
A lethal neonatal prominent genitourinary - jmg.bmj.com · Histologi-cally,...
5
57Med Genet 1997;34:520-524 A maternally transmitted lethal neonatal progeroid syndrome with prominent genitourinary and gastrointestinal features Martin B Delatycki, Maureen A Cleary, Agnes Bankier, Peter N McDougall, Jagjit S Ahluwalia, Chung W Chow, Claire M Cooke-Yarborough Victorian Clinical Genetics Service, Murdoch Institute, Flemington Road, Parkville 3052, Victoria, Australia M B Delatycki M A Cleary A Bankier Department of Pathology, Royal Children's Hospital, Flemington Road, Parkville 3052, Victoria, Australia C W Chow C M Cooke-Yarborough Department of Neonatology, Royal Children's Hospital, Flemington Road, Parkville 3052, Victoria, Australia P N McDougall J S Ahluwalia* *Present address: Neonatal Intensive Care Unit, Rosie Maternity Hospital, Robinson Way, Cambridge, UK. Correspondence to: Dr Delatycki. Received 15 October 1996 Revised version accepted for publication 14 January 1997 Abstract Twin brothers and their maternal uncle with a previously undescribed neonatal progeroid syndrome are presented. In addition to progeroid features, they had pseudo-obstruction of the urinary and gastrointestinal tracts, severe leucocyto- sis, liver dysfunction, and low complex III and IV in muscle but not in liver. Previ- ously described neonatal progeroid syn- dromes and syndromes featuring pseudo- obstruction are discussed. The two most likely aetiological mechanisms are an X linked single gene disorder or a mitochon- drial disorder. The evidence for these pos- sibilities is presented. (JMed Genet 1997;34:520-524) Keywords: progeroid; pseudo-obstruction; mitochon- dria Twin brothers and their maternal uncle presented with progeroid features and gas- trointestinal and urinary tract pseudo- obstruction and died in infancy. No specific diagnosis could be made pathologically or by comparison with previously described syn- dromes. Case reports PATIENT I Patient 1 (III.4, fig 1) was born at term to non-consanguineous parents. Weight was 2115 g (<3rd centile), length 43 cm (<3rd cen- tile), and occipitofrontal head circumference 1 2 11 III IV 1 2 3 4 4 5 Figure 1 Family pedigree. (OFC) 30 cm (<3rd centile). He was noted to have several dysmorphic features including hir- sutism with a low anterior hair line, a small jaw and mouth, flat philtrum, small upturned nose, folded down ears, undescended testes, micro- penis, and dorsiflexed halluces. The abdomen was distended and renal ultrasound showed grossly dilated kidneys and ureters. A small patent ductus arteriosus was present. Cranial ultrasound and ophthalmological examination were normal. He had an unexpected cardiores- piratory arrest on day 4 and died. No clinical photographs were taken of this baby. The karyotype was 46,XY Necropsy showed a markedly hypertrophied bladder with enlarged kidneys and dilated renal pelvis and ureters, but no obstructing lesion in the urethra. The large bowel had focal areas of necrotising enterocolitis. The liver showed marked intracanalicular cholestasis but no obvious obstructive lesion in the biliary tree was identified. PATIENTS 2 AND 3 Patients 2 and 3 (IV. 1 and IV.2, respectively, in fig 1, figs 2 and 3) were monochorionic, monoamniotic twin boys, born to the sister of patient 1. Patient 1 's sister and her husband are non-consanguineous and are of different cul- tural backgrounds. An ultrasound at 36 weeks' gestation showed hydronephrosis in both twins with one having hydroureter bilaterally as well. The bladder was reported as normal. Liquor volume was normal. At birth at 37 weeks' ges- tation, both infants were noted to be growth retarded, dysmorphic, and had early onset res- piratory distress requiring ventilation with 100% oxygen. Patient 2 had a birth weight of 2300 g (< 10th centile), length of 42.5 cm (< 10th centile), and OFC of 34.5 cm (50th centile). Multiple dysmorphic features were present and included prominent parietal bones, hair extending onto the forehead and cheeks, a very small an- teverted nose, flat philtrum, micrognathia, microstomia, and a markedly distended abdo- men. The anterior fontanelle was widely open. Formal ophthalmological examination did not show any definite pathology and in particular there were no cataracts. On day 1 he had a cardiovascular collapse associated with a marked metabolic acidosis (pH 6.77, pCO2 67 mm Hg, HCO, 9.3 mmol/l, base deficit >22 mmol/l). He responded to supportive measures of intravenous albumin, 520 on 3 September 2018 by guest. Protected by copyright. http://jmg.bmj.com/ J Med Genet: first published as 10.1136/jmg.34.6.520 on 1 June 1997. Downloaded from
Transcript of A lethal neonatal prominent genitourinary - jmg.bmj.com · Histologi-cally,...

