
Analyzing the Efficiency of Mn-(C H ) (M = Sc, Ti, Fe, Ni ... · 1 Analyzing the Efficiency of...
23
1 Analyzing the Efficiency of M n -(C 2 H 4 ) (M = Sc, Ti, Fe, Ni; n = 1, 2) Complexes as Effective Hydrogen Storage Materials Arindam Chakraborty,[a] Santanab Giri[a] and Pratim Kumar Chattaraj*[a] Department of Chemistry and Center for Theoretical Studies Indian Institute of Technology Kharagpur Kharagpur- 721302 E-mail: [email protected] Abstract: Hydrogen trapping ability of various metal – ethylene complexes has been studied at the B3LYP and MP2 level of theory using the 6-311+G(d,p) basis set. Different global and local reactivity descriptors and the associated electronic structure principles provide important insights into the associated interactions. There exist two distinct classes of bonding patterns, viz., a Kubas-type interaction between the metal and the H2 molecule behaving as a η2-ligand and an electrostatic interaction between the metal and the atomic hydrogens. Keywords: Density functional theory • Hydrogen storage • Metal-ethylene complex • Electrophilicity
Transcript of Analyzing the Efficiency of Mn-(C H ) (M = Sc, Ti, Fe, Ni ... · 1 Analyzing the Efficiency of...

1
Analyzing the Efficiency of Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) Complexes as
Effective Hydrogen Storage Materials
Arindam Chakraborty,[a] Santanab Giri[a] and Pratim Kumar Chattaraj*[a]
Department of Chemistry and Center for Theoretical Studies Indian Institute of Technology Kharagpur
Kharagpur- 721302
E-mail: [email protected]
Abstract:
Hydrogen trapping ability of various metal – ethylene complexes has been studied at the B3LYP and MP2 level of
theory using the 6-311+G(d,p) basis set. Different global and local reactivity descriptors and the associated
electronic structure principles provide important insights into the associated interactions. There exist two distinct
classes of bonding patterns, viz., a Kubas-type interaction between the metal and the H2 molecule behaving as a
η2-ligand and an electrostatic interaction between the metal and the atomic hydrogens.
Keywords: Density functional theory • Hydrogen storage • Metal-ethylene complex • Electrophilicity

2
Introduction
Hydrogen, the third most abundant element on Earth and perhaps the most abundant element on the
Universe is being conceived as a very promising alternative energy carrier as well as a future fuel reserve
for the transportation industry. In its molecular form hydrogen can be used directly as a fuel to drive a
vehicle, to heat water or indirectly to produce electricity for industrial, transport and domestic use. The
superiority of hydrogen over the so-called fossil fuels lies in its sheer “cleanliness”, a unique fuel that is
totally non-polluting and upon use produces water as a harmless by-product. Unlike petroleum, hydrogen
can be easily generated from renewable energy resources which further eliminate the production of oxides
of nitrogen and sulfur, greenhouse gases like carbon dioxide and methane as by-products thereby
eradicating further scopes of environmental pollution. But the storage of gaseous hydrogen in a practical
sense creates difficulties as the materials that can trap the same in large gravimetric and volumetric
quantities are really numbered. In liquid form, hydrogen can only be stored under cryogenic temperatures
which can never be a good option for day by day use. But scientists nevertheless have been somewhat
successful in designing novel materials that can store hydrogen at ambient conditions.[1-3] Numerous other
materials, like aluminum nitride (AlN) nanostructures[4], transition-metal doped boron nitride (BN)
systems[5], alkali-metal doped benzenoid[6] and fullerene clusters[7], bare as well as light metal and
transition-metal coated boron buckyballs, B80[8], and magnesium clusters[9] have been confirmed both
experimentally and theoretically to serve as potential hydrogen-storage materials. Further, based on a
theoretical study invoking the metastability of hydrogen-stacked Mgn clusters[10], Chattaraj et al[11] have
very recently demonstrated that a host of small to medium metal cluster moieties involving Li3+, Na3
+,
Mgn and Can (n = 8-10) cages have got a fair capability of trapping hydrogen in both atomic and
molecular forms. The stability of the aforesaid all-metal systems has been attributed to the existence of an
aromaticity criterion in the metallic rings which was assessed through the rationale of nucleus
independent chemical shift (NICS)[12]. Yildirim et al[13-18] along with a few other research groups[19,20]
have been quite successful in establishing that the C=C bond in an ethylene molecule, C2H4, like that of
fullerenes and other carbon-based nanostructures can form stable complexes with a transition metal,
Titanium (Ti) and the resulting Tin− C2H4 (n = 1, 2) complex in turn can bind up to ten H2 molecules
efficiently. The interaction between the hydrogen molecules and the transition metal is worth-mentioning
as it is somewhat intermediate between physical and chemical adsorption but, can best be explained from
the Kubas model[21] of hydrogen binding: (η2-H2) – metal interaction.

