
Ab Initio and Effective Fragment Potential Dynamics Heather M. Netzloff and Mark S. Gordon Iowa...
31
Ab Initio and Effective Fragment Potential Dynamics Heather M. Netzloff and Mark S. Gordon Iowa State University
-
Upload
tania-plush -
Category
Documents
-
view
218 -
download
1
Transcript of Ab Initio and Effective Fragment Potential Dynamics Heather M. Netzloff and Mark S. Gordon Iowa...

Ab Initio and Effective Fragment Potential Dynamics
Heather M. Netzloff and Mark S. Gordon
Iowa State University

Outline
Computer simulations– Treatment of liquids
Effective Fragment Potential Method Molecular Dynamics in GAMESS Applications Future Plans

Microscopic details
Macroscopic properties
Test theories and compare with experiments Simulate under extreme conditions
unattainable to experimentalists
Laws of Statistical Mechanics
Computer simulation:Motivation

Computer simulations:Condensed phase
Treatment of fluid-like states and solvation is essentially a many-particle problem…– Importance of accuracy and reliability of
the method used• Limited by the size of the system and
computational costs
GOAL: model CLUSTER to BULK behavior

Condensed phase For accuracy,
preference = ab initio methods, BUT…– Computationally expensive– Only realistic for small systems
Solvation Models:– Clusters:
• Ab initio, DFT, Effective Fragment Potential,…– Bulk:
• Continuum methods, TIPnP, SPC/E,…
Limited number of methods which span the clusterbulk gap well

The Effective Fragment Potential Method1,2
Treatment of discrete solvent effects
EFP1/HF: – Developed at Hartree Fock level of theory (DH(d,p))– Reproduces RHF/ab initio results…
Esystem = Eab initio + Einteraction
Standard Ab Initio Calculation
Effective Fragment Potential Calculation
1Day et. al. J. Chem. Phys. 105,1968(1996). 2Gordon et. al. J. Phys. Chem A. 105,2(2001).

EFP:Menshutkin Reaction
R3N + RX R4N+X-
– Reaction rate increases with polarity of solvent– Study process of solvation on ion formation
EFP test (Simon Webb): NH3 + CH3Br – Add EFP waters; AI solute/solvent (RHF/DZVP)
-20
-10
0
10
20
30
-20.0 -10.0 0.0 10.0 20.0 30.0 40.0
Reaction Coordinate (amu 1/2bohr)
all ab initio MEP
2 EFP water molecules MEP

EFP:Water Clusters
Monte Carlo Simulated Annealing with EFP (Paul Day, Grant Merrill, Ruth Pachter) – N = 6 - 20
• Water hexamer– Re-optimization with HF, MP2– CCSD(T) single points
(H2O)6 RELATIVE ENERGIES (KCAL/MOL)
Relative Energies(kcal/mol)
Prism Cage Book Bag Cyclic Boat
EFP 0.0 0.5 1.0 1.8 1.3 2.3RHF 0.0 0.4 0.4 1.3 -0.2 0.7MP2//MP2 0.0 0.7 1.7 2.6 2.5 3.9CCSD(T)//MP2 0.0 0.8 2.0 2.9 2.9 4.3

Effect of “solvent” molecules (fragments) added as one-electron terms to ab initio Hamiltonian, HAR
V = Fragment interaction potential
Effective Fragment Potential Method
Ab initio region
EFP region
Hsystem=HAR +V
€
V = V elec +V pol +V rep

Effective Fragment Potential Method
Interaction Potential Terms--EFP1/HF
Electrostatics: Coulomb interactions– Multipolar expansion up to octupoles– Screening to account for overlapping charge densities– Expansion points: nuclear centers and bond midpoints
Polarization: Dipole/induced dipole potential– Induced dipoles iterated to self-consistency– Centered on bonding and lone-pair localized molecular orbitals
Exchange-repulsion/charge transfer: Remainder term– EFP1: exponential functions optimized by fitting procedure
• Limited to water – Located at fragment atom centers and center of mass

Effective Fragment Potential Method
Solute explicitly treated with ab initio wavefunction of choice; remainder treated as effective fragments QM/MM method
Multipoles and polarizabilities – Determined from ab initio calculations on a single solvent molecule– Potential can be systematically improved!
Exchange repulsion/charge transfer term– EFP1/HF: Currently fit to functional form solvent specific– EFP2: generalize for any solvent
• NO FITS: Exchange repulsion from LMO intermolecular overlap and kinetic energy integrals
NEW!! EFP1/DFT (B3LYP)
All 3 terms are calculated independently from each other Only ONE-ELECTRON integrals are needed
– MUCH less CPU intensive than quantum mechanics

EFP/Molecular Dynamics
•Do i = 1, number of simulation steps
Calculate PE and gradients forces for particle at time t
Solve equations of motion for each particle to obtain KE at time t and new positions at time t + dt
•End do
First test of EFP to reproduce bulk behavior with its implementation with Molecular Dynamics
Basic MD Simulation loop:(user specifies time step size, dt, and length of simulation)
MD is dependent upon the accuracy and reliability of the potential used to generate gradients/forces…

