a. Vit. B12 injections monthly for lifeb. Iron Preparationsc. Folic Acid
2. Transfusion therapy3. Bed rest4. Mouth care5. Dietary teaching6. Teach about importance of lifelong Vitamin B12 therapy
• Provide a nutritious diet high in iron, protein, and vitamins (fish, meat, milk/milk products and eggs).• Avoid highly seasoned, coarse or very hot foods if client has a mouth sores.• Provide mouth care before and after meals using a soft toothbrush and non irritating rinses.
HEMOLYTIC ANEMIAS• increase rate of RBC destruction• short life span of RBC.
TYPES:• G6PD• Sickle cell anemia• Thalassemia• DIC• Transfussion incompatibilities
Sickle Cell Anemia• Most common inherited disease among black Americans.• Also found in Arabian, Mediterranean and Caribbean descent• Hgb S ( abnormal hemoglobin ), which has reduced oxygen carrying capacity, replaces all or part of the hemoglobin in the
RBC’s.• Life span is 6-20 days instead of 120, causing hemolytic anemia.• Death often occurs in early adulthood due to occlusion or infection.• During decreased O2 tension, lowered pH, dehydration and severe infections, RBC’s change from round to sickle or crescent
shape• Sickled cells don’t slide thru vessels as normal RBC’s do, causing clumping, thrombosis, arterial obstruction, increased blood
viscosity, hemolysis and eventual tissue ischemia and necrosis
Sickle Cell Crisis :• Cause : infection, dehydration, fever, cold exposure, hypoxia, strenuous exercise, extreme fatigue or extreme changes in
altitudeVASO-OCCLUSIVE CRISIS:
A. most common and most painful type of crisis• caused by stasis of blood with clumping of the cells in the microcirculation leading to ischemia & infarction.
B. signs include fever, pain, and tissue engorgementC. Treatment
– hydration, electrolyte replacement, bed rest, broad spectrum antibiotics, transfusions and oxygen therapy.
SPLENIC SEQUESTRATION:

A. Life - threatening crisis caused by the pooling of blood in the spleen. (from congestion of sickled cells)
B. signs include profound anemia, hypovolemia, and shock
C. treatment : blood transfusions and splenectomy
APLASTIC CRISIS:
A. Occurs infrequently and is caused by:• diminished production of RBC• increased destruction of RBC’s• triggered by a viral infection or the depletion of folic acid.
B. signs include profound anemia, pallor, and PANCYTOPENIA.
C. Treatment – Transfusion of packed RBC’s
• Frequent infection esp. with H. influenzae• Infants may have Dactylitis (hand – foot syndrome) symmetrical painful soft tissue swelling in the hands and feet in the
absence of trauma
ASSESSMENT• Signs and symptoms of anemia – pallor, weakness• Hepatospleenomegaly• Dactylitis (Symmetric swelling of the hands and feet) – called hand-foot syndrome• Other problems :
– CVA– MI– Growth retardation – initial manifestation– Decreased fertility– Priapism– Recurrent severe infections
MEDICAL MANAGEMENTA. Drug therapy > analgesic/narcotics to control pain
• Avoid meperidine (Demerol) due increased risk of seizures in children > antibiotics to control infection.B. Blood transfusionsC. Hydration:oral and IVD. Bed rest E. Surgery: splenectomy
INTERVENTIONS• Administer O2 & Blood Transfusion as Rx• Maintain adequate hydration• Avoid tight clothing that could impair circulation.• Keep wounds clean and dry.• Provide bed rest to decrease energy expenditure and oxygen use.• Encourage patient to eat foods high in calories, CHON, with folic acid supplementation.• Analgesics:
• Acetaminophen• Morphine• avoid aspirin as it enhances acidosis,which promotes sickling
• Avoid anticoagulants( sludging is not due to clotting ).• Antibiotics. • Avoid activities that require so much energy.• Keep arms and legs from extreme cold.• Decrease emotional stress.• Provide good skin care
THALASSEMIA MAJOR (Cooley’s anemia)
• B - thalassemia refers to an inherited hemolytic anemia, characterized by reduction or absence of the B-globulin chain in Hgb synthesis
• Fragile RBC & short life span• Autosomal recessive pattern of inheritance• Insufficient B-globulin chain synthesis allows large amounts of unstable chains to accumulate

