
CHRISTIAN LAFLAMME - · PDF filepaper or electronic formats. The author retains ownership of...
82
CHRISTIAN LAFLAMME IMPLICATION DE L'APOPTOSE DES LYMPHOCYTESBRONCHO- ALVÉOLAIRES HUMAINS DANS L'AL*OLITE ALLERGIQUE EXTRLNSEQUE. Mémoire présenté À la Facultd des études supérieures de l'université Laval pour l'obtention du grade de maître ès sciences (M.Sc.) FACULTÉ DE MJ?DECINE &DECINE EXPÉRIMENTALE UNIVERSITÉ LAVAL MAI 2000 O Christian Laflamme, 2000
Transcript of CHRISTIAN LAFLAMME - · PDF filepaper or electronic formats. The author retains ownership of...

CHRISTIAN LAFLAMME
IMPLICATION DE L'APOPTOSE DES LYMPHOCYTES BRONCHO-
ALVÉOLAIRES HUMAINS DANS L'AL*OLITE ALLERGIQUE
EXTRLNSEQUE.
Mémoire
présenté
À la Facultd des études supérieures
de l'université Laval
pour l'obtention
du grade de maître ès sciences (M.Sc.)
FACULTÉ DE MJ?DECINE
&DECINE EXPÉRIMENTALE
UNIVERSITÉ LAVAL
MAI 2000
O Christian Laflamme, 2000

National Library I*I .,,a Bibliotheque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques
395 Wellington Sveet 395, ~ i e Wdimgton OnawaûN KlAON4 O(lewa ON K1A ON4 Canada Canada
The author has granted a non- exclusive licence allowing the National Library of Canada to reproduce, loan, distribute or sel1 copies of this thesis in microform, paper or electronic formats.
The author retains ownership of the copyright in this thesis. Neither the thesis nor substantial extracts fiom it may be printed or otherwise reproduced without the author's permission.
L'auteur a accordé une licence non exclusive permettant à la Bibliothèque nationale du Canada de reproduire, prêter, distribuer ou vendre des copies de cette thèse sous la fome de microfiche/fdm, de reproduction sur papier ou sur format électronique.
L'auteur conserve la propriété du droit d'auteur qui protège cette thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation.

Résumé
L'aivéolite allergique extrinsèque ( A M ) est une maladie inflammatoire
pulmonaire caractérisée par une lymphocytose alvéolaire persistante. Les buts de l'étude
étaient : 1) évaluer si une diminution de l'apoptose des lymphocytes pulmonaires pouvait
expliquer la persistance des lymphocytes dans le poumon, 2) décrire les mécanismes de
cette apoptose. Deux catégories de sujets furent étudiées, AAE et normaux sans contact.
Le pourcentage de lymphocytes en apoptose était significativement plus élevé chez les
sujets normaux que chez les patients atteints de I'AAE. La proportion de lymphocytes
positifs pour le Fas (récepteur associé à l'apoptose) était environ 50% plus élevée chez
les lymphocytes des patients AAE que chez ceux des normaux. Le Fas soluble (Fas,) est
augmenté de plus de 300% dans le liquide du lavage bronchoalvéolaire (LBA) des
patients AAE. De plus, les lymphocytes des patients AAE produisent significativement
plus de Bcl-xL (protéine mi-apoptotique). Lorsqu'isolés et mis en culture pendant 24
hres, 4 1.2% des lymphocytes pulmonaires non stimulés des patients AAE sont morts par
apoptose comparativement à 12'9% chez les lymphocytes provenant des sujets contrôles
@=0,004). Les lymphocytes T des patients AAE ne sont pas sensibles à l'apoptose suite à
une stimulation à I'anti-Fas. Par contre, l'interleukine-2 (IL-2) inhibe I'apoptose de ces
celluIes @=0,07). En conclusion, l'apoptose des lymphocytes dans I'AAE est diminuée et
s'explique en partie par 1) une augmentation du Fas, (inhibe l'initiation de l'apoptose), 2)
une augmentation de Bcl-xL, 3) une augmentation de l'IL-2 dms le poumon et 4) une
défaillance dans Ie sentier Fas-FasL.
Christian ~ a f l & e B.SC.
Étudiant
~ y ~ v o n Cormier M.D.
Directeur de recherche

Ce mémoire est composé d'un article et d'une section de résultats
complémentaires à l'article. Le chapitre 3 présente la publication s'intitulant : Apoptosis
of bronchoalveolar lavage lymphocytes in hypersensitivity pneumonitis. (C. Laflamme, E.
Israël-Assayag, Y. Cormier). Cette étude a été soumise pour publication dans uThe
Journal of Immunology B. Le chapitre 4 est, quant à lui, formé d'une section de résultats
complémentaires (non-publiés) à l'article. Ii est important de prendre note que la
bibliographie, située à la fin de ce mémoire, englobe tous les chapitres sauf le numéro 3,
qui a sa propre section de références associées.
Je tiens tout d'abord à remercier le Dr Yvon Cormier, qui m'a accueilli au sein de
son équipe. Ses nombreux conseils et sa simplicité ont su modifier mon approche tant au
niveau scientifique que dans la vie de tous les jours. Merci h Évelyne Israël-Assayag,
1' assistante de recherche de notre laboratoire, pour son soutien technique et scientifique.
Évelyne est une personne indispensable, tant pour ses idées que pour ses connaissances.
Merci également à tout les gens des laboratoires de pneumologie et de
cardiologie. le pense entre autres à Nathaiie Pagé pour ses conseils techniques, au Dr
Guy Tremblay (père spirituel pour les congrès), à Martin Coulombe, mon grand ami,
pour tout le défoulement lors de nos parties de squash « fiil1 contact ),, à Francis Davoine,
pour nos conversations de tracteurs et à Sophie Lavigne pour la cytométrie en fiux.
Finalement, je ne peux passer sous silence mes parents Yvan et Blanche, mes
arnis(es) est bien sûr ma copine Stéphanie; leur soutien et leur amour sont source de
motivation et d'énergie.
Encore une fois merci!

TABLE DES MATI~RES
PAGE
RÉSUMÉ
AVANT-PROPOS
TABLE DES MATIÈRES
LISTE DES TABLEAUX
LISTE DES FIGURES
LISTE DES ABRÉVIATIONS
CHAPITRE 1 Introduction
1 . 1 L'alvéolite ailergique extrinsèque
1.2 L'inflammation pulmonaire
1.2.1 Cellules épithéliales des voies respiratoires
1.2.2 Macrophages alvéolaires
1.2.3 Neutrophiles
1.2.4 Lymphocytes
1.3 L'inflammation pulmonaire dans I'AAE
1.3.1 Prolifération locale
1.3.2 Augmentation du recrutement
1.3.3 Inhibition de l'apoptose
1.4 L'homéostasie cellulaire
1.4.1 Prolifération locale
1.4.2 Mort cellulaire
1.4.2.1 Nécrose
1.4.2.2 Apoptose
A. Le Fas :récepteur cellulaire de l'apoptose
B. Les gènes impliqués dans l'apoptose
B. 1 Les produits de gènes pro-apoptotiques
8 1.1 Bax et Bad
B.2 Les produits de gènes anti-apoptotiques

B.2.1 Bcl-2
B.2.2 Bcl-XL
C. IRs enzymes impliquées dans I'apoptose
D. Fragmentation de l'ADN
1.5 Sommaire
CHAPITRE 2 Hypothèses de travail
CHAPITRE 3 Apoptosis of bronchoalveolar lavage lymphocytes
in hypersensiîiviîy peurnonilis.
3.1 Résumé
3.2 Abstract
3.3 Introduction
3.4 Materials and Methods
3.5 Results
3.6 Discussion
3.7 Rrferences
3.8 Table and Figtrres
CHAPITRE 4 Augmentation et mécanismes de 19apoptose in vitro
des lymphocytes du lavage broncho-alvéolaire de
I'alvéolite allergique extrinsèque.
4.1 Mise en situation
4.2 Matériel et méthodes
4.2.1 Lavage broncho-alvéolaire
4.2.2 Purification des lymphocytes sur colonne laine-nylon
et culture cellulaire
4.2.3 TUNEL
4.2.4 Activité de la caspase 3
4.2.5 Analyse statistique
4.3 Résultats

4.3.1 Quantification de l'apoptose (test de TUNEL)
des lymphocytes T en culture non stimulés. 57
4.3.2 Mesure de l'activité de la caspase 3 chez les
lymphocytes T pulmonaires cultivés pendant 24 heures
sans stimulation. 58
4.3.3 Effet de I'anti-Fas et de I'IL-2 sur l'apoptose
des lymphocytes T cultivés pendant 24 heures (apoptose
quantifiée par TUNEL). 58
CHAPITRE 5 Conclusion générale 63
Bibliographie 67

LISTE DES TABLEAUX
PAGE
Tableau 1 . 1 : Maladies associées à un dérèglement du processus apoptotique. 11
Tableau 3.1 : BAL characteristics of HP patients and nomals subjects. 4 1

LISTE DES FIGURES
PAGE
Figure 3.1 :
Figure 3.2 :
Figure 3.3 :
Figure 3.4 :
Figure 3.5 :
Figure 3.6 :
Figure 4.1 :
Figure 4.2 :
Figure 4.3 :
Evaluation of BAL lymphocytes apoptosis. 43
Percentage of BAL lymphocytes for the surface
receptors Fas and FasL. 45
Quantification of ,Fas in BAL. 47
Correlation between BAL lymphocytosis and the concentration
of ,Fa5 in the BAL fluid and the percentage of Annexin+/PI-cells. 49
Proportion of BAL Iymphocytes positives for Bcl-2, 5 L
Quantification of the Bcl-xL protein in the BAL lymphocytes. 53
Dénombrement de la quantité & lymphocytes pulmonaires
cultivés sans stimulation et qui sont positifs pour le test de
TUNEL.
Activité de la caspase 3 en fonction du temps.
Effet de l'anti-Fas et de l'IL-2 sur l'apoptose des
lymphocytes T pulmonaires après 24 hres de culture.

AAE
ACV
APC
A l w n
BAL
BS A
DUTP
FasL
Fus
FBS
GM-CSF
HP
ICE
IL- 1
IL-2
IL-6
L-8
LBA
MA
MIP- 1 a
N
PBS
PI
PVDF
sF=
SR
SIDA
TBS
TdT
alvéolite allergique extrinsèque
Arrêt cardio-respiratoire
antigen presenting ce11
acide ribonucléique messager
bronchoalveolar lavage
bovine serum albumin
desoxy uridine tri-phosphate
Fas ligand
Fas soluble
Fœtal bovine serum
Granulocyte Macrophage Colony Srimulating Factor
hypersensitivity pneumonitis
interleukine-1 convening enzyme
interleukine 1
interleukine 2
interleukine 6
interleukine 8
lavage broncho-alvéolaire
macrophage alvéolaire
macrophage idanimatory protein- 1 a
normaux, normal
phosphate buffered saline
propidium iodine
polyvinylidene de fluoride
soluble Fus
Saccaropolyspora rectivirgula
S indrôme d' irnmuno-déficience acquise
tris buffered saline
terminal deoxinucleotidyl transferase

TNF Tumor Necrosis Factor
TUNEL Terminal Uridine Nick End Labelling


1.1 L'alvéolite allergique extrinsèaue
L'alvéolite allergique extrinsèque (AM) est une maladie pulmonaire interstitielle
inflammatoire résultant d'une réaction immunitaire d'hypersensibilité à des antigènes
dérivés de bactéries, de champignons microscopiques, de protéines animales et, plus
rarement, de substances chimiques réactives (1). Chez les patients, les symptômes varient
en fonction du stade de la maladie (aigu, subaigu ou chronique). Les manifestations les
plus courantes sont de la fièvre, de la dyspnée, une toux sèche et une perte de poids (2).
Dans certains cas plus graves, on constate des dommages pulmonaires irréversibles
comme la fibrose et l'emphysème (2).
Au niveau pulmonaire, cette pathologie est caractérisée par une augmentation
importante de cellules inflammatoires et de médiateurs pro-inflammatoires (cytokines)
(3). Dans l'établissement du diagnostique de l'AAE, un lavage broncho-alvéolaire (LBA)
est pratiqué. Cet examen de routine consiste 21 injecter et à retirer séquentiellement de
petits aliquots (50-60 ml) de saline physioIogique (0,996).
Chez un patient AAE en phase aiguë, le nombre de cellules dans le LBA est élevé
(100 à 150 x 106 cellules par lavage de 300 ml) et le pourcentage de lymphocytes T
dépasse souvent les 50% (se situe entre 30 et 70%) (2). Les nombres absolus de
neutrophiles et de macrophages sont aussi plus élevés. Par opposition, chez un sujet sain,
le nombre de cellules inflammatoires dans le LBA est d'environ 15 x 106 cellules par
lavage de 300 ml (2). Le LBA contient normalement, en moyenne, 89% de macrophages,
10% de lymphocytes et 1% de neutrophiles (4).
1.2 L'inflammation pulmonaire
De façon générale, l'inflammation est une réaction physiologique complexe et
multifactorielle par laquelle un tissu vascularisé, comme le poumon, répond à un stress de
nature étrangère ou à une blessure (5). Durant ce processus, toute une panoplie de