3
In this article we have made an attempt to study the binding of hydrogen with a host of transition metal
(M) – ethylene (C2H4) complexes [Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2)] on the basic premise of
conceptual density functional theory (CDFT)[22-25] and its various allied global reactivity descriptors like
electronegativity[26-28] (χ), hardness[29-31] (η), electrophilicity[32-34] (ω) and the local variants like atomic
charges[35] (Qk) and Fukui functions[36] (fk). The stability of the resulting hydrogen-bound [Mn-(C2H4) (M
= Sc, Ti, Fe, Ni; n = 1, 2)] complexes may be understood from the corresponding interaction energies
(∆E) and reaction electrophilicities (∆ω) of a number of plausible trapping reactions. Further, for most of
the hydrogen bound metal – ethylene complexes [(H2)x – Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2; x = 1-
8)], hydrogen prefers mostly to coordinate with the metal center (M) in its molecular (dihydrogen) form.
The H2 molecule therefore approximately behaves as a η2-type ligand and the complex subsequently
attains stability though a Kubas-type[21] interaction between the metal atom and the interacting dihydrogen
moieties.
Theoretical Background
The thermodynamic stability of molecular systems may be meaningfully justified in a quantitative manner
from a careful scrutiny of their chemical hardness (η) and electrophilicity (ω) values. This has been
further validated by the establishment of some associated molecular electronic structure principles like the
Principle of Maximum Hardness[37-39] (PMH) together with the Minimum Polarizability Principle[40,41]
(MPP) and Minimum Electrophilicity Principle[42,43] (MEP). These electronic structure principles serve as
major determinants towards assessing the stability and reactivity trends of chemical systems. For an N-
electron system, the electronegativity[26-28] (χ) and hardness[29-31] (η) can be defined as follows:
)(rvN
Er
⎟⎠⎞
⎜⎝⎛∂∂
−=−= μχ (1)
)(
2
2
rvNE
r⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=η (2)
Here E is the total energy of the N-electron system and μ and )(rv v are its chemical potential and external
potential respectively. The electrophilicity[32-34] (ω) is defined as:

4
ημω2
2
= ηχ2
2
= (3)
A finite difference approximation to Eqs. 1 and 2 can be expressed as:
2AI +
=χ (4)
and AI −=η (5)
where I and A represent the ionization potential and electron affinity of the system respectively and are
computed in terms of the energies of the N and N ± 1 electron systems. For an N-electron system with
energy E (N) they may be expressed as follows:
I = E (N–1) – E(N) (6)
and A = E(N) – E(N+1) (7)
The local reactivity descriptor, Fukui function[36] (FF) measures the change in electron density at a given
point when an electron is added to or removed from a system at constant ( )v rr . It may be written as:
( )
( )( )( )v r N
rf rN v rρ δμ
δ⎛ ⎞∂⎛ ⎞= =⎜ ⎟⎜ ⎟∂⎝ ⎠ ⎝ ⎠v
vv
v (8)
Condensation of this Fukui function, ( )f rv to an individual atomic site k in a molecule gives rise to the
following expressions in terms of electron population[44] qk
( 1) ( )k k kf q N q N+ = + − for nucleophilic attack (9a)
( ) ( 1)k k kf q N q N− = − − for electrophilic attack (9b)
[ ]( 1) ( 1) 2ok k kf q N q N= + − − for radical attack (9c)
Computational Details
The geometry optimization and subsequent frequency calculations of the different metal – ethylene
complexes Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) and their corresponding hydrogen-trapped analogues

