
18050954 rapid-review-pathology
480
ELSEVIER MOSBY CD-R0 with current US -typ questions and rationales • Book: Two 50-question tests with rationales at end of book" • Quick outline text covering USMLE topics 9 • High-Yield Margin Notes . • • • • • P I IP <1. • • • • - • `YE-Nr%.- • • • EDWARD F. GDLLJAN
Transcript of 18050954 rapid-review-pathology

ELSEVIERMOSBY
CD-R0 with current US -typquestions and rationales
• Book: Two 50-question tests
with rationales at end of book"
• Quick outline text covering
USMLE topics
9• High-Yield Margin Notes .
••••• P I IP <1.• • •
• -• `YE-Nr%.-•••
EDWARD F. GDLLJAN

Practical and easy to use, this innovative reviewtool is designed specifically for you! Includedwith this book, the Rapid Review CD-ROMprovides an electronic test-taking experiencejust like the USMLE.
Featuring figures from the book in full color, theCD-ROM includes hundreds of randomizedquestions, answers, and rationales—availablein both a Tutorial Mode and a Test Mode.
11110111
',m.o.:Ir.. number or quest., Gy t We.,.1tra, i F be displayer: relee answer ,e sr.ere,
Yee have owe, lure 0 Ore feaaFac+ r,were. fesling
• Yee wlIroure 60 re, re, cre=rersIe = err Morrwecr greeter, rf yo L, eer,,ere =
rens n, ,neee,.re ree c•re =r,ret
This unique CD-ROM makes Rapid Reviewthe most complete learning and studying toolavailable! Plus, you're sure to feel confident andmore prepared when you take the USMLEI
Imen...cr on. n to Mt c*Iture. a *are.cuadyted ■All IA.!. n hopatoc.,.
I 1 Hophophoglyteralc !Thosphogiycrtalcireelere 69kaaphac Inatm e I 61,31.0."rn
••
The Tutorial Mode allows you to answer aselected number of review questions andreceive rationales for correct and incorrectanswer choices. After you complete thequestions at your own pace, a diagnosticgraph reports your results—broken out byscience or by system.
The CD-ROM's Test Mode simulates theactual USMLE test experience by presenting50 randomized questions to be answered ina 60-minute time frame. Once you havecompleted all of the questions, a diagnosticgraph appears, giving you your results.

•S
RAPID REVIEW SERIES• PA% .1 0 0 GY•••••••••••••••••••••••••••

Visit our website at www.mosby.com

RAPID REVIEW SERIESSeries Editor
Edward F. Goljan, MD
JATHOLEIGY
Edward F. Goljan, MDProfessor and ChairDepartment of PathologyOklahoma State University Center for Health SciencesCollege of Osteopathic MedicineTulsa, Oklahoma

ELSEVIERMOSBY
The Curtis Center170 S Independence Mall W 300EPhiladelphia, Pennsylvania 19106
PATHOLOGY
Copyright © 2004, Mosby, Inc. All rights reserved.
No part of this publication may be reproduced or transmitted in any form or by any means,electronic or mechanical, including photocopying, recording, or any information storageand retrieval system, without permission in writing from the publisher.
Permissions may be sought directly from Elsevier's Health Sciences RightsDepartment in Philadelphia, PA, USA: phone: (+1) 215 239 3804, fax: (+1) 215 239 3805,e-mail: [email protected]. You may also complete your request on-linevia the Elsevier homepage (http://www.elsevier.com), by selecting 'Customer Support'and then 'Obtaining Permissions'.
NOTICE
Pathology is an ever-changing field. Standard safety precautions must be followed, but as newresearch and clinical experience broaden our knowledge, changes in treatment and drugtherapy may become necessary or appropriate. Readers are advised to check the most currentproduct information provided by the manufacturer of each drug to be administered to verifythe recommended dose, the method and duration of administration, and contraindications. Itis the responsibility of the licensed health care provider, relying on experience and knowledgeof the patient, to determine dosages and the best treatment for each individual patient.Neither the publisher nor the editor assumes any liability for any injury and/or damage topersons or property arising from this publication.
The Publisher
ISBN-13: 978-0-323-02393-1ISBN-10: 0-323-02393-2
Acquisitions Editor: William SchmittManaging Editor: Susan KellyDevelopmental Editors: Martha Cushman, Carol VartanianPublishing Services Manager: Patricia TannianSenior Project Manager: Anne AltepeterSenior Designer: Kathi GoscheCover Designer: Melissa WalterIllustrator: Matt Chansky
Printed in the United States of America
Last digit is the print number: 9 8 7 6 5 4 3

To my wife, Joyce. Without you, my life would truly be incomplete. -EFG


-7 IV••••••
••
••I•
• The Rapid Review Series is designed for today's busy medical student whohas completed basic science courses and has only a limited time to prepare
• for the United States Medical Licensing Examination (USMLE) Step 1. After
•conducting numerous focus groups throughout the United States, we werecommitted to responding to students' comments and to designing a productthat would help students prepare for the Step 1 examination. Each book in
•the Rapid Review Series offers a visually integrated approach to review and ispackaged with a CD-ROM, which serves as practice for the USMLE Step 1.
• Special Features• BOOK• • Two-color, easy to follow outline: concisely organized need-to-• know information that integrates basic and clinical sciences
• High-yield margin notes: recall topics most likely to be tested on• Step 1
•• Visual elements: 34 two-color schematics, 90 black and white im-
ages, 80 summary tables
• • Bold and color text: highlights key words and phrases• Practice examinations: two sets of 50 USMLE Step 1—type clinically
• oriented, multiple-choice questions (with 32 images); complete discus-sions (rationales) for all options
• Table of common laboratory values• CD-ROM• • Full-color: 350 USMLE Step 1—type clinically oriented, multiple-• choice questions, including 167 color images and 6 two-color schemat-
ics; complete discussions for all options• • Test mode: 60-minute timed test of 50-question block by topic,• system, or random selection
• Tutorial (review) mode: customize your review (questions and
• discussions) by topic, system, or random selection and receive immedi-ate feedback
0 • Bookmark capability• • Table of common laboratory values
• Scoring function: instant statistical analysis shows your strengths• and weaknesses; print capability
•• vii

11111011111110
Acknowledgmentof Reviewers
The publisher expresses sincere thanks to the medical students whoprovided many useful comments and suggestions for improving the textand questions. Our publishing program will continue to benefit from thecombined insight and experience provided by your reviews. For alwaysencouraging us to focus on our target, the USMLE Step 1, we thank:
Richard M. AwdehYale University School of Medicine
Joy A. BaldwinUniversity of Vermont College of Medicine
John CowdenYale University School of Medicine
Andrew DeakNortheastern Ohio Universities College of Medicine
Tracey DeLuciaLoyola University ChicagoStritch School of Medicine
Steven EngmanLoyola University ChicagoStritch School of Medicine
Michael HoffmanUniversity of Medicine and Dentistry New JerseyRobert Wood Johnson School of Medicine
James MassulloNortheastern Ohio Universities College of Medicine
Sarah SchlegelUniversity of Connecticut School of Medicine
Tina TranVirginia Commonwealth University School of Medicine
viii

,1111.1pimoAcknowledgments
This book is the culmination of more than 22 years of teaching medicalstudents and almost 10 years of teaching USMLE Step 1 board reviewcourses in pathology. Discussions with colleagues, mentors, and medicalstudents, both here and abroad, have contributed greatly to the writing ofthis book.
I especially thank Ivan Damjanov, who has been a constant source ofinspiration over the years that I have been a student and a teacher ofpathology. Many of his photographs are included in the book (and on theCD-ROM) and exemplify his incredible breadth and depth of knowledgeand experience in the field of pathology. Ivan, I truly value your friendshipand support.
Special thanks to Susan Kelly, Managing Editor, and her excellent teamof editors (Martha Cushman and Carol Vartanian). Thank you, Susan, forproviding me with valuable insights that often changed my gibberish(thoughts) into words that actually made sense.
I also thank Matt Chansky, illustrator, who translated my thoughts intofigures, and my valued friend Karlis Sloka, who helped me write many ofthe questions and discussions that are included in the book and on theCD-ROM.
Edward F. Goljan, MD
ix

Table of Contents
1 Cell Injury 1I. Tissue hypoxia 1
II. Consequences of hypoxic cell injury 3III. Free radical cell injury 3IV. Injury to cellular organelles 4V. Intracellular accumulations 5
VI. Adaptation to cell injury: growth alterations 7VII. Cell death 10
2 Inflammation and Repair 14I. Acute inflammation 14
II. Chronic inflammation 17III. Patterns of inflammation 19IV. Tissue repair 20V. Laboratory findings associated with inflammation 23
3 Immunopathology 24I. Cells of the immune system 24
II. Major histocompatibility complex 25III. Hypersensitivity reactions 25IV. Transplantation immunology 27V. Autoimmune diseases 29
VI. Immunodeficiency disorders 33VII. Amyloidosis 36
4 Fluid and Hemodynamic Disorders 38I. Edema 38
II. Thrombosis 39III. Embolism 40IV. Shock 42

Table of Contents xi
5 Genetic and Developmental Disorders 44I. Mutations 44
II. Mendelian disorders 45III. Chromosomal disorders 51IV. Other patterns of inheritance 55V. Disorders of sex differentiation 56
VI. Congenital anomalies 57VII. Selected perinatal and infant disorders 59
VIII. Diagnosis of genetic and developmental disorders 596 Environmental Pathology 60
I. Chemical injury 60II. Physical injury 63
III. Radiation injury 657 Nutritional Disorders 67
I. Protein-energy malnutrition 67II. Eating disorders and obesity 67
III. Fat-soluble vitamins 69IV. Water-soluble vitamins 72
8 Neoplasia 75I. Nomenclature 75
II. Properties of benign and malignant tumors 77III. Cancer epidemiology 81IV. Carcinogenesis 81V. Carcinogenic agents 84
VI. Clinical oncology 869 Vascular Disorders 90
I. Lipids 90II. Lipid disorders 90
III. Arteriosclerosis 91IV. Vessel aneurysms 92V. Venous system disorders 95
VI. Lymphatic disorders 96VII. Vascular tumors and tumor-like conditions 97
VIII. Vasculitic disorders 97IX. Hypertension 100
10 Heart Disorders 103I. Ventricular hypertrophy 103
II. Congestive heart failure 103III. Ischemic heart disease 105IV. Congenital heart disease 109V. Acquired valvular heart disease 112
VI. Myocardial and pericardial disorders 117VII. Cardiomyopathy 118
VIII. Tumors of the heart 119

Xii Table of Contents
11 Red Blood Cell Disorders 121I. Erythropoiesis 121
II. Extramedullary hematopoiesis 122III. Complete blood cell count and other studies 122IV. Microcytic anemias 125V. Macrocytic anemias 130
VI. Normocytic anemias with corrected reticulocytecount < 2% 133
VII. Normocytic anemias with corrected reticulocytecount > 3% 134
12 White Blood Cell Disorders 142I. Benign qualitative white blood cell disorders 142
II. Benign quantitative white blood cell disorders 143III. Neoplastic myeloid disorders 145IV. Lymphoid leukemias 151
13 Lymphoid Tissue Disorders 153I. Lymphadenopathy 153
II. Reactive lymphadenitis 154III. Non-Hodgkin's lymphoma 155IV. Hodgkin's lymphoma 156V. Plasma cell dyscrasias 158
VI. Langerhans' cell histiocytoses 159VII. Mast cell disorders 160
VIII. Disorders of the spleen 160
14 Coagulation Disorders 162I. Normal hemostasis 162
II. Laboratory findings associated with hemostasis 166III. Platelet disorders 168IV. Coagulation disorders 169V. Thrombotic disorders 172
15 Blood Transfusion Disorders 174I. ABO blood groups 174
II. Rh and non-Rh antigen systems 174III. Blood transfusion therapy 175IV. Hemolytic disease of the newborn 177
16 Respiratory Disorders 180I. Upper airway disorders 180
II. Atelectasis 181III. Respiratory infections 183IV. Vascular lung lesions 189V. Restrictive lung diseases 191
VI. Obstructive lung diseases 194VII. Neoplasms 198
VIII. Disorders of the pleura 200

Table of Contents xiii
17 Gastrointestinal Disorders 201I. Disorders of the oral cavity: mouth and jaw 201
II. Salivary gland tumors 202III. Disorders of the esophagus 203IV. Stomach disorders 207V. Disorders of the small and large bowels 210
VI. Anorectal disorders 221VII. Acute appendicitis 221
18 Hepatobiliary and Pancreatic Disorders 2221. Laboratory evaluation in liver cell injury 222
II. Viral hepatitis 223III. Other inflammatory hepatic disorders 227IV. Circulatory disorders of the liver 228V. Alcohol-related and drug- and chemical-induced liver disorders 230
VI. Cholestatic (obstructive) liver disease 230VII. Cirrhosis 231
VIII. Liver tumors 234IX. Gallbladder and biliary tract disorders 235X. Cystic fibrosis 236
XI. Pancreatic disorders 237
19 Kidney Disorders 2391. laboratory studies used to assess renal function 239
II. Congenital anomalies and cystic diseases of the kidney 240III. Glomerular disorders 242IV. Disorders affecting tubules and interstitium 251V. Chronic renal failure 254
VI. Vascular disorders 255VII. Obstructive disorders 256
VIII. Tumors of the kidney 256
20 Lower Urinary Tract and Male Reproductive Disorders 259I. Disorders of the urethra and bladder 259
II. Disorders of the penis 260III. Disorders of the scrotum, testis, and epididymis 261IV. Prostate disorders 262V. Male hypogonadism and erectile dysfunction 266
21 Female Reproductive Disorders and Breast Disorders 267I. Sexually transmitted diseases and other genital infections 267
II. Disorders of the vulva 269III. Disorders of the vagina 270IV. Disorders of the cervix 271V. Disorders of the uterus 272
VI. Fallopian tube disorders: PID 275VII. Disorders of the ovary 275
VIII. Gestational disorders 277IX. Breast disorders in females 279X. Breast disorders in males 283

XIV Table of Contents
22 Endocrine Disorders 284
I. Overview of endocrine disease 284II. Pituitary gland 284
III. Thyroid gland 287IV. Parathyroid glands 292V. Adrenal glands 294
VI. Pancreas 298
23 Musculoskeletal Disorders 302I. Bone disorders 302
II. Joint disorders 304III. Muscle disorders 309IV. Soft tissue disorders 310
24 Skin Disorders 311I. Terminology 311
II. Viral disorders 312III. Bacterial disorders 313IV. Fungal disorders 314V. Benign noninfectious disorders 315
VI. Benign melanocytic disorders 318VII. Neoplastic skin disorders 318
25 Nervous System Disorders 321I. Cerebral edema, herniation, and hydrocephalus 321
II. Developmental disorders 322III. Head trauma 324IV. CNS vascular disorders 325V. CNS infections 327
VI. Demyelinating disorders 329VII. Degenerative disorders 331
VIII. Toxic and metabolic disorders 334IX. CNS tumors 335X. Peripheral nervous system and pineal gland disorders 336
XI. Selected eye and ear disorders 337
Tests 339
Table of Common Laboratory Values 340Test 1 Questions 343Test 1 Answers and Discussions 357Test 2 Questions 389Test 2 Answers and Discussions 403
Index 437

Cell Injury
1. Tissue FlypoxiaA. Hypoxia: general term for disorders causing inadequate oxy-
genation of tissue• Several types of hypoxia produce oxygen (02)-related
changes reported with arterial blood gas measurements(Table 1-1).
B. Ischemia: reduction in arterial blood flow (e.g., occlusion ofarteries, such as coronary artery atherosclerosis)
C. Hypoxemia: decrease in the amount of 0 2 dissolved inplasma; caused by:1. Respiratory acidosis: due to carbon dioxide (CO 2) reten-
tion in the lungs2. Ventilation defect: impaired 02 delivery to the alveoli
a. Perfusion of alveoli without gas exchangeb. Produces intrapulmonary shunting of blood
3. Perfusion defect: absence of blood flow to alveoli (e.g.,pulmonary embolus)
4. Diffusion defect: inability of 02 to diffuse through thealveolar-capillary interface (e.g., interstitial fibrosis)
D. Hemoglobin (Hb)-related abnormalities1. Anemia: reduction in Hb concentration (normal 0 2 dis-
solved in the plasma of arterial blood, Pa02; andnormal arterial 0 2 saturation, Sa02)
2. Methemoglobinemia: excessive methemoglobin(metHb) in the blooda. Oxidized heme groups cannot bind 0 2, decreasing
5a02 without affecting Pa02.b. Caused by oxidizing agents (e.g., nitrite- or sulfur-
containing drugs, such as nitroglycerin and trimetho-prim—sulfamethoxazole) or a deficiency of metHbreductase (normally converts ferric iron, Fe 3+, toferrous iron, Fe2+)
Most commoncause of hypoxia:coronary arteryatherosclerosis
1

2 Pathology
TABLE 1-1 Terminology Associated With Oxygen Transport and Hypoxia
Term
Definition Contributing Factors
Significance
Pa02 Amount of 02dissolved in plasmaof arterial blood
Sa02 Average percentageof 02 bound to Hb
02 content Total amount of 02carried in blood
Percent 0 2 in inspiredair, atmosphericpressure, normal0 2 exchange
Pa02 and valence ofheme iron in eachof the four hemegroups
Fe' binds to 0 2 ; Fe3+does not
Hb concentration inred blood cells(most importantfactor), Pa0 2 , Sa02
Reduced in hypox-emia
Sa02 < 80% producescyanosis of skinand mucousmembranes
Hb is most importantcarrier of 02
Fee', ferrous iron; Fe', ferric iron; Hb, hemoglobin; 02, oxygen Pa02, partial pressure ofarterial oxygen; Sa02, arterial oxygen saturation
Patients with methemoglobinemia havechocolate-colored blood and cyanosis; their skincolor does not return to normal after administra-tion of 02 . Treatment is methylene blue (acti-vates metHb reductase) and ascorbic acid (re-duces Fe3+ to Fe2+).
3. Carbon monoxide (CO) poisoninga. Tissue hypoxia
(1) CO competes with 02 for binding sites on Hb,which decreases Sa02 without affecting Pa02.
(2) It inhibits cytochrome oxidase in the electrontransport chain.
(3) It causes a left shift in the 0 2-binding curve.b. Causes: automobile exhaust, smoke inhalation
4. Factors causing a left shift in the 02-binding curvea. Decreased 2,3-bisphosphoglycerateb. CO, alkalosis, metHb, fetal Hb, hypothermia
E. Abnormalities in oxidative phosphorylation: decreasedsynthesis of adenosine triphosphate (ATP) in the inner mito-chondrial membrane1. Defective oxidative phosphorylation
a. CO and cyanide (CN) inhibit cytochrome oxidase inthe electron transport chain, which normally trans-fers electrons to 02.
b. CN poisoning may result from drugs (e.g., nitroprus-side) and combustion of polyurethane products.
2. Uncoupling of oxidative phosphorylationa. Uncoupling proteins (e.g., thermogenin in brown fat
in newborns) carry protons pumped from the elec-tron transport chain into the mitochondrial matrix,
Treat CO poisoningwith 100% 02_
CO and CN inhibitcytochrome oxidase.

Chapter 1 Cell Injury 3
bypassing ATP synthase and decreasing ATPsynthesis.
Agents such as alcohol and salicylates act asmitochondrial toxins. They damage the innermitochondrial membrane, causing protons tomove into the mitochondrial matrix.
b. Oxidative energy is released as heat rather than asATP, increasing the danger of hyperthermia.
II. Consequences of Hypoxic Cell InjuryA. Decreased synthesis of ATP: reversible change
1. Anaerobic glycolysis is used for ATP synthesis and isaccompanied by:a. Activation of phosphofructokinase caused by
low citrate levels and increased adenosinemonophosphate
b. Decrease in intracellular pH caused by an excess oflactate
2. Impaired Ne, K+ -ATPase pump, resulting in diffusionof M.+ and H 20 into cells and causing cellular swelling
3. Impaired calcium (Ca 2+)-ATPase pump, resulting in in-creased cytosolic Ca2+
4. Decreased protein synthesis, resulting from the detach-ment of ribosomes from the rough endoplasmicreticulum
B. Increased cytosolic Ca2+, which leads to:1. Enzyme activation
a. Activates phospholipase: increases cell and organ-elle membrane permeability
b. Activates proteases: damages membrane and struc-tural proteins
c. Activates endonucleases: damages nuclear chro-matin, causing fading (karyolysis)
2. Reentry of Ca2+ into mitochondria: increases mitochon-drial membrane permeability, with release of cyto-chrome c (activates apoptosis)
[II. Free Radical Cell Injury• Free radicals are compounds with unpaired electrons in the
outer orbit.A. 02-derived free radicals
1. Superoxides (02.): neutralized by superoxide dismutase2. Hydroxyl ions (OH •): neutralized by glutathione
peroxidase3. Peroxides (H 202): neutralized by catalase (located in per-
oxisomes) and glutathione peroxidaseB. Drug and chemical free radicals: conversion to free radicals
occurs via the cytochrome P-450 system in the liver.
Lactate decreasesintracellular pHand denatures struc-tural and enzymeproteins.

4 Pathology
1. Free radicals from acetaminophen, which may be neu-tralized by glutathione peroxidase, lead to liver andkidney injury.
2. Carbon tetrachloride (CC14) is converted to CC13•,leading to liver cell necrosis with fatty change.
C. Consequences of free radical injury1. Lipid peroxidation of polyunsaturated fats, leading to
increased permeability of cells and organelles2. Irreversible injury to nuclear DNA and cytoskeletal
proteins3. Clinical correlations
a. Reperfusion injury in the heart after myocardialinfarction: 02i and cytosolic Ca 2+ irreversiblydamage previously injured cells on restoration ofblood flow.
b. 02 toxicity (levels > 50%): 0 2i may damage retinaltissue, causing blindness.
c. Iron overload (e.g., hemochromatosis): intracellulariron produces OH•, which damages parenchymalcells (e.g., cirrhosis).
IV. Injury to Cellular OrganellesA. Mitochondria: injury initiates apoptosis resulting from the
release of cytochrome c.B. Smooth endoplasmic reticulum (SER)
1. Initiation of the cytochrome P-450 system by drugs orchemicalsa. Caused by alcohol, barbiturates, phenytoin, and
nicotineb. Results in SER hyperplasia and increased drug de-
toxification, with lower-than-expected therapeuticdrug levels
2. Inhibition of the cytochrome P-450 systema. Caused by histamine receptor blockers (e.g.,
cimetidine) and proton pump inhibitors(e.g., omeprazole)
b. Results in decreased drug detoxification, withhigher-than-expected therapeutic drug levels
C. Lysosomes1. Primary lysosomes
a. Hydrolytic enzymes destined for primary lysosomesare marked with mannose 6-phosphate in theGolgi apparatus.
b. Marked enzymes are transferred to primarylysosomes.
SER hyperplasialeads to increaseddrug metabolism.

Chapter 1 Cell Injury 5
Inclusion (D-cell disease is a rare inherited con-dition in which lysosomal enzymes lack themannose 6-phosphate marker. Therefore, pri-mary lysosomes do not contain the hydrolyticenzymes necessary to degrade complex sub-strates, and undigested substrates accumulate aslarge inclusions in the cytosol. Symptoms in-clude psychomotor retardation and early death.
2. Secondary lysosomes (phagolysosomes): arise fromfusion of primary lysosomes with phagocytic vacuoles:defective in Chêdiak-Higashi syndrome
D. Cytoskeleton1. Mitotic spindle defects: drugs bind to tubulin in micro-
tubules (e.g., vinca alkaloids, colchicine).2. Mallory bodies: damaged keratin intermediate filaments
in alcoholic liver disease3. Rigor mortis: myosin heads become locked to actin fila-
ments due to a lack of ATP.
V. Intracellular Accumulations• These exogenous or endogenous substances are a sign of cell
injury (Table 1-2).A. Fatty change in the liver: cytosolic accumulation of
triacylglycerol1. Mechanisms of fatty change
a. Increased glycerol 3-phosphate(1) Reduced nicotinamide adenine dinucleotide
(NADH) is a product of alcohol metabolism.(2) Increased NADH accelerates enzyme conver-
sion of dihydroxyacetone phosphate to glycerol3-phosphate.
b. Increased fatty acid synthesis (e.g., increased pro-duction of acetyl coenzyme A, a product of alcoholmetabolism)
c. Decreased (3-oxidation of fatty acids (e.g., alcohol,diphtheria toxin)
d. Increased mobilization of fatty acids from adiposetissue (e.g., starvation, alcohol)
e. Decreased synthesis of apolipoprotein B-100 (e.g.,decreased protein intake in kwashiorkor)
f. Decreased hepatic release of very low density lipo-protein (e.g., alcohol)
2. Morphologya. Gross: normal or enlarged liver with a yellowish
discolorationb. Microscopic: clear space pushing the nucleus of the
hepatocyte to the peripheryB. Iron (see Table 1-2)
1. Ferritin: major soluble iron storage protein
Most commoncause of fattychange in the liver:alcohol

6 Pathology
TABLE 1-2 Intracellular Accumulations
Substance Clinical Significance
Endogenous AccumulationsCholesterol Xanthelasma: yellow plaque on eyelid; cholesterol in
macrophagesAtherosclerosis: cholesterol-laden smooth muscle cells and
macrophages are component of fibrofatty plaque
Glycogen Diabetes mellitus: increased glycogen in hepatocyte nucleiand renal tubule cells
Von Gierke's glycogenosis: deficiency of glucose-6-phosphatase; glycogen excess in hepatocytes and renaltubular cells
Melanin Nevus: benign pigmented melanocytic neoplasm of skin
Hemosiderin and ferritin
Bilirubin
Iron overload disorders (e.g., hemochromatosis): excesshemosiderin deposition in parenchymal cells, leading tofree radical damage and organ dysfunction (e.g., cirrho-sis); increase in serum ferritin
Iron deficiency: decrease in ferritin and hemosiderin
Kernicterus: fat-soluble unconjugated bilirubin derivedfrom Rh hemolytic disease of newborn; bilirubin entersbasal ganglia nuclei of brain, causing permanent damage
Coal worker's pneumoconiosis: phagocytosis of blackanthracotic pigment (coal dust) by alveolar macrophages("dust cells")
Lead poisoning: lead deposits in nuclei of proximal renaltubular cells (acid-fast inclusion) contribute to nephro-toxic changes in proximal tubule
Exogenous AccumulationsAnthracotic pigment
Lead
a. Primary storage sites: hepatocytes and bone marrowmacrophages
b. Small amounts circulate in serum: decreased serumferritin correlates with decreased ferritin stores inbone marrow macrophages.
2. Hemosiderin: product of ferritin degradation in lyso-somes; appears as golden-brown granules in tissue or asblue granules when stained with Prussian blue
C. Pathologic calcification1. Dystrophic calcification: deposition of calcium phos-
phate in necrotic tissuea. Normal serum calcium and phosphateb. Example: calcified atherosclerotic plaque
2. Metastatic calcification: deposition of calcium phos-phate in normal tissue (e.g., calcification of renal tubularbasement membranes); causes include:a. Hypercalcemia (primary hyperparathyroidism)b. Hyperphosphatemia: phosphate drives calcium into
normal tissue (renal failure).
Serum ferritin isdecreased In irondeficiency anemia

Chapter 1 Cell Injury 7
Figure 1-1 Left ventricular hypertrophy. shovvolg the thickened free Left ventricular wall (right
side) and the thickened interventricular septum The right ventricle wall (left side) is of normalthickness.
VI. Adaptation to Cell Injury: Growth AlterationsA. Atrophy: diminished cell size or loss of cells
1. Causes of diminished cell sizea. Decreased hormone stimulation (e.g., hypopituita-
rism causing atrophy of target organs, such as thethyroid)
b. Decreased activity (e.g., muscle atrophy followingloss of lower motor neurons in poliomyelitis)
c. Reduced blood flow (e.g., cerebral atrophy)d. Occlusion of secretory ducts
2. Cell loss is caused by apoptosis.3. Cell and organ effects of atrophy
a. Increased catabolism of cell organelles (e.g., mito-chondria) leads to a reduction in tissue mass andfunction.
b. Presence of vacuoles containing lipofuscin
Brown atrophy is a tissue discoloration thatresults from lysosomal accumulation of lipofus-cin ("wear and tear" pigment), which is associ-ated with free radical damage and tissue atrophy.Lipofuscin is an indigestible lipid derived fromlipid peroxidation of cell membranes.
B. Hypertrophy: increase in cell size due to increased demand1. Causes
a. Increased workload (e.g., increased peripheral resis-tance imposed on cardiac muscle in the left ventri-cle in essential hypertension; (Figure I-I)
b. Removal of an organ (e.g., removal of one kidneycauses hypertrophy of the remaining kidney)
2. Cell and organ effects of hypertrophy: increased syn-thesis of cell structural components and organelles leadsto an increase in organ size and function.
Pancreatic exocrinedeficiency in cysticfibrosis is due toatrophy of the exo-crine glands.

8 Pathology
C. Hyperplasia: increase in the number of normal cells1. Causes
a. Hypersecretion of a trophic hormone (e.g., excessrelease of growth hormone in acromegaly)
b. Chronic irritation (e.g., bronchial mucous gland hy-perplasia in smokers)
c. Chemical imbalance (e.g., hypocalcemia stimulatesparathyroid gland hyperplasia)
2. Hyperplasia (and hypertrophy) depends on the regener-ative capacity of different types of cells.a. Labile cells (stem cells) divide continuously and
mainly undergo hyperplasia as an adaptation to cellinjury (e.g., stimulation of red blood cell stem cellsby erythropoietin in blood loss).
b. Stable cells (resting cells) divide infrequently andundergo hyperplasia and/or hypertrophy (e.g., hyper-plasia of hepatocytes in liver injury; hyperplasia andhypertrophy of smooth muscle cells in the uterusduring pregnancy).
c. Permanent cells (nonreplicating cells) are highlyspecialized cells that undergo hypertrophy only (e.g.,cardiac and striated muscle).
3. Cell and organ effects of hyperplasiaa. Increase in organ size and functionb. Potential for developing cancer if not treated
D. Metaplasia: replacement of one fully differentiated tissue byanother1. Involves reprogramming stem cells in response to
signals, such as hormones (e.g., estrogen); vitamins(e.g., retinoic acid); or chemical irritants (e.g., cigarettesmoke); sometimes reversible
2. Types of metaplasiaa. Squamous: replacement of columnar epithelium by
squamous epithelium (e.g., squamous metaplasiaof mainstem bronchus from smoking tobacco)
b. Glandular: replacement of squamous epitheliumwith intestinal cells (e.g., goblet cells, mucus-secreting cells)
Glandular metaplasia of the distal esophagusresults from injury of the squamous epitheliumby gastric acid in gastroesophageal reflux disease(Barrett's esophagus). Persistence of acid refluxmay result in gastric adenocarcinoma of thedistal esophagus.
E. Dysplasia: abnormal tissue development1. Causes
a. Hyperplasia (e.g., endometrial hyperplasia caused byestrogen excess)
Hypertrophy andhyperplasia dependon the regener-ative capacity ofcells.
..110 • 'dr
Chapter 1 Cell Injury 9
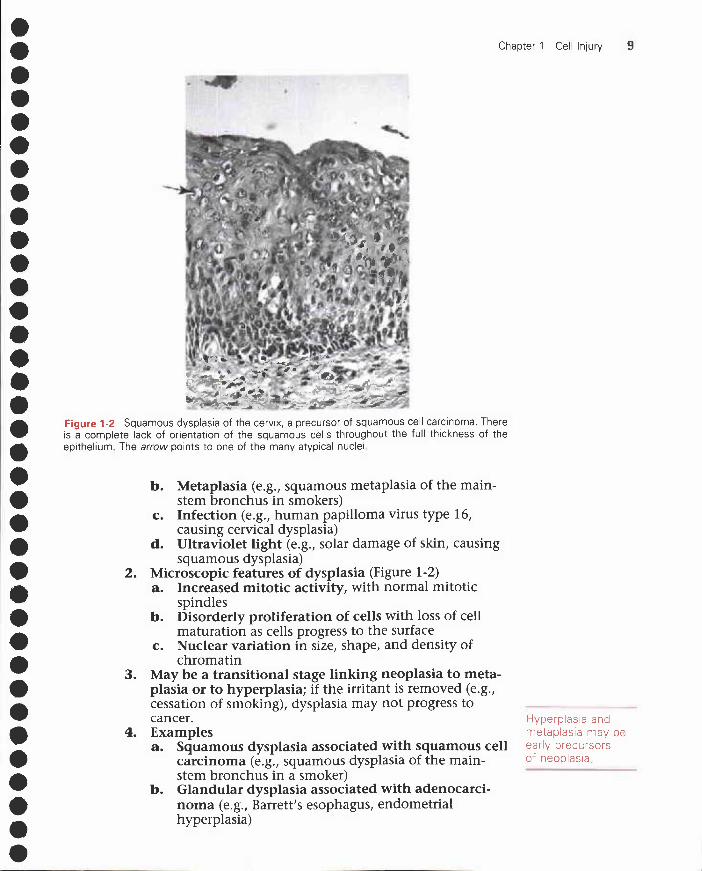
Figure 1-2 Squamous dysplasia of the cervix, a precursor of squamous cell carcinoma. Thereis a complete lack of orientation of the squamous cells throughout the full thickness of theepithelium. The arrow points to one of the many atypical nuclei
b. Metaplasia (e.g., squamous metaplasia of the main-stem bronchus in smokers)
c. Infection (e.g., human papilloma virus type 16,causing cervical dysplasia)
d. Ultraviolet light (e.g., solar damage of skin, causingsquamous dysplasia)
2. Microscopic features of dysplasia (Figure 1-2)a. Increased mitotic activity, with normal mitotic
spindlesb. Disorderly proliferation of cells with loss of cell
maturation as cells progress to the surfacec. Nuclear variation in size, shape, and density of
chromatin3. May be a transitional stage linking neoplasia to meta-
plasia or to hyperplasia; if the irritant is removed (e.g.,cessation of smoking), dysplasia may not progress tocancer.
4. Examplesa. Squamous dysplasia associated with squamous cell
carcinoma (e.g., squamous dysplasia of the main-stem bronchus in a smoker)
b. Glandular dysplasia associated with adenocarci-noma (e.g., Barrett's esophagus, endometrialhyperplasia)
••••••••••••••••S••••••••••••••••••
Hyperplasia andmetaplasia may beearly precursorsof neoplasia.