57MedGenet 1997;34:520-524
A maternally transmitted lethal neonatalprogeroid syndrome with prominent genitourinaryand gastrointestinal features
Martin B Delatycki, Maureen A Cleary, Agnes Bankier, Peter N McDougall,Jagjit S Ahluwalia, Chung W Chow, Claire M Cooke-Yarborough
Victorian ClinicalGenetics Service,Murdoch Institute,Flemington Road,Parkville 3052,Victoria, AustraliaM B DelatyckiM A ClearyA Bankier
Department ofPathology, RoyalChildren's Hospital,Flemington Road,Parkville 3052,Victoria, AustraliaC W ChowC M Cooke-Yarborough
Department ofNeonatology, RoyalChildren's Hospital,Flemington Road,Parkville 3052,Victoria, AustraliaP N McDougallJ S Ahluwalia*
*Present address: NeonatalIntensive Care Unit, RosieMaternity Hospital,Robinson Way, Cambridge,UK.
Correspondence to:Dr Delatycki.
Received 15 October 1996Revised version accepted forpublication 14 January 1997
AbstractTwin brothers and their maternal unclewith a previously undescribed neonatalprogeroid syndrome are presented. Inaddition to progeroid features, they hadpseudo-obstruction of the urinary andgastrointestinal tracts, severe leucocyto-sis, liver dysfunction, and low complex IIIand IV in muscle but not in liver. Previ-ously described neonatal progeroid syn-dromes and syndromes featuring pseudo-obstruction are discussed. The two mostlikely aetiological mechanisms are an Xlinked single gene disorder or a mitochon-drial disorder. The evidence for these pos-sibilities is presented.(JMed Genet 1997;34:520-524)
Keywords: progeroid; pseudo-obstruction; mitochon-dria
Twin brothers and their maternal unclepresented with progeroid features and gas-trointestinal and urinary tract pseudo-obstruction and died in infancy. No specificdiagnosis could be made pathologically or bycomparison with previously described syn-dromes.
Case reportsPATIENT IPatient 1 (III.4, fig 1) was born at termto non-consanguineous parents. Weight was2115 g (<3rd centile), length 43 cm (<3rd cen-tile), and occipitofrontal head circumference
1 2
11
III
IV
1 2 3 4
4 5
Figure 1 Family pedigree.
(OFC) 30 cm (<3rd centile). He was noted tohave several dysmorphic features including hir-sutism with a low anterior hair line, a small jawand mouth, flat philtrum, small upturned nose,folded down ears, undescended testes, micro-penis, and dorsiflexed halluces. The abdomenwas distended and renal ultrasound showedgrossly dilated kidneys and ureters. A smallpatent ductus arteriosus was present. Cranialultrasound and ophthalmological examinationwere normal. He had an unexpected cardiores-piratory arrest on day 4 and died. No clinicalphotographs were taken of this baby. Thekaryotype was 46,XYNecropsy showed a markedly hypertrophied
bladder with enlarged kidneys and dilated renalpelvis and ureters, but no obstructing lesion inthe urethra. The large bowel had focal areas ofnecrotising enterocolitis. The liver showedmarked intracanalicular cholestasis but noobvious obstructive lesion in the biliary treewas identified.
PATIENTS 2 AND 3Patients 2 and 3 (IV. 1 and IV.2, respectively, infig 1, figs 2 and 3) were monochorionic,monoamniotic twin boys, born to the sister ofpatient 1. Patient 1 's sister and her husband arenon-consanguineous and are of different cul-tural backgrounds. An ultrasound at 36 weeks'gestation showed hydronephrosis in both twinswith one having hydroureter bilaterally as well.The bladder was reported as normal. Liquorvolume was normal. At birth at 37 weeks' ges-tation, both infants were noted to be growthretarded, dysmorphic, and had early onset res-piratory distress requiring ventilation with100% oxygen.
