
A facile fabrication of porous PMMA as a potential bone substitute
7
A facile fabrication of porous PMMA as a potential bone substitute Jing Han a , Guiping Ma a , Jun Nie a,b, ⁎ a State Key Laboratory of Chemical Resource Engineering, The Key Laboratory of Beijing City on Preparation and Processing of Novel Polymer Materials, Beijing University of Chemical Technology, Beijing 100029, PR China b College of Chemistry and Molecular Science, Wuhan University, Wuhan 430072, PR China abstract article info Article history: Received 14 May 2010 Received in revised form 4 February 2011 Accepted 2 April 2011 Available online 16 April 2011 Keywords: Chitosan oligosaccharide Poly(methyl methacrylate) Composite Porous materials Bone substitute This study is aimed to develop porous poly(methyl methacrylate) (PMMA) as a potential bone substitute via a facile fabrication method. Composites consisting of water-soluble chitosan oligosaccharide (CSO) and PMMA were prepared by combining freeze-drying with radical polymerization. Open porous PMMA with controlled porosities were obtained after the CSO was extracted gradually from the composites. The CSO aqueous solutions with different concentrations were frozen and then freeze-dried to obtain interconnected porous framework. Methyl methacrylate with initiators and a crosslink agent was introduced into the porous framework and polymerized, resulting in two-continuous phase composites. The mechanical properties of the initial composites and porous materials after immersion in PBS for 8 weeks were investigated. Dynamic mechanical analysis was conducted to study the mechanical strength of the composite, compared with bulk PMMA. Porosity and morphology of porous PMMA were studied using the liquid displacement method and scanning electron microscopy, respectively. Thermogravimetric analysis indicated that composite exhibited better thermal stability than bulk PMMA. The composites became porous materials after extracting bioactive CSO component. The mechanical properties of porous materials were closer to those of cancellous bone. The generation of pores using CSO seems to be a promising method to prepare porous PMMA as a potential bone substitute. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Poly(methyl methacrylate) (PMMA) and its derivatives have been successfully used in orthopedic surgeries such as bone filler or vertebroplasty [1,2]. However, the disadvantages of PMMA as a biomaterial, such as its insufficient adhesion to bone surface [3], monomer toxicity [4] and high exothermic reaction temperature [5], cannot be ignored. Researchers have tried solving these problems by adding hydroxyapatite (HA) powder reinforcement [6], bone particles [7,8] and growth hormones [9]. The bioactivity and adhesion of PMMA- based materials have been improved through these modifications. Another significant issue of PMMA is the mechanical property. The Young's modulus of commonly used bone substitute materials based on bulk PMMA (2–3 GPa) is much higher than that of cancellous bone (5–800 MPa [10]). An increased fracture risk has been demonstrated for the adjacent vertebral bodies after reinforcement [11,12]. It is desirable to fabricate PMMA with low Young's modulus suitable for bone substitute. Fabricating porous materials with controlled modu- lus and porosity is a feasible solution. The earliest research on porous bone cements with a lower bulk modulus was conducted by De Wijin, who introduced non-miscible phases [13]. Klawitter et al. fabricated and characterized porous-rooted PMMA with two different porous root structures as dental implants, the large pore size intended to accept bone tissue ingrowth and a fine pore size intended to accept fibrous tissue ingrowth [14]. Nathanson et al. investigated the histologic response to porous PMMA implant materials and none of the implants was rejected or caused chronic inflammation [15]. Van Mullen et al. proposed the incorporation of carboxymethyl cellulose as biodegradable component to acrylic bone cement of formulations to create porous cement after the degradable phase leached during time [16]. Tricalcium phosphate as a powder or as an aqueous dispersion modified PMMA porous bone cement was fabricated by Beruto et al. [17]. Liu et al. investigated four types of porous implant materials and found that porous PMMA was biocompatible but possessed less osteogenic potential than hydroxyapatite [18]. Espi- gares et al. fabricated partially degradable and bioactive acrylic bone cements based on corn starch/cellulose acetate blends and ceramic fillers hydroxylapatite [19]. Bruens et al. prepared porous PMMA as bone substitute in the craniofacial area by using carboxymethyl cellulose gel [20]. Employing a commonly used porogen leaching technique, Shimko et al. fabricated highly porous PMMA scaffold with controllable elastic modulus and permeability for use in tissue grafting and tissue engineering applications [21]. Boger et al. prepared low modulus PMMA bone cement for osteoporotic bone by mixing Materials Science and Engineering C 31 (2011) 1278–1284 ⁎ Corresponding author at: State Key Laboratory of Chemical Resource Engineering, The Key Laboratory of Beijing City on Preparation and Processing of Novel Polymer Materials, Beijing University of Chemical Technology, Beijing 100029, PR China. Fax: +86 10 64421310. E-mail address: [email protected] (J. Nie). 0928-4931/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.msec.2011.04.001 Contents lists available at ScienceDirect Materials Science and Engineering C journal homepage: www.elsevier.com/locate/msec
Transcript of A facile fabrication of porous PMMA as a potential bone substitute

Materials Science and Engineering C 31 (2011) 1278–1284
Contents lists available at ScienceDirect
Materials Science and Engineering C
j ourna l homepage: www.e lsev ie r.com/ locate /msec
A facile fabrication of porous PMMA as a potential bone substitute
Jing Han a, Guiping Ma a, Jun Nie a,b,⁎a State Key Laboratory of Chemical Resource Engineering, The Key Laboratory of Beijing City on Preparation and Processing of Novel Polymer Materials,Beijing University of Chemical Technology, Beijing 100029, PR Chinab College of Chemistry and Molecular Science, Wuhan University, Wuhan 430072, PR China
⁎ Corresponding author at: State Key Laboratory of CThe Key Laboratory of Beijing City on Preparation andMaterials, Beijing University of Chemical TechnologFax: +86 10 64421310.