• Precipitates of alpha chains that form cause RBC’s to be rigid & easily destroyed, leading to severe hemolytic anemia = chronic hypoxia
• Skeletal deformities: pathologic fractures• Hemosiderosis – excess iron supply, which leads to iron deposits in the organ tissues leading to decreased function
CLINICAL MANIFESTATIONS• onset is usually insidious• Sx are primarily related to progressive anemia, expansion of marrow cavities of the bone & developmemnt of hemosiderosis• Early Sx often include progressive pallor, poor feeding & lethargy• Further signs: hemorrhage, bone pain, exercise intolerance, jaundice, & protuberant abdomen
DIAGNOSTIC EVALUATION• Decrease hemoglobin• RBC= increase in number• Hgb elctrophoresis
– elevated levels of HgF ( doesn’t hold O2 well )– limited amount of HgA
Management• Frequent and regular transfusion of packed RBC’s to maintain Hgb levels above 10 g/dL• Iron chelation therapy with deferoxamine (Desferal) – reduces toxic effects of excess iron & increases iron excretion thru urine
& feces• Splenectomy• Supportive management of symptoms• Bone marrow transplant• Prognosis and Survival rate is poor because of no known cure• Often fatal in late adolescence or early adulthood
Complications• Splenomegaly• Growth retardation in the second decade• Endocrine abnormalities :
– delayed development of secondary sex characteristics – most boys fail to undergo puberty, girls – menstruation problems
– DM – due to iron deposits in the pancreas– Hypermetabolic rates
• Skeletal complications– Frontal & parietal bossing (Enlargement)– Maxillary hypertrophy – leading to occlusion– Premature closure of epiphyses of long bones– Osteoporosis & pathologic fractures
• Cardiac problems: pericarditis & CHF – usual cause of death• Gallbladder disease
– Gallstones that often require surgery• Skin – bronze pigmentation caused by iron deposits in the dermis• Leg ulcers
ERYTHROBLASTOSIS FETALISRh Incompatibility
• Destruction of RBCs that result from Ag-Ab rxn• Characterized by hemolytic anemia or hyperbilirubinemia• Possibly caused by Rh incompatibility between the mother & the fetus (Ag & Ab reaction)• Sensitization of Rh (-) woman by transfusion of Rh (+) blood• Sensitization of Rh (-) woman by presence of Rh (+) RBCs from her fetus conceived with Rh (+) man• Approximately 65% of infants conceived by this combination of parents will be Rh (+)• Mother is sensitized by passage of Rh (+) RBCs thru placenta, either during pregnancy (break/leak in
membrane) or at the time of separation of the placenta after delivery.
RH INCOMPATIBILTY• FIRST PREGNANCY
- mother may become sensitized, baby rarely affectedINDIRECT COOMB’S TEST - Tests for anti-Rh(+) Ab in mother’s circulation - performed during pregnancy at first visit & again about 28 week’s gestation.RESULTS: - If (-) at 28 weeks, a small dose of (MicroRhogam) is given prophylactically to prevent sensitization in the 3rd trimester. - Rhogam may also be given after 2nd trimester amniocentesis - If (+), levels are titrated to determine potential effects on the fetus
DIRECT COOMBS’ TEST - Tests done on the cord blood at delivery to determine presence of (+) Ab on fetal RBCs

RESULTS - If both indirect & direct Coombs’ test is NEGATIVE & infant is Rh(+): - NEGATIVE: No formation of Anti-Rh (+) Ab - Rhogam (Rho[D] human immune globulin is given to the Rh(-) mother to prevent development of anti-Rh(+) Ab as the result of sensitization from present or just terminated pregnancy.
• In each pregnancy, an Rh(-) mother who carries an Rh (+) fetus receives Rhogam if both the mother and infant is (-) to both direct & indirect Coombs’ test.
• If mother is has been sensitized:- anti-Rh(+) Ab are present
- Rhogam is not indicated
• Rhogam must be injected into unsensitized mother’s system within 72 hours of delivery of Rh(+) infantCLINICAL FINDINGS
• Anemia• Jaundice that develops rapidly after birth and before 24 hours or that occurs within 24 - 36 hours• Enlarged placenta• Edema• Ascites
NURSING INTERVENTIONS• Determine blood type and Rh early in pregnancy.• Determine results of direct Coomb’s test early in pregnancy & again at 28 week’s.• Determine results of direct Coomb’s test on cord blood.
- type & Rh, Hgb, Hct• Implement phototherapy or exchange transfusion.• Administer Rh0 (D) immune globulin to the mother during the first 72 hrs. after delivery if the Rh(-) mother delivers an Rh
(+) fetus but remains unsensitized• Assist with exchange transfusion as prescribed.• The baby undergoes transfusion of blood to stop the destruction of the baby’s RBC
- the transfused blood is replaced with the baby’s own blood gradually• Reassure the mother that the newborn will suffer no untoward effects from the condition
MYELOPROLIFERATIVE DISORDER
POLYCYTHEMIA VERAChronic myeloproliferative d.o. involves bone marrow elements increase RBC mass & hgb
• Underlying cause is unknown• Hyperplasia of all bone marrow elements
> increase RBC mass> increase blood volume viscosity> decrease marrow iron reserve
> Splenomegaly
ASSESSMENT• Reddish purple hue of skin & mucosa, pruritus • Splenomegaly, hepatomegaly• Epigastric discomfort, abdominal discomfort• Painful fingers & toes from paresthesias• Altered mentation• Weakness, fatigue, night sweats, bleeding tendency• Hyperuricemia – from increased RBD formation and destruction
DX TESTS• CBC• BONE MARROW ASPIRATION & Biopsy
MANAGEMENT• HYPERVISCOSITY
= phlebotomy @ intervals determined by CBC results to decrease RBC mass=generally 250-500ml removal @ a time
• HYPERPLASIA= myelosuppressive therapy, = generally using hydroxyurea or IV radioactive phosphorus (32P), biologic response modifier, ie alpha interferon
• HYPERURICEMIA= allupurinol (Zyloprim)• PRURITUS = antihistamines (cimitidine), low dose acetyl salicylic acid; certain anti-depressants (paroxetin), phototherapy,
cholestyramineINTERVENTION