Patient 2 had a birth weight of2300 g (<10thcentile), length of 42.5 cm (< 10th centile), andOFC of 34.5 cm (50th centile). Multipledysmorphic features were present and includedprominent parietal bones, hair extending ontothe forehead and cheeks, a very small an-teverted nose, flat philtrum, micrognathia,microstomia, and a markedly distended abdo-men. The anterior fontanelle was widely open.Formal ophthalmological examination did notshow any definite pathology and in particularthere were no cataracts.On day 1 he had a cardiovascular collapse
associated with a marked metabolic acidosis(pH 6.77, pCO2 67 mm Hg, HCO, 9.3 mmol/l,base deficit >22 mmol/l). He responded tosupportive measures of intravenous albumin,
520
on 3 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.6.520 on 1 June 1997. D
ownloaded from
A maternally transmitted lethal neonatal progeroid syndrome
A C
*' *} v-,.; .rr
o j.X
t .s: st r*.B ......... ,!-:
r ..
.._
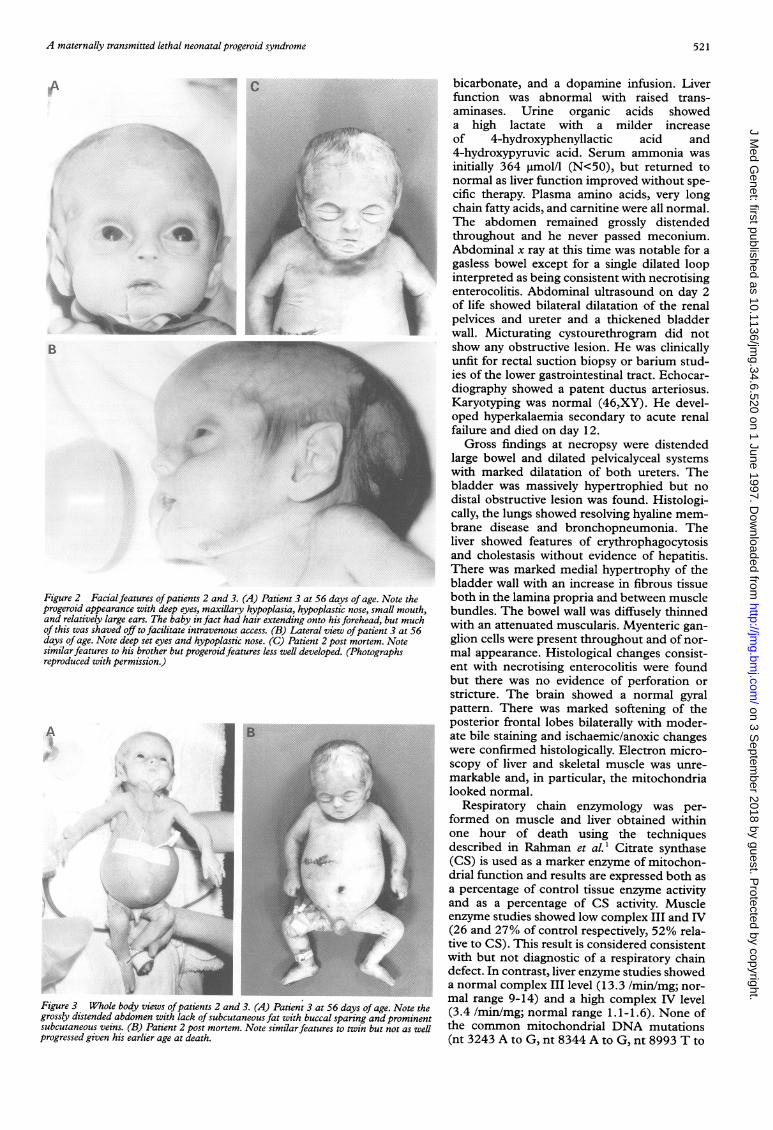
Figure 2 Facialfeatures ofpatients 2 and 3. (A) Patient 3 at 56 days of age. Noteprogeroid appearance with deep eyes, maxillary hypoplasia, hypoplastic nose, small mand relatively large ears. The baby in fact had hair extending onto his forehead, but n
of this was shaved off to facilitate intravenous access. (B) Lateral view ofpatient 3 atdays of age. Note deep set eyes and hypoplastic nose. (C) Patient 2 post mortem. Notesimilarfeatures to his brother but progeroidfeatures less well developed. (Photographsreproduced with permission.)
