
Drug Regulatory Overview Milan Smid, MD, PhD Technical Officer Quality Assurance and Safety:...
58
Drug Regulatory Drug Regulatory Overview Overview Milan Smid, MD, PhD Technical Officer Quality Assurance and Safety: Medicines Essential Medicines and Pharmaceutical Policies, World Health Organization [email protected]
-
Upload
reynold-griffin -
Category
Documents
-
view
221 -
download
1
Transcript of Drug Regulatory Overview Milan Smid, MD, PhD Technical Officer Quality Assurance and Safety:...

Drug Regulatory OverviewDrug Regulatory OverviewDrug Regulatory OverviewDrug Regulatory Overview
Milan Smid, MD, PhDTechnical OfficerQuality Assurance and Safety: MedicinesEssential Medicines and Pharmaceutical Policies, World Health [email protected]

2 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Principles of drug regulation
WHO regulatory support activities– Harmonization and worksharing– Assessment of regulatory functions– Trainings
Discussion

3 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Major health technologiesMajor health technologies
Medical devicesincluding
diagnostics
Medicines and
vaccines
Herbal and traditional medicines
Functional food
Nutraceuticals
Advanced therapies
Blood componentsTransplants

4 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Common features of health technologiesCommon features of health technologies
Used to prevent, diagnose or treat disease
Benefits from use must overweight the risks in order to improve health
To evaluate risk-benefit is frequently highly scientifically demanding
Not all health technologies bear comparable risks
Health technologies are subject of commerce and profit oriented behaviour

5 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Protection of public healthProtection of public health
To achieve acceptable level of risk related to use of health technologies and maximize their benefit, governments have to intervene
Governmental interventions interfere with rights of persons and companies and with business practices
Regulatory systems and scope of regulation differ worldwide
Health products are split into different categories
Regulation of medicines is currently one of most sophisticated and complicated regulatory functions globally

6 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Medicines regulation Medicines regulation is the totality of all
measures - legal, administrative and technical - which governments take to ensure the safety, efficacy and quality of medicines, as well as the relevance and accuracy of product information
Medicines are special goods for which also prices are frequently regulated by governments to achieve affordability for patients
Medicines are needed and different government incentives are used to develop and produce them

7 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Medicines regulationMedicines regulation
Models for regulation of medicines are determined by:
Public health needs
Organization of the state and state administration
Size of the pharmaceutical market
Presence and type of pharmaceutical industry
Availability of resources (human, scientific, financial)
Maturity of stakeholders
Participation in regulatory networks

8 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
substandard, counterfeit, harmful, useless medicines on the market
biased information about medicines circulates substandard practices in clinical testing,
manufacture and supply of medicines irrational prescription and consumption
Rationale for Government role
Consequences of weak drug regulatory capacity

9 | Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Consequences of over- or improper regulation
Rationale for Government role
lack of needed medicines or delayed access increased costs of medicines due to cost of
regulatory system lack of regulatory flexibility

10 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Regulation should
Regulatory basics
Keep patient in its centre Be evidence based Be risk oriented Bring added value Respect interests of stakeholders and real
possibilities Be transparent but respect confidentiality Be effective and flexible Be part of broader drug policy

11 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Parties involved in regulatory system Parties involved in regulatory system
Regulated subjects– Investigators and sponsors, members of ethics commitees– Manufacturers and importers– Wholesalers and distributors– Health professionals
Regulators– Regulatory authorities– Governments and political representations
Interested parties– General public and patients groups– Academia– Media

12 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Development of drug regulation
Quality
Safety
Efficacy
Information
Environment
Risk management and risk minimization

13 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Essential regulatory activities
R&D – data quality and ethics (GLP, GCP, bioethics)
Manufacture, import and supply – quality (GMP, GDP)
Market entry and existence on the market – Q, S, E, I, Env authorization/registration, maintenance, renewals
Special access schemes
Dispensation – safe and indicated use (GPhP)
Use – pharmacovigilance (GPhVP)
Promotion – unbiased, valid and understandable information (Conduct codes)
Environment – premarket review and environment monitoring
Regulatory and scientific advice