A
B
10 Pathology
Figure 1-3 Dry and wet gangrene of the feet. Dry gangrene (A) involving the second toe ofthe right foot shows coagulation necrosis The dark black area of gangrene is bordered by thelight-colored, parchment-like skin The tip of the third toe has early signs of gangrene. Wetgangrene (B) involving the hallux area of the left foot shows liquefactive necrosis caused by asuperimposed infection of anaerobic bacteria, usually Clostridium perfringens The taut skinand areas of ulceration extend from the metatarsal head to the lateral border of the big toe
•••••••••••••
VII. Cell Death• Cell death occurs when cells or tissues are unable to adapt to
injury.A. Necrosis: death of groups of cells, often accompanied by an
inflammatory infiltrate1. Coagulation necrosis: preservation of the structural
outline of dead cellsa. Mechanism: denaturation of enzymes and struc-
tural proteins by intracellular accumulation oflactate or heavy metals (e.g., lead, mercury); inacti-vation of intracellular enzymes prevents dissolu-tion (autolysis) of the cell.
b. Infarcts: gross manifestations of coagulation necrosissecondary to the sudden occlusion of a vessel(1) Usually wedge-shaped and occur when dichot-
omously branching vessels (e.g., pulmonaryartery) are occluded
(2) Pale (ischemic): increased density of tissue(e.g., heart, kidney, spleen) prevents red bloodcells from diffusing through necrotic tissue.
Dry gangrene of the toes in individualswith diabetes mellitus is a form of infarc-tion that results from ischemia. Coagula-tion necrosis is the primary type of necro-sis present in the dead tissue (Figure 1-3, A) .
(3) Hemorrhagic (red): loose-textured tissue (e.g.,lungs, small bowel) allows red blood cells todiffuse through necrotic tissue.
••••••••••••••••••••••

Chapter 1 Cell Injury 11
Figure 1-4 Acute myocardial infarction (MI) showing coagulation necrosis This section ofmyocardial tissue is from a 3-day-old acute MI The outlines of the myocardial fibers are intact;however, they lack nuclei and cross-striations A neutrophilic infiltrate is present betweensome of the dead fibers
c. Microscopic features (Figure 1-4)(1) Indistinct outlines of cells within dead tissue(2) Absent nuclei or karyolysis (fading of nuclear
chromatin)2. Liquefactive necrosis: necrotic degradation of tissue
that softens and becomes liquifieda. Central nervous system infarction: autocatalytic
effect of hydrolytic enzymes generated by neuroglialcells produces a cystic space.
b. Abscess in a bacterial infection: hydrolytic enzymesgenerated by neutrophils liquefy dead tissue.
Wet gangrene of the toes of individuals withdiabetes mellitus is a superimposed anaerobicinfection of dead tissue. Liquefactive necrosis isthe primary type of necrosis present in the deadtissue (Figure 1-3, B).
3. Caseous necrosis: variant of coagulation necrosis associ-ated with acellular, cheese-like (caseous) materiala. Gaseous material is formed by the release of lipid
from the cell walls of Mycobacterium tuberculosis andsystemic fungi (e.g., Histoplasma) after destruction bymacrophages.
b. Microscopic features(1) The acellular material in the center of a granu-
loma contains activated macrophages, helperT cells, and multinucleated giant cells (seeChapter 2).
(2) Some granulomas do not exhibit caseation (e.g.,sarcoidosis).
4. Enzymatic fat necrosis: peculiar to adipose tissue locatedaround an acutely inflamed pancreas
Most commoncause of caseousnecrosis:tuberculosis
Enzymatic fat ne-crosis is associatedwith acutepancreatitis.

12 Pathology
a. Mechanisms(1) Activation of pancreatic lipase (e.g., alcohol
excess): hydrolysis of triacylglycerol in fat cells(2) Conversion of fatty acids into soap (saponifi-
cation): combination of fatty acids and calciumb. Gross appearance: chalky yellow-white deposits
are primarily located in peripancreatic andomental adipose tissue.
c. Microscopic appearance: pale outlines of fat cellsfilled with basophilic-staining calcified areas
d. Distinction from traumatic fat necrosis: occurs infatty tissue (e.g., female breast tissue) as a result oftrauma; is not enzyme-mediated
5. Fibrinoid necrosis: limited to small muscular arteries, ar-terioles, venules, and glomerular capillariesa. Mechanism: deposition of pink-staining proteina-
ceous material in damaged vessel wallsb. Associated conditions: immune vasculitis (e.g.,
Henoch-SchOnlein purpura), malignant hypertensionB. Apoptosis: genetically controlled, enzyme-dependent death
of individual cells1. Events in apoptosis
a. Signals that initiate the process(1) Binding of tumor necrosis factor to its receptor(2) Injurious agents: viruses, radiation, free radicals
that damage DNA(3) Withdrawal of growth factors or hormones
b. Modulators that control cell response to the signal(1) TP53 (p53) suppressor gene: temporarily
arrests the cell cycle to repair DNA damage(aborts apoptosis) or promotes apoptosis if DNAdamage is too great by activating the BAXapoptosis gene
(2) BCL2 gene family: manufactures gene productsthat inhibit apoptosis by preventing mitochon-dria) leakage of cytochrome c into the cytosol
c. Enzymatic cell death beginning with activation ofthe caspases (group of cysteine proteases)(1) Activation of endonuclease leads to nuclear
pyknosis ("ink dot" appearance) andfragmentation.
(2) Activation of protease leads to the breakdownof the cytoskeleton.
d. Formation of cytoplasmic buds on the cell mem-brane, which contain nuclear fragments, mitochon-dria, and condensed protein fragments
e. Formation of apoptotic bodies by the breaking off ofcytoplasmic buds
f. Phagocytosis of apoptotic bodies by neighboringcells or macrophages
2. Microscopic appearancea. Cell detachment from neighboring cells
Caspases causeenzymatic cell deathin apoptosis.

Chapter 1 Cell Injury 13
TABLE 1-3 Enzyme Markers of Cell Death
Enzyme Diagnostic Use
Aspartate aminotransferase(AST)
Alanine aminotransferase (ALT)
Creatine kinase MB (CK-MB)
Amylase and lipase
Marker of diffuse liver cell necrosis (e.g., viralhepatitis)
Mitochondrial enzyme preferentially increased inalcohol-induced liver disease
Marker of diffuse liver cell necrosis (e.g., viralhepatitis)
More specific for liver cell necrosis than AST
Isoenzyme elevated in acute myocardial infarction ormyocarditis
Marker enzymes for acute pancreatitisLipase more specific than amylase for pancreatitisAmylase also increased in salivary gland inflamma-
tion (e.g., mumps)
b. Deeply eosinophilic-staining cytoplasmc. Pyknotic, fragmented, or absent nucleusd. Minimal or no inflammatory infiltrate surrounding
the cell3. Examples
a. Programmed destruction of cells during embryo-genesis (e.g., loss of mullerian structures in malefetus)
b. Hormone-dependent atrophy of tissue (e.g., endo-metrial cell breakdown after withdrawal of estro-gen and progesterone in the menstrual cycle)
c. Death of tumor cells by cytotoxic T cellsC. Enzyme markers of cell death
1. Tissues release certain enzymes that indicate the type oftissue involved and extent of injury.
2. Table 1-3 lists clinically significant enzyme markers.

2
Inflammationand Repair
I. Acute Inflammation• Transient and early response to injury (e.g., bacterial infection)
involves release of chemical mediators, causing stereotypic vesseland leukocyte responses.
A. Cardinal signs of inflammation1. Rubor (redness): histamine-mediated vasodilation of
arterioles2. Calor (heat): histamine-mediated vasodilation of
arterioles3. Tumor (swelling): histamine-mediated increase in per-
meability of venules4. Dolor (pain): caused by prostaglandin E2 and bradykinin
B. Vascular events1. Transient vasoconstriction of arterioles: lasts only
seconds2. Vasodilation of arterioles: mast cells release histamine,
which acts on vascular smooth muscle and causes in-creased blood flow.
3. Increased permeability of venules: histamine contractsendothelial cells of the vessels, causing movement of atransudate into interstitial tissue.
4. Swelling of tissue: outflow of fluid surpasses lymphaticability to remove fluid.
5. Reduced blood flow: caused by outflow of fluid fromblood vessels
C. Cellular events1. Margination: red blood cells (RBCs) aggregate into rou-
leaux ("stacks of coins") in venules, with the neutrophilspushed to the periphery.
2. Rolling: selectin molecules on the cell surfaces cause theneutrophils to "roll" along the endothelium or toadhere to it temporarily.
3. Adhesion: neutrophils adhere to endothelial cells.
Neutrophils are theprimary leuko-cytes in acuteinflammation
14

Chapter 2 Inflammation and Repai r 15
a. Activation of adhesion molecules on neutrophils(CD11/CD18 integrins) is mediated by C5a and leu-kotriene B 4 (LTB4).
b. Activation of adhesion molecules on endothelial cellsis mediated by interleukin-1 (IL-1) and tumor necro-sis factor (TNF).
4. Transmigration: neutrophils move through the base-ment membrane of venules and release type IV collage-nase, producing an exudate in the interstitial tissue.
Leukocyte adhesion molecule defect prevents neu-trophil adhesion and transmigration into tissue,causing failure of the umbilical cord to separate afterbirth. Histologic sections of the umbilical cord do notshow margination or transmigration by neutrophilsinto the connective tissue matrix.
5. Chemotaxis: neutrophils migrate toward bacteria.a. Chemical mediators (e.g., C5a, LTB4) bind to neu-
trophil receptors.b. Binding causes the release of calcium, increasing neu-
trophil motility.6. Phagocytosis: neutrophils ingest opsonized bacteria.
a. Opsonization: IgG and C3b attach to bacteria.b. Ingestion: neutrophils with membrane receptors for
IgG and C3b engulf and then trap bacteria inphagocytic vacuoles.
c. Phagolysosome formation: primary lysosomesempty hydrolytic enzymes into phagocytic vacuoles.
In Chediak-Higashi syndrome, a defect in mi-crotubule polymerization inhibits phagocytosisand chemotaxis. The lysosomes are packed withenzymes and appear as large azurophilic granulesin leukocytes.
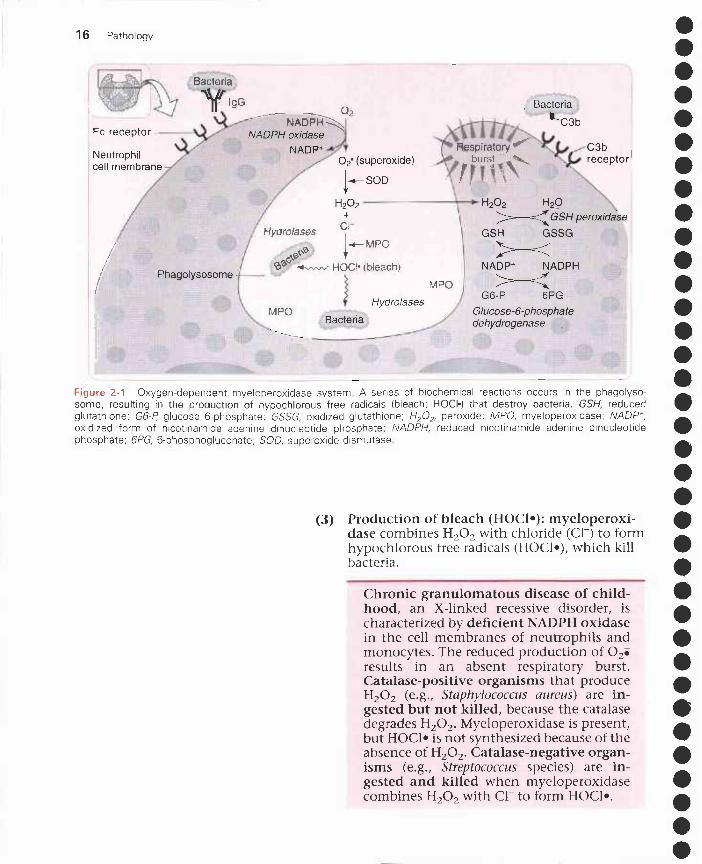
7. Bacterial killing by neutrophilsa. 02-dependent myeloperoxidase system (Figure 2-1)
(1) Production of superoxide free radicals (0 2i ):reduced nicotinamide adenine dinucleotidephosphate (NADPH) oxidase in the neutrophilcell membrane converts molecular 02 to 02iusing NADPH (pentose phosphate pathway);the resulting release of energy is called the res-piratory (oxidative) burst.
(2) Production of peroxide (H202): superoxidedismutase converts 02i to H202, which is neu-tralized by glutathione peroxidase.
••••••••••••••••••••••••••••••••••••
Opsonization is de-fective in Bruton'sagammaglobulinemia.
Most potent mIcro-bicidal system:02-dependentmyeloperoxidasesystem
C3breceptor '
Fc receptor
Neutrophilcell membrane
02
NADPH oxidase
NADP+02 (superoxide)
SOD
H202
Hydrolases
\esN'a'
Cl
MPO
HOC]• (bleach)Phagolysosome
MPO
Hydrolases
H202 F120peroxidase
, Bacteria
C3b
Respiratory Or.--burst
GSH GSSG
NADP+NADPH
G6-P 6PGGlucose-6-phosphatedehydrogenase
16 Pathology
Figure 2 - 1 Oxygen-dependent myeloperoxidase system A series of biochemical reactions occurs in the phagolyso-some, resulting in the production of hypochlorous free radicals (bleach; HOC• that destroy bacteria, GSH, reducedglutathione; G6-P, glucose 6-phosphate; GSSG, oxidized glutathione; H202, peroxide; MPO, myeloperoxidase;oxidized form of nicotinamide adenine dinucleotide phosphate; NADPH, reduced nicotinamide adenine dinucleotidephosphate; 6PG, 6-phosphogluconate; SOD, superoxide dismutase
(3) Production of bleach (HOC1 •): myeloperoxi-dase combines H202 with chloride (a-) to formhypochlorous free radicals (HOC1 •), which killbacteria.
Chronic granulomatous disease of child-hood, an X-linked recessive disorder, ischaracterized by deficient NADPH oxidasein the cell membranes of neutrophils andmonocytes. The reduced production of 02iresults in an absent respiratory burst.Catalase-positive organisms that produceH202 (e.g., Staphylococcus aureus) are in-gested but not killed, because the catalasedegrades H202 . Myeloperoxidase is present,but HOC1 • is not synthesized because of theabsence of H202 . Catalase-negative organ-isms (e.g., Streptococcus species) are in-gested and killed when myeloperoxidasecombines FI202 with to form HOC1•.

Chapter 2 Inflammation and Repair 17
Myeloperoxidase deficiency, an autosomal re-cessive disorder, differs from chronic granuloma-tous disease in that both 02: and H202 areproduced (normal respiratory burst). The ab-sence of myeloperoxidase prevents synthesis ofHOC1•.
b. 02-independent system (e.g., lysosomal enzymes;lactoferrin, which binds iron)
D. Chemical mediators (Table 2-1)1. Histamine: vasoactive amine; acts as a key mediator in
acute inflammation2. Serotonin: vasoactive mediator, with actions similar to
histamine3. Arachidonic acid mediators (Figure 2-2)
a. Arachidonic acid is released from membrane phos-pholipids by phospholipase A2 and is synthe-sized from linoleic acid (o)6 essential fatty acid).
b. Compounds synthesized from arachidonic acid:prostaglandins; thromboxane A2 (TXA 2); leukotrienes(LT B,, LTC 4, LTD4 , LTE4)
4. Nitric oxide (NO): free radical gas5. Cytokines: IL-1 and TNF6. Bradykinin7. Complement
E. Outcome of acute inflammation1. Complete resolution: only mild injury to labile and
stable cells2. Tissue destruction and scar formation: extensive injury
(e.g., abscess) or damage to permanent cells3. Progression to chronic inflammation
II. Chronic Inflammation• Inflammation of prolonged duration (weeks to years) most often
results from persistence of an injury-causing agent.A. Injurious agents include infectious diseases (e.g., hepatitis C,
tuberculosis) and alcohol.B. Morphology
1. Cell types: monocytes and/or macrophages, lympho-cytes and/or plasma cells, eosinophils, fibroblasts, endo-thelial cells
2. Necrosis: not as prominent a feature as in acuteinflammation
3. Destruction of parenchyma: loss of functional tissue,with repair by fibrosis
C. Granulomatous inflammation1. Causes
a. Infectious agents include tuberculosis and systemicfungal infection; usually associated with caseousnecrosis
Histamine is themain chemical medi-ator of acuteinflammation.
Bradykinin inducescough and angio-edema in patientstaking ACEinhibitors.
Monocytes and/ormacrophages arethe primary leuko-cytes in chronicinflammation.

Mediator
Histamine
Serotonin
Arachidonic acidmetabolites
Prostaglandins (PGs)
Thromboxane A2 (TXA2 ) PlateletsConverted from PGH, by thromboxane
synthase
Leukotrienes (LTs) Converted from arachidonic acid bylipoxygenase-mediated hydroxylation
Vasodilation, increased permeability
Vasodilation, increased permeability
PGE 2 : vasodilation, pain, feverPGI 2 : vasodilation; inhibition of platelet
aggregation
Vasoconstriction, platelet aggregation,bronchoconstriction
LTB 4 : chemotaxis and adhesion neutro-phil molecule activation
LTC 4 , LTD,, LTE 4 : vasoconstriction,increased vessel permeability,bronchoconstriction
Vasodilation, bactericidal
Initiate PGE 2 synthesis in anterior hypo-thalamus, leading to production offever
Activate endothelial cell adhesionmolecules
Increase liver synthesis of acute-phasereactants, such as ferritin, coagulationfactors (e.g., fibrinogen), andC-reactive protein
Increase release of neutrophils frombone marrow
Vasodilation, increased vessel perme-ability, bronchoconstriction, pain
C3a, C5a (anaphylatoxins): stimulatemast cell release of histamine
C3b: opsonizationC5a: activation of neutrophil adhesion
molecules, chemotaxisC5–C9 (membrane attack complex): cell
lysis
•••••••••••••••••••••••••••••••••••
18 Pathology
TABLE 2-1 Sources and Functions of Chemical Mediators
Source(s) Function(s)
Mast cells (primary cell), basophils,platelets
Mast cells, basophils, platelets
Leukocytes, endothelial cells, plateletsPGH 2 : major precursor of PGs and
thromboxanesPGG 2 : converted from arachidonic acid
by cyclooxygenase (COX)
Nitric oxide (NO)
Cytokines: interleukin-1,tumor necrosis factor
Bradykinin
Complement
Macrophages, endothelial cellsReleased during conversion of arginine
to citrulline by NO synthaseLymphocytes, macrophages, endothelial
cells
Product of kinin system activation byactivated factor XII
Synthesized in liver
b. Noninfectious causes include sarcoidosis andCrohn's disease; associated with noncaseatingnecrosis
2. Morphologya. Gross: pale, white nodule with or without central
caseationb. Microscopic (Figure 2 -3)
(1) Usually well-circumscribed
•

Chapter 2 Inflammation and Repair 19
Cell membrane phospholipids
,1,Phospholipase A2
Linoleic acid Arachidonic acid
5- Lipoxygenase Cyclooxygenase
LTB4 , LTC 4 , LTD 4 , LTE4 PGG2
Thromboxane synthase Prostacyclin synthaseTXA2 PGH2 PGI2
PGE2
Figure 2-2 Arachidonic acid metabolism Arachidonic acid is released from membranephospholipids It is converted into prostaglandins (PGs), thromboxane A2 (TXA2), andleukotrienes (LTs).
rt'
Figure 2-3 Caseous tuberculous granuloma in the lung showing the well-circumscribednature of the granuloma. Granular, acellular material in the center of the lesion representscaseous necrosis The arrow indicates a multinucleated giant cell The majority of cellsrepresent activated macrophages (epithelioid cells) and activated T H 1 cells.
(2) Cell types: epithelioid cells (activated macro-phages), mononuclear (round cell) infiltrate(CD4 helper T cells, or TH cells of the TH 1 type)
(3) Multinucleated giant cells: fusion of epitheli-oid cells; nuclei usually at the periphery
3. Pathogenesis of a tuberculous granuloma (Box 2-1)
[II. Patterns of InflammationA. Suppurative (purulent) inflammation
1. Localized proliferation of pus-forming organisms, such asStaphylococcus aureus (e.g., skin abscess)
2. S. aureus contains coagulase, which cleaves fibrinogeninto fibrin and traps bacteria and neutrophils.
B. Cellulitis1. Proliferation of bacteria (e.g., Streptococcus pyogenes)
through subcutaneous tissue (e.g., erysipelas)

20 Pathology
BOX 2-1 Sequence of Formation of a Tuberculous Granuloma
1. The tubercle bacillus Mycobacterium tuberculosis undergoes phagocytosisby alveolar macrophages (processing of bacterial antigen).
2. Macrophages present antigen to CD4 T cells in association with class IIantigen sites.
3. Macrophages release IL-12 (stimulates formation of TH 1 class cells) andIL-1 (causes fever; activates T H I cells).
4. T 1 1 cells release 1L-2 (stimulates T H 1 proliferation), 7-interferon(activates macrophages to kill tubercle bacillus; epithelioid cells), andmigration inhibitory factor (causes macrophages to accumulate).
5. Lipids from killed tubercle bacillus lead to caseous necrosis.6. Activated macrophages fuse and become multinucleated giant cells.
2. S. pyogenes contains hyaluronidase, which hydrolyzes theextracellular matrix (ECM), causing bacteria to spread.
C. Pseudomembranous inflammation• Bacterial toxin-induced damage of the mucosal lining, pro-
ducing a shaggy membrane composed of necrotic tissue(e.g., pseudomembranes associated with Clostridium difficilein pseudomembranous colitis)
D. Fibrinous inflammation• Increased vessel permeability, with deposition of fibrin-rich
exudate along serosal surfaces (e.g., fibrinous pericarditis)E. Ulceration
• Injurious agent (e.g., acid, microbial pathogen) producing adefect in the epithelial lining of skin or mucosa (e.g., pepticulcer disease caused by Helicobacter pylori–induced cyto-kine damage)
F. Fistula• Inflammatory reaction producing a passage between two
hollow organs (e.g., bowel-to-bowel fistulas in Crohn'sdisease)
IV. Tissue RepairA. Factors involved in tissue repair
1. Parenchymal cell regeneration2. Repair by connective tissue (fibrosis)
B. Parenchymal cell regeneration1. Depends on the ability of cells to replicate
a. Labile cells (e.g., stem cells in epidermis) and stablecells (e.g., hepatocytes, fibroblasts) are able to rep-licate (see Chapter 1).
b. Permanent cells are unable to replicate.(1) Cardiac and striated muscle are replaced by scar
tissue (fibrosis).(2) Neurons cannot be replaced.
2. Requires factors that stimulate parenchymal cell regenera-tion (e.g., growth factors, hormones, loss of tissue)
3. Restoration to normal (e.g., first-degree burn) requirespreservation of the basement membrane and a

Chapter 2 Inflammation and Repair 21
relatively intact connective tissue infrastructure(e.g., collagen, adhesive proteins).
C. Repair by connective tissue (fibrosis)1. Occurs when injury to parenchymal cells, basement
membranes, and connective tissue infrastructure is severeor persistent (e.g., third-degree burn)
2. Requires neutrophil transmigration to liquefy injuredtissue and then macrophage transmigration to removethe debris
3. Depends on the formation of granulation tissue inthe ECMa. Granulation tissue is highly vascular and composed
of newly formed blood vessels and fibroblasts.b. It requires fibronectin, whose functions include:
(1) Chemotaxis of fibroblasts, which synthesizecollagen, and endothelial cells, which formnew blood vessels (angiogenesis)
(2) Binding of collagen and other components toglycoproteins on the cell surface (integrins)
c. It accumulates in the ECM and eventually producesdense fibrotic tissue (scar).
4. Requires the initial production of type III collagena. Collagen is the major fibrous component of connec-
tive tissue.b. It is a triple helix of cross-linked a-chains; lysyl
oxidase cross-links points of hydroxylation (vitaminC—mediated) on adjacent a-chains.
c. Cross-linking increases the tensile strength ofcollagen.
d. Type I collagen in skin, bone, and tendons hasgreater tensile strength than type III collagen in theearly phases of tissue repair.
Ehlers-Danlos syndrome consists of a group ofmendelian disorders characterized by defects oftype I and type III collagen synthesis and struc-ture. Clinical findings include hypermobilejoints, aortic dissection (most common cause ofdeath), bleeding into the skin (ecchymoses), andpoor wound healing.
5. Dense scar tissue produced from granulation tissue mustbe remodeled.a. Remodeling increases the tensile strength of scar
tissue.b. Metalloproteinases (collagenases) replace type III
collagen with type I collagen, increasing tensilestrength to approximately 80% of the original.
An intact basementmembrane is es-sential for normalcell proliferation andrepair.
Granulation tissueis essential fornormal connectivetissue repair.

22 Pathology
BOX 2-2 Wound Healing by Primary and Secondary Intention
Primary intentionDay 1: fibrin clot (hematoma) develops. Neutrophils infiltrate thewound margins. There is increased mitotic activity of basal cells of squa-mous epithelium in the apposing wound margins.Day 2: squamous cells from apposing basal cell layers migrate under thefibrin clot and seal off the wound after 48 hours. Macrophages emi-grate into the wound.Day 3: granulation tissue begins to form. Initial deposition of type IIIcollagen begins but does not bridge the incision site. Macrophages replaceneutrophils.Days 4-6: granulation tissue formation peaks, and collagen bridges theincision site.Week 2: collagen compresses blood vessels in fibrous tissue, resulting inreduced blood flow. Tensile strength is about 10%.Month 1: collagenase remodeling of the wound occurs, with replace-ment of type III collagen by type I collagen. Tensile strength increases,reaching 80% within 3 months. Scar tissue is devoid of adnexal structures(e.g., hair, sweat glands) and inflammatory cells.
Secondary intention: typically, these wounds show:More intense inflammatory reaction than primary healingIncreased amount of granulation tissue formation than in primaryhealingWound contraction caused by increased numbers of myofibroblasts
6. Wound healing (Box 2 -2)a. Healing by primary intention: approximation of
wound edges by sutures; used for clean surgicalwounds
b. Healing by secondary intention: wound remainsopen; used for gaping or infected wounds
D. Factors that impair healing1. Persistent infection2. Metabolic disorders (e.g., diabetes mellitus): susceptibil-
ity to infection caused by impaired circulation and in-creased glucose
3. Nutritional deficiencies: decreased protein, vitamin Cdeficiency, trace metals (zinc; cofactor in type IIIcollagenase)
4. Glucocorticoids: interfere with collagen formation anddecrease tensile strength; occasionally used with antibiot-ics to prevent scar formation (e.g., bacterial meningitis)
Keloids, the raised scars caused by excessive synthesisof type III collagen, are common in African Ameri-cans and may occur as the result of third-degreeburns. Microscopically, keloids appear as irregular,thick collagen bundles that extend beyond the con-fines of the original injury.
Infections that in-terfere with healingare most com-monly caused byS auteus.

