Acute Motor Neuropathy
68
Disorders of the Peripheral Nervous System
-
date post
19-Oct-2014 -
Category
Documents
-
view
2.866 -
download
5
description
Transcript of Acute Motor Neuropathy

Disorders of the Peripheral Nervous System

Peripheral Nerve DisordersThe spectrum of peripheral nerve disorders includes
– Mononeuropathies (entrapment, trauma, etc)
– Mononeuritis multiplex (DM, vasculitis)
– Plexopathies (immune, neoplastic)
– Radiculopathies (discs, immune)
– Peripheral Neuropathies

Peripheral Neuropathies (PN)
Peripheral neuropathies may beoperationally defined as a “generalised disorder of peripheralmotor, sensory or autonomic nerves, but excluding single nerve lesionscaused by entrapment or trauma” (Warlow. 1991)

Peripheral Neuropathies (PN)Peripheral neuropathies are heterogeneous disorders
with variation in
peripheral nerve components involved,
clinical course,
pathology
and presumed aetiology
(Dyck et al. 1996; McLeod. 1995).

Peripheral Neuropathies (PN) >100 etiological factors have been identified in patients with PN
(Asbury and Thomas. 1995; McLeod. 1995)
Immune: GBS, CIDP, Paraneoplastic, VASCULITISVitamin deficiency: B1, B6, B12, Vit EToxins and metals: OP, lead, ALCOHOLEndocrine: diabetes, thyroid, parathyroidDrugs: Vincristine, Isoniazid, phenytoinGenetic: HMSN, HLPP, HMN, ALDIDIOPATHIC

Workup of a patient with suspected Peripheral Neuropathy
• History Time course (acute, subacute,chronic, episodic)Negative numbnessPostive tingling, painWeakness and loss of functionBalancePostural dizzinessPMH ?DMMedicationSocial, toxins, dietFamily history

Workup of a patient with suspected Peripheral Neuropathy
• ExaminationGait (foot drop, stepage, unsteady Romberg positive)Cranial Nerves (retinopathy; facial, bulbar, or neckweakness, tonic pupils)Limbs
Pseudoathetosis, Pes cavus, Clawing, Wasting, fasiculationFlaccidity, palpable nervesDistal weakness (radiculopathy)Reduced or absent DTRsGlove loss, allodynia (Small or large fibre)
Systemic rash, BP

Workup of a patient with suspected Peripheral Neuropathy
NeurophysiologyOften indicates nerve pathology
Demyelination (low velocities, latencies)Axonal change (low amp SNAPs and CMAPs)Neuronopathy (very low or absent NAP)
Special tests are needed to look at small fibrefunction

Workup of a patient with suspected Peripheral Neuropathy
Bloods
FBC, U&Es, Ca and LFTs, RBG, B12,
Folate, VDRL, TFTs, Autoantibodies (VLCFAs etc)
CSF (protein)
NERVE BIOPSY (Aetiology not existence of neuropathy)

Guilllain Barre Syndrome (GBS) GBS is defined as “a syndrome of acute weakness of the limbs and reduced or
absent reflexes, with or without sensory loss attributable to a disorder of the peripheral nerves not due to systemic disease” (Hughes. 1990).
GBS is a clinical diagnosis though there are frequently abnormal laboratory features including an elevated CSF protein and evidence of peripheral nerve demyelination (Hughes. 1994; Hartung et al. 1998).

Guilllain Barre Syndrome (GBS)GBS is a leading cause of neuromuscular paralysisworld-wide (UK annual incidence of 1.0-2.0 per 100,000, with age, and M>F)
In 1859 Landry described 10pts with an ascending paralysis caused by peripheral nerve dysfunction
1916 Guillain, Barre and Strohl described an acute, monophasic, benign, flaccid paralysis in French soldiersin the First World War with trench fever and noted anincrease in cerebrospinal fluid protein, though not cellsalbuminocytologic dissociation.