A JB
-6
lik
,4
--I
Figure 3 Whole body views ofpatients 2 and 3. (A) Patient 3 at 56 days of age. Nogrossly distended abdomen with lack of subcutaneous fat with buccal sparing and pronsubcutaneous veins. (B) Patient 2 post mortem. Note similarfeatures to twin but not aprogressed given his earlier age at death.
bicarbonate, and a dopamine infusion. Liverfunction was abnormal with raised trans-aminases. Urine organic acids showeda high lactate with a milder increaseof 4-hydroxyphenyllactic acid and4-hydroxypyruvic acid. Serum ammonia wasinitially 364 gmol/l (N<50), but returned tonormal as liver function improved without spe-cific therapy. Plasma amino acids, very longchain fatty acids, and carnitine were all normal.
| The abdomen remained grossly distendedthroughout and he never passed meconium.
\y;-;- Abdominal x ray at this time was notable for agasless bowel except for a single dilated loopinterpreted as being consistent with necrotising
L enterocolitis. Abdominal ultrasound on day 2of life showed bilateral dilatation of the renalpelvices and ureter and a thickened bladderwall. Micturating cystourethrogram did notshow any obstructive lesion. He was clinicallyunfit for rectal suction biopsy or barium stud-ies of the lower gastrointestinal tract. Echocar-diography showed a patent ductus arteriosus.Karyotyping was normal (46,XY). He devel-oped hyperkalaemia secondary to acute renalfailure and died on day 12.
Gross findings at necropsy were distendedlarge bowel and dilated pelvicalyceal systemswith marked dilatation of both ureters. Thebladder was massively hypertrophied but nodistal obstructive lesion was found. Histologi-cally, the lungs showed resolving hyaline mem-brane disease and bronchopneumonia. Theliver showed features of erythrophagocytosisand cholestasis without evidence of hepatitis.There was marked medial hypertrophy of thebladder wall with an increase in fibrous tissue
the both in the lamina propria and between muscleouth, bundles. The bowel wall was diffusely thinnednuch with an attenuated muscularis. Myenteric gan-t56e glion cells were present throughout and of nor-
mal appearance. Histological changes consist-ent with necrotising enterocolitis were foundbut there was no evidence of perforation orstricture. The brain showed a normal gyralpattern. There was marked softening of theposterior frontal lobes bilaterally with moder-ate bile staining and ischaemic/anoxic changeswere confirmed histologically. Electron micro-scopy of liver and skeletal muscle was unre-markable and, in particular, the mitochondrialooked normal.
Respiratory chain enzymology was per-formed on muscle and liver obtained withinone hour of death using the techniquesdescribed in Rahman et al.' Citrate synthase(CS) is used as a marker enzyme of mitochon-drial function and results are expressed both asa percentage of control tissue enzyme activity
| and as a percentage of CS activity. Muscleenzyme studies showed low complex III and IV(26 and 27% of control respectively, 52% rela-tive to CS). This result is considered consistentwith but not diagnostic of a respiratory chaindefect. In contrast, liver enzyme studies showeda normal complex III level (13.3 /min/mg; nor-mal range 9-14) and a high complex IV level
te thet (3.4 /min/mg; normal range 1.1-1.6). None ofZs well the common mitochondrial DNA mutations
(nt 3243 A to G, nt 8344 A to G, nt 8993 T to
521
I
!:.