5
are carried out at the B3LYP level of theory using the 6-311+G(d,p) basis set with the aid of the
GAUSSIAN 03 program package.[45] However, for the di-iron – ethylene complexes [Fe2-(C2H4)]
convergence could not be achieved owing to which the aforesaid molecules could not be considered for
hydrogen trapping/storage. The number of imaginary frequency (NIMAG) values of all the optimized
geometries are zero thereby confirming their existence at the minima on the potential energy surface
(PES). Single point calculations are further done to evaluate the energies of the N ± 1 electron systems by
adopting the geometries of the corresponding N-electron systems optimized at the B3LYP/6-311+G(d)
level of theory. The I and A values are calculated using a SCFΔ technique. The electrophilicity (ω) and
hardness (η) are computed using the eqs. 3 and 5 respectively. A Mulliken population analysis (MPA)
scheme is adopted to calculate the atomic charges (Qk) and the corresponding Fukui functions ( ( )f rv ) on
the metal centers. The frontier molecular orbital pictures are obtained through the GAUSSVIEW 03
package.[45]
Results and Discussion
The total energy (E, au) and the important global reactivity descriptors like electronegativity (χ), hardness
(η) and electrophilicity (ω) for all the interacting transition metals, hydrogen (atomic and molecular) and
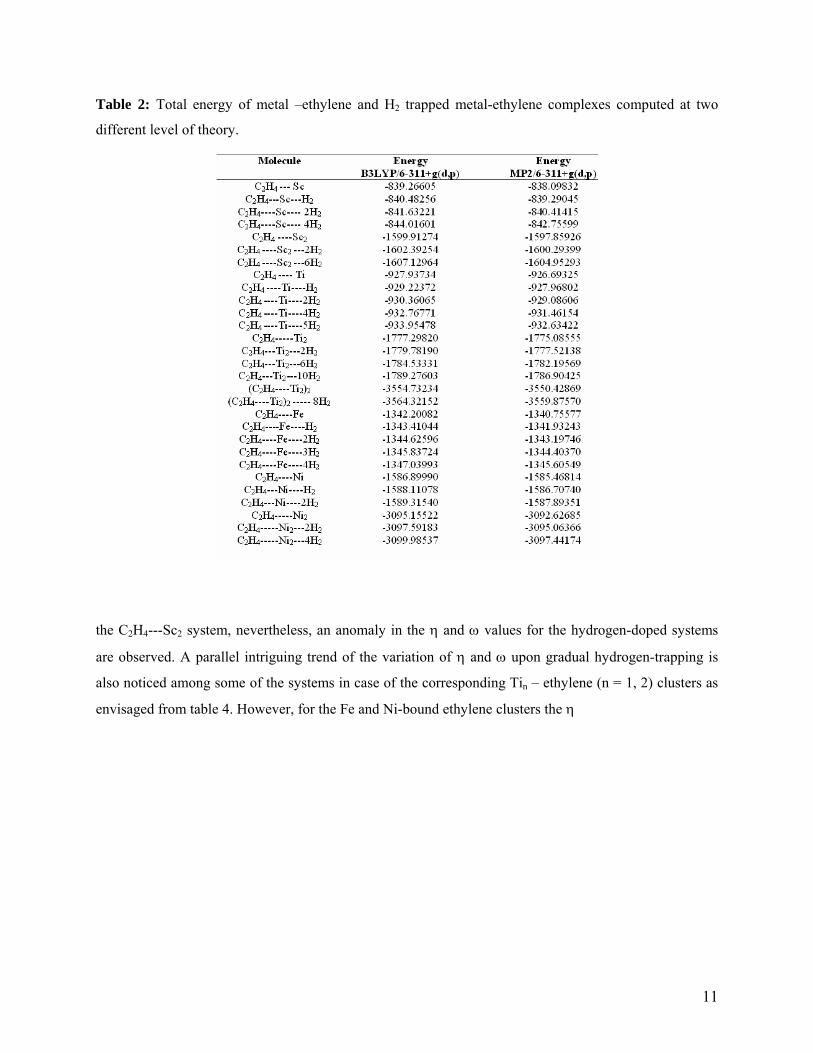
ethylene are shown in table 1. A detailed tabulation of the total energy (E, au) computed at two different
level of theory are tabulated in table 2. The above-mentioned global reactivity parameters of the various
metal – ethylene complexes Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) and their corresponding hydrogen-
trapped analogs [(H2)x – Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2; x = 1-8)] are presented in tables 3, 4, 5
and 6 for the metals Sc, Ti, Fe, and Ni respectively. However, for all the complexed moieties, we have
computed the local parameters like atomic charge (Qk) and Fukui functions (fk+, fk
-) using the Mulliken
population analysis (MPA) scheme for the metal site only as it is conceived that the metal center virtually
plays the key role towards binding the incoming hydrogen molecules during trapping reactions. A number
of plausible hydrogen trapping reactions between the metal – ethylene complexes Mn-(C2H4) (M = Sc, Ti,
Fe, Ni; n = 1, 2) and the incoming hydrogen molecules for the different transition metals are displayed in
tables 7-10. The molecular point groups (PG) of all the complex structures and the corresponding H – H
bond