14 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Regulatory toolkitRegulatory toolkit
Legislation Standards (GXPs, Pharmacopoeias, Q,S,E guidelines) Data assessment
– Submitted data– Collected data
Inspections Licensing (authorizations / registrations, products and
operations) Certification Laboratory control Provision of information Regulatory intelligence and co-operation

15 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Incentives to develop, manufacture and supply needy medicines
Incentives to develop, manufacture and supply needy medicines
Patents and supplementary patent protection
Data exclusivities
Market exclusivities
Limited data requirements
Regulatory advice
Regulatory fast tracks
Waivers of regulatory fees
Public-private partnership, stimulating price, guaranteed demand
16 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
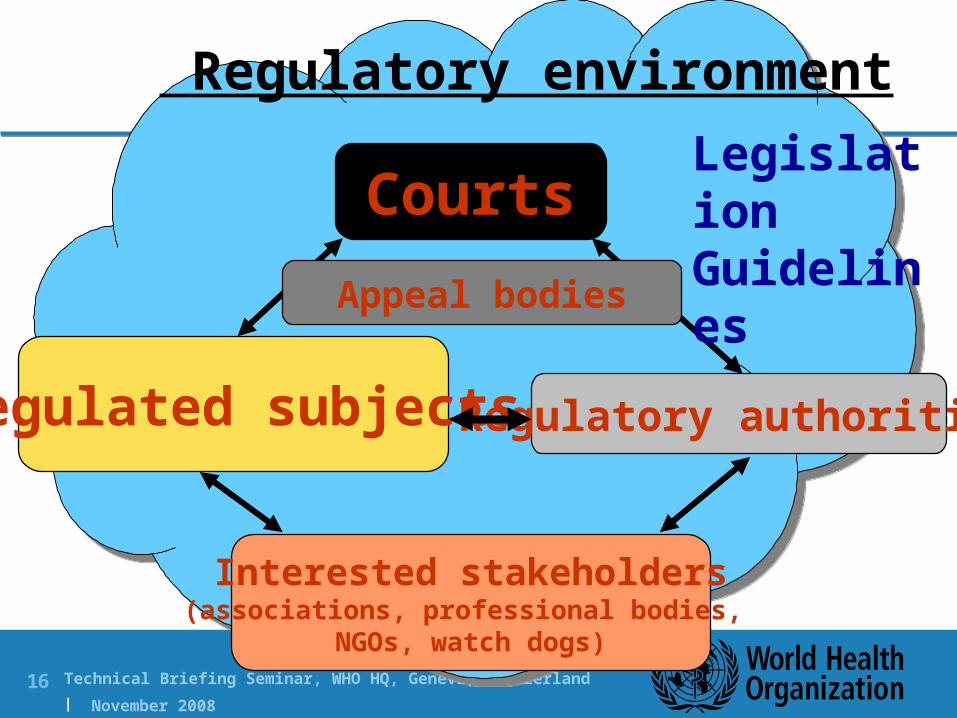
Regulatory environment
Courts
Regulated subjects Regulatory authorities
Interested stakeholders(associations, professional bodies,
NGOs, watch dogs)
LegislationGuidelines
Appeal bodies

17 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Responsibilities of parties involved in regulatory system
Responsibilities of parties involved in regulatory system
LegislatedInvestigators and sponsors Manufacturers and importersWholesalers and distributors Health professionals Regulatory authorities
Non-legislated Governments and political representationsPatients Media

18 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
DrugRegulatory
Cycle Emergency and crisis
management
Variations
Line extensions
Postregistrationcommitments
Registration
Renewals
PSUR
PSUR
PSUR
PSUR
PSURPSUR

19 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Principles applied in regulatory approvals Principles applied in regulatory approvals
Benefits prevail the risks at a time of regulatory approval and nothing indicates that benefits will not prevail also during use of product in normal medical practice
– Available data about quality (Dossier)– Available data about efficacy and safety or interchangeability (Dossier)– Available data are credible and were eticaly obtained
• Good practices (GLP, GCP, GPhVP, …)– Existing reassurance about production in stable quality and quality
assurance mechanisms• GMP
– Way of use of medicine characterized for physicians and patients • Data sheets, SPCs, PILs, package labeling
– Lack of knowledge is properly managed• Pharmacovigilance, risk management programmes,
commitments

