
Multifocal Motor N - AANEM Review... · INVITED REVIEW ABSTRACT: Multifocal motor neuropathy (MMN)...
20
Eduard No one i The id Mu do Nobile-O nvolved in th America deas and opin ultifo Orazio, MD e planning of Aut CM an Associatio 2621 S nions in this Jo cal M D, PhD,Alb f this CME act thors/faculty ME is availabl Copy on of Neurom Superior Dr N ournal Review represent th Motor N berto Capp PhD tivity has any have nothing le 3/15/2008 yright: 200 muscular and NW Rochest w are solely th hose of the AA Neuro ellari, MD y relevant fina g to disclose. - 3/15/2011 05 d Electrodiag ter, MN 559 those of the au ANEM opathy D,and Alber ancial relation gnostic Medi 01 uthor and do Produ y rto Priori, M nships to discl icine not necessari uct: JR03 MD, lose. ily
Transcript of Multifocal Motor N - AANEM Review... · INVITED REVIEW ABSTRACT: Multifocal motor neuropathy (MMN)...

Eduard
No one i
The id
Mudo Nobile-O
involved in th
America
deas and opin
ultifoOrazio, MD
e planning ofAut
CM
an Associatio
2621 S
nions in this Jo
cal MD, PhD,Alb
f this CME actthors/faculty
ME is availabl
Copy
on of Neurom
Superior Dr N
ournal Reviewrepresent th
Motor Nberto Capp
PhD
tivity has any have nothing
le 3/15/2008
yright: 200
muscular and
NW Rochest
w are solely thhose of the AA
Neuroellari, MD
y relevant finag to disclose.
- 3/15/2011
05
d Electrodiag
ter, MN 559
those of the auANEM
opathyD,and Alber
ancial relation
gnostic Medi
01
uthor and do
Produ
y rto Priori, M
nships to discl
icine
not necessari
uct: JR03
MD,
lose.
ily

CMPr CoMmredidythtradneInTheldi LeU
ReExexD AcThCoboon HoOpuvi
ME Informatioroduct: JR03 ‐
ourse DescriptMultifocal motomultifocal partiesponse to higiagnosis, pathoysimmune neuhe pathophysioreatments in Mddressed in teuropathy. ntended Audiehis course is ectrodiagnostisorders.
earning Objectpon conclusion
1. identify2. differe
Barré s3. recogn4. design
elease Date: 3xpiration Datexpiration date.uration/Comp
ccreditation anhe American Aouncil for Conoth sections ofnly the credit c
ow to Obtain Cnce you have urchased this pew a transcr
on Multifocal Mo
tion or neuropathy al motor condgh‐dose intravogenesis, and uropathies; theological basis MMN and the mthis review of
nce intended for ic medicine w
tives n of this progray the clinical anntiate betweesyndrome, chroize serological an appropriate
/15/2008 e: 3/15/2011. Y pletion Time: 2
nd DesignationAssociation of ntinuing Medicf this enduringcommensurate
CME reviewed the product. Answript of your
otor Neuropath
(MMN) is nowduction blocksvenous immuntherapy of the degree of CBof CB; the pamost effective f the main c
Neurologists,with the inten
am, participannd electrodiagn multifocal monic inflammatand imaging fe diagnostic an
Your request to
2 hours
n Statements Neuromuscul
cal Education tg material for ae with the exte
materials, youwer the questioCME by log
hy
w a well‐defines (CB), frequenoglobulin (IVIghis condition iB necessary forathogenetic roregimen; and tclinical, electro
, Physiatrists, t to improve
ts should be abnostic featuresmotor neuropatory demyelinaindings that sund therapeutic
o receive AMA
ar and Electroto provide cona combined mant of their part
u can obtain Cons and click sugging into ww
ed purely motont association g) therapy. Honcluding its nr the diagnosisle of antigangthe treatment ophysiological,
and others wthe quality o
ble to: s of multifocal athy and otherating polyneuroupport or refut strategy for p
PRA Category
odiagnostic Mntinuing medicaximum of 2 Aticipation in th
CME credit by ubmit. Once yww.aanem.org
or multineurowith anti‐GM
owever, severosological poss of MMN; theglioside antiboto be used in , immunologic
who practice of medical car
motor neuropr disorders witopathy and iscte the diagnosiatients with m
1 Credits™ mu
edicine (AANEcal education MA PRA Categhe activity.
clicking on thyour answers hg and clicking
pathy characteM1 IgM antiboral issues remasition and its e existence of odies; the mecunresponsive cal, and ther
neuromusculre to patients
pathy with conth conduction chemic (vasculiis of mulitfocalmultifocal moto
ust be submitte
EM) is accredifor physiciansgory 1 creditsTM
e link in the ehave been submg View Profi
erized by the podies, and usuain to be clarrelation to otan axonal formchanism of actpatients. Thesapeutic featu
ar, musculosks with muscle
duction block.block, includiitic) neuropathl motor neuropor neuropathy.
ed on or befor
ited by the Acs. The AANEMM. Physicians s
e‐mail receivedmitted, you wile and then
presence of ally a good rified in the her chronic m of MMN; tion of IVIg e issues are res of this
keletal, and and nerve
ng Guillain‐hy. pathy.
re the credit
ccreditation designates hould claim
d when you ll be able to My CME.

INVITED REVIEW ABSTRACT: Multifocal motor neuropathy (MMN) is now a well-definedpurely motor multineuropathy characterized by the presence of multifocalpartial motor conduction blocks (CB), frequent association with anti-GM1IgM antibodies, and usually a good response to high-dose intravenousimmunoglobulin (IVIg) therapy. However, several issues remain to be clar-ified in the diagnosis, pathogenesis, and therapy of this condition includingits nosological position and its relation to other chronic dysimmune neurop-athies; the degree of CB necessary for the diagnosis of MMN; the existenceof an axonal form of MMN; the pathophysiological basis of CB; the patho-genetic role of antiganglioside antibodies; the mechanism of action of IVIgtreatments in MMN and the most effective regimen; and the treatment to beused in unresponsive patients. These issues are addressed in this review ofthe main clinical, electrophysiological, immunological, and therapeutic fea-tures of this neuropathy.
Muscle Nerve 31: 663–680, 2005
MULTIFOCAL MOTOR NEUROPATHY: CURRENTCONCEPTS AND CONTROVERSIES
EDUARDO NOBILE-ORAZIO, MD, PhD,1 ALBERTO CAPPELLARI, MD,2
and ALBERTO PRIORI, MD, PhD2
1 Giorgio Spagnol Service of Clinical Neuroimmunology, Dino Ferrari Centre and Centre of Excellencefor Neurodegenerative Diseases, Department of Neurological Sciences, Milan University, IRCCS OspedaleMaggiore Policlinico, and Humanitas Clinical Institute, Via Manzoni 56, 20089 Rozzano, Milan, Italy2 Service of Clinical Neurophysiology, Dino Ferrari Centre, Department of Neurological Sciences,Milan University, IRCCS Ospedale Maggiore Policlinico, Milan, Italy
Accepted 21 December 2004
The term multifocal motor neuropathy (MMN) wasfirst introduced in the literature in 1988 by Pestronket al.,126 who reported two patients with a progressivepurely motor, predominantly distal, asymmetric neu-ropathy with multifocal persistent conduction blocks(CB) on motor but not sensory nerves. A few similarpatients had been reported some years before byseveral other investigators,37,122,123,137 but Pestronk etal.126 first highlighted two of the main features of thisneuropathy: its frequent association with anti-GM1IgM antibodies and response to immune therapies.Between 1992 and 1993, a number of investiga-
tors38,39,68,76,106 started independently to report theeffectiveness in these patients of high-dose intrave-nous immunoglobulin (IVIg) therapy, which is nowconsidered the “gold standard” for treatment ofMMN.156 More recently, diagnostic criteria for thisneuropathy have been proposed by various groups54,166
or associations such as the American Association ofElectrodiagnostic Medicine (AAEM).7,117 Althoughseveral points, including the identification of MMNas a separate nosological entity, have now been clar-ified and extensively reviewed,20,83,104 there are stillunsettled and controversial issues in the diagnosis,pathogenesis, and treatment of this neuropathy.
CLINICAL FEATURES
Clinical Presentation. MMN is a rare disorder prob-ably affecting no more than 1 or 2 individuals per100,000. It is more frequent in men than women,with an approximate ratio of 2.6:1.104 It usually af-fects young adults with a mean age of onset around40 years, and almost 80% of reported patientspresent their first symptoms at between 20 and 50years of age.104
MMN almost invariably presents with asymmetricweakness often related to the distribution of individ-
Available for Category 1 CME credit through the AANEM at www.aanem.org.
Abbreviations: AAEM, American Association of Electrodiagnostic Medicine;ALS, amyotrophic lateral sclerosis; CB, conduction block; CIDP, chronic in-flammatory demyelinating polyneuropathy; CSF, cerebrospinal fluid; CV, con-duction velocity; EMG, electromyography; GBS, Guillain–Barre syndrome;IVIg, intravenous immunoglobulin; MMN, multifocal motor neuropathy; MND,motor neuron disease; MR, magnetic resonance; MUAP, motor unit actionpotential; TD, temporal dispersionKey words: antibodies; axonal excitability; conduction block; GM1; intrave-nous immunoglobulin (IVIg); multifocal motor neuropathy; therapyCorrespondence to: E. Nobile-Orazio; e-mail: [email protected]
© 2005 Wiley Periodicals, Inc.Published online 15 March 2005 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/mus.20296
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 663

ual nerves. Arms are usually affected earlier andmore severely than legs, with more than 80% ofpatients initially affected in forearm or hand mus-cles. Occasional patients may present with moreproximal or even girdle weakness (approximately5%) or with symptoms in their legs (10%).104,148
Early in the disease it is not infrequent for patients tohave a typical crossed distribution (one arm andcontralateral leg). Cranial nerve involvement68,90,96,136
has been reported occasionally in the course of thedisease, often limited to the XIIth cranial nerve,although in one patient ophthalmoplegia was thepresenting symptom.133 A few patients have beenreported with unilateral or bilateral phrenic nervepalsy leading to respiratory failure that, in at leastone of them, was the presenting symptom.19,35,96
More than 50% of MMN patients report fascicula-tions and cramps104 outside the territory of the affectednerves, whereas myokymia is seldom reported.21,90,138
In most patients, tendon reflexes are reduced in apatchy way or diffusely,90,104,148 but may be normal oreven brisk in 20–30% of instances,90,104,126 highlight-ing the similarity of MMN with motor neuron disease(MND). Localized muscle atrophy is usually mild orirrelevant in the early stage of the disease, but wehave seen occasional patients with severe focal atro-phy a few months after onset of symptoms. Atrophyoften correlates with the electrophysiological findingof a marked reduction of distal compound muscleaction potential (CMAP) amplitude or of inexcit-able nerves and often predicts a poor response totherapy.22,106,166
One of the main clinical features of MMN is thepurely motor impairment. Sensory symptoms are,however, reported by some patients in the course ofthe disease, and minor sensory loss has been docu-mented in 20% of patients.104 It is not clear whatdegree of sensory impairment is acceptable for thediagnosis of MMN117 as opposed to multifocal demy-elinating (sensory and motor) neuropathy, whichwas reported by Lewis et al.,95 and is also known asLewis–Sumner syndrome.
Clinical Course and Natural History. After onset, themajority of patients have a slowly progressivecourse.153,170 Some patients, however, have a step-wise progression with rapid deteriorations followedby prolonged stabilization,106,111 or even spontane-ous remission on occasion.22,37 Asymmetry and pre-dominance of arm involvement may become lessevident during the course of the disease but usuallyremain present even after several years. Althoughsome patients have a spontaneously favorable func-tional long-term prognosis in the absence of effec-
tive therapy, most eventually become impaired intheir daily life,170 usually by a reduced dexterity inmanual activities or by fatigue.71 In our series, forinstance, the proportion of patients with a disabilityscore above 2 on the Rankin scale before startingeffective therapy was 12% at 5 years, 25% at 10 years,and 60% at 15 years, although most could still walkindependently.153 Only a few patients have been re-ported to have a fatal outcome after several years ofdisease,96,120 whereas in others death was related toconcomitant diseases,16,19 among them MND.4,80,125,173
A few patients have been reported with an acuteand monophasic presentation of weakness involvingthe four limbs, which was distinguished from Guillain–Barre syndrome (GBS) by the unusually protractedprogression of weakness, purely motor multineuro-pathic distribution, normal stretch reflexes, and pres-ence of persistent CB with normal motor and sensoryconduction velocities (CV).1,145,151,176,177,192 In some pa-tients the disease was preceded by Campylabacter jejuniinfections1,145,176 or therapeutic injection of a mix-ture of bovine gangliosides.151 Even if the investiga-tors originally considered this form an acute variantof MMN, it probably corresponds to the acute motorconduction block neuropathy that was recently pro-posed as a variant of GBS.26
Clinical Diagnostic Criteria and Differential Diagnosis.
Diagnostic criteria for this neuropathy have beenproposed by several groups.7,51,54,117,166 These crite-ria all share similar clinical features, which are wellrepresented by the AAEM117 (Table 1): (1) Weak-ness without objective sensory loss in the distributionof two or more named nerves (and not simply asym-metric weakness). During the early stages of symp-tomatic weakness, a history or physical finding ofdiffuse, symmetric weakness excludes multifocalmotor neuropathy. (2) The absence of each of thefollowing upper motor neuron signs: spastic tone,clonus, extensor plantar responses, and pseudobul-bar palsy. Additional clinical criteria (no more thanseven of eight affected limb regions, predominanceof weakness in the upper limbs, decreased or absenttendon reflexes, and age of onset between 20 and 65years) have also been proposed.166 These featureswere associated with a more frequent response toimmunoglobulin therapy, but it was unclear howthey influenced diagnostic accuracy, and the ab-sence of some of these features is not uncommon inpatients with otherwise typical MMN.
From the clinical point of view, MMN should bedifferentiated mainly from MND,17,37,50,53,101,123,128,173,174
particularly in patients with predominant or exclu-sive lower motor neuron impairment. MMN is usu-
664 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