Molecular Dynamics
Solve Newton’s equations of motion
– Require both ENERGY and GRADIENT from PE routine
– Integrate simultaneously for all atoms in the system to generate a trajectory for each atom
– Perform integration in SMALL time steps (usually 1-10 fs)
• Time step size Energy conservation
€
rF i = mi
d2r r i( t)dt2
r F i = −
∂V (r r i, ...
r r N )
∂r r i

Current MD Implementation in GAMESS
EFP fragment-fragment interaction (EFP-EFP MD)– Classical interaction terms– EFP: EFP1/HF, EFP1/HF, EFP2
Ab initio interaction (AI MD)– Ab initio interaction– Basis set and level of theory of choice and
availability in GAMESS

Current MD Implementation:EFP-EFP MD
Integration EFP treats water as a rigid molecule
– Center of mass motion:• Translational motion leapfrog integration algorithm• Rotational motion quaternions and modified leapfrog
integration algorithm
Water model parameters (calculated based on ab initio methods) are stored in GAMESS or given with input file– After each move all information must be
rotated/translated along with the fragment

Current MD Implementation:EFP-EFP MD
T0
T(t) T0 = desired/bath temperature
T(t) = temperature obtained from translational or rotational
kinetic energy at time t
Ensembles: NVE (constant energy)
NVT (constant temperature) : based on velocity scaling– Separate treatment for translational and rotational
velocity components– Rescale by:

Periodic Boundary Conditions
Minimum Image Convention
Current MD Implementation:EFP-EFP MD
Periodic boundary conditions – Introduced to minimize surface effects
Minimum image convention– Restrict infinite number of terms in force calculation to only
the closest N -1 periodic images– Based on center-of-mass (COM) positions

Current MD Implementation:AI MD
Each atom is treated independently– Leapfrog integration algorithm
NVE and NVT (velocity scaling) ensembles
No periodic boundary conditions or minimum image convention currently implemented

EFP-EFP MD ApplicationsPreparation of the system Heating:
– NVE ensemble: Initial target temp = 50 K• Initial quaternions chosen randomly• Initial translational velocities sampled from Maxwell-Boltzmann
distribution at 100 K; angular velocities start from zero– Use these coordinates to start simulation at 100 K– Same procedure to start 200 K and 300 K runs
Equilibration--TARGET TEMP = 300 K:– Coordinates taken from 300 K heating run; initial translational
velocities sampled from Maxwell-Boltzmann distribution at 600 K– NVT ensemble at 300K
• velocities rescaled only if they are outside 300 + 5 K (scaling factors not allowed to exceed 1.3)
Production:– Initial coordinates, velocities, and quaternions from equilibration– NVE and/or NVT ensemble: measure observables of interest

Compare with other water potentials…Example:
– SPC/E (Simple Extended Point Charge)4
• Point charges on O and H sites• Reparameterized SPC model to include polarization
• Values for O, O, and q (point charge) are determined by parameterization with experimental density and Evap as targets
EFP-EFP MD Applications
Vab =qiqje
2
rijij∑ +4εO
σO
rOO
⎛
⎝ ⎜
⎞
⎠ ⎟
12
−σO
rOO
⎛
⎝ ⎜
⎞
⎠ ⎟
6⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
+0.4238e+0.4238e
-2.0qH
4Berendsen et. al. J. Phys. Chem. 92,6269(1987).

EFP-EFP MD Applications
Radial Distribution Function (RDF):– Measures the number of atoms a distance r from a given atom
compared with the number at the same distance in an ideal gas at the same density
– Gives information on how molecules pack in ‘shells’ of neighbors, as well as average structure
– Can be measured spectroscopically (X-ray or neutron diffraction)
– Test theoretical models versus experimental results
3 SITE-SITE radial distribution functions for water:– gOO(r), gOH(r), gHH(r)