20 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Regulation of innovatory products and generics
For innovator products proof of QUALITY, SAFETY and EFFICACY is needed. Newly also plan of prospective risk-management.
For multisource products QUALITY, safety and efficacy data is referred to the originator, providing only evidence of INTERCHANGEABILITY (bioequivalence, clinical testing, in certain cases dissolution data).

21 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Proof of interchangeabilityProof of interchangeability
Administrative and summarizing data, including GMPAdministrative and summarizing data, including GMP
Pharmaceutical dataPharmaceutical data
Preclinical dataPreclinical data
Innovative medicineInnovative medicineExperimental data/ LiteratureExperimental data/ Literature
Generic medicineGeneric medicineMultisource interchangeableMultisource interchangeable
Clinical dataClinical data
Data required for regulatory approval
Pharmaceutical dataPharmaceutical data

22 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Scientific and regulatory complexity concerning medicinal products is growing
Scientific and regulatory complexity concerning medicinal products is growing
New technologies New therapeutic approaches Evidence based medicine Globalisation of commerce Telematic instruments Information and transparency
requirements Risk aversion in society New tasks Speed of change
Regulatory resources rarely reflect expectations of society and regulators themselves
Paradigms of drug regulation are evolving

23 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Capacity to make regulatory decisionsCapacity to make regulatory decisions
193 WHO Member States:
20% 50%
30% Developed
Varying
Limited

24 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Regulatory delays and backlogs
Lack of regulatory tools and guidelines
Lack of expertise
Lack of management skills
Regulatory pendulum
ChallengesChallenges

25 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Strategies used to cope with increasing demands?
Strategies used to cope with increasing demands?
Concentration on priority issues most relevant for public health
Co-operation with partners in order to increase regulatory capacity by elimination of duplicated activities
– Facilitated by comparable standards and administrative requirements
Increased effectivity of internal operations– Quality systems, international benchmarking

26 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Sharing of expertise vs. recognition of decisionSharing of expertise vs. recognition of decision
Acceptance of expertise is not equal to acceptance of decision
Acceptance of decision– is formal legal act, frequently requiring international treaties– may modify liabilities of involved parties and requires legal
specification of acceptance and non-acceptance
Acceptance of expertise– is sovereign and complex regulatory decision of NRA based on
scientific arguments and confidence – may be applied case to case– is followed by formal independent decision according to national
legislation and mandate of NRA

27 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Stimulating / initiating collaboration between regulators from various developing countries on various regulatory activities;
Employing internationally accepted guidelines adapted to suit local needs/circumstances;
Facilitating joint assessments between regulators through sub-regional approaches.
WHO approaches for considerationWHO approaches for consideration

28 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
13th ICDRA16-19 September 2008, Bern
13th ICDRA16-19 September 2008, Bern
Concentration on priority issues most relevant for public health
– Risk-based approaches: regulation of clinical trials, GMP, product licensing, safety monitoring, market oversight
Increased effectivity and efficiency of internal operations
– assessment of practices and performance by common toolkits

29 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
13th ICDRA16-19 September 2008, Bern
13th ICDRA16-19 September 2008, Bern
Co-operation with partners in order to increase regulatory capacity by elimination of duplicated work investments and optimising use of experts
– Harmonization: standards: terminology, guidelines, good practices, pharmacopoeias, format and data content of applications, safety monitoring practices,
– Collaboration and networking: inspections, market surveillance, counterfeits,
– Information sharing and common data sets, information availability: trial registries, inspection outcomes and assessment reports, licensing outcomes, pharmacovigilance
– Building mutual trust– New phenomenon: Major regulatory authorities (FDA, EMEA)
recognize need of collaboration with NRA in exporting countries concerning oversight of manufacturers and information exchange