••• 10••I • '13•••••••
Chapter 2 Inflammation and Repair 23
Figure 2 -4 Absolute leuko-cytosis with left shift. Arrowspoint to band (stab) neutrophils,which exhibit prominence of theazurophilic granules (toxic granu-lation) Vacuoles in the cytoplasmrepresent phagolysosomes
•• V. Laboratory Findings Associated With Inflammation
•A. Leukocytes
1. Acute inflammation (e.g., bacterial infection)
• (Figure 2-4)a. Absolute neutrophilic leukocytosis: accelerated
• release of neutrophils from the bone marrow; medi-ated by IL-1 and TNF
b. Left shift: > 10% band (stab) neutrophils or the pres-a ence of earlier precursors (e.g., metamyelocytes)c. Toxic granulation: prominence of azurophilic gran-
• ules (primary lysosomes)
•2. Chronic inflammation (e.g., tuberculosis): absolute
monocytosis• B. Erythrocyte sedimentation rate (ESR)• • ESR is the rate of settling of RBCs in a vertical tube in
mm/h.• 1. ESR is elevated in acute and/or chronic inflammation(e.g., multiple myeloma)
• 2. Plasma factor or RBC factors that promote rouleaux for-• mation increase the ESR.
a. Plasma factor: increase in fibrinogen (acute-phase• reactant) decreases negative charge in RBCs, promot-
•ing rouleaux formation.
b. RBC factors: anemia promotes rouleaux formation.• C. C-reactive protein: acute-phase reactant; general scavenger
molecule• 1. Sensitive indicator of acute inflammation (e.g., inflamma-
•tory atherosclerotic plaques, bacterial infection)
2. Monitor of disease activity (e.g., rheumatoid arthritis)•••

3Immunopathology
IFFP-w
I. Cells of the Immune System (Table 3-1)
TABLE 3-1 Types of Immune Cells
Cell Type
Derivation Location
Function
T cellsCD4 (helper)CD8 (cytotoxic)
B cells
Natural killer cells
Macrophages
Dendritic cells
Bone marrow stem cellsin thymus
Bone marrow stem cells
Bone marrow stem cells
Conversion of mono-cytes into macro-phages in connectivetissue
Bone marrow stem cells
Peripheral blood and bonemarrow, thymus, pare-cortex of lymph nodes,Peyer's patches
Peripheral blood and bonemarrow, germinal folliclesin lymph nodes, Peyer'spatches
Peripheral blood (largegranular lymphocytes)
Connective tissue; organs(e.g., alveolar macro-phages, lymph nodesinuses)
Skin (Langerhans' cells),germinal follicles
CD4 cells: secrete cytokines(IL-2 proliferation ofCD8 T cells; 7-interferon—> activation of macro-phages); help B cellsbecome antibody-producing plasma cells
CD8 cells: kill virus-infected,neoplastic, and donorgraft cells
Differentiate into plasmacells that produce im-munoglobulins to killencapsulated bacteria(e.g., Streptococcuspneumoniae)
Act as APCs that interactwith CD4 cells
Kill virus-infected andneoplastic cells
Involved in phagocytosisand cytokine production
Act as APCs
Act as APCs
APC, antigen-presenting cell; IL, interleukin
24

•Chapter 3 Immunopathology 25
• II. Major Histocompatibility Complex (MHC)
•A. Located on the short arm of chromosome 6B. Contains human leukocyte antigen (HLA) genes, which code
• for HLA proteins that are unique to each individual
•C. Class I MHC molecules
1. Coded by HLA-A, -B, and -C genes
•2. Present on the membranes of all nucleated cells3. Recognized by CD8 T cells and natural killer cells
• D. Class II MHC molecules• 1. Coded by HLA-DP, -DQ and -DR genes
2. Present on antigen-presenting cells (APCs): B cells, mac-
• rophages, dendritic cells3. Recognized by CD4 T cells
• E. HLA association with disease
•1. HLA-B27: ankylosing spondylitis2. HLA-DR3 and -DR4: type 1 diabetes mellitus
• 3. HLA-DR2: multiple sclerosis
•F. Uses for HLA testing
1. Transplantation workup: close matches of HLA-A, -B,
•and -D loci in both the donor and graft recipient increasethe chance of graft survival.
• 2. Determining disease risk (e.g., HLA-B27—positive indi-• viduals have an increased risk of ankylosing spondylitis)
• III. Hypersensitivity Reactions (Table 3-2)A. Type I (immediate) hypersensitivity: IgE antibody-medi-
• ated activation of mast cells (effector cells) produces an in-• flammatory reaction.
1. IgE antibody production (sensitization)a. Allergens (e.g., pollen, drugs) are first processed by
APCs (macrophages or dendritic cells).b. APCs interact with CD4 T H2 cells, causing interleu-
• kins (ILs) to stimulate B-cell maturation. IL-4causes plasma cells to switch from IgM to IgE
• synthesis.c. IgE antibodies are bound to mast cells.
2. Mast cell activation (reexposure)
• a. Allergens cross-link IgE antibodies specific for the al-lergen on mast cell membranes.
• b. IgE triggering causes mast cell release of preformed
•mediators (e.g., histamine, chemotactic factors), pro-ducing tissue swelling and bronchoconstriction.
• c. Late-phase reaction: mast cells synthesize and
•release prostaglandins and leukotrienes, enhancingtissue swelling.
•••••9
Desensitization therapy involves repeated in-jections of increasingly greater amounts of aller-gen, resulting in production of IgG antibodiesthat attach to allergens and prevent them frombinding to mast cells.

26 Pathology••••
Anaphylactic shockis a potentiallyfatal type I hyper-sensitivity reaction.
TABLE 3-2 Hypersensitivity Reactions
Reaction Pathogenesis Examples
Type I IgE-dependent activation Atopic disorders: hay fever, eczema, hives,of mast cells asthma, reaction to bee sting
Drug hypersensitivity: penicillin rash oranaphylaxis
Type II Antibody-dependent Complement-dependent reactionsreaction Lysis: ABO mismatch, Goodpasture's
syndrome, hyperacute transplantationrejection
Phagocytosis: warm (IgG) autoimmunehemolytic anemia, ABO and Rh hemolyticdisease of newborn
Antibody (IgG, IgE)-dependent cell-mediatedcytotoxicity: natural killer cell destruction ofneoplastic and virus-infected cells
Antibodies directed against cell surface recep-tors: myasthenia gravis, Graves' disease
Type Ill Deposition of antigen- Systemic lupus erythematosusantibody complexes (DNA-anti-DNA)
Serum sickness (horse antithymocyteglobulin-antibody)
Arthus reaction (farmer's lung)
Type IV Antibody - i ndependent Delayed type: contact dermatitis (e.g., poisonT cell-mediated ivy), tuberculous granulomareactions Cell-mediated cytotoxicity: killing of tumor
cells and virus-infected cells
3. Tests used to evaluate type I hypersensitivitya. Scratch test (best overall sensitivity): results in
histamine-mediated wheal-and-flare reaction in pa-tients with atopy
b. Radioimmunosorbent test: detects specific IgE anti-bodies against specific allergens
4. Clinical findings (see Table 3-2)B. Type II (cytotoxic) hypersensitivity: antibody-dependent
cytotoxic reactions1. Complement-dependent reactions
a. Lysis: antibody (IgG or IgM) directed against antigenon the cell membrane activates the complementsystem, leading to lysis by the membrane attackcomplex.
b. Phagocytosis: fixed macrophages (e.g., in spleen)phagocytose and destroy hematopoietic cells (e.g.,red blood cells; RBCs), coated by IgG antibodies andcomplement (C3b).
2. Antibody (IgG, IgE)-dependent cell-mediated cytotox-icity: leukocytes with receptors for IgG or IgE lyse butdo not phagocytose cells coated by antibodies withoutcomplement.
3. IgG autoantibodies directed against cell surface recep-tors (e.g., acetylcholine receptors)
••••••••••••••••••••••••••••••••

Chapter 3 Immunopathology 27
4. Tests used to evaluate type II hypersensitivitya. Direct Coombs' test: detects IgG and/or C3b
on RBCsb. Indirect Coombs' test: detects antibodies in serum
5. Clinical findings (see Table 3-2)C. Type III (immunocomplex) hypersensitivity: activation of
the complement system by circulating antigen-antibodycomplexes1. First exposure to antigen: synthesis of antibodies2. Second exposure to antigen
a. Deposition of antigen-antibody complexesb. Complement activation, producing C5a, which at-
tracts neutrophils that damage tissue3. Arthus reaction: localized immunocomplex reaction
(e.g., farmer's lung from exposure to thermophilic actino-mycetes, or antigens, in air)
4. Test used to evaluate type III hypersensitivity: immu-nofluorescent staining of tissue biopsies (e.g., glomer-uli in glomerulonephritis)
5. Clinical findings (see Table 3-2)D. Type IV hypersensitivity: antibody-independent
T cell-mediated reactions1. Delayed reaction: CD4 cells interact with macrophages
(APCs with MHC class II antigens), resulting in cyto-kine injury to tissue.
2. Cell-mediated cytotoxicity: CD8 T cells interact withaltered MHC class I antigens on neoplastic, virus-infected,or donor graft cells, causing cell lysis.
3. Test used to evaluate type IV hypersensitivity: patchtest to confirm contact dermatitis
4. Clinical findings (see Table 3-2)
IV. Transplantation ImmunologyA. Factors that enhance graft viability
1. ABO blood group compatibility between recipients anddonors
2. Absence of preformed anti-HLA cytotoxic antibodiesin recipients
3. Close matches of HLA-A, -B, and -D loci between recipi-ents and donors
B. Types of grafts1. Autograft: associated with best survival rate2. Syngeneic graft (isograft): between identical twins3. Allograft: between genetically different individuals of the
same species4. Xenograft: between two species (e.g., transplant of heart
valve from pig to human)C. Types of rejection: transplantation rejection involves a
humoral and/or cell-mediated host response against MHCantigens in the donor graft.1. Hyperacute rejection: irreversible; occurs within
minutes
Types I, II, and IIIhypersensitivity re-actions are all an-tibody mediated.
ABO blood groupcompatibility is themost importantrequirement forsuccessfultransplantation

28 Pathology
a. Blood vessel injury with thrombosisb. ABO incompatibility or action of preformed anti-
HLA antibodies in the recipient directed againstdonor antigens
c. Type II hypersensitivity reaction2. Acute rejection: reversible; occurs within days to weeks;
most common type of transplantation rejectiona. Cell-mediated reaction: type IV cell-mediated
hypersensitivity(1) CD4 T cells release cytokines, resulting in acti-
vation of host macrophages, proliferation ofCD8 T cells, and destruction of donor graftcells.
(2) Pathologic findings: extensive interstitial roundcell lymphocytic infiltrate in the graft, edema,endothelial cell injury
b. Antibody-mediated reaction: type II hyper-sensitivity(1) Cytokines from CD4 T cells promote B-cell dif-
ferentiation into plasma cells, producing anti-HLA antibodies that attack vessels in the donorgraft.
(2) Pathologic findings: vasculitis with intravascu-lar thrombosis, intimal thickening with obliter-ation of vessel lumens in older grafts
Acute rejection is potentially reversiblewith immunosuppressive agents, such ascyclosporine (blocks CD4 T-cell release ofIL-2) and corticosteroids (lymphotoxic).Immunosuppressive therapy is associatedwith an increased risk of cervical cancer,malignant lymphoma, and squamous cellcarcinoma of the skin.
3. Chronic rejection: irreversible; occurs over months toyearsa. Not well characterized; involves continued vascular
injury with ischemia to tissueb. Blood vessel damage with intimal thickening and
fibrosisD. Graft-versus-host (GVH) reaction
1. Potential complication in bone marrow and liver trans-plants and in blood transfusions given to patients withconditions associated with T-cell immunodeficiency
2. Donor T cells recognize host tissue as foreign and activatehost CD4 and CD8 T cells.
3. Clinical findings include bile duct necrosis (jaundice),gastrointestinal mucosa ulceration (bloody diarrhea),and dermatitis.
E. Types of transplants (Table 3-3)
I•I Chapter 3 Immunopathology 29
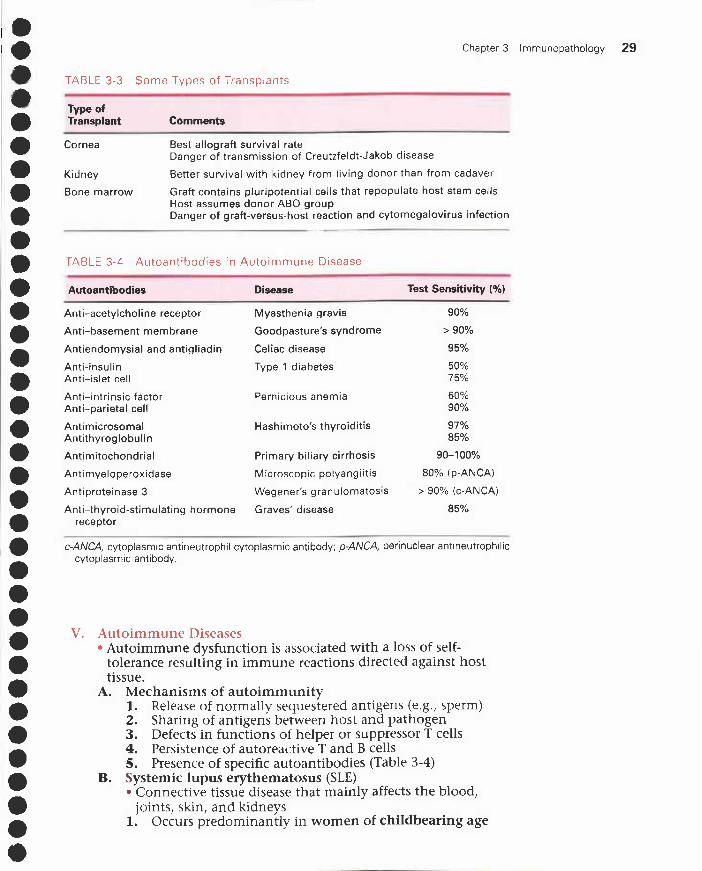
TABLE 3-3 Some Types of Transplants
•• Type of
Transplant Comments
•Cornea Best allograft survival rate
Danger of transmission of Creutzfeldt-Jakob disease
• Kidney Better survival with kidney from living donor than from cadaver
•Bone marrow Graft contains pluripotential cells that repopulate host stem cells
Host assumes donor ABO group
•Danger of graft-versus-host reaction and cytomegalovirus infection
•
•TABLE 3-4 Autoantibodies in Autoimmune Disease
• Autoantibodies Disease Test Sensitivity (%)
• Anti-acetylcholine receptor Myasthenia gravis 90%
•Anti-basement membrane Goodpasture's syndrome > 90%
•Antiendomysial and antigliadin
Anti-insulin
Celiac disease
Type 1 diabetes
95%
50%
•Anti-islet cell 75%
•Anti-intrinsic factorAnti-parietal cell
Pernicious anemia 60%90%
•AntimicrosomalAntithyroglobulin
Hashimoto's thyroiditis 97%85%
• Antimitochondrial Primary biliary cirrhosis 90-100%
•Antimyeloperoxidase Microscopic polyangiitis 80% (p-ANCA)
• Antiproteinase 3 Wegener's granulomatosis > 90% (c-ANCA)
Anti-thyroid-stimulating hormone Graves' disease 85%• receptor
• c-ANCA, cytoplasmic antineutrophil cytoplasmic antibody; p-ANCA, perinuclear antineutrophiliccytoplasmic antibody.
•
•••
V. Autoimmune Diseases• Autoimmune dysfunction is associated with a loss of self-
• tolerance resulting in immune reactions directed against hosttissue.
• A. Mechanisms of autoimmunity
•1. Release of normally sequestered antigens (e.g., sperm)2. Sharing of antigens between host and pathogen3. Defects in functions of helper or suppressor T cells
•4. Persistence of autoreactive T and B cells5. Presence of specific autoantibodies (Table 3-4)
•B. Systemic lupus erythematosus (SLE)
• Connective tissue disease that mainly affects the blood,• joints, skin, and kidneys
•1. Occurs predominantly in women of childbearing age
•

30 Pathology
Figure 3-1 Malar rash in sys-temic lupus erythematosusshowing the butterfly-wingdistribution.
2. Pathogenesis: polyclonal B-cell activation, sustained es-trogen activity, environmental triggers (e.g., sun,procainamide)
3. Clinical findingsa. Hematologic: autoimmune hemolytic anemia,
thrombocytopenia, leukopeniab. Lymphatic: generalized lymphadenopathy,
splenomegalyc. Musculoskeletal: small-joint inflammation (e.g.,
hands), absence of joint deformityd. Skin: immunocomplex deposition along basement
membrane (liquefactive degeneration), malar butter-fly rash (Figure 3-1)
e. Renal: diffuse proliferative glomerulonephritis (mostcommon glomerulonephritis)
f. Cardiovascular: pericarditis, Libman-Sacks endocar-ditis (sterile vegetations on mitral valve)
g. Respiratory: interstitial fibrosis of lungs, pleural effu-sion with friction rub
h. Pregnancy-related(1) Complete heart block in newborns, which is
caused by anti-SS-A (Ro) antibodies crossingthe placenta
(2) Recurrent spontaneous abortions4. Drug-induced lupus erythematosus
a. Associated drugs: procainamide, hydralazineb. Features that distinguish drug-induced lupus
from SLE(1) Antihistone antibodies(2) Low incidence of renal and central nervous
system (CNS) involvement
Most commoncardiac finding inSLE: fibrinous peri-carditis witheffusion
Most common drugassociated withdrug-induced lupus:procainamide

Chapter 3 Immunopathology 31
(3) Disappearance of symptoms when the drug isdiscontinued
5. Laboratory findings in SLEa. Positive serum antinuclear antibody (ANA) (almost
all cases)(1) Anti-double-stranded DNA antibodies and
anti-Sm antibodies: used to confirm the diag-nosis of SLE because they are highly specificfor the disease (i.e., few false-positive results)
(2) Anti-Ro antibodies: positive in 25-50% of casesb. Antiphospholipid antibodies (lupus anticoagulant
and/or anticardiolipin antibodies): damage vesselendothelium, producing vessel thrombosis andcausing an increased incidence of strokes
Anticardiolipin antibodies may produce a false-positive syphilis serology by cross-reacting withcardiolipin in the rapid plasma reagin (RPR) andVenereal Disease Research Laboratory (VDRL)tests.
c. Lupus erythematosus cell: neutrophil containingphagocytosed altered DNA; not specific for SLE
d. Decreased serum complemente. Immunocomplexes at the dermal-epidermal junc-
tion in skin biopsiesC. Systemic sclerosis (scleroderma)
• Excessive production of collagen that primarily targets theskin (scleroderma), gastrointestinal tract, lungs, and kidneys
1. Occurs predominantly in women of childbearing age2. Pathogenesis
a. Small-vessel endothelial cell damage produces bloodvessel fibrosis and ischemic injury.
b. T-cell release of cytokines results in excessive colla-gen synthesis.
3. Clinical findingsa. Skin
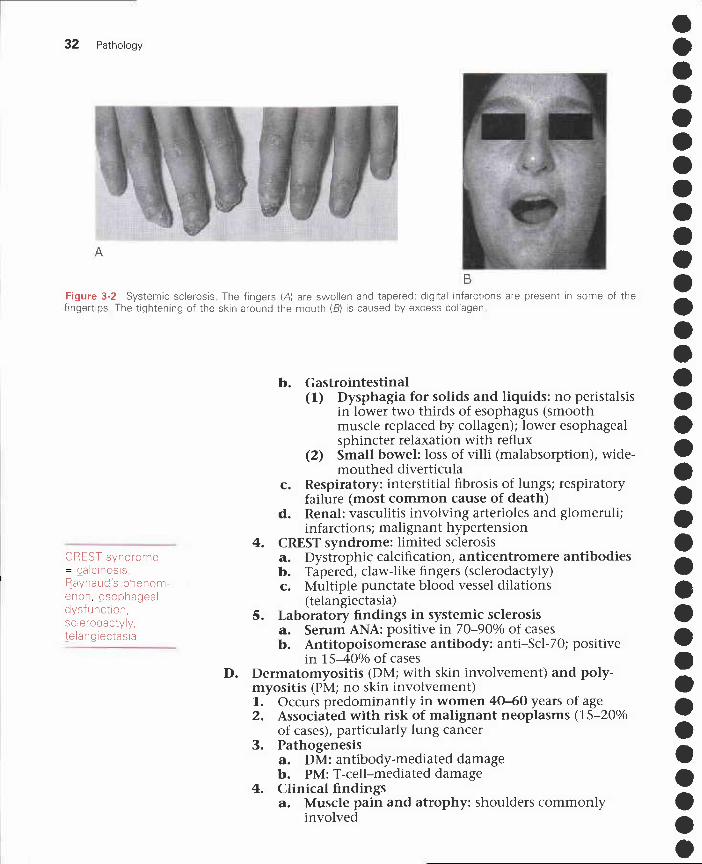
(1) Skin atrophy and tissue swelling beginning inthe fingers and extending proximally;parchment-like appearance; extensive dystro-phic calcification
(2) Raynaud's phenomenon: sequential colorchanges (normal to blue to red) caused bydigital vessel vasculitis and fibrosis; digitalinfarcts (Figure 3-2, A)
(3) Tightened facial features (e.g., radial furrowingaround the lips) (Figure 3-2, B)
Most common initialsign of systemicsclerosis: Raynaud'sphenomenon
32 Pathology
A
Figure 3-2 Systemic sclerosis. The fingers (A) are swollen and tapered; digital infarctions are present in some of thefingertips. The tightening of the skin around the mouth (B) is caused by excess collagen
b. Gastrointestinal(1) Dysphagia for solids and liquids: no peristalsis
in lower two thirds of esophagus (smoothmuscle replaced by collagen); lower esophagealsphincter relaxation with reflux
(2) Small bowel: loss of villi (malabsorption), wide-mouthed diverticula
c. Respiratory: interstitial fibrosis of lungs; respiratoryfailure (most common cause of death)
d. Renal: vasculitis involving arterioles and glomeruli;infarctions; malignant hypertension
4. CREST syndrome: limited sclerosisa. Dystrophic calcification, anticentromere antibodiesb. Tapered, claw-like fingers (sclerodactyly)c. Multiple punctate blood vessel dilations
(telangiectasia)5. Laboratory findings in systemic sclerosis
a. Serum ANA: positive in 70-90% of casesb. Antitopoisomerase antibody: anti-Scl-70; positive
in 15-40% of casesD. Dermatomyositis (DM; with skin involvement) and poly-
myositis (PM; no skin involvement)1. Occurs predominantly in women 40-60 years of age2. Associated with risk of malignant neoplasms (15-20%
of cases), particularly lung cancer3. Pathogenesis
a. DM: antibody-mediated damageb. PM: T-cell-mediated damage
4. Clinical findingsa. Muscle pain and atrophy: shoulders commonly
involved
CREST syndrome= calcinosis,Raynaud's phenom-enon, esophagealdysfunction,sclerodactyly,telangiectasia
•
•
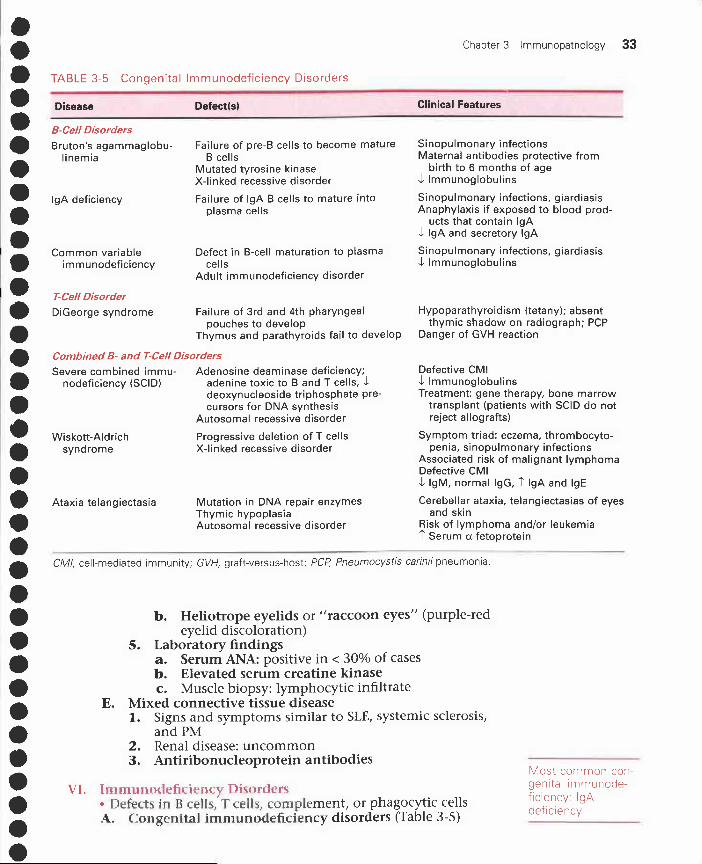
• TABLE 3-5 Congenital Immunodeficiency Disorders
• Disease Defect(s)
• B-Cell Disorders
Chapter 3 Immunopathology 33
Clinical Features
• Bruton's agammaglobu-linemia
•
• IgA deficiency
•
•
•
Common variableimmunodeficiency
• T-Cell Disorder
• DiGeorge syndrome
•
•• Severe combined immu-
nodeficiency (SCID)
•
• Wiskott-Aldrich
•syndrome
•
• Ataxia telangiectasia
••
Failure of pre-B cells to become matureB cells
Mutated tyrosine kinaseX-linked recessive disorder
Failure of IgA B cells to matureplasma cells
Defect in B-cell maturation to plasmacells
Adult immunodeficiency disorder
Failure of 3rd and 4th pharyngealpouches to develop
Thymus and parathyroids fail to develop
Adenosine deaminase deficiency;adenine toxic to B and T cells,deoxynucleoside triphosphate pre-cursors for DNA synthesis
Autosomal recessive disorder
Progressive deletion of T cellsX-linked recessive disorder
Mutation in DNA repair enzymesThymic hypoplasiaAutosomal recessive disorder
Sinopulmonary infectionsMaternal antibodies protective from
birth to 6 months of ageImmunoglobulins
Hypoparathyroidism (tetany); absentthymic shadow on radiograph; PCP
Danger of GVH reaction
Defective CMIImmunoglobulins
Treatment: gene therapy, bone marrowtransplant (patients with SCID do notreject allografts)
Symptom triad: eczema, thrombocyto-penia, sinopulmonary infections
Associated risk of malignant lymphomaDefective CMI
IgM, normal IgG, T IgA and IgE
Cerebellar ataxia, telangiectasias of eyesand skin
Risk of lymphoma and/or leukemiaT Serum a-fetoprotein
Combined B- and T-Cell Disorders
into Sinopulmonary infections, giardiasisAnaphylaxis if exposed to blood prod-
ucts that contain IgAL IgA and secretory IgA
Sinopulmonary infections, giardiasisImmunoglobulins
CMI, cell-mediated immunity; GVH, graft-versus-host; PCP Pneumocystis carinii pneumonia.
•••••••S••••
b. Heliotrope eyelids or "raccoon eyes" (purple-redeyelid discoloration)
5. Laboratory findingsa. Serum ANA: positive in < 30% of casesb. Elevated serum creatine kinasec. Muscle biopsy: lymphocytic infiltrate
E. Mixed connective tissue disease1. Signs and symptoms similar to SLE, systemic sclerosis,
and PM2. Renal disease: uncommon3. Antiribonucleoprotein antibodies
VI. Immunodeficiency Disorders• Defects in B cells, T cells, complement, or phagocytic cellsA. Congenital immunodeficiency disorders (Table 3-5)
Most common con-genital immunode-ficiency: IgAdeficiency

34 Pathology
1. B-cell disorders: recurrent encapsulated bacterial infec-tions (e.g., Streptococcus pneumoniae)
2. T-cell disorders: recurrent infections caused by intracel-lular pathogens (fungi, viruses, protozoa)
3. Combined B- and T-cell disordersB. Acquired immunodeficiency syndrome (AIDS)
1. Modes of transmissiona. Sexual transmission: > 75% of cases
(1) Homosexual transmission (anal intercoursebetween men): most common in the UnitedStates
(2) Heterosexual transmission: most common indeveloping countries
b. Intravenous drug abusec. Other modes of transmission
(1) Vertical transmission: transplacental route,blood contamination, breastfeeding
(2) Accidental needlestick: risk per accident is 0.3%.(3) Blood products: risk per unit of blood
< 0.0002%d. Body fluids containing HIV: blood, semen, breast
milk; the virus cannot enter intact skin or mucosa.2. Etiology: RNA retrovirus
a. HIV-1: most common cause in the United Statesb. HIV-2: most common cause in developing countries
3. Pathogenesisa. HIV envelope protein (gp120) attaches to the CD4
molecule of T cells.b. HIV infects CD4 T cells, causing direct cytotoxicity
and loss of cell-mediated immunity.c. Reverse transcriptase converts viral RNA into provi-
ral double-stranded DNA, which is integrated intothe host DNA.
4. HIV and AIDS testing (Table 3-6)5. Clinical findings
a. Acute phase: mononucleosis-like syndrome 3-6weeks after infection
b. Latent (chronic) phase: asymptomatic period 2-10years after infection(1) CD4 T-cell count > 500 cells/4(2) Viral replication in follicular dendritic cells
(reservoir cells)c. Early symptomatic phase
(1) CD4 T-cell count 200-500 cells/4(2) Generalized lymphadenopathy; non-AIDS-
defining infections, including hairy leukopla-kia, or Epstein-Barr virus (EBV)-caused glossitis;fever, weight loss, diarrhea
d. AIDS (Table 3-7)(1) Criteria: HIV-positive with CD4 T-cell count
200 cells/4 or with an AIDS-definingcondition
Most common ac-quired immunodefi-ciency diseaseworldwide: AIDS
Most commonCNS fungal infectionin AIDS:cryptococcosis

Chapter 3 Immunopathology 35
TABLE 3-6 Laboratory Tests Used in HIV and AIDS
Test Use Comments
ELISA Screening test Detects anti-gp120 antibodiesSensitivity - 100%Positive within 6-10 weeks
Western blot Confirmatory test Used if ELISA is positive or indeterminate-- 100% specificity
p24 Antigen Indicator of active viral Positive prior to seroconversion and whenreplication AIDS is diagnosed (two distinct peaks)
CD4 T-cell count Monitoring immune Useful in determining when to initiatestatus HIV treatment and when to administer
prophylaxis against opportunisticinfections
HIV viral load Detection of actively Most sensitive test for diagnosis of acutedividing virus HIV before seroconversion
ELISA, enzyme-linked immunoabsorbent assay
TABLE 3-7 Organ Systems Affected by AIDS
Organ System
Condition
Comments
Central nervoussystem (CNS)
AIDS-dementia complexPrimary CNS lymphomaCryptococcosisToxoplasmosisCMV retinitis
EsophagitisColitis
Biliary tract infectionFocal segmental
glomerulosclerosis
Pneumonia
Kaposi's sarcomaBacillary angiomatosis
Caused by HIVCaused by EBVCause of CNS fungal infectionCause of space-occupying lesionsCause of blindness
Caused by Candida, herpes, CMVCaused by Cryptosporidium
Caused by CMVCauses hypertension and nephrotic
syndrome
Caused by Pneumocystis carinii andStreptococcus pneumoniae
Caused by HSV-8Caused by Bartonella henselae
Gastrointestinal
Hepatobiliary
Renal
Respiratory
Skin
CMV, cytomegalovirus EBV, Epstein-Barr virus; HSV-8, herpes simplex virus type 8
(2) Most common AIDS-defining infections:Pneumocystis carinii pneumonia, systemic can-didiasis
(3) AIDS-defining malignancies: Kaposi's sarcoma(Figure 3-3), Burkitt's lymphoma (EBV), primaryCNS lymphoma (EBV)
(4) Causes of death: disseminated infections (cyto-megalovirus, Mycobacterium a yilun complex)
6. Immunologic abnormalities: Iymphopenia (low CD4T-cell count), cutaneous anergy (defect in cell-mediated
Most common ma-lignancy in AIDS:Kaposi's sarcoma
36 Pathology
Figure 3-3 Kaposi's sarcoma in HIV. Skin lesions are raised, red, and nonpruritic.
TABLE 3-8 Complement Disorders
Disorder Comments
Hereditaryangioedema
C6–C9 deficiency
Deficiency of Cl esterase inhibitor; continued Cl activationdecreases C2 and C4 and increases their cleavage products,which have anaphylatoxic activity
Normal C3Swelling of face and oropharynx
Increased susceptibility to disseminated Neisseria gonorrhoeaeor N. meningitidis infections
immunity), hypergammaglobulinemia (due to poly-clonal B-cell stimulation by EBV), CD4:CD8 ratio < 1
7. Pregnant women with AIDS: treatment with a reversetranscriptase inhibitor reduces transmission to newbornsto less than 8%.
C. Complement system disorders (Table 3-8)1. Complement pathways: classic and alternative
a. Cl esterase inhibitor inactivates the protease activ-ity of Cl in the classic pathway.
b. Membrane attack complex (C5–C9) is the finalcommon pathway for both the classic and alternativepathways.
2. Testing of the complement systema. A decrease in C4 or C2 indicates activation of the
classic pathway.b. A decrease in factor B indicates activation of the al-
ternative pathway.c. A decrease in C3 indicates activation of either
system.
VII. AmyloidosisA. Amyloid: fibrillar protein that forms deposits in interstitial
tissue, resulting in organ dysfunction1. Characteristics
a. Linear, nonbranching filaments in a (3-pleated sheet

Chapter 3 Immunopathology 37
TABLE 3-9 Common Types of Amyloidosis and Associated Clinical Findings
Type ofAmyloidosis Clinical Findings
Primary andsecondary
Senile cerebral
Nephrotic syndrome, renal failure (common cause of death)Arrhythmia, heart failureMacroglossia, malabsorptionHepatosplenomegalyCarpal tunnel syndrome
Dementia (Alzheimer's type) caused by toxic AP deposits in neuronsAmyloid precursor protein coded by chromosome 21Associated with Down syndrome
b. Apple-green—colored birefringence in polarizedlight with Congo red stain of tissue
c. Eosinophilic staining with H&E stain2. Types: derived from different proteins
a. Amyloid light chains (AL)b. Amyloid-associated (AA)c. (3-Amyloid (A(): derived from amyloid precursor
proteinB. Types of amyloidosis (Table 3-9)
1. Systemic: similar tissue involvement in both typesa. Primary: AL amyloid; associated with multiple
myeloma (30% of cases)b. Secondary (reactive): AA amyloid; associated with
chronic inflammation (e.g., rheumatoid arthritis,tuberculosis)
2. Localized: confined to a single organ (e.g., brain)3. Hereditary: autosomal recessive disorder involving AA
amyloid (e.g., familial Mediterranean fever)C. Techniques used to diagnose amyloidosis
1. Immunoelectrophoresis (to detect light chains) inprimary amyloidosis
2. Tissue biopsy (e.g., adipose, rectum)
13-Amyloid is asso-ciated with Alz-heimer's disease inDown syndrome.