ally distinguished by its mononeuritis multiplex–like,and not simply asymmetric, pattern of motor impair-ment, absence of bulbar involvement, and slowerprogression. It should also be distinguished fromentrapment neuropathies18 or even hereditary neu-ropathy with liability to pressure palsy. Often theseare characterized by a concomitant motor and sen-sory impairment (which makes it more of a con-founder for Lewis–Sumner syndrome than MMN), attimes a painful acute onset, and by the localization ofthe CB to the usual sites of nerve compression. It isnevertheless not infrequent, at least in our experi-ence, for patients with MMN to have received one ofthese diagnoses before they are diagnosed correctly.More subtle is the distinction from Lewis–Sumnersyndrome, which mainly relies on the absence inMMN of definite sensory impairment, although this
distinction may be difficult in patients with minorsensory symptoms. Whether these are two separatediseases94,115,143,167 or different aspects of the sameclinical spectrum100,122 is still debated. This distinc-tion may be of more than theoretical interest, be-cause some patients with Lewis–Sumner syndromeare reported to respond to steroids,95 which areineffective or even harmful in most MMN patients(see later). Another diagnosis to be considered ischronic inflammatory demyelinating polyneurop-athy (CIDP), particularly its purely motor variant; itsusually symmetrical, often proximal, polyneuro-pathic impairment and its more severe course areusually sufficient to distinguish it clinically fromMMN.
ELECTROPHYSIOLOGICAL FEATURES
Definition and Diagnosis of Partial Conduction Block in
MMN. The essential electrodiagnostic finding ofMMN is persistent, multifocal, partial CB (Fig. 1) ofmotor axons outside the usual sites of nerve com-pression.40,84,124 It is, however, important to under-line that even if CB is the hallmark for diagnosis ofMMN (and Lewis–Sumner syndrome), it is not spe-cific for these diseases as it can also be found inseveral other pathological conditions involving theperipheral nervous system including: (1) other ac-quired demyelinating neuropathies such as CIDP2
and GBS,24 wherein, unlike MMN, CB is associatedwith other prominent electrophysiological abnor-malities suggestive of demyelination; (2) acute com-pressive neuropathy or nerve entrapment, in whichthe CB is found at the usual sites of nerve compres-sion or entrapment; and (3) nerve ischemia, inwhich it is usually transient and reversible.63,65
Hence, the presence of a CB should be always inter-preted in the context of the clinical presentation andof other neurophysiological abnormalities.
CB has been defined as a reduction in the am-plitude or area of the CMAP on proximal comparedto distal stimulation, accompanied by no significantor only focal abnormal temporal dispersion(TD).24,78,150 Unfortunately, no uniformly acceptedcriteria exist for CB identification, as the reductionof CMAP amplitude or area required to be signifi-cant for CB has varied considerably among differentstudies (Table 2). A greater than 20% decline inpeak-to-peak amplitude with a less than 15% changein negative-peak duration of the CMAP has beenused as evidence of CB in GBS24 and CIDP.2 In thesediseases, however, the diagnosis of a demyelinatingneuropathy does not rely only on the presence of CBbut also on other abnormalities suggestive of demy-
Table 1. Criteria for the diagnosis of multifocal motor neuropathy.
Criteria for definite multifocal motor neuropathy:1. Weakness without objective sensory loss in the distribution
of two or more named nerves. During the early stages ofsymptomatic weakness, the historical or physical finding ofdiffuse, symmetric weakness excludes multifocal motorneuropathy.
2. Definite conduction block (see Table 3) is present in two ormore nerves outside of common entrapment sites.*
3. Normal sensory nerve conduction velocity across the samesegments with demonstrated motor conduction block.
4. Normal results for sensory nerve conduction studies on alltested nerves, with a minimum of three nerves tested. Theabsence of each of the following upper motor neuron signs:spastic tone, clonus, extensor plantar response, andpseudobulbar palsy.
Criteria for probable multifocal motor neuropathy1. Weakness without objective sensory loss in the distribution
of two or more named nerves. During the initial weeks ofsymptomatic weakness, the presence of diffuse, symmetricweakness excludes multifocal motor neuropathy.
2. The presence of either:(a) Probable conduction block in two or more motor nervesegments that are not common entrapment sites, or(b) Definite conduction block in one motor nerve segmentand probable conduction block in a different motor nervesegment, neither of which segments are commonentrapment sites.
3. Normal sensory nerve conduction velocity across the samesegments with demonstrated motor conduction block, whenthis segment is technically feasible for study (i.e., this is notrequired for segments proximal to axilla or popliteal fossa).
4. Normal results for sensory nerve conduction studies on alltested nerves, with a minimum of three nerves tested.
5. The absence of each of the following upper motor neuronsigns: spastic tone, clonus, extensor plantar response, andpseudobulbar palsy.
*Median nerve at wrist; ulnar nerve at elbow or wrist; peroneal nerve atfibular head.Reproduced from Olney et al.117
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 665

elination, including reduced conduction velocitiesand increased minimal F-wave latencies and distallatencies. This is not the case for MMN in which theelectrophysiological diagnosis relies only on CB, jus-tifying the need for more specific criteria. Most stud-ies on MMN have used a CMAP amplitude reductionof �50% with a duration increase �15% as indica-tive of proximal CB. The use of these criteria haslikely been influenced by the study of Rhee et al.135
on computer analysis of the effect of “interphasecancellation” on motor unit action potentials(MUAPs). This phenomenon is caused by the over-lap and cancellation of the positive and negative
components of different MUAPs that may occur inchronic axonal loss as a consequence of the in-creased polyphasia and reduced number of MUAPs,or in chronic demyelination due to the markedlyincreased range of conduction velocities. It may re-sult in a disproportionate proximal CMAP amplitudereduction mimicking a true CB.88,135,146 Through asimulation of phase interactions of individualMUAPs, these investigators showed that the reduc-tion of the CMAP area caused by interphase cancel-lation may reach 50% and that greater CMAP reduc-tion may therefore be caused by some degree ofCB.135 It remains unclear whether this model alsoapplies in vivo to human diseases. In addition, sub-stantial differences in the degree of CMAP ampli-tude reduction and duration increase on proximalstimulation have been reported among variousnerves in normal controls,149 so that different crite-ria may be required for the diagnosis of CB in armand leg nerves.6,30,107,147,166
Consensus criteria for the diagnosis of CB havebeen proposed recently by the AAEM (Table 3).7In this consensus, the proponents emphasized theneed to ensure suparamaximal stimulation ofnerves, particularly in obese individuals or in thepresence of limb edema. They also advised not toapply these criteria in the context of severe axonalloss, quantified as a distal CMAP amplitude of lessthan 20% of the lower normal limit. The criteriaproposed for both definite CB and probable par-tial CB were intentionally restrictive to avoid con-fusion between real CB and CMAP amplitude re-duction due to interphase cancellation, and variedwith different nerves, being more restrictive forthe radial, peroneal, and tibial nerves than for theulnar and median nerves. The use of these strin-gent criteria may, however, lead to the underdiag-nosis of MMN, a potentially treatable neuropa-thy.30,74 Although CB is considered to bepersistent,88,95 it is a dynamic entity that changesover time, and a CMAP reduction of �50% may bepreceded by a smaller decrease.30 This was exem-plified by one of our patients who initially pre-sented with a 24% reduction of proximal vs. distalCMAP amplitude without temporal dispersion(TD) in the right ulnar and median nerves inner-vating weak muscles, which, 2 years later, as thepatient became more severely affected, increasedto 70% and 88%, respectively.30 However, minordegrees of CB may be found in nerves to muscleswith normal strength and should be therefore in-terpreted cautiously. In some nerves, CB can bediagnosed, even if it does not attain the aforemen-tioned level, by stimulation at several sites as close
FIGURE 1. Conduction block in the right ulnar nerve in a patientwith multifocal motor neuropathy. The amplitude and area of thecompound muscle action potentials evoked by repeated stimula-tion at different sites along the nerve abruptly decrease between6 and 9 cm proximal to the wrist. A second area of amplitudedecrease with clear temporal dispersion is observed on moreproximal stimulation distal to the elbow, suggesting the presenceof a possible further segment with conduction impairment.
666 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

as 2–2.5 cm apart (“inching” technique).7,46,85 Us-ing this technique, the localization of an abrupt,focal reduction of CMAP amplitude to a relativelysmall region of nerve (Fig. 1) may allow the dis-tinction between focal CB47 and the pseudo-CBsometimes observed in chronic axonal loss andcharacterized by a gradual reduction in CMAPamplitude with increasing separation of stimula-tion sites.6
CB may be present in several different nervesand in several segments (Fig. 1) along the courseof the same nerve.6,161 Motor nerve stimulation upto the axilla fails to detect CB occurring in themost proximal segments. The absence of an F-wave, although suggestive, cannot be relied upon
as secure evidence of proximal CB.9 Recently,magnetic or transcutaneous cervical stimulationwas found useful in the localization of proximalCB in patients suspected of having MMN.9,33 Evenif this technique is useful when no CB can beidentified by more distal stimulation, it should beinterpreted with caution due to the difficulty inproducing supramaximal stimulation of demyelinatednerve roots, especially by magnetic stimulation.7 CBdistal to the most distal site of stimulation is suggestedby a low-amplitude CMAP recorded by a weak, but notwasted, muscle.29 Weakness of a muscle supplied by anerve with no evident motor CB raises the possibility ofanomalous innervation of the affected muscle by acontiguous nerve with CB.31
Table 2. Reported diagnostic criteria for partial conduction block (CB) in patients with multifocal motor neuropathy.
Referenceno.
No. ofpatients
Reduction of proximal to distal CMAP Increase of proximal todistal CMAP duration
(maximal TD)Amplitude Area
138 1 �20% 15%165 1 �20% 15%57 13 �30% 15%87 10 �50% �50% 30% (�30% CB � TD)68, 69 3 �40% �20%106 5 �40% 15%30 3 �15% median, �20% ulnar 15%
�35% peroneal, �50% tibial 25%39, 40 9 �50% �50% 15%11 5 �40% �40%51 4 �40%21, 90 24 �50% proximal to axilla �50% proximal to axilla 30%
�30% distal to axilla �30% distal to axilla136 8 �50% �30% (tibial �50%)162 6 �50% �50% 30%66 8 �50% �50% 30%74 16 (5 no CB) �50% �50% 30% (�30% possible CB)171 9 Definite: �50%; possible: �30% arm, 40% leg118 5 (no CB) �50% 15%164 7 (5) �50%14 2 �40%16 9 �50% �50% 15%101 12 �50% �50% 30%7 See Table 3148 46 Definite: �20% within 10 cm or �80% over 10 cm (except tibial);
probable: 50–79% within 10 cm (except tibial) or �75% for tibial;possible: 30–49% within 10 cm (except tibial) or 50–74% for tibial
56 16 �30% �30% 15%166 37 Definite: 50% area in long segment or 30% amplitude over 2.5 cm;
probable: 30% amplitude in an arm nerve(distal CMAP must be �1 mV)
75 9 (no CB) 30% in amplitude or area across any standard segment107* 23 (3 no CB) 30% arm, 40% leg or 30%
40% arm and 50% leg or 31–60%50% arm and 60% leg �60%
Criteria for definite and probable CB: same as American Association of Electrodiagnostic Medicine.7
*Criteria for possible CB.
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 667