• Encourage/assist ambulation• Assess for early S/Sx of thromboembolic complications : swelling of limbs, increased warmth, pain• Monitor CBC & assist with phlebotomy as ordered
Patient Education• Educate about risk of thrombosis; encourage patient to maintain normal activity pattern & avoid long periods of rest• Avoid hot showers• Report @ regular intervals for follow up blood
DISORDERS OF PLATELETS AND CLOTTING MECHANISM
HEMOPHILIA• Hereditary coagulation defect, usually transmitted to affected male by female carrier through sex – linked recessive gene,
resulting in prolonged clotting time.• Most common type is Hemophilia A or Classic Hemophilia - factor VIII deficiency (called Antihemophilic Factor / AHF)• Hemophilia B or Christmas Disease – factor IX deficiency (called the Christmas Factor)• Male inherits hemophilia from their mothers, and females inherit the carrier status from their fathers.
– Found predominantly, but not exclusive, in male offsprings• Bleeding occurs due to impaired ability to form fibrin clot

ASSESSMENT• Abnormal bleeding in response to trauma or surgery. (muscles/joints)• Joint bleeding causing pain, tenderness, swelling, and limited range of motion.• Tendency to bruise easily.• Epistaxis• Hemarthrosis (bleeding in joints causing pain, swelling and limited movement)
IMPLEMENTATION• Administer factor VIII concentrate.• Monitor for bleeding and maintain bleeding precautions.• Monitor for joint pain; IMMOBILIZE the affected extremity if joint pains occur.• Monitor urine for hematuria.• Instruct the parents regarding activities for the child, emphasizing the avoidance of contact sports.• Instruct the parents on how to control bleeding (direct/indirect pressure)• DDVAP (Desmopressin) – promotes the release of Factor VIII in hemophilia A• Use soft toothbrush and point out need for regular dental checkups• Refer to National Hemophilia Association• Emphasize avoidance of Aspirin • Provide diet information as excess weight places further stress on joints
R - RestI - ImmobilizeC - Cold CompressE - Elevate

DISSIMINATED INTRAVASCULAR COAGULATION• DIC is a disorder of diffuse activation of the clotting cascade that results in depletion of clotting factors in the blood.• occurs when the blood clotting mechanisms are activated all over the body instead of being localized to an area of injury. • grave coagulopathy resulting from overstimulation of clotting & anticlotting processess in response to disease & injury
• Small blood clots form throughout the body, and eventually the blood clotting factors are used up and not available to form clots at sites of tissue injury.
• Clot - dissolving mechanisms are also increased stimulated by many factors including infection in the blood & severe tissue injury – burns and head injury, reactions to blood transfusions, carcinomas and obstetrical complications such as retained placenta after delivery.
ASSESSMENT• purpura on lower extremities & abdomen• hemorrhagic bullae, acral cyanosis, focal gangrene in skin
Dx Tests:• marked decrease of blood platelets• low levels of fibrinogen & other clotting factors• prolonged prothrombin & partial thromboplastin times & abnormal erythrocyte morphologic characteristics
generalized intravascular clotting which in turn overstimulates fibrinolytic mechanisms
hypercoagulability
hypocoagulability
hemorrhage