i
on 3 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.6.520 on 1 June 1997. D
ownloaded from

Delatycki et al
Table 1 Results of investigations on the patients
Investigation Patient 1 Patient 2 Patient 3
Karyotype 46,XY 46,XY 46,XYUrine amino acids nd Normal Slightly raised pipecolic acidUrine organic acids nd Raised lactate, 4 Raised 4-hydroxyphenyllactic and
hydroxyphenyllactate, 4-hydroxyphenylpyruvic acids4-hydroxypyruvate
Respiratory chain enzymes in liver nd Raised complex IV ndRespiratory chain enzymes in muscle nd Decreased complex III, IV Decreased complex III, IVTotal carnitine (35-65) nd 43 12Free carnitine (30-60) nd 20 8Mitochondrial DNA nd No mutation detected No mutation detectedLactate maximum mmol/l (1.0-1.8) nd 23.5 3.0Very long chain fatty acids nd Normal NormalPlasma amino acids nd Normal ndWhite cell count maximum 1 0'/l
(4-11) 2.3 21.9 94.7AST U/ (20-80) nd 172 146Total protein g/l (50-71) nd 51 83Albumin g/l (29-45) nd 38 43Bilirubin conjugated tmol/l 80 167 79Blood ammonia maximum ltmol/l
(<50) nd 364 41Triglycerides mmol/l (0.9-2.0) nd nd 3.3Cholesterol mmol/l (<4.5) nd nd 5.4
nd: not done.
G, nt 8993 T to C) were present inlymphocytes or muscle.
Patient 3 had an identical appearance to hisbrother. His birth weight was 2360 g (5th cen-tile), length 43.5 cm (<3rd centile), and OFC34.5 cm (50th centile). He also had a grosslydistended abdomen and a thickened bladderwall was visualised on ultrasonography. A suc-tion rectal biopsy showed ganglion cells,excluding Hirschsprung disease. He developednecrotising enterocolitis which was managedconservatively with intravenous nutrition, anti-biotics, and nasogastric suction and drainage.Patient 3's phenotype evolved with time. Over aperiod of weeks he developed a progeroidappearance with deeply set eyes, prominentcheeks, and relative enlargement of the cra-nium, and marked reduction of subcutaneousfat with prominent superficial veins was seenthroughout including on the buttocks. Therewere no abnormal fatty deposits. The skin was
Table 2 Comparison of the clinicalfeatures in the patients described with those seen inWiedemann-Rautenstrauch syndrome
Wiedemann-RautenstrauchClinical sign Cases presented syndrome
Survival All dead by 75 days Prolonged survival recordedIUGR Yes YesPostnatal growth retardation Severe YesNeurodevelopmental progress None seen in longest Developmental delay in most
survivor casesSex All male Male and femalePseudohydrocephalus No YesFontanelles Wide anterior Wide anterior, delayed closureProminent scalp veins Yes YesHair Low anterior hair line SparseProgeroid face Yes YesEyes Deep set Deep setNose Very small Small, beakedMouth Small SmallNatal teeth No YesJaw Micrognathia ProminentLipoatrophy Yes YesSacral fat accumulation No May occur with timeSudanophilic leucodystrophy No YesPseudo-obstruction of GIT,GUT Yes No
Liver dysfunction Yes NoInheritance ?X linked, ?mitochondrial Autosomal recessive
IUGR: intrauterine growth retardation, GIT: gastrointestinal tract, GUT: genitourinary tract.
translucent and thin in appearence and texture.There was no evidence of neurodevelopmentalprogress.A muscle biopsy done in life showed no
diagnostic abnormality and in particular therewere no changes suggesting mitochondrialpathology, such as ragged red fibres or absenceof cytochrome C oxidase (COX) staining.He died aged 75 days. Necropsy identified
similar pathology of the gastrointestinal andurogenital tracts to his brother. Changes ofnecrotising enterocolitis were present with noevidence of perforation or stricture. The liverhistology was markedly abnormal with choles-tatic hepatitis with expanded portal tracts con-taining a proliferation of bile ductules. Earlyportal tract to portal tract bridging by fibroustissue containing proliferated bile ductules wasnoted. The extrahepatic biliary tree wasnormal. The neuropathology was essentiallynormal. Skeletal x rays done post mortem didnot show any significant features.The results of the investigations on the
patients are shown in table 1.
