
Explicitly-Correlated Electronic Structure Theory · 1 Explicitly-Correlated Electronic Structure...
9
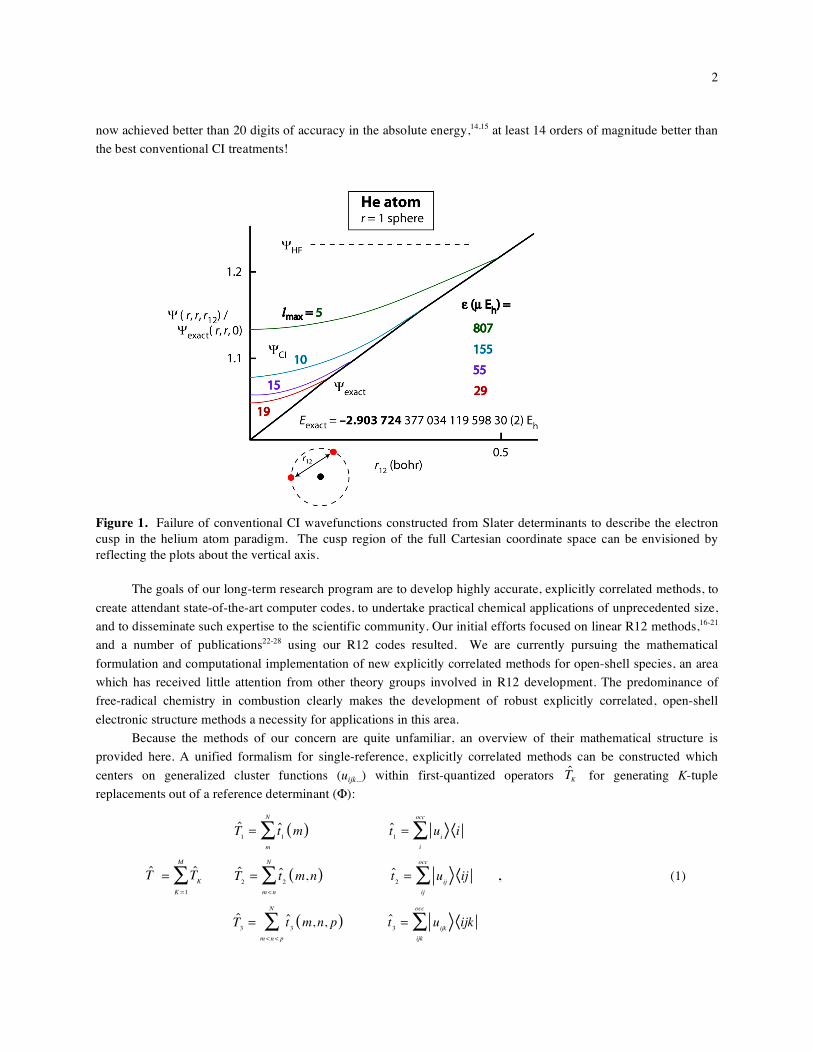
1 Explicitly-Correlated Electronic Structure Theory All ab initio wavefunction methods commonly used today in electronic structure theory in some way involve the orbital approximation, wherein antisymmetrized products (Slater determinants) of one-electron functions are used to represent many-electron wavefunctions of chemical systems. These current-generation methods have been increasingly refined over the past few decades to deliver reliable predictions of molecular energies, geometric structures, and an abundance of properties for diverse fields of chemistry. In addition to computational tractability, orbital-expansion methods have interpretive appeal, because they embody the traditional bonding concepts so thoroughly engrained in the chemical literature. However, there is a fundamental and troublesome flaw in current- generation approaches, namely, the mathematical inability of Slater determinants to properly describe the electron cusp region of the exact electronic wavefunction. 1-3 This flaw is directly manifested in an unacceptably protracted, and seemingly unphysical, dependence of computed electron correlation energies on high angular momentum functions in the one-particle (typically atomic orbital) basis set. Current-generation methods designed to deliver chemical accuracy or better ( < 1 kcal mol -1 ) rest on assumed cancellation of much larger errors in absolute energies of individual chemical species and/or empirical schemes to correct for methodological deficiencies. In order to establish the building blocks of the world’s thermochemical database, inter alia, next-generation electronic structure methods are needed that are capable of achieving subchemical accuracy (0.1-0.2 kcal mol –1 ). The saga of the heat of formation of the OH radical 4,5 is a striking example of the general need for definitive computational thermochemistry. It is remarkable that an error as large as 0.5 kcal mol -1 in one the most essential thermochemical parameters in all of chemistry [ f H 0 (OH) ] persisted until the year 2002, and it is sobering that comparable errors for other fundamental species certainly remain. In IUPAC panels 6 and elsewhere there is increasing recognition of the need for high-accuracy ab initio thermochemical methods, for use within the Active Thermochemical Tables, 7 for example. The HEAT approach 8 is one such high-accuracy scheme, but it is currently limited to rather small systems. Many chemical problems cannot be satisfactorily solved without thermochemical accuracy better than 1-2 kcal mol -1 . One excellent example comes from the ethyl + O 2 reaction prototype for alkyl radical oxidation. Specifically, for the critical transition state for concerted elimination of HO 2 from the ethylperoxy radical (CH 3 CH 2 OO), an change of 2 kcal mol -1 in the barrier causes the computed rate coefficient at T = 900 K to deviate from experiment by a factor of three. 9 We are undertaking theoretical development of explicitly-correlated wavefunction approaches 10,11 that solve the electron cusp problem and promise to mature into a widely-used new generation of methods of subchemical accuracy. A depiction of the electron-electron cusp in an essentially exact Hylleraas wavefunction of the helium atom ground state is shown in Figure 1. In the region of the electron coalescence point, the exact wavefunction is linear in the interelectronic distance (r 12 ). However, conventional quantum chemical wavefunctions effectively incorporate only even powers of r 12 , do not have sufficient flexibility to fully relax into the conical region of the cusp, and thus poorly account for the short-range electron correlation. In Figure 1, these deficiencies are clearly exhibited by the full configuration interaction wavefunctions ( CI ) that we have computed using special analytical and numerical techniques for extremely high angular momentum (l) cutoffs. 12 With a one-particle basis saturated through l = 5 (h functions), CI is too large by 14% at the coalescence point, and the corresponding error ( ) in the electronic energy (E CI ) is 807 μE h . Even after extending the basis set to an unprecedented l = 19, the errors in CI and E CI are still 4% and 29 μE h , respectively. The helium atom results demonstrate that converging absolute energies even to the mE h level is essentially impossible in practical computations with Slater determinants. In contrast, the early work of Hylleraas 13 showed the astounding superiority of explicitly-correlated methods, which directly incorporate the r 12 variable into the wavefunction. Hylleraas CI computations on the ground state of He have
Transcript of Explicitly-Correlated Electronic Structure Theory · 1 Explicitly-Correlated Electronic Structure...