MMN without Partial CB: An Unsettled Issue. Someinvestigators have questioned whether the presence ofCB should be considered a mandatory criterion for thediagnosis of MMN for patients with an otherwise typicalclinical presentation.74,118 In some of these patientswithout CB, a lesser degree of amplitude reductionthan required by the current diagnostic criteria waspresent, whereas in others no evidence of CB could befound, although some other minor features of demy-elination were often present. More recently, Katz etal.75 introduced the term “axonal MMN” to describenine patients with a clinically typical presentation ofMMN who had neither CB nor other features of demy-elination. Some of these patients also had anti-GM1antibodies and improved with IVIg therapy. However,it was unclear whether these patients ever or no longerhad CB. We and others have observed, for instance,patients in whom typical CB may decrease or evendisappear in some nerves after several years of thedisease because of a progressive reduction in distalCMAP amplitude.147,152,169 This could reflect eithersecondary axonal degeneration or the appearance of
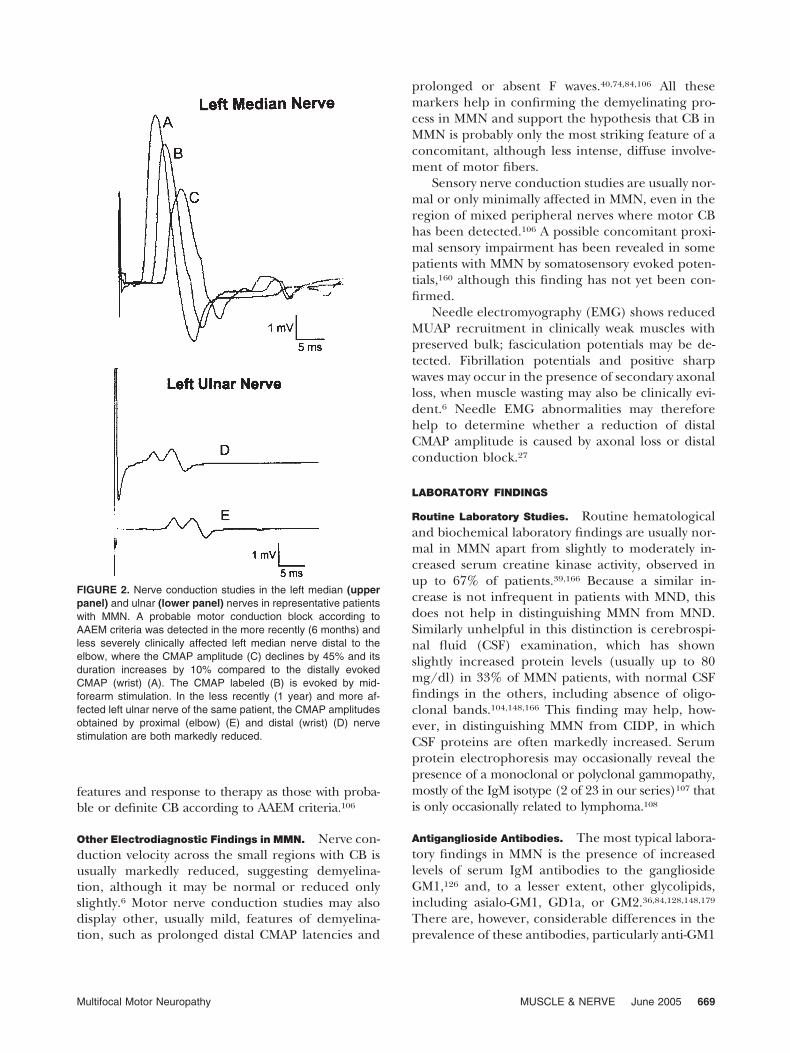
unrecognized, very distal CB. This may occur early inthe disease, as was probably the case in one of ourpatients presenting with a 1-year history of weakness inthe territory of the left ulnar nerve, resulting in severemuscle atrophy, followed by more recent involvementof muscles innervated by the left median nerve. At ourfirst evaluation, motor nerve conduction studies dis-closed a markedly reduced distal and proximal CMAPamplitude (0.5 mV) in the initially affected left ulnarnerve, and a 45% CB with minimal TD in the morerecently affected left median nerve (Fig. 2). This exam-ple clarifies how the same pathological process mayexpress itself in different nerves with electrodiagnosticfeatures consistent with either CB or axonal degener-ation. In any case, we prefer to consider CB an essentialelectrodiagnostic marker of MMN, although for thisdiagnosis we accept the less restrictive criteria of possi-ble CB (Table 2), defined as a 10% lesser degree ofCMAP reduction than that required by the AAEM forprobable CB.106 This approach is appropriate becausepatients with the typical clinical features of MMN and apossible CB had the same clinical and immunological
Table 3. Proposed criteria for partial conduction block.
Nerve segment (proximal/distalstimulation sites)
Minimal temporal dispersion (duration increased by 30% or less)
Moderate temporal dispersion(duration increased by 31% to
60%)
Definite partial conduction blockProbable partial conduction
blockProbable partial conduction
block
Amplitudereduction (%)
Area reduction(%)
Amplitudereduction (%)
Areareduction (%)
Amplitudereduction (%)
Areareduction (%)
MedianForearm (elbow/wrist) �50 �40 40–49 30–39 �50 �40Arm (axilla/elbow) �50 �40 40–49 30–39 �50 �40Proximal (EP/axilla) Not accepted* Not accepted* �40 �30 �50 �40
UlnarForearm (below elbow/wrist) �50 �40 40–49 30–39 �50 �40Across elbow (above/below) �50 �40 40–49 30–39 �50 �40Arm (axilla/above elbow) �50 �40 40–49 30–39 �50 �40Proximal (EP/axilla) Not accepted* Not accepted* �40 �30 �50 �40
RadialForearm (elbow/distal
forearm) Not accepted* Not accepted* �50 �40 �50 �40Arm (axilla/above elbow) Not accepted* Not accepted* �50 �40 �50 �40Proximal (EP/axilla) Not accepted* Not accepted* �50 �40 �50 �40
PeronealLeg (below fibular/ankle) �60 �50 50–59 40–49 �60 �50Across fibular head (above/
below) �50 �40 40–49 30–39 �50 �40Thigh (SN/above fibular) Not accepted* Not accepted* �50 �40 �60 �50
TibialLeg (knee/ankle) �60 �50 50–59 40–49 �60 �50Thigh (SN/knee) Not accepted* Not accepted* �50 �40 �60 �50
Reproduced from the American Association of Electrodiagnostic Medicine.7 EP, Erb’s point; SN, sciatic notch.*Reproduced with the permission from reference 1, which can also be consulted for the reason that definite partial conduction block is not accepted for thesesegments.
668 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005
features and response to therapy as those with proba-ble or definite CB according to AAEM criteria.106
Other Electrodiagnostic Findings in MMN. Nerve con-duction velocity across the small regions with CB isusually markedly reduced, suggesting demyelina-tion, although it may be normal or reduced onlyslightly.6 Motor nerve conduction studies may alsodisplay other, usually mild, features of demyelina-tion, such as prolonged distal CMAP latencies and
prolonged or absent F waves.40,74,84,106 All thesemarkers help in confirming the demyelinating pro-cess in MMN and support the hypothesis that CB inMMN is probably only the most striking feature of aconcomitant, although less intense, diffuse involve-ment of motor fibers.
Sensory nerve conduction studies are usually nor-mal or only minimally affected in MMN, even in theregion of mixed peripheral nerves where motor CBhas been detected.106 A possible concomitant proxi-mal sensory impairment has been revealed in somepatients with MMN by somatosensory evoked poten-tials,160 although this finding has not yet been con-firmed.
Needle electromyography (EMG) shows reducedMUAP recruitment in clinically weak muscles withpreserved bulk; fasciculation potentials may be de-tected. Fibrillation potentials and positive sharpwaves may occur in the presence of secondary axonalloss, when muscle wasting may also be clinically evi-dent.6 Needle EMG abnormalities may thereforehelp to determine whether a reduction of distalCMAP amplitude is caused by axonal loss or distalconduction block.27
LABORATORY FINDINGS
Routine Laboratory Studies. Routine hematologicaland biochemical laboratory findings are usually nor-mal in MMN apart from slightly to moderately in-creased serum creatine kinase activity, observed inup to 67% of patients.39,166 Because a similar in-crease is not infrequent in patients with MND, thisdoes not help in distinguishing MMN from MND.Similarly unhelpful in this distinction is cerebrospi-nal fluid (CSF) examination, which has shownslightly increased protein levels (usually up to 80mg/dl) in 33% of MMN patients, with normal CSFfindings in the others, including absence of oligo-clonal bands.104,148,166 This finding may help, how-ever, in distinguishing MMN from CIDP, in whichCSF proteins are often markedly increased. Serumprotein electrophoresis may occasionally reveal thepresence of a monoclonal or polyclonal gammopathy,mostly of the IgM isotype (2 of 23 in our series)107 thatis only occasionally related to lymphoma.108
Antiganglioside Antibodies. The most typical labora-tory findings in MMN is the presence of increasedlevels of serum IgM antibodies to the gangliosideGM1,126 and, to a lesser extent, other glycolipids,including asialo-GM1, GD1a, or GM2.36,84,128,148,179
There are, however, considerable differences in theprevalence of these antibodies, particularly anti-GM1
FIGURE 2. Nerve conduction studies in the left median (upperpanel) and ulnar (lower panel) nerves in representative patientswith MMN. A probable motor conduction block according toAAEM criteria was detected in the more recently (6 months) andless severely clinically affected left median nerve distal to theelbow, where the CMAP amplitude (C) declines by 45% and itsduration increases by 10% compared to the distally evokedCMAP (wrist) (A). The CMAP labeled (B) is evoked by mid-forearm stimulation. In the less recently (1 year) and more af-fected left ulnar nerve of the same patient, the CMAP amplitudesobtained by proximal (elbow) (E) and distal (wrist) (D) nervestimulation are both markedly reduced.
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 669

IgM, in MMN,104 with figures ranging in differentseries from 30% to 80%,3,12,21,34,39,74,82,139,148,164 al-though usually between 40% and 50%. These dis-crepancies have been variably attributed to technicaldifferences in the procedures adopted by differentlaboratories,96,172,180,186 or in the often unclear diag-nostic criteria adopted for MMN or in the controlsused to establish normal reference values.34,62 Still,the reasons for such a wide variation in the percent-age of abnormal findings remains to be elucidated.
The issue of the diagnostic relevance of theseantibodies is further complicated by the fact thatthese antibodies are not specific for MMN as theyhave also been reported, although less frequently, inother dysimmune neuropathies,64,86,139,181 in approx-imately 10% of patients with MND with exclusivesigns of lower motor neuron impairment and no CB,and in 5% of those with amyotrophic lateral sclerosis(ALS).11,79,105,140,147,158,172,183 Several attempts havebeen made, with conflicting results,34 to standardizethe anti-GM1 assay used among different laborato-ries,97,180,186 or to improve the sensitivity and speci-ficity for MMN of these antibodies using more so-phisticated enzyme-linked immunoassay (ELISA)techniques129 or agglutination immunossays.5 How-ever, there remains no consensus on the most effec-tive and reproducible ELISA procedure and, evenmore importantly, on the diagnostic relevance ofthis assay in the diagnosis of MMN.62,121 In the afore-mentioned diagnostic criteria of the AAEM,117 forinstance, the presence of these antibodies is not evenincluded as possibly supportive laboratory criteriafor this diagnosis, and in those proposed by others166
it was less relevant than normal or slightly increasedlevels of CSF protein. However, two large studies,including a meta-analysis on the diagnostic value ofanti-GM1 IgM antibodies, indicated that their pres-ence can help to distinguish MMN from MND.147,172
This assay therefore may be helpful for patients inwhom a definitive diagnosis cannot be establishedafter clinical and electrophysiological evaluation,even though the absence of these antibodies doesnot exclude the diagnosis of MMN.
PATHOLOGICAL FEATURES
Nerve biopsy is seldom if ever useful in the diagnosisof MMN because it is routinely performed on thesural or other sensory nerves, which are typicallyclinically and electrophysiologically normal in MMN.This may explain why pathological studies have ei-ther shown normal findings or mild axonal degen-eration or demyelination, or both.21,47,51,84,106,111,126
Even ultrastructural studies on these nerves may re-
veal only mild pathological abnormalities consistentwith a demyelinating process.47 Sural nerve biopsy istherefore not indicated in the diagnosis of MMNunless other diagnoses, such as CIDP, Lewis–Sumnersyndrome, or nerve vasculitis, are also suspected.Motor nerve biopsy has been proposed to differen-tiate motor neuropathy, including MMN, from mo-tor neuron disease, as it showed in the former asignificantly higher density of regenerative small my-elinated fibers associated in some patients with evi-dence of demyelination with thinly myelinated axonsand small onion-bulb formations.45 This procedureis available in only a few centers and is definitelymore invasive than sensory nerve biopsy. In two pa-tients in whom a motor or mixed nerve biopsy wasperformed adjacent to the site of CB,10,69 patholog-ical studies showed demyelination with onion bulbswithout inflammatory infiltrates. The presence ofdemyelination at the level of the motor CB in thebrachial plexus was also observed at autopsy in apatient in whom MMN was associated with an other-wise clinically typical and ultimately fatal MND un-responsive to immune therapies, and with the patho-logical features of MND in the spinal cord.173 Thesepathological findings at the site of CB were notconfirmed in a systematic study of seven patientswith typical MMM in whom fascicular nerve biopsy atthe site of the CB disclosed only multifocal fiberdegeneration and loss and clusters of regeneratingfibers without obvious sign of demyelination.149 Intwo patients with predominantly motor neuropathywith multifocal CB, the pathological features ofCIDP were reported with widespread segmental de-myelination, and generally prominent inflammatorycell infiltrates in the motor cranial nerves and motorroots114 or in the brachial plexus, where large onionbulbs were also observed.22 However, the markedlyreduced motor conduction velocities observed inboth patients, together with the diffuse althoughasymmetric motor impairment in one and the re-markable vibratory impairment with prominent sen-sory conduction abnormalities in the other, weremore reminiscent of CIDP or Lewis–Sumner syn-drome than of MMN.
MAGNETIC RESONANCE IMAGING
Magnetic resonance (MR) imaging studies of theforearm69 or brachial plexus171 reportedly show, insome patients with MMN, an asymmetrically in-creased signal intensity in T2-weighted images or inT1-weighted images after gadolinium enhancement,often colocalizing with CB, and thus confirming thefocality of the pathological process in MMN. Even if
670 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