Guilllain Barre Syndrome (GBS) GBS is considered an immune-mediated disorder,
strong supportive evidence includes – Frequent association with preceding infection or
vaccination
– Coexistent auto-immune disease
– Response to immunomodulatory therapy
– Activated T cells and humoral responses against neural
antigens

Guilllain Barre Syndrome (GBS) The typical clinical features of GBS reflect prominent involvement of motor nerves, and may progress for up to four weeks:
Weakness of all four limbs with a proximal bias, Bilateral facial paralysis, and Weakness of bulbar musclesWeakness of respiratory muscles (VC)Reflexes are gradually reduced and then lostSensory symptoms and signs (back pain)
Autonomic failure is seen in a few (morbidity and mortality)

Guilllain Barre Syndrome (GBS) The laboratory features of GBS also evolve with time-
Initial CSF protein is often normal, but becomes elevated within 3/52 in over 90% of patients.
Initial electrophysiological studies are often normal, but become abnormal within three weeks in the majority of patients (Abnormalities in motor conduction with proximal conduction block, prolonged distal motor latencies or generalised slowing of conduction are common; abnormalities of sensory nerve conduction are also common)

Guilllain Barre Syndrome (GBS) As GBS is a clinical syndrome it is important to exclude both other causes of neuropathy and other neurological disorders:
CNS spinal cord compression, brainstem disorders including locked in state
PNS myasthenia, polio, tetanus, botulism, buckthorn poisoning, malignant meningitis

Guilllain Barre Syndrome (GBS) Immunotherpy (Steroids, IVIG, Exchange)
Supportive carePain
Ventilation
Communication
Prognosis Monophasic, 10-25% significant disability at 1yr

The Anatomy of the Neuromuscular Junction
• Motor neurone terminates as a bouton or pre-synaptic nerve terminal separated from the muscle by a thin synaptic cleft (Motor endplate)
• The blood nerve barrier is relatively deficient at the NMJ
• Nerve and muscle are kept in close proximity by bridging protein (laminin), with release zones and the crests of post synaptic folds aligned
• The skeletal neuromuscular junction is the most studied and best understood synapse

Healthy Neuromuscular Junction

The Physiology of Neuromuscular transmission
• Neuronal Action potential invades the pre-synaptic nerve terminal
• Depolarisation triggers opening of VGCCs• Calcium influx triggers quantal release of ACh• ACh binds to post synaptic nAChRs• Ca and Na ions influx through nAChR triggering
muscle membrane depolarisation via VGSCs- CMAP and muscle contraction

Spontaneous and Nerve Evoked Endplate Responses

Myasthenia Gravis (MG)MG is the most common disorder of neuromuscular transmission Incidence 2-6 per 106 , prevalence 40 per 106 population
MG is an acquired autoimmune disease characterised by theformation of anti- nAChR antibodies
MG is common in young women, and older men
MG is characterized by fluctuating and fatigable weaknessWeakness may be limited to a few muscles, such as the extraocularmuscles, bulbar, limb or be generalised in fashion As the weakness is often worse with activity and improved by rest, it is often worse in the evening

Myasthenia Gravis (MG)Ocular features: ptosis, diplopia, ophthalmoplegia
Facial weakness esp ob oculi and oris (snarl)
Bulbar weakness: nasal speech, reduced gag, swallowingproblems, aspiration (silent), weak neck (dropped)
Limb weakness: proximal, fatiguable
Reflexes: normal
Respiratory weakness: diaphragm and intercostal

Myasthenia Gravis (MG) Sources of Diagnostic Confusion
Ocular thyroid, orbital myositis, cavernous sinus, III,IV,VI, brainstem lesions, botulism, MFS variant of GBS
Facial GBS, myopathies
Bulbar LMN MND, skull base, polymyositis
Neck MG, MND, FSH, IM, GBS
Limbs Myopathies, LEMS, Motor Neuropathies

Myasthenia Gravis (MG)
MG is a defect of neuromuscular transmission with
reduced efficacy of Acetyl Choline at the post synaptic
motor endplate due to pathogenic antibodies which • Block the nAChR, • Down regulate the nAChR• & cause complement dependent destruction of the motor
endplate

Myasthenia Gravis (MG)
The immunopathogenesis of MG is unclear but involves• Genetic factors (HLA B8)• Thymus
– Vast majority of young onset cases are autoimmune and associated with thymic hyperplasia
– Around 10% of patients with MG, often older patients) have an associated thymic tumour (oft striated muscle Abs)
• Seronegative (10% gen, 50% OMG)• Neonatal MG

Myasthenia Gravis (MG)
• Diagnosis – Typical clinical picture– Detection of anti-AChR antibodies in serum (90%)– Positive Tensilon test (atropine)– Repeptitive nerve stimulation at low frequency leads
to a decrement in compound muscle action potential amplitude