30 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Different initiatives to increase regulatory capacity and assemble needed scientific / inspection resources
Different initiatives to increase regulatory capacity and assemble needed scientific / inspection resources
WHO – standards, international expert teams, pharmacovigilance UMC ICH – common standards for developed markets, involvement of regulators
as well as industry experts EMEA - co-ordination of expertise available to EU/EEA Member StatesAustralia and New Zealand – merger of NRAs (currently frozen)Monaco, Liechtenstein – recognition of regulatory decisions of France resp.
Switzerland Canada, Switzerland, Japan, EU, Australia etc. – MRA on mutual acceptance
of GMP standards FDA, EMEA, WHO, TGA … etc. – MoU on sharing of informationPIC/S – share of standards and expertise on GMP, building confidenceRegional and subregional initiatives (ASEAN, GCC, EAC) – different extent
of co-operationNational initiatives – involvement of external experts to support NRAs,
recognition of expertise or decisions, co-operation with industry and learned societies

31 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Not to share expertise - If trustful expertise is available, not to consider it may be unjustified waste of resources needed elsewhere
Accepting the expertise of other regulators is complex regulatory and scientific decision, depending on
– legislation, mandate – capacities and competence available for given issue – scientific arguments related to benefit/risk for public health– priorities for protection of public health
Understanding to limitations of transfer of expertise and its proper management are needed when considering expertise of others
Targeted, risk-based approach may be appropriate in some situations

32 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Expertise in support of developing countries
Expertise in support of developing countries
WHO PQP - HIV/AIDS, TB, malaria, RH, paediatric therapy
US PEPFAR - HIV/AIDS
EU Article 58 – assessment of products for non-EU teritories
Canada's Access to Medicines Regime – assessment of products according to WHO Model List of Essential Medicines
Other - orphans, paediatric therapy

33 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Faster access to medicines through sharing of regulatory information
Faster access to medicines through sharing of regulatory information
WHO is working with regulators to find out how best to build confidence in regulatory decisions taken by other regulators, including
-- how to facilitate exchange of consolidated information about assessments.

34 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
WHO registration packageWHO registration package
Purpose:
For less-resourced agencies – in decision making process, to benefit from technical information developed by well-resourced ("mature") regulatory authorities;
To facilitate the "transfer of expertise";
To create a format for exchange of regulatory information.

35 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Different initiatives to increase regulatory capacity
Different initiatives to increase regulatory capacity
WHO proposed a framework based on what is publicly available:
– Summary Basis for Decision (Health Canada)– European Public Assessment Report (EMEA and EC)– Common Technical Document (US FDA)– WHO Public Assessment Report (WHOPAR) and
Public Inspection Report (WHOPIR)

36 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
WHO Pilot ProjectWHO Pilot Project
Supported by European Commission;
With participation of 5 (7) countries:
Ghana, Kenya, Nigeria, South Africa, Uganda, United Republic of Tanzania and Zimbabwe

37 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
WHO Pilot ProjectWHO Pilot Project
Purpose:
To field-test a model technical registration package;
To finalize the content of the package and the guidance for its effective implementation in countries;
To ensure that the package is developed and implemented through a wide consultation with existing and potential stakeholders;

38 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Field testing exercise
In order to have a maximum benefit from the field-test exercise, it was decided to provide all participating DRAs with the same drug application;
To ask them to develop their own registration package, based on the provided draft template;
One of the recently developed pharmaceutical products has served as a model application for the field-testing of the WHO model registration package.
WHO Pilot ProjectWHO Pilot Project

39 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
WHO Pilot ProjectWHO Pilot Project
The same application was submitted to EMEA experts for their opinion;
In the end of the project a follow up WHO consultative meeting was conducted;
The aim was to consider:
─ the results and lessons learnt from the field-testing exercise,
─ to finalize the contents of the model technical regulatory package,
─ to provide guidance for adoption of the package by national MRAs.