gOO(r)--EFP1/HF
0
0.5
1
1.5
2
2.5
3
3.5
0 1 2 3 4 5 6 7
gOO
(r)
r (Angstroms)
Initial structure: 62 EFP waters, 26 ps equilibration
Timestep size = 1 fs, Simulation = 5000 fs
EFP1/HF--NVEEFP1/HF--NVTSPC/E--NVTExp (THG)
Exp (THG): X-ray; Sorenson et. al. J. Chem. Phys. 113,9149(2000).
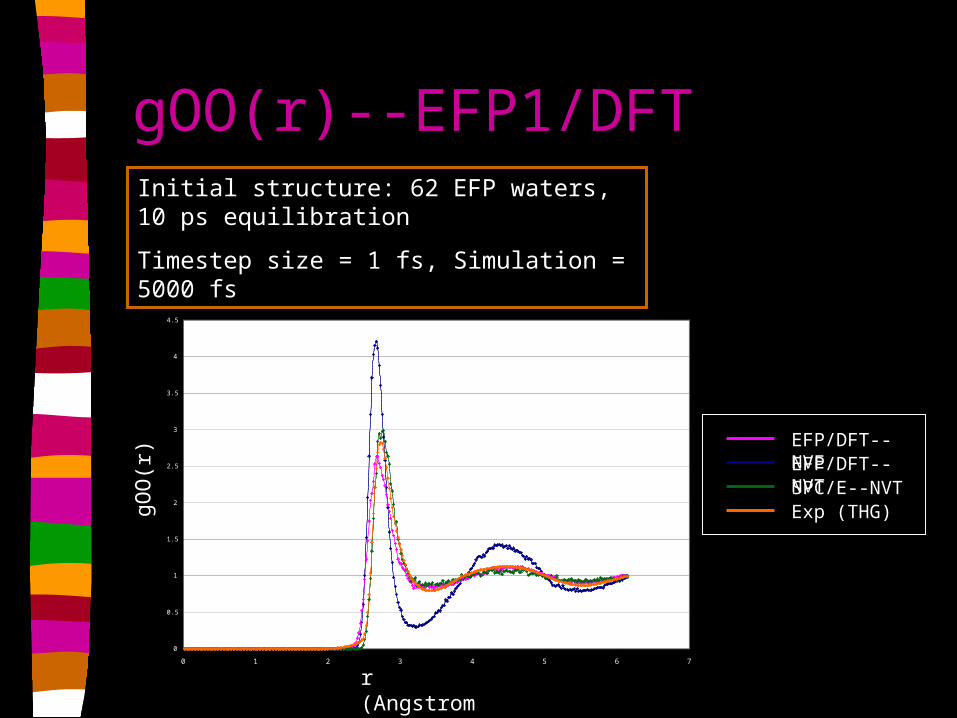
gOO(r)--EFP1/DFT
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
0 1 2 3 4 5 6 7
gOO
(r)
r (Angstroms)
EFP/DFT--NVEEFP/DFT--NVTSPC/E--NVTExp (THG)
Initial structure: 62 EFP waters, 10 ps equilibration
Timestep size = 1 fs, Simulation = 5000 fs

gOO(r) Error:62 water analysis
Err
or =
gO
O(e
xp)-
gOO
(X)
r (Angstroms)
-1.5
-1
-0.5
0
0.5
1
1.5
2
0 1 2 3 4 5 6 7
X
X
X
X
EFP/DFT--NVEEFP1/HF--NVTSPC/E--NVT
X Exp peak/valley locations

gOO(r)--EFP1/HF:62 water analysis with Monte Carlo
0
0.5
1
1.5
2
2.5
3
3.5
4
0 1 2 3 4 5 6 7
0
0.5
1
1.5
2
2.5
3
0 1 2 3 4 5 6 7
Initial structure: 62 EFP waters, 1.0 ps MD equilibration
RDF measured with MC accepted structures (2 criteria for EFP MC) 6790 accepted (100,000)
gOO
(r)
r (Angstroms)
gOO
(r)
EFP1/HF--MC
Exp (THG)
Polynomial Fit (4th order) to EFP

gOO(r)--EFP1/HF:512 water analysis
0
0.5
1
1.5
2
2.5
3
0 2 4 6 8 10 12 14
gOO
(r)
r (Angstroms)-1
-0.5
0
0.5
1
1.5
2
0 2 4 6 8 10 12 14
X
X
X
X
X
Initial structure: 512 EFP waters, 7.5 ps equilibration
Timestep size = 1 fs, Simulation = 5000 fs
r (Angstroms)
Err
or =
gO
O(e
xp)-
gOO
(X)
EFP1/HF--NVTSPC/E--NVTExp (THG)

gOH(r):EFP1/HF, EFP1/DFT, SPC/E62 waters
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0 1 2 3 4 5 6 7
EFP/DFT--NVEEFP/HF--NVTSPC/E--NVTExp (ND)
gOH
(r)
r (Angstroms)
Exp (ND): Neutron Diffraction; Soper et. al.

gHH(r):EFP1/HF, EFP1/DFT, SPC/E62 waters
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 1 2 3 4 5 6 7
EFP/DFT--NVEEFP/HF--NVTSPC/E--NVTExp (ND)
gHH
(r)
r (Angstroms)

Timings: Energy + Gradient calculation
Method*20 water molecules
62 water molecules
122 water molecules
512 water molecules
Ab initio** 3.19 hrs --- --- ~157 yrs***
EFP2 3.3 sec 26.1 sec 95.3 sec 26.8 min
EFP1/HF 0.2 sec 2.6 sec 5.1 sec 97.8 sec
SPC/E 0.02 sec 0.02 sec 0.1 sec 0.7 sec
* Run on 1200 MHz Athlon/Linux machine
**Ab initio: DZP basis set, *** Assuming N4 scaling

Future Plans Addition of minimum image convention to EFP2
implementation and AI MD– Coordinates, not only distances, must be manipulated
Treatment of long range forces– First layer: Ewald sum method for charge-charge interaction– Second layer: Fast Multipole Method– Use continuum versus minimum image convention/periodic
boundary conditions
Utilization/development of parallel algorithms– Utilize parallel AI GAMESS code for calculation of AI energy
and gradient– Parallelization of the EFP method

Acknowledgements
Jon Sorenson, Grant Merrill, Mark Freitag
$$$$$
DOE Computational Science Graduate Fellowship
Thank You!


















