
Good manufacturing practices- schedule
77
SCHEDULE M JIJO THOMAS M.PHARM PHARMACEUTICS COLLEGE OF PHARMACEUTICAL SCIENCES TRIVANDRUM
-
Upload
jijothomaschirayil -
Category
Education
-
view
191 -
download
5
Transcript of Good manufacturing practices- schedule

SCHEDULE M
J I JO THOMAS
M.PHARM PHARMACEUTICS
COLLEGE OF PHARMACEUTICAL SCIENCES
TRIVANDRUM

CONTENTS Introduction
Schedule M
Part I Part II
Schedule M I
Schedule M II
Schedule M III

INTRODUCTION
GMP is that part of Quality assurance which ensures that the products are consistently manufactured and controlled to the Quality standards appropriate to their intended use.
"GMP" - A set of principles and procedures which, when followed by manufacturers for therapeutic goods, helps ensure that the products manufactured will have the required quality

US FDA GMP Regulation : Section 21 of the CFR contains most regulations pertaining to food and drugs21 Code of Federal Regulations Part 210:Current Good Manufacturing Practice in Manufacturing Processing, packing, or Holding of Drugs.21 Code of Federal Regulations Part 211: Current Good Manufacturing Practice for Finished Pharmaceuticals.
ICH GMP Regulation:Q7 /Q&As Q7: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients

GMP REGULATIONS IN INDIA
• GMP regulations were introduced in the form of amended schedule M in 1988.
• Again amended by Drug & Cosmetics Rules 2001 and embraces rules 71,74,76&78 under the D&C rules 1945
• Each licensee shall evolve appropriate methodology systems and procedures, which shall be documented and maintained for inspection and reference.
• The manufacturing premises shall be used exclusively for production of drugs and not for any other manufacturing.

SCHEDULE M

PART -1
GMP for premises & materials
• Specific requirements for manufacture of sterile products, parenteral preparations, & sterile ophthalmic preparation
Part 1A• Specific requirements for manufacture of oral solid
dosage formsPart 1B• Specific requirements for manufacture of oral
liquidsPart 1C• Specific requirements for manufacture of topical
preparationsPart 1D• Specific requirements for manufacture of metered
dose inhalers(MDI)Part 1E• Specific requirements for manufacture of active
pharmaceutical ingredientsPart 1F

1. GENERAL REQUIREMENTS
1.1 . Location and surroundings:
The factory building(s) for manufacture of drugs shall be so situated that it avoid risk of contamination from external environmental including open sewage, drain, public lavatory
1.2. Building and premises :
• Buildings shall be designed, constructed, adapted and maintained to permit production of drugs under hygienic conditions
• They shall conform to the conditions laid down in the Factories Act, 1948

The premises used for manufacturing, processing, warehousing, packaging, labeling and testing purposes shall be
i. compatible with other drug manufacturing operations
ii. adequate working space to avoid the risk of mix-up between different materials & avoid the possibilities of contamination and cross contamination
iii. designed / constructed / maintained to prevent entry of insects, pests, birds, and rodents
iv. Well illuminated, effectively ventilated, with proper Air Handling Units

1.3 Water System:
• There shall be validated system for treatment of water in accordance with standards specified by the Bureau of Indian Standards or Local Municipality or Pharmacopoeial specification
• Water shall be stored in tanks, which do not adversely affect quality of water and ensure freedom from microbiological growth
• The tank shall be cleaned periodically and records maintained by the licensee in this behalf

1.4. Disposal of waste
• The disposal of sewage and effluents (solid, liquid and gas) be in conformity with the requirements of Environment Pollution Control Board
• All bio-medical waste shall be destroyed as per the provisions of the Bio-Medical Waste (Management and Handling) Rules, 1996
• Records shall be maintained for all disposal of waste
• Provisions shall be made for the proper and safe storage of waste materials awaiting disposal

2. WAREHOUSING AREA
2.1 Adequate areas to allow orderly warehousing of various categories of materials.
2.2 Good storage conditions
2.3 There shall be a separate sampling for active raw materials and excipients
2.4. Segregation shall be provided for the storage of rejected, recalled or returned materials or products
2.5 Highly hazardous, poisonous and explosive materials shall be stored in safe and secure areas

3. PRODUCTION AREA
3.1. Separate dedicated and self-contained facilities shall be made available for the production of sensitive pharmaceutical products
3.2. Orderly and logical positioning of equipment and materials and movement of personnel to minimize risk of omission or wrong application of any manufacturing and control measures
3.3. Services lines shall preferably be identified by colours and the nature of the supply and direction of the flow shall be marked/indicated

4. ANCILLARY AREAS
4.1 Rest and refreshment rooms shall be separate from other areas.
Facilities for changing, storing clothes and for washing and toilet purposes shall be adequate for the number of users.
4.2 Animal houses shall be those as prescribed in Rule 150-C(3) of the Drugs and Cosmetics Rules, 1945 which shall be adopted for production purposes.