these findings do not clarify the nature of the un-derlying pathological process except for demonstrat-ing the presence of an impaired blood–nerve bar-rier, they indicate that this noninvasive techniquemay have potential for diagnostic usefulness inMMN, particularly with regard to exploration ofproximal nerve segments.
PATHOPHYSIOLOGY OF MMN
Axonal Excitability Studies. Although the pathologi-cal basis of persistent partial CB is believed to befocal demyelination,55 with a consequent lack of con-duction of the nervous impulse through a “nude”axon, this hypothesis has been confirmed only rarelyin MMN by morphological studies,10,69,173 so that thepathophysiological mechanisms underlying MMNremain largely unclear.
In recent years, special neurophysiological tech-niques for assessing axonal excitability noninvasivelyin humans have expanded understanding of periph-eral nervous system pathophysiology. Although anexhaustive description is beyond the scope of thisreview and can be found elsewhere,25 these ap-proaches indirectly estimate the ionic mechanismsinvolved in impulse conduction along human axons.They have also prompted interesting hypothesesconcerning the differential functional involvementof specific regions in the axo-myelinic complex andmorphological–functional correlations of nervousimpulse conduction along peripheral myelinated fi-bers. Although relatively simple, the currently avail-able techniques for studying axonal excitability arestill essentially limited to research applications andtheir results should be interpreted with caution be-cause they derive from an indirect approach. Duringaxonal excitability testing, human nerve fibers arestimulated through the skin, subcutaneous tissues,and endoneurial space, and motor axon propertiesare estimated indirectly from changes in the muscleevoked responses. Hence, there is the need to takeinto account the resistive-capacitative properties ofnon-nerve tissue and the possible bias introduced bythe complex dynamics of the motor endplate.
Axonal Excitability Outside the Conduction Block.
Published data on axonal excitability in patients withMMN are rare, occasionally contradictory among thevarious groups, and often derive from small studysamples, mainly due to the rarity of this condition.Yokota et al.184 examined two patients with MMNand found in both an increased motor axonalthreshold also outside the CB. They hypothesizedeither impaired remyelination or a block of Na�
channels. Cappelen-Smith et al.27 measured axonalexcitability after median nerve stimulation outsidethe site of the CB in three patients with MMN, andfound that supernormality was comparable tohealthy controls, but the strength–duration curvetime-constant (chronaxie) was prolonged. In a laterstudy, Kiernan et al.77 measured multiple axonalexcitability variables in the median nerve of six pa-tients with MMN. They stimulated the median nervein the forearm just distal to a well-identified CB andrecorded the CMAP from the abductor pollicis bre-vis muscle. Although the strength–duration curvetime-constant almost matched control values, therheobase, threshold, stimulus–response slope, andsuperexcitability were significantly abnormal. Be-cause depolarizing current normalized these indicesof axonal excitability, they suggested that motor ax-ons distal to the CB are hyperpolarized in MMN,whereas motor axons at the site of the CB are depo-larized. They also hypothesized that the distal hyper-polarization might compensate for proximal depo-larization with an intraaxonal Na� flux going awayfrom the block. In addition, the abnormal increasein Na� influx at the site of the block might eventu-ally lead to axonal degeneration through an intraax-onal increase in Ca��.
We also assessed the strength–duration curveand its descriptors in 22 ulnar nerves from patientswith MMN and found that the strength–durationcurve time-constant was abnormally short.134 In ad-dition, the axonal threshold and rheobasic currentwere abnormally increased and abnormalities wereat least partly reversed soon after IVIg treatment(Fig. 3). These findings suggest that axons are hy-perpolarized and that nodal resting Na� conduc-tances are impaired in MMN, even outside the site ofthe CB. Yet, remarkably, several ulnar nerves had nodocumented conduction block up to the axilla.
In conclusion, despite some differences, thesestudies agree in reporting motor axonal hyperpolar-ization outside the site of the CB, probably arisingfrom impaired Na� conductances and involving clin-ically unaffected nerves as well. Determining how farthese subtle axonal abnormalities extend is impor-tant because hyperpolarization could be either com-pensatory to a hypothetical focal depolarization atthe site of the CB,77 or simply a primary membraneabnormality. Although the origin of hyperpolariza-tion is still unclear, the frequent observation of dif-fuse hyporeflexia or areflexia and cramps or fascic-ulation outside the territory innervated by nerveswith CB in patients with MMN argue in favor ofwidespread axonal involvement. Furthermore, it isunlikely that compensatory hyperpolarization ex-
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 671

tends for more than a few centimeters outside theCB.
Finally, one may wonder whether the observedchanges in motor axonal excitability are specific toMMN or are also common to other disorders involv-ing the motor axons. When we compared axonalexcitability in patients with MMN and MND, wefound that the latter group showed a distinctive pro-file of abnormalities.134 Conversely, patients withCIDP have a pattern of rheobasic and strength–duration abnormalities,28 qualitatively resemblingthat of patients with MMN. The similar pattern ofabnormalities in CIDP and the different pattern inMND suggest that, in MMN, abnormal ionic conduc-tances arise from an abnormal axomyelinic interac-tion, far more complex than a specific conductanceabnormality linked primarily to a pure axonal dys-function.
Axonal Excitability at the Site of the Conduction Block.
Assessing motor nerve abnormalities at the site ofthe CB is difficult for two reasons: first, the CB site isalmost inexcitable by an electrical shock (i.e., thethreshold is very high); and second, the effects of CBare indirectly estimated from changes in the CMAPelicited in a muscle several centimeters distal to thesite of the CB. These findings imply that the conduc-tion abnormalities in MMN might arise also fromother pathogenetic factors involving the axonalmembrane outside the block. Despite these limita-tions, studies involving stimulation outside the CB
have been used for indirectly studying the mem-brane properties at the site of the block.
In five patients with MMN, Kaji et al.71 studied theeffects of voluntary muscle activation on the degree ofCB and on the force developed by muscle twitch. Be-cause motor activity worsened the CB, they concludedthat CB accounts for the pronounced muscle fatigueseen in patients with MMN and that voluntary contrac-tion could represent a reliable method (i.e., a sort ofstress test) for improving the detection of CB in rou-tine electrodiagnostic examinations.
A number of indirect observations, often made inonly a few nerves, have been used to argue that theaxonal membrane is depolarized at the site of the CB,67
and that focal depolarization leads to CB (i.e., a depo-larizing block). When the rest of the membrane aboveand below the CB is hyperpolarized,77,134 the hyperpo-larization outside the block presumably compensatesfor the supposed depolarization at the site of CB. Evenif this issue is crucial for the pathophysiology of MMN,an ultimate proof for axonal depolarization at the siteof the block is still lacking, because, as mentionedearlier, both the threshold185 and the rheobasic cur-rent (personal observation) are markedly increased atthe site of the block, not allowing reliable local assess-ment of membrane properties. An important issuearguing against the hypothesis of focal depolarizationarises from the observation that activity-dependent hy-perpolarization worsens the CB in MMN,71 whereas itshould improve it if the membrane at the site of the CBis depolarized. CB could simply reflect an enhance-ment of the axonal hyperpolarization present outsidethe block with a complete inactivation of resting Na�
conductances accounting for the increased rheobaseand threshold at the site of the CB. Focal hyperpolar-ization would thus explain why a further activity-depen-dent polarization worsens, rather than improves, theCB.
In conclusion, in MMN motor axons are hyper-polarized, probably owing to a diffuse, but variableimpairment of nodal resting Na� conductances. Thedegree of CB in MMN is proportional to the recenthistory of impulse conduction along the motor axon:sustained voluntary activity can transiently worsenCB in MMN by eliciting hyperpolarization. A furtherfocal increase of axonal hyperpolarization, and pos-sibly of the other subtle physiological abnormalitiesobserved outside the CB, might help to explain theCB in MMN. Although the pathophysiological mech-anisms underlying CB are still largely unknown andprobably multiple, even in the same patient, theblock can be viewed as the “tip of the iceberg” ofaxo-myelinic abnormalities in MMN.
FIGURE 3. Motor axonal excitability in MMN. The time-constantof the strength–duration curve (�SD, left panel) and the rheobasiccurrent for eliciting a 50% CMAP (rh50%, right panel). Columnsare means, error bars are SD. Cross-hatched columns are con-trol nerves (n � 22), open columns are non–recently treatednerves (n � 9), and solid columns are recently treated nerves(n � 9). Whereas in recently treated nerves only the rheobasiccurrent was significantly abnormal, in non–recently treatednerves both the �SD and the rh50% differed significantly fromcontrol nerves. Hence, IVIg treatment seems to correct motoraxonal hyperpolarization and nodal abnormalities in MMN (fromPriori et al.134).
672 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

IMMUNOPATHOGENESIS
The frequent association of MMN with antiglycolipidantibodies, and the improvement observed in mostpatients after IVIg or other immune therapies sup-port the widespread opinion that the disease is im-munologically mediated21,82,104 and possibly causedby binding of anti-GM1 antibodies to neural struc-tures. Several pieces of data support this hypothesisbut some argue against it. As already mentioned,anti-GM1 IgM antibodies are significantly associatedwith MMN and reportedly decrease during clinicalimprovement, particularly after therapy with cyclo-phosphamide.57,126,127 There is, however, a consis-tent proportion of patients in most series (50%) notbearing these antibodies, making it unclear whatcauses the disease in such patients. Most of thesepatients also respond to immune thera-pies12,21,39,74,93,162,166 in a way similar to GM1-positivepatients. In addition, response to IVIg does not cor-relate with a consistent decrease in antibody titers. Ifthis lack of correlation seems to exclude the possi-bility that IVIg interferes with antibody synthesis orbinding to GM1 in the in vitro system used to testthem, it cannot exclude an effect on their binding tothe nerve in vivo.
The presence of high titers of anti-GM1 IgM in atleast 5–10% of patients with MND or other dysim-mune neuropathies, and the absence of a definitecorrelation between the fine specificity13,179 or reac-tivity with neural structures43,109,110 of anti-GM1 IgMand the different clinical syndromes, also make itunclear how similar antibodies cause different dis-eases. GM1 may be an ideal target for an immuneresponse causing CB, being highly represented inthe peripheral nervous system178 where it is localizedat the level of the nodes of Ranvier, compact orouter myelin, and motor endplate at the neuromus-cular junction.43,44,89,102,109,110,142,144,154,155 The higherconcentration of GM1 (and GD1a) in motor than insensory nerve myelin114 is also consistent with theselective motor impairment in MMN. IgM depositshave been detected at the level of the nodes ofRanvier in the sural nerve of a patient with MND,multifocal CB, and high titers of anti-GM1 IgM an-tibodies,141 although this finding has not been con-firmed in other patients and, most importantly, inmotor nerves. The concomitant presence of GM1 insensory structures and spinal motor neuron and graymatter leaves it unclear why these structures are notalso affected in MMN.43,102,109,110,154,155 Differencesin the distribution or expression of gangliosides113
or in their ceramide composition112 in motor andsensory fibers, or in the different susceptibility to
nerve injury or repair capability of motor and sen-sory fibers,146 may theoretically explain the selectivemotor impairment in MMN; however, the reason forthis discrepancy remains unknown, as does the al-most invariable predominant upper-limb involve-ment in these patients.
Similarly inconclusive are the results of experi-mental studies both in vitro and in vivo directed attesting the capacity of these antibodies to cause CB.Intraneural injection or exposure to sera from pa-tients with high anti-GM1 antibodies and MMN, butnot with MND, was able to induce focal CB invivo142,159 and in vitro.8 The latter results were notconfirmed, however, using purified anti-GM1 anti-bodies,60 even when binding of the antibody to thenodes of Ranvier and consequent complement acti-vation was demonstrated.119 In another study, a sim-ilar blocking effect on mouse distal motor nerveconduction was experimentally induced in vitro bysera from MMN patients with and without high anti-GM1 antibodies,136 suggesting that sera from pa-tients with MMN may indeed contain a soluble factorable to affect neural transmission in vitro, althoughthe role of anti-GM1 antibody in this blocking effectremains to be proven. A different mechanism foranti-GM1 antibodies was suggested70 by the selectivecapacity of mouse anti-GM1 antibodies to affect invitro the permeability of the blood–nerve barrier,possibly by interfering with the recognition or sig-naling modulation properties of glycosphingolip-ids.72 This possibility may be supported by the morepronounced disruption of the blood–nerve barrierobserved in the sural nerve of patients with antibod-ies vs. those without.73 It is unclear whether thismechanism may be also postulated in MMN, becausethe degree of gadolinium enhancement observed byMR, which is a marker of blood–nerve barrier dam-age, did not seem to correlate with the presence ofthese antibodies.171
If the possible role of anti-GM1 antibodies in thepathogenesis of MMN remains to be clarified, it iseven less clear what causes the production of theseantibodies in MMN. In GBS, high anti-GM1 IgGantibodies have been significantly associated with anantecedent infection by particular strains of the en-teropathogenic bacteria Campylobacter jejuni, whichhas a lipopolysaccharide that is responsible for theinduction of antiganglioside antibodies by a mecha-nism of molecular mimicry.64,181 A similar associa-tion has been raised in some patients who werebelieved to develop MMN and high titers of anti-GM1 antibodies after C. jejuni enteritis,1,145,177 even ifthe acute, monophasic, and symmetrical presenta-tion in most patients was more consistent with an
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 673