6
Figure 1. Sc-ethylene complex and its corresponding H2-trapped analogues.
distances (Å) for the H2 molecule(s) trapped on to the metal – ethylene clusters are shown in table 11. The
molecular geometries of all the metal – ethylene complexes Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) and
their corresponding hydrogen-bound analogues [(H2)x – Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2; x = 1-8)]
optimized at the B3LYP/6-311+G(d, p) level of theory are provided in figures 1-4 corresponding to the
central metal atom in the order of sequence as Sc, Ti, Fe and Ni. Figures 1 – 4 also depict the atomic
charges on

7
Figure 2: Ti-ethylene complex and its corresponding H2-trapped analogues.
the central metal atom as well as the hydrogen centers of the H2 molecules bound to the metal – ethylene
moiety either in the split, atomic or closely-bonded molecular form. The important frontier molecular
orbitals (FMOs) of all the complex clusters (free and H2-bound) are portrayed in figure 5. A scrutiny of
table 1 reveals that the global hardness (η) of the transition metals approximately decrease from Sc to Fe
with a subsequent increase in their respective electrophilicity (ω) values. The energies of the transition
metals

8
Figure 3: Fe-ethylene complex and its corresponding H2-trapped analogues
computed at the B3LYP/6-311+G(d, p) level of theory show an expected increasing trend from Sc to Fe
which is quite relevant from their increasing atomic numbers. A detailed analysis of the various metal –
ethylene complexes Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) and their corresponding trapping reactions to
form the hydrogen-bound analogues can be fruitfully materialized from tables 3 – 10. An analysis of the
global and local properties for the Sc – ethylene
Figure 4: Ni-ethylene complex and its corresponding H2-trapped analogues.