5. QUALITY CONTROL AREA.
5.1. QC Lab shall be independent of the production areas. Separate areas each for physico-chemical, biological, microbiological or radio-isotope analysis
5.2 Adequate space shall be provided to avoid mix-ups and proper storage for test samples, retained samples, reference standards, reagents and records.

6. PERSONNEL
6.1. Qualifications and practical experience in the relevant field
6.2. Written duties of technical and Quality Control personnel shall be laid and follow strictly
6.3. Number of personnel employed shall be adequate and in direct proportion to the workload

7. HEALTH, CLOTHING AND SANITATION OF WORKERS
7.1 Prior to employment, all personnel, shall undergo medical examination and a periodically examination is carried out, records are also maintained
7.2 Proper training shall be given to all employees to maintain personnel hygiene and adequate facilities for personal cleanliness
7.3 Smoking, eating, drinking, chewing , food, drink and personal medicines not permitted in production, laboratory, storage area

8. MANUFACTURING OPERATIONS AND CONTROLS
8.1 All manufacturing operations shall be carried out under the supervision of technical staff approved by the Licensing Authority
8.2. Precautions against mix-up and cross-contamination-
1. By proper air handling system, pressure differential, segregation, status labelling and cleaning. Proper records and SOP there of shall be maintained.
2. Processing of sensitive drugs and cytotoxic substances in segregated areas

3. Proper labeling of materials and equipments
4. Packaging lines shall be independent and adequately segregated
5. All printing and overprinting shall be authorized in writing
6. The manufacturing environment maintained at the required levels of temperature, humidity and cleanliness

9. SANITATION IN THE MANUFACTURING PREMISES
9.1 The manufacturing premises shall be cleaned and in an orderly manner
9.2 The manufacturing areas shall not be used for other operations
9.3 A routine sanitation program shall be drawn up which shall be properly recorded and which indicate–
a) specific areas to be cleaned and cleaning intervals
b) cleaning procedure to be followed
c) personnel assigned to and responsible for the cleaning operation.

10. RAW MATERIALS
10.1 Keeping an inventory of all raw materials to be used and maintain records as per Schedule U
10.2 Authorized staff who examine each consignment for its integrity10.3 Labeling with the following information:
a) Name of the product and the internal code reference and analytical reference number
b) Manufacturer’s name, address and batch number

c. The status of the contents (e.g. Quarantine, under test, released, approved, rejected)
d. The manufacturing date, expiry date and re-test date.

11. EQUIPMENT
11.1 Equipment shall be located, designed, constructed, adapted to suit the operations and log book is maintained
11.2 Equipment shall be calibrated and checked on a scheduled basis in accordance to SOP and maintain records
11.3 Equipment shall be inert and defective are removed and labeled

12. DOCUMENTATION AND RECORDS
Documentation is an essential part of the Quality assurance system and Its aim is to define the specifications for all materials, method of manufacture and control to release a batch of drug for sale

12.1 Documents shall be approved, signed and dated by authorized persons
12.2 Documents shall specify : the title, nature and purpose laid out in an orderly fashion & kept up to date
12.3 SOP shall be retained for at least one year after the expiry date of the finished product
12.3 Data may be recorded by electronic data processing systems but Master Formulae and detailed operating procedures relating to the system in use shall also be available in a hard copy to facilitate checking of the accuracy of the records

13. LABELS AND OTHER PRINTED MATERIALS
• Labels are necessary for identification of the drugs and their use. The Printing shall be done in bright colours and in a legible manner
• The label shall carry all the prescribed details about the product
13.1 All containers and equipment shall bear appropriate labels
13.2 Prior to release, all labels for containers shall be examined by the QC Department
13.3 Records of receipt of all labeling and packaging materials shall be maintained and unused coded, damaged labels and packaging materials shall be destroyed and recorded.