acute motor CB neuropathy.26 The possible role ofC. jejuni in MMN was also supported by the reactivityof anti-GM1 antibodies from patients with chronicmotor neuropathies with the lipopolysaccharide ofC. jejuni132,182 and by the induction in vivo of theseantibodies by the lipopolysaccharide of C. jejuni.91
Only 1 of our 20 patients with MMN had highanti–C. jejuni antibodies,151 however, making it un-likely that C. jejuni is responsible for the disease inmost patients.
In conclusion, even if all available data are con-sistent with the hypothesis that MMN is an immune-mediated neuropathy, the effector mechanisms andthe antigenic targets of this immune response stillremain to be fully elucidated.
THERAPY OF MMN
Since the original report by Pestronk et al.127 on theresponse to intravenous cyclophosphamide in twopatients with MMN, a number of immune therapieshave been used on the assumption that MMN is animmune-mediated disease.
Steroids and Plasma Exchange. The vast majority ofpatients with MMN fail to respond to ste-roids,12,39,42,49,50,51,57,66,68,81,84,106,124,126,127,133,163,165,175
even when given in high doses intravenously133,163; in-deed, almost 20% have been reported to worsen, evendramatically, when on this therapy.51,57,68,90,106,163,175 Forinstance, we had a patient with recurrent but consis-tent worsening during the spring, which disappearedafter he stopped using steroid-containing nasal sprayfor allergic asthma.
Plasma exchange,39,57,68,124 immunoadsorption,58
and CSF filtration59 are similarly ineffective in mostreported patients with MMN and marginally effec-tive in a few of them.18,84,133,163 In occasional pa-tients, plasma exchange was rapidly followed by se-vere clinical worsening and by the appearance of CBin previously clinically unaffected motor nerves.33,41,133
These findings highlight the fact that the distinctionbetween MMN and CIDP or Lewis–Sumner syn-drome is not purely of theoretical interest, becausesteroids and plasma exchange, which are usuallyeffective in CIDP and Lewis–Sumner syndrome, areineffective or even dangerous in MMN.
IVIg. After initial reports on the frequent and con-sistent improvement of MMN after IVIg,38,39,68,76,106
this therapy has been used widely in MMN. Al-most 80% of patients with MMN respond toIVIg,21,42,51,66,74,90,107,111,118,166,175,185 which has an ef-
ficacy that has been now confirmed in four random-ized, double-blind, placebo-controlled trials on a to-tal of 46 patients.11,56,93,161 Response to treatmentwas more frequent in patients with than in thosewithout anti-GM1 antibodies,12,42,93,166 although thishas not been confirmed by all investigators.21,40,104
IVIg induces a rapid improvement, which often oc-curs within 1 week of treatment, and is usually moreevident in recently affected limb regions with minoror no effect on stabilized deficits. This correlateswith the fact that, although responding patientsmore frequently have definite or probable CB,107,166
nonresponding patients, at least in our experience,usually have a longer disease duration before ther-apy, increased severity of the disease, and more fre-quent signs of axonal degeneration that may havecaused the disappearance of the CB.107,152 Clinicalimprovement is variably associated with reduction orresolution of motor CB in some but not all of thenerves involved,29,40,42,68,99,106,162,164 yet does not con-sistently correlate with a reduction of antigangliosideantibody titers.14,21,42,106 Only a few patients havepersistent improvement after a single or a fewcourses of therapy; in the vast majority, the effect ofIVIg only lasts for a few weeks and has to be main-tained by periodic IVIg infusions for long periods, ifnot indefinitely.12,99,164,169
IVIg therapy in MMN patients is usually started atthe standard dose of 2 g/kg on 2–5 consecutive days.This is followed by a variable regimen of mainte-nance infusions ranging from 0.4 g/kg once aweek164 to 1–2 g/kg on 2–5 days monthly12 or at thetime of or immediately before clinical worsening,152
which usually occurs after 3–8 weeks. Most patientsbecome progressively less responsive to IVIg after afew years or even a few months of treatment,52 how-ever, and require an increasing dosage (in case ofinsufficient response) or frequency (in case of re-duced duration of the response) of IVIg to maintainimprovement.152,169 Nevertheless, this response toincreasing IVIg dose or frequency tends to declineafter several years concurrently with the progressivereduction of distal CMAP amplitude, which revealsthe development of secondary axonal degenera-tion.151,168 Still, it has been reported recently thatearly use of larger maintenance doses of IVIg mightprevent the development of axonal degenerationand promote reinnervation.176
IVIg is usually as safe and well tolerated in MMNas it is in other diseases; even if minor side effects,including headache, nausea, fever, chills, flushing,malaise, rash, and itching occur after IVIg infusions,they are usually transient and remit spontaneously.Major complications, such as acute renal failure,
674 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

congestive heart failure, aseptic meningitis, neutro-penia, coagulopathy, anaphylaxis, and venous or ar-terial thromboembolism including stroke, are rareand are mostly associated with preexisting risk fac-tors such as renal or heart disease, IgA deficiency,migraine, increased serum viscosity, stroke risk fac-tors, or increased age.16,23,32,48,116 Transmission ofhepatitis C virus does not seem to represent a prob-lem since the introduction of new virucidal steps. Ina review of the complications of IVIg in 88 neuro-logical patients,23 major complications were ob-served in only 4 patients (4.5%), always occurring inpatients with preexisting heart or renal disease andin a bedbound-state. Some patients may developeven severe allergic dermatitis after IVIg. This reac-tion is often caused not by the Ig but by residualproducts of sterilization and are usually prevented bychanging the brand of Ig.
Other Immune Therapies. Less clear is how to pro-ceed in MMN patients not responding or becomingresistant to IVIg. After the original report by Pes-tronk et al.,126 cyclophosphamide given intrave-nously at high doses, followed by oral cyclophosph-amide as maintenance therapy, was reported to beeffective in approximately 50% of MMN patientsassessed,39,57,84,127 whereas low-dose oral cyclophos-phamide alone was seldom effective.68,133 This drugmay have side effects, especially when given at highdoses or for a long period of time, and may thereforebe unsuitable for less severely affected patients, es-pecially if young. Occasional patients improve orstabilize with azathioprine,21,61,124 whereas conflict-ing results were reported with mycophenolatemofetil.15,157 Two patients were recently reported toimprove substantially with cyclosporine, but fol-low-up of these patients was relatively short.103 Inter-feron-�1a was also effective in a few patients withMMN,98,168 some of whom had failed to respond toother therapies, including IVIg. Some patients re-sponded to this therapy better than to IVIg, whereasothers deteriorated, probably because of interrup-tion of previously effective IVIg therapy.168 Morerecently, a positive effect was reported with the anti-CD20 monoclonal antibody rituximab, directedagainst B-lymphocytes, even if the mean improve-ment in muscle strength was quite modest and de-layed in time (1 year).131 These positive results werenot confirmed in two other patients with purelymotor chronic demyelinating neuropathy, includingone patient with MMN and a declining response toIVIg, who had no response to treatment with ritux-imab during the following year.137 For all these ther-apies, the small numbers of reported patients and
the lack of controlled studies do not allow any firmconclusion to be drawn on their possible efficacy inMMN,156 so they should be offered only to compro-mised MMN patients resistant or responding poorlyto IVIg.
Similar consideration can be extended to the useof immunosuppressants as adjunctive therapy to re-duce the frequency or even suspend costly infusionsof IVIg in responsive MMN patients. Low-dose oralcyclophosphamide permitted the frequency of IVIginfusions to be delayed progressively, and sometimestheir prolonged, although ultimately temporary, sus-pension.12,100 A similar sparing effect on IVIg wasrecently reported with mycophenolate mofetil.15
Both drugs were associated in some patients withside effects requiring their suspension. We believethat, until the cost–benefit ratio of the association ofthese immunosuppressive agents with IVIg in MMNis properly addressed in randomized trials, their useshould be discouraged in patients responding toIVIg.
CONCLUSION: OPEN ISSUES IN MMN
MMN is one of the few recently described neurolog-ical disorders whose identification was followed soonafter by the discovery of an effective, although prob-ably not curative, treatment. However, several issuesremain concerning this disease. As briefly men-tioned, although typically characterized by the pres-ence of a CB, the pathophysiological abnormalitiesin MMN probably extend far beyond it, suggestingthat CB detected by standard electrodiagnostic tech-niques represents the “tip of the iceberg” of morewidespread involvement of motor fibers. This mightalso explain why patients with otherwise clinicallytypical MMN but no detectable CB also improve withIVIg. It remains unclear whether the recently re-ported axonal MMN should be considered a variantof typical MMN with CB or may simply represent apossible evolution of the same disease. Similarly,because the presence of antiganglioside antibodiesin MMN seems to represent a marker of an immuneinvolvement in MMN that is of possible diagnosticrelevance rather than the real effector of neuraldamage, this and the target of the immune responsein MMN have yet to be clarified. It also remainsunclear what causes this immune response and therole, if any, of cellular immunity in the disease.Finally, after more that 10 years of experience withIVIg in MMN, it is now evident that, although thistherapy can improve the neuropathy for severalyears, it does not ultimately cure the condition, mak-
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 675