1
Explicitly-Correlated Electronic Structure Theory All ab initio wavefunction methods commonly used today in electronic structure theory in some way involve
the orbital approximation, wherein antisymmetrized products (Slater determinants) of one-electron functions are
used to represent many-electron wavefunctions of chemical systems. These current-generation methods have been
increasingly refined over the past few decades to deliver reliable predictions of molecular energies, geometric
structures, and an abundance of properties for diverse fields of chemistry. In addition to computational tractability,
orbital-expansion methods have interpretive appeal, because they embody the traditional bonding concepts so
thoroughly engrained in the chemical literature. However, there is a fundamental and troublesome flaw in current-
generation approaches, namely, the mathematical inability of Slater determinants to properly describe the electron
cusp region of the exact electronic wavefunction.1-3 This flaw is directly manifested in an unacceptably protracted,
and seemingly unphysical, dependence of computed electron correlation energies on high angular momentum
functions in the one-particle (typically atomic orbital) basis set. Current-generation methods designed to deliver
chemical accuracy or better ( < 1 kcal mol-1) rest on assumed cancellation of much larger errors in absolute energies
of individual chemical species and/or empirical schemes to correct for methodological deficiencies.
In order to establish the building blocks of the world’s thermochemical database, inter alia, next-generation
electronic structure methods are needed that are capable of achieving subchemical accuracy (0.1-0.2 kcal mol–1).
The saga of the heat of formation of the OH radical4,5 is a striking example of the general need for definitive
computational thermochemistry. It is remarkable that an error as large as 0.5 kcal mol-1 in one the most essential
thermochemical parameters in all of chemistry [
f H0 (OH) ] persisted until the year 2002, and it is sobering that
comparable errors for other fundamental species certainly remain. In IUPAC panels6 and elsewhere there is
increasing recognition of the need for high-accuracy ab initio thermochemical methods, for use within the Active
Thermochemical Tables,7 for example. The HEAT approach8 is one such high-accuracy scheme, but it is currently
limited to rather small systems. Many chemical problems cannot be satisfactorily solved without thermochemical
accuracy better than 1-2 kcal mol-1. One excellent example comes from the ethyl + O2 reaction prototype for alkyl
radical oxidation. Specifically, for the critical transition state for concerted elimination of HO2 from the ethylperoxy
radical (CH3CH2OO), an change of 2 kcal mol-1 in the barrier causes the computed rate coefficient at T = 900 K to
deviate from experiment by a factor of three.9
We are undertaking theoretical development of explicitly-correlated wavefunction approaches10,11 that solve
the electron cusp problem and promise to mature into a widely-used new generation of methods of subchemical
accuracy. A depiction of the electron-electron cusp in an essentially exact Hylleraas wavefunction of the helium
atom ground state is shown in Figure 1. In the region of the electron coalescence point, the exact wavefunction is
linear in the interelectronic distance (r12). However, conventional quantum chemical wavefunctions effectively
incorporate only even powers of r12, do not have sufficient flexibility to fully relax into the conical region of the
cusp, and thus poorly account for the short-range electron correlation. In Figure 1, these deficiencies are clearly
exhibited by the full configuration interaction wavefunctions ( CI) that we have computed using special analytical
and numerical techniques for extremely high angular momentum (l) cutoffs.12 With a one-particle basis saturated
through l = 5 (h functions), CI is too large by 14% at the coalescence point, and the corresponding error ( ) in the
electronic energy (ECI) is 807 μEh. Even after extending the basis set to an unprecedented l = 19, the errors in CI
and ECI are still 4% and 29 μEh, respectively. The helium atom results demonstrate that converging absolute
energies even to the mEh level is essentially impossible in practical computations with Slater determinants. In
contrast, the early work of Hylleraas13 showed the astounding superiority of explicitly-correlated methods, which
directly incorporate the r12 variable into the wavefunction. Hylleraas CI computations on the ground state of He have
2
now achieved better than 20 digits of accuracy in the absolute energy,14,15 at least 14 orders of magnitude better than
the best conventional CI treatments!
Figure 1. Failure of conventional CI wavefunctions constructed from Slater determinants to describe the electron cusp in the helium atom paradigm. The cusp region of the full Cartesian coordinate space can be envisioned by reflecting the plots about the vertical axis.
