
Differentiation of vascular smooth muscle cells from local ... of vascular smooth muscle cells from...
6
Differentiation of vascular smooth muscle cells from local precursors during embryonic and adult arteriogenesis requires Notch signaling Linda Chang a,b , Michela Noseda c , Michelle Higginson a , Michelle Ly a , Alexandre Patenaude a,c , Megan Fuller a,c , Alastair H. Kyle d , Andrew I. Minchinton d , Mira C. Puri e,f , Daniel J. Dumont e,f , and Aly Karsan a,b,c,g,1 a Genome Sciences Centre, d Integrative Oncology Department, and g Cancer Genetics Laboratory, British Columbia Cancer Agency, Vancouver, BC, Canada V5Z 1L3; b Experimental Medicine Program and c Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada V6T 1Z4; e Sunnybrook Research Institute, Sunnybrook Health Sciences Centre, Toronto, ON, Canada M4N 3M5; and f Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 2M9 Edited by Clifford J. Tabin, Harvard Medical School, Boston, MA, and approved March 20, 2012 (received for review November 14, 2011) Vascular smooth muscle cells (VSMC) have been suggested to arise from various developmental sources during embryogenesis, de- pending on the vascular bed. However, evidence also points to a common subpopulation of vascular progenitor cells predisposed to VSMC fate in the embryo. In the present study, we use binary transgenic reporter mice to identify a Tie1 + CD31 dim vascular endo- thelial (VE)-cadherin - CD45 - precursor that gives rise to VSMC in vivo in all vascular beds examined. This precursor does not rep- resent a mature endothelial cell, because a VE-cadherin promoter- driven reporter shows no expression in VSMC during murine de- velopment. Blockade of Notch signaling in the Tie1 + precursor cell, but not the VE-cadherin + endothelial cell, decreases VSMC invest- ment of developing arteries, leading to localized hemorrhage in the embryo at the time of vascular maturation. However, Notch signaling is not required in the Tie1 + precursor after establishment of a stable artery. Thus, Notch activity is required in the differen- tiation of a Tie1 + local precursor to VSMC in a spatiotemporal fashion across all vascular beds. vascular development | dominant-negative Mastermind-like V ascular smooth muscle cells (VSMC) play an important role in vascular homeostasis in the adult and the embryo. Iden- tification of VSMC progenitors in the embryo may define the source of new VSMC seen during arteriogenesis and athero- sclerotic plaque formation in the adult. However, the devel- opmental origin of VSMC is still under investigation. From lineage-tracing experiments using either a genetic reporter or cross-species transplants, embryonic tissues such as the neural crest, the proepicardium, and the somites have been shown to be sources of VSMC during development (1, 2). Although numer- ous cells from the somites migrate to the site of dorsal aorta formation during avian development, only a small percentage of these migrating cells actually integrate into the dorsal aorta (3), suggesting that further differentiation is required to specify a VSMC fate. A few studies have been performed to identify immediate VSMC precursors localized to nascent vessels. Mesoangioblasts are progenitor cells isolated from dorsal aorta of embryonic day (E)9.5 mouse embryos and can be expanded in culture as clones expressing both endothelial and mesenchymal markers (4). Mesoangioblasts have been shown to differentiate into VSMC in vitro and in mouse–chick chimeras (4). Endothelial cells (EC) also have been suggested to be a possible source of VSMC through the process of endothelial–mesenchymal transdifferentiation in an avian developmental model and in pathological arterial neo- intimal thickening in pulmonary hypertension (5, 6). However, no studies have examined whether the endothelium is a source of VSMC precursor cells during mammalian development. The Notch signaling pathway has been shown to induce en- dothelial-to-mesenchymal transdifferentiation during cardiac cushion formation and in mature EC in vitro (7, 8). Disruption of Notch signaling during embryonic development leads to cardiac malformations as well as to vascular defects (9). A decrease in aortic VSMC coverage has been observed in some Notch mutants; however, the cellular mechanism of this disruption has not been characterized (10). Blockade of Notch signaling in specifically mature mouse VSMC does not result in embryonic vascular defects, suggesting that mature VSMC do not require Notch during embryogenesis (11). However, Notch activation in murine Pax3 + neural crest-derived cells has been shown to be required for VSMC development of the branchial arch arteries and vessels derived from the outflow tract (12), and EC expression of the ligand Jagged1 has been suggested to provide the Notch-activa- tion signal (13). The requirement of Notch signaling from sources other than the neural crest for VSMC differentiation in mammalian systems has not been investigated, and where VSMC Notch activity is required in the path from neural crest to branchial arch also is not clear. To determine whether endothelial–mesenchymal transdiffer- entiation is involved in the development of VSMC in a mam- malian system, we used a binary tetracycline-inducible transgenic system to follow EC during murine vascular development. Two endothelial promoters were used: one that is endothelial specific, vascular endothelial (VE)-cadherin, and another that is more promiscuous, Tie1. We found that mature endothelium is not a source of VSMC during murine development. We also observed that the Tie1 promoter is active in a VSMC precursor that gives rise to VSMC following localized Notch activation in vivo. Our results suggest that Notch signaling is essential for the differen- tiation of VSMC from local Tie1 + CD31 dim VE-cadherin − CD45 − precursor cells. Results VSMC Are Derived from a Tie1 + CD31 dim VE-Cadherin - CD45 - Precursor Cell. To determine whether EC can transdifferentiate into VSMC during mammalian development, we used a tetracycline-in- ducible (Tet-off) binary transgenic system to mark EC-derived cells. Mice expressing the tetracycline transcription activator (tTA) under an EC-specific promoter were crossed with re- sponder mice expressing a nuclear-localized β-gal reporter under the tTA-activated tetracycline-responsive promoter tetOS in the absence of tetracycline (Fig. S1A) (14). Both EC promoter driver lines, Tie1-tTA and VE-cadherin-tTA (VECtTA) were active in Author contributions: L.C., M.N., and A.K. designed research; L.C., M.H., M.L., A.P., and M.F. performed research; A.H.K., A.I.M., M.C.P., and D.J.D. contributed new reagents/ analytic tools; L.C., M.H., M.L., A.P., and A.K. analyzed data; and L.C. and A.K. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1118512109/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1118512109 PNAS | May 1, 2012 | vol. 109 | no. 18 | 6993–6998 DEVELOPMENTAL BIOLOGY
Transcript of Differentiation of vascular smooth muscle cells from local ... of vascular smooth muscle cells from...

Differentiation of vascular smooth muscle cells fromlocal precursors during embryonic and adultarteriogenesis requires Notch signalingLinda Changa,b, Michela Nosedac, Michelle Higginsona, Michelle Lya, Alexandre Patenaudea,c, Megan Fullera,c,Alastair H. Kyled, Andrew I. Minchintond, Mira C. Purie,f, Daniel J. Dumonte,f, and Aly Karsana,b,c,g,1
aGenome Sciences Centre, dIntegrative Oncology Department, and gCancer Genetics Laboratory, British Columbia Cancer Agency, Vancouver, BC, Canada V5Z1L3; bExperimental Medicine Program and cDepartment of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada V6T1Z4; eSunnybrook Research Institute, Sunnybrook Health Sciences Centre, Toronto, ON, Canada M4N 3M5; and fDepartment of Medical Biophysics, Universityof Toronto, Toronto, ON, Canada M5G 2M9
Edited by Clifford J. Tabin, Harvard Medical School, Boston, MA, and approved March 20, 2012 (received for review November 14, 2011)
Vascular smooth muscle cells (VSMC) have been suggested to arisefrom various developmental sources during embryogenesis, de-pending on the vascular bed. However, evidence also points toa common subpopulation of vascular progenitor cells predisposedto VSMC fate in the embryo. In the present study, we use binarytransgenic reporter mice to identify a Tie1+CD31dimvascular endo-thelial (VE)-cadherin−CD45− precursor that gives rise to VSMCin vivo in all vascular beds examined. This precursor does not rep-resent a mature endothelial cell, because a VE-cadherin promoter-driven reporter shows no expression in VSMC during murine de-velopment. Blockade of Notch signaling in the Tie1+ precursor cell,but not the VE-cadherin+ endothelial cell, decreases VSMC invest-ment of developing arteries, leading to localized hemorrhage inthe embryo at the time of vascular maturation. However, Notchsignaling is not required in the Tie1+ precursor after establishmentof a stable artery. Thus, Notch activity is required in the differen-tiation of a Tie1+ local precursor to VSMC in a spatiotemporalfashion across all vascular beds.