••■• .....
•• • Fluid and....
HemodynamicDisorders
I. EdemaA. Total body water is 60% of the body weight.
1. Total body water is distributed between two compart-ments: intracellular fluid (ICF; 40% of body weight) andextracellular fluid (ECF; 20% of body weight).
2. ECF compartment is subdivided into two minor compart-ments: interstitial fluid and blood plasma.
B. Edema is the presence of increased fluid in the interstitialspace or body cavities.1. Body cavity effusions are named according to their loca-
tion (e.g., hydrothorax).2. Fluid in the peritoneal cavity is called ascites.
C. Pathophysiology of edema1. Increased vascular hydrostatic pressure (e.g., pulmo-
nary edema in left-sided heart failure, peripheral pittingedema in right-sided heart failure)
2. Decreased vascular plasma oncotic pressure (hypoal-buminemia) (e.g., peripheral edema and ascites in thenephrotic syndrome, or protein loss in the urine)
3. Increased vascular permeability (e.g., tissue swellingfollowing a bee sting)
4. Renal retention of sodium and water: increases hydro-static pressure (increased plasma volume) and decreasesoncotic pressure (dilutional effect on albumin)a. Primary retention of sodium and water (e.g., in-
creased sodium intake in a patient with renal failure)b. Secondary retention of sodium and water
(1) Decreased cardiac output (e.g., left-sided heartfailure) decreases renal blood flow and activatesthe renin-angiotensin-aldosterone system.
38
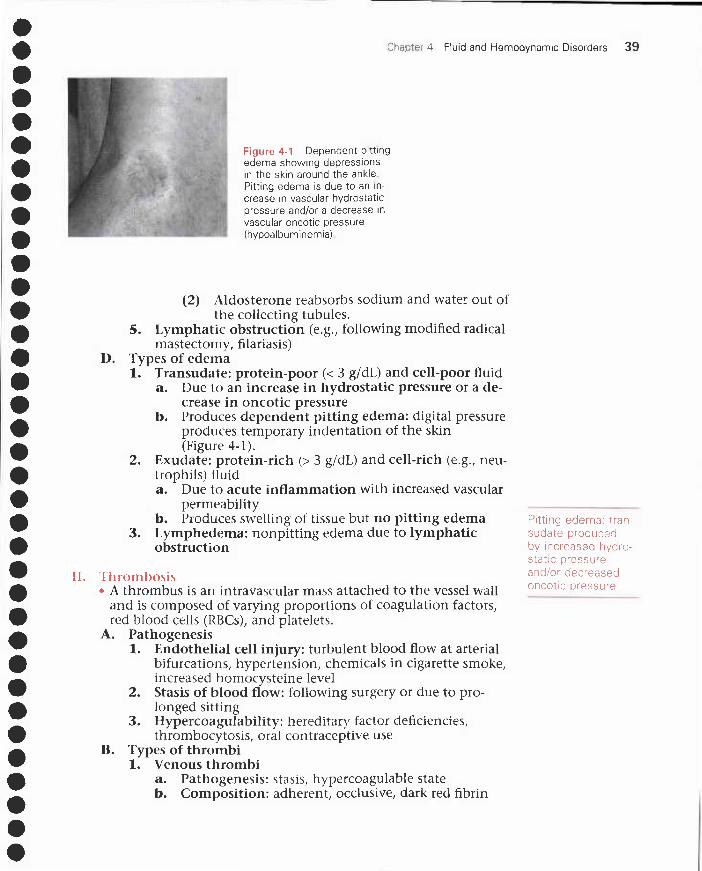
Chapter 4 Fluid and Hemodynamic Disorders 39
Figure 4-1 Dependent pittingedema showing depressionsin the skin around the anklePitting edema is due to an in-crease in vascular hydrostaticpressure and/or a decrease invascular oncotic pressure(hypoalbuminemia)
(2) Aldosterone reabsorbs sodium and water out ofthe collecting tubules.
5. Lymphatic obstruction (e.g., following modified radicalmastectomy, filariasis)
D. Types of edema1. Transudate: protein-poor (< 3 g/dL) and cell-poor fluid
a. Due to an increase in hydrostatic pressure or a de-crease in oncotic pressure
b. Produces dependent pitting edema: digital pressureproduces temporary indentation of the skin(Figure 4-1).
2. Exudate: protein-rich (> 3 g/dL) and cell-rich (e.g., neu-trophils) fluida. Due to acute inflammation with increased vascular
permeabilityb. Produces swelling of tissue but no pitting edema
3. Lymphedema: nonpitting edema due to lymphaticobstruction
II. Thrombosis• A thrombus is an intravascular mass attached to the vessel wall
and is composed of varying proportions of coagulation factors,red blood cells (RBCs), and platelets.
A. Pathogenesis1. Endothelial cell injury: turbulent blood flow at arterial
bifurcations, hypertension, chemicals in cigarette smoke,increased homocysteine level
2. Stasis of blood flow: following surgery or due to pro-longed sitting
3. Hypercoagulability: hereditary factor deficiencies,thrombocytosis, oral contraceptive use
B. Types of thrombi1. Venous thrombi
a. Pathogenesis: stasis, hypercoagulable stateb. Composition: adherent, occlusive, dark red fibrin
Pitting edema: tran-sudate producedby increased hydro-static pressureand/or decreasedoncotic pressure

40 Pathology
clot with entrapped RBCs, white blood cells, andplatelets
c. Sites(1) The most common site is the deep vein in the
lower extremity.(2) Other sites include superficial saphenous,
hepatic, and renal veins.d. Venous clots in the deep veins of the lower
extremity(1) Extend (propagate) toward the heart(2) Danger of pulmonary artery embolization
e. Heparin and warfarin inhibit formation of venousclots.
2. Arterial thrombia. Pathogenesis: turbulent blood flow with endothelial
cell injuryb. Composition: adherent mass of platelets held to-
gether by fibrin strandsc. Sites: overlie atheromatous plaques in elastic and
muscular arteries (e.g., coronary, internal carotid,and superior mesenteric arteries)
d. Clinical findings: infarction (e.g., acute myocardialinfarction, stroke), embolization
e. Aspirin prevents formation of arterial thrombi:inhibits platelet cyclooxygenase and subsequentproduction of thromboxane A 2 (platelet aggregator)
3. Postmortem clota. Fibrin clot of plasma (resembles chicken fat) without
entrapped cellsb. It is not attached to the vessel wall.
[II. Embolism• An embolus is a detached mass (e.g., clot, fat, gas) carried
through the blood to a distant site.A. Pulmonary thromboembolism
1. Most emboli originate from the femoral veins; othersoriginate from the pelvic veins.
2. Only a few result in a pulmonary infarction.B. Arterial embolism: about 80% of arterial emboli originate
from the left side of the heart (e.g., mural thrombus, valvevegetation, atrial myxoma).1. Atrial fibrillation predisposes to atrial clot formation
and embolization.2. Sites of embolism
a. Lower extremities (most common)b. Brain (via the middle cerebral artery)c. Small bowel (via the superior mesenteric artery)d. Spleen and kidneys
3. Clinical findingsa. Pale infarcts in the digits, spleen, and kidneysb. Hemorrhagic infarcts in the brain and small bowel
(see Chapter 1)
Prevent venousthrombus withheparin or warfarin
Prevent arterialthrombus withaspirin

Chapter 4 Fluid and Hemodynamic Disorders 41
4. Paradoxical emboli: arise in the venous system and passthrough an atrial septal defect into the systemiccirculation
C. Fat embolism: occurs following traumatic fracture of thelong bones (e.g., femur) and pelvis1. Pathogenesis
a. Microglobules of fat from the bone marrow andadipose lodge in the microvasculature throughoutthe body.
b. Fatty acids damage vessel endothelium, resulting information of platelet thrombi.
2. Clinical findings: usually delayed until 24-72 hours aftertraumaa. Central nervous system damage: ischemia,
hemorrhageb. Respiratory failure: fat microglobules and platelet
thrombi in the pulmonary capillaries interferewith gas exchange, causing hypoxemia.
c. Thrombocytopenia: due to platelet consumption inthrombi
D. Amniotic fluid embolism: maternal complication duringdelivery1. Pathogenesis
a. Tear in the placental membranesb. Infusion of amniotic fluid with fetal squamous cells
and lanugo into the maternal circulation and pulmo-nary vessels
2. Clinical findingsa. Sudden onset of dyspnea, cyanosis, and hypotensive
shockb. Disseminated intravascular coagulation (DIC)
commonly occurs.c. Maternal death in 80% of cases
E. Decompression sickness (caisson disease)1. Pathogenesis
a. Complication of deep-sea divingb. Atmospheric pressure increases by 1 for every 33 feet
of descent into water.c. Under increased atmospheric pressure, nitrogen gas
moves from the alveoli, through the blood, anddissolves in tissues.
d. Rapid ascent: forces nitrogen gas bubbles to developin tissue and the lumen of blood vessels
2. Clinical findingsa. The "bends" (pain in joints, muscles, and bones)b. Hemiparesis and bladder and bowel dysfunctionc. Pneumothorax
(1) Complication of a sudden rise to the surface(2) Rupture of a subpleural bleb causes dyspnea
and pleuritic chest pain.d. Pulmonary embolus
(1) Can develop while diving in - 60 feet of water
Pneumothorax andpulmonary embo-lism are complica-tions of deep-sea diving

42 Pathology
TABLE 4-1 Types of Shock
Type CO PVR LVEDV
Hypovolemic
Cardiogenic
Endotoxic (septic)
I
I
T
T
T
I
I
T
1
CO, cardiac output; LVEDV, left-ventricular end-diastolic volume; PVR, peripheral vascularresistance.
(2) Increased pressure on the veins in the lowerextremities produces stasis and thrombusformation.
(3) Pulmonary thromboembolism develops withdyspnea and pleuritic chest pain.
e. Chronic changes: aseptic necrosis in bones (femur,tibia, humerus) from bone infarctions
IV. Shock
• Shock is reduced perfusion of tissue, which results in impairedoxygenation of tissue.
A. Types of shock1. Hypovolemic shock: excessive fluid loss (e.g., blood,
sweat) (Table 4-1)a. Hemorrhage: loss of > 20% of blood volume
(- 1000 mL) results in shock.b. Changes in hemoglobin and hematocrit
(1) No initial effect from loss of whole blood(2) Normal body response: plasma is replaced first;
plasma replacement reveals the RBC deficitwithin hours to days.
(3) Infusion of 0.9% saline to increase bloodpressure immediately indicates the RBCdeficit.
c. Decreased cardiac output: less blood in the leftventricle
d. Decreased left ventricular end-diastolic volume(LVEDV)
e. Increased peripheral vascular resistance (PVR): dueto vasoconstriction of arterioles from catechola-mines and angiotensin II, which are released in re-sponse to the decreased cardiac output
f. Clinical findings(1) Cold, clammy skin (from vasoconstriction of
skin vessels)(2) Hypotension; rapid, weak pulse (compensa-
tory response to decreased cardiac output)2. Cardiogenic shock, such as acute myocardial infarction
(see Table 4-1)a. Decreased cardiac output: decreased force of con-
traction in the left ventricle
Hypovolemic shock:cardiac output,LVEDV, T PVR
Cardiogenic shock-I cardiac output,T LVEDV, T PVR

•
•Chapter 4 Fluid and Hemodynamic Disorders 43
• b. Increased LVEDV: blood accumulates in the leftventricle.
c. Increased PVR: same mechanism as in hypovolemic• shock
•d. Clinical findings: similar to those in hypovolemic
shock
•3. Septic shock (see Table 4-1)
a. Cause: usually Escherichia coli
• b. Pathogenesis
•(1) Endotoxins of gram-negative bacteria (e.g.,
E. coli) directly damage endothelial cells, releas-• ing nitric oxide (vasodilator) and prostaglan-
din 12 (vasodilator).• (2) Endotoxins activate the alternative complement
•pathway, releasing anaphylatoxins (C3a andC5a), further enhancing vasodilation of the pe-ripheral resistance arterioles.
•(3) Interleukin-1 and tumor necrosis factor re-
leased from macrophages: activate neutrophiladhesion molecules, causing neutrophil adher-ence to pulmonary capillaries.
• c. Cardiac output
•(1) Initially increased because of excessive blood
flow through dilated peripheral resistance arteri-• oles, causing increased return of blood to the
heart• (2) Tissue is unable to remove oxygen because of
the increased blood flow.d. Decreased LVEDV: due to neutrophil emigration
• from pulmonary capillaries into alveoli Septic shock T car-• e. PVR: decreased due to vasodilation of peripheral output,diacre-
sistance arterioles I LVEDV, I PVR
•f. Clinical findings
(1) Warm skin: from vasodilation of skin vessels• (2) Increased cardiac output
•(3) Acute respiratory distress syndrome: due to
neutrophil emigration into alveoli
• (4) DIC: due to activation of the intrinsic and ex-trinsic coagulation system
• B. Complications associated with shock
•1. Ischemic acute tubular necrosis
a. Proximal tubules in the cortex and cells in the thick• ascending limb of the medulla are most often
affected.b.b. Tubular cells undergo coagulation necrosis, leading
•to renal failure.
2. Multiorgan dysfunction: most common cause of death• 3. Metabolic acidosis: lactic acidosis due to tissue hypoxia
••••

aOk‘ 4411%ienetic and
DevelopmentalDisorders
I. MutationsA. Mutations refer to a permanent change in DNA.B. Point mutations are changes in a single nucleotide base
within a gene.1. Silent mutation: altered DNA codes for the same amino
acid without changing the phenotypic effect.2. Missense mutation: altered DNA codes for a different •
amino acid, which changes the phenotypic effect.
In both sickle cell trait and sickle cell disease, amissense mutation occurs when adenine replacesthymidine, causing valine to replace glutamic acid inthe sixth position of the 3-globin chain. As a result,red blood cells spontaneously sickle in the peripheralblood if the amount of sickle hemoglobin is greaterthan 60%. •
••••••••
3. Nonsense mutation: altered DNA codes for a stop codonthat causes premature termination of protein synthesis.
In p-thalassemia major, a nonsense mutation pro-duces a stop codon that causes premature termina-tion of DNA transcription of the P-globin chain.Consequently, there is a marked decrease in thesynthesis of hemoglobin A (a2(32), resulting in amicrocytic anemia.
•••••44
•••••••••••••••••••••
•

Chapter 5 Genetic and Developmental Disorders 45
A
7 II A 0Normal Affected Heterozygous Normal
Affected Heterozygous
male male male female
female female
Figure 5-1 Pedigree of an autosomal recessive disorder Both parents must have the mutantgene to transmit the disorder to their children On average, 25% of the children ofheterozygous parents are normal, 50% are asymptomatic heterozygous carriers, and 25%have the disorder
C. Frameshift mutation1. Insertion or deletion of one or more nucleotides shifts
the reading frame of the DNA strand.2. Example: in Tay-Sachs disease, a four-base insertion
results in the synthesis of a defective lysosomal enzyme(hexosaminidase).
D. Trinucleotide repeat disorders1. Errors in DNA replication cause amplification of a se-
quence of three nucleotides (e.g., CAG), which disruptsgene function.
2. Anticipation is the increasing severity of clinical diseasein each successive generation.a. Anticipation is caused by the addition of more tri-
nucleotide sequences during gametogenesis.b. Female carriers may be symptomatic if they have
more paternally (than maternally) derived X chromo-somes with trinucleotide repeats.
3. Examples: fragile X syndrome, Huntington's disease,Friedreich's ataxia, myotonic dystrophy
II. Mendelian Disorders• Usually single-gene mutationsA. Autosomal recessive disorders
1. Inheritance pattern (Figure 5-1)a. Individuals must be homozygous for the mutant
recessive gene (aa) to express the disorder; they aresymptomatic early in life.
b. Heterozygous individuals (Aa) are asymptomaticcarriers; the dominant gene (A) overrides the mutantrecessive gene (a).
c. Both parents must be heterozygous to transmit thedisorder.
Most common typeof mendelian dis-order: autosomalrecessive

46 Pathology
TABLE 5-1 Protein Defects Associated With Some Mendelian Disorders
Protein Type Specific Protein
Disorder
Inheritance Pattern
Enzyme
Structural
Cl esterase inhibitordeficiency
Glucose-6-phosphatedehydrogenase
Sickle hemoglobinSpectrin
Dystrophin
Hereditary angio-edema
Glucose-6-phosphatedehydrogenasedeficiency
Sickle cell diseaseCongenital sphero-
cytosisDuchenne's muscular
dystrophy
Cystic fibrosis
Familial hypercholes-terolemia
Neurofibromatosis
Hemophilia A
Autosomal dominant
X-linked recessive
Autosomal recessiveAutosomal dominant
X-linked recessive
Autosomal recessive
Autosomal dominant
Autosomal dominant
X-linked recessive
Transport Cystic fibrosis trans-membrane regu-lator
Receptor Low-density lipopro-tein receptor
Growth Neurofibrominregulating
Hemostasis Factor VIII
Example: Aa x Aa ---> AA, Aa, Aa, as (25% withoutdisorder; 50% asymptomatic carriers; 25% withdisorder)
Cystic fibrosis (CF) is an autosomal recessivedisease with a carrier rate of 1/25. To calculatethe prevalence of CF in the population, thenumber of couples at risk of having a child withCF (1/25 x 1/25, or 1/625) is multiplied by thechance of having a child with CF (1/4).
Prevalence of CF = 1/625 x 1/4, or 1/2500
2. Protein defects (Table 5-1)3. Inborn errors of metabolism (Table 5-2): metabolic dis-
orders characterized by an enzyme deficiencya. These disorders involve the metabolism of amino
acids (e.g., phenylketonuria, PKU); carbohydrates(e.g., galactosemia); ammonia (arginase deficiency);or organic acids (e.g., pyruvate dehydrogenasedeficiency).(1) Substrate and intermediates proximal to the
enzyme block increase.(2) Intermediates and the end-product distal to the
enzyme block decrease.
Most autosomal re-cessive disordersinvolve enzymedeficiencies.
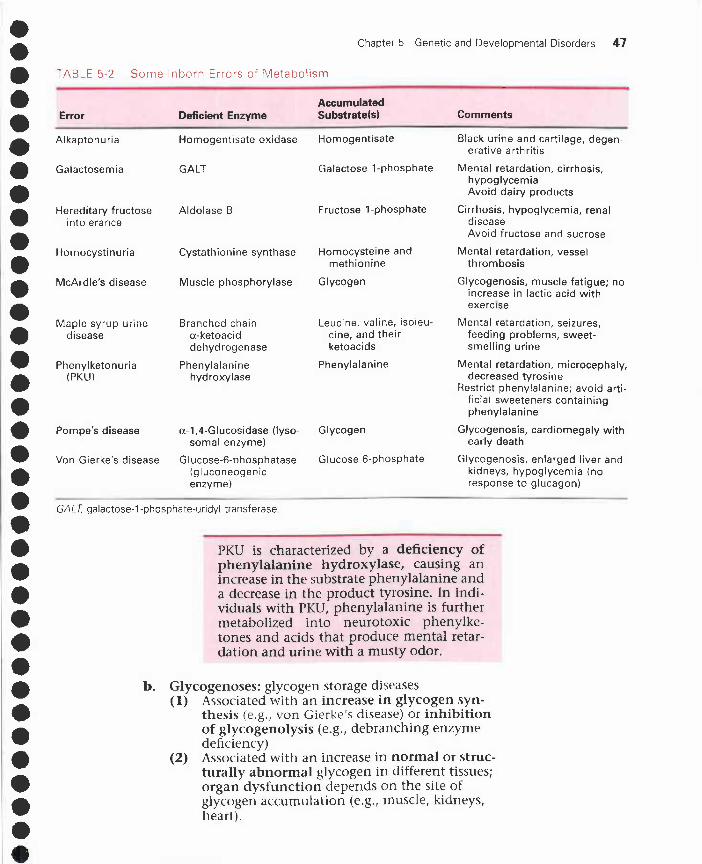
Chapter 5 Genetic and Developmental Disorders 47
TABLE 5-2 Some Inborn Errors of Metabolism
Error Deficient EnzymeAccumulatedSubstrate(s) Comments
Alkaptonuria Homogentisate oxidase Homogentisate Black urine and cartilage, degen-erative arthritis
Galactosemia GALT Galactose 1-phosphate Mental retardation, cirrhosis,hypoglycemiaAvoid dairy products
Hereditary fructoseintolerance
Aldolase B Fructose 1-phosphate Cirrhosis, hypoglycemia, renaldiseaseAvoid fructose and sucrose
Homocystinuria Cystathionine synthase Homocysteine andmethionine
Mental retardation, vesselthrombosis
McArdle's disease Muscle phosphorylase Glycogen Glycogenosis, muscle fatigue; noincrease in lactic acid withexercise
Maple syrup urinedisease
Branched chaina-ketoacid
Leucine, valine, isoleu-cine, and their
Mental retardation, seizures,feeding problems, sweet-
dehydrogenase ketoacids smelling urine
Phenylketonuria(PKUI
Phenylalaninehydroxylase
Phenylalanine Mental retardation, microcephaly,decreased tyrosine
Restrict phenylalanine; avoid arti-ficial sweeteners containingphenylalanine
Pompe's disease a-1,4-Glucosidase (lyso-somal enzyme)
Glycogen Glycogenosis, cardiomegaly withearly death
Von Gierke's disease Glucose-6-phosphatase(gluconeogenicenzyme)
Glucose 6-phosphate Glycogenosis, enlarged liver andkidneys, hypoglycemia (noresponse to glucagon)
GALT, galactose-1-phosphate-uridyl transferase.
PKU is characterized by a deficiency ofphenylalanine hydroxylase, causing anincrease in the substrate phenylalanine anda decrease in the product tyrosine. In indi-viduals with PKU, phenylalanine is furthermetabolized into neurotoxic phenylke-tones and acids that produce mental retar-dation and urine with a musty odor.
Glycogenoses: glycogen storage diseases(1) Associated with an increase in glycogen syn-
thesis (e.g., von Gierke's disease) or inhibitionof glycogenolysis (e.g., debranching enzymedeficiency)
(2) Associated with an increase in normal or struc-turally abnormal glycogen in different tissues;organ dysfunction depends on the site ofglycogen accumulation (e.g., muscle, kidneys,heart).
b.
48 Pathology
TABLE 5-3 Some Lysosomal Storage Disorders
AccumulatedDisorder Deficient Enzyme
Substrate
Clinical Findings
Gaucher's disease Glucocerebrosidase Glucocerebroside Hepatosplenomegaly;(adult type) fibrillar appearing
macrophages in liver,spleen, and bonemarrow
Hurler's syndrome a-L-Iduronidase
Niemann-Pick Sphingomyelinasedisease
Tay-Sachs disease Hexosaminidase
Dermatan andheparan sulfate
Sphingomyelin
GM 2 ganglioside
Mental retardation,coarse facial features,corneal clouding,coronary arterydisease
X-linked recessive form(Hunter's syndrome)is milder
Mental retardation,hepatosplenomegaly,foamy macrophages
Mental retardation,muscle weakness,cherry-red macula,blindness
(3) Fasting hypoglycemia may result from inter-ference with gluconeogenesis (e.g., glucose6-phosphatase deficiency) or liver glycogenoly-sis (e.g., liver phosphorylase deficiency).
c. Lysosomal storage diseases (Table 5-3)(1) Enzyme deficiencies lead to accumulation of
undigested substrates (e.g., glycosaminoglycans,sphingolipids) in lysosomes.
(2) Metabolites accumulate where the substrate ismost active (e.g., sphingomyelin in brain; glu-cocerebrosides in macrophages in liver, bonemarrow, and spleen).
4. Other autosomal recessive disorders: cc,-antitrypsin de-ficiency, 21-hydroxylase deficiency, Wilson's disease
B. Autosomal dominant disorders1. Inheritance pattern
a. One dominant mutant gene (A) is required toexpress the disorder.(1) Heterozygotes (Aa) express the disorder.(2) Homozygotes (AA) are often spontaneously
aborted.(3) Example: Aa x aa —> Aa, Aa, aa, aa (50% with
disorder; 50% without disorder)b. Some disorders arise by new mutations involving
either an egg or a sperm.2. Protein defects (see Table 5-1); enzyme deficiencies are
relatively uncommon.
Most commonautosomal reces-sive disorder:hemochromatosis

Chapter 5 Genetic and Developmental Disorders 49
A
4n b 6 a
B
06
q 0 0
a
Normal
Affected
Normal
Affected
ti male
male
female
female ,
Figure 5-2 Pedigrees showing complete and reduced penetrance in an autosomal dominantdisorder. Complete penetrance (A) means that all individuals with the mutant gene express thedisorder. Reduced penetrance (8) means that an individual has the mutant gene but does notexpress the disorder (arrow) The unaffected father has transmitted the disorder to his son
3. Characteristicsa. Delayed manifestations of disease: symptoms and
signs may not occur early in life (e.g., develop-ment of cysts between 10-20 years of age in adultpolycystic kidney disease).
b. Penetrance(1) Complete penetrance (Figure 5-2, A): all indi-
viduals with the mutant gene express the disor-der (e.g., familial polyposis coli).
(2) Reduced penetrance (Figure 5-2, B): individualswith the mutant gene are phenotypicallynormal but are able to transmit the disorder(e.g., Marfan syndrome).
c. Variable expressivity: all individuals with themutant gene express the disorder but at differentlevels of severity.
4. Other autosomal dominant disorders: Huntington'sdisease, osteogenesis imperfecta
•••••••••••••••••••••••••••••••••••
Most commonautosomal dominantdisorder: vonWillebrand's disease

50 Pathology
■ 0 ■
Normal Affected Normal Heterozygousmale male female female
Figure 5-3 Pedigree of an X-linked recessive disorder. The affected male transmits theabnormal X chromosome to both of his daughters and none of his sons. Both daughters areasymptomatic heterozygous carriers of the mutant gene The daughter with four children hastransmitted the mutant gene to 50% of her sons.
C. X-linked recessive disorders1. Inheritance pattern (Figure 5-3)
a. Males must have the mutant recessive gene on theX chromosome to express the disorder.
b. Affected males transmit the mutant gene to all oftheir daughters, who are usually asymptomaticcarriers.Example: XY x XX —> XX, XX, XY, XY
c. Asymptomatic female carriers of the mutant genetransmit the disorder to 50% of their male offspring.Example: XX x XY —> XX, XX, XY, XY
d. Symptomatic female carriers have maternallyderived X chromosomes (without the mutant gene)that are preferentially inactivated; only paternallyderived X chromosomes with the mutant generemain.
e. Although most X-linked recessive disorders are inher-ited in mendelian fashion, some may arise as newmutations.
2. Protein defects (see Table 5-1); enzymes are the mostcommon type of protein affected in X-linked recessivedisorders.
3. Fragile X syndrome: trinucleotide repeat disordera. Clinical findings
(1) Mental retardation: most common mendeliandisorder that causes mental retardation; 50%of female carriers may develop mentalretardation.
(2) Phenotypic changes: long face, large mandible,everted ears
(3) Macro-orchidism (enlarged testes) at pubertyb. Diagnosis: DNA analysis to identify trinucleotide
repeats; fragile X chromosome study
Most commonX-linked disorder:fragile X syndrome

__L 4 a
Chapter 5 Genetic and Developmental Disorders 51
II 0 0Normal Affected Normal Affected
male male female female
Figure 5-4 Pedigree of an X-linked dominant disorder In these rare disorders, female carriersand males with the mutant dominant gene express the disorder The distribution is similar tothat of X-linked recessive disorders, except that carrier females are symptomatic.
4. Lesch-Nyhan syndromea. Deficiency of hypoxanthine-guanine phosphori-
bosyltransferase, which is normally involved in sal-vaging the purines hypoxanthine and guanine
b. Clinical findings: mental retardation, hyperurice-mia, self-mutilation
5. Other X-linked recessive disorders: testicular feminiza-tion, chronic granulomatous disease, Bruton'sagammaglobulinemia
D. X-linked dominant disorders1. Inheritance pattern: same as X-linked recessive except
that the dominant mutant gene causes disease in bothmales and females (Figure 5-4)
2. Vitamin D—resistant rickets: defect in renal and gastro-intestinal reabsorption of phosphate, causing defectivebone mineralization
3. Alport's syndrome: hereditary glomerulonephritis withnerve deafness
III. Chromosomal DisordersA. General considerations
1. Most human cells are diploid; cells contain 46 chromo-somes, with 22 pairs of autosomes and 1 pair of sexchromosomes (XX in females and XY in males).
2. Gametes, the products of meiosis, are haploid andcontain 23 chromosomes.
3. Lyon hypothesisa. One of the two X chromosomes in females is ran-
domly inactivated early in the embryonic period,forming a Barr body.
b. A Barr body is attached to the nuclear membraneand is visible in squamous cells obtained in a buccalsmear.
c. Normal females have one Barr body, and normalmales have none.
Number of Barrbodies = number ofX chromosomes – 1

52 Pathology
d. The inactivation accounts for the parental derivationof the X chromosomes in females: - 50% are pater-nal and - 50% are maternal.
B. Chromosomal alterations: numeric or structural abnormali-ties of autosomes or sex chromosomes1. Nondisjunction: unequal separation of chromosomes
in the first phase of meiosis, resulting in 22 or 24 chro-mosomes; this may lead to Down syndrome, ortrisomy 21.
2. Mosaicism: nondisjunction of chromosomes duringmitotic division in the early embryonic perioda. Two chromosomally different cell lines are
derived from a single fertilized egg.b. Most cases of mosaicism involve sex chromosomes
(e.g., Turner's syndrome).3. Translocation: transfer of chromosome parts between
nonhomologous chromosomesa. Balanced translocation: translocated fragment is
functional.b. Robertsonian translocation: balanced translocation
between two acrocentric chromosomes (e.g., chro-mosomes 14 and 21)
In a form of Down syndrome, the mother of anaffected child has 45 (not 46) chromosomesbecause of a Robertsonian translocation be-tween the long arms of chromosomes 21 and 14,producing one long chromosome (14;21). Themother also has one chromosome 14 and onechromosome 21, and the father has the normal46 chromosomes. The affected child has 46 chro-mosomes with three functional 21 chromo-somes: a chromosome (14;21), a chromosome 21from the mother, and a chromosome 21 fromthe father.
4. Deletion: loss of a portion of a chromosomeC. Disorders involving autosomes
1. Down syndrome: trisomy 21a. Causes: nondisjunction (95% of cases), Robertso-
nian translocation (4% of cases), mosaicism (1% ofcases)
b. Increased maternal age is a risk factor; Down syn-drome occurs in 1 in 25 live births in women olderthan 45 years of age.
c. Clinical findings(1) General: mental retardation, epicanthic folds
and flat facial profile, simian crease(2) Combined atrial and ventricular septal
defects (cushion defects); major factor affectingsurvival in early childhood
Most commongenetic cause ofmental retardation.Down syndrome