14. QUALITY ASSURANCEIt is a wide-ranging concept concerning all matters that individually or collectively influence the quality of a product
It shall ensure that: -
• The pharmaceutical products are designed and developed in a way that takes account of the requirement of GMP ,GLP and GCP
• Adequate controls on starting materials, intermediate products, and other in-process controls, calibrations, and validations are carried out
• Products are released after authorized persons have certified

15. SELF INSPECTION AND QUALITY AUDIT
It is for the assessment of all or part of a system with the specific purpose of improving it
15.1 The program is designed to detect shortcomings in the implementation of GMP and to recommend the necessary corrective actions.
• Self-inspections shall be performed routinely and on specific occasions
• The team responsible for self-inspection shall consist of independent, experienced, qualified persons from within or outside the company who can evaluate the implementation of GMP objectively; all recommendations for corrective action shall be implemented.

16. QUALITY CONTROL SYSTEM
• Quality control shall be concerned with sampling, specifications, testing, documentation, release procedures
• Materials are not released for use, nor products released for sale or supply until their quality has been judged to be satisfactory
15.2 The procedure for self-inspection shall be documented indicating self-inspection results; evaluation, conclusions and recommended corrective actions with effective follow up program.

16.1 QC lab should have qualified and experience staff
16.2 QC lab may be divided into Chemical, Instrumentation, Microbiological and Biological testing
16.3 The QC department shall conduct stability studies of the products to ensure and assign their shelf life. All records of such studies shall be maintained
16.4 All instruments shall be calibrated and validated before adopted for routine testing
16.5 Pharmacopoeia, reference standards, working standards, references spectra, other reference materials and technical books, as required, shall be available in the QC Laboratory

17. SPECIFICATION17.1 For raw materials and packaging materials
They shall include:
a) The designated name and internal code reference
b) Reference, if any, to a pharmacopoeial monograph
c) Qualitative and quantitative requirements with acceptance limits
d) Name and address of manufacturer or supplier and original manufacturer of the material
e) Specimen of printed material
f) Directions for sampling and testing or reference to procedures
g) Storage conditions and
h) Maximum period of storage before re-testing

17.2 For finished products. Appropriate specifications for finished products shall include:
a) The designated name of the product and the code reference
b) The formula or a reference to the formula and the pharmacopoeial reference
c) Directions for sampling and testing or a reference to procedures
d) A description of the dosage form and package details
e) The storage conditions and precautions, where applicable
f) The shelf-life

18. MASTER FORMULA RECORDS• There shall be Master Formula records relating to all
manufacturing procedures for each product and batch size
• These shall be prepared and endorsed by head of production and quality control
• The master Formula shall include:
a) the name of the product together with product reference code relating to its specifications
b) the patent or proprietary name of the product along with the generic name, a description of the dosage form, strength, composition of the product and batch size
c) name, quantity, and reference number of all the starting materials to be used

d. A statement of the processing location and the principal equipment to be used
e. detailed stepwise processing instructions and the time taken for each step
f. the instructions for in-process control with their limits
g. the requirements for storage conditions of the products, including the container, labeling and special storage conditions where applicable
h. any special precautions to be observed and
i. packing details and specimen labels

19. PACKING RECORDS
There shall be authorised packaging instructions for each product, pack size and type
These shall include or have a reference to the following:
a) Name of the product
b) Description of the dosage form, strength and composition
c) The pack size expressed in terms of the number of doses, weight or volume of the product in the final container
d) Special precautions to be observed, including a careful examination of the area and equipment in order to ascertain the line clearance before the operations begin.

20. BATCH PACKAGING RECORDS• A batch packaging record shall be kept for each batch or
part batch processed
• It shall be based on the relevant parts of the packaging instructions, and the method of preparation of such records shall be designed to avoid transcription errors
21. BATCH PROCESSING RECORDS• There shall be Batch Processing Record for each product
• It shall be based on the relevant parts of the currently approved Master Formula
• The method of preparation of such records included in the Master Formula shall be designed to avoid transcription errors

22. STANDARD OPERATING PROCEDURES (SOPs) There shall be written SOP and records for the :
• Receipt of each delivery of raw, primary and printed material
• Internal labeling, quarantine and storage of starting materials, packaging materials and other materials, as appropriate
• For each instrument and equipment
• Sampling which include the person(s) authorized to take the samples
• Describing the details of the batch numbering which ensure that each batch of intermediate, bulk or finished product is identified with a specific batch number
• Testing materials and products at different stages of manufacture, describing the methods and equipment to be used