Repetitive Nerve Stimulation (Supramaximal 2Hz)

Myasthenia Gravis (MG)Treatment
– Symptomatic (pyridostigmine oft with probatheline)
– Thymectomy • Hyperplasia (trans-sternal approach), • Thymoma (locally invasive)
– Immunotherapy • steroids, and other agents including Azathioprine• plasma exchange, • IVIG

Lambert Eaton Myasthenic syndrome (LEMS)
• A defect of neuromuscular transmission with reduced quantal release of Acetyl Choline from the presynaptic nerve terminal
• Pathogenic antibodies directed against voltage gated calcium channels (VGCCS) expressed at the NMJ and autonomic ganglia
• 2/3 patients with LEMS have cancer, most commonly Small cell lung Ca (express VGCCs)

Lambert Eaton Myasthenic syndrome (LEMS)
• Clinical features– Dry mouth
– Fatigable weakness of proximal muscles (like MG)
– Wasting of proximal muscles (X MG)
– Depressed reflexes (X MG)
– Ocular and bulbar weakness rare (X MG)

Lambert Eaton Myasthenic syndrome (LEMS)
Diagnosis
Typical clinical picture
Detection of anti-VGCC antibodies in serum
Positive Tensilon test (like MG)
Repeptitive nerve stimulation at low frequency leads to a decrement in compound muscle action potential amplitude (like MG)
Repeptitive nerve stimulation at high frequency leads to a increment in compound muscle action potential amplitude (X MG)

Repetitive Nerve Stimulation (Supramaximal 2Hz)

Lambert Eaton Myasthenic syndrome (LEMS)
Treatment
• Treating the underlying lung tumour improves LEMS
• Treatment for LEMS per se– Symptomatic (mestinon, 3-4 DAP)
– Immunotherapy (steroids, plasma exchange, IVIG)

Muscle Disease
Does the patient
(i) have a muscle disorder and
(ii) is it acquired or inherited?

Inherited muscle disease : Congenital Myopathies
• Congenital myopathies– Lack of structural proteins e.g. Merosin
deficient– Ultrastructural appearance
• Central Core• Nemaline Rod

Inherited muscle disease : Dystrophy• Lack of structural proteins
– Dystrophinopathies (Becker, Duchenne)– Sarcoglycanopathies (4 forms of LGMD)
• Alteration in muscle enzymes– Calpain 3– Myotonic dystrophy (DMPK)

Inherited muscle disease : Other• Defects in muscle energy metabolism
– glyco(geno)lysis e.g. McArdles– lipid oxidation
– CPT 1, CPT 2– carnitine deficiency
– mitochondrial defects

Acquired muscle disease (i)
Drugs (statins, diuretics)
Endocrine (PTH,thyroid)
Inflammatory (PM, DM, IBM)
Infective (pyomyositis, viral myositis)
Toxic (venom)
Paraneoplastic (DM)

Acquired muscle disease (ii)
The spectrum of inflammatory myopathies
•Polymyositis
–Idiopathic
–Drug induced
–Collagen vascular disease
–Jo-1 and other antibodies
–Sarcoidosis
•Dermatomyositis
–Childhood
–Adult
–Malignancy associated
Inclusion Body Myositis
•Others–Fascitis
–Focal myositis
–Orbital

Clinical features of myopathies• Muscle pain
• Muscle weakness (limb, bulbar, respiratory)
• Breathlessness (respiratory or cardiovascular)
• Palpitations (cardiomyopathy or arrthymia)
• Skin rash and other systemic features
• Myoglobinuria

Muscle Pain• Muscle disorders associated with contractures (glycolytic
myopathies)
• Conditions associated with cramps (idiopathic, neurogenic etc)
• Muscle disorders associated with muscle stiffness (myotonias, PMR, FM)
• Muscle disorders causing localised or diffuse myalgia (IM, PMR, FM, Mitochondrial)
(Kissel and Miller 1999)

Causes of muscle pain (i)
• Drug or toxin induced– alcohol
– Via hypokalaemia: diuretics
– Other mechanisms: statins, heroine
• Secondary to systemic disease– defects of calcium metabolism: hyper or hypo PTH
– defects of thyroid function: hyper or hypo thyroidism
– connective tissue disease:SLE, MTCD, SS, etc