Nursing Interventions / Treatment1. To determine the underlying cause of DIC and provide treatment for it.2. Replacement therapy of the coagulation factors is achieved by transfusion of fresh frozen plasma. Cryoprecipitates may also be used if fibrinogen is significantly decreased. Platelet transfusions if platelets are diminished3. Heparin, a medication used to prevent thrombosis, is sometimes used in combination with replacement therapy. ( still controversial )4. Prevent further injury
a. avoid IM injectionsb. apply pressure to bleeding sitesc. turn patient frequently and gentlyd. provide mouth care – soft bristled toothbrush
5. Teach patient the importance of avoiding aspirin.
IDIOPATHIC THROMBOCYTOPENIA PURPURA• Increased destruction of platelets with resultant platelet count of less that 100,000/mm3 characterized by petechiae and
ecchymoses of the skin.• Exact cause unknown; may be autoimmune.• Spleen is the site for destruction of platelets• often triggered by URTI or Childhood communicable disease – Measles & chickenpox
ASSESSMENT:• Petechiae• Ecchymosis• Blood in any body secretions, bleeding form mucous membranes, nosebleeds.• Decreased platelet count• Anemia• easy bruising• blood in stool or urine• CBC reveals platelet count below 20,000/mm3• Bone marrow aspiration done to rule out leukemia
MEDICAL MANAGEMENT:• Drug therapy:
• Prednisone – decreases anti-platelet antibodies (monitor for infection)• IVIG (Intravenous Immune Globulin) – helps to effectively increase platelet count• Anti-D Antibody – one dose treatment
• Given to pt’s 1 year but less than 19 years old• Normal WBC and hemoglobin• no active bleeding present• no concurrent infection• Diphenhydramine and hydrocortisine are made ready for possible allergic reactions to the medication
• Platelet transfusion• Splenectomy
INTERVENTION• Prevent, control and minimize bleeding.• Prevent bruising• Provide support to client and be sensitive to change in body image.• Protect from infection.• Administer analgesics (acetaminophen) as ordered; avoid aspirin.• administer meds orally, rectally, or I.V. rather than I.M.
BLOOD AND BLOOD PRODUCTS
Whole Blood• Contents
– RBC’s– WBC’s– Platelets– Plasma– Clotting factors
• Indications– Acute loss of whole blood
Packed Cells• Contents
– RBC’s– 20% Plasma
• Indications– Replace O2 carrying capacity with less volume– Severe anemia, slow blood loss, CHF

Granulocytes• Contents
– WBC’s– 20% Plasma
• Indications– Life-threatening decreases in WBC count
Plasma• Contents
– Clotting factors– Fibrinogen– Prothrombin– Albumin– Globulins
• Indications– Clotting factor deficiency– Volume expansion
Plasma Protein Fraction• Contents
– 5% Albumin/Globin in saline• Indications
– Expand volume in burns– Hemorrhage– Hypoproteinemia
Albumin• Contents
– 5% or 25% albumin• Indications
– Replace volume in shock– Burns– Hypoproteinemia
Cryoprecipitate• Contents
– Factors VIII and XIII, Fibrinogen• Indications
– Hemophilia A– Fibrinogen deficiency– Factor XIII deficiency
Prothrombin• Contents
– Factors II, VII, IX, and X• Indications
– Hemophilia B– Liver disease
Blood Transfusion• ABO Antigens
– A Antigen Type A– B Antigens Type B– A and B Antigens Type AB– No Antigens Type O
TRANSFUSION COMPLICATIONSAllergic Reactions
• Signs/Symptoms– Itching– Uticaria– Chills– Fever– Facial edema– Wheezing– Anaphylactic shock
• Management– Oxygen– IV fluids

– Epinephrine– Antihistamines
Hemolytic Reaction• Signs/Symptoms
– Chills, fever– Low back pain– Headache– Chest pain– Dyspnea– Cyanosis– Restlessness, anxiety – Hypotension– Red urine
• Management– Stop transfusion– Treat shock– Volume replacement– Mannitol
Volume Overload• Signs/Symptoms
– Cough– Chest pain– Dyspnea– Distended neck veins– Rales– Frothy sputum
• Management– Slow infusion– Diuretics– Vasodilators
Transfusion Complications• Coagulation Disturbances
– Platelet/Clotting factor deterioration• Citrate Intoxication
– Hypocalcemia– Metabolic Alkalosis
• Hyperkalemia– RBC’s Lyse/Release K+
• Hypothermia– Inadequate warming during transfusion
• Viral Hepatitis– Risk rises with each unit
Blood Transfusion• IV catheter 18g or larger• No fluid other than saline
– D5W lyses RBC’s– LR contains calcium/triggers clotting
• Two persons confirm ABO/Rh• Blood filter in administration set• Infusion pumps
– Excessive pressure can cause hemolysis• Rewarming above 380C can cause hemolysis• Never add medications directly
Plasma Clotting FactorsI FibrinogenII ProthrombinIII Tissue ThromboplastinIV CalciumV ProacelerinVII ProconvertinVIII Antihemophilic FactorIX Christmas FactorX Stuart – Prower FactorXI Plasma Thromboplastin AntecedentXII Hageman FactorXIII Fibrin Stabilizing Factor



