23. REFERENCE SAMPLES
23.1 Each lot of every active ingredient, in a quality sufficient to carryout all the tests, except sterility and pyrogens / Bacterial Endotoxin Test, shall be retained for a period of 3 months after the date of expiry of the last batch produced from that active ingredient
23.2. Samples of finished formulations shall be stored in the same or simulated containers in which the drug has been actually marketed

24. REPROCESSING AND RECOVERIES:• Where reprocessing is necessary, written procedures
shall be established & approved by quality assurance department
• Recovery of product residue may be carried out , if permitted, in the master production
25.DISTRIBUTION RECORDS: Records of distribution shall be maintained in a manner such that finished batch of a drug can be traced to the retain level to facilitate prompt and complete recall of the batch, if and when necessary

26. VALIDATION AND PROCESS VALIDATION: • The process employed has been optimized , so that data
generated may be considered credible & evaluated for consistency as well as relevance
• Validation studies shall conducted as per the pre-defined protocols
• These shall include validation of processing, testing and cleaning procedures
27. PRODUCT RECALLS: • A prompt and effective product recall system of defective
products shall be devised for timely information of all concerned stockiest, wholesalers, suppliers, up to the retail level within the shortest period
• The licensee may make use of both print and electronic media in this regard

28. COMPLAINTS AND ADVERSE REACTIONS:• All complaints thereof concerning product quality
shall be carefully reviewed and recorded according to written procedures
• Reports of serious adverse drug reactions resulting from the use of a drug along with comments and documents shall be reported to the concerned licensing authority

29. SITE MASTER FILE: • It contains specific and factual GMP about the production
and/or control of pharmaceutical manufacturing preparations carried out in the licensed premises
• It shall contain the following:-General Information Personnel PremisesEquipmentSanitationDocumentationProductionQuality ControlLoan license manufacture and LicenseDistribution, Complaints and Product recallSelf Inspection & Export of Drugs

• General : Sterile products being very critical & sensitive in
nature, a high degree of precautions, prevention & preparations are needed
Prescribed standards for supply of water, air, active materials, maintenance of hygienic conditions
• Buildings and civil works:
Built of standardized material to avoid cracks in critical areas
Manufacturing area clearly separated into support areas, preparation areas, change areas and aseptic areas
PART 1A: sterile products

Aseptic area:
Walls, floors and ceilings should be impervious, non-shedding & non-cracking
Walls shall be flat & ledges and recesses can be avoided
Ceilings shall be solid & joints shall be sealed
No sinks & drains in grade A & grade B areas
Doors made of aluminum or steel material & shall open towards high pressure area
Separate exit space from the aseptic areas is advisable
Change rooms to aseptic areas shall be clearly demarcated into black, grey & white rooms
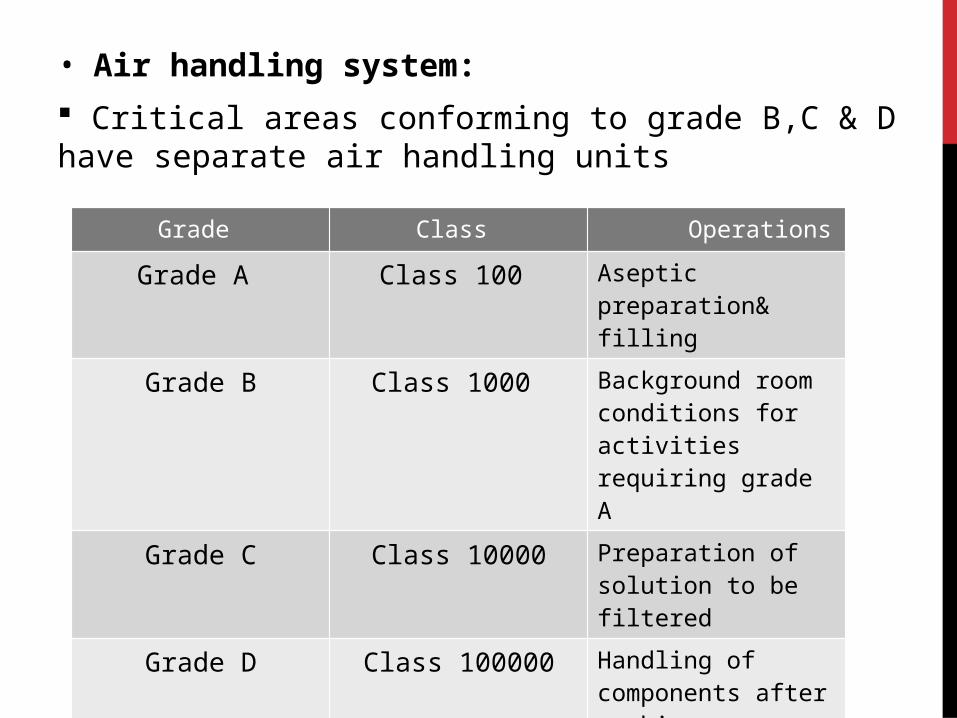
• Air handling system:
Critical areas conforming to grade B,C & D have separate air handling units
Grade Class Operations
Grade A Class 100 Aseptic preparation& filling
Grade B Class 1000 Background room conditions for activities requiring grade A
Grade C Class 10000 Preparation of solution to be filtered
Grade D Class 100000 Handling of components after washing