40 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Conclusions (1)Conclusions (1)
The main objective for DRAs should be to get the relevant information for the country, while avoiding collection of large amounts of the country-specific data that does not add value;
Sharing of information can help with best use of available resources, reduce workload and improve overall regulatory performance;
Available tools can be packaged for ease of reference and information exchange;

41 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Conclusions (2)Conclusions (2)
Procedures can be streamlined and processes can be improved (e.g., Fast Track or Priority Review for those products that address unmet medical needs);
Expert knowledge can be pooled and resources directed to functions that can improve public health and facilitate access to essential medicines.

42 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Regulatory support core functionsRegulatory support core functions
Developing evidence on the situation of Medicines Regulatory Systems worldwide
Providing Country support for strengthening medicines regulation
Providing Regional support for strengthening medicines regulation
Facilitate communication and promote harmonization among medicines regulatory authorities
Develop and continuously improve internal supporting tools, mechanism and capacities

43 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Developing evidence Developing evidence
To develop and maintain a comprehensive database on DRAs
To assess of medicine regulatory systems– Perform assessment to identify needs– Provide evidence for various supporting activities (Financial,
Training, Consultancy,Equipment) – Develop institutional plan
To promote self-assessment as a tool for analysis and continuous improvement
– Two training events performed for DRAs
Tool for harmonization purposes– 5 Pacific Island Countries / WPRO– 3 EAC Countries / AFRO

44 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Developing evidenceDeveloping evidence
44 Assessments performed on 40 Regulatory systems (with the involvement of HQ)
– AFRO - 21 COUNTRIES / 24 ASSESSMENTS– EURO - 2 COUNTRIES / 2 ASSESSMENTS– EMRO - 4 COUNTRIES / 5 ASSESSMENTS– SEARO - 4 COUNTRIES / 4 ASSESSMENTS– WPRO - 7 COUNTRIES / 7 ASSESSMENTS– PAHO - 2 COUNTRIES / 2 ASSESSMENTS

45 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Developing evidenceDeveloping evidence
2008
2007
2006
2004
2003
No
2003

46 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Country supportCountry support
Develop and organize training opportunities– Strengthen information management capacity - SIAMED– Strengthen inspection capacity– Strengthen QC laboratory capacity– Strengthen marketing authorization capacities
Promoting good regulatory practices by providing guidelines, tools and technical assistance
In close collaboration with capacity building team within the PQ Program
In cooperation with IVB Vaccines on clinical trials

47 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Regional support Regional support
Provision of technical assistance to harmonization initiative and supporting participation of regulators
– SADC, EAC, PIC, CARICOM,…
Financial support for technical secretariat (EAC, SADC and UEMOA)
WHO/EAC joined assessment of DRAs
Promoting and facilitating networking– Network of DRAS (EAC, SADC)

48 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Introduction to the grading modelIntroduction to the grading model
Preliminary work made by Eshetu Wondemagegnehu– Possible approaches to developing drug regulation in:Effective Drug Regulation: what can countries do?
Based on Capability Maturity Model (CMM)– first described by W. Humphrey in Managing the Software Process
Maturity model for quality management system– introduced by P. Crosby in his book 'Quality is Free'.
Generic approach of maturity of organization as a learning curve applied to medicines regulatory
processes
Not to give a mark