médiateurs solubles et de composantes cellulaires iravaillent de concert afin de réguler, le
plus finement possible, I'blimination de l'agent causal. L'infIamrnation est un phénomène
crucial au niveau immunitaire pour maintenir l'organisme en santé. Cependant, cet
équilibre est fragile et lorsque l'inflammation est peu contrôlée, il en résulte des
pathologies.
De façon traditionnelle, l'inflammation est divisée en deux catégories.
Dépendemment des auteurs, on parlera d'immunité cellulaire et d'immunité humorde ou
d'inflammation aiguë et d'inflammation chronique (5). Quoi qu'il y ait certaines
différences entre les deux terminologies, les bases conceptuelles sont ii peu près les
mêmes. L'inflammation aiguë est de courte durée (minutes B jours). Elle est caractérisée
par l'accumulation de fluide, de protéines plasmatiques et de leucocytes phagocytants, le
plus souvent des neutrophiles (5). En contrepartie, il y a la forme chronique qui est de
longue durée et est caractérisée par un influx de lymphocytes (T et B) et de macrophages
( 5 ) .
Au niveau pulmonaire, le contrôle de l'inflammation est vitai. En effet, il est
essentiel de conserver intacte et stérile l'interface entre les alvéoles et l'air ambiant, et ce
afin d'assurer l'efficacité des échanges gazeux dans le processus respiratoire (6). il
devient dors important, dans ce milieu fragile, de neutraliser et d'éliminer les antigènes
respirés le plus rapidement et le plus discrètement possible. Dans le cas d'une inhalation
massive et répétée d'antigène (comme dans L'AM), le processus inflammatoire s'engage
laissant place aux différentes cellules inflammatoires et aux médiateurs solubles.
Mentionnons toutefois que seulement une faible proporcion de gens exposés à des
antigenes causant I'AAE vont développés la maladie. La raison à ce fait est jusqu'à
présent inconnu.
Plusieurs types de cellules régulent l'inflammation pulmonaire. Parmi celles-ci,
mentionnons les ceIlules épithéliales bronchiques et alvéolaires, les cellules dendntiques,
les neuuophiles, les macrophages alvéolaires (MA), les éosinophiles, les mastocytes, les
lymphocytes, etc. Chacune d'entre elles joue un rôle clé dans le processus inflammatoire

et cela aussi bien par leurs actions (phagocytose, présentation d'antig6nes. ..) que par le
biais de la sécrétion d'un patron de médiateurs solubles qui est propre à chacun des
différents types cellulaires (7).
1.2.1 Cellules épithéliales des voies resoiratoires
Les cellules épithéliales des voies respiratoires peuvent participer à la régulation
de Ia fonction des cellules inflammatoires en synthétisant et en sécrétant différentes
cytokines. Parmi celles-ci mentionnons certaines chimiokines (l'interleukine4 (ILS), le
Growth regrrlaring oncogene-alpha (Gro-a), le Gro- y), l ' L I , l'IL-6, le Tumor Necrosis
Factor (TNF), différents Colony Stimulating Factor (CSF) comme le Grandocyre
Macrophage - CSF (GM-CSF) qui vont promouvoir la survie, l'activation et la
différentiation des macrophages, des neutrophiles et des éosinophiles, et bien d'autres
cytokines (8). Dépendemrnent de la pathologie et du type de stress inflammatoire, les
cellules épithéliales bronchiques sécrèteront différents médiateurs solubles.
1.2.7 Macro~hages alvéolaires
Le MA occupe une position cxuciale dans l'immunité pulmonaire. En effet, il agit
comme première ligne de défense, afin de conserver les épithéliums alvéolaires et
pulmonaires exempts de particules dtrangères (ex : poussière, matières organiques,
bactéries, etc.) (9). Lorsque des antigènes sont inhalés, les MA phagocytent ces
substances, ce qui les stimulent. iis relâchent alors des médiateurs pro-inflammatoires à
large spectre de stimulation, comme l'IL-1 et le TNF (10). Ces cytokines ont pour effet
direct d'avertir les cellules avoisinantes de la présence d'un étranger et de promouvoir le
recrutement des leucocytes de la circulation sanguine en augmentant la perméabilité
vasculaire (1 1). De plus, ces médiateurs sont responsables de la libération d'autres
cytokines contribuant à l'activation du processus inflammatoire comme l'IL-8 et l'IL-6.
1.2.3 Neutrophiles

Une des réponses les plus précoces et crucides dans l'inflammation est le
recrutement de neutrophiles du sang périphérique au site inflammatoire (5). Ceci se fait
sous l'influence d'agents chimiotactiques comme l'IL-8. Les neutrophiles recrutés au site
de l'inflammation sont dors activés et phagocytent les particdes étrangères et
pathogènes (12). Ils vont ainsi digérer ces particules à l'aide de leurs granules
intracellulaires contenant des oxydants endogènes et des enzymes protéolytiques (12).
La majorité des antigènes sont éliminés de cette façon. Cependant, dépendemrnent
de Ia nature et de la quantité d'antigènes présents dans le poumon, ii arrive parfois que les
cellules phagocytantes ne suffisent pas à la tâche et il faut dors enclencher une réaction
inflammatoire plus spécifique menant à l'inflammation chronique.
Une augmentation de la concentration de certaines cytokines, dont I'IL-1, au site
de l'inflammation va servir de chimioattractant et de stimulant pour les lymphocytes. Les
lymphocytes T-helper (Th) activés par certaines cytokines vont enclencher une réponse
Th1 ou Th2. Certaines lymphokines seront alors synthétisées (13). Parmi ces
lymphokines, l'IL-2 semble jouer un rôle important dans l'activation et la survie des
lymphocytes dans le poumon (14). Les lymphocytes T activés vont ensuite stimuler les
lymphocytes B pour la production d'immunoglobulines spécifiques à l'antigène à
éliminer. Certaines cytokines produites subséquemment par les lymphocytes
contribueront à la régulation et au retour à l'homéostasie lors de la résolution de la
réaction inflammatoire.
1.3 L'inflammation ~ulma~iaire dans l'ME
L'AAE est caractérisée par une réponse irnmunologique anormale suite à une
exposition à des antigènes environnementaux non pathogènes. En effet, chez certaines
personnes exposées à ces antigènes, il se développe une réaction d'hypersensibilité face à

ceux-ci. L'augmentation du nombre de lymphocytes T dans le LBA des patients AAE
constitue une particulmit6 de cette maladie (2). Les lymphocytes T ont, en génhl, un
profi1 CD8 positif (lymphocytes T suppresseurs/cytotoxiques) (2). De plus. ces cellules
sont activées et sécrktent des facteurs de croissance reconnus pour occasionner des
dommages tissulaires importants, voire même irréversibles (ex : fibrose, emphysème)
( 12).
Une propriété importante de cette lymphocytose pulmonaire est qu'elle est
persistante dans le temps, parfois même jusqu'à deux ans après la fin du contact
antigénique (15). il devient alors tout ii fait pertinent de tenter d'expliquer l'étiologie de
ce débalancement homéostasique si dommageable pour le tissu pulmonaire. D'emblée,
deux mécanismes généraux sont retenus : 1) une prolifdration locale (l6,17,18) et 2) une
augmentation du recrutement de ces cellules (19,20). C'est deux processus
immunologiques ont déjà fait l'objet d'études plus approfondies. Cependant, un troisième
mécanisme, celui-là hypothétique, n'a jamais été évdué. ii s'agit d'une augmentation de
la survie par l'inhibition de l'apoptose normale. Cette hypothèse sera vérifiée dans ta
présente étude. ii est raisonnable de penser que les trois mécanismes puissent agir de
concert.
1.3.1 Prolifération locale
Plusieurs rapports ont déjà fait état d'évidences sur l'état prolifératif des
lymphocytes pulmonaires dans I'AAE. L'un d'eiles a démontré que les MA et le
surfactant des patients atteints d ' M E ne sont plus capables d'inhiber l'activation des
lymphocytes T pulmonaires, suggérant que ces ceiiules sont en prolifération (16). De
plus, il a été démontré que les lymphocytes T du LBA des patients AAE expriment
davantage de marqueurs d'activation, comme le récepteur p75, associé à l'IL-2, le VLA-1
et l'antigène du HLA-Dr comparativement aux lymphocytes du LBA de sujets contrôles
(17). Ces observations sont donc des indices sur l'état prolifératifs des lymphocytes T
pulmonaires dans 1'AAE.

Une autre étude a démontré que le niveau d'IL-2, un facteur important dans
l'expansion clonale et la survie (par inhibition de l'apoptose) des lymphocytes T, est
augmenté dans le liquide du LBA des patients AAE (18). De plus, ces cellules proliférent
de façon dose-dépendante suite à une stimulation avec cette cytokines (18). Ces résultats
suggèrent que l'IL-2 joue un rôle important dans I'AAE en stimulant la prolifération des
lymphocytes et peut-être permet leur persistance dans le poumon. L'évaluation de ce
dernier item fait partie de ce rapport.
1.3.2 Augmentation du recrutement
D'autres études ont vérifié la possibilité d'une augmentation du recrutement des
lymphocytes au site de l'inflammation. L'une d'elles a remarqué l'augmentation des
ARN, des chirniokines RANTES et macrophage inflammatory protein-1-alpha (MIP-1-
a) chez les cellules inflammatoires du LBA des patients AAE (19). De plus, il a été
démontré que des cellules épithéliales pulmonaires (A549) stimulées avec l'antigène
Saccharopolyspora rectivergula (SR), responsable de ta maladie du poumon de fermier
(une forme d'AM), expriment (ARN,) et sécrètent (protéine) l'IL-8 en réponse à ce
stress (20). 11 est connu que RANTES, MIP-la et l'IL-8 sont des facteurs
chirnioattractants pour les lymphocytes (21). Pris ensemble, ces résultats suggèrent une
augmentation du recrutement des lymphocytes dans I'AAE et donc l'influence de ce
phénomène dans le maintien de la lymphocytose.
1.3.3 inhibition de I'awvtose
A notre connaissance, aucune étude n'a été publiée concernant un rôle potentiel
de I'apoptose des lymphocytes T pulmonaires dans le maintien de l'inflammation dans
I'AAE. Cependant, on retrouve dans la Littérature des indices nous permettant de poser
comme hypothèse qu'une diminution de I'apoptose des lymphocytes T pulmonaires
pourrait être impliquée dans I'AAE. Tout d'abord, tel que mentionné plus haut, le niveau
d'IL-2 (un facteur anti-apoptotique pour les lymphocytes T) est augmenté dans I'AAE et
les lymphocytes T pulmonaires sont aptes à répondre à cette cytokine grâce, entre autres,

à leur récepteur p75 qui est surexprimé (17). De plus, ces cellules proliferent de manière
dose-dépendante en réponse à L'IL-2 (18).
Un autre indice intéressant est issu de la recherche dans le domaine du systeme de
costimulation B7-CD28. Ce sentier est impliqué spécifiquement dans l'activation et la
survie des lymphocytes. Le CD28 est une molécule portée par les lymphocytes et est un
ligand pour le B7-1 (CD80) et B7-2 (CD86) qui sont des récepteurs associés aux cellules
présentatrices d'antigènes (22). La costimulation B7-CD28 est connue pour augmenter la
production d'IL-2 par les lymphocytes permettant ainsi à ces cellules de résister
davantage à I'apoptose (23). De plus, la costimulation B7-CD28 amplifie, de façon
substantielle, l'expression du Bcl-xL (protéine anti-apoptotique) chez les lymphocytes
123).
Dans L'AAE, l'expression du 87 (1 et 2) est augmentée sur les macrophages
alvéolaires des patients atteints de la maladie (24). De plus, le CD28 est fortement
exprimé chez tous les lymphocytes pulmonaires des patients AAE (24). Prises ensemble,
ces données suggèrent que les lymphocytes du LBA des patients AAE sont susceptibles
de produire davantage d'iL-2 et de Bcl-xL, deux facteurs de survie pour ces cellules.
Une observation intéressante a été faite par le groupe de Agostini et al. qui a
démontré que les lymphocytes pulmonaires des patients AAE expriment davantage de
Fas (récepteur connu pour induire l'apoptose chez les lymphocytes) à leur surface
comparativement aux lymphocytes pulmonaires de sujets normaux (25). Cet élément
d'information va à l'encontre de l'hypothèse d'une diminution de l'apoptose des
lymphocytes dans I'AAE puisqu'une augmentation de l'expression cellulaire du Fas
corrèle habituellement avec une augmentation de l'apoptose.
En résumé, I'AAE est caractérisée par une forte inflammation pulmonaire. La
grande majorité des cellules inflammatoires retrouvées dans le poumon sont des
lymphocytes T. Or, il semble que cette lymphocytose pulmonaire soit persistante dans le
temps. L'hypothèse d'une diminution de I'apoptose des lymphocytes dans I'AAE n'a pas

été évaluée. Cet élément pourrait peut-être expliquer la lymphocytose persistante dans
cette pathologie où l'homéostasie cellulaire est affectée.
1.4 L'homéostasie cellulaire
On peut définir l'homéostasie cellulaire comme un état d'équilibre entre les
ceilules en prolifération, celles en différentiation et celles qui meurent (26). Ce contrôle
des populations cellulaires chez les êtres vivants est un phénomène vital pour maintenir
un organisme en santé.
Si l'un ou l'autre des différents états est dérégulé, cet équilibre est rompu. Les
réactions physiologiques découlant directement de cette homéostasie deviennent dors
affectées à différents degrés. L'inflammation est un phénomène de débalancement
homéostasique et la résorption de l'inflammation peut être vue comme le retour 21
l'homéostasie par l'élimination des cellules inflammatoires accumulées.
1.4.1 Prolifération cellulaire
La proIifération cellulaire est un phénomène par lequel un clone cellulaire entre en
division cellulaire. Le phénomène a déjii été abordé plus haut (section 1.3.1).
1.4.3 Mort cellulaire
II existe deux types distincts de mort cellulaire, soient la nécrose et l'apoptose.
Des deux catégories de mon cellulaire, seule I'apoptose est impliquée dans le maintien
physiologique de l'homéostasie cellulaire, constituant ainsi une mort cellulaire naturelle
(27).