• Environment monitoring:Recommended periodic monitoring include:
Particulate monitoring in air - 6 monthlyHEPA filter integrity testing – yearlyAir change rates – 6 monthlyAir pressure differentials – dailyTemperature & humidity – dailyMicrobiological monitoring – daily
• Garments: Made of non-shedding & tight weave materialOutdoor clothing shall not be brought into sterile areas
• Sanitation:Employees carrying out sanitation shall be trained Different sanitizing agents used in rotation Records of rotational use these agents to be maintained

• Include component washing machines, steam sterilizers, membrane filters, liquid & powder filling machine , sealing & labelling machines etc
Equipment
• Potable water – microbiological specification(<500cfu/ml) &
• Absence of individual pathogens per 100ml used for preparation of purified water
• Steam coming in contact with products primary containers shall be sterile & pyrogen free
Water and steam system
• Bulk raw materials monitored for bio-burden periodically
• A limit of not more than 100cfu/ml is recommended
• Finished products shall be filled in continuous operation with great care
Manufacturing process
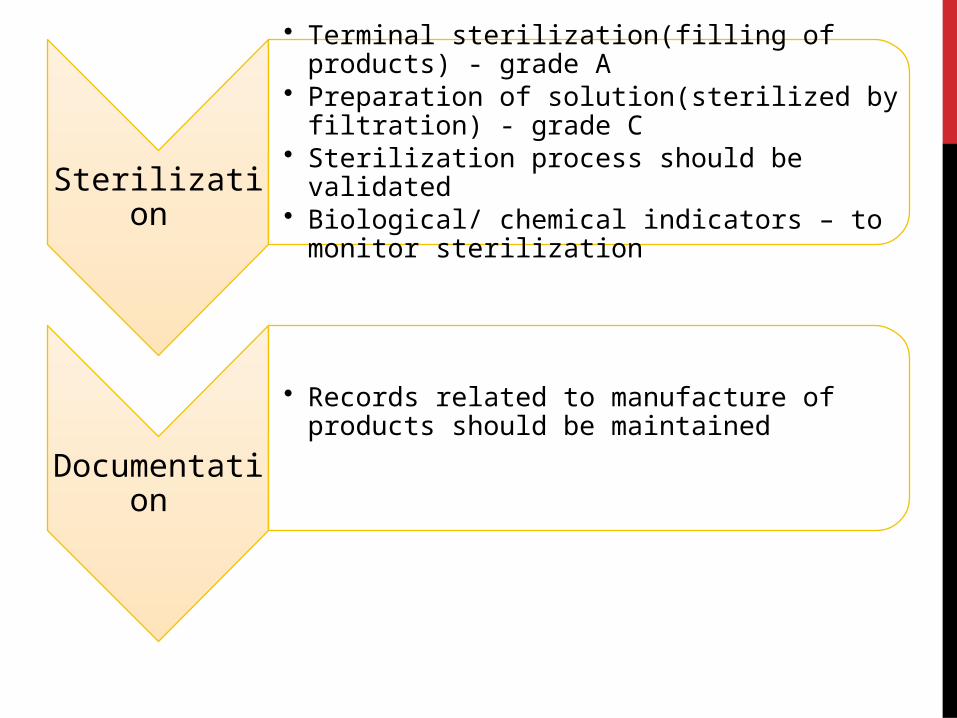
Sterilization
• Terminal sterilization(filling of products) - grade A• Preparation of solution(sterilized by filtration) -
grade C• Sterilization process should be validated• Biological/ chemical indicators – to monitor
sterilization
Documentation
• Records related to manufacture of products should be maintained