vascular development | dominant-negative Mastermind-like
Vascular smooth muscle cells (VSMC) play an important rolein vascular homeostasis in the adult and the embryo. Iden-
tification of VSMC progenitors in the embryo may define thesource of new VSMC seen during arteriogenesis and athero-sclerotic plaque formation in the adult. However, the devel-opmental origin of VSMC is still under investigation. Fromlineage-tracing experiments using either a genetic reporter orcross-species transplants, embryonic tissues such as the neuralcrest, the proepicardium, and the somites have been shown to besources of VSMC during development (1, 2). Although numer-ous cells from the somites migrate to the site of dorsal aortaformation during avian development, only a small percentage ofthese migrating cells actually integrate into the dorsal aorta (3),suggesting that further differentiation is required to specify aVSMC fate.A few studies have been performed to identify immediate
VSMC precursors localized to nascent vessels. Mesoangioblastsare progenitor cells isolated from dorsal aorta of embryonic day(E)9.5 mouse embryos and can be expanded in culture as clonesexpressing both endothelial and mesenchymal markers (4).Mesoangioblasts have been shown to differentiate into VSMCin vitro and in mouse–chick chimeras (4). Endothelial cells (EC)also have been suggested to be a possible source of VSMC throughthe process of endothelial–mesenchymal transdifferentiation inan avian developmental model and in pathological arterial neo-intimal thickening in pulmonary hypertension (5, 6). However,no studies have examined whether the endothelium is a source ofVSMC precursor cells during mammalian development.The Notch signaling pathway has been shown to induce en-
dothelial-to-mesenchymal transdifferentiation during cardiaccushion formation and in mature EC in vitro (7, 8). Disruption of
Notch signaling during embryonic development leads to cardiacmalformations as well as to vascular defects (9). A decrease inaortic VSMC coverage has been observed in some Notch mutants;however, the cellular mechanism of this disruption has not beencharacterized (10). Blockade of Notch signaling in specificallymature mouse VSMC does not result in embryonic vasculardefects, suggesting that mature VSMC do not require Notchduring embryogenesis (11). However, Notch activation in murinePax3+ neural crest-derived cells has been shown to be requiredfor VSMC development of the branchial arch arteries and vesselsderived from the outflow tract (12), and EC expression of theligand Jagged1 has been suggested to provide the Notch-activa-tion signal (13). The requirement of Notch signaling fromsources other than the neural crest for VSMC differentiation inmammalian systems has not been investigated, and where VSMCNotch activity is required in the path from neural crest tobranchial arch also is not clear.To determine whether endothelial–mesenchymal transdiffer-
entiation is involved in the development of VSMC in a mam-malian system, we used a binary tetracycline-inducible transgenicsystem to follow EC during murine vascular development. Twoendothelial promoters were used: one that is endothelial specific,vascular endothelial (VE)-cadherin, and another that is morepromiscuous, Tie1. We found that mature endothelium is nota source of VSMC during murine development. We also observedthat the Tie1 promoter is active in a VSMC precursor that givesrise to VSMC following localized Notch activation in vivo. Ourresults suggest that Notch signaling is essential for the differen-tiation of VSMC from local Tie1+CD31dimVE-cadherin−CD45−
precursor cells.
ResultsVSMC Are Derived from a Tie1+CD31dimVE-Cadherin−CD45− PrecursorCell. To determine whether EC can transdifferentiate into VSMCduring mammalian development, we used a tetracycline-in-ducible (Tet-off) binary transgenic system to mark EC-derivedcells. Mice expressing the tetracycline transcription activator(tTA) under an EC-specific promoter were crossed with re-sponder mice expressing a nuclear-localized β-gal reporter underthe tTA-activated tetracycline-responsive promoter tetOS in theabsence of tetracycline (Fig. S1A) (14). Both EC promoter driverlines, Tie1-tTA and VE-cadherin-tTA (VECtTA) were active in
Author contributions: L.C., M.N., and A.K. designed research; L.C., M.H., M.L., A.P., andM.F. performed research; A.H.K., A.I.M., M.C.P., and D.J.D. contributed new reagents/analytic tools; L.C., M.H., M.L., A.P., and A.K. analyzed data; and L.C. and A.K. wrotethe paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118512109/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1118512109 PNAS | May 1, 2012 | vol. 109 | no. 18 | 6993–6998