E-mail address: [email protected] (J. Nie).
0928-4931/$ – see front matter © 2011 Elsevier B.V. Adoi:10.1016/j.msec.2011.04.001
a b s t r a c t
a r t i c l e i n f oArticle history:Received 14 May 2010Received in revised form 4 February 2011Accepted 2 April 2011Available online 16 April 2011
Keywords:Chitosan oligosaccharidePoly(methyl methacrylate)CompositePorous materialsBone substitute
This study is aimed to develop porous poly(methyl methacrylate) (PMMA) as a potential bone substitute via afacile fabrication method. Composites consisting of water-soluble chitosan oligosaccharide (CSO) and PMMAwere prepared by combining freeze-drying with radical polymerization. Open porous PMMA with controlledporosities were obtained after the CSO was extracted gradually from the composites. The CSO aqueoussolutions with different concentrations were frozen and then freeze-dried to obtain interconnected porousframework. Methyl methacrylate with initiators and a crosslink agent was introduced into the porousframework and polymerized, resulting in two-continuous phase composites. Themechanical properties of theinitial composites and porous materials after immersion in PBS for 8 weeks were investigated. Dynamicmechanical analysis was conducted to study the mechanical strength of the composite, compared with bulkPMMA. Porosity and morphology of porous PMMA were studied using the liquid displacement method andscanning electron microscopy, respectively. Thermogravimetric analysis indicated that composite exhibitedbetter thermal stability than bulk PMMA. The composites became porous materials after extracting bioactiveCSO component. The mechanical properties of porous materials were closer to those of cancellous bone. Thegeneration of pores using CSO seems to be a promising method to prepare porous PMMA as a potential bonesubstitute.
hemical Resource Engineering,Processing of Novel Polymer
y, Beijing 100029, PR China.
ll rights reserved.
© 2011 Elsevier B.V. All rights reserved.
1. Introduction
Poly(methyl methacrylate) (PMMA) and its derivatives have beensuccessfully used in orthopedic surgeries such as bone filler orvertebroplasty [1,2]. However, the disadvantages of PMMA as abiomaterial, such as its insufficient adhesion to bone surface [3],monomer toxicity [4] and high exothermic reaction temperature [5],cannot be ignored. Researchers have tried solving these problems byadding hydroxyapatite (HA) powder reinforcement [6], bone particles[7,8] and growth hormones [9]. The bioactivity and adhesion of PMMA-based materials have been improved through these modifications.
Another significant issue of PMMA is the mechanical property. TheYoung's modulus of commonly used bone substitute materials basedon bulk PMMA (2–3 GPa) is much higher than that of cancellous bone(5–800 MPa [10]). An increased fracture risk has been demonstratedfor the adjacent vertebral bodies after reinforcement [11,12]. It isdesirable to fabricate PMMA with low Young's modulus suitable forbone substitute. Fabricating porous materials with controlled modu-lus and porosity is a feasible solution. The earliest research on porous
bone cements with a lower bulk modulus was conducted by DeWijin,who introduced non-miscible phases [13]. Klawitter et al. fabricatedand characterized porous-rooted PMMA with two different porousroot structures as dental implants, the large pore size intended toaccept bone tissue ingrowth and a fine pore size intended to acceptfibrous tissue ingrowth [14]. Nathanson et al. investigated thehistologic response to porous PMMA implant materials and none ofthe implants was rejected or caused chronic inflammation [15]. VanMullen et al. proposed the incorporation of carboxymethyl celluloseas biodegradable component to acrylic bone cement of formulationsto create porous cement after the degradable phase leached duringtime [16]. Tricalcium phosphate as a powder or as an aqueousdispersion modified PMMA porous bone cement was fabricated byBeruto et al. [17]. Liu et al. investigated four types of porous implantmaterials and found that porous PMMA was biocompatible butpossessed less osteogenic potential than hydroxyapatite [18]. Espi-gares et al. fabricated partially degradable and bioactive acrylic bonecements based on corn starch/cellulose acetate blends and ceramicfillers hydroxylapatite [19]. Bruens et al. prepared porous PMMA asbone substitute in the craniofacial area by using carboxymethylcellulose gel [20]. Employing a commonly used porogen leachingtechnique, Shimko et al. fabricated highly porous PMMA scaffold withcontrollable elastic modulus and permeability for use in tissuegrafting and tissue engineering applications [21]. Boger et al. preparedlow modulus PMMA bone cement for osteoporotic bone by mixing

1279J. Han et al. / Materials Science and Engineering C 31 (2011) 1278–1284
commercially available PMMA cement and sodium hyaluronatepolymer solution [22]. In this research, we fabricated porous PMMAby adding bioactive chitosan oligosaccharide (CSO) as a framework orporogen.