• General requirements:
Enclosed dust control manufacturing system shall be employed
Suitable environmental conditions maintained by installation of air- conditioning
Effective air-extraction system shall be provided with discharge
Filters to retain dust to protect the local environment
• Sifting, mixing & granulation:
All equipments shall be fitted with dust extractors
Critical operating parameters like time &temperature shall be specified in master formula
PART 1B: oral solid dosage forms

Monitored during processing & recorded in batch records
Granulation & coating solution- made, stored & used in a manner to minimize contamination
Tablet compression:
Compressing machine – effective dust control facilities
Suitable labelling to prevent mix up of granules & tablets on compression machinery
Tablets examined for appearance, wt. variation, disintegration, hardness, friability & thickness
Tablet coating:
Air provided with suitable exhaust system & environmental control
Air supplied to coating pans shall be filtered & of suitable quantity

• Stored in conditions which ensure safety from effects of excessive heat & moisture
Filling of hard gelatin capsules
• Special care must be taken to avoid product mix-up during printing
• Done using edible colors & suitable printing ink
Printing(tablets & Capsules)
• Before packaging operation all ‘rogue’ tablets & capsules are removed
• Strips - inspected for defects such as misprint, cut on foil, missing tablets & improper sealing
Packaging

Layout & design of manufacturing area – minimize risk of contamination & mix-up
Manufacturing area – entry through double door air-lock facility
Parts of equipment – made of stainless steel
Quality of purified water used shall be specified & monitored routinely
Manufacturing personnel shall wear non- shedding clothing
Primary packaging area – air supply filtered through 5 micron filters & temperature not exceed 30ºC
PART 1C : oral liquid dosage
forms

Suitable air-lock shall be provided at the entrance
Manufacturing area – air shall be filtered & air conditioned
These areas to be fitted with exhaust systems
Equipments designed to prevent product contamination
For cleaning or drying no rags or clusters to be used
Water used in compounding – Purified Water IP
Powders used must be properly sieved
Separate packaging section – for primary packaging
PART 1D : topical preparations

General:Manufacture of MDI - minimum microbial & particulate contamination
Building & civil works:Located on a solid foundation to reduce risk of cracking walls & floors
Environmental condition:Where products or clean components are exposed, the area shall be supplied with filtered air of grade C
Equipment:Manufacturing equipment – closed system where vessels & supply lines are stainless steel
PART 1E : MDIs

Manufacture:Approved master formula records are required for manufacture of MDI
Primary packaging material – cleaned by compressed air filtered through 0.2 micron filter
Filled containers shall be quarantined for a suitable period to detect leaking containers
Documentation :Temperature & humidity in the manufacturing area
Periodic filled weights of formulation
Records of rejections

PART 1F : APIs
Buildings & civil works: Manufacture of antibiotics, steroids & cytotoxic
substances – carried out in confined areas to prevent contamination of other manufactured drugs
Air filtration system – includes pre-filters & particulate matter retention air filters
Sterile products:
Filling of such products must be done aseptically
Manufacture of sterile API must include- filtration, crystallization & lyophilization

Utilities / Services: Equipments used – serviced, cleaned & maintained at
appropriate intervals
So as to prevent mal- function or contamination of drug product
Equipment design, size & location:
Equipment used in manufacture, processing, packaging or holding of API shall be adequate size, appropriate design &
Suitably located to facilitate operations for its intended use & for its cleaning & maintenance
Equipments - cleaned between successive batches

In- process control:
For chemical reactions include:Reaction timeReaction mass appearance Reaction temperatureConcentration of reactantAssay or purity of the product
For physical operations include:Appearance & colorUniformity of blendTemperature of a processConcentration of solutionProcessing rate or time

Product containers & closures:
• Should comply with pharmacopoeial specification
• Containers & closures shall be non-reactive, additive, adsorptive or leachable
• To an extent that affects quality or purity of the drug