DiscussionClearly the three males in this family have thesame condition. This association of featureshas not been previously described.A number of neonatal progeroid syndromes
have been reported. The best known of these isthe Wiedemann-Rautenstrauch neonatal prog-eroid syndrome."A In this syndrome the typicalprogeroid appearance is associated with aprominent jaw and natal teeth. By contrast, thepatients we presented had micrognathia andnatal teeth were absent. The prominentgastrointestinal and urinary tract features ofour cases are not featured in the reportsof Wiedemann-Rautenstrauch syndromewhich is autosomal recessive.5 There have beenreports of sudanophilic leucodystrophy inWiedemann-Rautenstrauch syndrome.6 Thiswas not the case in our patients. Table 2 com-pares this syndrome with the patients pre-sented.
522
on 3 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.6.520 on 1 June 1997. D
ownloaded from

A maternally transmitted lethal neonatal progeroid syndrome
Hagadorn et a/ reported a girl with featuresof a neonatal progeroid syndrome with ab-dominal distension and vesicoureteric reflux.She differed from the cases we have describedin that her gastrointestinal distension resolvedwith nasogastric feeding and urinary tract dila-tation was limited to the renal pelvices. Theirpatient had hypercalciuria and renal parenchy-mal calcification. Hypercalciuria was also seenin the patient reported by Bitoun et at but wasnot looked for in our patients.The family described by de Martinville et all'
shows some features similar to the cases in thisreport. Male monozygotic twins with a neona-tal progeroid syndrome were presented. Anolder sib with dysmorphic features died at 50days. There are no details of the child's sex orclinical features other than talipes equinovarus.The children's mother also had two miscar-riages. One twin died at 10 minutes and detailsare limited regarding its ante- and postmortemfeatures. This child is said to have had the samedysmorphic features as his twin brother whichincluded typical progeroid features as well asmicrognathia, sparse hair, eyelashes, and eye-brows, hypoplastic finger nails and absent nailson the fifth finger bilaterally, camptodactyly,and no natal teeth. The longer surviving twinhad gastrointestinal problems, the exact natureof which were not reported apart from severefailure to thrive. Significant changes werepresent in the skin and its appendages. He diedat 6 months. Necropsy was not performed. It ispossible that these three sibs had the samecondition as the patients we describe, but thereare some differences between the cases. Thepatients of de Martinville et al'0 had prominentdigital changes and there was no description ofurinary or gastrointestinal pseudo-obstruction.The pattern in our family is strongly sugges-
tive of inheritance through the maternal line.Neither a mitochondrial nor an X linkedneonatal progeroid syndrome has previouslybeen described.
In view of the prominence of gastrointestinaland genitourinary pseudo-obstruction in ourpatients, we reviewed other conditions in whichsuch features are mentioned. Intestinalpseudo-obstruction is seen in a number ofconditions. Myoneurogastrointestinal en-cephalopathy syndrome (MNGIE) haspseudo-obstruction as one of its features and insome cases results from mitochondrial DNAdeletions." This condition has not been seen inneonates. An X linked form of intestinalpseudo-obstruction has recently been mappedto Xq28,12 but here specific histological abnor-malities are present in the bowel. These werenot specifically looked for in our cases but therewas no pyloric stenosis, malrotation, or shortgut. Harris et al'3 reported brothers with intes-tinal pseudo-obstruction and natal teeth whoboth died before 6 months. No mention of uri-nary tract dilatation or progeroid features wasmade and the relevance to our cases is unclearbut is of interest given the presence of natalteeth in Wiedemann-Rautenstrauch syndrome.A mitochondrial aetiology is possible with
the maternal inheritance pattern observed inthe cases described. The results of mitochon-
drial investigations have not given convincingevidence for a mitochondrial cause, however.Although inconsistency in respiratory chainenzyme activities between tissues is oftenobserved in respiratory chain disorders, theabnormal levels are usually observed in theclinically affected organ. In this case, complexIII activity was normal in the liver, yet liverfunction was abnormal, but reduced in muscle,and complex IV was raised in the liver ofpatient 2. The results cannot be regarded asdiagnostic and are at best consistent withprimary mitochondrial pathology.An X linked recessive aetiology would also
be consistent with the inheritance patternobserved. It is possible that a mutation in agene on the X chromosome, which encodes fora protein involved in the respiratory chain, ispresent. None of the subunits encodingrespiratory chain enzymes is known to resideon the X chromosome; however, many nuclearencoded genes are yet to be mapped. '"Complex III contains 11 polypeptide subunits,10 of which are nuclear encoded, the otherbeing encoded by a mitochondrial DNA gene.Complex IV is composed of 13 polypeptidesubunits of which three are mitochondriallyencoded and the remainder nuclear."'An alternative mechanism is that there is a
gene locus on the X chromosome which has aneffect on the mitochondrial genome. The firstreport of linkage for a nuclear gene predispos-ing to mitochondrial deletions has recentlybeen published.'5
In conclusion, we have presented a neonatalprogeroid syndrome with pseudo-obstructionof the urinary and gastrointestinal tracts affect-ing monozygous twin boys and their maternaluncle. Their features differ significantly fromany cases previously described. These patientsadd weight to the proposition of Hagadorn etat that there is clinical heterogeneity amongconditions diagnosed as a "neonatal progeroidsyndrome".