1.4.2.1 Nécrose
La nécrose est une mort ceildaire non physiologique résultant d'un événement
fortuit comme par exemple une privaiion subite d'oxygène, une force mécanique
localisée, une élévation rapide du taux d'acidité, etc, (28). Ce phénomène ne nécessite
pas une dépense d'énergie (sous forme (L'ATP) pour la cellule (29). Lors de la nécrose, la
membrane cellulaire est brisée et le contenu cytoplasmique est alors déversé dans
I'environnement immédiat de la cellule, induisant ainsi une inflamrnatiori tissulaire
localisée (30).
En 1972, Kerr et al. publièrent un nouveau type de mort cellulaire qu'ils ont
nommé apoptose, terme désignant, en Grec, la chute des feuilles à l'automne (31).
L'apoptose ou mort cellulaire programmée, est un phénomène hautement régulé de
suicide cellulaire. Son rôle dans l'homéostatie est axé principalement sur l'élimination
des cellules qui ont été produites en excès (ou accumulées), qui se sont mal développées
ou qui ont subi des dommages génétiques lors de leur développement (30).
Une cellule qui entre en apopiose subit des changements morphologiques
caractéristiques. Elle se rétrécit légèrement et démontre subséquemment une chromatine
dense, le noyau se fragmente, le cytoplasme bourgeonne et, finalement, la cellule se
subdivise en petits morceaux que l'on appelle des corps apoptotiques. Ces corps
apoptotiques seront alors phagocytés, évitant ainsi que le contenu cellulaire ne soit
déversé dans l'environnement (28). De ce fait, l'apoptose est un processus n'induisant
pas d'inflammation.
L'apoptose affecte les cellules de façon individuelle et précise, et ce sans atteindre
les cellules saines. À l'opposé, la nécrose est beaucoup moins singularisée et est piutôt un
événement de masse. L'apoptose est en quelque sorte l'étape finale dans le
renouvellement des populations de celiules. Dans la majorité des cas, une cellule naît
d'une division cellulaire, se différencie et finalement meurt par apoptose.

Des évidences récentes suggèrent qu'une altération de l'apoptose normale de
différents types cellulaires est la cause d'une panoplie de pathologies et de dérèglements
physiologiques. Par exemple, une suactivation de l'apoptose serait impliquée dans des
maladies neuro-dégénératives, comme la maladie de Parkinson ou d'Alzheimer, alors
qu'une apoptose inhibée serait à la base de certains cancers et de maladies auto-immune
(voir tableau 1.1) (32).
Tableau 1.1 Maladies associées à un dérèglement du processus apoptotique.
Catégories Exemples APptose
Suractivée Inhibée
Maladies neuro- Alzheimer + Dégénératives Parkinson + Désordres Maladies auto- + Immunitaires immunes
SIDA + Diabète + Thyroïdi te +
Ischémie Infarctus du myocarde + Reperfusion ACV + Néoplasies Lymphomes +
Astrocytomes + Hépatomes + Mélanomes + Autres +
Divers Vieillissement
Alopécie
L'apoptose peut être activée chez les cellules de mammiferes par une multitude de
facteurs intrinsèques et extrinsèques ailant des radiations ionisantes aux rayons
ultraviolets, aux infections virales en passant par les cytokines, l'expression d'une vaste

gamme de gènes faisant partie de la famiHe du Bcl-2 et le contact cellule/cellule via les
systèmes ligands-récepteurs (comme par exemple celui du Fas/FasL) (30).
Au niveau immunitaire, I'apoptose des cellules est généralement initiée par des
systèmes de ligands-récepteurs. En effet, c'est un contact cellule/cellule via certains
récepteurs et leurs ligands physiologiques qui contrôle en bonne partie le déclenchement
de cette mort cellulaire. L'implication d'une multitude de gènes est également
déterminante dans l'initiation et la régulation du phénomène de mort cellulaire
programmée. De plus, l'environnement dans tequel résident les cellules est aussi un
facteur ayant beaucoup d'influence.
A. Le Fas : récerxeur cellulaire de I'awotose
Le plus documenté des récepteurs cellulaires associés à I'apoptose est sans
contredit le Fas (aussi connu sous le nom de CD95 ou de APO-1). Le Fas fait partie de la
famille de récepteurs du TNF. Chez l'humain, ce récepteur est constitué de 325 acides
aminés (45 KDa) et est classé parmi les prothes membranaires de type 1 (33). 1 est
présent chez une variété de tissus du corps humain, comme le thymus, le foie, les
poumons, les reins, les ovaires, etc. Certaines cellules, comme les lymphocytes T
matures, expriment de façon constitutive le Fas (33). L'activation de ces cellules par un
antigène a pour effet d'augmenter l'expression de ce récepteur à leur surface. Ainsi, les
lymphocytes T sont généraiement plus sensibles B I'apoptose via le sentier Fas/FasL (34).
Le ligand physiologique du Fas, le Fas ligand (FasL), est également une mdécule
faisant partie de la famille du TNF. La ligation du FasL au Fas induit normalement
I'apoptose. Dans des conditions nonnales, il y a seulement un nombre très réduit de
cellules qui expriment le FasL et l'expression de cette molécule de surface est faible (35).
Le FasL est exprimé principalement chez les cellules T activées (35). Le sentier Fas/FasL
est un mécanisme très utilisé par le système immunitaire afin de contrôler les populations
de lymphocytes T activés par des antigènes et ainsi de s'assurer qu'il n'y ait pas un

surplus de ces cellules stimuldes (35). De plus, ce système initiateur de l'apoptose est
employé pour éliminer les cellules infectées par des virus (35).
Une forme soluble du Fas peut être produite à partir de I'ARN, du Fas par
épissage alternatif. Le Fas soluble (Fe) est similaire au Fas cellulaire, exception faite
qu'il lui manque le domaine transmembranaire. Le Fas, a la propriété d'inhiber
l'initiation de l'apoptose par le sentier Fas/FasL. En effet, le Fas, se lie au FasL (molécule
limitée en nombre) présent à la surface des cellules, bloquant ainsi le site actif et
l'empêchant de livrer son message de suicide celIulaire (36).
B. Les gènes impliaués dans f'amotose
Les gènes influencent beaucoup le devenir cellulaire. Au niveau de l'apoptose, il
existe une balance entre l'expression de gènes anti-apoptiques et de gènes pro-
apoptotiques. La génétique de l'apoptose a été intensément étudiée grjse au modèle du
nématode Camohabditis elegans. L'avantage d'un tel modèle est que la séquence de
division et de mort cellulaire durant le développement est connue. En effet, suc 671
cellules formées au cours de son développement, il y a exactement 1 13 d'entre elles qui
meurent par apoptose et ce, il des temps précis (37). Trois gènes régulent cette mort
programmée, soient ced-3, ced-4 et ced-9. Les gènes ced-3 et ced-4 sont essentiels pour
la mort cellulaire programmée durant le développement des nématodes tandis que ced-9
peut prévenir leurs actions (37).
Chez les cellules de mammifères, les gènes régulant l'apoptose font partie de la
famille Bcl-2. À ce jour, pas moins de 15 protéines de cette famille ont été identifiées et
cela sans compter les protéines virales apparentées (38). Les membres de la famille Bc1-2
sont localisés dans la membrane externe des mitochondries. Ils peuvent se pairer les uns
aux autres dans des combinaisons variées. Par exemple, il est possible de retrouver un
homodymère Bcl-2:Bcl-2 ou un hétérodymères Bci-2:Bax. Ces dimères forment des
canaux ioniques dans la membrane mitochondnale et contrôlent ainsi le transfert d'ions
entre le cytoplasme et la mitochondrie (particulièrement le cytochrome c) (39). Depuis

quelques années, la régulation mitochondriale a pris une place considérable dans l'étude
et la description de la mort cellulaire programmée. Elle semble être impliquée dans deux
phases distinctes de I'apoptose, soient la phase de contrôle et la phase effectrice (39).
B. 1 Les produits de gènes pro-apoptotiques
B.l.l Bax et Bad
Bax et Bad sont les deux protéines pro-apoptotiques de la famille Bcl-2 les plus
documentées. Une surexpression de ces gènes cause l'apoptose rapide des cellules (38).
B.2 Les uroduits de gènes anti-aw~totiaues
Le gène bcI-2 est le premier gène de la famille à avoir dté découvert (40). Le
produit du gène bcl-2 (la protéine Bcl-2) est un homologue structural et fonctionnel du
gène c d 9 (chez le nématode). il est situé sur un site de translocation entre les
chromosomes 14 et 18. Le gène bel-2 est reconnu pour permettre aux lymphocytes
quiescents (mémoire) de survivre sur une longue période de temps (40). De plus, une
surexpression de ce gène prévient l'initiation de I'apoptose chez la cellule en réponse à
une vaste gamme de stimulations. Ainsi, une grande quantité de la protéine Bcl-2 chez la
cellule protège celle-ci de I'apoptose induite par les rayons uliraviolets, la privation de
cytokines, certains conicostéroïdes, etc. (41). De plus, une concentration élevée de cette
protéine est reconnue pour protéger la cellule de la mort cellulaire programmée induite
par le Fas chez de nombreux types cellulaires dont les lymphocytes T (42).
La protéine Bcl-xL est issue du gène Bcl-x. Ce peptide est composé de 233 acides
aminés. Tout comme le Bcl-2, la protéine Bcl-xL est reconnue pour augmenter la

résistance cellulaire au processus apoptotique. La différence enue les deux protéines est
que le Bcl-2 est présent davantage chez les ceiiules T naïves (constitutif), alors que le
Bcl-xL est surtout pksent chez Les ceiiules T activées (inductible) (43). Il a été demontré,
chez des cellules MCF7 (cellules épithéliales tumorales) exprimant le Fas, qu'une
surexpression de Bcl-xL rend ces cellules résistantes ii l'apoptose induite par le Fas (44).
C. Les enzymes impliquées dans l'am~tose
La vaste gamme de signaux initiateurs (intra et extracellulaires) de l'apoptose
chez Ies cellules de mammifères convergent inévitablement vers un effecteur commun,
les caspases. La compréhension de la phase exécutrice de L'apoptose a débutée lorsque
ced-3 a été démontré comme faisant partie d'une nouvelle famille de protéases
(maintenant appelées caspases) (45). et où l'activation séquentielle et le clivage de
protéines cibles, en aval de la régulation par les gènes de la famille Bd-2, mène à la mort
de la cellule par apoptose (45). Les caspases sont des cystéines protdases de la famille de
l'interleukine-1 converting entyme (ICE) et sont très conservées chez les cellules de
mammiferes (46). ElIes clivent leurs substrais protéiques (en général une autre caspase) à
des sites contenant un résidu aspartate, activant ainsi une nouvelle caspase (45). Ceci
initie une cascade protéolytique létale menant à l'exécution de l'apoptose. Ainsi, les
caspases activées clivent les autres caspases subséquentes en des formes matures et
actives d'enzymes. Une fois que la cascade des caspases est activée, les changements
morphoIogiques cellulaires de'butent et i'apoptose devient à ce moment un processus
irréversible (45). Notons que leurs actions dans l'apoptose sont universelles (ex : la
caspase 3 est retrouvée dans tous les modèles d'apoptose chez les cellules de
mammiferes) (47). Jusqu'à maintenant, douze caspases sont connues chez les cellules de
rnammiferes (45).
D. La framentation de l'ADN
L'apoptose en phase finale se caractérise par la fragmentation de l'ADN
génomique en morceaux de 180 paires de bases (48). Dès les premiers stades du

processus, une endonucléas endogène ca2+ et M~'' ddpndante s'active et coupe l'ADN
entre Les nuciéosomes (situes à tous les 180-200 paires des bases dans le génome) (48).
Ces fragments sont en fait des complexes ADN-octamères d'histone. Cette segmentation
de l'ADN est à la base de plusieurs tests de détection de l'apoptose. En effet, la
dégradation de l'ADN peut être ditectée par electrophorèse sur gel d'agame (49). De
plus, cette fragmentation d'ADN peut être détectée in situ dans la cellule (en
microscopie) par le test de TUNEL (Terminal Uridinie Nick End Lubelling), méthode qui
a été mise au point par Graviel et al. en 1992 (50).
1.5 Sommaire
L'apoptose est donc un phénoméne hautement régule tant au niveau de son
initiation (FasEasL) que de son contrôle ginétique (Bcl-2. Bçl-xL, Bax ...) et de son
éxécution (cascade des caspases, Eragmentation de l'ADN.. .). Nous proposons d'étudier
l'apoptose des lymphocytes T pulmonaires dans le contexte de 1'AAE et de caractériser
ce phénomène.


Tel que mentionné précédemment, I'AAE est une maladie inflammatoire
caractérisée par une lymphocytose pulmonaire persistante. Deux mécanismes pouvant
expliquer la lymphocytose alvéolaire ont été avancés et étuàiés. Nous proposons
d'étudier une troisième possibilité, celle-là hypothétique, selon laquelle les lymphocytes
T pulmonaires de I'AAE auraient une apoptose diminuée. Ceci pourrait expliquer leur
persistance et leur accumulation dans le poumon. L'évaluation de certains paramètres de
I'apoptose (Fas, FasL, Fas,, Bcl-2 et Bcl-xL) font également partie de cette étude de
quantification et de caractérisation des mécanismes. Les données obtenues pour les
patients atteints d 'AM seront comparées avec celles obtenues chez des volontaires sains
(sans contact avec des antigènes causant I'AAE). Le chapitre 3 est voué à la résolution de
cette première hypothèse et présente l'article : Apoptosis of bronchoalveolar lavage
lymphocytes in hypersensitivity pneumoniris.
Dans un deuxième temps, le travail est axé davantage sur la mécanistique de
l'apoptose des lymphocytes dans I'AAE. Plus précisément, les lymphocytes T
pulmonaires des deux groupes de sujets à l'étude seront comparés dans un système de
culture cellulaire. L'effet de la stimulation avec l'IL-2 et avec I'anti-Fas (jouant le rôle de
FasL) sur I'apoptose de ces cellules sera évalué. L'hypothèse est que l'IL-2, diminue
l'apoptose des lymphocytes de I'AAE en culture et que l'anti-Fas fait augmenter cette
apoptose (comparativement dans les deux cas aux lymphocytes non stimulés). Le chapiue
4 intitulé : Augmentation et mécanismes de I'apoptose in vitro des lymphocytes T du
lavage broncho-alvéolaire de I'alvéolite allergique exinns2que » est consacré à
l'évaluation de cette deuxième hypothèse.

CHAPITRE 3
APOPTOSIS OF BRONCHOALVEOLAR LAVAGE LYMPHOCYTES IN
HYPERSENSITMTY PNEWMONITIS.
Article soumis pour publication dans : Am J Respir Ce11 Mol Biol.