ing it necessary to search for more effective andpossibly curative treatments for this disease.
This study was supported by the Associazione Amici Centro DinoFerrari and IRCCS Ospedale Maggiore Policlinico, Milan, Italy.The authors thank Dr. F. Terenghi, Dr. G. Ardolino, and Dr. B.Bossi for their kind cooperation.
REFERENCES
1. Abruzzese M, Reni L, Schenone A, Mancardi GL, PrimaveraA. Multifocal motor neuropathy with conduction block afterCampylobacter jejuni enteritis. Neurology 1997;48:544.
2. Ad Hoc Subcommittee of the American Academy of Neurol-ogy AIDS Task Force. Research criteria for diagnosis ofchronic inflammatory demyelinating polyneuropathy(CIDP). Neurology 1991;41:617–618.
3. Adams D, Kuntzer T, Burger D, Chofflon M, Magistris MR,Regli F, et al. Predictive value of anti-GM1 ganglioside anti-bodies in neuromuscular diseases: a study of 180 sera. J Neu-roimmunol 1991;32:223–230.
4. Adams D, Kuntzer T, Steck AJ, Lobrinus A, Janzer RC, RegliF. Motor conduction block and high titres of anti-GM1 gan-glioside antibodies: pathological evidence of motor neurop-athy in a patient with motor neuron syndrome. J NeurolNeurosurg Psychiatry 1993;56:982–987.
5. Alaedini A, Wirguin I, Latov N. Ganglioside agglutinin im-munoassay for rapid detection of autoantibodies in immune-mediated neuropathy. J Clin Lab Anal 2001;15:96–99.
6. Amato AA, Dumitru D. Acquired neuropathies. In: DumitruD, Amato AA, Zwarts MJ, editors. Electrodiagnostic medi-cine, 2nd ed. Philadelphia: Hanley & Belfus; 2002. p 937–1041.
7. American Association of Electrodiagnostic Medicine. Con-sensus criteria for the diagnosis of partial conduction block.Muscle Nerve 1999;8(suppl):S225–S229.
8. Arasaki, K, Kusunoki S, Kudo N, Kanazawa I. Acute conduc-tion block in vitro following exposure to anti-gangliosidesera. Muscle Nerve 1993;16:587–593.
9. Arunachalam R, Osei-Lah A, Mills KR. Transcutaneous cer-vical root stimulation in the diagnosis of multifocal motorneuropathy with conduction block. J Neurol Neurosurg Psy-chiatry 2003;74:1329–1331.
10. Auer RN, Bell RB, Lee MA. Neuropathy with onion bulbformations and pure motor manifestations. Can J Neurol Sci1989;16:194–197.
11. Azulay J-P, Blin O, Pouget J, Boucraut J, Bille-Turc F, CarlesG, et al. Intravenous immunoglobulin treatment in patientswith motor neuron syndromes associated with anti-GM1 an-tibodies. Neurology 1994;44:429–432.
12. Azulay J-P, Rihet P, Pouget J, Cador F, Blin O, Boucraut J, etal. Long-term follow-up of multifocal motor neuropathy withconduction block under treatment. J Neurol Neurosurg Psy-chiatry 1997;62:391–394.
13. Baba H, Daune GC, Ilyas AA, Pestronk A, Cornblath DR,Chaudhry V, et al. Anti-GM1 ganglioside antibodies withdiffering fine specificities in patients with multifocal motorneuropathy. J Neuroimmunol 1989;25:143–150.
14. Bech E, Andersen H, Orntoft TF, Jakobsen J. Association ofIgM type anti-GM1 antibodies and muscle strength inchronic acquired demyelinating polyneuropathy. Ann Neu-rol 1998;43:72–78.
15. Benedetti L, Grandis M, Nobbio L, Beronio A, Ghiglione E,Manzino M, et al. Mycophenolate mofetil in dysimmuneneuropathies: a preliminary study. Muscle Nerve 2004;29:748–749.
16. Bertorini TE, Nance AM, Horner LH, Greene W, GelfandMS, Jaster JH. Complications of intravenous gammaglobulinin neuromuscular and other diseases. Muscle Nerve 1996;19:388:391.
17. Bentes C, de Carvalho M, Evangelista T, Sales-Luis ML.Multifocal motor neuropathy mimicking motor neuron dis-ease: nine cases. J Neurol Sci 1999;169:76–79.
18. Beydoun SR. Multifocal motor neuropathy with conductionblock misdiagnosed as multiple entrapment neuropathies.Muscle Nerve 1998;21:813–815.
19. Beydoun SR, Copeland D. Bilateral phrenic neuropathy as apresenting feature of multifocal motor neuropathy with con-duction block. Muscle Nerve 2000;23:556–559.
20. Biessels GJ, Franssen H, van den Berg LH, Gibson A, Kap-pelle LJ, Venables GS, Wokke JHJ. Multifocal motor neurop-athy. J Neurol 1997;244:143–152.
21. Bouche P, Moulonguet A, Younes-Chennoufi AB, Adams D,Bauman N, Meininger V, et al. Multifocal motor neuropathywith conduction block: a study of 24 patients. J NeurolNeurosurg Psychiatry 1995;59:38–44.
22. Bradley WG, Bennett RK, Good P, Little B. Proximal chronicinflammatory polyneuropathy with multifocal conductionblock. Arch Neurol 1988;45:451–455.
23. Brannagan TH, Nagle KJ, Lange DJ, Rowland LP. Compli-cations of intravenous immunoglobulin treatment in neuro-logic disease. Neurology 1996;47:674–677.
24. Brown WF, Feasby TE. Conduction block and denervation inGuillain–Barre polyneuropathy. Brain 1984;107:219–239.
25. Burke D, Kiernan MC, Bostock H. Excitability of humanaxons. Clin Neurophysiol 2001;112:1575–1585.
26. Capasso M, Caporale CM, Pomilio F, Gandolfi P, Lugaresi A,Uncini A. Acute motor conduction block neuropathy. An-other Guillain–Barre syndrome variant. Neurology 2003;61:617–622.
27. Cappelen-Smith C, Kuwabara S, Lyn CSY, Mogyoros I, BurkeD. Activity-dependent hyperpolarization and conductionblock in chronic inflammatory demyelinating polyneurop-athy. Ann Neurol 2000;48:826–832.
28. Cappelen-Smith C, Kuwabara S, Lin SYC, Mogyoros I, BurkeD. Membrane properties in chronic inflammatory demyeli-nating polyneuropathy. Brain 2001;124:2439–2447.
29. Cappellari A, Nobile-Orazio E, Meucci N, Scarlato G, Bar-bieri S. Multifocal motor neuropathy: a source of error inthe serial evaluation of conduction block. Muscle Nerve1996;19:666–669.
30. Cappellari A, Nobile-Orazio E, Meucci N, Scarlato G, Bar-bieri S. Criteria for early detection of conduction block inmultifocal motor neuropathy (MMN): a study based on con-trol populations and follow-up of MMN patients. J Neurol1997;244:625–630.
31. Cappellari A, Barbieri S. Erroneous evaluation of conduc-tion block due to anomalous innervation of the extremities.Muscle Nerve 1997;20:396–397.
32. Caress JB, Cartwright MS, Donofrio PD, Peacock JE. Theclinical features of 16 cases of stroke associated with admin-istration of IVIg. Neurology 2003;60:1022–1828.
33. Carpo M, Cappellari A, Mora G, Pedotti R, Barbieri S, Scar-lato G, et al. Deterioration of multifocal motor neuropathyafter plasma exchange. Neurology 1998;50:1480–1482.
34. Carpo M, Allaria S, Scarlato G, Nobile-Orazio E. Marginallyimproved detection of GM1 antibodies by Covalink ELISA inmultifocal motor neuropathy. Neurology 1999;53:2206–2207.
35. Cavaletti G, Zincone A, Marzorati L, Frattola L, Molteni F,Navalesi P. Rapidly progressive multifocal motor neuropathywith phrenic nerve paralysis: effect of nocturnal assistedventilation. J Neurol 1998;24:613–616.
36. Cavanna B, Carpo M, Pedotti R, Meucci N, Allaria S, ScarpiniE, et al. Anti-GM2 IgM antibodies: clinical correlates andreactivity with a human neuroblastoma cell line. J Neuroim-munol 1999;94:157–164.
37. Chad DA, Hammer K, Sargent J. Slow resolution of multifo-cal weakness and fasciculation: a reversible motor neuronsyndrome. Neurology 1986;36:1260–1263.
676 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

38. Charles N, Benoit P, Vial C, Bierme T, Moreau T, Bady B.Intravenous immunoglobulin treatment in multifocal motorneuropathy. Lancet 1992;340:182.
39. Chaudhry V, Corse A, Cornblath DR, Kuncl RW, DrachmanDB, Freimer ML, et al. Multifocal motor neuropathy: re-sponse to human immune globulin. Ann Neurol 1993;33:237–242.
40. Chaudhry V, Corse A, Cornblath DR, Kunkl RW, FreimerML, Griffin JW. Multifocal motor neuropathy: electrodiag-nostic features. Muscle Nerve 1994;17:198–205.
41. Claus D, Specht S, Zieschang M. Plasmapheresis in multifo-cal motor neuropathy: a case report. J Neurol NeurosurgPsychiatry 2000;68:533–535.
42. Comi G, Amadio S, Galardi G, Fazio R, Nemni R. Clinicaland neurophysiological assessment of immunoglobulin ther-apy in five patients with multifocal motor neuropathy. J Neu-rol Neurosurg Psychiatry 1994;57(suppl):35–37.
43. Corbo M, Quattrini A, Lugaresi A, Santoro M, Latov N, HaysAP. Patterns of reactivity of human anti-GM1 antibodies withspinal cord and motor neurons. Ann Neurol 1992;32:487–493.
44. Corbo M, Quattrini A, Latov N, Hays AP. Localization ofGM1 and Gal(�1-3)GalNAc antigenic determinants in pe-ripheral nerve. Neurology 1993;43:809–814.
45. Corbo M, Abouzahr MK, Latov N, Iannaccone S, QuattriniA, Nemni R, et al. Motor nerve biopsy studies in motorneuropathy and motor neuron disease. Muscle Nerve 1997;20:15–21.
46. Cornblath DR, Sumner AJ, Daube J, Gilliat RW, Brown WF,Parry GJ, et al. Conduction block in clinical practice. MuscleNerve 1991;14:869–871.
47. Corse AM, Chaudry W, Crawford TO, Cornblath DR, KunclRW, Griffin JW. Sural nerve pathology in multifocal motorneuropathy. Ann Neurol 1996;39:319–325.
48. Dalakas MC, Clark WM. Strokes, thromboembolic eventsand IVIg. Rare incidences blemish an excellent safetyrecord. Neurology 2003;60:1736–1737.
49. de Carvalho M, Luis ML. Relapsing chronic low-dose corti-costeroid-responsive multifocal motor neuropathy with con-duction block. Electromyogr Clin Neurophysiol 1997;37:95–97.
50. Di Bella P, Logullo F, Dionisi L, Danni M, Scarpelli M,Angeleri F. Chronic multifocal demyelinating neuropathysimulating motor neuron disease. Ital J Neurol Sci 1991;12:113–118.
51. Donaghy M, Mills KR, Boniface SJ, Simmons J, Wright I,Gregson N, et al. Pure motor demyelinating neuropathy:deterioration after steroid treatment and improvement withintravenous immunoglobulin. J Neurol Neurosurg Psychia-try 1994;57:778–783.
52. Elliot JL, Pestronk A. Progression of multifocal motor neu-ropathy during apparently successful treatment with humanimmunoglobulin. Neurology 1994;44:967–968.
53. Ellis CM, Leary S, Payan J, Shaw C, Hu M, O’Brien M, LeighPN. Use of human intravenous immunoglobulin in lowermotor neuron syndromes. J Neurol Neurosurg Psychiatry1999;67:15–19.
54. European Neuromuscular Center. 79th International Work-shop: multifocal motor neuropathy. Neuromuscul Disord2001;11:309–314.
55. Feasby TH, Brown WF, Gilbert JJ, Hahn AF. The pathologi-cal basis of conduction block in human neuropathies. J Neu-rol Neurosurg Psychiatry 1985;48:239–244.
56. Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE.Multifocal motor neuropathy improved by IVIg. Random-ized, double-blind, placebo-controlled, study. Neurology2000;55:1257–1262.
57. Feldman EL, Bromberg MB, Albers JW, Pestronk A. Immu-nosuppressive treatment in multifocal motor neuropathy.Ann Neurol 1991;30:397–401.
58. Finsterer J, Derfler K. Immunoadsorption in multifocal mo-tor neuropathy. J Immunother 1999;22:441–442.
59. Finsterer J, Schwerer B, Bittner RE, Mamoli B. Cerebrospinalfluid filtration and immunoglobulins in multifocal motorneuropathy. Clin Neuropathol 1999;18:31–36.
60. Harvey GK, Toyka KV, Zielasek J, Kiefer R, Simonis C, Har-tung HP. Failure of anti-GM1 IgG or IgM to induce conduc-tion block following intraneural transfer. Muscle Nerve1995;18:388–394.
61. Hausmanowa-Petrusewicz I, Rowisnka-Marcinska K, KopecK. Chronic acquired demyelinating motor neuropathy. ActaNeurol Scand 1991;84:40–45.
62. Holloway RG, Feasby T. To test or not to test?: that is thequestion in neurology. Neurology 1999;53:1905–1907.
63. Homberg V, Reiners K, Toyka K. Reversible conductionblock in human ischemic neuropathy after ergotamineabuse. Muscle Nerve 1992;15:467–470.
64. Hughes RAC, Hadden RDM, Gregson NA, Smith KJ. Patho-genesis of Guillain–Barre syndrome. J Neuroimmunol 1999;100:74–97.
65. Jamieson PW, Giuliani MJ, Martinez AJ. Necrotizing angiop-athy presenting with multifocal conduction blocks. Neurol-ogy 1991;41:442–444.
66. Jaspert A, Claus D, Grehl H, Neundorfer B. Multifocal motorneuropathy: clinical and electrophysiological findings.J Neurol 1996;243:684–692.
67. Kaji R. Physiology of conduction block in multifocal motorneuropathy and other demyelinating neuropathies. MuscleNerve 2003;27:285–286.
68. Kaji R, Shibasaki H, Kimura J. Multifocal demyelinatingmotor neuropathy: cranial nerve involvement and immuno-globulin therapy. Neurology 1992;42:506–509.
69. Kaji R, Nobuyuki O, Tsuji T, Mezaki T, Nishio T, Akiguchi I,et al. Pathological findings at the site of conduction block inmultifocal motor neuropathy. Ann Neurol 1993;33:152–158.
70. Kaji R, Hirota N, Oka N, Kohara N, Watanabe T, Nishio T, etal. Anti-GM1 antibodies and impaired blood–nerve barriermay interfere with remyelination in multifocal motor neu-ropathy. Muscle Nerve 1994;17:108–110.
71. Kaji R, Bostock H, Kohara N, Murase N, Kimura J, ShibasakiH. Activity-dependent conduction block in multifocal motorneuropathy. Brain 2000;123:1602–1611.
72. Kanda T, Iwasaki T, Yamawaki M, Tai T, Mizusawa H. Anti-GM1 antibody facilitates leakage in an in vitro blood–nervebarrier model. Neurology 2000;55:585–587.
73. Kanda T, Yamawaki M, Iwasaki T, Mizusawa H. Glycosphyn-golipid antibodies and blood–nerve barrier in autoimmunedemyelinative neuropathy. Neurology 2000;54:1459–1464.
74. Katz JS, Wolfe GI, Bryan WW, Jackson CE, Amato AA,Barohn RJ. Electrophysiologic findings in multifocal motorneuropathy. Neurology 1997;48:700–707.
75. Katz JS, Barohn RJ, Kojan S, Wolfe GI, Nations SP, Saper-stein DS, et al. Axonal multifocal motor neuropathy withoutconduction block or other features of demyelination. Neu-rology 2002;58:615–620.
76. Kermode AG, Laing BA, Carroll WM, Mastaglia FL. Intrave-nous immunoglobulin for multifocal motor neuropathy.Lancet 1992;340:920–921.
77. Kiernan MC, Guglielmi JM, Kaji R, Murray NMF, Bostock H.Evidence for axonal membrane hyperpolarization in multi-focal motor neuropathy with conduction block. Brain 2002;125:664–675.
78. Kimura J. Principles and pitfalls of nerve conduction studies.Ann Neurol 1984;16:415–429.
79. Kinsella LJ, Lange DJ, Trojaborg W, Sadiq SA, Younger DS,Latov N. Clinical and electrophysiologic correlates of ele-vated anti-GM1 antibody titers. Neurology 1994;44:1278–1282.
80. Kituchi H, Kawano Y, Dohura K, Kawamura T, Taniwaki T,Yamada T, et al. An autopsy case with lower motor neurondisease showing a transient appearance of anti-GM1 anti-body and an improvement of conduction block after gam-ma-globulin administration (clinical conference). NoShinkei 1999;51:455–464.
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 677