Chapter 5 Genetic and Developmental Disorders 53
Figure 5 -5 Turner's syndrome is characterized by a webbed neck Other findings includeshort stature, primary amenorrhea, and delayed secondary sex characteristics (e.g., underde-veloped breasts).
(3) Increased risk of Hirschsprung's disease andduodenal atresia
(4) Increased risk of acute megakaryocytic leuke-mia (< 3 years of age) or acute lymphoblasticleukemia (> 3 years of age)
(5) Alzheimer's disease by 35 years of age in mostcases; major factor affecting survival in olderindividuals
(6) Sterility in all males; females have a 50%chance of having a child with Down syndrome.
2. Edwards' syndrome: trisomy 18, with mental retarda-tion, clenched hands with overlapping fingers, ventricu-lar septal defect (VSD), and rocker bottom feet; resultsin early death
3. Patau's syndrome: trisomy 13, with mental retardation,cleft lip and/or palate, polydactyly, VSD, and cystickidneys; results in early death
4. Cri du chat syndromea. Loss of the short arm of chromosome 5b. Clinical findings: mental retardation, cat-like
cry, VSDD. Disorders involving sex chromosomes
1. Turner's syndromea. Etiology
(1) Nondisjunction resulting in a 45,X karyotype(no Barr body) in - 60% of cases
(2) Mosaicism resulting in a 45,X/46,XX karyotypein - 40% of cases
b. Clinical and laboratory findings(1) Short stature: cardinal finding; normal growth
hormone and insulin-like growth factor(2) Lymphedema in hands and feet in infancy;
webbed neck is caused by dilated lymphaticchannels (cystic hygroma) (Figure 5-5).
(3) Congenital heart disease: preductal coarcta-tion and bicuspid aortic valve
Advanced maternalage increases therisk of bearing chil-dren with trisomysyndromes
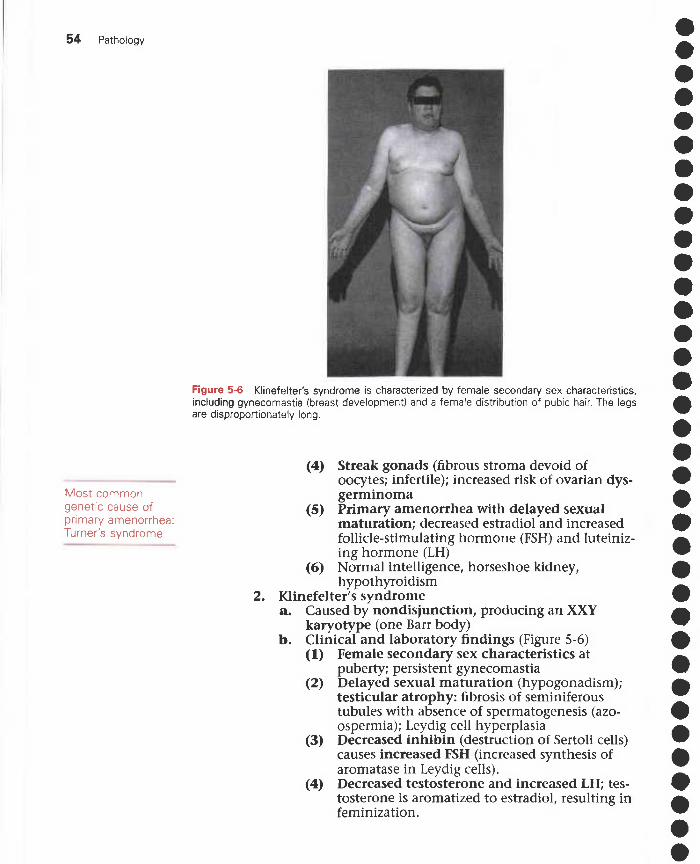
54 Pathology
Figure 5-6 Klinefelter's syndrome is characterized by female secondary sex characteristics,including gynecomastia (breast development) and a female distribution of pubic hair. The legsare disproportionately long.
(4) Streak gonads (fibrous stroma devoid ofoocytes; infertile); increased risk of ovarian dys-germinoma
(5) Primary amenorrhea with delayed sexualmaturation; decreased estradiol and increasedfollicle-stimulating hormone (FSH) and luteiniz-ing hormone (LH)
(6) Normal intelligence, horseshoe kidney,hypothyroidism
2. Klinefelter's syndromea. Caused by nondisjunction, producing an XXY
karyotype (one Barr body)b. Clinical and laboratory findings (Figure 5-6)
(1) Female secondary sex characteristics atpuberty; persistent gynecomastia
(2) Delayed sexual maturation (hypogonadism);testicular atrophy: fibrosis of seminiferoustubules with absence of spermatogenesis (azo-ospermia); Leydig cell hyperplasia
(3) Decreased inhibin (destruction of Sertoli cells)causes increased FSH (increased synthesis ofaromatase in Leydig cells).
(4) Decreased testosterone and increased LH; tes-tosterone is aromatized to estradiol, resulting infeminization.
Most commongenetic cause ofprimary amenorrhea:Turner's syndrome

( ■ 0 •Normal Affected Normal Affected
, male male female female i
Figure 5-7 Pedigree showing transmission of mitochondria) DNA Affected females transmitthe disorder to all their children, whereas affected males do not
(5) Disproportionately long legs, learningdisabilities
3. XYY syndrome: caused by paternal nondisjunction; asso-ciated with aggressive (sometimes criminal) behavior; nohypogonadism
IV. Other Patterns of InheritanceA. Multifactorial (polygenic) inheritance
1. Caused by a combination of multiple minor gene muta-tions and environmental factors (e.g., associationbetween neural tube defects and diets low in folate)
2. Risk of expressing the disorder correlates with thenumber of mutant genes involved.
3. Examples: cleft lip or palate, congenital heart disease,coronary artery disease, gout, type 2 diabetes mellitus,essential hypertension
B. Mitochondrial DNA disorders (Figure 5-7)1. Mitochondrial DNA primarily codes for enzymes that are
involved in mitochondrial oxidative phosphoryla-tion reactions.
2. Affected females transmit the mutant gene to all theirchildren (ova contain mitochondria).
3. Affected males do not transmit the mutant gene to anyof their children (sperm lose their mitochondria duringfertilization).
4. Examples: Leber's hereditary optic neuropathy, myo-clonic epilepsy
C. Genomic imprinting1. Inheritance of a trait depends on whether the mutant
gene is of maternal or paternal origin.2. Examples: Prader-Willi syndrome and Angelman's
syndromea. Maternal chromosome 15 during gametogenesis:
the Prader-Willi gene is inactivated by methyla-tion (imprinted), and the Angelman's gene is de-methylated (activated).
•••••••••••••••••••••••••••••••••••
Chapter 5 Genetic and Developmental Disorders 55
Polygenic disordersare more commonthan mendelian andchromosomaldisorders.
Disorders involvingmitochondrial DNAare associatedwith maternalinheritance
•

56 Pathology
b. Paternal chromosome 15 during gametogenesis:the Prader-Willi gene is activated, and the Angel-man's gene is imprinted (inactivated).
c. Microdeletion of the entire gene site on paternalchromosome 15 causes Prader-Willi syndrome,because the maternal Prader-Willi gene is inactive;clinical findings include mental retardation, obesity,and hypogonadism.
d. Microdeletion of the entire gene site on maternalchromosome 15 causes Angelman's syndrome,because the paternal Angelman gene is inactive;clinical findings include mental retardation, ataxicgait, and inappropriate laughter ("happy puppet"syndrome).
V. Disorders of Sex DifferentiationA. Normal sex differentiation
1. Absence of the Y chromosome: germinal tissue differen-tiates into ovaries; wolffian (mesonephric) duct struc-tures undergo apoptosis.
2. Presence of the Y chromosome: germinal tissue differen-tiates into testes; mullerian inhibitory factor causes mul-lerian tissue to undergo apoptosis.a. Fetal testosterone develops the wolffian duct struc-
tures (epididymis, seminal vesicles, vas deferens)b. The enzyme 5a-reductase converts testosterone to
dihydrotestosterone (DHT).c. Fetal DHT develops the prostate and external male
genitalia (genitalia is phenotypically female beforeDHT is produced).
B. True hermaphrodite: fetus has both male and femalegonads; karyotype is usually 46,XX.
C. Pseudohermaphrodite: phenotype and genotype do notmatch.1. Male pseudohermaphrodite: genotypic male (XY with
testes); phenotypic female (e.g., testicular feminization).2. Female pseudohermaphrodite: genotypic female (XX
with ovaries); phenotypic male (e.g., virilization in adre-nogenital syndrome).
D. Testicular feminization: X-linked recessive disorder with adeficiency of androgen receptors1. Fetal DHT and testosterone are unable to function
without a receptor.2. Clinical and laboratory findings
a. Testicles in the inguinal canal or abdominal cavityb. No mullerian structures (e.g., absent fallopian tubes,
uterus, cervix, upper vagina): mullerian inhibitoryfactor is present.
c. No male accessory structures: normal male levels oftestosterone and DHT; no testosterone effect on thewolffian duct structures
The Y chromosomedetermines thegenetic sex of anindividual.

d. External genitalia remain female: no DHT effect;vagina ends as a blind pouch.
e. Estrogen activity is unopposed, because estrogen re-ceptors are present.
•••••
VI. Congenital Anomalies• Defects present at birth that may or may not have a genetic basisA. Malformations: disturbances in the morphogenesis of an
organ; usually multifactorial (e.g., drugs, infection)1. Pathogenesis
a. Most sensitive period for malformations to occur isduring the fourth and fifth week of embryogenesis.
b. Teratogens interfere with formation of the mitoticspindle, production of adenosine triphosphate,or with genes regulating morphogenesis (Table 5-4).
Pregnant women should not be treated for acneI with retinoic acid. Retinoic acid disrupts the function of the HOX gene, leading to craniofacial
and cardiovascular malformations in the fetus.
•S••••••••••••••
c. Some diseases in pregnant women predispose thenewborn to malformations.(1) Diabetes mellitus: a newborn has an increased
risk of neural tube defects and congenital heartdisease.
(2) Systemic lupus erythematosus: a newbornmay develop congenital heart block if themother has anti-Ro antibodies.
Most commoncause of congenitalmalformations:alcohol
Alcohol
Cocaine
DES
Phenytoin
Retinoic acid
Thalidomide
Tobacco
Valproate
Warfarin
••
Mental retardation, microcephaly, atrial septal defect
Microcephaly, renal agenesis, congenital heart disease
Vaginal and/or cervical clear cell carcinoma, mtillerian defects
Nail and distal phalanx hypoplasia, cleft lip and/or palate
Craniofacial, central nervous system, and cardiovascular defects
Limb defects, amelia (absent limbs), phocomelia (seal-like limbs)
IUGR, low birth weight
Neural tube defects
Nasal hypoplasia, agenesis corpus callosum
••••••••••••
Chapter 5 Genetic and Developmental Disorders 57
TABLE 5-4 Teratogens Associated With Congenital Defects
Teratogen
Defect
DES, diethylstilbestrol; IUGR, intrauterine growth retardation

58 Pathology
TABLE 5-5 Congenital Infections Associated With Congenital Defects
Infection
Transmission Clinical Findings
Toxoplasmosis Transplacental Blindness, CNS calcification(basal ganglia)
Pregnant woman should avoidcat litter
Syphilis Transplacental Occurs after 20 weeks' gesta-tion
Hepatitis, saddle nose, blind-ness, peg teeth
Varicella Transplacental Limb defects, mental retarda-tion, blindness
Rubella Transplacental Deafness, patent ductus arteri-osus
Cytomegalovirus Transplacental Deafness, IUGR, CNS calcifica-tion (periventricular)
Culture urine (best fluid toculture)
Urine cytology (intranuclearinclusions)
Herpes simplex type 2
Birth canal IUGR
CNS, central nervous system; IUGR, intrauterine growth retardation.
2. Congenital infections causing malformations(Table 5-5)a. Increase in cord blood IgM: IgM normally is not
synthesized in the fetus unless there is a congenitalinfection.
b. Vertical transmission: transplacental, birth canal,breastfeeding
B. Deformations: extrinsic disturbances in fetal development;usually occur in the last 24 weeks of gestation, after fetalorgans have developed
Oligohydramnios (decreased amniotic fluid) from de-creased production of fetal urine (e.g., cystic disease of thekidneys) restricts fetal movement in the uterine cavity. Asa result, newborns have flat facial features (Potter's fa-cies) and underdevelopment of the chest wall.
C. Agenesis: complete absence of an organ resulting fromabsence of the anlage (primordial tissue) (e.g., renal agenesis)
D. Aplasia: anlage is present but never develops (e.g., lungaplasia with tissue containing rudimentary ducts and connec-tive tissue).
E. Hypoplasia: anlage develops incompletely, but the tissue ishistologically normal (e.g., microcephaly).
F. Atresia: incomplete formation of a lumen (e.g., duodenalatresia)
Most common con-genital infection:cytomegalovirus
TORCH syndrome =toxoplasmosis,other, rubella, cyto-megalovirus,herpes simplex virus

•• Chapter 5 Genetic and Developmental Disorders 59
• VII. Selected Perinatal and Infant Disorders• A. Stillbirth
1. Birth of a dead child• 2. Most often caused by abruptio placentae (e.g., prema-• ture separation of the placenta because of a retroplacental
blood clot)O B. Spontaneous abortion
•1. Termination of a pregnancy before 20 weeks2. Caused by a fetal karyotypic abnormality (usually
• trisomy 16) in about 50% of cases3. Predisposing factors: advanced maternal age; infections• (e.g., Streptococcus agalactiae, Listeria monocytogenes);
tobacco and alcohol useC. Sudden infant death syndrome (SIDS)
• 1. Sudden and unexpected death of an infant younger than
•1 year of age whose death remains unexplained afterautopsy
• 2. Pathogenesis: no single cause; maternal factors (e.g.,smoking, young age); infant factors (e.g., prematurity,
• sleeping prone, neural developmental delay)
O3. Autopsy findings: thickened pulmonary arteries, pete-
chiae on the pleura and epicardium; microscopic changes
• of hypoxia in the brainstem
• VIII. Diagnosis of Genetic and Developmental Disorders
•A. Amniocentesis: used to identify genetic defects prenatallyB. Ultrasound: used to rule out neural tube defects
• C. Maternal screening tests: triple marker screen
•1. a-Fetoprotein (AFP): increased in neural tube defects
(causally related to folate deficiency)
• 2. Human chorionic gonadotropin (hCG): levels vary withgestational age
• 3. Urine for unconjugated estriol: excellent marker offetal, placental, or maternal dysfunction
D. Genetic analysis• 1. Chromosome karyotyping: used to identify numeric
and structural abnormalities• 2. DNA polymerase chain reaction: amplifies DNA frag-
•3. ments harboring abnormal gene loci
Restriction fragment length polymorphism (RFLP):• used to identify abnormal gene when the exact site is• unknown
••••
Triple marker forDown syndrome:AFP decreased,hCG increased,urine unconjugated,estriol decreased
••

klEnvironmentalPathology
I. Chemical Injury• The two leading causes of morbidity and mortality in the United
States are tobacco and alcohol use.A. Tobacco use
1. Tobacco is associated with approximately 20% of alldeaths.
2. The rate of cigarette smoking is increasing in females anddecreasing in males.
3. Chemical components of tobaccoa. Nicotine: rapidly absorbed; most addictive chemical
in tobacco smokeb. Polycyclic hydrocarbons: primary carcinogens
4. Use of smokeless tobacco (e.g., chewing tobacco) cancause nicotine addiction and cancer.
5. Passive smoke inhalation has the greatest impact onchildren and is associated with increased risk of respira-tory and middle ear infections and cancer.
6. Cancers most commonly caused by tobaccoa. Respiratory: lung (squamous cell carcinoma, small
cell carcinoma); larynx (squamous cell carcinoma)b. Gastrointestinal: oropharynx (squamous cell carci-
noma); esophagus (squamous cell carcinoma),pancreas (adenocarcinoma)
c. Urogenital: kidneys (adenocarcinoma); bladder(transitional cell carcinoma)
7. Other conditions commonly associated withtobacco usea. Cardiovascular: acute myocardial infarction (MI),
peripheral vascular diseaseb. Respiratory: chronic bronchitis, emphysemac. Gastrointestinal: gastroesophageal refluxd. Central nervous system (CNS): atherosclerotic
stroke
Tobacco is theleading cause ofpremature death inthe US.
60

Chapter 6 Environmental Pathology 61
B. Alcohol use1. Absorption occurs in the stomach and small intestine.2. Metabolism occurs in the liver, resulting in reduced nic-
otinamide adenine dinucleotide, acetate, and acetyl co-enzyme A.a. Alcohol dehydrogenase is the rate-limiting enzyme.b. Alcohol induction of the cytochrome P-450 enzyme
system increases alcohol metabolism, which in-creases the tolerance for alcohol.
3. Legal intoxication: blood alcohol > 100 mg/dL4. Risk factors
a. The risk of acquiring alcohol-related disease increasesas the amount of alcohol consumed within a givenperiod increases.
b. Other risk factors are female sex (low gastric alcoholdehydrogenase levels) and genetic susceptibility.
5. Conditions most commonly caused by alcohol abusea. Vitamin deficiencies
(1) Thiamine deficiency: Wernicke's syndrome(confusion, ataxia, nystagmus), Korsakoff'spsychosis (memory deficits), cardiomyopathy
(2) Folate deficiency: macrocytic anemiab. Acquired sideroblastic anemiac. Mallory-Weiss syndrome (tear of distal esophagus),
Boerhaave's syndrome (rupture of distal esophagus),esophageal varices (caused by portal vein hyper-tension in alcoholic cirrhosis)
d. Fatty change, cirrhosis, pancreatitise. Fetal alcohol syndrome (mental retardation)
6. Laboratory findings in alcohol abusea. Fasting hypoglycemiab. Increased anion gap metabolic acidosis caused by
lactic acidosis and P-hydroxybutyric ketoacidosisc. Hyperuricemia, hypertriglyceridemia, serum aspar-
tate aminotransferase > serum alanine aminotransfer-ase, increased serum y-glutamyltransferase
C. Other drugs of abuse: sedatives, stimulants, hallucinogens(Table 6-1)1. CNS effects of long-term drug abuse: damage to neuro-
transmitter receptor sites, cerebral atrophy (e.g., alcohol)2. Complications of intravenous drug use: hepatitis B,
HIV, infective endocarditis (caused by Staphylococcusaureus)
D. Adverse effects of therapeutic drug use (Table 6-2)1. Acetaminophen: conversion to free radicals in the liver
may result in damage to the liver (fulminant hepatitis)and kidneys (renal papillary necrosis).
2. Aspirin (acetylsalicylic acid) overdosea. General symptoms such as tinnitus, vertigo, change
in mental status (confusion, seizures), and tachypneab. Acid-base disorders
(1) Respiratory alkalosis may occur initially (within
Thiamine deficiencycauses Wernicke'ssyndrome andKorsakoff'spsychosis.
Most common sys-temic complica-tion of intravenousdrug use: hepatitis B

62 Pathology
Marijuana THC-containing psycho-(Cannabis)* active stimulant
MPTP
By-product of synthesisof meperidine
Mydriasis, tachycardia, hypertensionAssociated risk of acute MI, CNS infarc-
tion, perforation of nasal septum (intra-nasal use)
Physical dependenceMiotic pupils, noncardiogenic pulmonary
edema (frothing from mouth), focalsegmental glomerulosclerosis
Granulomatous reactions in skin andlungs from material used to "cut"(dilute) drug
Red conjunctiva, euphoria, delayed reac-tion time
Irreversible Parkinson's disease (cytotoxicto neurons in nigrostriatal dopami-nergic pathways)
TABLE 6-1 Selected Drugs of Abuse and Their Effects
Drug
Description
Toxic Effects
••••••••••••••
Cocaine
Stimulant
Heroin
Opiate
*Used medically to decrease nausea and vomiting associated with chemotherapy and todecrease intraocular pressure in glaucoma.
CNS, central nervous system; MI, myocardial infarction; MPTP 1-methyl-4-phenyl-1, 2, 3,6-tetrahydropyridine; THC, e-tetrahydrocannabinol.
12-24 hours) and involves direct stimulationof the respiratory center.
(2) A shift to metabolic acidosis with an increasedanion gap occurs more often in children.
(3) Mixed primary respiratory alkalosis and met-abolic acidosis are common in adults.
c. Hyperthermia(1) Salicylates damage the inner mitochondrial
membrane.(2) Oxidative energy is released as heat, not as
adenosine triphosphate.d. Hemorrhagic gastritis, fulminant hepatitis
3. Exogenous estrogen without progestin may result in:a. Cancer: endometrium, breastb. Venous thromboembolism: estrogen decreases anti-
thrombin III (normally neutralizes coagulationfactors) and increases synthesis of factors I (fibrino-gen), V, and VIII.
c. Intrahepatic cholestasisd. Cardiovascular effects: MI, stroke
4. Oral contraceptives: estrogen-progestin combinationsmay result in:a. Cancer: breast, cervixb. Venous thromboembolismc. Folate deficiency (decreases jejunal reabsorption)d. Hypertension (increased synthesis of angio-
tensinogen)
Both acetaminophenand aspirin causefulminant hepatitis.
Most commoncause of hyperten-sion in youngwomen: oralcontraceptives
••••••••••••••••••••••

Chapter 6 Environmental Pathology 63
TABLE 6-2 Adverse Reactions Associated with Therapeutic Drug Use
Reaction Drug(s)
Blood DyscrasiasAplastic anemiaHemolytic anemiaMacrocytic anemia
Platelet dysfunctionThrombocytopenia
CardiacCongestive cardiomyopathy
Central Nervous System
Tinnitus, vertigo
Cutaneous
AngioedemaMaculopapular rashPhotosensitive rashUrticaria
Gastrointestinal
Hemorrhagic gastritis
Hepatic
CholestasisFatty changeHepatic adenomaLiver necrosis
PulmonaryAsthma Aspirin, other NSAIDsInterstitial fibrosis Bleomycin, busulfan, nitrofurantoin, methotrexate
SystemicDrug-induced lupus Procainamide, hydralazine
ACE, angiotensin-converting enzyme; NSAID, nonsteroidal anti-inflammatory drug.
e. Hepatic adenoma (risk of intraperitoneal hemor-rhage), intrahepatic cholestasis with jaundice, choles-terol gallstones
E. Injuries caused by environmental chemicals (Table 6-3)
II. Physical InjuryA. Mechanical injury
1. Typesa. Contusion (bruise): blunt force injury to blood
vessels with subsequent escape of blood into tissueb. Abrasion: superficial excoriation of the epidermisc. Laceration: jagged tear with intact bridging blood
vessels, nerves, and connective tissued. Incision: wound with sharp margins with severed
bridging blood vessels
••••••
•••••••••••••••••••••••••••••
Chloramphenicol, alkylating agentsPenicillin, methyldopa, quinidinePhenytoin, oral contraceptives, methotrexate,
5-fluorouracilAspirin, other NSAIDsHeparin, quinidine
Doxorubicin, daunorubicin
Salicylates
ACE inhibitorsPenicillinTetracyclinePenicillin
Iron, salicylates
Oral contraceptives, estrogen, anabolic steroidsAmiodarone, tetracycline, methotrexateOral contraceptivesAcetaminophen, isoniazid, salicylates, halothane, iron

64 Pathology
TABLE 6-3 Environmental Chemicals and Associated Toxic Effects
Chemical
Source Toxic Effects
Pesticides, animal dips
Insulation, roofing material
Solvent
Automobile exhaust, housefires
House fires
Antifreeze
End-product: oxalic acid
Rubbing alcoholEnd-product: acetone
Lead-based paint, batteries,metal casting
Mercury Fish, insecticides
Methanol Window-washer fluid
End-product: formic acid
Organophosphates Pesticides
Polyvinyl chloride Plastics industry
Diarrhea, transverse bands in nails,convulsions
Squamous cell carcinoma of skin,liver angiosarcoma, lung cancer
Primary lung cancer, mesothelioma
Acute leukemia, aplastic anemia
Headache (first sign), cherry-redskin, coma
Decreased oxygen saturationSeizures
Increased anion gap metabolicacidosis
Acute renal failure
Deep coma
Microcytic anemia with coarsebasophilic stippling, nephrotox-icity in proximal tubule
Diarrhea, constricted visual fields,nephrotoxicity in proximal tubule
Increased anion gap metabolicacidosis
Blindness
Miotic pupils, paralysisLow serum and red blood cell
cholinesterase levels
Liver angiosarcoma
Arsenic
Asbestos
Benzene
Carbon monoxide
Cyanide
Ethylene glycol
Isopropyl alcohol
Lead
2. Gunshot wounds: penetrating (only enters body) or per-forating (exits body)a. Contact wounds are stellate-shaped and contain
soot and gunpowder (fouling).b. Intermediate-range wounds cause powder tattooing
(stippling) of the skin around the entrance site.c. Long-range wounds cause no powder tattooing.d. Exit wounds are typically larger and more irregular
than entrance wounds.3. Motor vehicle collisions often cause mechanical injury
and are frequently alcohol-related.B. Thermal injury
1. Burnsa. First-degree: painful partial-thickness burns (e.g.,
sunburn) that heal without scarringb. Second-degree: painful partial-thickness burns
(1) Damage to entire epidermis; blister formation(2) Usually heal without scarring
c. Third-degree: painless full-thickness burns
Most commoncause of accidentaldeath in peopleages 1-39 years:motor vehiclecollisions

Chapter 6 Environmental Pathology 65
TABLE 6-4 Heat Injuries*
Type of Injury Temperature
Heat cramps 37.0°C (98.6°F)
Heat exhaustion > 37.8°C (> 100°F)
Heat stroke > 40°C (> 104°F)
Symptoms
Volume depletion caused by loss of salt andwater, muscle spasms
Sweating, volume depletion, hyperventilation
Anhidrosis (absence of sweating), delirium,seizures, hyperventilation; volume depletion
*Heat injury is exacerbated by high humidity.
(1) Extensive necrosis of epidermis and adnexa(2) Scarring is inevitable.(3) Healing begins at residual epithelium at burn
margins and at adnexal structures.d. Complications include infection and Curling's
ulcers (stomach).2. Heat injuries (Table 6-4)3. Frostbite
a. Pathogenesis: localized tissue injury caused by directdamage (e.g., ice crystallization in cells) or indirectdamage (e.g., vasodilation, thrombosis)
b. Clinical findings: loss of pain sensation, waxyappearance
C. Electrical injury1. Alternating current (AC) is more dangerous than direct
current (DC); AC produces tetanic contractions, and DCproduces a single shock.
2. Wet skin decreases resistance, which increases current;dry skin increases resistance, which decreases current.
3. Tissue damage increases with increased voltage and dura-tion of exposure.
4. Current moving from the left arm to the right leg is themost dangerous because it affects the heart. Death resultsfrom cardiorespiratory arrest.
III. Radiation InjuryA. Ionizing radiation injury (e.g., x-rays, y-rays)
1. Pathophysiologya. Injury correlates with type of radiation, cumulative
dose, and amount of surface area exposed.b. Direct or indirect DNA injury occurs via hydroxyl
free radicals.2. Tissue susceptibility
a. Most radiosensitive tissues (highest mitotic activ-ity): lymphoid tissue (most sensitive), bone marrow,mucosa of gastrointestinal tract, germinal tissue
b. Least radiosensitive tissues: bone (least sensitive),brain, muscle, skin
Most commoncause of death inpatients with post-burn Infections:sepsis caused byPseudomonasaerugtnosa

66 Pathology
3. Radiation effects in different tissuesa. Hematopoietic: lymphopenia (first change), throm-
bocytopenia, bone marrow hypoplasiab. Vascular: thrombosis (early), fibrosis (late), ischemic
damagec. Epidermal: acute effects are erythema, edema, blis-
tering; chronic effect is radiodermatitis with po-tential for squamous cell carcinoma.
d. Gastrointestinal: acute effect is diarrhea; chroniceffects are adhesions with potential for bowelobstruction.
4. Cancers caused by radiation: acute leukemia, papillarycarcinoma of the thyroid, osteogenic sarcoma
B. Nonionizing radiation1. Ultraviolet light B (UVB) is most damaging.
a. Pathogenesis: pyrimidine dimers distort the DNAhelix, inactivating the TP53 (p53) suppressor geneand activating the RAS oncogene.
b. General effects: sunburn, actinic keratosis (precursorof squamous cell carcinoma), corneal burns (skiing)
c. Cancers: basal cell carcinoma, squamous cell carci-noma, malignant melanoma
2. Effects of other types of radiationa. Laser radiation: third-degree burnsb. Microwave radiation: skin burns, cataracts, sterilityc. Infrared radiation: skin burns, cataracts
Most common UVBlight—related skincancer: basal cellcarcinoma
Type of cancer mostfrequently causedby radiation:acute leukemia
••••••••••••o•••••••••••••••••••s•••

7
Nutritional Disorders
o
I. Protein-Energy MalnutritionA. Protein stores
1. Somatic protein: muscle protein2. Visceral proteins: protein in liver and other organs
B. Kwashiorkor1. Inadequate protein intake; adequate caloric intake con-
sisting mainly of carbohydratesa. Visceral protein is decreased.b. Somatic protein is relatively unchanged.
2. Clinical findings (Figure 7-1, A)a. Pitting edema and ascites, caused by hypoalbumin-
emia and loss of plasma oncotic pressureb. Fatty liver, caused by decreased synthesis of
apoproteinsc. Diarrhea (loss of brush border enzymes); anemia and
defects in cell-mediated immunity (CMI)C. Marasmus
1. Dietary deficiency of both protein and calories; de-crease in somatic protein
2. Clinical findings (Figure 7-1, B)a. Extreme muscle wasting ("broomstick extremi-
ties") caused by breakdown of muscle protein forenergy and loss of subcutaneous fat; growthretardation
b. Anemia, defects in CMI
II. Eating Disorders and ObesityA. Anorexia nervosa
1. Self-induced starvation leading to protein-energymalnutrition
2. Clinical findingsa. Secondary amenorrhea
(1) Decreased gonadotropin-releasing hormonecaused by loss of body fat and weight
Pitting edema ischaracteristic ofkwashiorkor.
Extreme musclewasting is commonin marasmus.
Most commoncause of death inanorexia nervosa:ventricular arrhyth-mia precipitatedby hypokalemia
67
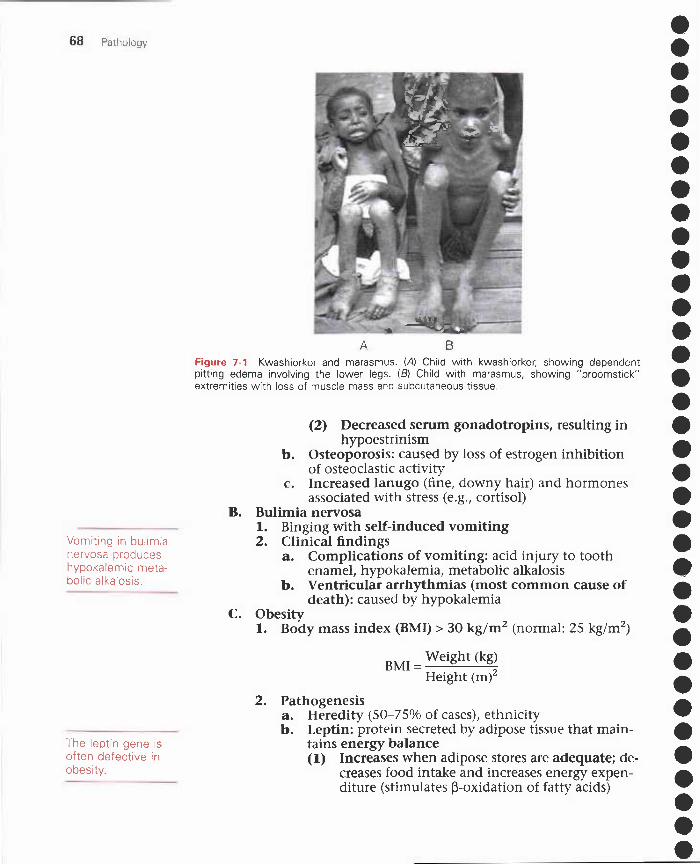
68 Pathology
A
Figure 7-1 Kwashiorkor and marasmus. (A) Child with kwashiorkor, showing dependentpitting edema involving the lower legs. (B) Child with marasmus, showing "broomstick"extremities with loss of muscle mass and subcutaneous tissue.
(2) Decreased serum gonadotropins, resulting inhypoestrinism
b. Osteoporosis: caused by loss of estrogen inhibitionof osteoclastic activity
c. Increased lanugo (fine, downy hair) and hormonesassociated with stress (e.g., cortisol)
B. Bulimia nervosa1. Binging with self-induced vomiting2. Clinical findings
a. Complications of vomiting: acid injury to toothenamel, hypokalemia, metabolic alkalosis
b. Ventricular arrhythmias (most common cause ofdeath): caused by hypokalemia
C. Obesity1. Body mass index (BMI) > 30 kg/
Vomiting in bulimianervosa produceshypokalemic meta-bolic alkalosis.
m2 (normal: 25 kg/m2)
BMI - Weight (kg)
Height (m)2
2. Pathogenesisa. Heredity (50-75% of cases), ethnicityb. Leptin: protein secreted by adipose tissue that main-
tains energy balance(1) Increases when adipose stores are adequate; de-
creases food intake and increases energy expen-diture (stimulates 13-oxidation of fatty acids)
The leptin gene isoften defective inobesity.