The authors would like to thank Dr David Thorburn for hishelpful comments regarding this manuscript and Ms LaurenceDubourg for translating the paper by de Martinville et allo intoEnglish.
1 Rahman S, Blok RB, Dahl HHM, et al. Leigh syndrome:clinical features and biochemical and DNA abnormalities.Ann Neurol 1996;39:343-51.
2 Rautenstrauch T, Snigula F, Krieg T, Gay S, Muller PK.Progeria: a cell culture study and clinical report of familialincidence. Eur_ Pediatr 1977;124: 101-11.
3 Wiedemann HR. An unidentified neonatal progeroidsyndrome: follow-up report. EurJ'Pediatr 1979;130:65-70.
4 Snigula F, Rautenstrauch R. A new neonatal progeroid syn-drome. EurJPediatr 1981;136:325.
5 Devos EA, Leroy JG, Frijns JP, Van den Berghe H. TheWiedemann-Rautenstrauch or neonatal progeroidsyndrome: report of a patient with consanguineous parents.EurJrPediatr 198 1;136:245-8.
6 Martin Jn, Ceuterick CM, Leroy JG, Devos EA, Roelens JG.The Wiedemann-Rautenstrauch or neonatal progeroidsyndrome: neuropathological study of a case. Neuropediat-rics 1984;15:43-8.
7 Rudin C, Thommen L, Fliegel C, Steinmann B, Biihler U.The neonatal pseudo-hydrocephalic progeroid syndrome(Wiedemann-Rautenstrauch). Report of a new patient andreview of the literature. EurJ Pediatr 1988;147:433-88.
8 Hagadorn JI, Wilson WG, Hogge WA, Callicott JH, BealeEF. Neonatal progeroid syndrome: more than one disease?AmJMed Genet 1990;35:91-4.
9 Bitoun P, Lachassine E, Sellier N, Sauviun S, Gaudelus J.The Wiedemann-Rautenstrauch neonatal progeroid
523
on 3 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.6.520 on 1 June 1997. D
ownloaded from

Delatycki et al
syndrome: a case report and review of the literature. ClinDysmorphol 1995;4:239-45.
10 de Martinville B, Sorin M, Briard ML, Frezal J. Progeria deGilford-Hutchinson a debut neonatal chez des jumeauxmonozygotes. Arch Fr Pediatr 1980;37:679-81.
11 Johns DR, Threlkeld AB, Miller AB, Hurko 0. Multiplemitochondrial DNA deletions in myo-neuro-gastrointestinal encephalopathy syndrome. Am Ophthal-mol 1993;115:108-9.
12 Auricchio A, Brancolini V, Casari G, et al. The locus for anovel syndromic form of neuronal intestinal pseudoob-struction maps to Xq28. Am Hum Genet 1996;58:743-8.
13 Harris DJ, Ashcraft KW, Beatty EC, Holder TM, LeonidasJC. Natal teeth, patent ductus arteriosus and intestinalpseudo-obstruction: a lethal syndrome in the newborn.Clin Genet 1976;9:479-82.
14 Shoffner JM, Wallace DC. Oxidative phosphorylationdiseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The metabolic and molecular bases of inherited disease. 7th ed.New York: McGraw Hill, 1995.
15 Suomalainen A, Kaukonen J, Amati P, et al. An autosomallocus predisposing to deletions ofmitochondrial DNA. NatGenet 1995;9:146-51.
524
on 3 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.34.6.520 on 1 June 1997. D
ownloaded from


