Chitosan, the deacetylated derivative of chitin, composed ofβ-(1,4)-2-amine-2-deoxy-D-glucopyranose units and small amountof N-acetyl-D-glucosamine residues, has been utilized in biomedicalfield due to its excellent biocompatibility, biodegradability andantibacterial activity. Chitosan powder or nanoparticles have beenincorporated into bone cement aiming to improve its properties [6,23].However, poor solubility in physiology conditions makes chitosandifficult to use in food and biomedical applications. Recently, re-searchers have become interested in partially hydrolyzed chitosan andchitosan oligosaccharide (CSO). CSO is readily soluble inwater due to itsshorter chain lengths and free amino groups in D-glucosamine units.CSO, like chitosan, has positive chargeswhich allow it to bind strongly tonegatively charges and had some properties including biologicalactivity, non-toxicity, biodegradability and biocompatibility. Previouswork has demonstrated that CSO possesses versatile functionalproperties such as antitumor activity [24], antimicrobial activity [25]and free radical scavenging activity [26,27]. Recent interest has focusedon chitosan oligosaccharides as a bone-inducing substance for use asbone graft material [28]. Li et al. found that low molecular weightchitosan (LMWC) inhibited osteoclast formation and the resorbingactivity of osteoclasts, as a result, LMWC prevented decreases in bonedensity after ovariectomy [29]. Iwata et al. also demonstrated thatchitosan oligosaccharides prevented a decrease in cancellous bonemass, which suggested that chitosan oligosaccharidesmight be useful inpreventing bone loss associated with postmenopausal osteoporosis[28].
The purpose of the present study was to develop porous PMMA asa potential bone substitute material derived from CSO/PMMAcomposites. Chitosan oligosaccharide as a bone-inducing biopolymerwas utilized as a framework or porogen. The mechanical propertiesand thermal stability of composites were characterized. Due to thewater-solubility of chitosan oligosaccharide, the CSO componentdissolved gradually from the composites and left the PMMA asinterconnected porous material with an appropriate compressionmodulus and strength, which would be an ideal bone substitute.
2. Experimental
2.1. Materials
Chitosan oligosaccharide (molecular weight of 3000 g/mol, about85% deacetylated) was purchased from Zhejiang Golden-ShellBiochemical Co., Ltd. (Zhejiang, China). Methyl methacrylate (MMA,Tianjin Fuchen Chemical Co.) was purified by vacuum distillation.Triethyleneglycol dimethacrylate (TEGDMA) was kindly supplied bySartomer Company (Guangzhou, China) and used without furtherpurification. Benzoyl peroxide (BPO) and N,N-bis(2-hydroxyethyl)-p-toluidine (BHET) were obtained from Xilong Chemical Co., Ltd.(Shantou, China) and Fluka Chemical Co., respectively. Benzoyl
Fig. 1. Schematic illustration for prepa
peroxide was purified by fractional precipitation from a chloroformsolution, using methanol as precipitant.
2.2. Preparation of CSO/PMMA composites
The fabrication process of CSO/PMMA composites and porousPMMA is illustrated in Fig. 1. First, the cylinder chitosan oligosaccha-ride frames were fabricated via a freeze-drying method. Chitosanoligosaccharide solutions with different concentrations were pre-pared by dissolving CSO in deionizedwater. The solutions were placedinto cylinder molds, maintained at −40 °C for 24 h, and thenlyophilized in a freeze-dryer until dried. Secondly, two kinds ofmethyl methacrylate solution were prepared: one MMA solutioncontaining 0.5 wt.% BPO as an initiator while the other containing0.5 wt.% BHET as a co-initiator and 5 wt.% TEGDMA as a crosslinkagent. Finally, two kinds of MMA solutionsweremixed in a 1/1 weightratio and injected into the dried porous CSO framework quickly.Radical polymerization of MMA and TEGDMA took placed in a porousframework at room temperature. After 48 h, columned CSO/PMMAcomposites with different weight ratios were obtained. For compar-ison, bulk PMMA without chitosan oligosaccharide was also preparedat the same condition. CSO could be extracted gradually from thecomposites through immersion in water, leaving the PMMA compo-nent as an interconnected porous material. Table 1 illustrates thesample codes and their compositions.