3.1 Résumé
L'alvéolite allergique extrinsèque (AAE) est une maladie pulmonaire caractérisde par
l'accumulation d'une grande quantité & lymphocytes dans le poumon. Cette étude a été
faite dans le but d'évaluer le rôle potentiel de l'apoptose (mort cellulaire programmée)
des lymphocytes broncho-alv6olaires dans cette pathologie. Deux groupes de sujets ont
été étudiés: patients AAE et normaux sans contact. Le pourcentage des lymphocytes
apoptotiques était plus faible chez les patients de AAE que chez les sujets N (37.4%
contre 56.5% pour le test Annexine/iodure de propidium (p=û,004) et 0,4% contre 1.0%
pour les analyses de TUNEL (pû.033). La proportion de lymphocytes positifs pour Fas
était 42% plus élevée chez les sujets AAE que chez les sujets normaux (71.7% contre
50.4%). Cependant, aucune différence significative n'a été trouvée pour la proportion de
lymphocytes positifs pour le FasL. Les niveaux de Fas soIuble dans le liquide du lavage
bronchoalvéolaire ( D A ) des patients AAE et des sujets normaux étaient de 80,s pg/rnl et
de 23.2 pg/rnl respectivement (p=0,0001). Une condlation positive a été trouv6e entre le
pourcentage de lymphocytes et la concentration de Fas dans le LBA. La concentration
intracellulaire de Bcl-xL (prot6ine anti-apoptotique) était augmentée de 2,6 fois chez les
lymphocytes pulmonaires des patients AAE comparativement aux normaux. Aucune
différence significative n'a été trouv6e concernant le pourcentage des lymphocytes du
LBA positifs pour le Bci-2 (protéine anti-apoptotique constitutive). En conclusion,
I'apoptose des lymphocytes pulmonaires est diminuée chez les sujets AAE
comparativement aux sujets normaux. Cette diminution d'apoptose peut s'expliquer, du
moins en partie, par une augmentation du Fass et par l'augmentation de la protéine h l -
xL.

APOPTOSIS OF BRONCHOALVEOLAR LAVAGE LYMPHOCYTES IN
HYPERSENSUMTY PNEUMOMTIS.
Christian Laflamme, Evelyne Israël-Assayag and Yvon Cormier.
Unité de recherche, Centre de Pneumologie, Hôpital Laval, institut de cardiologie et de
pneumologie de l'université Lavai.
Running title : Apoptosis of BAL Lymphocytes in Hypersensitivity Pneumonitis.
Keyword: h g , inflammation, apoptosis.
Please send correspondence to:
Dr. Yvon Cormier, Hôpital Laval
2725, Chemin Ste-Foy
S te-Fo y, Québec, Canada, G 1V 4G5
Tel: 4 18-656-4747
FAX: 4 18-656-4762
E-mail: yvon.cormier8med.ulaval.ca

3.2 Abstract
Hypersensitivity pneumonitis (HP) is a pulmonary disease characterized by the
accumulation of large quantities of lymphocytes in the lung. This snidy was done to
evaluate a possible role of apoptosis (programmed ce11 death) of bronchoalveolar lavage
(BAL) Iymphocytes in HP. Two groups were studied: HP patients and nomal 0
unexposed controls. The percentage of apoptotic lymphocytes was signiflcantly lower in
HP patients than in N subjects (37.4% vs 56.5% for AnnexinJpropidium iodine test
(p=0,0041 and 0.4% vs 1 .O% for the TUNEL assays @=0,033). The proportion of BAL
lymphocytes positive for Fas was 42% higher in HP than in N (71.7% vs 50.4%).
However, no significant difference was found for the proportion of BAL lymphocytes
positive for FasL between the two groups of subjects. Soluble Fas (Jas) levels in the
BAL fluid of the HP patients and N subjects were: 80.5 p g h i and 23.2 p g h i
respectively @=û,0001). A positive correlation was found between the percentage of
BAL lymphocytes and the concentration of ,Fa. The intracellular quantity of the
inductible anti-apoptotic gene Bcl-xL product was increased 2.6 times in the pulmonary
Iymphocytes of HP patients compared to N. No difference was found for the percentage
of BAL tyrnphocytes positive for %cl-2 (constitutive anti-apoptotic protein). In
conclusion, the apoptosis of pulmonary lymphocytes is lower in HP than in N subjects.
This lower apoptosis can be explained, at least in part, by an increase of ,Fa5 and by the
intracelIular increase of the Bcl-xL protein.

3.3 Introduction
Although the lung is contiauously exposed to antigens, sometirnes to massive
loads, local immune responses are under tight regdations and usualiy prevent
inflammation and tissue damage. Hypersensitivity pneumonitis (HP), an antigen-induced
inflammatory lung disease, represents a clinical situation where the immune regulation is
overcome, leading to the accumulation of large numbers of lymphocytes in the lung (1).
There are three possible mechanisms, not mutually exclusive, by whicb lymphocytes can
accumulate in the lung in HP: 1) increased recruitment (2), 2) local proliferation (3,4),
and 3) increased survival (decreased apoptosis). This study addresses the issue of
lymphocyte removal by apoptosis in HP.
During an immune response against foreign antigens, cional expansion of
immunocompetent cells is necessary to mount an effective protection. These cells must
eventually die off in order to maintain tissue homeostasis (5). Regulation of ce11 survival
is therefore crucial to lirnit the accumulation of potentially harmful cells in the lung.
Apoptosis is a physiological, genetically controlled, cellular response to externd and
interna1 stimuli whose purpose is to eliminate unwanted cells while preventing damage to
surrounding celIs or tissue (6). Regulation of apoptosis is achieved by several gene
products as well as by exulnsic factors. Apoptosis is often associated with the cross-
linking between Fas antigen (Fas or CD95) and its physiologie ligand (FasL or CD95L)
through cell-ce11 interactions (7). These interactions are often implicated in the regulation
of inflamrnatory ce11 death, particularly the lymphocytes (8). This pathway can be
inhibited by the soluble fonn of Fas molecule (3s) (9).

The Bcl-2 gene family regulates the apoptotic process through the balance of pro-
apoptotic (Bax, Bcl-xS) and anti-apoptotic products (Bcl-2, Bcl-xL) (10). Several
cytokines can also affect apoptosis. For example, interleukin-2 (IL-2) c m inhibit
inflarnrnatory ce11 apoptosis (1 1). IL-2 is increased in the BAL fluid of HP patients and
could therefore contribute to decrease lymphocyte apoptosis (12).
Another mechanism implied in lymphocytes swvivai of the lymphocytes is the
B7-CD28 costimulation pathway- CD28, a molecule camied by the lymphocytes, is a
ligand for B7-1 and B7-2 receptors on antigen presenting ce11 (APCs) like the alveolar
macrophage (13). CD28 stimulation increases the expression of Bcl-xL and IL-2
production by lymphocytes (14). We recently showed that aiveolar macrophages of
patients with active HP have an up-regulated expression of B7 molecules (15). Moreover.
in an animal mode1 of HP, a treatment with the fusion protein CTLA4-Ig, a high affinity
ligand for the 87 receptor without the effect of costimulation, decreases pulmonary
lymphocytosis in mice exposed to the HP antigen (16). Thus, B7-CD28 costimulation
pathway could contribute to lymphocytes accumulation in HP by an up-regdation of
certain lymphocyte specific anti-apoptotic factors.
Pulmonary lymphocytes are not oniy increased in HP but are also highly activated
and reside in a milieu containing high levels of anti-apoptotic cytokines. We hypothesize
therefore that lymphocyte accumulation could result from decreased apoptosis. The
present study was done to quantify the apoptosis of lung lymphocytes recovered from
BAL of normal subjects and from HP patients and characterize this process.

3.4 Materials and Methods
Subjects. Thirty nine patients with non treated HP and 40 non-smoking normal 0
subjects (not exposed to HP antigens) were studied. The diagnosis of HP was based on
previously published criteria which include exposure to an appropriate environment, the
presence of inspiratory crackles on physical examination, intestitial infiltration on chest
X rays, dtered lung functions, and an abnomai number of lymphocytes in
bronchoalveolar lavage (BAL) (>3O % lymphocytes) (1). BAL was done in al1 patients
as part of their clinical diagnostic evaiuation. Our Institution's Ethics Committee
approved the snidy and al1 subjects signed an infonned consent form.
BAL. BAL was obtained by standard bronchoscopy. For the patients, a total of 300 ml of
sterile 0.9% saline solution (warmed at 37°C) was instilled in the middle lobe or the
lingula in 50 mi aliquot with a fiberoptic bronchoscope. Since normal subjects have fewer
BAL lymphocytes, they had a double lavage (240 mI in right middle lobe and 240 ml in
the lingula). Each instilled aliquot was aspirated and the recovered volume determined.
BAL cells were washed with HBSS solution, resuspended in RPMI 1640 medium
(Canadian Life Techndogy, Burlington, Ont.) supplemented with 10% fatal bovine
serum (FM) and 1% penicillin-streptavidin, and counted with a hemacytometer. Ce11
viability was done using trypan blue exclusion dye. BAL cells viability ranged between
90% to 99% (average of 97.2%). Differentiai counts for the different inflarnmatory cells
(lymphocytes, macrophages, neutrophils and eosinophils) was perfonned using a glass
cover technique (17) stained with Diff-Quik (Baxter Diagnostics, Mississauga, Ont). The
isolation of BAL Iymphocytes was done using the standard method of nylon-wwl

column (18). Using this technique, an average of 92 % pure lymphocytes was recovered
(range between 85 % to 95 '$6).
Annexin Vfpropidium iodine (PI) assays. The BAL cells were stained using Apoptosis
Detection kit ( M D System, Minneapolis, Min) according to the manufacturer instruction
with minor modifications. Briefly, celis were washed in phosphate buffered saline (PBS)
(pH 7.4) and suspended at a concentration of 106 cellslml. One hundred microliters of
suspension (10' cells) were added to a 5-ml cytometry tube with 2 pl of fluorescein-
conjugated annexin V and 2 pl of propidium iodine (PI). The mixture was gently
vortexed and incubated for 15 minute at room temperature. At the end of the incubation,
300 pl of binding buffer (IOmM HEPESNaOH (pH 7.4) + 140rnM NaCl + 2.5mM
CaC1,) were added to stop the reaction. Cytometry anaiysis of the cells was done
immediately using an EPICS" ELïTE ESP flow cytometer (Beckman-coulter. Miami,
FL). The appropriate controls were done. Cells that were annexin positive and PI
negative were counted as apoptotic.
Analysis of in situ DNA fragmentation. Paraformaldehyde-fixed air-dned cytospin
preparations were stained by the terminal deoxinucleotidyl transferase (TdT)-mediated
dUTP nick end labeling technique (TUNEL). For the assay, the cytospin preparation was
fixed 30 min in fresh solution of 4% paraformaldehyde in PBS (pH 7.4). The slides were
then washed twice with PBS and incubated in a permeabilisation solution (0.1% Triton
X- LOO in O. 1% sodium citrate) for 2 min on ice (4°C). After another lavage with PBS, the
area around the sample was dried and a drop of 20 pl of enzyme solution (0.3 U rTdT

(Canadian Life Technologies), 5x10''~ M biotin-16-dUTP (Roche Diagnostics Canada,
Laval, Que.), 0.1 M potassium cacodylate (pH 7.2), 2x10" M C0Cl2, 2x10' M DTT) was
added and a cover slip was placed on the sample. The preparation was incubated for 60
min at 37°C in a humidified chamber. After three lavages in PBS, 13.5 nM of conjugated
fluorescent dye sueptavidine-Aiexa 4 8 P (Molecular probes, Eugene, OR) was added in
a drop of 20 pl and kept in the dark for 20 min at 4°C. ProlongTM Antifade kit (Molecular
probes, Eugene, OR) was used (according to the manufacturer protocol) to mount the
slides and to avoid fluorescence quenching. For negative control, rTdT was replaced with
enzyme buffer and, for positive control, a pre-incubation with 5 U of Dnase 1 (Arnersham
Pharmacia Biotech, Piscataway, NJ) was done immediately before the incubation with
enzyme solution. The count was done using a epifluorescence microscope (Nikkon
Eclipse 6600) with the appropriate filter (Nikkon filter B2-A, ex: 450490, em: 505). A
minimum of 300 cells was count for each siide. Two criteria were used to qualify a
positive ce11 : bright green fluorescence on nuclei and morphologic feature of a ce11 in
apoptosis.
Antibodies. Bcl-21100 (mouse IgGi anti-human Bcl-2-FiTC), Dx2 (mouse IgGl, anti-
human CD95-PE (Fas)), NOK-1 (mouse IgGi ami-human CD95L-PE (FasL)), 2H12
(mouse anti-human Bcl-xL), HIT3a (mouse IgG1 anti-human CD3-PerCP) and the
isotype control (mouse IgGi-FFTC, mouse IgGI-PE and mouse IgGi-PerCP) were
purchased from Phanningen (San Diego, CA).

Flow cytometry for Fas, FasL and Bcl-2. 2.0~106 BAL ccils were resuspended in 200
pl of PBS + bovine serum aibumin @SA) 1%. A batch of cells (5xld ceW50 ~ 1 ) was
labelle with 2 pl of anti-CD3 or 2 pl of isotype control and put on ice (4'C) for 45 min.
The same protocol was used to label a sample of the ceUs 1.0~106 with anti-CD95 (2 VI)
and mti-CD95L (2 pl) and spccific isotype conmls. The remaining celis (5xld a U in
50 pl) were permeabilized using a commercial kit (Perm Kit, Pharmingen) M o r e the
incubation with anti-Bcl-2 antibodies and its specific isotype control (2 pl of the
antibodies, 45 min on ice).
Evaluation of ,Fas in BAL fiuid. The quantification of the soluble form of Fas in BAL
fluids was done by a sandwich type ELISA using OptElA Human Fas kit (Pharmingen)
according to the protocol provided by the manufacturer.
Preparation of the samples for Western hl& 1x10~ BAL Lymphocytes purified on
nylon-wooi columns were resuspended in 20 pl of lysis buffer (10mM Tris-HCl (pH 7.4).
150rnM NaCI, 2mM EDTA, 2% viton X-100, 1% of mixture of protease inhibitor (2 mM
EDTA, 2 mM PMSF, Ip@d of aprotinin, lpglml of leupeptine, aü from Sigma)) and
?Op1 of 2x loading buffer (200 rnM Tris HC1 (pH 6.8), 20% glycerol, 2% SDS, 0.00058
bromophenol blue, 10% Zmercapto-ethanol). The mixture was denatured by a passage at
IO0 O C during 5 min and then centrifuged at 10 OOOg for 1 min in order to precipitate
phospholipids present in tbe samples. The supernatant (40~1) was fiozen at -70°C until
used.