The goals of our long-term research program are to develop highly accurate, explicitly correlated methods, to
create attendant state-of-the-art computer codes, to undertake practical chemical applications of unprecedented size,
and to disseminate such expertise to the scientific community. Our initial efforts focused on linear R12 methods,16-21
and a number of publications22-28 using our R12 codes resulted. We are currently pursuing the mathematical
formulation and computational implementation of new explicitly correlated methods for open-shell species, an area
which has received little attention from other theory groups involved in R12 development. The predominance of
free-radical chemistry in combustion clearly makes the development of robust explicitly correlated, open-shell
electronic structure methods a necessity for applications in this area.
Because the methods of our concern are quite unfamiliar, an overview of their mathematical structure is
provided here. A unified formalism for single-reference, explicitly correlated methods can be constructed which
centers on generalized cluster functions (uijk...) within first-quantized operators TK for generating K-tuple
replacements out of a reference determinant ( ):
T1= t
1m( )
m
N
t1= u
i
i
occ
i
T = TK
K =1
M
T2= t
2m,n( )
m< n
N
t2= u
ij
ij
occ
ij
T3= t
3m,n, p( )
m< n< p
N
t3= u
ijk
ijk
occ
ijk
, (1)

3
where (m, n, p) run over the N electrons, and (i, j, k) index the spin orbitals occupied (occ) in . Correlated
wavefunctions ( ) are derived by application of the composite excitation operator T to according to a chosen
ansatz, such as = eT for single-reference coupled-cluster theory, or = (1 + T ) for configuration interaction
computations. Exact integro-differential equations arise for the cluster functions by placing the correlated
wavefunction ansatz into the Schrödinger equation. For a closed-shell Hartree-Fock reference, the general form of
such equations is
F 1( )i[ ]ui 1( ) = G
1{
i},{u
ijk ...}; (1)[ ]
F 1( ) + F 2( )i j[ ]uij
s 1, 2( ) = Q 1( )Q 2( )i j[ ]
s
r12
+ G2
{i},{u
ijk ...}; (1, 2)[ ]
F 1( ) + F 2( ) + F 3( )i j k[ ]uijk 1, 2, 3( ) = G
3{
i},{u
ijk ...}; (1, 2, 3)[ ]
... ...
, (2)
where F is the customary Fock operator with orbital energies i, Q = 1 P , P is the projection operator onto the
Hartree-Fock occupied space, the i are spatial molecular orbitals, and the GK are generalized functionals depending
on K electrons. The parameter s = (0, 1) denotes (singlet, triplet) pairing of electron spins, and the square brackets
give symmetric or antisymmetric combinations:
1, 2( )[ ]s1
21 + 1 2s( ) ˆ
12[ ] 1, 2( ) , (3)
where 1, 2( ) is any two-particle function and ˆ12
permutes electrons 1 and 2. The two-body functions uijs 1, 2( )
are of special significance, and they are often called pair correlation functions.
The derivation of the GK functionals is rather involved. Bukowski, Jeziorski, and Szalewicz29 have reported
formulas for G1 and G2 in the CCSD case, but first-quantized equations for CCSDT and many other levels of N-
particle theory have not been published to our knowledge. It is important to note that the GK functionals generally
couple the differential equation for one cluster function with those of all the rest, so that the system of equations
must be solved iteratively to self-consistency. Moreover, the exact equations for the cluster functions do not assume
any orbital-product expansion. Equation (2) does involve the occupied orbitals of the system, but only because the
reference wavefunction ( ) is written as a Slater determinant; the usual expansion in terms of virtual orbitals is
completely absent. If one does approximate the cluster functions by such an expansion, then the traditional spin-
orbital equations of each electronic structure method are recovered, but at the expense of failing to describe the
requisite electron cusp behavior.
To be specific and as simple as possible, let us consider the closed-shell MP2 method, where T gives the
first-order perturbation wavefunction. In this special case, ui(1) = 0 and G2(1,2) = 0, so that
F 1( ) + F 2( )i j[ ]uij
s 1, 2( ) = Q 1( )Q 2( )i j[ ]
s
r12
. (4)
In intermediate normalization, we have the auxiliary constraint uij
s 1, 2( ) [ i j ]s = 0 . The resulting second-order
energy is rigorously
E2= f
ij
0
i j
occ
+ fij
1
i< j
occ
, (5)
where the singlet- and triplet-pair energies are evaluated as single electron-repulsion integrals:
fij
s=2s + 1( )
1 +ij( ) i j[ ]
s
1
r12
uij
s 1, 2( ) + ui1( )u
j2( )[ ]
s . (6)

4
If robust analytical or numerical techniques were available to solve Eq. (4) for instantaneous repulsions of a single
electron pair within a field of distributed nuclei and average interactions with the remaining electrons, then exact
MP2 correlation energies could be determined without resort to a one-particle basis set.
Existing electron correlation methods can be categorized according to the ansatz adopted to approximate the
cluster functions. For the all-important pair correlation functions, we can write
uij
s r1, r
2( ) = c
ab
ij , s
a b[ ]sa b
vir
+ wij
s r1, r
2( ) , (7)
where the first term is the usual orbital-product expansion involving double excitations into the virtual (vir) space
with adjustable coefficients cabij , s
, and the second term is some correction function. Conventional methods simply set wij
s r1, r
2( ) = 0 . In contrast, explicitly correlated methods choose a nonzero wij
s r1, r
2( ) that depends on the
interelectronic distance r12, the most common form being the orbital-invariant ansatz30,31
wij
s r1, r
2( ) =
1
2R 1( ) R 2( )g r
12( ) d
kl
ij , s
k l[ ]sk l
occ
, (8)
where g(r12) is the correlation factor, the sum over the occupied orbitals involves additional variable parameters dkl
ij , s, and the projection operator R is preferably32 Q = 1 P . Linear R12 methods16-21 invoke the simplest cusp-
satisfying function, g(r12) = r12. Thus, the pair correlation functions in linear R12 theory assume Eqs. (7) and (8) and
a simple correlation factor r12, with the coefficients cabij , s
and dkl
ij , s determined from the fundamental Eq. (2).