External preparation:
Basic installation area: 30sq.m
Ancillary area: 10sq.m
Equipments include:
• Mixing & storage tank(stainless steel)• Jacketed kettle(stainless container)• Mixer• A colloidal mill or suitable emulsifier• A triple roller mill• Liquid filling equipment• Tube filling equipment
PART II

Oral liquid preparation:
Basic installation area:30sq.m
Ancillary area:10sq.m
Equipments include:
• Mixing & storage tanks• Jacketed kettle• Portable stirrer• A colloidal mill• Filtration equipment• Semi automatic filling machine• Pilfer proof cap sealing machine• Water distillation unit deionizer• Clarity testing inspection unit
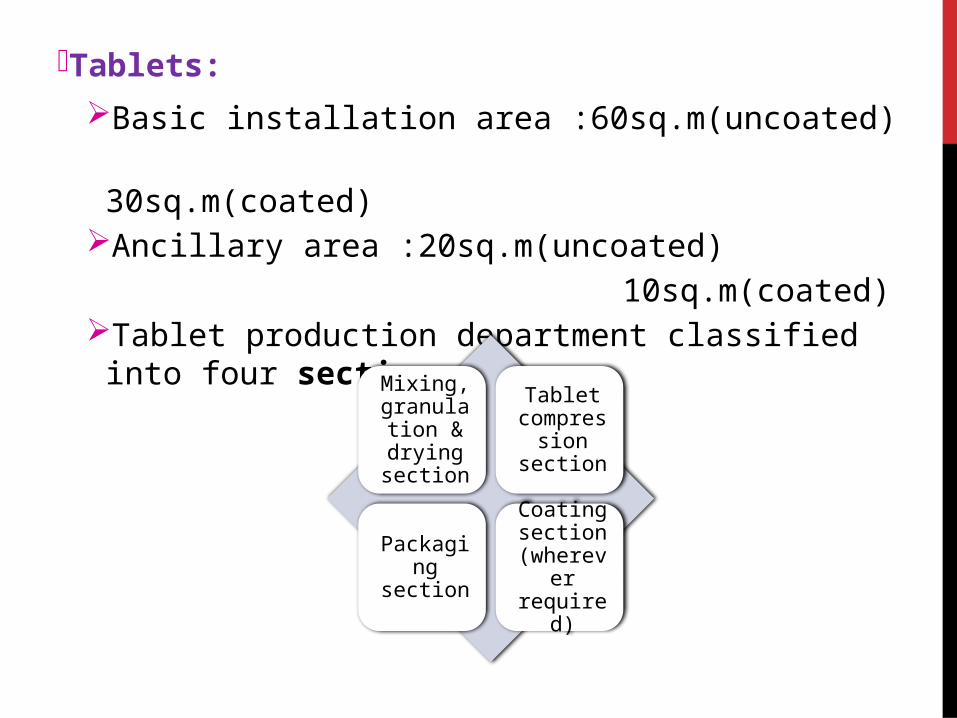
Tablets:
Basic installation area :60sq.m(uncoated)
30sq.m(coated) Ancillary area :20sq.m(uncoated)
10sq.m(coated)Tablet production department classified into four
sections:
Mixing, granulation & drying
section
Tablet compression section
Packaging section
Coating section
(wherever required)

Mixing, granulation & drying section:
• Disintegrator & sifter• Powder mixer• Granulator wherever required• Thermostatically controlled hot air oven• Weighing machines
Compression section:
• Tablet compression machine• Punch & die storage cabinets• Tablet de-duster• Tablet inspection unit• Dissolution test apparatus• In-process testing apparatus• Air conditioning & dehumidifier

Packaging section:
• Strip /blister packaging machine• Leak test apparatus• Tablet counters • Air-conditioner & dehumidifier
Coating section:
• Jacketed kettle• Coating pan• Polishing pan• Exhaust system• Air conditioner & dehumidifier• Weighing balance

Powders:
Basic installation area: 30sq.m
Equipments include:
• Disintegrator • Mixer (electrically operated)• Sifter• Stainless steel vessels• Filling equipment
Capsules:
Basic installation area: 25sq.m
Ancillary area:10sq.m
Equipments include:
• Mixing & blending equipment• Capsule filling unit• Capsule counters &• Polishing equipment

Surgical dressing:
Basic installation area:30sq.m
Equipments include:
• Rolling & trimming machine• Cutting equipment• Folding & pressing machine for gauze• Mixing tanks for processing medicated dressing• Hot air dry oven• Steam or dry heat sterilizer
Ophthalmic preparations :
Basic installation area : 25sq.m
Equipments include:
• Hot air oven• Kettle• Colloidal mill• Tube filling equipment