DEV
ELOPM
ENTA
LBIOLO
GY
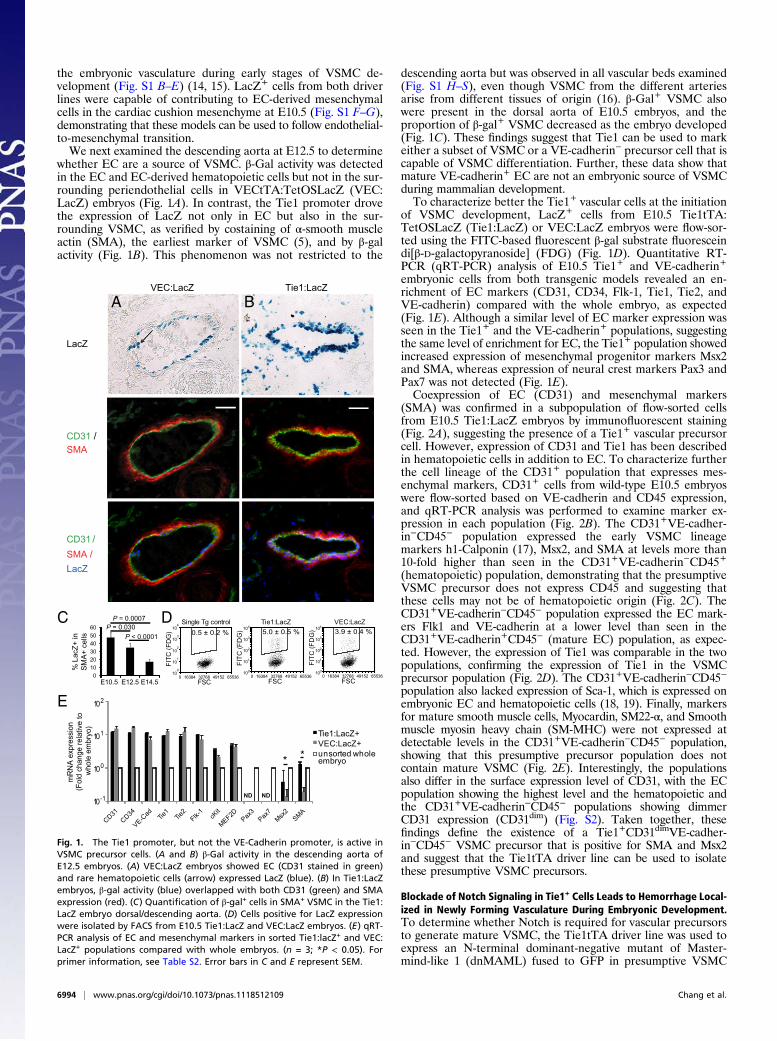
the embryonic vasculature during early stages of VSMC de-velopment (Fig. S1 B–E) (14, 15). LacZ+ cells from both driverlines were capable of contributing to EC-derived mesenchymalcells in the cardiac cushion mesenchyme at E10.5 (Fig. S1 F–G),demonstrating that these models can be used to follow endothelial-to-mesenchymal transition.We next examined the descending aorta at E12.5 to determine
whether EC are a source of VSMC. β-Gal activity was detectedin the EC and EC-derived hematopoietic cells but not in the sur-rounding periendothelial cells in VECtTA:TetOSLacZ (VEC:LacZ) embryos (Fig. 1A). In contrast, the Tie1 promoter drovethe expression of LacZ not only in EC but also in the sur-rounding VSMC, as verified by costaining of α-smooth muscleactin (SMA), the earliest marker of VSMC (5), and by β-galactivity (Fig. 1B). This phenomenon was not restricted to the
descending aorta but was observed in all vascular beds examined(Fig. S1 H–S), even though VSMC from the different arteriesarise from different tissues of origin (16). β-Gal+ VSMC alsowere present in the dorsal aorta of E10.5 embryos, and theproportion of β-gal+ VSMC decreased as the embryo developed(Fig. 1C). These findings suggest that Tie1 can be used to markeither a subset of VSMC or a VE-cadherin− precursor cell that iscapable of VSMC differentiation. Further, these data show thatmature VE-cadherin+ EC are not an embryonic source of VSMCduring mammalian development.To characterize better the Tie1+ vascular cells at the initiation
of VSMC development, LacZ+ cells from E10.5 Tie1tTA:TetOSLacZ (Tie1:LacZ) or VEC:LacZ embryos were flow-sor-ted using the FITC-based fluorescent β-gal substrate fluoresceindi[β-D-galactopyranoside] (FDG) (Fig. 1D). Quantitative RT-PCR (qRT-PCR) analysis of E10.5 Tie1+ and VE-cadherin+
embryonic cells from both transgenic models revealed an en-richment of EC markers (CD31, CD34, Flk-1, Tie1, Tie2, andVE-cadherin) compared with the whole embryo, as expected(Fig. 1E). Although a similar level of EC marker expression wasseen in the Tie1+ and the VE-cadherin+ populations, suggestingthe same level of enrichment for EC, the Tie1+ population showedincreased expression of mesenchymal progenitor markers Msx2and SMA, whereas expression of neural crest markers Pax3 andPax7 was not detected (Fig. 1E).Coexpression of EC (CD31) and mesenchymal markers
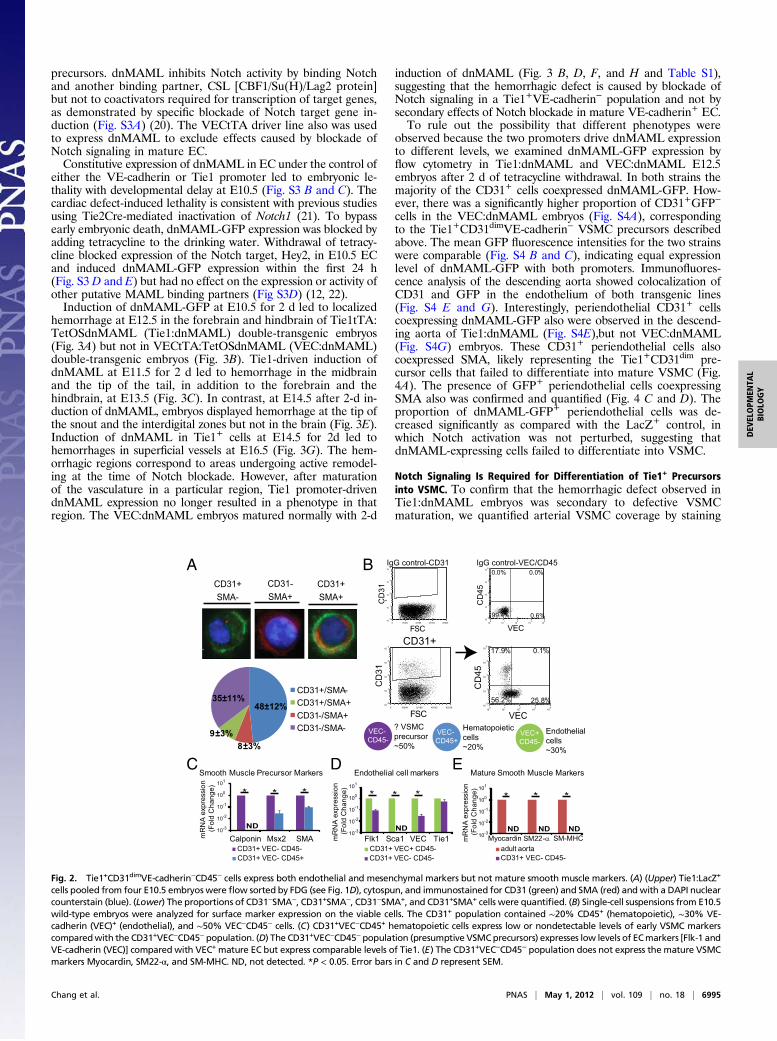
(SMA) was confirmed in a subpopulation of flow-sorted cellsfrom E10.5 Tie1:LacZ embryos by immunofluorescent staining(Fig. 2A), suggesting the presence of a Tie1+ vascular precursorcell. However, expression of CD31 and Tie1 has been describedin hematopoietic cells in addition to EC. To characterize furtherthe cell lineage of the CD31+ population that expresses mes-enchymal markers, CD31+ cells from wild-type E10.5 embryoswere flow-sorted based on VE-cadherin and CD45 expression,and qRT-PCR analysis was performed to examine marker ex-pression in each population (Fig. 2B). The CD31+VE-cadher-in−CD45− population expressed the early VSMC lineagemarkers h1-Calponin (17), Msx2, and SMA at levels more than10-fold higher than seen in the CD31+VE-cadherin−CD45+
(hematopoietic) population, demonstrating that the presumptiveVSMC precursor does not express CD45 and suggesting thatthese cells may not be of hematopoietic origin (Fig. 2C). TheCD31+VE-cadherin−CD45− population expressed the EC mark-ers Flk1 and VE-cadherin at a lower level than seen in theCD31+VE-cadherin+CD45− (mature EC) population, as expec-ted. However, the expression of Tie1 was comparable in the twopopulations, confirming the expression of Tie1 in the VSMCprecursor population (Fig. 2D). The CD31+VE-cadherin−CD45−
population also lacked expression of Sca-1, which is expressed onembryonic EC and hematopoietic cells (18, 19). Finally, markersfor mature smooth muscle cells, Myocardin, SM22-α, and Smoothmuscle myosin heavy chain (SM-MHC) were not expressed atdetectable levels in the CD31+VE-cadherin−CD45− population,showing that this presumptive precursor population does notcontain mature VSMC (Fig. 2E). Interestingly, the populationsalso differ in the surface expression level of CD31, with the ECpopulation showing the highest level and the hematopoietic andthe CD31+VE-cadherin−CD45− populations showing dimmerCD31 expression (CD31dim) (Fig. S2). Taken together, thesefindings define the existence of a Tie1+CD31dimVE-cadher-in−CD45− VSMC precursor that is positive for SMA and Msx2and suggest that the Tie1tTA driver line can be used to isolatethese presumptive VSMC precursors.