Western blot for Bcl-XI,. The samples (106 purified BAL lymphocytes) were sepmied
on a 12 % polyacrylamide gel (SDS-PAGE) and transferred on a polyvinylidene
difluonde (PVDF) membrane. The blot was blocked for 1 h at m m temperature with
blocking buffer (5% nonfat dry milk, 0.05% tween 20 in tris buffered saline (TBS)) and
probed for 1 h with a monoctonal antibody to Bc1-xi. used at a dilution ot' 1: 1000 in the
blocking buffer. The secondary probe (l:lûûû, in blocking buffer) with a goat IgG anti-
mouse IgG HRPO (Cedariane, Ont) was detected by cherniluminescence using ECL
(Arnersham). BAL alveolar macrophages were used as positive control. The
densitornetric anaiysis was performed using Scion image (beta 3b,
htt~:llwww.scioncom.corn~.
Sîatistical analysis. Results were expressed as mean value f SEM. Al1 data were
analyzed using t-test except for Fig. 4A and 4B where a Spearman rank correlation test
was used and for Fig. 6B where a Mann-Whitney U test was applied. Ail reported P-
value were declareci significant atp < 0.05.

3.5 Results
BAL characteristics of HP patients and normal subjects.
A total of 39 HP patients and 40 normai subjects tmk part in the snidy (table 1).
There was no difference between the proportion of men and women in the two groups of
subjects. However. the average age of the HP subjects was higher than that of the normal
subjects. The percentage and number of the various BAL cells for each group of subjects
was similas to that previously published (15) (table 1).
BAL lymphocytes apoptosis.
The BAL lymphocytes of HP subjects were less Annexin+/PI- than those of
normal subjects (37.4% I 3.4 (n=lS) vs 56.5% I 5.5 (n=15) respectively, p d . 0 0 4 , Fig.
IA). To confirm this result, cytospins of BAL cdls were tested by TUNEL in a subgroup
of our subjects. This technique allows the quantification of cells in their final phase of
apoptosis, at the stage where the genomic DNA breaks-up ( 19). Fewer BAL lymphocytes
of HP patients were TUNEL positive than those from hedthy subjects (0.4% 2 0.1
(n=9)vs 1 .O% I: 0.2 (n=7), p=0.033, Fig 1 B).
No correlation was found between the nurnber of apoptotic lung Iymphocyte
(tested with AnnexidPI and TUNEL wsay) and the age of the subject in the study
(patient and control) (Fig 1C).
F m receptor.
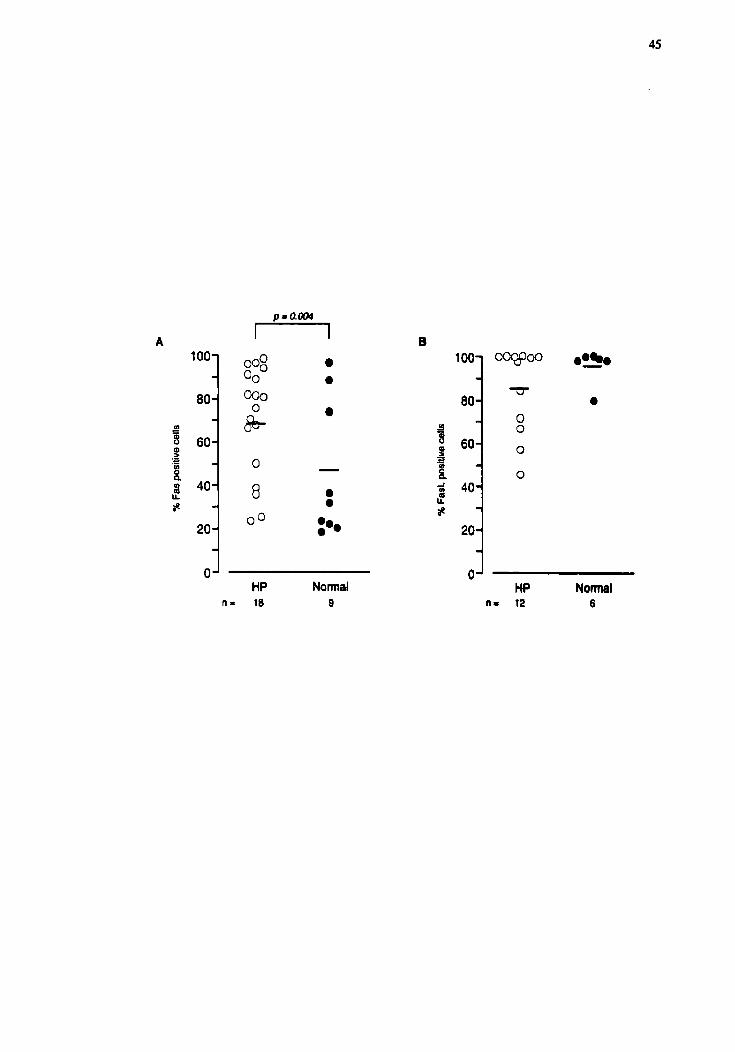
As shown in the Fig.2A, the percentage of positive lymphocytes for Fas was
approximately 50% higher in HP patients han in normai subjects (68.8 î 5.4 (n=18) and
46.7% r 9.0 (n=9) respectively, pd.004).

The proportion of positive lymphocytes for FasL was also determined. As show
in the Fig. 2B, no signifiant difference was found in the proportion of BAL lymphocytes
positive for FasL (84.9 î 5.7 (n=12) and 95.6 î 3.2 (n=6), @,23) respectively for HP
patients and normal subjects.
SolrtbIe Fus.
The quantity of sFas present in the BAL fluid was higher by more than 3-folds in
HP patients cornpared to the normals (80.5 pghl I 8.5 (n=23) vs 23.2 pglrnl I 3.1
(n=14), p=0.0001, Fig 3). A positive correlation was obse~ed between the percentage of
BAL 1 ymp hoc ytes and the concentration of $as in the BAL fluid (R' = 0.38, p=0.000 1,
Fig 4A). A negative correlation (R' = 0.24, p=0.0058, Fig 4B) was found between the
percentage of lymphocytes and the percentage of the cells in apoptosis (AnnexinPI test).
However no correlation (data not show) was found between the concentration of ,Fas and
the percentage of cells in apoptosis (AnnexinPI test).
Bcl-2 prorein and intracellular concentration of Bcl-xl in BAL lymphocytes.
In order to further explain the reduction in the apoptosis of pulmonary
lymphocytes within the framework of HP, the proportion of BAL positive lymphocytes
for the anti-apoptotic protein Bcl-2 was obtained by flow cytometry and the expression of
Bcl-xL, another anti-apoptotic protein, was quantified by Western blot.
No significant difference was found in the proportion of positive lymphocytes for
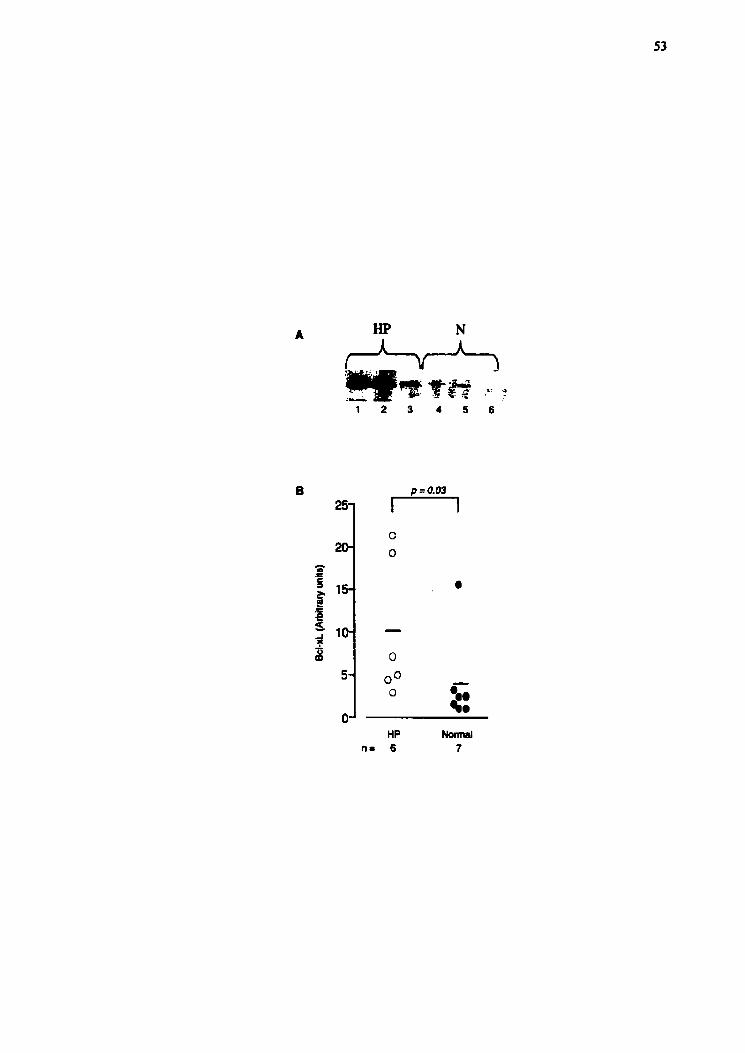
Bcl-2 protein (74,4.% I: 7.71 (n=6) vs 93.1% * 2,8 (n=5), p=0.066, Fig. 5). However, the
intracellular level of Bcl-xL (Fig. 6A,B) was increased by more than 2.5 times in the

BAL lymphocytes of HP compared to N (9.9 t 3.3 (n=6) vs 3.8 * 1.9 (n=7), pd.03, Fig.
6B). Fig 6A show representative results on western blot.

3.6 Discussion
This study shows, by two different techniques, that the apoptosis of BAL
lymphocytes is decreased in HP comparai to BAL lymphocytes of normal subjects. The
persistent lymphocytosis characteristic of HP could therefore be explained, at Ieast partly,
by a reduction in the apoptosis of the pulmonary lymphocytes.
As mentioned in the introduction, there are tfuee mechanisms likely to influence
lymphocyte persistence in HP: Iwal proliferation, an increase in cellular recniitment, and
a reduction in the apoptosis of the pulmonary infiammatory cells. Previous reports
demonstrated an increased recruitment of the lymphocytes in HP. The mRNA of
RANTES (2)- MIP-1-alpha (2) and IL-8 (20) are upregulated in the BAL lung cells of HP
patient, t h e potent chemokine for the recruitment of lymphocytes. In contmt to
alveolar macrophages and surfactant of normal subjects, alveolar macrophages and
surfactant of HP patients do not inhibit the activation of pulmonary T-cells (3),
suggesting that these ceIls are more proliferative in HP. Moreover, BAL lymphocytes of
HP patients express more markers of activation like the p75 chah (receptor associated
with IL-21, the VLA-1 and the HLA-Dr antigen, an index of the state of activation and
proliferatian of lymphocytes (4). The results of the current study add another potential
mechanism to explain the alveolar accumulation of lymphocytes in HP. Our results
cannot quantify the relative importance of apoptosis in the maintenance of pulmonary
inflammation in HP but suggest it could be a signif~cant one. Although al1 tests were not
done on the BAL ceIls of al1 subjects of each groups, the marked differences between the
groups is clearly shown by the studies performed.

The presence of fewer apoptotic cells for lymphocytes of which there is an
increase in the number that are Fas positive at Fust seems contradictory. However, this
resuit is in agreement with the data of Agosrini and al. (21) who showed that the Fas is
expressed at a lower level on the surface of pulmonary lymphocytes of normal subjects
compared to those from infiammatory alveolar diseases like HP and sarcoidosis.
However, this result seems to be paradoxal since, as shown in the figure 1 (A and B),
BAL lymphocytes of HP patients were less apoptotic than the BAL lymphocytes of
normal subjects, the opposite of what would be expected in the presence of more Fas.
The anticipated higher percentage of apoptosis assosiated with Fas expression
was probably overcompensated by the increased concentration of the soluble Fas and by
the increased intracellular Bcl-xL. The ligation of FasFasL is a c e k e l l interaction often
implicated in the initiation of apoptosis. $as binds to FasL therefore limiting the number
of this pro-apoptotic receptor on activated lymphocytes (22). An increase of ,Fis
molecules was expected in the presence of a higher nurnber of BAL lymphocytes since
these cells are major producers of this molecule. An increase of 3 a s is an inflammatory
mechanism by which the immune system cm prolong the viability of lung lymphocytes;
this is what seems to happen in HP. This affirmation is support by a positive corelation
seen between the percentage of BAL lymphocytes and the concentration of ,Fa in the
BAL fluid (Fig. 4A).
We cannot elirninate the possibility of a failure in the initiation of the apoptosis by
the FasFasL. Other studies are necessq to verify if the Fas/FasL pathway is
dysfunctionai by other mechanisms. For example, an overproduction of the Bcl-2 protein
in lymphocytes in culture (of Jurkat type) inhibits the Fas-induced apoptosis (23). Also an