9
complex and hydrogen-trapped analogues as presented in table 3 explains that while the electronegativity
(χ) values of the different Scn – ethylene (n = 1, 2) complexes do not show any viable change upon
transformation from the free, unbound stage to the hydrogen-trapped [(H2)x – Scn-(C2H4) (n = 1, 2; x = 1,
2, 4, 6)] form, the hardness (η) and electrophilicity (ω) exhibit a mixed trend. The η
C2H4_HOMO C2H4_LUMO C2H4-Sc_4H2 HOMO C2H4-Sc_4H2 LUMO
C2H4-Sc2_2H2
HOMO
C2H4-Sc2_2H2
LUMO C2H4-Sc2_6H2 HOMO C2H4-Sc2_6H2 LUMO
C2H4-Ti_4H2
HOMO
C2H4-Ti_4H2
LUMO C2H4-Ti_5H2 HOMO C2H4-Ti_5H2 LUMO
C2H4-Ti2_10H2
HOMO
C2H4-Ti2_10H2
LUMO (C2H4-Ti2)2 _8H2 HOMO
(C2H4-Ti2)2 _8H2
LUMO
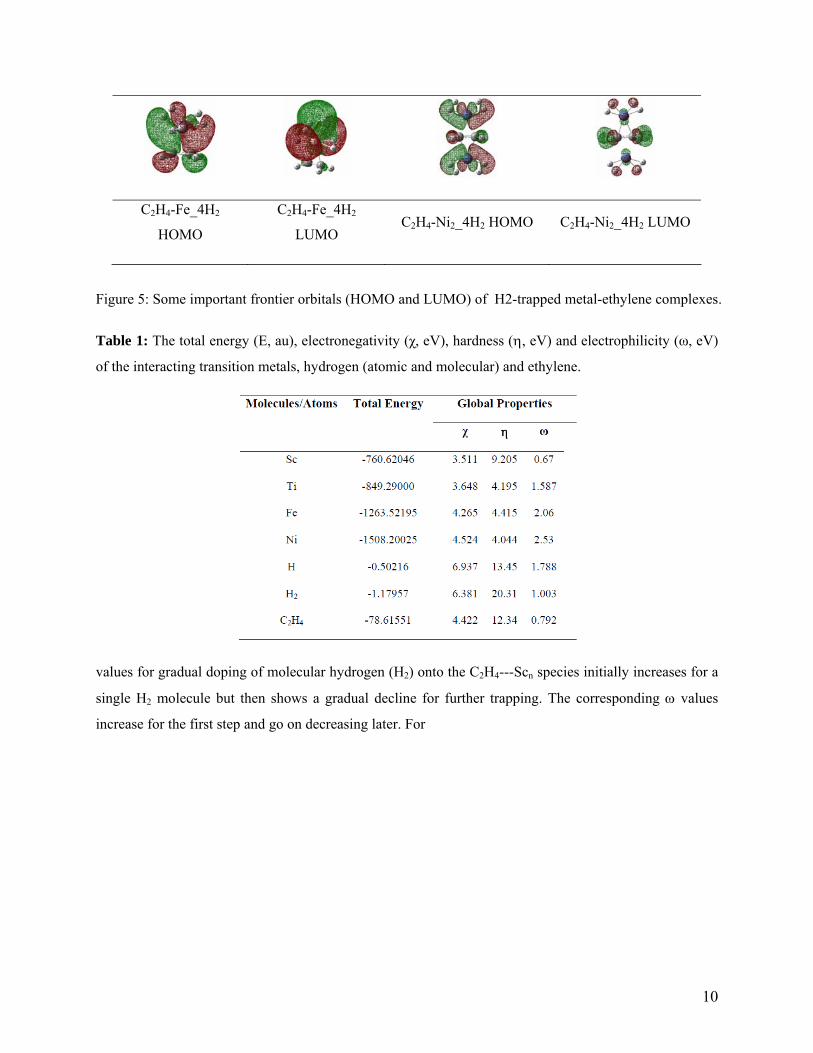
10
C2H4-Fe_4H2
HOMO
C2H4-Fe_4H2
LUMO C2H4-Ni2_4H2 HOMO C2H4-Ni2_4H2 LUMO
Figure 5: Some important frontier orbitals (HOMO and LUMO) of H2-trapped metal-ethylene complexes.
Table 1: The total energy (E, au), electronegativity (χ, eV), hardness (η, eV) and electrophilicity (ω, eV)
of the interacting transition metals, hydrogen (atomic and molecular) and ethylene.
values for gradual doping of molecular hydrogen (H2) onto the C2H4---Scn species initially increases for a
single H2 molecule but then shows a gradual decline for further trapping. The corresponding ω values
increase for the first step and go on decreasing later. For
11
Table 2: Total energy of metal –ethylene and H2 trapped metal-ethylene complexes computed at two
different level of theory.
the C2H4---Sc2 system, nevertheless, an anomaly in the η and ω values for the hydrogen-doped systems
are observed. A parallel intriguing trend of the variation of η and ω upon gradual hydrogen-trapping is
also noticed among some of the systems in case of the corresponding Tin – ethylene (n = 1, 2) clusters as
envisaged from table 4. However, for the Fe and Ni-bound ethylene clusters the η

12
Table 3: Total energy (E, au), electronegativity (χ, eV), hardness (η, eV) and electrophilicity (ω, eV) of
the Sc-ethylene system and its H2-trapped analogs along with the atomic charge (Qk) and Fukui functions
for the metal site only.
values are found to correlate pretty nicely with that of ω as relevant from tables 5 and 6 respectively. For
the Fe and Ni-bonded complexes the hardness (η) increases uniformly with a gradual decrease in the
consecutive (ω) values thereby corroborating the associated principles of maximum hardness37-39 and
minimum electrophilicity42, 43 which justify further stability of the clusters upon higher-order trapping. A
glance at tables 7, 8, 9 and 10 for some plausible trapping reactions conceived between the metal –
ethylene complexes Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) and hydrogen

13
Table 4: Electronegativity (χ, eV), hardness (η, eV) and electrophilicity (ω, eV) of the Ti-ethylene system
and its H2-trapped analogs along with the atomic charge (Qk) and Fukui functions for the metal site only.
molecule for the metals Sc, Ti, Fe and Ni respectively reveals that for all the reactions, the corresponding
interaction energies (IE) and reaction electrophilicities (∆ω) are negative. This is quite enthusing as a
negative IE or ∆ω value for a chemical process affirms considerable thermodynamic feasibility to the
same, thereby, rendering ample stability to the resultant H2-trapped complexes as well. The average
dissociative chemisorption energies (∆ECE)

14
Table 5: Electronegativity (χ, eV), hardness (η, eV) and electrophilicity (ω, eV) of the Fe-ethylene
system and its H2-trapped analogs along with the atomic charge (Qk) and Fukui functions for the metal
site only.
computed for the trapping of hydrogen molecule onto the metal – ethylene system is positive which
favors the incoming H2 species to bind with the metal site with formation of new bonds. However, with
increasing number of hydrogen atoms being adsorbed, the
Table 6: Electronegativity (χ, eV), hardness (η, eV) and electrophilicity (ω, eV) of the Ni-ethylene
system and its H2-trapped analogs along with the atomic charge (Qk) and Fukui functions for the metal
site only.