Blockade of Notch Signaling in Tie1+ Cells Leads to Hemorrhage Local-ized in Newly Forming Vasculature During Embryonic Development.To determine whether Notch is required for vascular precursorsto generate mature VSMC, the Tie1tTA driver line was used toexpress an N-terminal dominant-negative mutant of Master-mind-like 1 (dnMAML) fused to GFP in presumptive VSMC
LacZ
CD31 /SMA
CD31 /SMA /LacZ
Tie1:LacZVEC:LacZ
BA
C Single Tg control
FSC
FITC
(FD
G)
0 16384 32768 49152 6553610
0
101
102
103
104
0.5 ± 0.2 %Tie1:LacZ
FSC
FITC
(FD
G)
0 16384 32768 49152 65536100
101
102
103
104
5.0 ± 0.5 %VEC:LacZ
FSC
FITC
(FD
G)
0 16384 32768 49152 65536100
101
102
103
104
3.9 ± 0.4 %D
E
Tie1:LacZ+VEC:LacZ+unsorted whole embryo
mR
NA
expr
essi
on(F
old
chan
ge re
lativ
e to
w
hole
em
bryo
)
110
210
010
-110
* *
ND ND
0102030405060
E10.5 E12.5 E14.5
% L
acZ+
in
SM
A+
cells
P = 0.0007
P < 0.0001P = 0.030
Fig. 1. The Tie1 promoter, but not the VE-Cadherin promoter, is active inVSMC precursor cells. (A and B) β-Gal activity in the descending aorta ofE12.5 embryos. (A) VEC:LacZ embryos showed EC (CD31 stained in green)and rare hematopoietic cells (arrow) expressed LacZ (blue). (B) In Tie1:LacZembryos, β-gal activity (blue) overlapped with both CD31 (green) and SMAexpression (red). (C) Quantification of β-gal+ cells in SMA+ VSMC in the Tie1:LacZ embryo dorsal/descending aorta. (D) Cells positive for LacZ expressionwere isolated by FACS from E10.5 Tie1:LacZ and VEC:LacZ embryos. (E) qRT-PCR analysis of EC and mesenchymal markers in sorted Tie1:lacZ+ and VEC:LacZ+ populations compared with whole embryos. (n = 3; *P < 0.05). Forprimer information, see Table S2. Error bars in C and E represent SEM.
6994 | www.pnas.org/cgi/doi/10.1073/pnas.1118512109 Chang et al.
precursors. dnMAML inhibits Notch activity by binding Notchand another binding partner, CSL [CBF1/Su(H)/Lag2 protein]but not to coactivators required for transcription of target genes,as demonstrated by specific blockade of Notch target gene in-duction (Fig. S3A) (20). The VECtTA driver line also was usedto express dnMAML to exclude effects caused by blockade ofNotch signaling in mature EC.Constitutive expression of dnMAML in EC under the control of
either the VE-cadherin or Tie1 promoter led to embryonic le-thality with developmental delay at E10.5 (Fig. S3 B and C). Thecardiac defect-induced lethality is consistent with previous studiesusing Tie2Cre-mediated inactivation of Notch1 (21). To bypassearly embryonic death, dnMAML-GFP expression was blocked byadding tetracycline to the drinking water. Withdrawal of tetracy-cline blocked expression of the Notch target, Hey2, in E10.5 ECand induced dnMAML-GFP expression within the first 24 h(Fig. S3 D and E) but had no effect on the expression or activity ofother putative MAML binding partners (Fig S3D) (12, 22).Induction of dnMAML-GFP at E10.5 for 2 d led to localized
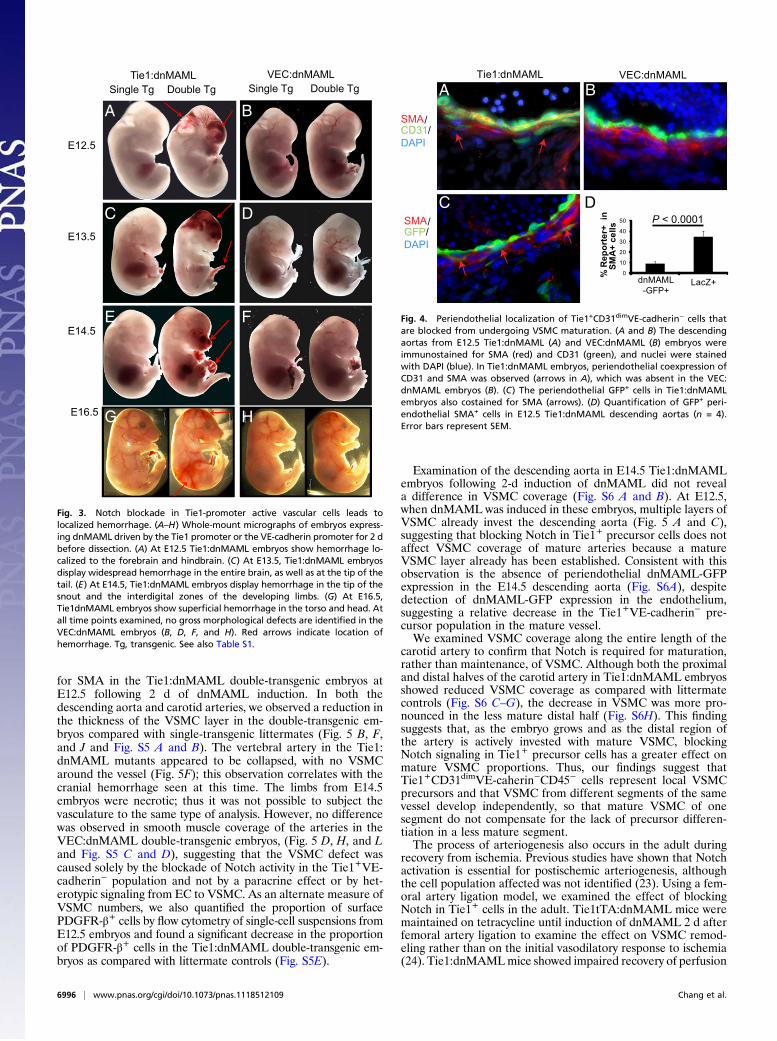
hemorrhage at E12.5 in the forebrain and hindbrain of Tie1tTA:TetOSdnMAML (Tie1:dnMAML) double-transgenic embryos(Fig. 3A) but not in VECtTA:TetOSdnMAML (VEC:dnMAML)double-transgenic embryos (Fig. 3B). Tie1-driven induction ofdnMAML at E11.5 for 2 d led to hemorrhage in the midbrainand the tip of the tail, in addition to the forebrain and thehindbrain, at E13.5 (Fig. 3C). In contrast, at E14.5 after 2-d in-duction of dnMAML, embryos displayed hemorrhage at the tip ofthe snout and the interdigital zones but not in the brain (Fig. 3E).Induction of dnMAML in Tie1+ cells at E14.5 for 2d led tohemorrhages in superficial vessels at E16.5 (Fig. 3G). The hem-orrhagic regions correspond to areas undergoing active remodel-ing at the time of Notch blockade. However, after maturationof the vasculature in a particular region, Tie1 promoter-drivendnMAML expression no longer resulted in a phenotype in thatregion. The VEC:dnMAML embryos matured normally with 2-d
induction of dnMAML (Fig. 3 B, D, F, and H and Table S1),suggesting that the hemorrhagic defect is caused by blockade ofNotch signaling in a Tie1+VE-cadherin− population and not bysecondary effects of Notch blockade in mature VE-cadherin+ EC.To rule out the possibility that different phenotypes were
observed because the two promoters drive dnMAML expressionto different levels, we examined dnMAML-GFP expression byflow cytometry in Tie1:dnMAML and VEC:dnMAML E12.5embryos after 2 d of tetracycline withdrawal. In both strains themajority of the CD31+ cells coexpressed dnMAML-GFP. How-ever, there was a significantly higher proportion of CD31+GFP−
cells in the VEC:dnMAML embryos (Fig. S4A), correspondingto the Tie1+CD31dimVE-cadherin− VSMC precursors describedabove. The mean GFP fluorescence intensities for the two strainswere comparable (Fig. S4 B and C), indicating equal expressionlevel of dnMAML-GFP with both promoters. Immunofluores-cence analysis of the descending aorta showed colocalization ofCD31 and GFP in the endothelium of both transgenic lines(Fig. S4 E and G). Interestingly, periendothelial CD31+ cellscoexpressing dnMAML-GFP also were observed in the descend-ing aorta of Tie1:dnMAML (Fig. S4E),but not VEC:dnMAML(Fig. S4G) embryos. These CD31+ periendothelial cells alsocoexpressed SMA, likely representing the Tie1+CD31dim pre-cursor cells that failed to differentiate into mature VSMC (Fig.4A). The presence of GFP+ periendothelial cells coexpressingSMA also was confirmed and quantified (Fig. 4 C and D). Theproportion of dnMAML-GFP+ periendothelial cells was de-creased significantly as compared with the LacZ+ control, inwhich Notch activation was not perturbed, suggesting thatdnMAML-expressing cells failed to differentiate into VSMC.