• Mixing & storage tanks• Seitz filter or sintered glass filters• Liquid filling equipment• Autoclaves
Pessaries & suppositories:
Basic installation area:20sq.m
Equipments include:
• Mixing & pouring equipment• Molding equipment• Weighing devices
Inhalers :
Basic installation area: 20sq.m
Equipments include:
• Mixing equipment• Graduated delivery equipment• Sealing equipment of medicament during filling

Repacking of drugs & pharmaceutical chemicals:
Basic installation area: 30sq.m
Equipments include:
• Sifter • Stainless steel vessels• Weighing & measuring equipment• Filling equipment• Electrical sealing machine

Parenteral preparation:
Basic installation area: 60sq.m(partitioned into suitable sized cubicles with air lock arrangement)
Process of manufacture divided into:Preparation of containers & closuresPreparation of solutionFilling & sealingSterilization & testing
Equipments include:Manufacturing area:• Storage equipment• Washing & drying equipment

• Dust proof storage cabinet• Water still• Preparation & mixing tanks and equipments• Filtering equipment• Hot air sterilizer
Aseptic filling & sealing room:• Benches for filling & sealing • Bacteriological filters• Filling & sealing unit under laminar flow
General room:• Inspection table• Leak testing equipment• Labelling & packaging unit• Storage equipment including cold storage & refrigeration

SCHEDULE M-I Requirements of factory premises of homoeopathic preparations
1. General Requirements: Location and Surroundings; Buildings; Water supply Health, Clothing and Sanitation of Workers; Medical Services, container management etc. comes under this.
2. Requirements of Plant and Equipment:
a) Mother tinctures and Mother solution Section- An area of 55 sq. meters is recommended for basic installations
b) Potentisation Section- Method of potentisation will be adopted as specified in Homoeopathic Pharmacopoeia of India Vol.I

SCHEDULE M-II : Requirement of premises for manufacture of cosmetics
• General Requirements: Location and Surroundings; Buildings; Water supply; Health, Clothing and Sanitation of Workers; Medical Services etc
• Requirements of Plant and Equipment: Specifications are given for only certain cosmetic preparations like
a) Powers
b) Creams, lotions, emulsions, pastes, cleansing milks, shampoos, pomade, brilliantine, shaving creams, and hair-oils etc
c) Nail Polishes and Nail lacquers
d) Lipsticks and lip-gloss
e) Depilatories
f) Preparations used for Eyes
g) Aerosol
h) Alcoholic Fragrance Solutions
i) Hair Dyes
j) Toilet Soaps
k) Tooth powders and toothpastes

SCHEDULE M-III Requirements of factory premises for manufacture of medical devicesGeneral Requirements: Location and Surroundings; Buildings; Water supply; Health, Clothing and Sanitation of Workers; Medical Services etc comes under this
Requirements for Manufacture Of Medical Devices: Shall be conducted bat the licensed premises, and the process of manufacture is divided into following separate operations/Sections-
a) Moulding
b) Assembling
c) Raw Materials
d) Storage Area
e) Washing, drying and sealing area
f) Sterilization
g) Testing facilities

cGMP
• What is cGMP ? : Usually see “cGMP” – where c = current, to emphasize that the expectations are dynamic
• In India it is established in 2000 & amended from July 2004
• It is periodically reversed and updated

CONCLUSION
• GMP is an important tool in quality control of drug products
• Legal regulation of GMP is important to control the quality of drugs manufactured with in the country

REFERENCES
1. Manual on Drugs and Cosmetics; sixth edition; 2009
2. Schedule M: Good Manufacturing Practices And Requirements Of Premises, Plant And Equipment For Pharmaceutical Products. Gazette Of India Extraordinary, Part II -section 3, Sub-section (I) Ministry Of Health And Family Welfare (Department Of Health)
3. Ansel’s Pharmaceutical Dosage forms and Drug Delivery system ;L. V. Allen et al. ; eight edition; Current good manufacturing practices and current good compounding practices;67-91
4. Quality Assurance of Pharmaceuticals; Good manufacturing practices and inspection; Volume – 2; WHO
5. Encyclopedia of pharmaceutical technology; third edition; volume – 1;Good Manufacturing Practices(GMPs): An overview;1941-1994



