In the closed-shell MP2 case [Eqs. (4)-(6)], after a rather involved derivation, the expression for the pair
energies reduces to
fij
s= e
ij
s 2s + 1( ) V ij , s( )T
G ij , s( )1
V ij , s 1 +ij( )
1
, (9)
in which eijs is the pair energy given by conventional MP2 theory, whereas the explicitly-correlated correction
involves vector V ij , s and matrix G ij , s
, each with dimensions ( Ns ) given in terms of the number of occupied spatial
orbitals (nocc) by Ns = nocc nocc + 1 2s( ) / 2 . The key point is that V ij , s and G ij , s
in Eq. (9) contain three- and four-
electron integrals over the operators r12r231 , r12r23 , r23T12 , r12r23r34
1 , and r12r23r241 , where the commutator
T12 = [r12 , 1
2+ 2
2 ] / 2 . The appearance of such many-electron integrals is characteristic of explicitly correlated
methods, regardless of the ansatz for the correlated wavefunction ( ). The approach to the many-electron integral
problem pioneered by Kutzelnigg, Klopper, and co-workers16-21 entails a resolution of the identity (RI) within the
integrals to allow their approximation as sums over products of nonstandard, but tractable, two-electron integrals.
For example, the RI scheme evaluates three-electron integrals over products of two-body operators as
pqs g12h23p q s = pq g
12p U Us h
23q s
U
all
, (10)
where the generic operators gmn
and hmn for electrons m and n are either rmn , rmn1, or Tmn . The one-particle
functions U comprise the RI basis, which is best chosen as an expansive auxiliary basis (ABS) distinct from the
smaller atomic orbital set used to represent the molecular orbitals.32,33
The power of explicitly-correlated electronic structure methods in chemical applications is illustrated in one
of our papers24 on the silicon dicarbide saga.34 The SiC2 molecule has a history of conundrums. In 1926 uncataloged
bands near 5000 Å were discovered in the spectra of several carbon-rich stars by Merrill and Sanford at Mt. Wilson
Observatory.35,36 Notwithstanding subsequent detailed investigations, the carrier of the electronic transitions was not
identified until Kleman37 reproduced the stellar bands in the laboratory some 30 years later by heating silicon in the
graphite tube of a King furnace to optimal temperatures of 2300-2400° C. On the basis of the complexity of the
band system, the experimental conditions, and a partial vibrational analysis, the blue-green emission spectrum was
tentatively ascribed to an Si-C-C species, assumed to be linear in analogy with the C3 prototype. The strongest band
5
at 4977 Å was assigned to the (000)-(000) transition. In 1984, the Smalley group38 achieved full rotational resolution
of the 4977 Å band system of SiC2 using resonant two-photon ionization. Prompted by suggestive ab initio
computations,39 the Merrill-Sanford bands were reassigned to a 1B2 1A1 transition of a T-shaped SiC2 ring. A
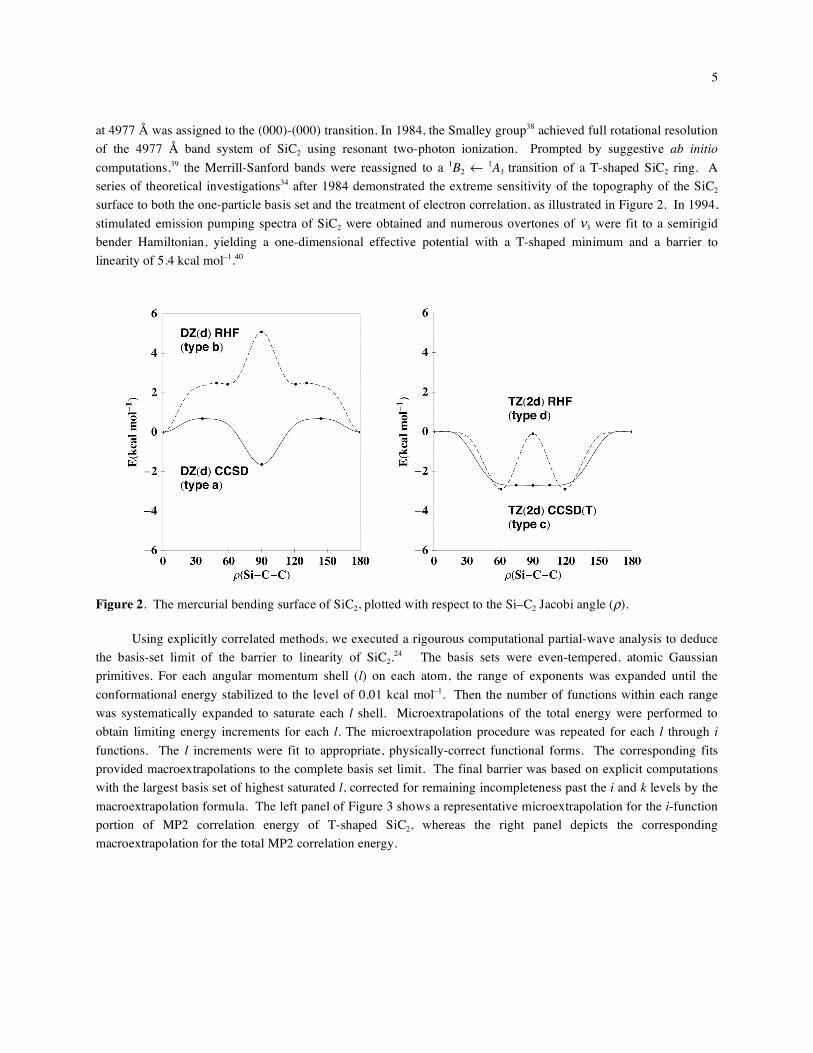
series of theoretical investigations34 after 1984 demonstrated the extreme sensitivity of the topography of the SiC2
surface to both the one-particle basis set and the treatment of electron correlation, as illustrated in Figure 2. In 1994,
stimulated emission pumping spectra of SiC2 were obtained and numerous overtones of 3 were fit to a semirigid
bender Hamiltonian, yielding a one-dimensional effective potential with a T-shaped minimum and a barrier to
linearity of 5.4 kcal mol–1.40
Figure 2. The mercurial bending surface of SiC2, plotted with respect to the Si–C2 Jacobi angle ( ).