81. Komiyama A, Toda H, Hasegawa O, Kuroiwa K. Multifocalmotor neuropathy. Neurology 1998;50:314.
82. Kornberg AJ, Pestronk A. The clinical and diagnostic role ofanti-GM1 antibody testing. Muscle Nerve 1994;17:100–104.
83. Kornberg AJ, Pestronk A. Chronic motor neuropathies: di-agnosis, therapy and pathogenesis. Ann Neurol 1995;37(suppl 1):S43–S50.
84. Krarup C, Stewart JD, Sumner AJ, Pestronk A, Lipton SA. Asyndrome of asymmetric limb weakness with motor conduc-tion block. Neurology 1990;40:118–127.
85. Kusunoki S, Chiba A, Tai T, Kanazawa I. Localization of GM1and GD1b antigens in the human peripheral nervous system.Muscle Nerve 1993;16:752–756.
86. Lamb NL, Patten BM. Clinical correlations of anti-GM1 an-tibodies in amyotrophic lateral sclerosis and neuropathies.Muscle Nerve 1991;14:1021–1027.
87. Lange DJ, Trojaborg W, Latov N, Hays AP, Younger DS,Uncini A, et al. Multifocal motor neuropathy with conduc-tion block: is it a distinct clinical entity? Neurology 1992;42:497–505.
88. Lange DJ, Trojaburg W, McDonald TD, Blake DM. Persistentand transient ‘conduction block‘ in motor neuron disease.Muscle Nerve 1993;16:896–903.
89. Latov N, Hays AP, Donofrio PD, Liao J, McGinnis S, Man-oussos K, et al. Monoclonal IgM with a unique specificity togangliosides GM1 and GD1b and to lacto-N-tetralose associ-ated with human motor neuron disease. Neurology 1988;33:763–768.
90. Le Forestier N, Chassande B, Moulonguet A, Maisonobe T,Schaeffer S, Birouk N, et al. Neuropathies motrice multifo-cales avec blocs de conduction. 39 cas. Rev Neurol (Paris)1997;153:579–586.
91. Lee G, Jeong Y, Wirguin I, Hays AP, Willison HJ, Latov N.Induction of human IgM and IgG anti-G1 antibodies intransgenic mice in response to lipopolysaccharides fromCampylobacter jejuni. J Neuroimmunol 2004;146:63–75.
92. Lefaucheur JP, Gregson NA, Gray I, von Raison F, BertocchiM, Creange A. A variant of multifocal motor neuropathywith acute, generalized presentation and persistent conduc-tion blocks. J Neurol Neurosurg Psychiatry 2003;74:1555–1561.
93. Leger J-M, Chassande B, Musset L, Meininger V, Bouche P,Bauman N. Intravenous immunoglobulin therapy in multi-focal motor neuropathy: a double-blind placebo-controlledstudy. Brain 2001;124:145–153.
94. Lewis RA. Multifocal motor neuropathy and Lewis–Sumnersyndrome. Two distinct entities. Muscle Nerve 1999;22:1738–1739.
95. Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocaldemyelinating neuropathy with persistent conduction block.Neurology 1982;32:958–964.
96. Magistris M, Roth G. Motor neuropathy with multifocal per-sistent conduction blocks. Muscle Nerve 1992;15:1056.
97. Marcus DM, Latov N, Hsi BP, Gillard BK, and participatinglaboratories. Measurement and significance of antibodiesagainst GM1 ganglioside. Report of a workshop, 18/4/1989,Chicago, USA. J Neuroimmunol 1989;25:255–259.
98. Martina ISJ, van Doorn PA, Schmitz PIM, Meulstee J, van derMeche FGA. Chronic motor neuropathies: response to in-terferon-�1a after failure of conventional therapies. J NeurolNeurosurg Psychiatry 1999;66:197–201.
99. Meucci N, Cappellari A, Barbieri S, Scarlato G, Nobile-Ora-zio E. Long term effect of intravenous immunoglobulins andoral cyclophosphamide in multifocal motor neuropathy.J Neurol Neurosurg Psychiatry 1997;63:765–769.
100. Mezaki T, Kaji R, Kimura J. Multifocal motor neuropathyand Lewis–Sumner syndrome. A clinical spectrum. MuscleNerve 1999;22:1739–1740.
101. Molinuevo JL, Cruz-Martinez A, Graus F, Serra J, Ribalta T,Valls-Sole J. Central motor conduction time in patients withmultifocal motor conduction block. Muscle Nerve 1999;22:926–932.
102. Nardelli E, Anzini P, Moretto G, Rizzuto N, Steck AJ. Patternof nervous tissue immunostaining by human anti-glycolipidantibodies. J Neurol Sci 1994;122:220–227.
103. Nemni R, Santuccio G, Calabrese E, Galardi G, Canal N.Efficacy of cyclosporine treatment in multifocal motor neu-ropathy. J Neurol 2003;250:1118–1120.
104. Nobile-Orazio E. Multifocal motor neuropathy. J Neuroim-munol 2001;115:4–18.
105. Nobile-Orazio E, Legname G, Daverio R, Carpo M, GiulianiA, Sonnino S, et al. Motor neuron disease in a patient witha monoclonal IgMk directed against GM1, GD1b and high-molecular-weight neural-specific glycoproteins. Ann Neurol1990;28:190–194.
106. Nobile-Orazio E, Meucci N, Barbieri S, Carpo M, Scarlato G.High dose intravenous immunoglobulin therapy in multifo-cal motor neuropathy. Neurology 1993;43:537–544.
107. Nobile-Orazio E, Meucci N, Carpo M, Terenghi, Bersano A,Cappellari A, et al. Multifocal motor neuropathy: clinicaland immunological features and response to IVIg in relationto the presence and degree of motor conduction block.J Neurol Neurosurg Psychiatry 2002;72:761–766.
108. Noguchi M, Mori K, Yamazaki S, Suda K, Sato N, Oshimi K.Multifocal motor neuropathy caused by a B-cell lymphomaproducing a monoclonal IgM autoantibody against periph-eral nerve myelin glycolipids GM1 and GD1b. Br J Haematol2003;123:600–605.
109. O’Hanlon GM, Paterson GJ, Wilson G, Doyle D, McHardie P,Willison HJ. Anti-GM1 ganglioside antibodies cloned fromautoimmune neuropathy patients show diverse binding pat-terns in the rodent nervous system. J Neuropathol Exp Neu-rol 1996;55:184–195.
110. O’Hanlon GM, Paterson GJ, Veitch J, Wilson G, Willison HJ.Mapping immunoreactive epitopes in the human peripheralnervous system using monoclonal anti-GM1 ganglioside an-tibodies. Acta Neuropathol 1998;95:605–616.
111. O’Leary CP, Mann AC, Lough J, Willison HJ. Muscle hyper-trophy in multifocal motor neuropathy is associated withcontinuous motor unit activity. Muscle Nerve 1997;20:479–485.
112. Ogawa-Goto K, Funamoto N, Abe T, Nagashima K. Differentceramide compositions of gangliosides between human mo-tor and sensory nerves. J Neurochem 1990;55:1486–1493.
113. Ogawa-Goto K, Funamoto N, Ohta Y, Abe T, Nagashima K.Myelin gangliosides of human peripheral nervous system: anenrichment of GM1 in the motor nerve myelin isolated fromcauda equina. J Neurochem 1992;59:1844–1849.
114. Oh SJ, Claussen GC, Odabasi Z, Palmer CP. Multifocal de-myelinating motor neuropathy: pathological evidence of ’in-flammatory demyelinating polyradiculoneuropathy.’ Neu-rology 1995;45:1828–1832.
115. Oh SJ, Claussen GC, Kim DS. Motor and sensory demyeli-nating mononeuropathy multiplex (multifocal motor andsensory demyelinating neuropathy): a separate entity or avariant of chronic inflammatory demyelinating polyneurop-athy? J Periph Nerv Syst 1997;2:362–369.
116. Okuda D, Flaster Frey J, Sivakumar K. Arterial thrombosisinduced by IVIg and its treatment with tPA. Neurology 2003;60:1825–1826.
117. Olney RK, Lewis RA, Putnam TD, Campellone JV. Consensuscriteria for the diagnosis of multifocal motor neuropathy.Muscle Nerve 2003;27:117–121.
118. Pakiam ASI, Parry GJ. Multifocal motor neuropathy withoutovert conduction block. Muscle Nerve 1998;21:243–245.
119. Paparounas K, O’Hanlon GM, O’Leary CP, Rowan EG, Wil-lison HJ. Anti-ganglioside antibodies can bind peripheralnerve nodes of Ranvier and activate the complement cascadewithout inducing acute conduction block in vitro. Brain1999;122:807–816.
120. Parry GJ. Motor neuropathy with multifocal persistent con-duction blocks. Muscle Nerve 1992;15:1057.
678 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005