15
∆ECE values decrease for all the metal – ethylene Mn-(C2H4) (M = Sc, Ti, Fe, Ni; n = 1, 2) molecules. This
reveals that with an increase in H-crowding around the metal – ethylene core, the driving force for H2
dissociation may further go on a decline.46 A careful scrutiny of
Table 7: Plausible H2-trapping reactions for Sc-ethylene complexes showing their interaction energy (IE),
interaction energy per H2 molecule (IE/H2), reaction electrophilicity (∆ω) and dissociative chemisorption
energy (∆ECE)
figure 1 shows that for the Sc-bound ethylene complexes, the hydrogen atoms are linked to the central
metal atom in both the atomic and molecular forms. An in-depth study of the H – H bond distances from
table 11 and the corresponding atomic charges on the H-centers of the trapped H2 molecules from figure 1
unveils something really unique which further gives a better impetus

16
Table 8: Plausible H2-trapping reactions for Ti-ethylene complexes showing their interaction energy (IE),
interaction energy per H2 molecule (IE/H2), reaction electrophilicity (∆ω) and dissociative chemisorption
energy (∆ECE)
towards understanding the driving force responsible for the possible feasibility of hydrogen storage in
such metal – ethylene systems. From figure 1 it is quite transparent that the H2 moieties bound to the
central metal site mostly in its molecular form bear a slight positive charge on the respective H-centers.
The H – H bond length, however, increases than that of its original molecular form thereby rendering the
H2 system to behave nonetheless as a ligand. The increment in the H – H distance upon complexation
(rather trapping) can be compared with an analogous increase in the C = C bond length in C2H4 that has
already been explained in literature on the backdrop of

17
Table 9: Plausible H2-trapping reactions for Fe-ethylene complexes showing their interaction energy (IE),
interaction energy per H2 molecule (IE/H2), reaction electrophilicity (∆ω) and dissociative chemisorption
energy (∆ECE)
the Dewar-Chatt-Duncanson (DCD) model of molecular binding. A similar behavior exhibited by the
incoming H2 molecule can be attributed to the Kubas-model21 of metal – dihydrogen interaction. The H2
molecule acting as a η2- ligand system is bonded to the
Table 10: Plausible H2-trapping reactions for Ni-ethylene complexes showing their interaction energy
(IE), interaction energy per H2 molecule (IE/H2), reaction electrophilicity (∆ω) and dissociative
chemisorption energy (∆ECE)
transition metal and thus stabilized. However, the H2 molecules that are intended to be bound to the
transition metal site mostly in its atomic form are found to split wide-apart with large H – H distances as
compared to the free, unbound molecular form. The atomic charges on the corresponding sites of the lone
hydrogen atoms are noticeably high compared to that of the η2-H2 ligands. Thus the interaction between
the central transition metal and the atomic hydrogens are supposed to be fairly electrostatic in nature. So,
the chemical binding of molecular hydrogen onto the metal – ethylene moiety to form stable H2-trapped

18
analogues have got two facets – a Kubas-type binding between the metal center and the η2-H2 ligands
which further sustain their molecular nature in the trapped form, and,
an electrostatic model of binding between the comparably highly charged atomic hydrogens (split wide-
open) and the central metal atom. The important frontier molecular orbitals (FMOs) of some
representative metal – ethylene complexes displayed in figure 5 reveals the dominance of a σ-antibonding
character in most of the HOMO contours. Nevertheless, the presence of a bulge of electron density near
the metal sites provides ample evidence of chemical binding between the metal center and ethylene
moiety. The stability of the M - η2-H2 linkage might be due to some favorable interactions between the
metal-d-orbitals and the σ-antibonding orbitals of hydrogen. The overall electron density distribution
pattern in the frontier orbitals of different hydrogen-trapped complexes reveals that the electrons are
delocalized over the entire molecular skeleton.