Using explicitly correlated methods, we executed a rigourous computational partial-wave analysis to deduce
the basis-set limit of the barrier to linearity of SiC2.24 The basis sets were even-tempered, atomic Gaussian
primitives. For each angular momentum shell (l) on each atom, the range of exponents was expanded until the
conformational energy stabilized to the level of 0.01 kcal mol–1. Then the number of functions within each range
was systematically expanded to saturate each l shell. Microextrapolations of the total energy were performed to
obtain limiting energy increments for each l. The microextrapolation procedure was repeated for each l through i
functions. The l increments were fit to appropriate, physically-correct functional forms. The corresponding fits
provided macroextrapolations to the complete basis set limit. The final barrier was based on explicit computations
with the largest basis set of highest saturated l, corrected for remaining incompleteness past the i and k levels by the
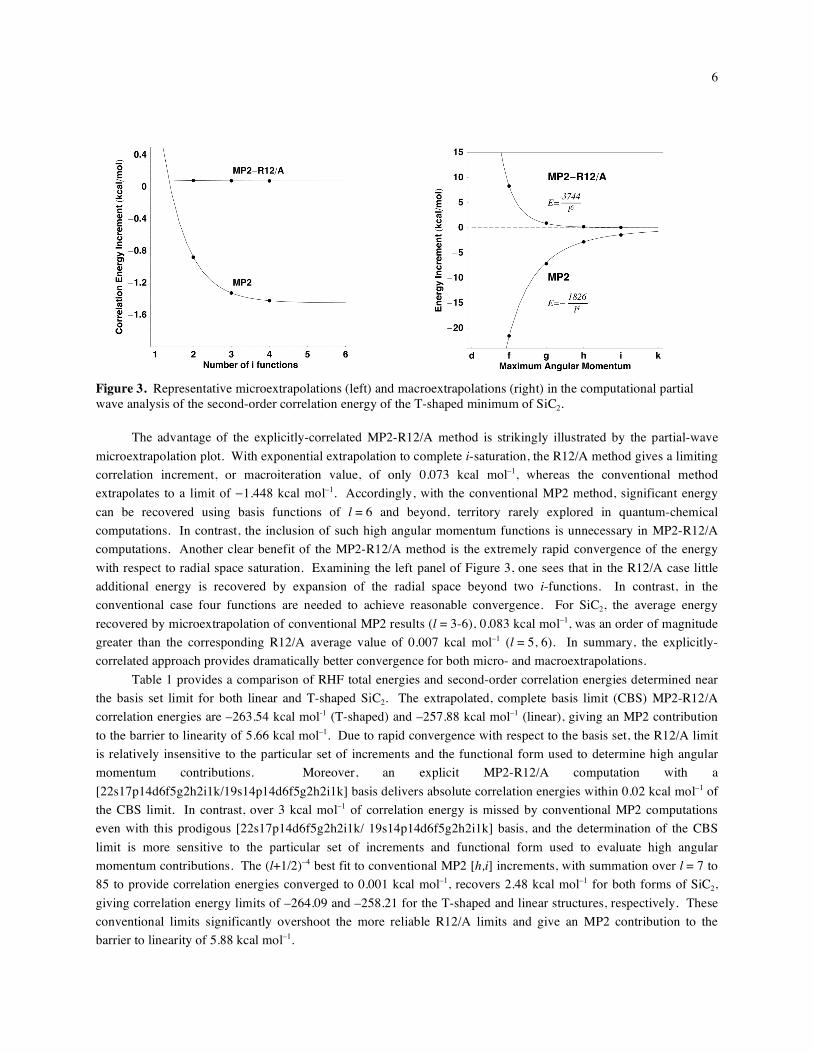
macroextrapolation formula. The left panel of Figure 3 shows a representative microextrapolation for the i-function
portion of MP2 correlation energy of T-shaped SiC2, whereas the right panel depicts the corresponding
macroextrapolation for the total MP2 correlation energy.
6
Figure 3. Representative microextrapolations (left) and macroextrapolations (right) in the computational partial wave analysis of the second-order correlation energy of the T-shaped minimum of SiC2.
The advantage of the explicitly-correlated MP2-R12/A method is strikingly illustrated by the partial-wave
microextrapolation plot. With exponential extrapolation to complete i-saturation, the R12/A method gives a limiting
correlation increment, or macroiteration value, of only 0.073 kcal mol–1, whereas the conventional method
extrapolates to a limit of 1.448 kcal mol–1. Accordingly, with the conventional MP2 method, significant energy
can be recovered using basis functions of l = 6 and beyond, territory rarely explored in quantum-chemical
computations. In contrast, the inclusion of such high angular momentum functions is unnecessary in MP2-R12/A
computations. Another clear benefit of the MP2-R12/A method is the extremely rapid convergence of the energy
with respect to radial space saturation. Examining the left panel of Figure 3, one sees that in the R12/A case little
additional energy is recovered by expansion of the radial space beyond two i-functions. In contrast, in the
conventional case four functions are needed to achieve reasonable convergence. For SiC2, the average energy
recovered by microextrapolation of conventional MP2 results (l = 3-6), 0.083 kcal mol–1, was an order of magnitude
greater than the corresponding R12/A average value of 0.007 kcal mol–1 (l = 5, 6). In summary, the explicitly-
correlated approach provides dramatically better convergence for both micro- and macroextrapolations.