121. Parry GJ. Antiganglioside antibodies do not necessarily playa role in multifocal motor neuropathy. Muscle Nerve 1994;17:97–99.
122. Parry GJ. Are multifocal motor neuropathy and Lewis–Sum-ner syndrome distinct nosological entities? Muscle Nerve1999;22:557–559.
123. Parry GJ, Clarke S. Pure motor neuropathy with multifocalconduction block masquerading as motor neuron diseaseMuscle Nerve 1985;8:167.
124. Parry GJ, Clarke S. Multifocal acquired demyelinating neu-ropathy masquerading as motor neuron disease. MuscleNerve 1988;11:103–107.
125. Pelliccioni G, Scarpino O. Amyotrophic lateral sclerosis andmultifocal motor neuropathy with conduction block. J Neu-rol Sci 1997;152:109–110.
126. Pestronk A, Cornblath DR, Ilyas A, Baba H, Quarles RH,Griffin JW, et al. A treatable multifocal motor neuropathywith antibodies to GM1 ganglioside. Ann Neurol 1988;24:73–78.
127. Pestronk A, Adams RN, Kuncl RW, Drachman DB, ClawsonLL, Cornblath DR. Differential effects of prednisone andcyclophosphamide on autoantibodies in human neuromus-cular disorders. Neurology 1989;39:628–633.
128. Pestronk A, Chaudhry V, Feldman EL, Griffin JW, CornblathDR, Denys EH, et al. Lower motor neuron syndromes de-fined by patterns of weakness, nerve conduction abnormal-ities, and high titers of antiglycolipid antibodies. Ann Neurol1990;27:316–326.
129. Pestronk A, Choksi R. Multifocal motor neuropathy. SerumIgM anti-GM1 ganglioside antibodies in most patients de-tected using covalent linkage of GM1 to ELISA plates. Neu-rology 1997;49:1289–1292.
130. Pestronk A, Cornblath DR, Ilyas A, Baba H, Quarles RH,Griffin JW, et al. A treatable multifocal motor neuropathywith antibodies to GM1 ganglioside. Ann Neurol 1988;24:73–78.
131. Pestronk A, Florence J, Miller T, Choksi R, Al-Lozi MT,Levine TD. Treatment of IgM associated polyneuropathiesusing rituximab. J Neurol Neurosurg Psychiatry 2003;74:485–489.
132. Prendergast MM, Willison HJ, Moran AP. Human monoclo-nal immunoglobulin M antibodies to ganglioside GM1 showdiverse cross-reactivities with lipopolysaccharides of Campy-lobacter jejuni strains associated with Guillain–Barre syn-dromes. Infect Immun 1999;67:3698–3671.
133. Pringle CE, Belden J, Veitch JE, Brown WF. Multifocal motorneuropathy presenting as ophthalmoplegia. Muscle Nerve1997;20:347–351.
134. Priori A, Cinnante C, Pesenti A, Carpo M, Cappellari A,Nobile-Orazio E, et al. Distinctive abnormalities of motoraxonal strength-duration properties in multifocal motorneuropathy and in motor neuron disease. Brain 2002;125:2481–2490.
135. Rhee EK, England JD, Sumner AJ. A computer stimulationof conduction block: effects produced by actual versus inter-phase cancellation. Ann Neurol 1990;28:146–156.
136. Roberts M, Willison HJ, Vincent A, Newsom-Davis J. Multi-focal motor neuropathy human sera block distal motornerve conduction in mice. Ann Neurol 1995;38:111–118.
137. Rojas-Garcia R, Gallardo E, de Andres I, de Luna N, JuarezJ, Juarez C, et al. Chronic neuropathy with IgM anti-gangli-oside antibodies: lack of long term response to rituximab.Neurology 2003;61:1814–1816.
138. Roth G, Rohr J, Magistris MR, Ochsner F. Motor neuropathywith proximal multifocal persistent conduction block, fas-ciculations and myokymia. Eur Neurol 1986;25:416–423.
139. Sadiq SA, Thomas FP, Kilidireas K, Protopsaltis MS, Hays AP,Lee K-W, et al. The spectrum of neurological disease associ-ated with anti-GM1 antibodies. Neurology 1990;40:1067–1072.
140. Salazar-Grueso EF, Routbort MJ, Martin J, Dawson G, RoosRP. Polyclonal IgM anti-GM1 ganglioside antibody in pa-
tients with motor neuron disease and variants. Ann Neurol1990;27:558–563.
141. Santoro M, Thomas FP, Fink ME, Lange DJ, Uncini A, WadiaNH, et al. IgM deposits at the node of Ranvier in a patientwith amyotrophic lateral sclerosis, anti-GM1 antibodies andmultifocal conduction block. Ann Neurol 1990;28:373–377.
142. Santoro M, Uncini A, Corbo M, Staugaitis SM, Thomas FP,Hays AP, et al. Experimental conduction block induced byserum from a patient with anti-GM1 antibodies. Ann Neurol1992;31:385–390.
143. Saperstein DS, Amato AA, Wolfe GI, Katz JS, Nations SP,Jackson CE, et al. Multifocal acquired demyelinating sensoryand motor neuropathy: the Lewis–Sumner syndrome. Mus-cle Nerve 1999;22:560–566.
144. Schluep M, Steck AJ. Immunostaining of motor nerve ter-minals by IgM M-protein with activity against gangliosidesGM1 and GD1b from a patient with motor neuron disease.Neurology 1988;38:1890–1892.
145. Sugie K, Murata K, Ikoma K, Suzumura A, Takayanagi T. Acase of acute multifocal motor neuropathy with conductionblock after Campylobacter jejuni enteritis. Rinsho Shinkeigaku1998;38:42–45.
146. Sumner AJ. Separating motor neuron diseases from puremotor neuropathies. Multifocal motor neuropathy with per-sistent conduction block. Adv Neurol 1991;56:399–403.
147. Taylor BV, Wright RA, Harper CM, Dyck PJ. Natural historyof 46 patients with multifocal motor neuropathy with con-duction block. Muscle Nerve 2000;23:900–908.
148. Taylor BV, Gross LA, Windebank AJ. The sensitivity andspecificity of anti-GM1 antibody testing. Neurology 1996;47:951–955.
149. Taylor BV, Dyck PJB, Engelstand J, Gruener G, Grant I, DyckPJ. Multifocal motor neuropathy: pathologic alterations atthe site of conduction block. J Neuropathol Exp Neurol2004;63:129–137.
150. Taylor PK. CMAP dispersion, amplitude decay, and areadecay in a normal population. Muscle Nerve 1993;16:1181–1187.
151. Terenghi F, Allaria S, Scarlato G, Nobile-Orazio E. Multifo-cal motor neuropathy and Campylobacter jejuni reactivity.Neurology 2002;59:282–284.
152. Terenghi F, Cappellari A, Bersano A, Carpo M, Barbieri S,Nobile-Orazio E. How long is IVIg effective in multifocalmotor neuropathy? Neurology 2004;62:666–668.
153. Terenghi F, Meucci N, Nobile-Orazio E. Long-term disabilityin multifocal motor neuropathy and its relation to IVIgtherapy. J Periph Nerv Syst 2003;8(Suppl 1):67.
154. Thomas FP, Adapon PH, Goldberg GP, Latov N, Hays AP.Localization of neural epitopes that bind to IgM monoclonalautoantibodies (M-proteins) from two patients with motorneuron disease. J Neuroimmunol 1989;23:167–174.
155. Thomas FP, Thomas JE, Sadiq SA, van den Berg LH, RoelofsRI, Latov N, et al. Human monoclonal IgM anti-Gal(�1-3)GalNAc autoantibodies bind to the surface of bovine spi-nal motorneurons. J Neuropathol Exp Neurol 1990;49:89–95.
156. Umapathi T, Hughes RAC, Nobile-Orazio E, Leger JM. Im-munosuppressive treatment for multifocal motor neuropa-thy. The Cochrane Library 2002, Issue 3. Oxford: UpdateSoftware.
157. Umapathi T, Hughes R. Mycophenolate in treatment resis-tant inflammatory neuropathies. Eur J Neurol 2002;9:683–685.
158. Uetz-von Allmen E, Sturzenegger M, Rieben R, Rihs F,Frauenfelder A, et al. Antiganglioside GM1 antibodies andtheir complement activating capacity in central and periph-eral nervous system disorders and in controls. Eur Neurol1998;39:103–110.
159. Uncini A, Santoro M, Corbo M, Lugaresi A, Latov N. Con-duction abnormalities induced by sera of patients with mul-tifocal motor neuropathy and anti-GM1 antibodies. MuscleNerve 1993;16:610–615.
Multifocal Motor Neuropathy MUSCLE & NERVE June 2005 679

160. Valls-Sole J, Cruz Martinez A, Graus F, Saiz A, Arpa J, GrauJM. Abnormal sensory conduction in multifocal demyelinat-ing neuropathy with persistent conduction block. Neurology1995;45:2024–2028.
161. Van Asseldonk JTH, Van den Berg LH, Van den Berg-VosRM, Wieneke GH, Wokke JHJ, Franssen H. Demyelinationand axonal loss in multifocal motor neuropathy: distributionand relation to weakness. Brain 2003;126:186–198.
162. Van den Berg LH, Kerkhoff H, Oey PL, Franssen H, MolleeI, Vermeulen M, et al. Treatment of multifocal motor neu-ropathy with high dose intravenous immunoglobulins: adouble blind, placebo controlled study. J Neurol NeurosurgPsychiatry 1995;59:248–252.
163. Van den Berg LH, Lockhorst H, Wokke JHJ. Pulsed high-dose dexamethasone is not effective in patients with multi-focal motor neuropathy. Neurology 1997;48:1135.
164. Van den Berg LH, Franssen H, Wokke JHJ. The long-termeffect of intravenous immunoglobulin treatment in multifo-cal motor neuropathy. Brain 1998;121:421–428.
165. Van den Bergh P, Logigian E, Kelly JJ. Motor neuropathywith multifocal conduction block. Muscle Nerve 1989;11:26–31.
166. Van den Berg-Vos RM, Franssen H, Wokke JHJ, Van Es HV,Van den Berg LH. Multifocal motor neuropathy: diagnosticcriteria that predict the response to immunoglobulin. Neu-rology 2000;48:919–926.
167. Van den Berg-Vos RM, Van den Berg LH, Franssen H,Vermeulen M, Witkamp TD, Jansen GH, et al. Multifocalinflammatory demyelinating neuropathy. A distinct clinicalentity? Neurology 2000;54:26–32.
168. Van den Berg-Vos RM, Van den Berg LH, Franssen H, VanDoorn PA, Merkies ISJ, Wokke JHJ. Treatment of multifocalmotor neuropathy with interferon-�1A. Neurology 2000;54:1518–1521.
169. Van den Berg-Vos RM, Franssen H, Wokke JHJ, Van den BergLH. Multifocal motor neuropathy: long-term clinical and elec-trophysiological assessment of intravenous immunoglobulinmaintenance treatment. Brain 2002;125:1875–1886.
170. Van den Berg-Vos RM, Franssen H, Visser J, de Visser M, deHaan RJ, Wokke, JHJ, et al. Disease severity in multifocalmotor neuropathy and its association with the response toimmunoglobulin treatment. J Neurol 2002;249:330–336.
171. Van Es HW, Van den Berg LH, Franssen H, Witkamp TD,Ramos LMP, Notermans NC, et al. Magnetic resonance im-aging of the brachial plexus in patients with multifocal mo-tor neuropathy. Neurology 1997;48:1218–1224.
172. van Schaik IN, Bossuyt PMM, Brand A, Vermeulen M. Diag-nostic value of GM1 antibodies in motor neuron disordersand neuropathies: a meta analysis. Neurology 1995;45:1570–1577.
173. Veugelers B, Theys P, Lammens M, Van Hees J, RobberechtW. Pathological findings in a patient with amyotrophic lat-eral sclerosis and multifocal motor neuropathy with conduc-tion block. J Neurol Sci 1996;136:64–70.
174. Visser J, van den Berg-Vos RM, Franssen H, van den BergLH, Vogels OJ, Wokke JHJ, et al. Mimic syndromes in spo-radic cases of progressive spinal muscular atrophy. Neurol-ogy 2002;58:1593–1596.
175. Vrethem M, Lindvall B, Kihlstrand S, Backman E, Brismar T,Fredman P, et al. High dose intravenous immunoglobulintherapy improved muscle strength in a patient with multifo-cal motor neuropathy and antibodies against LK1. EurJ Neurol 1996;3:156–159.
176. Vucic S, Black KR, Chong PS, Cros D. Multifocal motorneuropathy. Decrease in conduction blocks and reinnerva-tion with long-term IVIg. Neurology 2004;63:1264–1269.
177. White JR, Sachs GM, Gilchrist JM. Multifocal motor neurop-athy with conduction block and Campylobacter jejuni. Neurol-ogy 1996;46:562–563.
178. Wiegandt H. Gangliosides. In: Wiegandt H, editor. Glycolip-ids. Amsterdam: Elsevier, 1985. p 199–260.
179. Willison HJ, Paterson G, Kennedy PGE, Veitch J. Cloning ofhuman anti-GM1 antibodies from motor neuropathy pa-tients. Ann Neurol 1994;35:471–478.
180. Willison HJ, Veitch J, Swan AV, Bauman N, Comi G, GregsonNA, et al. Inter-laboratory validation of an ELISA for thedetermination of serum anti-ganglioside antibodies. EurJ Neurol 1999;6:71–77.
181. Willison HJ, Yuki NM. Peripheral neuropathies and anti-glycolipid antibodies. Brain 2002;125:2591–2625.
182. Wirguin I, Suturkova-Milosevic Lj, Della-Latta P, Fisher T,Brown RH, et al. Monoclonal IgM antibodies to GM1 andasialo-GM1 in chronic neuropathies cross-react with Campy-lobacter jejuni lipopolysaccharides. Ann Neurol 1994;35:698–703.
183. Wolfe G, El-Feky W, Katz J, Bryan W, Wians J, Barohn R.Antibody panels in idiopathic polyneuropathy and motorneuron disease. Muscle Nerve 1997;20:1275–1283.
184. Yokota T, Saito Y, Yuki N, Tanaka H. Persistent increasedthreshold of electrical stimulation selective to motor nervein multifocal motor neuropathy. Muscle Nerve 1996;19:823–828.
185. Yuki N, Yamazaki M, Kondo H, Suzuki K, Tsuji S. Treatmentof multifocal motor neuropathy with a high dosage of intra-venous immunoglobulin. Muscle Nerve 1993;16:220–221
186. Zielasek J, Ritter G, Magi S, Hartung HP, Toyka KV, andparticipating laboratories. A comparative trial of anti-glyco-conjugate antibody assays: IgM antibodies to GM1. J Neurol1994;241:475–480.
680 Multifocal Motor Neuropathy MUSCLE & NERVE June 2005


