Notch Signaling Is Required for Differentiation of Tie1+ Precursorsinto VSMC. To confirm that the hemorrhagic defect observed inTie1:dnMAML embryos was secondary to defective VSMCmaturation, we quantified arterial VSMC coverage by staining
CD31-SMA+
CD31+SMA+
CD31+SMA-
B
VEC- CD45+
Hematopoieticcells~20%
VEC- CD45-
? VSMCprecursor~50%
VEC+CD45-
Endothelialcells~30%
A
ECSmooth Muscle Precursor Markers
mR
NA
exp
ress
ion
(Fol
d C
hang
e)
CD31+ VEC- CD45-CD31+ VEC- CD45+
ND
100
10-2
101
10-1
10-3
Calponin Msx2 SMA
* * *
CD31+/SMA-
CD31-/SMA+CD31+/SMA+
CD31-/SMA-
48±12%35±11%
9±3%8±3%
FSC0 16384 32768 49152 65536
CD
31
010
110
210
310
410
CD31+
VEC0
101
102
103
104
10
CD
45
010
110
210
310
410 17.9% 0.1%
25.8%56.2%
DEndothelial cell markers
mR
NA
exp
ress
ion
(Fol
d C
hang
e)
CD31+ VEC- CD45-CD31+ VEC+ CD45-
*
NDFlk1 Sca1 Tie1VEC
* *100
10-2
101
10-1
10-3
Mature Smooth Muscle Markers
mR
NA
exp
ress
ion
(Fol
d C
hang
e)
CD31+ VEC- CD45-adult aorta
ND NDNDMyocardin SM22-α SM-MHC
* * *100
10-2
101
10-1
10-3
FSC0 16384 32768 49152 65536
CD
31
010
110
210
310
410
IgG control-CD310.0% 0.0%
0.6%99.4%
VEC0
101
102
103
104
10
CD
45
010
110
210
310
410
%
IgG control-VEC/CD45
Fig. 2. Tie1+CD31dimVE-cadherin−CD45− cells express both endothelial and mesenchymal markers but not mature smooth muscle markers. (A) (Upper) Tie1:LacZ+
cells pooled from four E10.5 embryos were flow sorted by FDG (see Fig. 1D), cytospun, and immunostained for CD31 (green) and SMA (red) andwith a DAPI nuclearcounterstain (blue). (Lower) The proportions of CD31−SMA−, CD31+SMA−, CD31−SMA+, and CD31+SMA+ cells were quantified. (B) Single-cell suspensions from E10.5wild-type embryos were analyzed for surface marker expression on the viable cells. The CD31+ population contained ∼20% CD45+ (hematopoietic), ∼30% VE-cadherin (VEC)+ (endothelial), and ∼50% VEC−CD45− cells. (C) CD31+VEC−CD45+ hematopoietic cells express low or nondetectable levels of early VSMC markerscomparedwith the CD31+VEC−CD45−population. (D) The CD31+VEC−CD45−population (presumptive VSMCprecursors) expresses low levels of ECmarkers [Flk-1 andVE-cadherin (VEC)] compared with VEC+ mature EC but express comparable levels of Tie1. (E) The CD31+VEC−CD45− population does not express the mature VSMCmarkers Myocardin, SM22-α, and SM-MHC. ND, not detected. *P < 0.05. Error bars in C and D represent SEM.
Chang et al. PNAS | May 1, 2012 | vol. 109 | no. 18 | 6995
DEV
ELOPM
ENTA
LBIOLO
GY
for SMA in the Tie1:dnMAML double-transgenic embryos atE12.5 following 2 d of dnMAML induction. In both thedescending aorta and carotid arteries, we observed a reduction inthe thickness of the VSMC layer in the double-transgenic em-bryos compared with single-transgenic littermates (Fig. 5 B, F,and J and Fig. S5 A and B). The vertebral artery in the Tie1:dnMAML mutants appeared to be collapsed, with no VSMCaround the vessel (Fig. 5F); this observation correlates with thecranial hemorrhage seen at this time. The limbs from E14.5embryos were necrotic; thus it was not possible to subject thevasculature to the same type of analysis. However, no differencewas observed in smooth muscle coverage of the arteries in theVEC:dnMAML double-transgenic embryos, (Fig. 5 D, H, and Land Fig. S5 C and D), suggesting that the VSMC defect wascaused solely by the blockade of Notch activity in the Tie1+VE-cadherin− population and not by a paracrine effect or by het-erotypic signaling from EC to VSMC. As an alternate measure ofVSMC numbers, we also quantified the proportion of surfacePDGFR-β+ cells by flow cytometry of single-cell suspensions fromE12.5 embryos and found a significant decrease in the proportionof PDGFR-β+ cells in the Tie1:dnMAML double-transgenic em-bryos as compared with littermate controls (Fig. S5E).
Examination of the descending aorta in E14.5 Tie1:dnMAMLembryos following 2-d induction of dnMAML did not reveala difference in VSMC coverage (Fig. S6 A and B). At E12.5,when dnMAML was induced in these embryos, multiple layers ofVSMC already invest the descending aorta (Fig. 5 A and C),suggesting that blocking Notch in Tie1+ precursor cells does notaffect VSMC coverage of mature arteries because a matureVSMC layer already has been established. Consistent with thisobservation is the absence of periendothelial dnMAML-GFPexpression in the E14.5 descending aorta (Fig. S6A), despitedetection of dnMAML-GFP expression in the endothelium,suggesting a relative decrease in the Tie1+VE-cadherin− pre-cursor population in the mature vessel.We examined VSMC coverage along the entire length of the
carotid artery to confirm that Notch is required for maturation,rather than maintenance, of VSMC. Although both the proximaland distal halves of the carotid artery in Tie1:dnMAML embryosshowed reduced VSMC coverage as compared with littermatecontrols (Fig. S6 C–G), the decrease in VSMC was more pro-nounced in the less mature distal half (Fig. S6H). This findingsuggests that, as the embryo grows and as the distal region ofthe artery is actively invested with mature VSMC, blockingNotch signaling in Tie1+ precursor cells has a greater effect onmature VSMC proportions. Thus, our findings suggest thatTie1+CD31dimVE-caherin−CD45− cells represent local VSMCprecursors and that VSMC from different segments of the samevessel develop independently, so that mature VSMC of onesegment do not compensate for the lack of precursor differen-tiation in a less mature segment.The process of arteriogenesis also occurs in the adult during
recovery from ischemia. Previous studies have shown that Notchactivation is essential for postischemic arteriogenesis, althoughthe cell population affected was not identified (23). Using a fem-oral artery ligation model, we examined the effect of blockingNotch in Tie1+ cells in the adult. Tie1tTA:dnMAML mice weremaintained on tetracycline until induction of dnMAML 2 d afterfemoral artery ligation to examine the effect on VSMC remod-eling rather than on the initial vasodilatory response to ischemia(24). Tie1:dnMAMLmice showed impaired recovery of perfusion
Tie1:dnMAML VEC:dnMAML Single Tg Double Tg Single Tg Double Tg
E12.5
E13.5
E14.5
A
F
D
B
C
E
E16.5 G H
Fig. 3. Notch blockade in Tie1-promoter active vascular cells leads tolocalized hemorrhage. (A–H) Whole-mount micrographs of embryos express-ing dnMAML driven by the Tie1 promoter or the VE-cadherin promoter for 2 dbefore dissection. (A) At E12.5 Tie1:dnMAML embryos show hemorrhage lo-calized to the forebrain and hindbrain. (C) At E13.5, Tie1:dnMAML embryosdisplay widespread hemorrhage in the entire brain, as well as at the tip of thetail. (E) At E14.5, Tie1:dnMAML embryos display hemorrhage in the tip of thesnout and the interdigital zones of the developing limbs. (G) At E16.5,Tie1dnMAML embryos show superficial hemorrhage in the torso and head. Atall time points examined, no gross morphological defects are identified in theVEC:dnMAML embryos (B, D, F, and H). Red arrows indicate location ofhemorrhage. Tg, transgenic. See also Table S1.