2.3. Mechanical compression testing
The compression modulus and compression strength of bulkPMMA and CSO/PMMA composites were determined using an Instron4505 mechanical tester (Instron, High Wycombe, England) with a10 kN load cell. The specimens were circular discs of 8 mm indiameter and 12 mm in thickness. The crosshead speed of the Instrontester was set at 5 mm/min and the load was applied until a 30%reduction in specimen height was achieved. Five samples were testedfor each composition. To analyze the long termmechanical propertiesof various samples, they were immersed in PBS (pH=7.4) 8 weeks,and their compression modulus and compression strength werecompared with the initial compression modulus and compressionstrength determined at the beginning of the experiment.
2.4. Dynamic mechanical analysis
The dynamic mechanical thermal analysis of the materials in theform of circular discs (diameter=8 mm, thickness=4 mm) wascarried out with a dynamic mechanical analyzer (NETZSCH DMA242C, Germany), operating in the compression mode. The scans wereperformed on samples maintained under a nitrogen atmosphere atfrequency of 1 Hz, temperature range of−50 to 300 °C with 5 °C/minheat rate.
ration of CSO/PMMA composites.

Table 1Sample codes and composite properties.
Sample code Concentration of CSOsolution (w/v %)
CSO content(%)
Porosity(%)
CSO10/PMMA 10 10.6 13.17CSO15/PMMA 15 16.3 18.07CSO20/PMMA 20 19.2 21.12
Fig. 2. Images of (a) bulk PMMA, (b) CSO10/PMMA composite and (c) porous PMMAafter leaching CSO.
1280 J. Han et al. / Materials Science and Engineering C 31 (2011) 1278–1284
2.5. Thermogravimetric analysis
The thermal stability of the bulk PMMA and composites wasexamined using a thermogravimetric analyzer (NETZSCH TG 209 F1analyzer, Germany). The experiments were performed at a heatingrate of 20 °C/min from room temperature to 500 °C under 50 mL/minnitrogen flow.
2.6. XRD
X-ray diffraction (XRD)measurements of CSO, bulk PMMAand CSO/PMMA composite were conducted by a Rigaku D/Max2500VB2+/Pcdiffractometer (Rigaku Company, Tokyo, Japan) with 40 kV and 50 mAusing Cu Kα radiation (λ=0.154 nm). The scanning scope of 2θwas 5°to 50° and the scanning rate was 5°/min.
2.7. Determination of CSO content and porosity for porous PMMA
The porosities of different porous PMMA samples were measuredby the liquid displacement method [30]. In order to leach CSO fromthe samples rapidly, composites were cut into discs with a diameter of6 mm and a thickness of 3 mm. The weight (W1) and volume (V) ofthe composites were measured, respectively. To leach the CSOcomponent, the samples were immersed in deionized water, whichwas changed daily. After 1 week, the CSO dissolved and thecomposites became porous PMMA. The porous PMMA, which wassaturated with water, was weighed and noted as W2. Finally, thesamples were dried at 60 °C overnight and then weighed (W3). TheCSO content in composites and porosity of resulting porous PMMAcould be calculated according to the following equations (ρ representsthe density of water).
CSO %ð Þ = W1−W3
W1× 100
Porosity %ð Þ = W2−W3
ρV× 100
2.8. Morphologies of porous PMMA
The microstructure of specimens was observed with a scanningelectron microscope (Hitachi S-4700, Hitachi Company, Japan). Thespecimens were fractured in liquid nitrogen, and then fixed on stubswith sputter coating of gold before observation.
3. Results and discussions
3.1. Fabrication process
Chitosan oligosaccharide component as a porous framework orscaffold was fabricated with a freeze-drying method, and then thepoly(methyl methacrylate) component was prepared via radicalpolymerization. The formulations included methyl methacrylate(MMA), crosslink agent TEGDMA, benzoyl peroxide (BPO) as aninitiator and amine BHET as an accelerator. Acrylic bone cement orPMMA resins have been used with three main activation types:
heating curing, self-curing and light-curing [4]. In the present study,the polymerization reaction of methacrylate monomer and crosslinkagent was initiated by the activation reaction of BPO, with BHET as anamine accelerator at room temperature. All the samples wereformulated with 0.5 wt.% BPO and 0.5 wt.% BHET concentration. Theratio of BPO/BHET was kept 1/1 because it was demonstrated thatboth less andmore amine contents results in less complete conversionof monomer [31].
Fig. 2 presents the images of bulk PMMA, CSO/PMMA compositeand porous PMMA after leached CSO component. Bulk PMMA istransparent, while CSO/PMMA composite exhibits yellow brownbecause chitosan oligosaccharide is yellow powder. Porous PMMAderived from the composite by leached CSO component appearsmilk white.