overproduction of Bcl-xL in MCF7 cells (thurnorai epitheliai cells) expressing Fas,
makes these cells resistant to Fas induced apoptosis (24).
Although we can confirm that puimonary lymphocytes in HP express more Bcl-
xL than normal ones, we do not know if the intracellular level of Bcl-2 is increased. Since
our results only indicate a percentage of positive lymphocytes, we canot say if the
intracellular levels of Bci-2 are increased in the BAL lymphocytes of HP patients. A
positive ce11 cm contain an unknown quantity of Bcl-2 (limit of detection of the test to a
maximum concentration). Western blot technique will be needed to further evaluate the
real implication of the Bcl-2 protein in the reduction of apoptosis in HP.
The increase in the intracellular levels of Bcl-xL of the BAL lymphocytes in HP
could be related to the increase in costimulation via the B7-CD28 pathway. Indeed. as
previously mentioned, this pathway is unregulated in HP. An increase in costimulation
B7-CD28 is known to increase resisrance of lymphocyte apoptosis, this via the
intracellular increase of the Bcl-xL protein and by the increased in the expression of IL-2,
a survival factor for these cells (14).
An interesting question raised by the results obtained in this study is whether the
reduction in the apoptosis of the BAL lymphocytes in HP is due to an intrinsic
modulation of the cells themselves andor to the pulmonary microenvironment from
where they were collected. It is possible that other anti-apoptotic factors, other than Bcl-
xL and ,Fa, present in the pulmonary environment, could participate to decreased
lymphocytes apoptosis. For example, it is known that IL-2 is an anti-apoptotic factor for
lymphocytes and that the concentration of this cytokine is increased in the BAL fluid of
HP patients (12). Moreover, the HP pdmonary lymphocytes proliferate in a dose-

dependent fashion during stimulation with IL-2 (12). Thus, this result suggests that IL-2
plays a role in HP by producing a stimulus for the accumulation of lymphocytes and iheir
persistence in the lung.
Further studies are needed to elucidate the mechanisms involved in the control of
lymphocyte survival in HP. The effect of the microenvironment would require in vitro
studies looking at the effect of anti-apoptotic cytokines on BAL lymphocytes. For
exarnple, purified BAL lymphocytes of HP patients and controls subjects could be
cultured with Fas agonist and IL-2 in order to evaluate these different stimulations on the
lymphocyte apoptosis in time. Our hypothesis is that in HP a Fas agonist stimulation
would have no effect on lymphocyte apoptosis while iL-2 would inhibit this p r o g r m e d
ceIl death. A better comprehension of these phenornena could be useful to understand
whether an intrinsic factor in the ce11 itself andlor the pulmonary microenvironment are
responsible for the conuol of lung inflarnmatory ce11 accumulation seen in HP or both
similar disease.
3.7 Conclusions
This study shows that a decrease in lymphocyte apoptosis may be involved in the
dveolar lymphocytosis seen in HP. Potential mechanisms for the increased survival of
these lymphocytes include high levels of Zas, enhanced Bcl-xL and the presence of anti-
apoptotic cytokines in the lungs.

We thank Dr Guy Tremblay and Dr Michel Laviolette for helpful discussion.
This work was support by the medical research council of Canada Grant (MT-15170) and
by the J. D. Bégin Foundarion.

3.9 References
1. Cormier, Y., and Schuyler, M. 1992. Part M : interstitial lung disease. In
Hypersensitivity Pneumonitis Texbook of Pulnionary Medecine, Vol 2, R.C. Bone ed.,
Mosby-year book, St Louis. 1.
2. Oshima M., Maeda A., Ishioka S., Hiyama K., Yamakido M. 1999. Expression of C-
C chemokines in bronchoaiveolar lavages cells from patient with granulomatous lung
diseases. Llrng. 177:229-240.
3. Israël-Assayag E., Cormier Y., 1997. Surfactant modifies the lymphoproliferative
activity of macrophage in hypersensitivity pneumonitis. Am. J. Physiol. 273:1258-
1264.
4. Trentin L., Migone N., Zarnbello R., Di Celle P.F., Aina F., Feruglio C., Bulian P.,
Masciarelli M., Agostini C., Cipriani A., Marcer G., Foa R., Pizzolo G., Semenzato
G. 1990. Mechanisms accounting for lyrnphocytic alveolitis in hypersensitivity
pneumonitis. J. Immunol. l45:2 147-2 154.
5. Hetts S.W. 1998. To die or not to die. An overview of Apoptosis and its role in
disease. JAMA. 279:300-306.
6. Thompson C.B. 1995. Apoptosis in the pathogenesis and treatment of disease.
Science. 267: 1456- 1462.
7. Nagata S., Goistein P. 1995. The Fas death factor. Science. 267: 1449-1456
8. Ju S.T., Panka D.J., Cui H., Ettinger R., El-Khatib M., Sherr D.H., Stranger B.Z.,
Marshak-Rothstein A. 1995. Fas(CD95)EasL interactions required for programmed
ce11 death after T-ce11 activation. Nature. 65 13:444-448.

9. Cheng J., Zhou T., Liu C., Shapiro J.P., Brauer M.J., Kiefer M.C., Barr P.J., Mountz
J.D. 1994. Protection from Fas-mediated apoptosis by a soluble form of the Fas
molecule. Science. 5 15 : 1759-1762.
10. M. Adams J., Cory S., 1998. The Bcl-2 protein farnily : arbiters of ceil sunrival.
Sciences. 28 1 : 1322- 1326.
11. Boise L.H., Minn A.J., Thompson C.B. 1995. Receptors that regulate T-ce11
susceptibility to apoptotic ce11 death. Ann. N. Y. Acad. Sci. 766:70-80.
12. Dakhama A., Israël-Assayag E., Cormier Y. 1998. Role of interleukin-2 in the
developrnent and persistence of lymphocytic aiveolitis in farmer's h g . Eur. Respir.
J. 11:1281-1286.
13. Harris N.L., Ronchese F., 1999. The role of B7 costimulation in T-ce11 irnrnunity.
Immirnol. Cell. Biol. 77:304-3 1 1.
14. Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson
C.B. 1995. CD28 costimulation can promote T-ce11 survival by enhancing the
expression of Bcl-xL. Immuniry. 3:87-98.
15. Israël-Assayag E., Dakhama A., Lavigne S., Laviolette M. and Cormier Y. 1999.
Expression of costirnulatory molecules on alveolar macrophages in hypersensitivity
pneumonitis. Am. J. Respir. Crit. Cure Med. 159 : 1830- 1834.
16. Israël-Assayag E., Fournier M. and Cormier Y. 1999. Blockage of T-ctll
costimulation by CTLA4-Ig inhibit lung inflammation in mutine hypersensitivity
pneumonitis. J. Immunol. 163:6794-6799.

17. Laviolette M., Camau M., Coulombe R. 1988. Bronchoalveolar lavage ceiI
differential on microscope glass cover. A simple and accwate technique. Am. Rev.
Repir. Dis. 138:45 1-457.
18. Eisen S.A., Wedner H.J., Parker C.W. 1972. Isolation of pure human peripheral blood
T-lymphocytes using nylon wool columns. Immunol. Commun. 157 1-577.
19. Gavrieli Y., Sherman Y., Ben-Sasson S.A., 1992. Identification of programmed ce11
death in situ via specific labeling of nuclear DNA fragmentation. J. Cell. Biol.
1 l9:493-5O 1.
20. Gudmundsson G., Hunninghake G.W., 1999. Repiratory epithelial cells reiease
interleukine-8 in response to a thermophilic bacteria that causes hypersensitivity
pneumonitis. Exp. Lung. Res. 252 17-228.
2 1. Agostini C., Zambello R., Sancetta R., Cerutti A., Milani A., Tassinari C., Facco M.,
Cipriani A., Trentin L., Semenzato G. 1996. Expression of tumor necrosis factor-
receptor Superfamily members by lung T lymphocytes in interstitial lung disease. Am.
1. Respir. Crit. Cure Med. 153: 1359-1367.
22. Tanaka M., Itai T., Adachi M., Nagata S., 1998. Downregualtion of Fas ligand by
shedding. NUI. Med. 1:3 1-36.
23. Kawahara A., Kobayashi T., Nagata S. 1998. inhibition of Fas-induced apoptosis by
Bcl-2. Oncogene. 17:2549-2554.
24. Medema J.P., S c a d i C., Krammer P.H., Peter M.E. 1998. Bcl-xL acts downstream
of caspase-8 activation by the CD95 death-inducing signaling cornplex. J. Biol.
Chem. 273:3388-3393.

Table 3.1 BAL characteristics of HP patients and nomals subjects.
Subjects
HP
Normal
n
39
40
Sexe (M-F)
25-14
26-14
Average ags
48.85
25.75
% lymphocytes
56.58
9.89
Celldml (x103)
734.80
102.11
% macrophages
34.1 9
87.89
% neutrophils
7.1 1
1.65

Figure 3.1 The BAL lymphocytes of HP patients (n=18) were Less AnnMPI- (smailer
population in apoptosis) than those of N subjects (n=15, @.004) (A). The results
obtained with the TUNEL test. [n the final phase of apoptosis assessed by this method,
BAL lymphocytes in HP (n=7) have less positive nuclei for DNA fragmentation thm
those of the conuol subjects (n=9, pr0.033) (B). No correlation was found between the
age of the patient en the percentage of lymphocyte apoptosis (using annexin/PI assay)
(C).
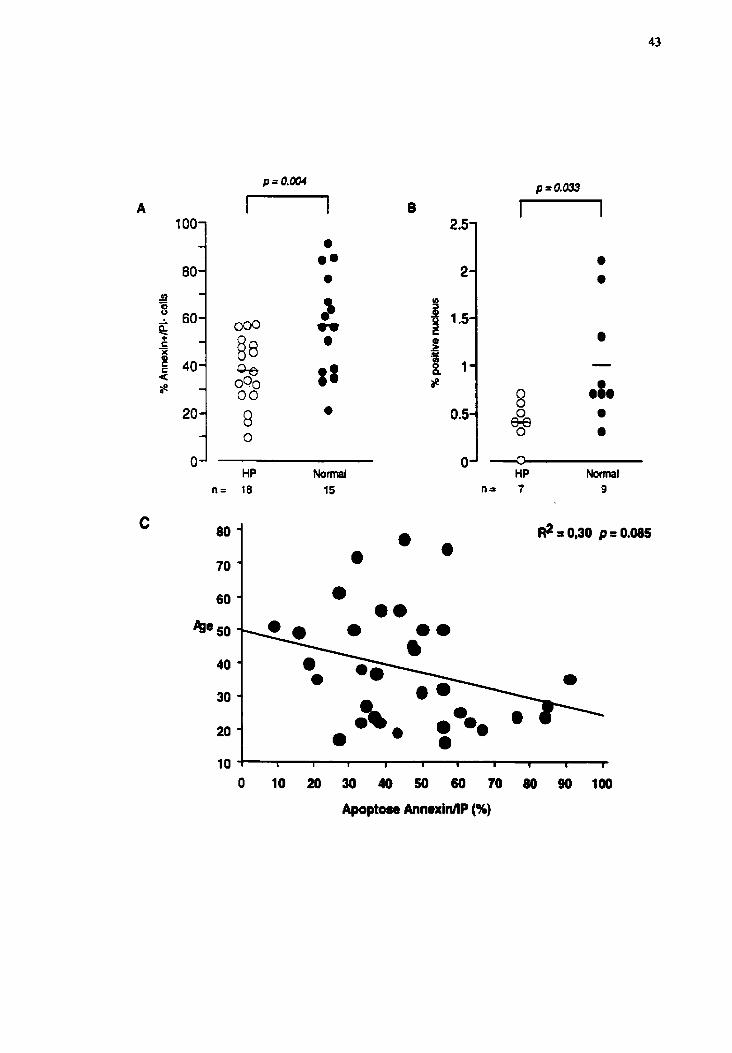

Figure 3.2 Percentage of BAL lymphocytes positive for the surface receptors Fm (A) and
FasL (B). The proportion of positive Iymphocytes for Fas was approximately 50% higher
in HP (n=18) than in N (n=9, pd.001) (A). No signifiant difference was noted in the
percentage of positive lymphocytes for FmL between the two groups (n=12 for HP
patient and n=6 for control subject) (B).
0J HP Normal
n a 18 9 HP Normal
n r 12 6

Figure 3.3 Quantification of ,Fas in BAL. BAL fluid ,Fas level was much higher in HP
patient (n=23) than in N subjects (n=14, p=0.0001).


Figure 3.4 Correlation between BAL lymphoçytosis and the concentration of ,Fas in the
BAL fluid {A) and (B) the percentage of annexintP1 cells. A positive correlation exist
between the percentage of BAL lymphocytes and the concentration of ,Fa in the BAL
fluid (A) (R' = 0.38, @.0001). A negative correlation is observed between the
percentage of BAL lymphocytes and the proportion of apoptotic BAL lymphocytes (B)
(R' = 0.24, p=û.006) suggesting that $as have a significant role to play in the reduction
of BAL Iymphocyte ripoprosis in HP.
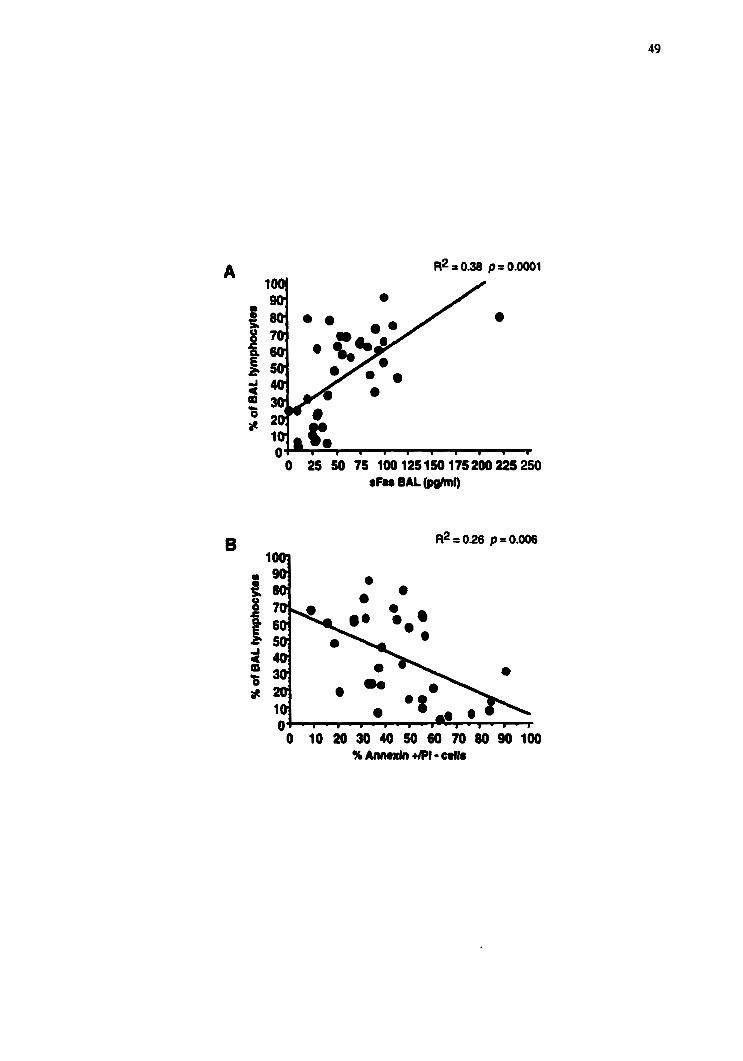
O 25 50 75 100125150175200225250 sFn BAL -1)

Figure 3.5 Proportion of BAL lymphocytes positive for Bcl-2. No significant difierence
was noted between the HP patients (n=6) and N subjects (n=S) for the percentage of
pulmonary lymphocytes positive for the Bcl-2 protein.