Tie1:dnMAML VEC:dnMAML
SMA/CD31/DAPI
SMA/GFP/DAPI
P < 0.0001
A B
C D
0
10
20
30
40
50
dnMAML-GFP+
LacZ+
% R
epor
ter+
in
SMA
+ ce
lls
Fig. 4. Periendothelial localization of Tie1+CD31dimVE-cadherin− cells thatare blocked from undergoing VSMC maturation. (A and B) The descendingaortas from E12.5 Tie1:dnMAML (A) and VEC:dnMAML (B) embryos wereimmunostained for SMA (red) and CD31 (green), and nuclei were stainedwith DAPI (blue). In Tie1:dnMAML embryos, periendothelial coexpression ofCD31 and SMA was observed (arrows in A), which was absent in the VEC:dnMAML embryos (B). (C) The periendothelial GFP+ cells in Tie1:dnMAMLembryos also costained for SMA (arrows). (D) Quantification of GFP+ peri-endothelial SMA+ cells in E12.5 Tie1:dnMAML descending aortas (n = 4).Error bars represent SEM.
6996 | www.pnas.org/cgi/doi/10.1073/pnas.1118512109 Chang et al.
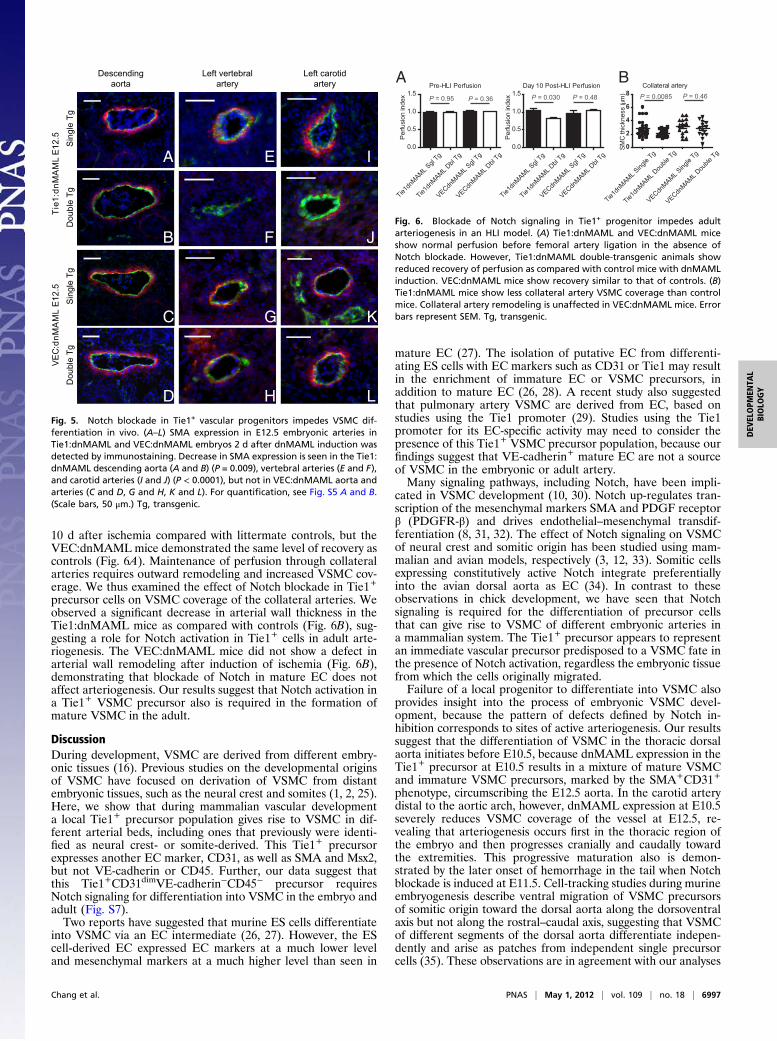
10 d after ischemia compared with littermate controls, but theVEC:dnMAML mice demonstrated the same level of recovery ascontrols (Fig. 6A). Maintenance of perfusion through collateralarteries requires outward remodeling and increased VSMC cov-erage. We thus examined the effect of Notch blockade in Tie1+
precursor cells on VSMC coverage of the collateral arteries. Weobserved a significant decrease in arterial wall thickness in theTie1:dnMAML mice as compared with controls (Fig. 6B), sug-gesting a role for Notch activation in Tie1+ cells in adult arte-riogenesis. The VEC:dnMAML mice did not show a defect inarterial wall remodeling after induction of ischemia (Fig. 6B),demonstrating that blockade of Notch in mature EC does notaffect arteriogenesis. Our results suggest that Notch activation ina Tie1+ VSMC precursor also is required in the formation ofmature VSMC in the adult.