3.2. Mechanical properties
The mechanical properties of materials play an influential role indetermining the long-term stability of biomaterials. The mechanicaltesting of composites without immersion showed that the compres-sion modulus was significantly influenced by the CSO content in thecomposites as illustrated in Fig. 3(a). The compression modulusimproved gradually with the increase of CSO weight ratio. When theCSO weight ratio was 10%, the compression modulus was lower thanthat of bulk PMMA; when the CSO weight ratio increased to 20%, thecompression modulus reached to 2275 MPa. Fig. 3(a) also shows anobvious decrease in the compression modulus of various CSO/PMMAcomposites after 8 weeks of immersion in PBS. Calculating the weightdifference between initial samples and immersed samples revealedthat the composites lost 60–80% of the CSO component. The differencein the compression modulus was due to the removal of the CSOcomponent. Although the CSO content was low, its contribution to thecompression modulus could not be ignored. CSO, as a rigid and semi-crystallized oligomer played an important role in improving mechan-ical strength of two-continuous phase composites.
Fig. 3(b) shows the initial compression strength of bulk PMMA andCSO/PMMA composites, compared with those after immersion. Thecompression strength and compression modulus followed a similartrend. The compression strength of all composites was lower than thatof bulk PMMA because of the inhomogeneous structure. However, thecompression strength of composites was improved with the increaseof CSO loading, which was attributed to its uniform dispersion inPMMA matrix and the CSO reinforcement effect. After 8 weeks ofimmersion in PBS, compression strength of bulk PMMA deceased byabout 10%, while all CSO/PMMA composite decreased by approxi-mately 40%. The lowest compression strength (56 MPa) was observedfor CSO10/PMMA. The mechanical strength of composites fromfreshly prepared materials was significantly different from themechanical strength of composites from materials that had beenimmersed in PBS for 8 weeks. In conclusion, the mechanical tests
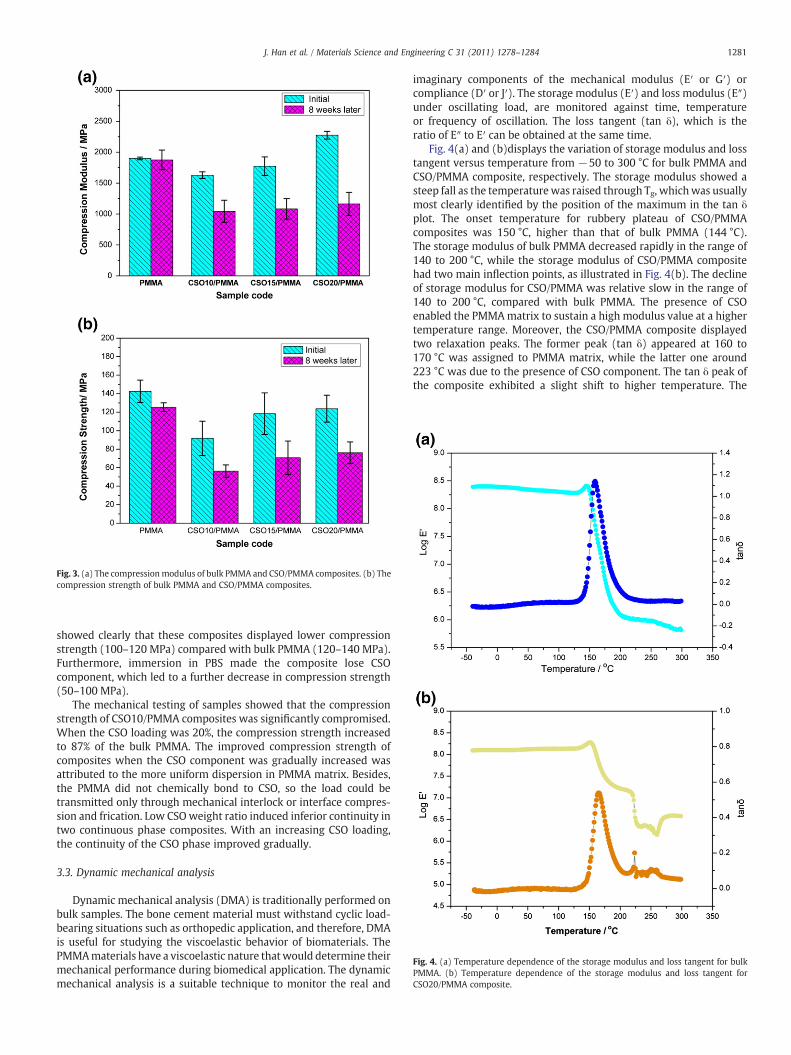
Fig. 3. (a) The compressionmodulus of bulk PMMA and CSO/PMMA composites. (b) Thecompression strength of bulk PMMA and CSO/PMMA composites.
Fig. 4. (a) Temperature dependence of the storage modulus and loss tangent for bulkPMMA. (b) Temperature dependence of the storage modulus and loss tangent forCSO20/PMMA composite.
1281J. Han et al. / Materials Science and Engineering C 31 (2011) 1278–1284
showed clearly that these composites displayed lower compressionstrength (100–120 MPa) compared with bulk PMMA (120–140 MPa).Furthermore, immersion in PBS made the composite lose CSOcomponent, which led to a further decrease in compression strength(50–100 MPa).