Chapter 7 Nutritional Disorders 69
TABLE 7-1 Clinical Findings Associated with Obesity
Clinical Finding Comments
Increased incidence of estrogen-related cancers (e.g., endo-metrial, breast) because of increased aromatization ofandrogens to estrogens in adipose
Increased incidence of cholecystitis and cholesterol stones;bile is supersaturated with cholesterol
Increased adipose down-regulates insulin receptor synthesis;hyperinsulinemia
Weight reduction up-regulates insulin receptor synthesis
Hyperinsulinemia increases sodium retention, leading toincrease in plasma volume
Left ventricular hypertrophy and stroke complicate hyperten-sion
Hypertriglyceridemia decreases serum high-density lipopro-tein levels, increasing risk of coronary artery disease
Hypercholesterolemia predisposes to coronary arterydisease
Degenerative arthritis in weight-bearing joints (e.g., femoralheads)
Cancer
Cholelithiasis
Diabetes mellitus,type 2
Hypertension
Hypertriglyceridemia
Increased low-densitylipoprotein levels
Osteoarthritis
(2) Decreases when adipose stores are inadequate;increases food intake and decreases energy ex-penditure (inhibits p-oxidation of fatty acids)
3. Clinical findings (Table 7-1)
[II. Fat-Soluble Vitamins• Vitamins A, D, E, and K are fat soluble.A. Vitamin A
1. Retinol, which is derived from dietary 3-carotenes andretinol esters, is the main transport and storage form ofvitamin A.
An excess of 13-carotenes in the diet causes the skin toturn yellow, but unlike in jaundice, the sclera re-mains white. 3-Carotenes also have antioxidant ac-tivity (neutralize free radicals).
2. Retinal (product of the oxidation of retinol) is a compo-nent of the visual pigment rhodopsin.
3. Functions of vitamin Aa. Normal vision in reduced lightb. Potentiating differentiation of mucus-secreting
epitheliumc. Stimulating the immune systemd. Growth and reproductione. Treatment of acne (e.g., isotretinoin) and acute pro-
myelocytic leukemia

70 Pathology
TABLE 7-2 Fat-Soluble Vitamins: Clinical Findings in Deficiency and Toxicity
Vitamin Deficiency Toxicity
A Impaired night vision, blindnessFollicular hyperkeratosis, pneumonia,
growth retardation, renal calculi
D Pathologic fractures, excess osteoid,bow legsChildren: rickets; craniotabes (soft
skull bones); rachitic rosary(defective mineralization andovergrowth of epiphyseal carti-lage in ribs)
Adults: osteomalaciaContinuous muscle contraction
(tetany)E Hemolytic anemia
Peripheral neuropathy, degenerationof posterior column (poor jointsensation) and spinocerebellartract (ataxia)
K Newborns: hemorrhagic diseaseof newborn (CNS bleeding,ecchymoses)
Adults: gastrointestinal bleeding,ecchymoses; prolongedprothrombin time
Papilledema, seizures, hepatitis
Hypercalcemia with metastaticcalcification, renal calculi
Decreased synthesis of vitaminK-dependent procoagulantfactors, synergistic effect withwarfarin anticoagulation
Hemolytic anemia and jaundice innewborns if mother receivesexcess vitamin K
CNS, central nervous system
4. Causes of deficiency: diet lacking sufficient yellow andgreen vegetables, fat malabsorption (e.g., celiac disease)
5. Causes of toxicity: eating bear liver, treatment withisotretinoin
6. Clinical findings in vitamin A deficiency and toxicity(Table 7-2)
B. Vitamin D1. Metabolism (Figure 7-2)
a. Preformed vitamin D in the diet consists of chole-calciferol (fish) and ergocalciferol (plants).
b. Endogenous synthesis of vitamin D in the skinoccurs by photoconversion of 7-dehydrocholesterolvia sunlight.
c. Reabsorption occurs in the small intestine.d. Liver hydroxylation to 25-hydroxyvitamin D
(25-OH-D) occurs in the cytochrome P-450 system.e. Kidney hydroxylation by 1 a-hydroxylase produces
1,25-(OH) 2-D (active form of vitamin D).f. Vitamin D increases reabsorption of calcium and
phosphorus from the intestine and calcium from thedistal renal tubules.
Night blindness isthe first sign ofvitamin A deficiency.

Chapter 7 Nutritional Disorders 71
Sunlight(7-Dehydrocholesterol)\ Liver
) Kidney
Diet 25-Hydroxylation 1-a-Hydroxylation
25-(OH)-D
1,25-(OH)2-D
Receptor y
Target organ
Figure 7-2 Vitamin D metabolism.
2. Functionsa. Maintenance of serum calcium and phosphorusb. Required for mineralization of epiphyseal cartilage
and osteoid matrix3. Causes of deficiency
a. Renal failureb. Inadequate exposure to sunlightc. Fat malabsorptiond. Chronic liver disease, enzyme induction of the cyto-
chrome P-450 system (e.g., alcohol, phenytoin)4. Megadoses may cause toxicity.5. Clinical findings in vitamin D deficiency and toxicity
(see Table 7-2)C. Vitamin E
1. Serves as an antioxidant2. Deficiency is uncommon; it occurs mainly in children
with fat malabsorption (e.g., cystic fibrosis) and inabetalipoproteinemia.
3. Megadoses may cause toxicity.4. Clinical findings in vitamin E deficiency and toxicity
(see Table 7-2)D. Vitamin K
1. Derived from endogenous bacteria and green vegetables2. Activated by the liver microsomal enzyme epoxide
reductase• The anticoagulant effect of coumarin derivatives
results from the inhibition of epoxide reductase.3. Function
a. y-Carboxylates glutamate residues in coagulantfactors II (prothrombin), VII, IX, and X that are syn-thesized in the liver
b. y-Carboxylation is required for proper functioning ofthe vitamin K—dependent coagulant factors.
4. Causes of deficiencya. Broad-spectrum antibiotics destroy bacterial syn-
thesis of vitamin K.b. Newborns lack the bacterial colonization of the
bowel necessary for endogenous synthesis ofvitamin K.
Most commoncause of vitamin Ddeficiency. renalfailure
Most commoncause of vitamin Kdeficiency:broad-spectrumantibiotics

72 Pathology
TABLE 7-3 Water-Soluble Vitamins: Clinical Findings in Deficiency
Vitamin Clinical Findings
Thiamine (vitamin B 1 ) Dry beriberi: peripheral neuropathy (demyelination)Wernicke's syndrome: ataxia, confusion, nystagmus,
mamillary body hemorrhageKorsakoff's syndrome: antegrade and/or retrograde
amnesia; demyelination in limbic systemWet beriberi: congestive cardiomyopathy with biventricular
failure
Riboflavin (vitamin B 2 ) Corneal neovascularization, glossitis, cheilosis (crackedlips), angular stomatitis (fissuring at angles of mouth)
Niacin (vitamin B 3 ) Pellagra: diarrhea, dermatitis (hyperpigmentation insun-exposed areas), dementia
Pyridoxine (vitamin B 6 ) Sideroblastic anemia (microcytic anemia with ringedsideroblasts), convulsions, peripheral neuropathy
Cobalamin (vitamin B 12 ) Megaloblastic anemia, neurologic disease (posteriorcolumn and lateral corticospinal tract demyelination),glossitis
Folic acid Megaloblastic anemia, with no neurologic disease (unlikevitamin B 12 ), glossitis
Biotin Dermatitis, alopecia, lactic acidosis
Ascorbic acid (vitamin C) Weak capillaries and venules, skin ecchymoses, perifol-licular hemorrhage (ring of hemorrhage around hairfollicles), hemarthrosis, bleeding gums, anemia(combined iron and folate deficiency)
Loosened teeth, glossitis, poor wound healing
(1) Neonates must receive vitamin K at birth toprevent hemorrhagic disease of the newborn.
(2) Breast milk is deficient in vitamin K.c. Coumarin derivativesd. Fat malabsorption
5. Toxicity caused by excessive intake of vitamin K isuncommon.
6. Clinical findings in vitamin K deficiency and toxicity(see Table 7-2)
[V. Water-Soluble Vitamins• The B complex vitamins, folic acid, biotin, and vitamin C are
water soluble.A. Thiamine (vitamin B1)
1. Function: cofactor in biochemical reactions that produceadenosine triphosphate (ATP) (e.g., pyruvate dehydro-genase-catalyzed conversion of pyruvate to acetyl CoA)
2. Causes of deficiencya. Chronic alcoholism (in the United States)b. Diet of nonenriched rice (in developing countries)c. Signs and symptoms mainly result from ATP
deficiency.3. Clinical findings in thiamine deficiency (Table 7-3)
Most commoncause of thiaminedeficiency in US:chronic alcoholism

Chapter 7 Nutritional Disorders 73
B. Riboflavin (vitamin B2)1. Active forms include flavin adenine dinucleotide and
flavin mononucleotide.2. Deficiency is caused by severe malnourishment.3. Clinical findings in riboflavin deficiency (see Table 7-3)
C. Niacin (vitamin B3, nicotinic acid)1. Functions
a. Active forms of niacin are oxidized nicotinamideadenine dinucleotide (NAD+) and oxidized nicotin-amide adenine dinucleotide phosphate (NADP+).
b. NAD+ and NADP+ are cofactors in oxidation-reduction reactions.
2. Causes of deficiency (pellagra) are diets deficient inniacin or the essential amino acid tryptophan, which isused to synthesize niacin. Corn-based diets are defi-cient in tryptophan and niacin.
3. Clinical findings in niacin deficiency (see Table 7-3)4. Excessive intake of niacin leads to flushing caused by
vasodilation; this reaction is an adverse effect of nicotinicacid, a lipid-lowering drug.
D. Pyridoxine (vitamin B6)1. Functions: required for transamination, heme synthesis,
and neurotransmitter synthesis2. Causes of deficiency: isoniazid, goat's milk, chronic
alcoholism3. Clinical findings in pyridoxine deficiency (see Table 7-3)
E. Cobalamin (vitamin B 12) (see Chapter 11)1. Present only in animal products (eggs, meat, dairy
products)2. Requires intrinsic factor for reabsorption in the terminal
ileum3. Functions: DNA synthesis, propionate (odd-chain fatty
acid) metabolism4. Causes of deficiency: strict vegan diet, pernicious
anemia, terminal ileal disease (e.g., Crohn's disease),bacterial overgrowth
5. Clinical findings in vitamin B 12 deficiency (see Table 7-3)F. Folic acid (see Chapter 11)
1. Present in most foods2. Function: DNA synthesis3. Causes of deficiency: dietary deficiency (e.g., elderly in-
dividuals, goat's milk); drugs (e.g., alcohol, methotrex-ate); malabsorption
4. Clinical findings in folic acid deficiency (see Table 7-3)G. Biotin
1. Cofactor in carboxylase reactions (e.g., pyruvate carboxy-lase–catalyzed conversion of pyruvate to oxaloacetate)
2. Causes of deficiency: eating raw eggs and takingantibiotics
3. Clinical findings in biotin deficiency (see Table 7-3)
Three Ds of pella-gra: dermatitis, diar-rhea, dementia
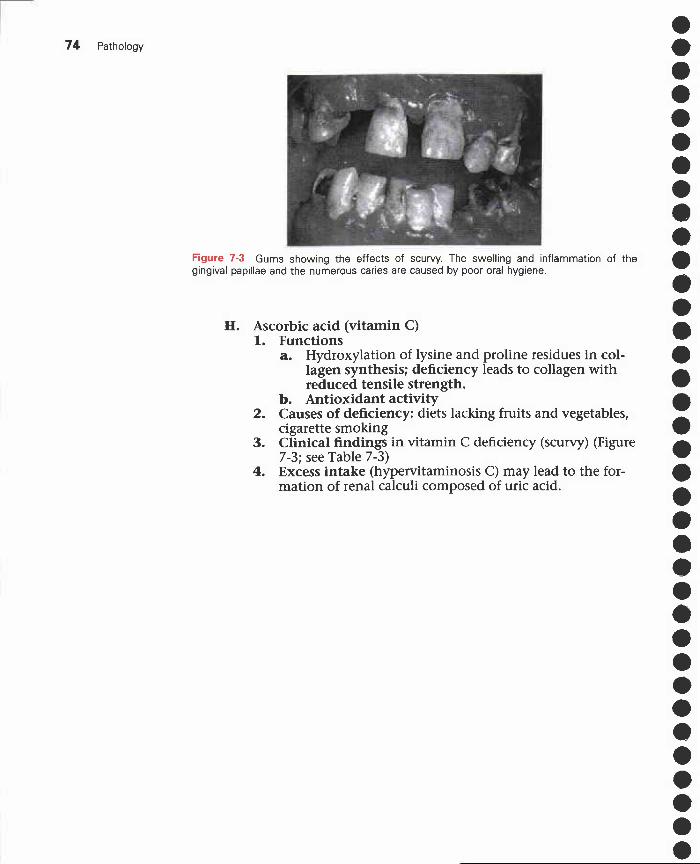
74 Pathology
Figure 7-3 Gums showing the effects of scurvy. The swelling and inflammation of thegingival papillae and the numerous caries are caused by poor oral hygiene.
H. Ascorbic acid (vitamin C)1. Functions
a. Hydroxylation of lysine and proline residues in col-lagen synthesis; deficiency leads to collagen withreduced tensile strength.
b. Antioxidant activity2. Causes of deficiency: diets lacking fruits and vegetables,
cigarette smoking3. Clinical findings in vitamin C deficiency (scurvy) (Figure
7-3; see Table 7-3)4. Excess intake (hypervitaminosis C) may lead to the for-
mation of renal calculi composed of uric acid.

I. NomenclatureA. Benign tumors
1. Suffix "oma" generally indicates a benign tumor.2. Benign tumors of epithelial origin arise from ectoderm
or endoderm (e.g., adenomas arise from glands).3. Benign tumors of connective tissue origin arise from
mesoderm (e.g., fibroma from fibroblast).
The most common benign tumor in women is aleiomyoma, which arises from the smooth muscle ofthe myometrium of the uterus. The most commonbenign tumor in men is a lipoma, which derivesfrom subcutaneous adipose tissue.
4. Tumors that are usually benigna. Mixed tumors: neoplastic cells have two different
morphologic patterns but derive from the samegerm cell layer (e.g., pleomorphic adenoma of theparotid gland).
b. Teratomas (germ cell tumors) derive from morethan one germ cell layer.(1) Sites: ovaries, testes, and midline sites (e.g.,
pineal gland, anterior mediastinum)(2) Characteristics: may contain hair, teeth, bone,
muscle, glandular epithelium, and neural tissue(Figure 8-1)
B. Malignant tumors (cancer)1. Carcinomas derive from epithelial tissue (squamous,
glandular, transitional).a. Sites of squamous cell carcinoma: oropharynx,
larynx, upper to middle esophagus, lung, cervix, skin(Figure 8-2)
b. Sites of adenocarcinoma (glandular epithelium):lung, distal esophagus to rectum, pancreas, liver,
Three variants ofcarcinoma: squa-mous cell carci-noma, adenocarci-noma, transitionalcell carcinoma
75
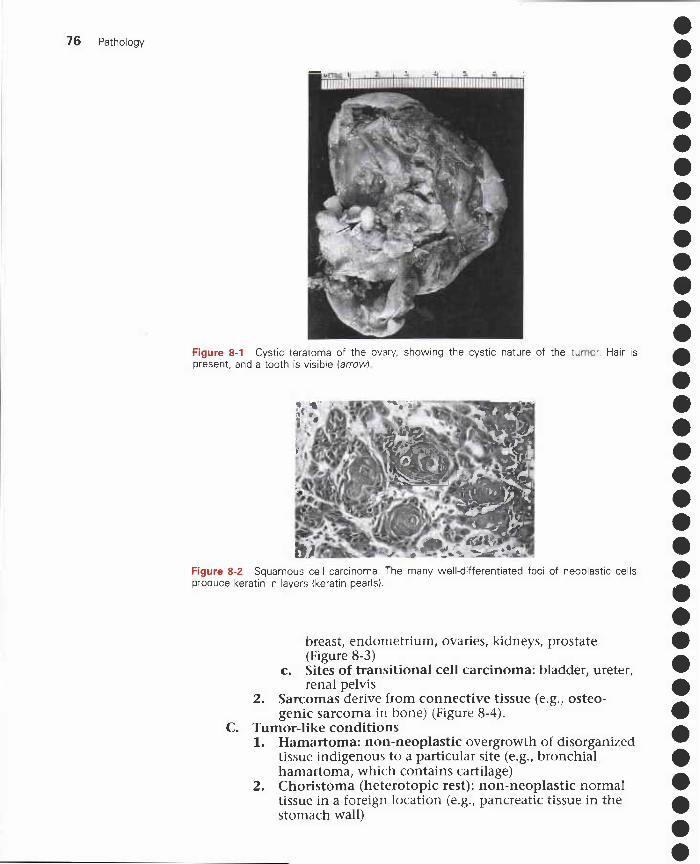
76 Pathology
Figure 8-1 Cystic teratoma of the ovary, showing the cystic nature of the tumor. Hair ispresent, and a tooth is visible (arrow)
•••••••••••••••••••••••
Figure 8-2 Squamous cell carcinoma The many well-differentiated foci of neoplastic cellsproduce keratin in layers (keratin pearls).
breast, endometrium, ovaries, kidneys, prostate(Figure 8-3)
c. Sites of transitional cell carcinoma: bladder, ureter,renal pelvis
2. Sarcomas derive from connective tissue (e.g., osteo-genic sarcoma in bone) (Figure 8-4).
C. Tumor-like conditions1. Hamartoma: non-neoplastic overgrowth of disorganized
tissue indigenous to a particular site (e.g., bronchialhamartoma, which contains cartilage)
2. Choristoma (heterotopic rest): non-neoplastic normaltissue in a foreign location (e.g., pancreatic tissue in thestomach wall)
•••
••••••••••
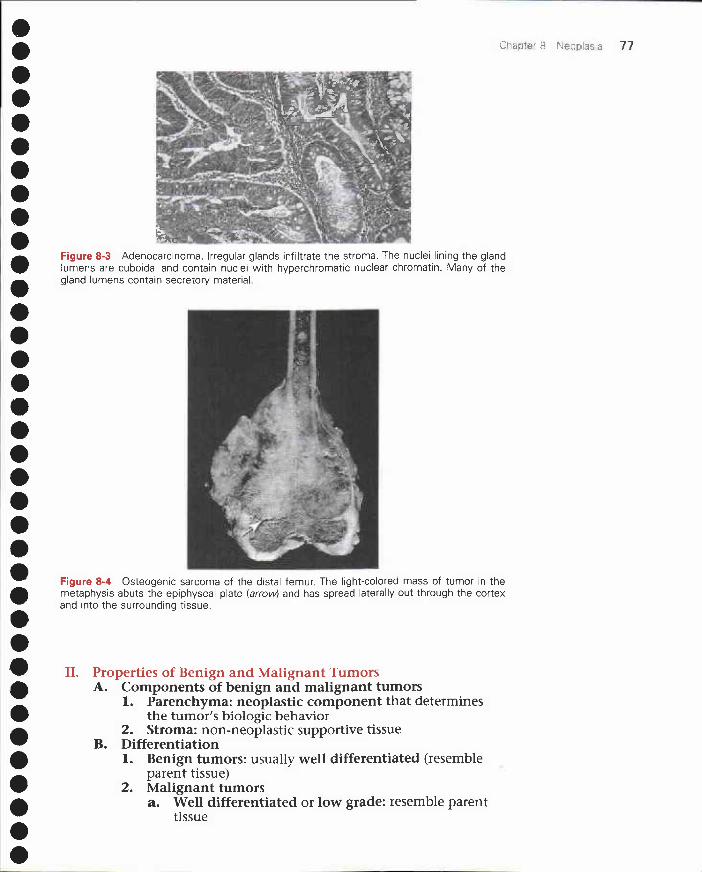
Chapter 8 Neoplasa 77
Figure 8-3 Adenocarcinoma. Irregular glands infiltrate the stroma. The nuclei lining the glandlumens are cuboidal and contain nuclei with hyperchromatic nuclear chromatin. Many of thegland lumens contain secretory material
Figure 8-4 Osteogenic sarcoma of the distal femur. The light-colored mass of tumor in themetaphysis abuts the epiphyseal plate (arrow) and has spread laterally out through the cortexand into the surrounding tissue.
•••••••••••••••••••••••••••• II. Properties of Benign and Malignant Tumors• A. Components of benign and malignant tumors
1. Parenchyma: neoplastic component that determines• the tumor's biologic behavior
2. Stroma: non-neoplastic supportive tissueB. Differentiation
1. Benign tumors: usually well differentiated (resembleparent tissue)
2. Malignant tumors
•a. Well differentiated or low grade: resemble parent
tissue•
•

78 Pathology
b. Poorly differentiated, high grade, or anaplastic:have no differentiating features
c. Intermediate grade: features are between low- andhigh-grade cancer.
C. Nuclear features1. Benign tumors
a. Nuclear:cytoplasmic ratio is close to normal.b. Mitoses have normal mitotic spindles.
2. Malignant tumorsa. Nuclear:cytoplasmic ratio is increased, and nucleoli
are prominent.b. Mitoses have normal and atypical mitotic spindles.
D. Growth rate1. Benign tumors usually have a slow growth rate.2. Malignant tumors have a variable growth rate that corre-
lates with the degree of differentiation (e.g., anaplas-tic cancers have an increased growth rate).
E. Monoclonality1. Benign and malignant tumors derive from a single pre-
cursor cell.2. Non-neoplastic proliferations derive from multiple cells
(polyclonal).
The monoclonal origin of neoplasms has beenshown by studying glucose-6-phosphate dehydroge-nase (G6PD) isoenzymes A and B in selected neo-plasms (e.g., leiomyoma of the uterus). All the neo-plastic smooth muscle cells in uterine leiomyomashave either the A or the B G6PD isoenzyme. Non-neoplastic smooth muscle proliferations in the uterus(e.g., pregnant uterus) have some cells with the Aisoenzyme and others with the B isoenzyme, indicat-ing their polyclonal origin.
F. Telomerase activity1. Telomerase function: preserves the length of telomeres
(sequences of nontranscribed DNA at the ends of chro-mosomes), which prevents gene loss after multiple celldivisions
2. Benign tumors have normal telomerase activity.3. Malignant tumors have increased telomerase activity
(do not lose genetic material after multiple cell divisions).G. Local invasion
1. Benign tumors do not invade and are usually enclosedby a fibrous capsule.• Uterine leiomyomas (10 not have a fibrous tissue
capsule.2. Malignant tumors invade tissue.3. Some tissues resist invasion (e.g., mature cartilage,
elastic tissue in arteries).
Basal cell carcino-mas of the skininvade tissue but donot metastasize.

Chapter 8 Neoplasta 79
4. Sequence of invasiona. Loss of intercellular adherence
• E-cadherin (intercellular adhesion agent) is notproduced.
b. Cell invasion occurs.(1) Cell receptors attach to laminin (glycoprotein
in the basement membrane).(2) Cells release type IV collagenase (metallopro-
teinase containing zinc), which dissolves thebasement membrane.
(3) Cell receptors attach to fibronectin in the un-derlying connective tissue.
(4) Cells produce cytokines (stimulate locomotion)and proteases (dissolve connective tissue).
(5) Cells produce factors that stimulate angio-genesi s.
H. Metastasis1. Benign tumors do not metastasize.2. Malignant tumors metastasize; pathways of dissemina-
tion include:a. Lymphatic spread to lymph nodes: usual mecha-
nism of dissemination of carcinomas
Regional lymph nodes are the first line of de-fense against the spread of a carcinoma. How-ever, if the nodal architecture is destroyed, ma-lignant cells enter the efferent lymphatics, whichempty into the bloodstream. In the bloodstream,malignant cells metastasize to distant organ sites(e.g., liver, lungs, bone).
b. Hematogenous spread: usual mechanism of dissemi-nation for sarcomas(1) Cells entering the portal vein metastasize to
the liver.(2) Cells entering the vena cava metastasize to the
lungs.
Some carcinomas have both lymphaticand hematogenous spread. Renal adeno-carcinomas commonly invade the renalvein, where the tumor has the potential forextending into the vena cava and the rightside of the heart. Hepatocellular carcinomasinvade the portal vein and the hepatic vein.Tumor obstruction of either vein producesportal hypertension, splenomegaly, anda scites .
Extranodal metas-tasis (e g., liver) hasgreater prognosticsignificance thannodal metastasis.
Routes of metasta-sis: lymphatic,hematogenous,seeding of bodycavities

80 Pathology
Figure 8-5 Metastasis to the liver. The liver contains multiple nodules that have a depressedcentral area ("umbilicated") and stellate-shaped borders.
c. Seeding: malignant cells exfoliate from a surfaceand implant and invade tissue in a body cavity; forexample:(1) Primary ovarian cancers (e.g., serous cystad-
enocarcinoma) commonly seed the omentumin the peritoneal cavity.
(2) Peripherally located lung cancers seed the pa-rietal and visceral pleura in the pleural cavity.
3. Bone metastasisa. Vertebral column
(1) Most common metastatic site in bone(2) Due to the Batson paravertebral venous
plexus, which has connections with the venacava and the vertebral bodies
b. Osteoblastic metastases(1) Radiodensities are seen on radiographs (e.g.,
prostate cancer).(2) Increased serum alkaline phosphatase indi-
cates reactive bone formation.c. Osteolytic metastases
(1) Radiolucencies are seen on radiographs (e.g.,lung cancer).
(2) Tumor may produce substances, such as pros-taglandin E, or parathyroid hormone (PTH)–related peptide (e.g., squamous cell carcinomain the lung, renal adenocarcinoma), that ac-tivate osteoclasts: danger of pathologic frac-tures and hypercalcemia
d. Pain in bone metastasis is treated with local radia-tion therapy.
4. In a given organ, the most common malignant tumor isa metastasis rather than a primary tumor (e.g., metas-tasis to the liver is more common than a primary hepato-cellular carcinoma) (Figure 8-5).
Bone metastasismay be osteoblastic(radiodense) or os-teolytic (radiolucent)

Chapter 8 Neoplasia 81
Cancer EpidemiologyA. Incidence
1. Cancers in children (in decreasing order)a. Acute lymphoblastic leukemiab. Central nervous system tumors (e.g., cerebellar
astrocytoma)c. Burkitt's lymphoma
2. Cancers in men (in decreasing order): prostate, lung,colorectal
3. Cancers in women (in decreasing order): breast, lung,colorectal
B. Mortality1. Cancer mortality in men (in decreasing order): lung,
prostate, colorectal2. Cancer mortality in women (in decreasing order): lung,
breast, colorectalC. Heredity
1. Autosomal dominant: retinoblastoma, familial adeno-matous polyposis (colon cancer)
2. Autosomal recessive: xeroderma pigmentosum3. X-linked recessive: Wiskott-Aldrich syndrome (malig-
nant lymphoma)4. Chromosomal disorders: Down syndrome (acute
leukemia)5. Familial: inactivation of BRCA1/BRCA2 suppressor genes
(breast cancer)D. Geographic area
1. Worldwide: malignant melanoma is increasing at themost rapid rate.
2. Southeast China: nasopharyngeal carcinoma secondaryto Epstein-Barr virus (EBV)
3. Japan: stomach adenocarcinoma due to smoked foods4. Southeast Asia: hepatocellular carcinoma due to hepa-
titis B virus plus aflatoxins (produced by Aspergillus)in food
5. Africa: Burkitt's lymphoma due to EBV and Kaposi'ssarcoma due to herpes simplex virus 8
E. Acquired preneoplastic disorders (Table 8-1)
IV. Carcinogenesis• Cancer is a multistep process involving gene mutations, telo-
merase activation, angiogenesis (formation of blood vessels),invasion, and metastasis.
A. Types of genes1. Regulatory genes
a. Proto-oncogenes(1) Involved in normal growth and repair; stimu-
late cells to enter the cell cycle(2) Proto-oncogene protein products: growth
factors (e.g., platelet-derived growth factor),growth factor receptors, signal transducers,nuclear transcribers
••••••••••••••••••••••••••••••••••••
Most commoncause of cancerdeath in adults: lungcancer
Actinic (solar) kera-tosis is a precur-sor of squamouscell carcinoma

82 Pathology
TABLE 8-1 Acquired Preneoplastic Disorders*
Precursor Lesion Cancer
Actinic (solar) keratosisAtypical hyperplasia of ductal epithelium of breastChronic irritation at sinus orifice, third-degree burn scarsChronic ulcerative colitisComplete hydatidiform moleDysplastic nevusEndometrial hyperplasiaGlandular metaplasia of esophagus (Barrett's esophagus)Glandular metaplasia of stomach (Helicobacter pylon)Myelodysplastic syndromeRegenerative nodules in cirrhosisScar tissue in lungSquamous dysplasia of oropharynx, larynx, bronchus, and
cervixTubular adenoma of colonVaginal adenosis (diethylstilbestrol exposure)Villous adenoma of rectum
Squamous cell carcinomaAdenocarcinomaSquamous cell carcinomaAdenocarcinomaChoriocarcinomaMalignant melanomaAdenocarcinomaAdenocarcinomaAdenocarcinomaAcute leukemiaAdenocarcinomaAdenocarcinomaSquamous cell carcinoma
AdenocarcinomaAdenocarcinomaAdenocarcinoma
*Metaplastic and hyperplastic cells become dysplastic before progressing to cancer
b. Suppressor genes (antioncogenes): protect againstunregulated cell growth
c. Apoptosis genes: regulate programmed cell death2. DNA repair genes
a. DNA repair genes produce protein products (repairenzymes) that repair nonlethal damage in regulatoryand nonregulatory genes.
b. Cells containing genes with excessive DNA damageundergo apoptosis, which prevents proliferation ofcells with multiple mutational defects.
c. Inherited defects involving DNA repair enzymesallow cells with nonlethal damage to proliferate,which increases the risk of cancer.
B. Types of regulatory gene mutations1. Point mutations: most common type of mutation2. Balanced translocations3. Other mutations: deletion, gene amplification (multiple
copies of a gene), overexpression (increase in baselinegene activity)
C. Mutations involving proto-oncogenes (Table 8-2)• Mutations activate proto-oncogenes, becoming oncogenes
(cancer-producing genes) and causing unregulated prolifera-tion of cells.
D. Mutations involving suppressor genes (Table 8-3)1. Mutations (usually point mutations) inactivate suppres-
sor genes and cause unregulated proliferation of cells.2. Retinoblastoma suppressor gene (RB)
a. Inactivation of the RB suppressor gene produces aretinoblastoma in children.
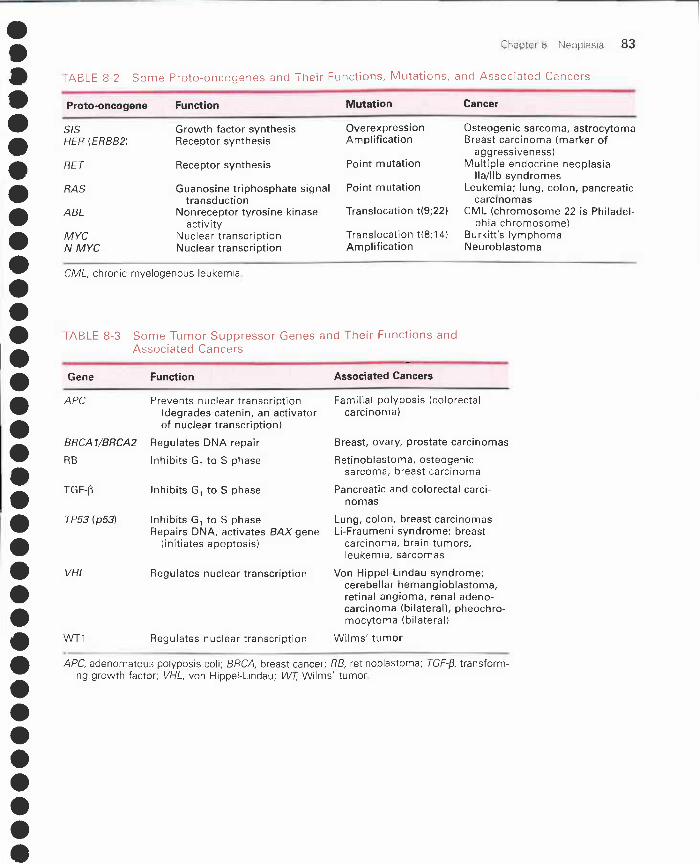
Chapter 8 Neoplasia 83
TABLE 8-2 Some Proto-oncogenes and Their Functions, Mutations, and Associated Cancers
Proto-oncogene Function Mutation Cancer
SIS Growth factor synthesis Overexpression Osteogenic sarcoma, astrocytomaHER (ERBB2) Receptor synthesis Amplification Breast carcinoma (marker of
aggressiveness)RET Receptor synthesis Point mutation Multiple endocrine neoplasia
Ilaillb syndromesRAS Guanosine triphosphate signal
transductionPoint mutation Leukemia; lung, colon, pancreatic
carcinomasABL Nonreceptor tyrosine kinase
activityTranslocation t(9;22) CML (chromosome 22 is Philadel-
phia chromosome)MYC Nuclear transcription Translocation t(8;14) Burkitt's lymphomaN- MYC Nuclear transcription Amplification Neuroblastoma
CML, chronic myelogenous leukemia
TABLE 8-3 Some Tumor Suppressor Genes and TheirAssociated Cancers
Functions and
Gene Function Associated Cancers
APC Prevents nuclear transcription(degrades catenin, an activatorof nuclear transcription)
BRCA1/BRCA2 Regulates DNA repairRB Inhibits G, to S phase
TGF-)3
Inhibits G, to S phase
TP53 (p53)
Inhibits G, to S phaseRepairs DNA, activates BAX gene
(initiates apoptosis)
VHL
Regulates nuclear transcription
WT1
Regulates nuclear transcription
Familial polyposis (colorectalcarcinoma)
Breast, ovary, prostate carcinomas
Retinoblastoma, osteogenicsarcoma, breast carcinoma
Pancreatic and colorectal carci-nomas
Lung, colon, breast carcinomasLi-Fraumeni syndrome: breast
carcinoma, brain tumors,leukemia, sarcomas
Von Hippel-Lindau syndrome:cerebellar hemangioblastoma,retinal angioma, renal adeno-carcinoma (bilateral), pheochro-mocytoma (bilateral)
Wilms' tumor
APC, adenomatous polyposis coli; BRCA, breast cancer; RB, retinoblastoma; TGF-I3, transform-ing growth factor; VHL, von Hippel-Lindau; VVT, Wilms' tumor