DiscussionDuring development, VSMC are derived from different embry-onic tissues (16). Previous studies on the developmental originsof VSMC have focused on derivation of VSMC from distantembryonic tissues, such as the neural crest and somites (1, 2, 25).Here, we show that during mammalian vascular developmenta local Tie1+ precursor population gives rise to VSMC in dif-ferent arterial beds, including ones that previously were identi-fied as neural crest- or somite-derived. This Tie1+ precursorexpresses another EC marker, CD31, as well as SMA and Msx2,but not VE-cadherin or CD45. Further, our data suggest thatthis Tie1+CD31dimVE-cadherin−CD45− precursor requiresNotch signaling for differentiation into VSMC in the embryo andadult (Fig. S7).Two reports have suggested that murine ES cells differentiate
into VSMC via an EC intermediate (26, 27). However, the EScell-derived EC expressed EC markers at a much lower leveland mesenchymal markers at a much higher level than seen in
mature EC (27). The isolation of putative EC from differenti-ating ES cells with EC markers such as CD31 or Tie1 may resultin the enrichment of immature EC or VSMC precursors, inaddition to mature EC (26, 28). A recent study also suggestedthat pulmonary artery VSMC are derived from EC, based onstudies using the Tie1 promoter (29). Studies using the Tie1promoter for its EC-specific activity may need to consider thepresence of this Tie1+ VSMC precursor population, because ourfindings suggest that VE-cadherin+ mature EC are not a sourceof VSMC in the embryonic or adult artery.Many signaling pathways, including Notch, have been impli-
cated in VSMC development (10, 30). Notch up-regulates tran-scription of the mesenchymal markers SMA and PDGF receptorβ (PDGFR-β) and drives endothelial–mesenchymal transdif-ferentiation (8, 31, 32). The effect of Notch signaling on VSMCof neural crest and somitic origin has been studied using mam-malian and avian models, respectively (3, 12, 33). Somitic cellsexpressing constitutively active Notch integrate preferentiallyinto the avian dorsal aorta as EC (34). In contrast to theseobservations in chick development, we have seen that Notchsignaling is required for the differentiation of precursor cellsthat can give rise to VSMC of different embryonic arteries ina mammalian system. The Tie1+ precursor appears to representan immediate vascular precursor predisposed to a VSMC fate inthe presence of Notch activation, regardless the embryonic tissuefrom which the cells originally migrated.Failure of a local progenitor to differentiate into VSMC also
provides insight into the process of embryonic VSMC devel-opment, because the pattern of defects defined by Notch in-hibition corresponds to sites of active arteriogenesis. Our resultssuggest that the differentiation of VSMC in the thoracic dorsalaorta initiates before E10.5, because dnMAML expression in theTie1+ precursor at E10.5 results in a mixture of mature VSMCand immature VSMC precursors, marked by the SMA+CD31+
phenotype, circumscribing the E12.5 aorta. In the carotid arterydistal to the aortic arch, however, dnMAML expression at E10.5severely reduces VSMC coverage of the vessel at E12.5, re-vealing that arteriogenesis occurs first in the thoracic region ofthe embryo and then progresses cranially and caudally towardthe extremities. This progressive maturation also is demon-strated by the later onset of hemorrhage in the tail when Notchblockade is induced at E11.5. Cell-tracking studies during murineembryogenesis describe ventral migration of VSMC precursorsof somitic origin toward the dorsal aorta along the dorsoventralaxis but not along the rostral–caudal axis, suggesting that VSMCof different segments of the dorsal aorta differentiate indepen-dently and arise as patches from independent single precursorcells (35). These observations are in agreement with our analyses
Descendingaorta
Left carotidartery
Left vertebralartery
Sin
gle
TgD
oubl
e Tg
Tie1
:dnM
AM
LE1
2.5
VEC:
dnM
AM
LE1
2.5
B F
D
A E
C G K
I
J
H L
Sin
gle
TgD
oubl
e Tg
Fig. 5. Notch blockade in Tie1+ vascular progenitors impedes VSMC dif-ferentiation in vivo. (A–L) SMA expression in E12.5 embryonic arteries inTie1:dnMAML and VEC:dnMAML embryos 2 d after dnMAML induction wasdetected by immunostaining. Decrease in SMA expression is seen in the Tie1:dnMAML descending aorta (A and B) (P = 0.009), vertebral arteries (E and F),and carotid arteries (I and J) (P < 0.0001), but not in VEC:dnMAML aorta andarteries (C and D, G and H, K and L). For quantification, see Fig. S5 A and B.(Scale bars, 50 μm.) Tg, transgenic.
Pre-HLI Perfusion
Tie1dn
MAML Sgl
Tg
Tie1dn
MAML Dbl
Tg
VECdnMAML S
gl Tg
VECdnMAML D
bl Tg
0.0
0.5
1.0
1.5
Perfu
sion
inde
x
P = 0.95 P = 0.36
Perfu
sion
inde
x
Day 10 Post-HLI Perfusion
Tie1dn
MAML Sgl
Tg
Tie1dn
MAML Dbl
Tg
VECdnMAML S
gl Tg
VECdnMAML D
bl Tg
0.0
0.5
1.0
1.5P = 0.030 P = 0.48
Collateral artery
Tie1dn
MAML Sing
le Tg
Tie1dn
MAML Dou
ble Tg
VECdnMAML S
ingle
Tg
VECdnMAML D
ouble
Tg0
2
4
6
8
SMC
thic
knes
s (μ m
)
P = 0.0085 P = 0.46
A B
Fig. 6. Blockade of Notch signaling in Tie1+ progenitor impedes adultarteriogenesis in an HLI model. (A) Tie1:dnMAML and VEC:dnMAML miceshow normal perfusion before femoral artery ligation in the absence ofNotch blockade. However, Tie1:dnMAML double-transgenic animals showreduced recovery of perfusion as compared with control mice with dnMAMLinduction. VEC:dnMAML mice show recovery similar to that of controls. (B)Tie1:dnMAML mice show less collateral artery VSMC coverage than controlmice. Collateral artery remodeling is unaffected in VEC:dnMAML mice. Errorbars represent SEM. Tg, transgenic.
Chang et al. PNAS | May 1, 2012 | vol. 109 | no. 18 | 6997
DEV
ELOPM
ENTA
LBIOLO
GY

suggesting that activation of Notch in a local VSMC precursor isrequired to generate mature VSMC in a coordinated fashionduring vascular development.In summary, we demonstrate that a local Tie1+CD31dimVE-
cadherin−CD45− precursor cell differentiates into VSMC in allvascular beds and requires Notch activation. Further, the Tie1:dnMAML transgenic system potentially can be used to map theprocess of embryonic arteriogenesis through development.
Materials and MethodsMice. Tie1-tTA and TetOSLacZ mice were maintained on a CD1 background.VECtTA mice were a gift from L. Benjamin (Harvard University, Cambridge,MA) and were maintained on the FVB/NJ background. Mice subsequentlywere backcrossed onto the C57BL/6J background for five generations. TetOS-dnMAML mice were generated by pronuclear injections of linearized DNAencoding the tetOS promoter followed by cDNA of the dnMAML-GFP fusionconstruct. dnMAML transgenic mice were identifiedwith genotyping primers5′-CAT GCC ATG GAT GGT GAG CAA GGG CGA G-3′ and 5′-CCA TCG ATT TACTTG TAC AGC TCG TCC A-3′. All animal experiments were approved by andconform to guidelines of the Animal Care Committee of the University ofBritish Columbia. For tetracycline treatment, timed-mated and pluggedfemales were provided with drinking water containing 50 mg/L tetracycline.The treated water was replaced daily and removed at the times specified.
Flow Cytometry. Mouse embryos at specified stages were dissected free ofyolk sac tissue, minced, and digested in a solution containing 1% (wt/vol)BSA, 550 U/mL collagenase type II, 550 U/mL collagenase type IV, and 100 U/mLDNase I (Sigma-Aldrich) in PBS for 15 min in a 37 °C water bath. The digestedembryos were treated with red blood cell lysis buffer, and single-cell suspen-sions were analyzed for GFP expression, stained with FDG, or immunostainedwith CD31 or PDGFRβ (BD Pharmingen) Ab followed by a secondary goatanti-rat Alexa Fluor 647 Ab (Invitrogen). Alternatively, phycoerythrin-conju-gated CD31 Ab (BD Pharmingen), FITC-conjugated CD45 Ab (eBioscience), or
allophycocyanin-conjugated VE-cadherin Ab (eBioscience) were used. FCSExpress (De Novo) software was used to analyze flow cytometry data.
β-Gal Detection. For whole-mount X-Gal staining, embryos were fixed,washed, and incubated with β-gal staining solution [1 mg/mL X-Gal (Invi-trogen), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide in β-galwash solution] at 37 °C for 2–4 h. The embryos were postfixed in 2% (wt/vol)paraformaldehyde and embedded in optimum cutting temperature com-pound for cryosectioning. Immunostaining was carried out as described (36).For flow cytometry analysis, single-cell suspensions from embryos were in-cubated with 0.5 mM FDG (Invitrogen) in DMEM at 37 °C for 2 min followedby 30 min at 4 °C.