The mechanical testing of samples showed that the compressionstrength of CSO10/PMMA composites was significantly compromised.When the CSO loading was 20%, the compression strength increasedto 87% of the bulk PMMA. The improved compression strength ofcomposites when the CSO component was gradually increased wasattributed to the more uniform dispersion in PMMA matrix. Besides,the PMMA did not chemically bond to CSO, so the load could betransmitted only through mechanical interlock or interface compres-sion and frication. Low CSOweight ratio induced inferior continuity intwo continuous phase composites. With an increasing CSO loading,the continuity of the CSO phase improved gradually.
3.3. Dynamic mechanical analysis
Dynamic mechanical analysis (DMA) is traditionally performed onbulk samples. The bone cement material must withstand cyclic load-bearing situations such as orthopedic application, and therefore, DMAis useful for studying the viscoelastic behavior of biomaterials. ThePMMAmaterials have a viscoelastic nature thatwould determine theirmechanical performance during biomedical application. The dynamicmechanical analysis is a suitable technique to monitor the real and
imaginary components of the mechanical modulus (E′ or G′) orcompliance (D′ or J′). The storage modulus (E′) and loss modulus (E″)under oscillating load, are monitored against time, temperatureor frequency of oscillation. The loss tangent (tan δ), which is theratio of E″ to E′ can be obtained at the same time.
Fig. 4(a) and (b)displays the variation of storage modulus and losstangent versus temperature from −50 to 300 °C for bulk PMMA andCSO/PMMA composite, respectively. The storage modulus showed asteep fall as the temperaturewas raised through Tg, whichwas usuallymost clearly identified by the position of the maximum in the tan δplot. The onset temperature for rubbery plateau of CSO/PMMAcomposites was 150 °C, higher than that of bulk PMMA (144 °C).The storage modulus of bulk PMMA decreased rapidly in the range of140 to 200 °C, while the storage modulus of CSO/PMMA compositehad two main inflection points, as illustrated in Fig. 4(b). The declineof storage modulus for CSO/PMMA was relative slow in the range of140 to 200 °C, compared with bulk PMMA. The presence of CSOenabled the PMMAmatrix to sustain a high modulus value at a highertemperature range. Moreover, the CSO/PMMA composite displayedtwo relaxation peaks. The former peak (tan δ) appeared at 160 to170 °C was assigned to PMMA matrix, while the latter one around223 °C was due to the presence of CSO component. The tan δ peak ofthe composite exhibited a slight shift to higher temperature. The

Fig. 6. XRD patterns of bulk PMMA (a), CSO20/PMMA composite (b) and CSO (c).
1282 J. Han et al. / Materials Science and Engineering C 31 (2011) 1278–1284
incorporation of CSO into PMMA matrix influenced the PMMA glasstransition because the chain segmental motion of PMMA could beretarded more or less by the rigid and semi-crystal CSO molecules[32]. In brief, the introduction of CSO delayed the onset of molecularmotions of PMMA and enabled CSO/PMMA to sustain relative highmodulus at a high temperature range, compared with bulk PMMA.
3.4. Thermal degradation
The thermal behavior of methyl methacrylate polymers cross-linked with TEGDMAwas investigated by thermogravimetric analysis(TGA). Fig. 5 shows the TGA and DTG curves of bulk PMMA andCSO20/PMMA composite, respectively. Bulk PMMA had an onsetdegradation temperature at 326 °C and lost nearly all weight at400 °C. It is well known that the degradation of PMMA occurs throughan unzipping mechanism. It consists of three stages. The first stage(150–200 °C) corresponds to a degradation initiation due to thebreaking of weak head to head linkage resulting from termination bycombination. The second (250–300 °C) stage is due to the degradationinitiated by the unsaturated ends resulting from termination bydisproportionation. The last stage (350 °C) is caused by the randomscission of the chain [33].
The degradation behavior of CSO/PMMA composite was differentfrom that of bulk PMMA. The composite had two obvious degradationstages at 226 °C and 395 °C, respectively. The former was ascribed tothe part degradation of CSO; the latter corresponded to thedegradation of PMMA. The presence of CSO offered a stabilizing effectfor PMMA since the onset of degradation occurred at a hightemperature, compared with bulk PMMA. The residual mass at500 °C for bulk PMMA was nearly 0%, while that for composite wasstill 15.68%. The data implied that the presence of CSO hindered theunzipping of the PMMA [32]. The retardation effectsmight attribute tothe interaction between CSO andmacro-radicals generated during thedegradation process.