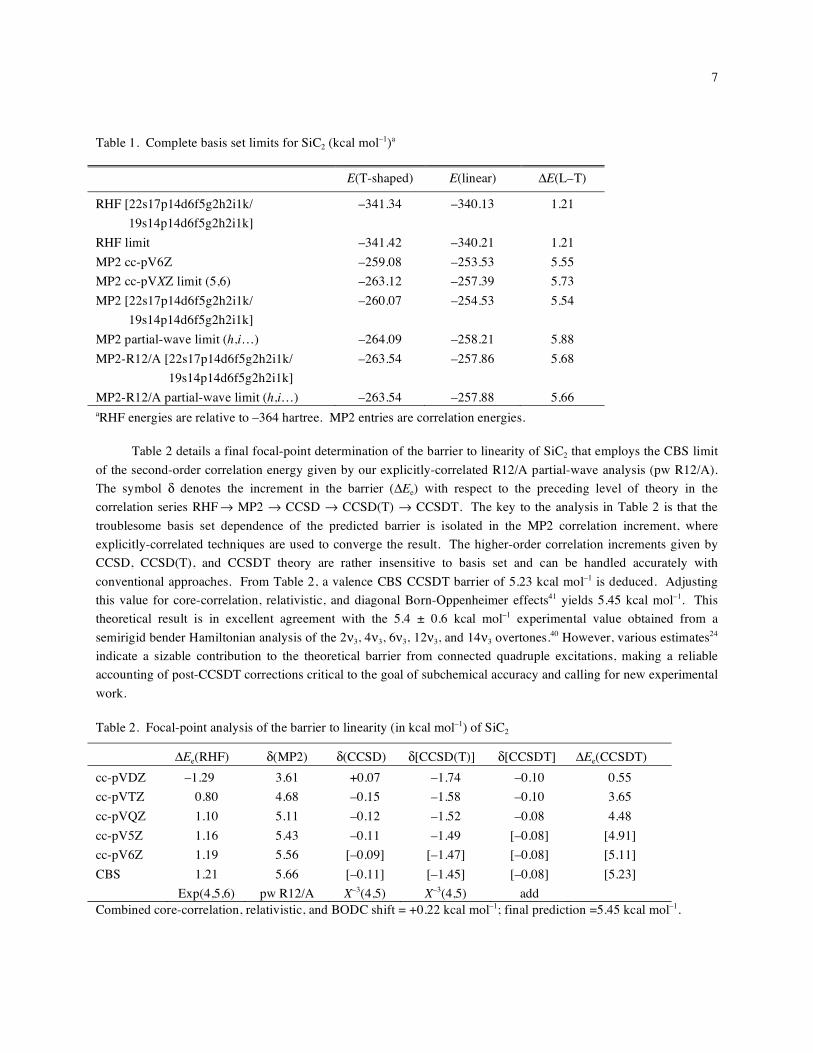
Table 1 provides a comparison of RHF total energies and second-order correlation energies determined near
the basis set limit for both linear and T-shaped SiC2. The extrapolated, complete basis limit (CBS) MP2-R12/A
correlation energies are –263.54 kcal mol-1 (T-shaped) and –257.88 kcal mol–1 (linear), giving an MP2 contribution
to the barrier to linearity of 5.66 kcal mol–1. Due to rapid convergence with respect to the basis set, the R12/A limit
is relatively insensitive to the particular set of increments and the functional form used to determine high angular
momentum contributions. Moreover, an explicit MP2-R12/A computation with a
[22s17p14d6f5g2h2i1k/19s14p14d6f5g2h2i1k] basis delivers absolute correlation energies within 0.02 kcal mol–1 of
the CBS limit. In contrast, over 3 kcal mol–1 of correlation energy is missed by conventional MP2 computations
even with this prodigous [22s17p14d6f5g2h2i1k/ 19s14p14d6f5g2h2i1k] basis, and the determination of the CBS
limit is more sensitive to the particular set of increments and functional form used to evaluate high angular
momentum contributions. The (l+1/2)–4 best fit to conventional MP2 [h,i] increments, with summation over l = 7 to
85 to provide correlation energies converged to 0.001 kcal mol–1, recovers 2.48 kcal mol–1 for both forms of SiC2,
giving correlation energy limits of –264.09 and –258.21 for the T-shaped and linear structures, respectively. These
conventional limits significantly overshoot the more reliable R12/A limits and give an MP2 contribution to the
barrier to linearity of 5.88 kcal mol–1.
7
Table 1. Complete basis set limits for SiC2 (kcal mol–1)a
E(T-shaped) E(linear) E(L–T)
RHF [22s17p14d6f5g2h2i1k/
19s14p14d6f5g2h2i1k] –341.34 –340.13 1.21
RHF limit –341.42 –340.21 1.21
MP2 cc-pV6Z –259.08 –253.53 5.55
MP2 cc-pVXZ limit (5,6) –263.12 –257.39 5.73
MP2 [22s17p14d6f5g2h2i1k/
19s14p14d6f5g2h2i1k] –260.07 –254.53 5.54
MP2 partial-wave limit (h,i…) –264.09 –258.21 5.88
MP2-R12/A [22s17p14d6f5g2h2i1k/
19s14p14d6f5g2h2i1k] –263.54 –257.86 5.68
MP2-R12/A partial-wave limit (h,i…) –263.54 –257.88 5.66
aRHF energies are relative to –364 hartree. MP2 entries are correlation energies. Table 2 details a final focal-point determination of the barrier to linearity of SiC2 that employs the CBS limit
of the second-order correlation energy given by our explicitly-correlated R12/A partial-wave analysis (pw R12/A).