84 Pathology
b. Sporadic type of retinoblastoma: both normalalleles on chromosome 13 must be inactivated by apoint mutation to produce a retinoblastoma (two-hittheory).
c. Autosomal dominant type of retinoblastoma(1) One allele is inactivated in the germ cells, and
the other is normal.(2) After birth, only a single point mutation on the
remaining allele is necessary to produce a reti-noblastoma (one-hit theory).
E. Mutations involving anti-apoptosis genes1. Mutations overexpress anti-apoptosis genes, leading to
cell immortality.2. B-cell follicular lymphoma
a. BCL2 genes (chromosome 18) produce gene prod-ucts that prevent mitochondrial leakage ofcytochrome c (signal for apoptosis).
b. Translocation t(14;18) causes overexpression of theBCL2 protein product, which prevents apoptosisof B lymphocytes.
F. Hereditary DNA repair of gene defects1. Hereditary nonpolyposis syndrome (Lynch syndrome):
autosomal dominanta. Due to inactivation of DNA mismatch genes,
which normally correct mismatches of nucleotidebases
b. Associated with colorectal cancer2. Xeroderma pigmentosum: autosomal recessive
a. Defect in DNA repair of ultraviolet (UV)light–damaged skin
b. Covalent joining of two adjacent pyrimidines(usually thymines) produces pyrimidine dimers thatcannot be excised.
c. Associated with basal cell and squamous cell carci-nomas and malignant melanoma
3. Chromosome instability syndromes: autosomalrecessivea. Chromosomes are susceptible to damage by irradia-
tion or drugsb. Ataxia telangiectasia: increased incidence of malig-
nant lymphomac. Bloom syndrome and Fanconi's syndrome:
increased incidence of acute leukemia
V. Carcinogenic AgentsA. Chemical carcinogens (Table 8-4)
1. Polycyclic hydrocarbons in tobacco smoke are theprimary carcinogens in the United States.
2. Mechanismsa. Direct-acting carcinogens: contain electron-
deficient atoms that react with electron-rich atoms inDNA (e.g., alkylating agents)
Key translocations:Burkitt's lym-phoma, t(8;14);CML, t(9;22); follicu-lar lymphoma,t(14;18)
Major cancer genes:TP53 (p53) sup-pressor gene, RASoncogene
Tobacco is the agentmost responsiblefor cancer andcancer mortality inthe United States

Chapter 8 Neoplasia 85
TABLE 8-4 Chemical Carcinogens
Carcinogen Associated Cancer
Aflatoxin (from Aspergillus)
Alcohol
Alkylating agentsArsenic
AsbestosBenzeneBerylliumChromiumCyclophosphamideDiethylstilbestrolp-Naphthylamine (aniline dyes)NickelNitrosamines*Oral contraceptivesPolycyclic hydrocarbons
Silica
Hepatocellular carcinoma in association with hepa-titis B virus
Squamous cell carcinoma of oropharynx and upper/middle esophagus, pancreatic and hepatocellularcarcinomas
Malignant lymphomaSquamous cell carcinoma of skin, lung cancer, liver
angiosarcomaBronchogenic carcinoma, pleural mesotheliomaAcute leukemiaBronchogenic carcinomaBronchogenic carcinomaTransitional cell carcinoma of urinary bladderClear cell carcinoma of vaginaTransitional cell carcinoma of urinary bladderBronchogenic carcinomaStomach carcinomaBreast, cervical carcinomasSquamous cell carcinoma: oral cavity,
midesophagus, larynx, lungAdenocarcinoma: distal esophagus, pancreasTransitional cell carcinoma: urinary bladder, renal
pelvisBronchogenic carcinoma
*Nitrosamines are produced when amines in food combine with sodium nitrite, a foodpreservative.
b. Indirect-acting carcinogens: activated by the cyto-chrome P-450 system in the liver (e.g., polycyclichydrocarbons)
3. Sequence of chemical carcinogenesisa. Initiation: irreversible mutationb. Promotion: promoters (e.g., estrogen) stimulate
mutated cells to enter the cell cycle.c. Progression: development of tumor heterogeneity
(e.g., production of cells that invade or metastasize)B. Microbes
1. Viruses (Table 8-5)2. Bacteria: stomach cancer and low-grade malignant lym-
phoma of the stomach mucosa due to Helicobacter pylori3. Parasites
a. Schistosoma hematobium: squamous cell carcinoma ofthe urinary bladder
b. Clonorchis sinensis and Opisthorchis viverrini: cholan-giocarcinoma of the bile ducts
C. Irradiation1. Ionizing irradiation-induced cancers
a. Mechanism: hydroxyl free radical injury to DNAb. Acute or chronic myelogenous leukemia: seen in
radiologists and in individuals exposed to irradiationin nuclear reactors
Most commoncancer due to ioniz-ing radiation: acuteor chronic mye-logenous leukemia

RNA Viruses
HCVHTLV-1
Produces postnecrotic cirrhosisActivates TAX gene, stimulates
polyclonal T cell proliferation,inhibits TP53 (p53) suppressorgene
DNA Viruses
EBV Promotes polyclonal B-cellproliferation, which increasesrisk for t(8;14) translocation
TABLE 8-5 Oncogenic RNA and DNA Viruses
Hepatocellular carcinomaT-cell leukemia and lymphoma
Virus Mechanism
Associated Cancer
Burkitt's lymphoma, CNSlymphoma in AIDS, mixedcellularity Hodgkin's disease,nasopharyngeal carcinoma
HBV Activates proto-oncogenes, inac- Hepatocellular carcinomativates p53 suppressor gene
HPV types 16 Type 16: E6 gene product
Squamous cell carcinoma ofand 18 inhibits TP53 (p53) suppressor vulva, vagina, cervix, and
gene anus (associated with analType 18: E7 gene product
intercourse)
inhibits RB suppressor geneHSV-8 Acts via cytokines released from
Kaposi's sarcoma in AIDS
HIV and HSV
CNS, central nervous system; EBV, Epstein-Barr virus; HBV, hepatitis B virus; HCV, hepatitis Cvirus; HPV, human papillomavirus; HSV, herpes simplex virus; HTLV, human T-cell lympho-tropic virus
c. Papillary thyroid carcinoma; lung and breast cancers,liver angiosarcoma (radioactive thorium dioxideused to visualize the arterial tree), osteogenicsarcoma
2. UV light–induced cancersa. Mechanism: formation of pyrimidine dimers, which
distort DNAb. Basal cell carcinoma, squamous cell carcinoma, ma-
lignant melanoma (Figures 8-6 and 8-7)D. Physical injury
• Squamous cell carcinoma may develop in third-degreeburn scars and at the orifices of chronically draining sinuses(e.g., chronic osteomyelitis).
VI. Clinical OncologyA. Host defense against tumors
1. Humoral: via antibodies and complement2. Type IV cellular immunity
a. Cellular immunity is the most efficient mechanismfor killing cancer cells.
b. Cytotoxic CD8 T cells recognize altered class I anti-gens on neoplastic cells and destroy them.
3. Natural killer cells: direct killing and indirect killingthrough type II hypersensitivity
4. Macrophages: activated by 7-interferon
86 Pathology
Most commoncancer due to ex-cessive UV light ex-posure: basal cellcarcinoma
••••••••••••••••••000•••••••••••••••
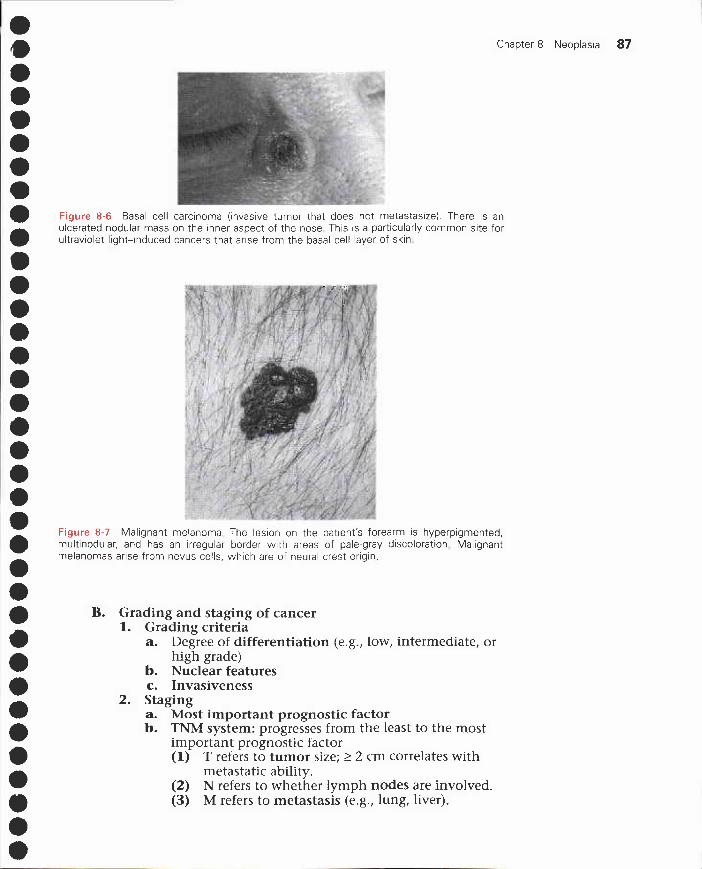
Chapter 8 Neoplasia 87
Figure 8 -6 Basal cell carcinoma (invasive tumor that does not metastasize). There is anulcerated nodular mass on the inner aspect of the nose This is a particularly common site forultraviolet light-induced cancers that arise from the basal cell layer of skin
Figure 8 -7 Malignant melanoma The lesion on the patient's forearm is hyperpigmented,multinodular, and has an irregular border with areas of pale-gray discoloration Malignantmelanomas arise from nevus cells, which are of neural crest origin.
•••••••••••••••••••••••••• B. Grading and staging of cancer
1. Grading criteria• a. Degree of differentiation (e.g., low, intermediate, or
high grade)b. Nuclear features
• c. Invasiveness
•2. Staging
a. Most important prognostic factor
• b. TNM system: progresses from the least to the mostimportant prognostic factor
• (1) T refers to tumor size; � 2 cm correlates withmetastatic ability.
(2) N refers to whether lymph nodes are involved.
•(3) M refers to metastasis (e.g., lung, liver).
••

88 Pathology
C. Cancer effects on the host1. Cachexia (wasting disease)
a. Irreversible catabolic reactionb. Tumor necrosis factor-a is secreted from host mac-
rophages and cancer cells; effects include:(1) Suppression of the appetite center(2) Decreased 13-oxidation of fatty acids for fuel
2. Anemiaa. Anemia of chronic disease: most commonb. Iron deficiency: due to gastrointestinal blood lossc. Macrocytic anemia: due to folate deficiency from
rapid tumor growthd. Myelophthisic anemia
(1) Anemia related to metastasis to bone(2) Immature hematopoietic elements (e.g., nucle-
ated red blood cells, myeloblasts) are forced intothe peripheral blood (leukoerythroblasticsmear).
3. Coagulation abnormalitiesa. Hypercoagulable state (e.g., thrombocytosis)b. Disseminated intravascular coagulation
4. Fever: usually due to infection (gram-negative sepsisfrom Escherichia coli or Pseudomonas aeruginosa)
5. Paraneoplastic syndromes: distant effects of a tumorthat are unrelated to metastasisa. Occur in 10- 15% of cancer patients: may predate
the onset of metastasisb. General features: involve multiple organ systems
and mimic metastatic diseasec. Examples: Table 8-6 lists endocrinopathies, and
Table 8-7 lists other clinical syndromes.D. Tumor markers: biologic markers used to identify tumors,
estimate tumor burden, and detect recurrence (Table 8-8)
Most commoncause of death incancer: gram-negative sepsis
Most commonparaneoplastic syn-dromes: hypercalce-mia, Cushing'ssyndrome, nonbac-terial thromboticendocarditis
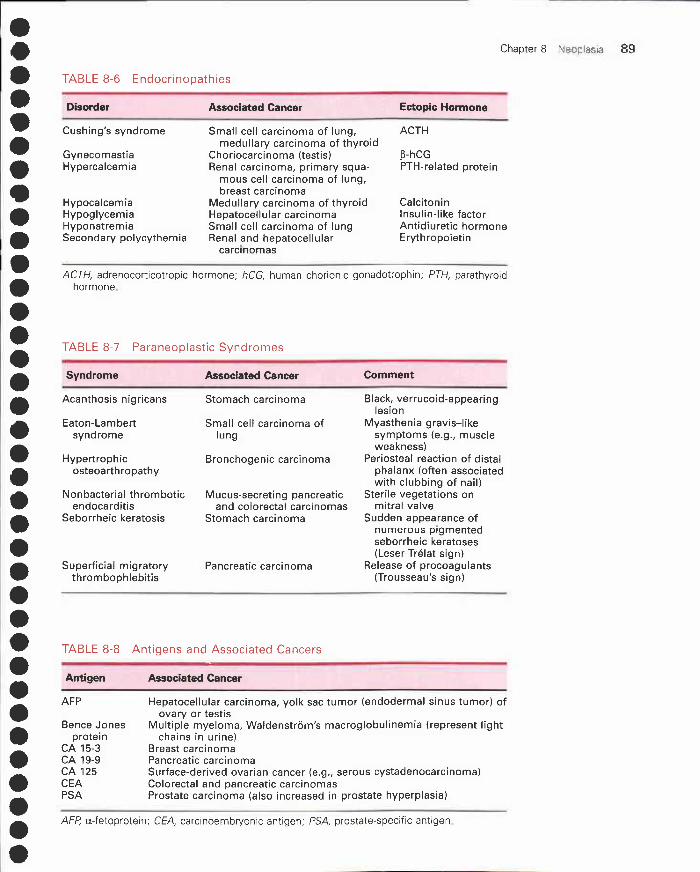
•
••••••I •
Chapter 8 Neoplasia 89
TABLE 8-6 Endocrinopathies
Disorder Associated Cancer Ectopic Hormone
Small cell carcinoma of lung, ACTHmedullary carcinoma of thyroid
Choriocarcinoma (testis) 6-hCGRenal carcinoma, primary squa- PTH-related protein
mous cell carcinoma of lung,breast carcinoma
Medullary carcinoma of thyroid
CalcitoninHepatocellular carcinoma
Insulin-like factor
Small cell carcinoma of lung
Antidiuretic hormoneRenal and hepatocellular
Erythropoietin
carcinomas
Cushing's syndrome
GynecomastiaHypercalcemia
HypocalcemiaHypoglycemiaHyponatremiaSecondary polycythemia
ACTH, adrenocorticotropic hormone; hCG, human chorionic gonadotrophin; PTH, parathyroidhormone
TABLE 8-7 Paraneoplastic Syndromes
Syndrome Associated Cancer Comment
Acanthosis nigricans Stomach carcinoma
Black, verrucoid-appearinglesion
Eaton-Lambert Small cell carcinoma of
Myasthenia gravis–likesyndrome lung symptoms (e.g., muscle
weakness)Hypertrophic Bronchogenic carcinoma Periosteal reaction of distal
osteoarthropathy phalanx (often associatedwith clubbing of nail)
Nonbacterial thrombotic Mucus-secreting pancreatic Sterile vegetations onendocarditis and colorectal carcinomas mitral valve
Seborrheic keratosis Stomach carcinoma Sudden appearance ofnumerous pigmentedseborrheic keratoses(Leser-Trêlat sign)
Superficial migratory Pancreatic carcinoma Release of procoagulantsthrombophlebitis (Trousseau's sign)
TABLE 8-8 Antigens and Associated Cancers
Antigen
Associated Cancer
AFP
Hepatocellular carcinoma, yolk sac tumor (endodermal sinus tumor) ofovary or testis
Bence Jones Multiple myeloma, WaldenstrOm's macroglobulinemia (represent lightprotein chains in urine)
CA 15-3
Breast carcinomaCA 19-9 Pancreatic carcinomaCA 125
Surface-derived ovarian cancer (e.g., serous cystadenocarcinoma)CEA
Colorectal and pancreatic carcinomasPSA
Prostate carcinoma (also increased in prostate hyperplasia)
AFP, a-fetoprotein; CEA, carcinoembryonic antigen; PSA, prostate-specific antigen
•••••••••••••••••••••••••

4 ,01Vascular Disorders
LipidsA. Lipoproteins are composed of cholesterol, kriacylglycerol,
and phospholipids.B. Lipoprotein fractions
1. Chylomicron: transports diet-derived triacylglycerol(surfaced by apolipoprotein B-48); chylomicrons areabsent during fasting.
2. Very low density lipoprotein (VLDL): transportstriacylglycerol synthesized in the liver
3. Low-density lipoprotein (LDL): transports cholesterol(surfaced by apolipoprotein B-100); derived fromcomplete hydrolysis of VLDL by capillary lipoproteinlipase
4. High-density lipoprotein (HDL): "good cholesterol";transports apolipoproteins and removes cholesterolfrom plaques
II. Lipid DisordersA. Fredrickson's classification of selected acquired and
genetic lipoprotein disorders1. Type II: familial hypercholesterolemia
a. Pathogenesis: absent or defective LDL receptors;increase in serum cholesterol
b. Lipid deposits: Achilles tendon xanthoma (diag-nostic); xanthelasma (yellow plaque on the eyelid)
2. Type III: familial dysbetalipoproteinemia ("remnantdisease")a. Pathogenesis: deficiency of apolipoprotein E;
accumulation of hydrolyzed chylomicron andintermediate-density lipoprotein remnants in theblood
b. Increase in serum cholesterol and triacylglycerol3. Type IV: familial hypertriglyceridemia; also associated
with alcoholism
Familial hypercho-lesterolemia:deficiency of LDLreceptors
90

Chapter 9 Vascular Disorders 91
a. Pathogenesis: increased synthesis or decreased ca-tabolism of VLDL
b. Clinical findings: eruptive xanthomas (yellow,papular lesions)
B. Apolipoprotein B deficiency (abetalipoproteinemia)1. Pathogenesis: deficiency of apolipoprotein B-48 and
apolipoprotein B-100, leading to deficiency of chylomi-crons, VLDL, and LDL
2. Clinical findingsa. Lipid profile: very low cholesterol and triacylglyc-
erol levelsb. Malabsorption: chylomicrons accumulate in villi
and prevent reabsorption of micelles.c. Central nervous system (CNS) disease (ataxia, reti-
nitis pigmentosa); hemolytic anemia
III. Arteriosclerosis• Arteriosclerosis is thickening and loss of elasticity of arterial
walls.A. Medial calcification (Monckeberg medial sclerosis)
1. Dystrophic calcification in the wall of muscular arteries(e.g., uterine, radial)
2. No clinical consequence unless associated withatherosclerosis
B. Atherosclerosis1. Pathogenesis: endothelial cell damage in muscular and
elastic arteries with eventual formation of a fibrousplaquea. Endothelial cell injury: risk factors include hyper-
tension, smoking tobacco, homocysteine, infec-tions (e.g., Chlamydia pneumoniae).
b. Macrophages and platelets respond by adhering todamaged endothelium and releasing cytokines.
c. Smooth muscle cells multiply, migrate to thetunica intima, form foam cells with macrophages,and produce extracellular matrix.
d. Development of fibrous cap (plaque)(1) Pathognomonic lesion of atherosclerosis(2) Components: smooth muscle, foam, and in-
flammatory cells; extracellular matrixe. Development of a complicated atheromatous
plaque: fibrous plaque becomes dystrophically cal-cified and ulcerated.
f. Platelet thrombi overlie complicated plaques.(1) Platelets adhere to areas of ulceration.(2) Plaques with a significant inflammatory com-
ponent often rupture and extrude thrombo-genic material into the lumen, leading tovessel thrombosis.
Most commonhyperlipopro-teinemia: familialhypertriglyceridemia
C-reactive protein:excellent marker ofinflammatory fibrousplaques

92 Pathology
Serum C-reactive peptide is increased inpatients with inflammatory plaques.Plaques rupture and produce vesselthrombosis, which leads to acute myo-cardial infarction (MI). C-reactive pro-tein may be a stronger predictor of cardio-vascular events than LDL.
2. Complications of atherosclerosisa. Vessel weakness (aneurysms)b. Thrombosis: acute MI (coronary artery), stroke (in-
ternal carotid artery), small bowel infarction (su-perior mesenteric artery)
c. Hypertension: proximal renal artery atherosclerosiswith activation of the renin-angiotensin-aldosterone system
d. Peripheral vascular disease(1) Increased risk of gangrene(2) Aortoiliac atherosclerosis often leads to im-
potence, atrophy of calf muscles, and pain inthe buttocks and when walking (claudication).
e. Cerebral atrophy: due to atherosclerosis involvingthe circle of Willis vessels and/or the internalcarotid artery
C. Arteriolosclerosis1. Hyaline arteriolosclerosis
a. Pathogenesis: increased protein is deposited in thevessel wall and occludes the lumen.
b. Associated conditions(1) Diabetes mellitus: nonenzymatic glycosyla-
tion of proteins in the basement membrane(2) Hypertension: increased intraluminal pres-
sure pushes plasma proteins into the wall ofthe arteriole.
2. Hyperplastic arteriolosclerosisa. Pathogenesis: acute increase in blood pressure
(e.g., malignant hypertension) causes smoothmuscle cell hyperplasia and basement membraneduplication in renal arterioles.
b. Arterioles have an "onion skin" appearance.
IV. Vessel Aneurysms• Vessel aneurysms are due to weakening of the vessel wall, fol-
lowed by dilation and a tendency to rupture.A. Abdominal aortic aneurysm
1. Usually located below the renal artery orifices2. Pathogenesis
a. Atherosclerosis: weakens vessel wall; lumenfills with atheromatous debris and blood clots(Figure 9-1).
Most commonaneurysm in men> 55 years of age:abdominal aorticaneurysm
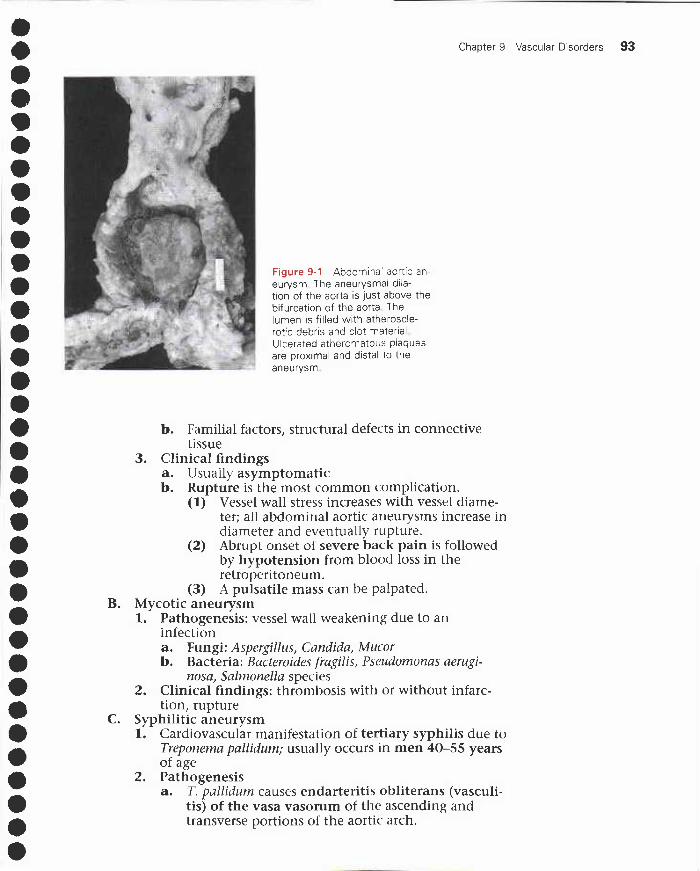
Chapter 9 Vascular Disorders 93
Figure 9 -1 Abdominal aortic an-eurysm The aneurysmal dila-tion of the aorta is just above thebifurcation of the aorta Thelumen is filled with atheroscle-rotic debris and clot materialUlcerated atheromatous plaquesare proximal and distal to theaneurysm
b. Familial factors, structural defects in connectivetissue
3. Clinical findingsa. Usually asymptomaticb. Rupture is the most common complication.
(1) Vessel wall stress increases with vessel diame-ter; all abdominal aortic aneurysms increase indiameter and eventually rupture.
(2) Abrupt onset of severe back pain is followedby hypotension from blood loss in theretroperitoneum.
(3) A pulsatile mass can be palpated.B. Mycotic aneurysm
1. Pathogenesis: vessel wall weakening due to aninfectiona. Fungi: Aspergillus, Candida, Mucorb. Bacteria: Bacteroides fragilis, Pseudomonas aerugi-
nosa, Salmonella species2. Clinical findings: thrombosis with or without infarc-
tion, ruptureC. Syphilitic aneurysm
1. Cardiovascular manifestation of tertiary syphilis due toTreponema pallidum; usually occurs in men 40-55 yearsof age
2. Pathogenesisa. T. pallidum causes endarteritis obliterans (vasculi-
tis) of the vasa vasorum of the ascending andtransverse portions of the aortic arch.