Causes of muscle pain (ii) Primary muscle diseasesInflammatory myopathies:
• polymyositis • dermatomyositis • IBM
Defects in muscle energy metabolism:• glycogenolysis• lipid oxidation• mitochondrial defects
Dystrophies:• dystrophinopathy• facioscapulohumeral dystrophy
Others• PROMM

Symptoms of muscle weaknessClassically muscle disorders produce proximal weaknesswith difficulties in
– Reaching for high objects– Combing hair– Getting up from low chairs– Getting up stairs– Getting up on to a bus
Bulbar symptoms- dysphagia, dysarthria
Breathlessness (RS)

History of muscle weakness often distinguishes acquired or inherited myopathies
– Pre-natal (foetal movements)– Peri-natal (delivery, flat, floppy infant)– Motor milestones (sit, crawl, walk, run)– School (level of sporting achievement)– Occupational hository Armed forces

Muscle Weakness (i)• Manual muscle testing and MRC scores
• Giveway, pain, and fatigue
• Functional assessments, squat, timed Gowers
• Hand held dynamometry
• Quantitative myometry

Muscle Weakness (ii)
Classically muscle disorders present with a proximal or limb girdle pattern of weakness,
BUT
1. Proximal weakness may be due to peripheral neuropathies
2. Myasthenia produces proximal weakness but not wasting
3. Some myopathies produce distal weakness

Examination of the musculoskeletal system may distinguish acquired or inherited myopathies
– Atrophy– Hypertrophy (pseudo)– Contractures– Rigid spine

Myopathy: bulbar features
• Speech may be nasal
• Swallowing problems
• Aspiration symptoms
• Beware “silent” aspiration
• Neck weakness

Neurological causes of neck flexion weaknessMyopathic disorders
• FSH• Myotonic Dystrophy• Inflammatory myopathies
Neuropathic disorders• GBS • CIDP
Anterior horn cell disorders• MND• SMA
Neuromuscular disorders• Myasthenia Gravis

Myoglobinuria
• Inflammatory myopathies
• Glyco(geno)lysis
• Lipid oxidation defect
• Mitochondrial disorder
• Brody’ syndrome
• Drugs (heroin)

Respiratory Involvement in Neuromuscular Disease (i)
• Respiratory problems occur because of – Reduced central drive (myotonic dystophy)– Weak intercostal muscles– Weak diaphragmatic muscles (acid maltase)– Involvement of phrenic nerves (GBS)– Involvement of neuromuscular junctions of
intercostal and diaphragmatic muscles

Respiratory Involvement in Neuromuscular Disease (ii)
• Respiratory symptoms of Neuromuscular Disease include– Breathlessness on exertion and on lying flat
(diaphragmatic splinting)– Nocturnal hypoventilation may result in
daytime hypersomnolence and early morning headache

Respiratory Involvement in Neuromuscular Disease (iii)
• Neuromuscular Disease reduce lung compliance and cause restrictive and NOT obstructive defects in lung function tests– VCs and not Peak flow are the measure of choice
• Sniff pressures and diaphragmatic screening• • Nocturnal oximetry and ear lobe blood gases

Serum Creatine kinase: value and limitationsThe most sensitive enzyme for muscle disease
BUT1. Normal: does not exclude muscle disease
(mitochondrial, lipid oxidation disorders, ?IM)
2. CK level only approximates the clinical course
3. Slightly abnormal are seen with intramuscular injections, liver disease, neurogenic disorders
4. High CK does not equal Inflammatory myopathy

EMG : value and limitations• EMG (unlike NCS) requires value judgments and is
investigator dependent
• EMG is crucial in excluding neuropathic disorders
• A myopathic EMG occurs in a variety of muscle disorders
• The significance of EMG findings always depends on the clinical context

Muscle biopsy : value and limitations• Biopsy is straightforward but processing is technically demanding
• Choice of muscle to biopsy (weak, avoid atrophied muscles-endstage pathology)
• Like EMG muscle biopsy evaluation requires value judgments and is investigator dependent
• Biopsy needs to be interpreted within the clinical context– Avoid over interpretation: ragged red fibre, type II atrophy– If normal on review consider repeating– Inflammatory myopathy or myopathy with inflammation– Polymyositis v dermatomyositis– Polymyositis v Inclusion body myositis