Figure 3.6 Quantification of the Bcl-xL protein in the BAL lymphocytes. The protein (A
representative result of Western blot are shown in A. for. The bands were then evaluated
by densiometry software (Scion hage)(B). A significantly higher concentration was
found in HP patients (n=6) than in N subjects (n=7, w.03).


4.1 Mise en situation
Dans le chapitre 3, il est démontré qu'une inhibition du processus ripoptotique
normal des lymphocytes est un des mécanismes impliqués dans l'accumulation des
lymphocytes dans 1'AA.E. En d'autres termes, le maintien de l'inflammation locale est
dû, en partie, à une mauvaise élimination des lymphocytes. Tel que vu dans le chapitre 3,
malgré une diminution de l'apoptose des lymphocytes dans L'AAE, ces cellules du LBA
de patients surexpriment le Fas. Cette surexpression devrait normalement s'associer à une
augmentation de l'apoptose des cellules en question. Ce paradoxe peut s'expliquer, en
partie, par une augmentation du Fas dans le liquide du LBA de ces patients. Le Fas,
inhibe la liaison FasIFasL, processus essentiel pour initier l'apoptose. ii est toutefois
posssible que le sentier FasIFasL soit disfonctionnel.
Ce chapitre est axé sur la clarification des mécanismes impliqués dans ce
dérèglement de I'apoptose dans I'AAE. L'objectif ultime est de déterminer si la
diminution de la mon cellulaire des lymphocytes obtenus du LBA dans I'AAE est due à
une défaillance intrinsèque des cellules elles-mêmes et/ou au micro-environnement
pulmonaire d'où elles proviennent (riche en facteurs anti-apoptotiques).
Une façon de tester cette hypothèse est de stimuler les lymphocytes du LBA avec
de l'IL-2 (facteur ami-apoptotique pour les lymphocytes T), de regarder l'apoptose dans
le temps et de comparer les données à des lymphocytes non stimulés du LBA de sujets
sains et de patients AAE. De la même façon, la fonctionnalité du sentier Fas/FasL sera
évaluée par une stimulation avec un anticorps anti-Fas (jouant le rôle du FasL) et
l'apoptose sera évaluée dans le temps avec les mêmes comparaisons que pour l'IL-2.
1.2 Matériel et méthodes
4.2.1 LBA
Voir chapitre 3 (section BAL dans « Materiais and Methods m).

4.2.2 Purification des lvm~hocvtes T sur colonne laine-nvlon et culture cellulaire
Les cellules totales du LBA ont été mises en culture dans du RPMI l m , 1 heure
à 37°C. 5% COz dans des plaques de culture 24 puits (Falcon) 5 une concentration de 106
macrophages/ml. Le but de cette étape était de faire adhérer les macrophages et ainsi de
récolter les cellules non adhérentes qui sont en majorité des lymphocytes. Ensuite, les
plaques ont été lavées trois fois avec le milieu de culture et les cellules non adhérentes
ont été passées dans une colonne laine-nylon. Après 1 hre incubation à 37°C les
colonnes ont été lavées avec 30 mi de RPMI, afin de recueillir les cellules non adhérentes
(lymphocytes T) de la colonne. Les lymphocytes T ainsi purifiés (pureté > 95%) ont été
ensuite incubés dans un système de culture cellulaire (toujours dans le RPMI 1640)
schématisé ici-bas, afin d'évaluer leur potentiel apoptotique en présence de différents
agents.
Lymphocytes seuls
Lymphocytes + L 2 * 1 Incubation O hre, 4 hres, 24 hres.
Lymphocytes + anti-Fas (2pg/mi)**
* La concentration optimale de stimulation était de lnglml.
** Concentration recommandé par le manufacturier (Phanningen).
Aux divers temps d'incubation, un échantillon cellulaire a été prélevé et les taux
d'apoptose ont été mesurés par deux tests : 1) TUNEL et 2) essai mesurant l'activité de la
caspase 3.
4.2.3 TUNEL
Voir chapitre 3 (section Analysis of in situ DNA fragmentation dans Materials and
Methods »).

4+2.4 Activité de la casDase 3
Un total de 2,5x1oS lymphocytes T purifiés a été resuspendu dans un tampon de
lyse à une concentration de 10x10~ ceiiuledd (25 pl). Le lysat a ensuite été déposé en
entier dans un des puits d'une plaque à 96 puits. Ensuite 200 pl de e p n HEPES IX a
été ajouté. Finalement, 5 pi de substrat de la caspase 3 a été ajoutés (Ac-DEVD-AMC).
Le contrôle suivant a éte effectué en duplicata : tampon HEPES (200 pi) + tampon lyse
(25 pi) + substrat de la caspase 3 (5 pi). On incube le tout 1 heure 37°C dans le noir.
Après l'incubation, la plaque a éti lue en fluoroscopie avec le filtre d'excitation 380 nM
et le filtre d'émission entre 420-460 nM. Les vdeurs obtenues ont été soustraites de la
valeur du puits contrôle.
Le test statistique de « student » a ét6 utilisé pour les figures 4.1 et 4.3. Dans le
cas de la figure 4.1, Le test est basé sur des données non-pairées (unpaired t-test) et pour
la figure 4.3, des analyses pairées et non-pairées ont été effectuées (paired t-test,
rrnpaired t-test). Les analyses ont été faite avec le logiciel Stat View 5.0.
Pour la figure 4.2, les courbes ont été comparées statistiquement par un test
d'mdyse de profil. Le test statistique a été effectué avec le logiciel SAS par le
biostatisticien de notre centre de recherche (Serge Sirnard). Toutes les données sont
présentées en moyenne h SEM.
4.3 Résultats
4.3.1 Quantification de l'amtose (test TUNEL) des lvm~hocytes T en culture non
stimulés.
Les lymphocytes T pulmonaires isolés des deux groupes de sujets à l'étude ont été
mis en culture sans stimülation. Les cellules ont été récoltées aux temps 0, 4 hres et 24
hres. L'apoptose a été quantifiee à ces différents temps par le test de TUNU.

Au temps 0, c'est-à-dire au moment où les cellules sont obtenues du LBA, ii y a
significativement moins de cellules ayant des noyaux TUNEL positifs chez les sujets
AAE que chez les N (voù figure 3.1 8). Cependant, lorsque les lymphocytes T de I'AAE
sont placés in vitro, en culture, on assiste à une apoptose massive de ces cellules. Comme
on peut le voir à la figure 4.1, après 4 hres de culture, il y a significativement plus
d'apoptose chez les lymphocytes T des sujets AAE que chez ceux des sujets contrôles
( 1 1'33% vs 4'38%' p=0,005). L'écart entre AAE et normaux augmente davantage après
24 hres de culture (4 1'17% vs 12.93%' p=0,004, fig.4.1).
4.3.2 Mesure de l'activité de la caspase 3 chez des ivm~hocvtes T ~ulmonaires cultivés
pendant 24 hres sans stimulation.
Les résultats démontrés à la figure 4.1 ont été confirmés par la mesure de
l'activité de la caspase 3 chez ces mêmes lymphocytes. En effet, une tendance
intéressante @=0,09 avec le test d'analyse de profil, Fig 4.2) montre que les lymphocytes
pulmonaires des sujets AAE ont une activité de la caspase 3 plus grande (pour tous les
temps : 0, 4 et 24 h m ) que chez les lymphocytes pulmonaires de sujets normaux.
Cependant, le nombre de sujets dans le groupe des normaux est trop faible pour faire des
statistiques vaiables (n=2).
4.3.3 Effet de I'anti-Fas et de l'IL-2 sur I'apo~tose des lymbhocvtes T cultivés mndant
24 hres (auoutose ~uantifiée par TUNEL).
De la figure 4.3, il se dégage quatre messages. Le premier est similaire à ce que
l'on voit à la figure 4.1, c'est-à-dire qu'il y a significativement plus d'apoptose après 24
hres chez les lymphocytes pulmonaires non stimulés des patients AAE que chez les
lymphocytes des sujets contrôles. Le deuxième est qu'une stimulation à I'anti-Fas n'a
aucun effet sur les taux d'apoptose de lymphocytes pulmonaires des sujets AAE. Ceci
laisse présager une défaillance dans l'initiation de I'apoptose par le sentier Fas/FasL. Le
troisième message est que l'IL-2 diminue les taux d'apoptose des lymphocytes
pulmonaires de I'AAE. La tendance est intéressante malgré le fait qu'elle n'est pas

significative (p=0,07). Ce demier résultat suggère fortement un rôle de cette cytokine in
vivo. Ensuite, le quatrième message est que la stimulation à l'IL-2 ramène les taux
d'apoptose des lymphocytes des patients AAE au même aiveau que ceux des sujets
contrôles stimulés Li l'IL-2, ce qui sugghe que les lymphocytes des patients AAE sont
dépendants de L'IL-2 pour leur survie.

T IAAE
Fieure 4.1 : Dénombrement de la quantité de lymphocytes pulmonaires cultivés sans
stimulation et qui sont positif pour le test de TUNEL. Les lymphocytes du LBA des
patients AAE et des sujets normaux ont ét6 mis en culture pendant 24 heures. Les cellules
ont ensuite été récoltées aux temps 0, 4 hres et 24 hres et l'ampleur de I'apoptose a été
mesurée par le test de TUNEL. Au temps O, il y avait significativement plus de
lymphocytes (exprimés en moyenne f SEM) ayant des noyaux TUNEL positifs
provenant des sujets normaux que des patients AAE @ = 0,03). Cependant, après 4 hres
et 24 hres, la situation est inverse et il y a significativement plus d'apoptose chez les
lymphocytes des sujets M E que chez les sujets normaux. ( p = 0,005 pout T = 4hres et p
= 0,004 pour T = 24 hres).

O 4 24 Temps (Hm)
Fimire 4.2 : Activité de la caspase 3 en fonction du temps, La mesure de l'activité de
la caspase 3 a ét6 faite dans le temps chez les lymphocytes pulmonaires des patients AAEi
et des sujets normaux. Une mesure a été prise aux temps 0 ,4 hres et 24 hres. A tous les
temps, les lymphocytes puIrnonaires des patients AAE ont démontré une activité plus
élevée de la caspase 3. Un test d'analyse de profil rivele une différence presque
significative entre les deux types de sujets et ce, pour tout temps confondus @ = 0,W)
malgré Ie faible nombre de sujets dans le groupe des normaux (n = 2).

AAE O Normaux
Sans Anîi-F as 11-2 stimulation
Figure 4.3 : Effet de I'anti-Fas et de l'IL-2 sur I'apoptose des lymphocytes T
pulmonaires après 24 heures de culture. Tel que déjh présenté à la figure 4.1, sans
stimulation. après 24 hres de culture, il y a davantage de lymphocytes pulmonaires
provenant des patients AAE qui sont apoptotiques comparativement à ceux provenant de
sujets normaux. Également, ce graphique montre qu'une stimulation h l'ami-Fas n'a
aucun effet sur I'apoptose des lymphocytes des sujets AAE (41,17% vs 38'95, p = 0.17).
Par contre, l'IL-2 diminue tes taux d'apoptose chez tes lymphocytes des patients AAE
comparativement à ces mêmes lymphocytes mais non stimulés (41,178 vs 15'94%' p =
0.07). Les taux d'apoptose des lymphocytes AAE stimdés à l'IL-2 sont équivalents aux
taux d'apoptose des lymphocytes pulmonaires des sujets normaux aussi stimulés à l'IL-2.

CHAPITRE 5
CONCLUSIONS GÉNERALES

La première partie de l'étude (objectif 1) a démontré, par deux techniques
distinctes (annexine Vliodure de propidium et TUNEL), que l'apoptose des lymphocytes
du LBA des patients AAE est diminuée. Deux mécanismes sont proposés pour expliquer
cette diminution. Tout d'abord, les quantités de Fas, sont augmentées dans le liquide du
LBA des patients AAE. Cette molécule a la propriété d'inhiber I'apoptose induite par le
sentier Fas-FasL. Ensuite, des concentrations élevées de Bcl-xL ont été retrouvées chez
les lymphocytes T des patients AAE (2'6 fois plus que chez les normaux). Cette protéine
est reconnue pour inhiber l'apoptose des cellules qui l'expriment.
On se rappellera que l ' M E est caractérisée par une lymphocytose persistante.
Deux mécanismes généraux ont été proposés p o ~ expliquer ce phénomène: 1) une
prolifération locale soutenue et 2) une augmentation du recrutement. Les deux premiers
principes ont déjà fait l'objet d'études plus poussées. il a déjà été démontré que la
prolifération lyrnphocytaire dans le poumon, ainsi que l'augmentation de molécules et de
médiateurs pour le recrutement lymphocytaire puisse expliquer, en partie, la persistance
lymphocytaire dans I'AAE. Nous avions posé comme hypothèse que la diminution de
I'apoptose des lymphocytes T pulmonaires pournit être un troisième mécanisme
impliqué. La présente étude apporte donc un nouvel élément dans l'élucidation du
mécanisme de la persistance lyrnphocytaire en démontrant une diminution de l'apoptose
des lymphocytes T.
Tel que mentionné dans l'introduction (Chapitre 1). un rapport a déjà démontré
(sans quantifier) que l'expression du Fas était augmentée à la surface des lymphocytes du
LBA dans I'AAE. Ce résultat a été reproduit et quantifié. Cependant, celui-ci est
paradoxal si l'on tient compte qu'une diminution de l'apoptose a été trouvée chez ces
mêmes lymphocytes. Ce paradoxe est en bonne partie résolu par le fait qu'il y a pIus de
Fas, dans le liquide du LBA des patients AAE. De plus, il a déjà été rapporté chez des
cellules MCF7 (cellules épithéliaies tumorales) exprimant le Fas qu'une surexpression du
Bcl-xL rend ces cellules résistantes à l'apoptose induite par le Fas. il est donc possible
que le même phénomène puisse s'appliquer chez les lymphocytes T du LBA des patients
AAE qui eux aussi surexpriment le Bcl-xL.