19
Table 11: The molecular point groups (PG) and the H – H bond distances (Å) of the H2 ligands trapped in
different forms (atomic and molecular) onto the metal – ethylene complexes.
Tables 3 – 6 also demonstrate that the Mulliken charges computed for the central metal sites show an
unusual variation upon gradual complexation with the incoming H2 molecules. The metal atoms that are
known to be an electropositive species are found to bear a negative charge upon increasing H2 trapping. It

20
has been observed by other researchers as well.47 It is assumed to be due to the drawback of the Mulliken
as well as the natural population analysis schemes. In this study it is also observed that a negative charge
is gradually piled up onto the metal atom with gradual loading of hydrogen. Again, most of the incoming
H2 molecules are bonded to the metal atom in the Kubas fashion augmenting the same obtained through
the back donation from ethylene as envisaged from figures 1 – 4 which prompts us to suggest a rigorous
charge transfer that
occurs from the η2-H2 ligands to the metal atom which allows the latter to sustain a formal negative
charge. The cumulative effect of charge donation by a number of H2-ligands in the Kubas fashion renders
the metal center formally negative. The bulge of electron density around the metal sites as evident from
the frontier orbitals also indirectly justifies the fact.
Conclusion
A comprehensive study of the binding of hydrogen onto some metal – ethylene complexes, Mn-(C2H4) (M
= Sc, Ti, Fe, Ni; n = 1, 2) have been performed using conceptual DFT based reactivity descriptors. The
efficiency of such metal complexes towards trapping of molecular hydrogen is quite unequivocal. The
stability of the trapped complexes has been understood both from the viewpoint of increasing hardness
(η) trends and a subsequent decrease in the electrophilicity (ω) in most cases upon gradual hydrogen
loading as well as on the basis of the Kubas-model. The ability of transition metals other than Sc or Ti to
act as potential storage stuffs for hydrogen in the presence of a π- system (like ethylene) has been
demonstrated. Presumably the existence of a negative charge on the metal sites upon gradual H2-binding
stems from an increasingly favorable orbital interaction between the incoming η2- dihydrogen ligands and
the central metal atom. The binding mode of molecular hydrogen to the metal site through this Kubas-
type interaction is established. Nonetheless, the usage of other transition metals associated with π-
systems other than ethylene that may serve as effective materials for hydrogen storage has opened an area
that deserves to be widely cultivated. Further work is in progress.
Acknowledgements
We thank Indo- EU (HYPOMAP) project for financial assistance. One of the authors (AC) would like
to acknowledge the CTS, IIT Kharagpur for a Visitors’ Fellowship.

21
References
[1] R. Coontz, B. Hanson, Science 2004, 305, 957.
[2] G. W. Crabtree, M. S. Dresselhaus, M. V. Buchanan, Phys. Today. 2004, 57, 39.
[3] A. Zuttel, Mater. Today. 2003, 6, 24.
[4] Q. Wang, Q. Sun, P. Jena, Y. Kawazoe ACS Nano. 2009, 3, 621.
[5] S. A. Shevlina,; Guo, Z. X. Appl. Phys. Lett. 2006, 89, 153104.
[6] Srinivasu, K.; Chandrakumar, K.R.S.; Ghosh, S. K. Phys. Chem. Chem. Phys. 2008, 10, 5832 and refs 2-12 therein.
[7] Peng, Q.; Chen, G.; Mizuseki, H.; Kawazoe, Y. J. Chem. Phys. 2009, 131, 214505.
[8] Wu, G.; Wang, J.; Zhang, X.; Zhu, L. J. Phys. Chem. C 2009, 113, 7052.
[9] Wagemans, R. W. P.; van Lenthe, J. H.; de Jongh, P. E.; van Dillen, A. J.; de Jong, K. P. J. Am. Chem. Soc. 2005, 127, 16675.
[10] McNelles, P.; Naumkin, F. Y. Phys. Chem. Chem. Phys. 2009, 11, 2858.
[11] Giri, S.; Chakraborty, A.; Chattaraj, P. K. J. Mol. Model. 2010, (in press)
[12] Schleyer, P. V. R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N. J. R. V. E. J. Am. Chem. Soc. 1996, 118, 6317.; Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer, P. v. R. Chem. Rev. 2005, 105, 3842.
[13] Yildirim, T.; Ciraci, S. Phys. Rev. Lett. 2005, 94, 175501.
[14] Yildirim, T.; Iniguez, J.; Ciraci, S. Phys. Rev. B. 2005, 72, 153403.
[15] Dag, S.; Ozturk, Y.; Ciraci, S.; Yildirim, T. Phys. Rev. B. 2005, 72, 155404.
[16] Durgun, E.; Ciraci, S.; Zhou, W.; Yildirim, T. Phys. Rev. Lett. 2006, 97, 226102.
[17] Akman, N.; Durgun, E.; Yildirim, T.; Ciraci, S. J. Phys. Condens. Matter. 2006, 18, 9509.
[18] Zhou, W.; Yildirim, T.; Durgun, E.; Ciraci, S. Phys. Rev. B. 2007, 76, 085434.
[19] Zhao, Y.; Kim, Y.-H.; Dillon, A. C.; Heben, M. J.; Zhang, S. B. Phys. Rev. Lett. 2005, 94, 155504.
[20] Kiran, B.; Kandalam, A. K.; Jena, P. J. Chem. Phys. 2006, 124, 224703.
[21] Kubas, G. J. Ed. Metal Dihydrogen and Bond Complexes—Structure, Theory and Reactivity, Kluwer Academic/Plenum, New York, 2001.
[22] Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules, Oxford University Press: New York, 1989.