Polymyositis (PM) -Clinical Features
• Adult onset inflammatory myopathy, very rare<16 years
• Female>male
• Subacute presentation (weeks to months) onset oft unclear
• Initial myalgia muscle tenderness
• Proximal > distal weakness
• Dysphagia, neck and occ respiratory muscle weakness
• No rash

Dermatomyositis (DM) - Clinical Features
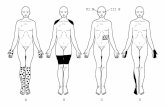
• Childhood and adult onset inflammatory myopathy• Female>male• Rash prior to or with muscle weakness (amyopathic), skin features
can include – Heliotrope rash on eyelids Facial erythema – Erythema of trunk and exposed areas – Erythema of knuckles with scaly eruption- Gottron rash– Dilated capillary loops at base of fingernails– Cracked, dirty lateral borders of hands “Mechanics” hands
• Acute or subacute presentation (few weeks)• Initial myalgia muscle tenderness• Proximal > distal weakness • Dysphagia, neck and occ respiratory muscle weakness

PM and DM-Associations• Connective tissue disorders
– PM: SLE, Sjogren’s, RA – DM: Scleroderma, MCTD (true overlap)
• Interstitial lung disease (ILD) DM>PM– Autoimmune-anti-Jo-1– Drug induced methotrexate
• Cardiac disease– Myocarditis (rare)– Steroids hypertension heart failure– ILD pulmonary hypertension heart failure
• Vasculitis (very rare, DM>PM)
• Systemic features weight loss, fever, malaise DM>PM

Polymyositis and Dermatomyositis-Laboratory
features• Normal or elevated CK (X5-50)
• Raised ESR-but non-specific
• Myopathic EMG, “irritable”- myogenic denervation
• Autoantibodies including Jo-1, ANA, in some patients but non-specific
• Muscle biopsy necrosis, inflammation

PM and DM –Immunopathology (i)Strong circumstantial evidence that PM and DM are
autoimmune
• More common in women
• A range of autoantibodies are commonly detected
• Myositis may arise or fluctuate in pregnancy
• Strong assoc with other both organ and non-organ specific AI disorders PBC, Psoriasis, MG
• Can be induced by D-Penicillamine, like MG

PM and DM – Immunopathology (ii)
PM Muscle biopsies provide evidence of T cell mediated
cytotoxic process directed against unknown muscle
antigens
DM Muscle biopsies provide evidence of Humorally
Mediated microangiopathy directed against unknown
antigens expressed by muscle capillary endothelium

Treatment of Polymyositis and Dermatomyositis (i)
• Prednisolone – 1mg/kg day 60-100mg/day– Response in 4-8 weeks, then taper dose and
change to alternate day regime– Problems include
• Treatment failure
• Steroid side effects
• Steroid myopathy (EMG, muscle biopsy)

Treatment of Polymyositis and Dermatomyositis (ii)
Immunosuppressants other than steroids are useful when• Steroids relatively contraindicated (IDDM etc)• “Steroid-sparing" effect required because of steroid
complications• Repeated relapses on attempts to reduce steroid dosage; • Steroids at an adequate dose ineffective after a 2- to 3-
month period • Rapidly progressive disease with severe weakness and
respiratory failure

Treatment of Polymyositis and Dermatomyositis (iii)
Choice of Immunosuppressants other than steroids is largely empirical
• Azathioprine (1.5-2.0 and rarely 3 mg/kg day)• Methotrexate (benefit?, lung fibrosis, RCT underway)• Cyclophosphamide and cyclosporin are both effective• Others (Tacrolimus, chlorambucil)
Immunomodulation with hIVIG or plasma exchange can
be useful in rapidly progressive disease (short lived)

Treatment of Polymyositis and Dermatomyositis (iv)
Assessing the response to treatment
• Pain subjective, arthralgia as well as myalgia, CTD• Well being steroid effects• ESR non specific• CK chasing, the chemical cure• Weakness clinical exam and myometry• (repeat EMG and muscle biopsy)

Treatment of Polymyositis and Dermatomyositis (v)
• Whilst prognostication is difficult poor prognostic features can include
– Cardiac involvement– Acute onset, very chronic course– Prominent systemic features (fever, arthritis,
vasculitis)– Lung fibrosis