La seconde partie de l'étude (objectif 2) a démontré que les lymphocytes T du
LBA des patients AAE, lorsque mis en culture, meurent (par apoptose) plus rapidement
que les lymphocytes des sujets contrôles et cela sans aucune stimulation. Ce résultat a été
quantifié par le test de TUNEL et a été c o n h é grâce à l'essai mesurant l'activité de la
caspase 3. Ainsi, ces données montrent bien l'implication du milieu pulmonaire
relativement au maintien de la survie des lymphocytes T dans 1'AAE.
Le milieu pulmonaire spécifique à 1'AAE est riche en facteurs anti-apoptotiques
de toutes sortes (Fas,, IL-2). Une fois retirés de ce milieu, les lymphocytes hautement
activés meurent rapidement par apoptose. De plus, il a été démontré que les lymphocytes
du LBA des patients AAE ne répondent pas à une stimulation à l'anti-Fas. Le type de
stimulation utilisé est un anticorps anti-Fas mimant le FasL. Ce résultat suggère une
défaillance dans l'initiation de l'apoptose par le sentier Fas/FasL. En d'autres ternes. le
message de mort cellulaire ne se transmet pas. Ceci explique donc, en partie, pourquoi on
retrouve moins d'apoptose chez les lymphocytes de I'AAE même s'ils expriment plus de
Fas que les lymphocytes provenant de sujets conuôles.
De façon à évaluer le rôle potentiel de l'IL-2 dans le maintien de la lymphucytose
dans I'AAE, les lymphocytes T des patients AAE et des sujets normaux ont été stimulés
avec des doses physiologiques de cette cytokine. Chez les sujets contrôles, l'IL-2 a peu
ou pas d'effet sur l'apoptose. Par contre, si l'on compare les lymphocytes T non stimulés
des patients AAE avec ceux stimulés l'IL-2, on se rend compte que cette cytokine
diminue de plus de 2,s fois l'apoptose. Cela vient confirmer que l'IL-2 est capable de
maintenir artificiellement en vie les lymphocytes T pulmonaires. De plus, la stimulation à
l'IL-2 des lymphocytes T des patients AAE donne des taux d'apoptose comparatifs à
ceux provenant des sujets nonnaux stimulés avec cette cytokine. Ceci suggère donc que
les lymphocytes de I'AAE sont dépendants de U-2 pour leur survie.
En résumé, cette étude a démontré que les lymphocytes du LBA des patients AAE
ont une apoptose diminuée par rapport aux lymphocytes de LBA des sujets normaux.
Cette diminution d'apoptose implique plusieurs mécanismes. Tout d'abord, le milieu

pulmonaire dans I'AAE joue un cale important puisque des niveaux élevés de Fas, et
d' IL-2 sont détectés. Ces deux médiateurs sont des facteurs anti-apoptiques pur les
lymphocytes T. De plus, les lymphocytes des sujets AAE manifestent des différences par
rapport aux lymphocytes normaux, en surexprimant le Bcl-xL et en ayant une disfonction
au niveau du Fas cellulaire étant donné que la stimulation à i'anti-Fas n'a aucun effet. Il
est clair ici que la diminution de l'apoptose est un mécanisme majeur dans le maintien de
Ia lymphocytose alvéolaire caractéristique à I'AAE. Cette étude apporte un nouvel
élément d'information dans l'établissement de la pathologie de 1'AA.E.

1. Kaltreider, B.H. Hypetsensitiviry pneurnonitis. West J Med. 159: 570-578, 1993.
2. Cormier, Y., Schuyler, U Part M : interstial lung disease. in hypersensitivity
pneumonitis. Textbook of Pulrnonary Medecine, Vol 2, R.C. Bonc ed., Mosby-year
book, St-Louis : 1-9, 1992.
3. Denis, M. Proinfiammatory cytokines in hypersensitivity pneumonitis. Am J Respir
Crit Care Med. 151: 164-169, 1995.
4. James, D.G., Rizzato, G. and Sharma, O.P. Bronchopulmonary lavage (BAL). A
window of the lungs. Sarcoidosis. 9: 3-14, 1992.
5. Rosenberg, H.F. and Gallin, JJ. Inflammation. Fundamental Immunology. 32:
1051-1066, 1999.
6. Reynolds, H.Y. Lung inflammation : Normal host defense or a complication of some
diseases? Ann Rev Med. 38: 295-323, 1987.
7. Shelhamer, J.H., Levine, SJ., Wu, T., Jacoby, D.B., Kiaiiner, MA. and Rennard,
S.I. Airway inflammation. Ann Intem Med. 123: 288-304, 1995.
8. Nicod, L.P. Pulmonary defence mechanisrns. Respiration. 66(1): 2-1 1, 1999.
9. Reynolds, A.Y. Respiratory host defenses-surface immunity. Immunobiology. 191:
402-4 12, 1994.
IO. Lohmann-Matthes, M L , SteUunuller, C. and Franke-Ullniann, G. Puimonaty
macrophages. Eur Respir J. 7: 1678- 1689,1994.

1 1. Tonnel, A.B., Gosseî, P., Molet, S., Tilie-Leblood, i., Jeannin, P. and Joseph, M.
Interactions between endothelid celis and effector celis in aliergic inflammation. Ann
N Y Acad Sci. 796 9-20, 1996.
12. Stockley, RA. Role of inflammation in respiratory tract infections. Am J Med.
99(6B): 8s-13S, 1995.
13. Mosmann, T.R. and Sad, S. The expanding unisense of T-cell subjects : Thl, Th2
and more. Immunol. 19: 138-146, 1996.
14. Pforte, A., Brunner, A., Gais, P., Striibel, M., Burger, G., Breyer, G.,
Haussinger, K, and Ziegler-Heitbrock, L. increased levels of soluble serum
interleukin-2 receptor in extrinsic allergic alveolitis correlate with interleukin-2
receptor expression alveolar macrophages. J Allergy Clin Immunol. 94: 1057-1064,
1994.
15. Reynold, H.Y. Concepts of pathogenesis and lung reactivity in hypersensitivity
pneumonitis. Ann N Y Acad Sci. 465: 587-603, 1986.
16. Israël-Assayrg, E. and Cormier, Y. Surfactant modifies the lyrnpho proliferative
activity of macrophage in hypersensitivity pneumonitis. Am J Physiol. 273: 125-131,
1997.
17. Trentin, L., Migaone, N., Zambello, R., Di Celle, P.F., Aina, F., Feruglim, C.,
Bulian, P., MasciareIli, M., Agostini, C., Cipriani, A, Marcer, G., Foa, R.,
Pizzolo, G. and Semenzato, G. Mecanisms accounting for lymphocytic aiveolitis in
hypersensitivity pneumonitis. J Immunol. 145: 2147-2152, 1990.
18. Dakhama, A., Israël-Assayag, E. and Cormier, Y. Role of interleukine-2 in the
development and persistence of lymphocytic aiveolitis in farmer's lung. Eur Respir J.
11: 128 l-I285,1998.
19. Oshima, M., Maeda, A., Ishioka, S., Hiyama, K. and Yamakido, M. Expression of
C-C chemokines in bronchoalveolar lavages cells from patient with granulomutous
lung diseases. h n g . 177: 229-234, 1999.

20. Gundmundsson, G. and Hunninghake, G.W. Respiratory epithelial cells release
interleukine-8 in response to a thermophilic bacteria that causes hypersensitivity
pneumonitis. Exp Lung Res. 25: 217-225, 1999.
2 1. Baggiolini, M., Loetscher, P. and Moser, B. Interleukin-8 and the chemokine
farnily. Int J Immunophannacol. 17: 103-108, 1995.
22. Harris, N.I. and Ronchese, F. The role of B7 costimulation in T-ceIl immunity.
Immltnol Ce11 Biol. 77: 304-320, 1999.
23. Boise, L.H., Minn, AJ., Noel, PJ., June, C.H., Accauitâi, M.A., Lindsten, T. and
Thompson, C.B. CD28 costirnulation compromote T-ceIl survivai by enhancing the
expression of Bcl-xL. Immuniiy. 3:87-94, 1995.
24. Israël-Assayag, E., Dakharna, A., Lavigne, S., Laviolette, M. and Cormier, Y.
Expression of costimulation molecules on aiveolar macrophages in hypersensitivity
pneumonitis. Am J Resp Crit Cure Med. 159: 1830-1836, 1999,
25. Agostini, C., Zambello, R., Sincetta, R., Cerutti, A., Milani, A., Tassinari, C.,
Facco, M., Cipriani, A., Trentin, L. and Semenzato, G. Expression of tumor
necrosis factor receptor super family members by lung T lymphocytes in interstial
lung disease. Am J Respir Crit Cure Med. 153: 1359- 1366, 1996.
26. Colten, H.R. Tissue-specific regulation of inflammation. J Appl Physiol. 72: 1-7,
1992.
27. Leach, A.P. Apoptosis : molecular mechanism for physiologk ce11 death. Clin La6
Sci. 11: 346-349. 1998.
28. Buja, L.M., Eigenbrodt, M.L. and Eigenbrodt, E.H. Apoptosis and necrosis. Basic
types and mechanisms of celi death. Arch Pathol Lab Med. 117: 1208-1214.
29. Nicotera, P., Leist, M. and Ferrando-May, E. Intracellular ATP, a switch in the
decision between apoptosis and necrosis. Toxicol Lett. 102- 103: 139-142, f 998.

30. Cohen, J J. Apoptosis : Mechanisms of life and death in the immune stystem. J
Allergy Clin Immunol. 103: 548-554, 1999.
31. Kerr, J.F., Wyllie, A.H. and Currie, A.R. Apoptosis : a basic biological
phenornenon with wide-ranging implications in tissue kinetics. Br J Cancer. 26: 239-
257, 1972.
32. Hetts, S.W. To dies or not to die. An overview of apoptosis and its role in disease.
JAMA. 279: 300-320. 1998.
33. Kuwano, K. and Ham, N. Signal transduction pathways of apoptosis and
inflammation induced by the tumor necrosis factor receptor family. Am J Respir Cell
Mol Biol. 22: 147- 149.2000.
34. Akbar, A.N. and Salmon, M. Cellular environments and apoptosis : tissue
microenvironments control activated T-ce11 death. lmmunoiogy Today. 18: 72-76,
1997.
35. Ju, S.T., Panka, PJ., Cui, H., Ettinger, R., El-Khayib, M., Sherr, D.H., Strager,
B.Z. and Marshak-Rothstein, A. Fas (CD95)FasL interactions required for
programmed ce11 death after T-cell activation. Nature. 65 13: W 8 , 1995.
36. Cheng, J., Zhou, T. Liu, C., Shapiro, J.P., Braver, MJ., Kiefer, M.C., Barr, PJ.
and Mountz, J.D. Protection from Fas-Mediated apoptosis by a soluble fonn of the
Fas molecule. Science. 5 15: 1759- 1765, 1994.
37. Williams, G.T. and Smith, C.A. Molecular regulation of apoptosis : Genetic
controls on ce11 death. Cell. 74: 777-779, 1993.
38. Tsujimoto, Y. and Shimizu, S. Bcl-2 farnily : Life-or-death switch. FEBS. 466: 6-
10.2000.
39. Yang, J., Liu, X., Bhalla, K., Kim, C.N., Ibrado, A.M., Cai, J., Peng, TJ., Jones,
D.P. and Wang, X. Prevention of apoptosis by Bcl-2 : Release of cytochrome c from
mitochondria blocked. Science. 275: 1 129- 1 13 1, 1997.

40. Steller, 8. Mechanisms and genes of cellular suicide. Science. 267: 1445-1449, 1995.
4 1 . Golstein, P. Controlling ce11 death. Science. 275: 108 1-1082, 1997.
42. Kawahara, A., Kobayashi, T. and Nagata, S. inhibition of Fas-induced apoptosis
by Bcl-2. Oncogene. 17: 2549-2554, 1998.
43. Adams, MJ. and Cory, S. The Bcl-2 protein family : arbiter of celi survivd.
Sciences. 28 1: 1322-1326, 1998.
44. Medema, J.P., Scaffidi, C., Krammer, P.H. and Peter, M.E. Bcl-xL act down
Stream of caspase-8 activation by the CD95 death-inducing signaiing comptex. J Bi01
Chem. 273: 3388-3394, 1998.
45. Mignon, A., Rouquet, N. and Joulin, V. Les caspases, les protéases à cystéine de
l'apoptose : un enjeu thérapeutique pour demain? M/S. 14: 9-17, 1998.
46. Miura, M., Hisahara, S., Araki, T. and Okano, H. Execution mechanisms of
programmed ce11 death by caspase (ICWCED-3) family proteases. Hearr Vessels. 12:
66-70, 1997.
47. Slee, E.A., Adrain, C. and Martin, SJ. Serial killers : ordering caspase activation
events in apoptosis. Ce11 Death Differ. 6: 1067-1074, 1999.
48. Vaux, D.L. and Strasser, A. The molecular biology of apoptosis. Proc Nat1 Acad Sci
USA. 93: 2239-2244, 1996.
49. Saraste, A. and Pulkki, K. Morphologie and biochemical hailmarks of apoptosis.
Cm-diovasc Res. 45: 528-537,2000.
50. Gravieli, Y, Sheman, Y. and Ben-Sasson, S.A. Identification of programmed ce11
death in situ via specific labelling of nuclear DNA fragmentation. J Ce11 Biol. 119:
493-497, 1992.


