49 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
For what purposes can it be used?For what purposes can it be used?
At country level– To build a strategy– To visualize the stage of development/maturity– To visualize and demonstrate progress– To assist process of harmonization– To identify areas of priority support
At sub-regional level– To organize sharing and exchange of expertise– To develop process of harmonization, cooperation or collaboration
At international level– To map regulatory systems– To build strategy/approach– To document results and planned activities
50 |
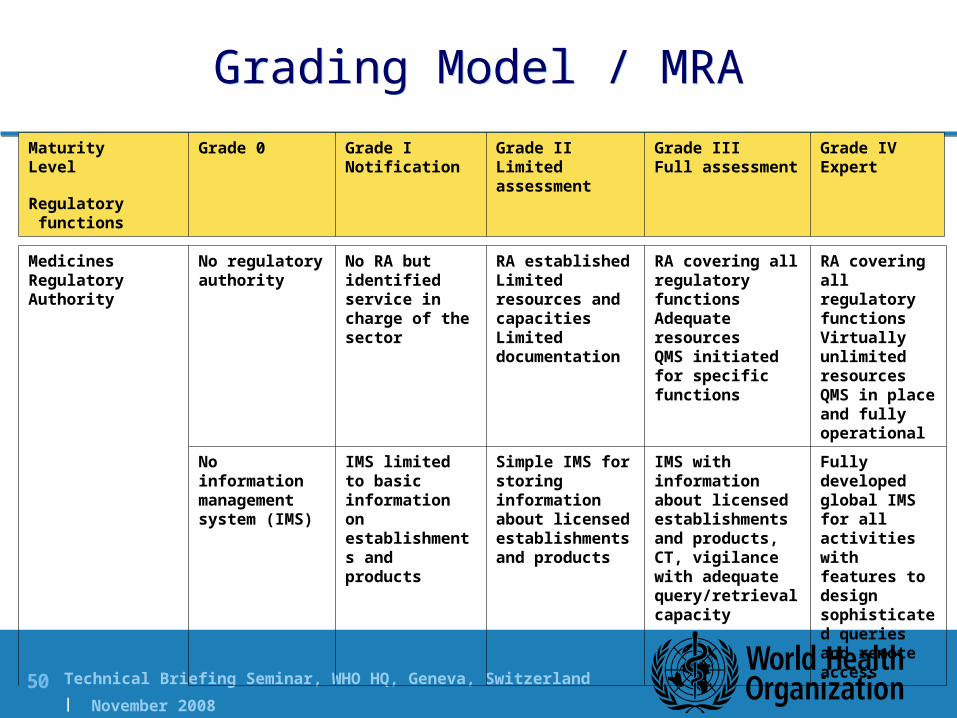
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Grading Model / MRAGrading Model / MRA
Medicines Regulatory Authority
No regulatory authority
No RA but identified service in charge of the sector
RA establishedLimited resources and capacitiesLimited documentation
RA covering all regulatory functionsAdequate resourcesQMS initiated for specific functions
RA covering all regulatory functionsVirtually unlimited resourcesQMS in place and fully operational
No information management system (IMS)
IMS limited to basic information on establishments and products
Simple IMS for storing information about licensed establishments and products
IMS with information about licensed establishments and products, CT, vigilance with adequate query/retrieval capacity
Fully developed global IMS for all activities with features to design sophisticated queries and remote access
MaturityLevel
Regulatory functions
Grade 0 Grade INotification
Grade IILimited assessment
Grade IIIFull assessment
Grade IVExpert

51 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Grading Model / Marketing authorizationGrading Model / Marketing authorization
Marketing authorisation
No MA system in place
Notification for all categories of products
No assessment performed
MA based on:- Information provided by DRA of other countries- WHO-type certificate- Evidence of MA in reference countries
List of products on the market
MA of generic products based on full assessment of applications
MA of generic products based on full assessment of applications
MA of generic products based on full assessment of applications
MA of new active substances (NAS) based on:
-own assessment of quality part and
- Information provided by DRA of other countries
Registration of NAS based on full assessment of application
Registration of new products based on full assessment of application
MA of biological & complex products (e.g. biotech) based on information provided by other DRA
MA of biological & complex products (e.g. biotech) based on full assessment of applications
Maturity level
Regulatory functions
Grade 0 Grade INotification
Grade IILimited assessment
Grade IIIFull assessment
Grade IVExpert

52 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Fictitious example of evaluation outcomesFictitious example of evaluation outcomes
Regulatory functions
Country A Country B Country C Country D Country E
Licensing Level 3 Level 3 Level 3 Level 4 Level 3
Registration Level 2 Level 3 Level 3 Level 4 Level 3
Inspections Level 2 Level 3 Level 3 Level 4 Level 3
QC Laboratory Level 2 Level 3 Level 3 Level 4 Level 3
Vigilance Level 2 Level 2 Level 1 Level 3 Level 2
Clinical Trials Level 3 Level 3 Level 1 Level 4 Level 3
Drug Promotion Level 3 Level 3 Level 3 Level 4 Level 3
Harmonisation : easier to achieve for Licensing & Control of PromotionPriority: To assist country A through collaboration with other countriesFollow-up: identify reasons behind country C’s low result on clinical trials and vigilance