The symbol denotes the increment in the barrier ( Ee) with respect to the preceding level of theory in the
correlation series RHF MP2 CCSD CCSD(T) CCSDT. The key to the analysis in Table 2 is that the
troublesome basis set dependence of the predicted barrier is isolated in the MP2 correlation increment, where
explicitly-correlated techniques are used to converge the result. The higher-order correlation increments given by
CCSD, CCSD(T), and CCSDT theory are rather insensitive to basis set and can be handled accurately with
conventional approaches. From Table 2, a valence CBS CCSDT barrier of 5.23 kcal mol–1 is deduced. Adjusting
this value for core-correlation, relativistic, and diagonal Born-Oppenheimer effects41 yields 5.45 kcal mol–1. This
theoretical result is in excellent agreement with the 5.4 ± 0.6 kcal mol–1 experimental value obtained from a
semirigid bender Hamiltonian analysis of the 2 3, 4 3, 6 3, 12 3, and 14 3 overtones.40 However, various estimates24
indicate a sizable contribution to the theoretical barrier from connected quadruple excitations, making a reliable
accounting of post-CCSDT corrections critical to the goal of subchemical accuracy and calling for new experimental
work.
Table 2. Focal-point analysis of the barrier to linearity (in kcal mol–1) of SiC2
Ee(RHF) (MP2) (CCSD) [CCSD(T)] [CCSDT] Ee(CCSDT)
cc-pVDZ –1.29 3.61 +0.07 –1.74 –0.10 0.55
cc-pVTZ 0.80 4.68 –0.15 –1.58 –0.10 3.65
cc-pVQZ 1.10 5.11 –0.12 –1.52 –0.08 4.48
cc-pV5Z 1.16 5.43 –0.11 –1.49 [–0.08] [4.91]
cc-pV6Z 1.19 5.56 [–0.09] [–1.47] [–0.08] [5.11]
CBS 1.21 5.66 [–0.11] [–1.45] [–0.08] [5.23]
Exp(4,5,6) pw R12/A X–3(4,5) X–3(4,5) add Combined core-correlation, relativistic, and BODC shift = +0.22 kcal mol–1; final prediction =5.45 kcal mol–1.

8
References 1 T. Kato, “On the eigenfunctions of many-particle systems in quantum mechanics,” Commun. Pure Appl. Math.
10, 151-177 (1957). 2 R. T. Pack and W. B. Brown, “Cusp conditions for molecular wavefunctions,” J. Chem. Phys. 45, 556-559
(1966). 3 W. Kutzelnigg and J. D. Morgan III, “Rates of convergence of the partial-wave expansions of atomic
correlation energies,” J. Chem. Phys. 96, 4484-4508 (1992). 4 B. Ruscic, D. Feller, D. A. Dixon, K. A. Peterson, L. B. Harding, R. L. Asher, and A. F. Wagner, “Evidence
for a lower enthalpy of formation of hydroxyl radical and a lower gas-phase bond dissociation energy of water,” J. Phys. Chem. A 105, 1-4 (2001).
5 B. Ruscic, A. F. Wagner, L. B. Harding, R. L. Asher, D. Feller, D. A. Dixon, K. A. Peterson, Y. Song, X. Qian, C.-Y. Ng, J. Liu, W. Chen, and D. W. Schwenke, “On the enthalpy of formation of hydroxyl radical and gas-phase dissociation energies of water and hydroxyl,” J. Phys. Chem. A 106, 2727-2747 (2002).
6 B. Ruscic, J. E. Boggs, A. Burcat, A. G. Császár, J. Demaison, R. Janoschek, J. M. L. Martin, M. L. Morton, M. J. Rossi, J. F. Stanton, P. G. Szalay, P. R. Westmoreland, F. Zabel, and T. Bérces, “IUPAC critical evaluation of thermochemical properties of selected radicals. Part I,” J. Phys. Chem. Ref. Data 34, 573-656 (2005).
7 B. Ruscic, R. E. Pinzon, M. L. Morton, G. von Laszavski, S. J. Bittner, S. G. Nijsure, K. A. Amin, M. Minkoff, and A. F. Wagner, “Introduction to Active Thermochemical Tables: Several key enthalpies of formation revisited,” J. Phys. Chem. A 108, 9979-9997 (2004).
8 A. Tajti, P. G. Szalay, A. G. Császár, M. Kállay, J. Gauss, E. F. Valeev, B. A. Flowers, J. Vázquez, and J. F. Stanton, “HEAT: High accuracy extrapolated ab initio thermochemistry,” J. Chem. Phys. 121, 11599-11613 (2004).
9 J. A. Miller and S. J. Klippenstein, “The reaction between ethyl and molecular oxygen. II: Further analysis,” Int. J. Chem. Kinet. 33, 654-668 (2001).
10 W. Klopper, in The Encyclopedia of Computational Chemistry, edited by P. v. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A. Kollmann, H. F. Schaefer, and P. R. Schreiner (Wiley, Chichester, 1998), pp. 2351-2375.
11 A. G. Császár, W. D. Allen, Y. Yamaguchi, and H. F. Schaefer III, in Computational Molecular Spectroscopy, edited by P. R. Bunker and P. Jensen (Wiley, New York, 2000), pp. 15-68.