3.5. XRD analysis
Fig. 6 illustrates the XRD patterns of CSO, bulk PMMA and CSO/PMMA composites. The CSO presented four typical peaks at 8.56°,11.84°, 18.97° and 23.81°. Unlike chitosan with high molecularweight, which exhibited obvious peaks, CSO's peaks were broad.The diffraction pattern of bulk PMMA was consistent with a previousreport [34], showing broad peak at 14.2° and 29.9°, indicating theamorphous state of PMMA. For CSO/PMMA composite, the diffractionpattern was the same as for bulk PMMA. The peaks assigned to CSO
Fig. 5. TGA and DTG curves of bulk PMMA and CSO20/PMMA composite.
disappeared in composites, due to the dissolving and freeze-drying ofCSO in the fabrication process. Moreover, the diffraction pattern ofcomposites also indicated that the existence of CSO has no significantinfluence on the amorphous state of PMMA.
3.6. Composition and porosity
The porosities of various porous PMMA ranged from 10% to 20%, asillustrated in Table 1. After long immersion in water, water-solubleCSO gradually dissolved and the composites became porousmaterials.The porosities of PMMA depended on the initial concentration of thechitosan oligosaccharide solution. In the other words, initial compo-sitions determined the porosities after removing the CSO component.The CSO component and PMMA do not have chemical bonds, so CSOcan dissolve gradually from the composites, leaving composites asporous PMMA. The porosities of various materials were a little higherthan the concentration of corresponding initial CSO solutions due todifferent densities of components. Moreover, there might be somedefects in the composites due to the limited miscibility, which led tohigher porosities.
3.7. Morphology
Fig. 7 exhibits the interconnected structure of porous PMMAderived from different CSO/PMMA composites. Fig. 7(a), (b) and (c)shows the cross-sectional microstructure of the porousmaterials afterleaching of the CSO component, respectively (Magnification ×5000).PMMA exhibited interconnected but irregular pore structure. Theweight ratio of CSO had no obvious influence on their morphologies,though the porosity was determined by the original CSO weight ratioin the composites, as explained in an above section. A lowmagnification (×100) SEM image given in Fig. 7(d) showed generalityof porous structure of PMMA derived from CSO10/PMMA composites.
The porous PMMA is a kind of attractive bone substitute because ofcontrollable mechanical property and interconnected structure fortransportation. Although many methods have been proposed tofabricate porous PMMA, some fabrication methods limit poreinterconnectivity. Our fabrication method ensures the interconnec-tivity of porous structure because the CSO component is a continuousphase in composites. In the composites, CSO component is water-soluble and degradable, while PMMA is a nondegradable polymericmaterial. CSO not only plays an important role as porogen, but alsomakes a great contribution to mechanical strength in initial stage.After extraction of the CSO component, compact composites becameporous materials with a lower but suitable modulus as a bone

Fig. 7. SEM morphologies of porous PMMA derived from different CSO/PMMA composites: (a) CSO10/PMMA, (b) CSO15/PMMA, (c) CSO20/PMMA, (d) CSO10/PMMA.
1283J. Han et al. / Materials Science and Engineering C 31 (2011) 1278–1284
substitute, providing interconnective pores for transporting nutrientsand oxygen. Although PMMA is a nondegradable material, the factthat no adverse effects result from the long term implantation ofPMMA makes it an acceptable as bone substitute. The SEMobservation indicated that the composites could provide porousspaces for osteoconduction after the chitosan oligosaccharide wasdissolved. In the long run, the porous PMMA would be filled andsurrounded by functional and structurally effectively bone and newlyformed bone/PMMA composites.
4. Conclusions
A series of CSO/PMMA composites with two-continuous phasestructure was fabricated through freezing–drying and radical poly-merization. The incorporation of CSO altered the mechanicalproperties of the materials. Dynamic mechanical analysis of bothCSO/PMMA composite and bulk PMMA showed the obvious differencedue to the existence of CSO. The composites became interconnectedporous PMMA after CSO was dissolved. The extraction of CSO led tolower compression modulus and strength, which were close to themechanical properties of cancellous bone and therefore the materialswould be considered as an ideal bone substitute. The porosities ofPMMA were determined by the initial content of CSO. The porousmicrostructure of PMMA was confirmed by the SEM images. Theintroduction of CSO couldmodulate themechanical strength of PMMAmatrix. The fabrication process also provides a facile way to obtaininterconnected porous material with certain porosity, which haspotential in biomedical field as a bone substitute.
Acknowledgments
The Program for Changjiang Scholars and Innovative ResearchTeam in University is gratefully acknowledged. This study was also
supported by the Key Laboratory for Green Chemical Process ofMinistry of Education, Wuhan Institute of Technology, Wuhan430073, P.R. China.
References
[1] S.S. Haas, G.M. Brauer, G. Dickson, J. Bone Joint Surg. Am. 57 (1975) 380.[2] H. Deramond, C. Depriester, P. Galibert, D. Le Gars, Radiol. Clin. North Am. 36 (1998)
533.[3] M.J. Dalby, L. Di Silvio, E.J. Harper, W. Bonfield, Biomaterials 23 (2002) 569.[4] B.Vazquez,C. Elvira, B. Levenfeld, B. Pascual, I.Goni,M.Gurruchaga,M.P. Ginebra, F.X.