22
[23] Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. 2003, 103, 1793.
[24] Chattaraj, P. K. Ed. Chemical Reactivity Theory: A Density Functional View; Taylor & Francis/CRC Press: Florida, 2009.
[25] Chattaraj, P. K.; Giri, S. Ann. Rep. Prog. Chem. Sect. C: Phys. Chem. 2009, 105, 13.
[26] Sen, K. D.; Jorgenson, C. K. Eds. Structure and Bonding, Vol. 66: Electronegativity; Springer: Berlin, 1987.
[27] Chattaraj, P. K. J. Indian. Chem. Soc. 1992, 69, 173.
[28] Parr, R. G.; Donnelly, R. A.; Levy, M.; Palke, W. E. J. Chem. Phys. 1978, 68, 3801.
[29] Sen, K. D.; Mingos, D.M. P. Eds. Structure and Bonding, Vol. 80: Chemical Hardness; Springer: Berlin, 1993.
[30] Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512.
[31] Pearson, R. G. Chemical Hardness: Applications from Molecules to Solids; Wiley-VCH: Weinheim, 1997.
[32] Parr, R. G.; Szentpaly, L. V.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922.
[33] Chattaraj, P. K.; Sarkar, U.; Roy, D. R. Chem. Rev. 2006, 106, 2065.
[34] Chattaraj, P. K.; Roy, D. R. Chem. Rev. 2007, 107, PR46.
[35] Mulliken, R. S. J. Chem. Phys. 1955, 23, 1833.
[36] Parr, R. G.; Yang, W. J. Am. Chem. Soc. 1984, 106, 4049.
[37] Pearson, R. G. J. Chem. Edu. 1987, 64, 561.
[38] Parr, R. G.; Chattaraj, P. K. J. Am. Chem. Soc. 1991, 113, 1854.
[39] Ayers, P. W.; Parr, R. G. J. Am. Chem. Soc. 2000, 122, 2010.
[40] Chattaraj, P. K.; Sengupta, S. J. Phys. Chem. 1996, 100, 16126.
[41] Fuentealba, P.; Simon-Manso, Y.; Chattaraj, P. K. J. Phys. Chem. A. 2000, 104, 3185.
[42] Chamorro, E.; Chattaraj, P. K.; Fuentealba, P. J. Phys. Chem. A. 2003, 107, 7068.
[43] Parthasarathi, R.; Elango, M.; Subramanian, V.; Chattaraj, P. K. Theo. Chem. Acc. 2005, 113, 257.
[44] Yang, W.; Mortier, W. J. J. Am. Chem. Soc. 1986, 108, 5708.
[45] GAUSSIAN 03, revision B.03, Gaussian, Inc., Pittsburgh, PA
[45] Chen, L.; Cooper, A. C.; Pez, G. P.; Cheng, H. J. Phys. Chem. C. 2007, 111, 5514.

23
[46] Orlova, G.; Scheiner, S. Organometallics 1998, 17, 4362; Kim, G.; Jhi, S. H.; Lim, S.; Park, N. Phys. Rev. B 2009, 79, 155437.


