53 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Contribution of PQ to capacity buildingContribution of PQ to capacity building
Organization of trainings– general and problem specific (HIV/AIDS, TB and antimalarial
products, pediatric dosage forms, BE, BE/BCS)
– Trainings of NRA staff and manufacturers frequently combined
Involvement of assessors from NRAs into PQ assessment
Involvement of inspectors from NRAs into PQ inspections
3 months rotations of experts from NRAs in WHO HQ – PQT
Technical Briefing Seminars
54 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
0
2
4
6
8
10
12
14
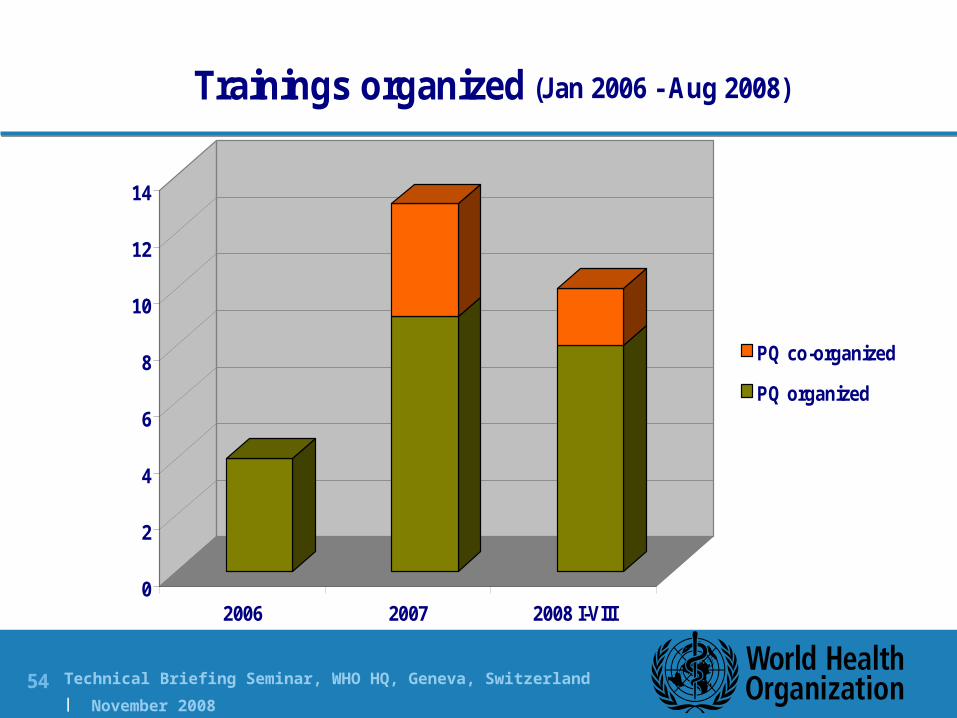
2006 2007 2008 I-VIII
Trainings organized (Jan 2006 - Aug 2008)
PQ co-organized
PQ organized

55 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Topics of workshops in 2006 – I-VIII 2008 Topics of workshops in 2006 – I-VIII 2008
3
9
33
3
2
3 1
Prequalification advocacyPrequalification requirementsGood manufacturing practiceQuality controlBioequivalence/BCS and GCPAssessment of medicinesPharmaceutical developmentGeneral

56 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Focus on specific medicinesFocus on specific medicines
1
2
13
3
HIV/AIDS medicines Antimalarials
TB medicines RH products
Pediatric formulations

57 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
0
100
200
300
400
500
600
2007 2008 I-VIII
Participants in trainings
Others
QCL staff
Regulators
Manufacturers

58 |
Technical Briefing Seminar, WHO HQ, Geneva, Switzerland | November 2008
Thank you for the attention


