12 E. F. Valeev, W. D. Allen, and H. F. Schaefer, unpublished results. 13 E. A. Hylleraas, Z. Phys. 54, 347-366 (1929). 14 V. I. Korobov, Phys. Rev. A 61, 064503 (2000). 15 J. S. Sims and S. A. Hagstrom, “High precision Hy-CI variational calculations for the ground state of neutral
helium and heliumlike ions,” Int. J. Quantum Chem. 90, 1600-1609 (2002). 16 W. Kutzelnigg, “r12-Dependent terms in the wave function as closed sums of partial wave amplitudes for large
l,” Theor. Chim. Acta 68, 445-469 (1985). 17 W. Kutzelnigg and W. Klopper, “Wave functions with terms linear in the interelectronic coordinates to take
care of the correlation cusp. I. General theory,” J. Chem. Phys. 94, 1985-2001 (1991). 18 W. Klopper and W. Kutzelnigg, “Wave functions with terms linear in the interelectronic coordinates to take
care of the correlation cusp. III. Second-order Møller-Plesset (MP2-R12) calculations on molecules of first row atoms,” J. Chem. Phys. 94, 2020-2030 (1991).
19 W. Klopper, R. Röhse, and W. Kutzelnigg, “CID and CEPA calculations with linear-R12 terms,” Chem. Phys. Lett. 178, 455-461 (1991).
20 J. Noga and W. Kutzelnigg, “Coupled-cluster theory that takes care of the correlation cusp by inclusion of linear terms in the interelectronic coordinates,” J. Chem. Phys. 101, 7738-7762 (1994).
21 J. Noga, W. Klopper, and W. Kutzelnigg, in Modern Ideas in Coupled-Cluster Methods, edited by R. J. Bartlett (World Scientific, Singapore, 1997), pp. 1-48.
22 E. F. Valeev and H. F. Schaefer, “Evaluation of two-electron integrals for explicit r12 theories,” J. Chem. Phys. 113, 3990-3995 (2000).
23 E. F. Valeev, W. D. Allen, H. F. Schaefer, and A. G. Császár, “The second-order Møller-Plesset limit for the barrier to linearity of water,” J. Chem. Phys. 114, 2875-2878 (2001).

9
24 J. P. Kenny, W. D. Allen, and H. F. Schaefer, “Complete basis set limit studies of conventional and R12 correlation methods: the silicon dicarbide (SiC2) barrier to linearity,” J. Chem. Phys. 118, 7353-7365 (2003).
25 E. F. Valeev, W. D. Allen, R. Hernandez, C. D. Sherrill, and H. F. Schaefer, “On the accuracy limits of orbital expansion methods: explicit effects of k-functions on atomic and molecular energies,” J. Chem. Phys. 118, 8594-8610 (2003).
26 M. S. Schuurman, S. R. Muir, W. D. Allen, and H. F. Schaefer, “Toward subchemical accuracy in computational thermochemistry: Focal point analysis of the heat of formation of NCO and [H,N,C,O] isomers,” J. Chem. Phys. 120, 11586-11599 (2004).
27 M. S. Schuurman, W. D. Allen, and H. F. Schaefer, “The Ab Initio Limit Quartic Force Field of BH3,” J. Comput. Chem. 26, 1106-1112 (2005).
28 M. S. Schuurman, W. D. Allen, P. v. R. Schleyer, and H. F. Schaefer, “The highly anharmonic BH5 potential energy surface characterized in the ab initio limit,” J. Chem. Phys. 122, 104302 (2005).
29 R. Bukowski, B. Jeziorski, and K. Szalewicz, “Gaussian geminals in explicitly correlated coupled cluster theory including singe and double excitations,” J. Chem. Phys. 110, 4165-4183 (1999).
30 W. Klopper, “Orbital-invariant formulation of the MP2-R12 method,” Chem. Phys. Lett. 186, 583-585 (1991). 31 W. Klopper, “Limiting values for Møller-Plesset second-order correlation energies of polyatomic systems: A
benckmark study on Ne, HF, H2O, N2, and He...He,” J. Chem. Phys. 102, 6168-6179 (1995). 32 W. Klopper and C. C. M. Samson, “Explicitly correlated second-order Møller-Plesset methods with auxiliary
basis sets,” J. Chem. Phys. 116, 6397-6410 (2002). 33 E. F. Valeev, “Improving the resolution of the identity in linear R12 ab initio theories,” Chem. Phys. Lett. 395,
190-195 (2004). 34 I. M. B. Nielsen, W. D. Allen, A. G. Császár, and H. F. Schaefer III, “Toward resolution of the silicon
dicarbide (SiC2) saga: Ab initio excursions in the web of polytopism,” J. Chem. Phys. 107, 1195-1211 (1997). 35 R. F. Sanford, “Two Bands in Spectra of Class N,” Publ. Astron. Soc. Pac. 38, 177-179 (1926). 36 P. W. Merrill, “Note on the Spectrum of UV Aurigae,” Publ. Astron. Soc. Pac. 38, 175-176 (1926). 37 B. Kleman, “Laboratory Excitation of the Blue-green Bands Observed in the Spectra of N-type Stars,”
Astrophys. J. 111, 262 (1956). 38 D. L. Michalopoulos, M. E. Geusic, P. R. R. Langridge-Smith, and R. E. Smalley, “Visible Spectroscopy of
Jet-cooled SiC2: Geometry and Electronic Structure,” J. Chem. Phys. 80, 3556 (1984). 39 R. S. Grev and H. F. Schaefer, “An Energetically Low-lying Silacyclopropyne Isomer of SiC2,” J. Chem. Phys.
80, 3552-3555 (1984). 40 S. C. Ross, T. J. Butenhoff, E. A. Rohlfing, and C. McMichael Rohlfing, “SiC2: A Molecular Pinwheel,” J.
Chem. Phys. 100, 4110-4126 (1994). 41 A. G. Császár, W. D. Allen, and H. F. Schaefer, “In pursuit of the ab initio limit for conformational energy
prototypes,” J. Chem. Phys. 108, 9751-9764 (1998).


