Hindlimb Ischemia Model. Tie1:dnMAML and VEC:dnMAML adults weregenerated by mating Tie1tTA and VECtTA mice with TetOSdnMAML mice.Mice were provided doxycycline or tetracycline (50 mg/L) in the drinkingwater until 2 d after hindlimb ischemia (HLI). HLI was performed as describedwhen the mice were 8 wk old (23). Blood flow was monitored with a laserDoppler (PeriScan PIM II; Perimed) before ligation and on day 10 after HLI.The level of reperfusion was calculated by taking the ratio of blood flow inthe ischemic leg over blood flow in the contralateral nonischemic leg usingLDPI 2.6 (Perimed). Mice were killed, and hindlimbs were perfusion-fixed forcryosectioning and immunostaining.
Statistical Analysis. Statistical analysis was performed with GraphPad Prism(GraphPad Software). P values were determined by Student’s t test.
ACKNOWLEDGMENTS. We thank Fred Wong, Denise McDougal, and theBritish Columbia Cancer Research Center Terry Fox Laboratory Flow Core forflow cytometry analysis and cell sorting and Patricia Umlandt for assistancein animal work. This work was supported by a grant from the Heart andStroke Foundation of British Columbia and the Yukon and by Grant MOP64354 from the Canadian Institutes of Health Research (both to A.K.). L.C.,A.P., and A.K. are supported by awards from the Michael Smith Foundationfor Health Research.
1. Pouget C, Pottin K, Jaffredo T (2008) Sclerotomal origin of vascular smooth musclecells and pericytes in the embryo. Dev Biol 315:437–447.
2. Wasteson P, et al. (2008) Developmental origin of smooth muscle cells in the de-scending aorta in mice. Development 135:1823–1832.
3. Sato Y, et al. (2008) Notch mediates the segmental specification of angioblasts insomites and their directed migration toward the dorsal aorta in avian embryos. DevCell 14:890–901.
4. Minasi MG, et al. (2002) The meso-angioblast: A multipotent, self-renewing cell thatoriginates from the dorsal aorta and differentiates into most mesodermal tissues.Development 129:2773–2783.
5. DeRuiter MC, et al. (1997) Embryonic endothelial cells transdifferentiate into mes-enchymal cells expressing smooth muscle actins in vivo and in vitro. Circ Res 80:444–451.
6. Arciniegas E, Ponce L, Hartt Y, Graterol A, Carlini RG (2000) Intimal thickening in-volves transdifferentiation of embryonic endothelial cells. Anat Rec 258:47–57.
7. Timmerman LA, et al. (2004) Notch promotes epithelial-mesenchymal transitionduring cardiac development and oncogenic transformation. Genes Dev 18:99–115.
8. Noseda M, et al. (2004) Notch activation results in phenotypic and functional changesconsistent with endothelial-to-mesenchymal transformation. Circ Res 94:910–917.
9. Phng LK, Gerhardt H (2009) Angiogenesis: A team effort coordinated by notch. DevCell 16:196–208.
10. Morrow D, et al. (2008) Notch and vascular smooth muscle cell phenotype. Circ Res103:1370–1382.
11. Proweller A, et al. (2007) Notch signaling in vascular smooth muscle cells is required topattern the cerebral vasculature. Proc Natl Acad Sci USA 104:16275–16280.
12. High FA, et al. (2007) An essential role for Notch in neural crest during cardiovasculardevelopment and smooth muscle differentiation. J Clin Invest 117:353–363.
13. High FA, et al. (2008) Endothelial expression of the Notch ligand Jagged1 is requiredfor vascular smooth muscle development. Proc Natl Acad Sci USA 105:1955–1959.
14. Sarao R, Dumont DJ (1998) Conditional transgene expression in endothelial cells.Transgenic Res 7:421–427.
15. Sun JF, et al. (2005) Microvascular patterning is controlled by fine-tuning the Aktsignal. Proc Natl Acad Sci USA 102:128–133.
16. Majesky MW (2007) Developmental basis of vascular smooth muscle diversity. Arte-rioscler Thromb Vasc Biol 27:1248–1258.
17. Miano JM, Olson EN (1996) Expression of the smooth muscle cell calponin gene marksthe early cardiac and smooth muscle cell lineages during mouse embryogenesis. J BiolChem 271:7095–7103.
18. Sata M, et al. (2002) Hematopoietic stem cells differentiate into vascular cells thatparticipate in the pathogenesis of atherosclerosis. Nat Med 8:403–409.
19. de Bruijn MF, et al. (2002) Hematopoietic stem cells localize to the endothelial celllayer in the midgestation mouse aorta. Immunity 16:673–683.
20. Weng AP, et al. (2003) Growth suppression of pre-T acute lymphoblastic leukemiacells by inhibition of notch signaling. Mol Cell Biol 23:655–664.
21. Limbourg FP, et al. (2005) Essential role of endothelial Notch1 in angiogenesis. Cir-culation 111:1826–1832.
22. McElhinny AS, Li JL, Wu L (2008) Mastermind-like transcriptional co-activators:Emerging roles in regulating cross talk among multiple signaling pathways. Onco-gene 27:5138–5147.
23. Limbourg A, et al. (2007) Notch ligand Delta-like 1 is essential for postnatal arterio-genesis. Circ Res 100:363–371.
24. Mees B, et al. (2007) Endothelial nitric oxide synthase activity is essential for vasodi-lation during blood flow recovery but not for arteriogenesis. Arterioscler ThrombVasc Biol 27:1926–1933.
25. Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM (2000) Fate of the mam-malian cardiac neural crest. Development 127:1607–1616.
26. Ema M, et al. (2003) Combinatorial effects of Flk1 and Tal1 on vascular and hema-topoietic development in the mouse. Genes Dev 17:380–393.
27. Hill KL, et al. (2010) Human embryonic stem cell-derived vascular progenitor cells capableof endothelial and smooth muscle cell function. Exp Hematol 38(3):246–257 e1.
28. Marchetti S, et al. (2002) Endothelial cells genetically selected from differentiatingmouse embryonic stem cells incorporate at sites of neovascularization in vivo. J CellSci 115:2075–2085.
29. Morimoto M, et al. (2010) Canonical Notch signaling in the developing lung is re-quired for determination of arterial smooth muscle cells and selection of Clara versusciliated cell fate. J Cell Sci 123:213–224.
30. Yoshida T, Owens GK (2005) Molecular determinants of vascular smooth muscle celldiversity. Circ Res 96:280–291.
31. Noseda M, et al. (2006) Smooth Muscle alpha-actin is a direct target of Notch/CSL. CircRes 98:1468–1470.
32. Jin S, et al. (2008) Notch signaling regulates platelet-derived growth factor re-ceptor-beta expression in vascular smooth muscle cells. Circ Res 102:1483–1491.
33. Ben-Yair R, Kalcheim C (2008) Notch and bone morphogenetic protein differentiallyact on dermomyotome cells to generate endothelium, smooth, and striated muscle.J Cell Biol 180:607–618.
34. Ohata E, Tadokoro R, Sato Y, Saito D, Takahashi Y (2009) Notch signal is sufficient todirect an endothelial conversion from non-endothelial somitic cells conveyed to theaortic region by CXCR4. Dev Biol 335:33–42.
35. Esner M, et al. (2006) Smooth muscle of the dorsal aorta shares a common clonalorigin with skeletal muscle of the myotome. Development 133:737–749.
36. Niessen K, et al. (2008) Slug is a direct Notch target required for initiation of cardiaccushion cellularization. J Cell Biol 182:315–325.
6998 | www.pnas.org/cgi/doi/10.1073/pnas.1118512109 Chang et al.


