Gil, J.A. Planell, P.A. Liso, M. Rebuelta, J. San Roman, J. Biomed. Mater. Res. 34 (1997)129.
[5] J.X. Lu, Z.W. Huang, P. Tropiano, B. Clouet D'Orval, M. Remusat, J. Dejou, J.P. Proust,D. Poitout, J. Mater. Sci. Mater. M. 13 (2002) 803.
[6] S.B. Kim, Y.J. Kim, T.L. Yoon, S.A. Park, I.H. Cho, E.J. Kim, I.A. Kim, J.W. Shin,Biomaterials 25 (2004) 5715.
[7] K.R. Dai, Y.K. Liu, J.B. Park, C.R. Clark, K. Nishiyama, Z.K. Zheng, J. Biomed. Mater.Res. 25 (1991) 141.
[8] Y.K. Liu, J.B. Park, G.O. Njus, D. Stienstra, J. Biomed. Mater. Res. 21 (1987) 247.[9] C.J. Goodwin,M.Braden, S.Downes,N.J.Marshall, J. Biomed.Mater. Res. 34 (1997)47.
[10] X. Banse, T.J. Sims, A.J. Bailey, J. Bone Miner. Res. 17 (2002) 1621.[11] P.F. Heini, U. Berlemann, M. Kaufmann, K. Lippuner, C. Fankhauser, P. van Landuyt,
Eur. Spine J. 10 (2001) 164.[12] A. Polikeit, L.P. Nolte, S.J. Ferguson, Spine 28 (2003) 991.[13] J.R. de Wijn, J. Biomed. Mater. Res. 10 (1976) 625.[14] J.J. Klawitter, A.M. Weinstein, L.J. Peterson, J. Dent. Res. 56 (1977) 385.[15] D. Nathanson, L. Gettleman, P. Schnitman, G. Shklar, J. Biomed.Mater. Res. 12 (1978)
13.[16] P.J. van Mullem, J.R. de Wijin, J.M. Vaandrager, Ann. Plas. Surg. 21 (1988) 576.[17] D.T. Beruto, S.A. Mezzasalma, M. Capurro, R. Botter, P. Cirillo, J. Biomed. Mater. Res.
49 (2000) 498.[18] Y.L. Liu, J. Schoenaers, K. de Groot, J.R. de Wijn, E. Schepers, J. Mater. Sci. Mater.
Med. 11 (2000) 711.[19] I. Espigares, C. Elvira, J.F. Mano, B. Vazquez, J.S. Raman, R.L. Reis, Biomaterials
23 (2002) 1883.[20] M.L. Bruens, H. Pieterman, J.R. deWijn, J.M. Vaandrager, J. Craniofac. Surg. 14 (2003)
63.[21] D.A. Shimko, E.A. Nauman, J. Biomed, Mater. Res. B Appl. Biomater. 80B (2007) 360.[22] A. Boger, M. Bohner, P. Heini, S. Verier, E. Schneider, J. Biomed. Mater. Res. B Appl.
Bimater. 86 (2008) 474.

1284 J. Han et al. / Materials Science and Engineering C 31 (2011) 1278–1284
[23] Z. Shi, K.G. Neoh, E.T. Kang, W. Wang, Biomaterials 27 (2006) 2440.[24] C. Qin, Y. Du, L. Xiao, Z. Li, X. Gao, Int. J. Biol. Macromol. 31 (2002) 111.[25] H.K. No, N.Y. Park, S.H. Lee, S.P. Meyers, Int. J. Food Microbiol. 67 (2002) 65.[26] P.J. Park, J.Y. Je, S.K. Kim, Carbohyd. Polym. 55 (2004) 17.[27] J.Y. Je, P.J. Park, S.K. Kim, Food Chem. Toxicol. 42 (2004) 381.[28] H. Iwata, S. Yana, M. Nasu, T. Yosue, Oral Radiol. 21 (2005) 19.[29] H. Li, T. Miyahara, Y. Tezuka, M. Watanabe, N. Nemoto, H. Seto, S. Kadota,
Phytomedicine 65 (1999) 305.
[30] Y. Zhang, M. Zhang, J. Biomed. Mater. Res. 55 (2001) 304.[31] M.P. Ginebra, F.X. Gil, J.A. Planell, B. Pascual, I. Goni, M. Gurruchaga, B. Levenfeld, B.
Vázouez, J. San Roman, J. Mater. Sci. Mater. Med. 7 (1996) 375.[32] T.M. Don, S.C. Hsu, W.Y. Chiu, J. Polym. Sci. A Polym. Chem. 39 (2001) 1646.[33] M. Cochez, M. Ferriol, J.V. Weber, P. Chaudron, N. Oget, J.L. Mieloszynski, Polym.
Degrad. Stabil. 70 (2000) 455.[34] Y. Li, B. Zhang, X. Pan, Composites Sci. Technol. 68 (2008) 1954.


