94 Pathology
b. Vessel ischemia of the medial tissue leads to dila-tion of the aorta and aortic valve ring.
3. Clinical findingsa. Aortic valve regurgitation: increased left ventricu-
lar end-diastolic volume (LVEDV) results in:(1) Left ventricular dilation and hypertrophy(2) Increased stroke volume: increased LVEDV
increases cardiac contraction (Frank-Starlingmechanism); bounding pulses.
(3) Congestive heart failureb. Brassy cough: left recurrent laryngeal nerve is
stretched by the aneurysm.c. Rupture leads to rapid death.
D. Aortic dissection1. Epidemiology
a. Men with a mean age of 40-60 years with anteced-ent hypertension (most common group)
b. Young patients with a connective tissue disorder(e.g., Marfan syndrome, Ehlers-Danlos syndrome)
Marfan syndrome is an autosomal dominantdisorder resulting in the production of weakelastic tissue. Cardiovascular abnormalitiesdominate. Dilation of the ascending aorta mayprogress to aortic dissection and/or aortic re-gurgitation. Mitral valve prolapse is the mostcommon valvular defect and is often associatedwith conduction defects causing sudden death.Skeletal defects include eunuchoid proportions(lower body length > upper body length, armspan > height) and arachnodactyly (spiderhands).
2. Pathogenesisa. Hypertension: increases vessel wall stressb. Cystic medial degeneration
(1) Pools of matrix collected between cells andtissues of the tunica media
(2) Elastic tissue fragmentationc. Intimal tear
(1) Hypertension and underlying structural weak-ness in the media
(2) Usually occurs within 10 cm of the aorticvalve
(3) Column of blood dissects under arterial pres-sure through the areas of weakness andprogresses proximally and/or distally.
Most commoncause of death inMarfan syndromeaortic dissection

S••
•
• Chapter 9 Vascular Disorders 95
• 3. Clinical findings
a a. Acute onset of severe retrosternal chest pain radi-
•ating to the back
b. Aortic valve regurgitation: due to aortic valve ring• dilation; a radiograph or echocardiogram shows
widening of the aortic valve root.• c. Loss of the upper extremity pulse: compression of
the subclavian arteryd. Rupture: usually into the pericardial sac (tampon-
•ade most common cause of death), pleural cavity,or peritoneal cavity
V. Venous System DisordersA. Overview of the venous system in the lower extremities
• 1. Superficial veins drain blood into the deep veins via
•penetrating branches; valves prevent reversal of bloodflow into the superficial system except around the
• ankles, where blood flows back to the superficial system.2. Deep veins direct blood to the heart.
B. Varicose veins: abnormally distended, lengthened, and tor-tuous veins1. Locations: superficial saphenous veins (most common
site); distal esophagus (due to portal hypertension);anorectal region (e.g., hemorrhoids)
• 2. Superficial varicosities: usually due to sentinel valve
•incompetence in the femoral vein, exacerbated bypregnancy or prolonged standing
lb C. Phlebothrombosis: thrombosis of a vein withoutinflammation1. Pathogenesis: stasis of blood flow (most common
11/cause), hypercoagulability (e.g., antithrombin IIIdeficiency)
it 2. Location: usually occurs in the deep vein of the calf
•3. Clinical findings associated with deep vein
thrombosis
• a. General: swelling, pain on dorsiflexion of foot andcompression of calf, pitting edema distal to the
• thrombosisb. Pulmonary thromboembolism: usually originates
from the femoral vein
• c. Deep venous insufficiency: postphlebiticsyndrome( 1) Stasis dermatitis: orange discoloration (he-
mosiderin) around the ankles caused byrupture of the penetrating branches Stasis dermatitis:
• (2) Varicosities develop in the superficial sign of deep vein
•system: blood flows back into the superfi-cial system.
thrombosis
I

96 Pathology
D. Thrombophlebitis: pain and tenderness along the course ofa superficial vein1. Pathogenesis
a. Intravenous cannulation of veinsb. Infection (Staphylococcus aureus)c. Carcinoma of the pancreatic head (superficial mi-
gratory thrombophlebitis): releases thrombogenicsubstances
2. Clinical findings: tender and palpable cord witherythema and edema of the overlying skin and subcu-taneous tissue
E. Superior vena cava syndrome1. Extrinsic compression of the superior vena cava from a
primary lung cancer (90% of cases): usually a smallcell carcinoma of the lung
2. Clinical findings: "puffiness" and blue to purple discol-oration of the face, arms, and shoulders; retinal hem-orrhage; stroke
F. Thoracic outlet syndrome1. Pathogenesis: compression of the neurovascular com-
partment in the neck by a cervical rib, spastic ante-rior scalene muscles, or positional change in the neckand arms
2. Clinical findingsa. Vascular (arm "falls asleep" while person is sleep-
ing) and nerve root signs (numbness, paresthesias)b. Positive Adson's test: pulse disappears when the
arm is outstretched and the patient looks to theside of the outstretched arm.
VI. Lymphatic DisordersA. Acute lymphangitis
• Usually due to cellulitis caused by Streptococcus pyogenesB. Lymphedema
1. Obstructive lymphedema: radiation damage followingradical mastectomy
2. Turner's syndrome (see Chapter 5)a. Lymphedema of hands and feet in newborns
caused by defective lymphaticsb. Dilated lymphatic channels in the neck (cystic
hygroma) produce webbed neck.
Thoracic outlet syn-drome: commonamong weight lifters

VII. Vascular Tumors and Tumor-like Conditions (Table 9-1)
TABLE 9-1 Vascular Tumors and Tumor - like Conditions
Capillary hemangio-mas in newbornsregress with age.
•••••
Chapter 9 Vascular Disorders 97
•••••••••••••, ••••••••••••••••
Tumor/Condition Clinical Findings
Angiosarcoma
Liver angiosarcoma associated with exposure topolyvinyl chloride, arsenic, thorium dioxide
Bacillary angiomatosis Benign capillary proliferation involving skin and/or
visceral organs in AIDS patientsCaused by Bartonella henselae, a gram-negative
bacillus
Capillary hemangioma Facial lesion in newborns that regresses with age
Cystic hygroma Lymphangioma in the neck associated with Turner'ssyndrome
Glomus tumor
Painful red subungual nodule in a digit
Hereditary telangiectasia (AD)
Dilated vessels on skin and mucous membranes inmouth and GI tract
Chronic iron deficiency anemia
Kaposi sarcoma Raised, red-purple discoloration (skin, GI tract)
Malignancy arises from endothelial cells or primitivemesenchymal cells associated with herpes simplexvirus type 8
Spider telangiectasia
Arteriovenous fistula (disappears when compressed)Associated with hyperestrinism (e.g., cirrhosis)
Sturge-Weber syndrome (AD)
Nevus flammeus ("birthmark") on face in distribu-tion of ophthalmic branch of cranial nerve V(trigeminal)
Von Hippel–Lindau Cavernous hemangiomas in cerebellum and retina
syndrome (AD)
Increased incidence of pheochromocytoma and bilat-eral renal cell carcinomas
AD, autosomal dominant; GI, gastrointestinal
VIII. Vasculitic DisordersA. Pathogenesis
1. Type III hypersensitivity (immunocomplex): Henoch-SchOnlein purpura
2. Type II hypersensitivity (antigen-antibody): Goodpas-ture's syndrome (anti–basement membrane antibodies)
3. Antineutrophil cytoplasmic antibodies (ANCA) acti-vate neutrophils: causes release of their enzymes andtoxic free radicals, resulting in vessel damagea. Cytoplasmic (c-ANCA): antibodies are directed
against proteinase 3 in cytoplasmic granules(e.g., Wegener's granulomatosis).
b. Perinuclear (p-ANCA): antibodies are directedagainst myeloperoxidase (e.g., microscopicpolyangiitis).
4. Direct invasion: all classes of microbial pathogensB. Clinical findings (Table 9-2)
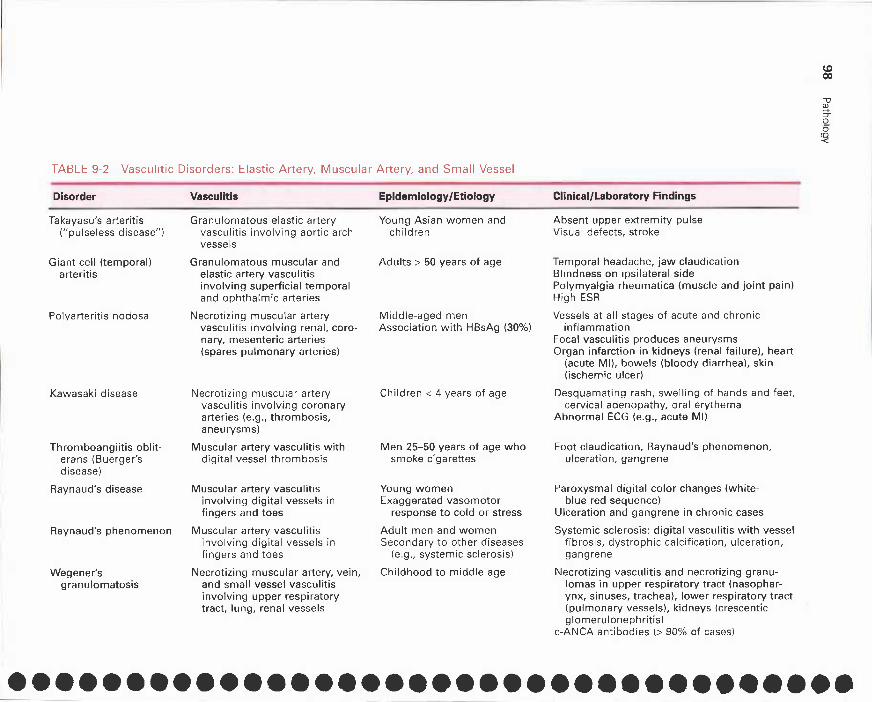
TABLE 9-2 Vasculitic Disorders: Elastic Artery, Muscular Artery, arid Small Vessel
Disorder
Vasculitis
Epidemiology/Etiology
Clinical/Laboratory Findings
co03
OO
-o
Takayasu's arteritis("pulseless disease")
Giant cell (temporal)arteritis
Polyarteritis nodosa
Kawasaki disease
Thromboangiitis oblit-erans (Buerger'sdisease)
Raynaud's disease
Raynaud's phenomenon
Wegener'sgranulomatosis
Granulomatous elastic arteryvasculitis involving aortic archvessels
Granulomatous muscular andelastic artery vasculitisinvolving superficial temporaland ophthalmic arteries
Necrotizing muscular arteryvasculitis involving renal, coro-nary, mesenteric arteries(spares pulmonary arteries)
Necrotizing muscular arteryvasculitis involving coronaryarteries (e.g., thrombosis,aneurysms)
Muscular artery vasculitis withdigital vessel thrombosis
Muscular artery vasculitisinvolving digital vessels infingers and toes
Muscular artery vasculitisinvolving digital vessels infingers and toes
Necrotizing muscular artery, vein,and small vessel vasculitisinvolving upper respiratorytract, lung, renal vessels
Young Asian women andchildren
Adults > 50 years of age
Middle-aged menAssociation with HBsAg (30%)
Children < 4 years of age
Men 25-50 years of age whosmoke cigarettes
Young womenExaggerated vasomotor
response to cold or stress
Adult men and womenSecondary to other diseases
(e.g., systemic sclerosis)
Childhood to middle age
Absent upper extremity pulseVisual defects, stroke
Temporal headache, jaw claudicationBlindness on ipsilateral sidePolymyalgia rheumatica (muscle and joint pain)High ESR
Vessels at all stages of acute and chronicinflammation
Focal vasculitis produces aneurysmsOrgan infarction in kidneys (renal failure), heart
(acute MI), bowels (bloody diarrhea), skin(ischemic ulcer)
Desquamating rash, swelling of hands and feet,cervical adenopathy, oral erythema
Abnormal ECG (e.g., acute MI)
Foot claudication, Raynaud's phenomenon,ulceration, gangrene
Paroxysmal digital color changes (white-blue-red sequence)
Ulceration and gangrene in chronic cases
Systemic sclerosis: digital vasculitis with vesselfibrosis, dystrophic calcification, ulceration,gangrene
Necrotizing vasculitis and necrotizing granu-lomas in upper respiratory tract (nasophar-ynx, sinuses, trachea), lower respiratory tract(pulmonary vessels), kidneys (crescenticglomerulonephritis)
c-ANCA antibodies (> 90% of cases)
•••••••••0•••••11•••••••••••.••0.1111••0
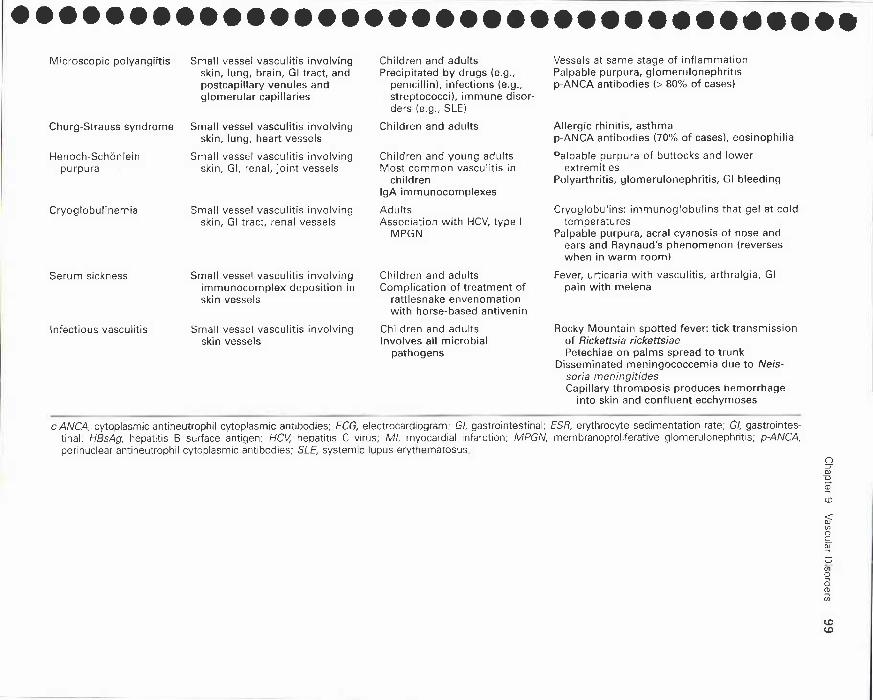
• • • • • • • • • • • • • • • • • • • • • • • • • • • ••• • 0 ••• •Microscopic polyangiitis
Serum sickness
Infectious vasculitis
Small vessel vasculitis involvingskin, lung, brain, GI tract, andpostcapillary venules andglomerular capillaries
Small vessel vasculitis involvingskin, lung, heart vessels
Small vessel vasculitis involvingskin, GI, renal, joint vessels
Small vessel vasculitis involvingskin, GI tract, renal vessels
Small vessel vasculitis involvingimmunocomplex deposition inskin vessels
Small vessel vasculitis involvingskin vessels
Children and adultsPrecipitated by drugs (e.g.,
penicillin), infections (e.g.,streptococci), immune disor-ders (e.g., SLE)
Children and adults
Children and young adultsMost common vasculitis in
childrenIgA immunocomplexes
AdultsAssociation with HCV, type I
MPGN
Children and adultsComplication of treatment of
rattlesnake envenomationwith horse-based antivenin
Children and adultsInvolves all microbial
pathogens
Vessels at same stage of inflammationPalpable purpura, glomerulonephritisp-ANCA antibodies (> 80% of cases)
Allergic rhinitis, asthmap-ANCA antibodies (70% of cases), eosinophilia
Palpable purpura of buttocks and lowerextremities
Polyarthritis, glomerulonephritis, GI bleeding
Cryoglobulins: immunoglobulins that gel at coldtemperatures
Palpable purpura, acral cyanosis of nose andears and Raynaud's phenomenon (reverseswhen in warm room)
Fever, urticaria with vasculitis, arthralgia, GIpain with melena
Rocky Mountain spotted fever: tick transmissionof Rickettsia rickettsiaePetechiae on palms spread to trunk
Disseminated meningococcemia due to Neis-seria meningitidesCapillary thrombosis produces hemorrhage
into skin and confluent ecchymoses
Churg-Strauss syndrome
Henoch-SchOnleinpurpura
Cryoglobulinemia
c-ANCA, cytoplasmic antineutrophil cytoplasmic antibodies; ECG, electrocardiogram; GI, gastrointestinal; ESR, erythrocyte sedimentation rate; GI, gastrointes-tinal; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus; MI, myocardial infarction; MPGN, membranoproliferative glomerulonephritis; p-ANCA,perinuclear antineutrophil cytoplasmic antibodies; SLE, systemic lupus erythematosus
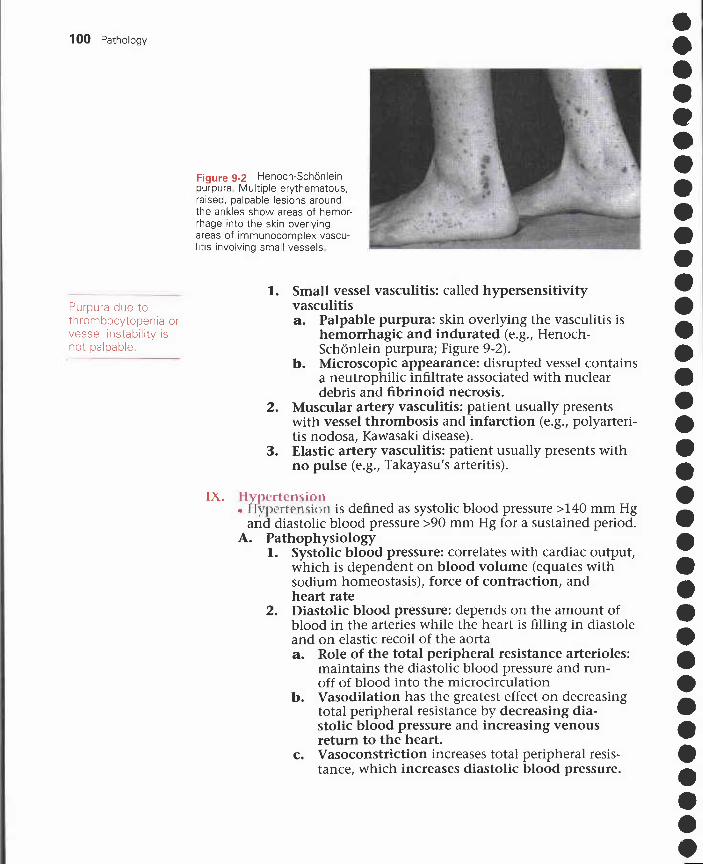
100 Pathology
Figure 9-2 Henoch-SchOnleinpurpura Multiple erythematous,raised, palpable lesions aroundthe ankles show areas of hemor-rhage into the skin overlyingareas of immunocomplex vascu-litis involving small vessels
1. Small vessel vasculitis: called hypersensitivityvasculitisa. Palpable purpura: skin overlying the vasculitis is
hemorrhagic and indurated (e.g., Henoch-SchOnlein purpura; Figure 9-2).
b. Microscopic appearance: disrupted vessel containsa neutrophilic infiltrate associated with nucleardebris and fibrinoid necrosis.
2. Muscular artery vasculitis: patient usually presentswith vessel thrombosis and infarction (e.g., polyarteri-tis nodosa, Kawasaki disease).
3. Elastic artery vasculitis: patient usually presents withno pulse (e.g., Takayasu's arteritis).
IX. HypertensionHypertension is defined as systolic blood pressure >140 mm Hgand diastolic blood pressure >90 mm Hg for a sustained period.
A. Pathophysiology1. Systolic blood pressure: correlates with cardiac output,
which is dependent on blood volume (equates withsodium homeostasis), force of contraction, andheart rate
2. Diastolic blood pressure: depends on the amount ofblood in the arteries while the heart is filling in diastoleand on elastic recoil of the aortaa. Role of the total peripheral resistance arterioles:
maintains the diastolic blood pressure and run-off of blood into the microcirculation
b. Vasodilation has the greatest effect on decreasingtotal peripheral resistance by decreasing dia-stolic blood pressure and increasing venousreturn to the heart.
c. Vasoconstriction increases total peripheral resis-tance, which increases diastolic blood pressure.
Purpura due tothrombocytopenia orvessel instability isnot palpable.

Chapter 9 Vascular Disorders 101
3. Role of sodium in hypertensiona. Excess sodium increases plasma volume, which
increases stroke volume and systolic blood pressure.b. Excess sodium enters arteriole smooth muscle cells
and opens calcium channels, causing vasocon-striction (increases diastolic blood pressure).
B. Essential hypertension1. Accounts for 95% of cases of hypertension2. Pathogenesis: genetic factors reduce renal sodium ex-
cretion; unknown factors cause vasoconstriction of arte-rioles; obesity; stress
Reduced renal sodium excretion is the primarymechanism of essential hypertension in AfricanAmericans and the elderly. Increased plasma vol-ume suppresses renin release from the juxtaglomer-ular apparatus.
C. Secondary hypertension1. Renovascular hypertension
a. Pathogenesis(1) Decreased renal arterial blood flow activates
the renin-angiotensin-aldosterone system.(2) Angiotensin II vasoconstricts peripheral-
resistance arterioles.(3) Aldosterone increases sodium retention.
b. Pathologic features(1) Elderly men: atherosclerotic plaque partially
blocks blood flow at the renal artery orifice.(2) Young to middle-aged women: fibromuscu-
lar hyperplasia occurs in multifocal areas ofthe renal artery and produces a "beaded" ap-pearance on an arteriogram.
c. Severe, uncontrollable hypertension: elevatedplasma renin activity(1) Involved kidney has increased plasma renin
activity in the renal vein.(2) Uninvolved kidney has decreased plasma
renin activity (suppressed by angiotensin IIand aldosterone) in the renal vein.
d. Epigastric bruit: due to turbulence of blood flowthrough the narrow renal artery
2. Other causes of secondary hypertension: pheochro-mocytoma, oral contraceptives, Graves' disease
D. Complications of hypertension1. Cardiovascular: left ventricular hypertrophy (most
common), acute myocardial infarction, atherosclerosis2. CNS: stroke due to an intracerebral hematoma or
rupture of a berry aneurysm
Most common typeof hypertension:essentialhypertension
Most commoncause of secondaryhypertension:renovascularhypertension

102 Pathology
3. Kidneys: nephrosclerosis (kidney of hypertension),malignant hypertension (sudden increase in bloodpressure >240/>100 mm Hg)
4. Retina: hypertensive retinopathy with arteriovenousnicking, hemorrhage of retinal vessels, exudates,papilledema (swelling of the optic disk due to increasedcerebral pressure)

lot-NOHeart Disorders
I. Ventricular HypertrophyA. Contraction against increased resistance (afterload):
causes concentric thickening of the ventricular wall1. Concentric left ventricular hypertrophy (LVH)
caused by:a. Essential hypertension (most common)b. Aortic stenosis
2. Concentric right ventricular hypertrophy (RVH)caused by:a. Pulmonary hypertensionb. Pulmonary artery stenosis
B. Volume overload (increased preload): causes dilation andhypertrophy of the ventricular wall1. Dilation and hypertrophy of the left ventricle
caused by:a. Mitral valve or aortic valve regurgitationb. Left-to-right shunting of blood, such as a ventricu-
lar septal defect (VSD)2. Dilation and hypertrophy of the right ventricle
caused by:a. Tricuspid valve regurgitationb. Pulmonary valve regurgitation
C. Consequences of ventricular hypertrophy1. Left-sided and/or right-sided heart failure2. Angina (primarily LVH)3. S4 heart sound: caused by blood entering a noncompli-
ant ventricle in late diastole
II. Congestive Heart Failure (CHF)A. Left-sided heart failure
1. Forward failure: left side of the heart is unable to ejectblood into the aorta, resulting in backup of blood intothe lungs.
Ventricular hyper-trophy is due to in-creased afterloador increasedpreload.
Left-sided heartfailure = forwardfailure –4 pulmonaryedema
103

2. Pathogenesisa. Decreased ventricular contraction (systolic dys-
function): ischemia, myocardial fibrosis, myocardi-tis, ventricular aneurysm
b. Noncompliant ventricle (diastolic dysfunction):restricted filling due to infiltration of musclewith amyloid, iron, or glycogen; concentrichypertrophy
Systolic dysfunction is characterized by a lowejection fraction (EF). The EF equals the strokevolume divided by the left ventricular end-diastolic volume. The normal value rangesfrom 55% to 70%. Diastolic dysfunction ischaracterized by normal to high EF (stiff ventri-cle) and an S4 gallop due to increased resistanceto filling in late diastole.
•••S
•••••••••
c. Increased workload: increased afterload (resis-•
tance) or preload (volume)3. Gross and microscopic findings •a. Gross: lungs are congested and exude a frothy pink
transudate (edema). •b. Microscopic: alveolar macrophages contain hemo-
siderin ("heart failure" cells).4. Clinical findings •
a. Dyspnea: difficulty breathingb. Pulmonary edema: bibasilar rales (crepitance due •
to fluid in the alveoli) •c. Left-sided S 3 heart sound: caused by volume over-
load of left ventricle; first cardiac finding in left- •sided heart failure
d. Mitral valve regurgitation: caused by stretching of •the valve ring; pansystolic murmur that increases •with intensity on expiration
e. Paroxysmal nocturnal dyspnea: choking sensa- •tion at night due to increased venous return to theheart; relieved by placing pillows under the head •(pillow orthopnea) •B. Right-sided heart failure
1. Backward failure: right side of the heart is unable to •pump blood from the venous system into the lungs, •causing blood to accumulate in the venous system.
2. Pathogenesis •a. Decreased contraction (e.g., right ventricular
infarction) or noncompliant right ventricle •(e.g., RVH) •b. Increased afterload (left-sided heart failure mostcommon cause) or increased preload (e.g., tri- •cuspid valve regurgitation) •
104 Pathology
Right-sided heartfailure = backwardfailure increase invenous hydrostaticpressure
•

Chapter 10 Heart Disorders 105
3. Clinical findingsa. Prominence of jugular veins due to increased hy-
drostatic pressureb. Right-sided 53 heart sound due to volume overloadc. Tricuspid valve regurgitation: caused by stretch-
ing of the valve ring; pansystolic murmur thatincreases in intensity with inspiration
d. Congestive hepatomegaly ("nutmeg liver")e. Dependent pitting edema and ascites
III. Ischemic Heart DiseaseA. Coronary artery blood flow
1. Oxygenates the hearta. Vessels fill in diastole.b. Increased heart rate (> 180 bpm) decreases filling
time, leading to ischemia.2. Left anterior descending coronary artery supplies the
anterior portion of the left ventricle and anterior twothirds of the interventricular septum.
3. Right coronary artery supplies the posteroinferior partof the left ventricle, posterior third of the interven-tricular septum, right ventricle, posteromedial papillarymuscle in left ventricle, and both atrioventricular andsinoatrial nodes.
B. Epidemiology1. Major cause of death in the United States; more
common in men2. Risk factors
a. Age: men 45 years, women � 55 yearsb. Family history of premature coronary artery
disease or strokec. Lipid abnormalities: low-density lipoprotein > 160
mg/dL, high-density lipoprotein < 35 mg/dLd. Smoking tobacco, hypertension, diabetes mellitus
C. Angina pectoris1. Stable angina: most common variant
a. Pathogenesis: atherosclerotic coronary arterydisease, subendocardial ischemia
b. Clinical findings: exercise-induced substernalchest pain lasting 1-15 minutes; relieved by restingand/or nitroglycerin; stress test shows STdepression.
2. Prinzmetal's anginaa. Pathogenesis: intermittent coronary artery vaso-
spasm at restb. Clinical findings: stress test shows ST elevation
(transmural ischemia).3. Unstable angina
a. Pathogenesis: severe, fixed, multivessel atheroscle-rotic disease; inflammatory plaques with or withoutplatelet nonocclusive thrombi
Most commonmanifestation of cor-onary artery disease:angina pectoris

106 Pathology
b. Clinical findings: frequent bouts of chest pain atrest or with minimal exertion; may progress toacute myocardial infarction (MI)
D. Chronic ischemic heart disease: progressive CHF resultingfrom long-term ischemic damage to myocardial tissue
E. Sudden cardiac death1. Unexpected death within 1 hour after onset of symp-
toms; diagnosis of exclusion after other causes areruled out
2. Pathogenesisa. Severe atherosclerotic coronary artery diseaseb. Absence of occlusive vessel thrombus (> 80% of
cases)c. Cause of death is ventricular fibrillation.
F. Myocardial infarction1. Pathogenesis
a. Platelet thrombus develops in response to throm-bogenic material exposed following rupture of aninflammatory atheromatous plaque.
b. Less common causes are vasculitis (polyarteritisnodosa, Kawasaki disease), cocaine use, and emboli-zation of plaque material.
2. Types of myocardial infarctiona. Transmural infarction (Q-wave infarction): in-
volves the full thickness of the myocardium;new Q waves develop in an electrocardiogram(ECG).
b. Subendocardial infarction (non-Q wave infarc-tion): involves the inner third of the myocar-dium; Q waves are absent.
3. Reperfusion injury: contraction band necrosis result-ing from hypercontraction of myofibrils in dying cellsdue to the influx of Ca2+ and 02i
4. Gross and microscopic findingsa. 0-24 hours: no gross changes until 24 hours after
MI; coagulation necrosis without neutrophil infil-trate is evident within 12-24 hours.
b. 1-3 days: pallor; myocyte nuclei disappear; neutro-phils lyse dead myocardial cells.
c. 4-7 days: red granulation tissue surrounds area ofinfarction; macrophages begin removal of ne-crotic debris for replacement by granulationtissue containing blood vessels and fibroblasts(Figure 10-1).
d. 7-10 days: necrotic area is bright yellow; granula-tion tissue and collagen formation are welldeveloped.
e. 2 months: most infarcted tissue has been com-pletely replaced by white, patchy, noncontractilescar tissue.
Rupture ofinflammatoryplaque --4 plateletthrombus --> MI

••••••••••
Chapter 10 Heart Disorders 107
Figure 10-1 Acute myocardialinfarction (day 6) in the posteriorwall of the left ventricle. Thepale area (arrow) is surroundedby a rim of dark granulationtissue
5. Clinical findingsa. Sudden onset of severe retrosternal pain
(1) Lasts > 30-45 minutes and is not relieved bynitroglycerin
(2) Radiates down the left arm into the shoul-ders or into the jaw or epigastrium; associ-ated with sweating (diaphoresis), anxiety, andhypotension
b. "Silent" acute MIs may occur in the elderly and inindividuals with diabetes mellitus.
6. Complicationsa. Arrhythmias: ventricular premature contractions
(most common); the most common cause of deathis ventricular fibrillation.
b. CHF: usually occurs within the first 24 hoursc. Rupture: most commonly occurs between days 3
and 7 (rarely occurs after day 7)(1) Anterior wall rupture with cardiac tampon-
ade is associated with thrombosis of the leftanterior descending coronary artery.
(2) Posteromedial papillary muscle rupture ordysfunction is associated with right coronaryartery thrombosis, acute onset of mitral valveregurgitation, and left-sided heart failure.
(3) Interventricular septum rupture is associatedwith left anterior coronary artery thrombosisand left-to-right shunt, causing right-sidedheart failure.
d. Mural thrombus is associated with left anterior de-scending coronary artery thrombosis; danger ofembolization.
e. Fibrinous pericarditis with or without effusion(1) Days 1-7 of transmural MI: precordial fric-
tion rub is heard because of increased vesselpermeability in the pericardium.
(2) Dressler's syndrome: autoimmune pericar-ditis that develops 6-8 weeks after an MI; au-toantibodies are directed against pericardialantigens.
•••••■••••••••••••••••••••
Most commoncause of death inacute MI: ventricularfibrillation
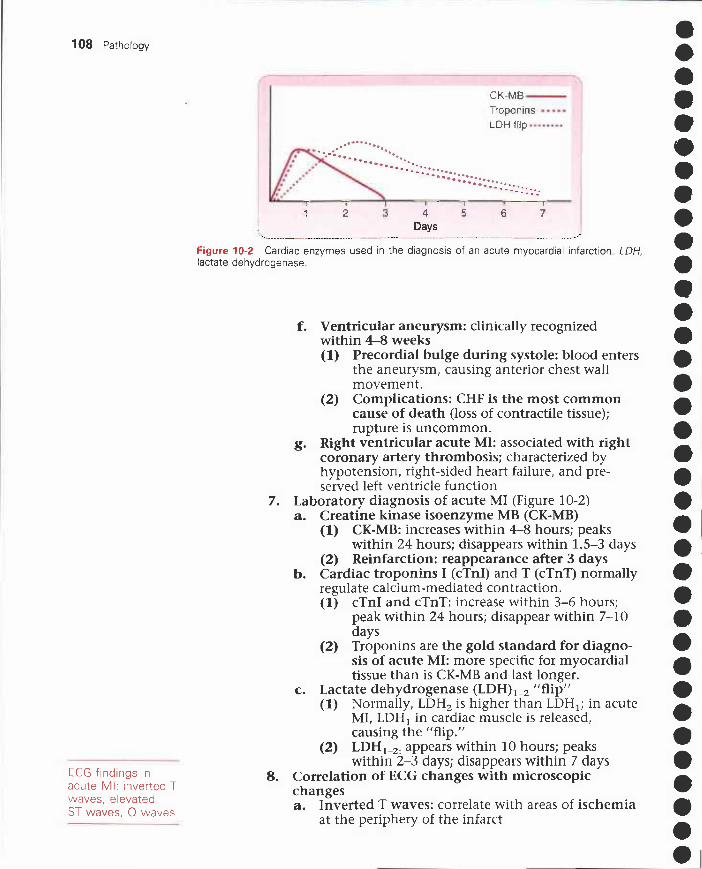
CK-MB —TroponinsLDFi rip
....... •••........ . ..............
...................... .. .
108 Pathology
1 2 3 4 5 6 7Days
Figure 10-2 Cardiac enzymes used in the diagnosis of an acute myocardial infarction LDH,lactate dehydrogenase.
f. Ventricular aneurysm: clinically recognizedwithin 4-8 weeks(1) Precordial bulge during systole: blood enters
the aneurysm, causing anterior chest wallmovement.
(2) Complications: CHF is the most commoncause of death (loss of contractile tissue);rupture is uncommon.
g. Right ventricular acute MI: associated with rightcoronary artery thrombosis; characterized byhypotension, right-sided heart failure, and pre-served left ventricle function
7. Laboratory diagnosis of acute MI (Figure 10-2)a. Creatine kinase isoenzyme MB (CK-MB)
(1) CK-MB: increases within 4-8 hours; peakswithin 24 hours; disappears within 1.5-3 days
(2) Reinfarction: reappearance after 3 daysb. Cardiac troponins I (cTnI) and T (cTnT) normally
regulate calcium-mediated contraction.(1) cTnI and cTnT: increase within 3-6 hours;
peak within 24 hours; disappear within 7-10days
(2) Troponins are the gold standard for diagno-sis of acute MI: more specific for myocardialtissue than is CK-MB and last longer.
c. Lactate dehydrogenase (LDH) 1 _2 "flip"(1) Normally, LDH2 is higher than LDH 1 ; in acute
MI, LDH, in cardiac muscle is released,causing the "flip."
(2) appears within 10 hours; peakswithin 2-3 days; disappears within 7 days
8. Correlation of ECG changes with microscopicchangesa. Inverted T waves: correlate with areas of ischemia
at the periphery of the infarct
ECG findings inacute MI: inverted Twaves, elevatedST waves, Q waves
••••••••••••••••••••••••••••••••••••

•••••••••••••••S•••••••••••••••••••
Chapter 10 Heart Disorders 109
b. Elevated ST waves: correlate with injured myocar-dial cells surrounding the area of necrosis
c. New Q waves: correlate with the area of coagula-tion necrosis
IV. Congenital Heart DiseaseA. Fetal circulation
1. Chorionic villus in the placenta: primary site for gasexchange in the fetus
2. Umbilical vein: vessel with the highest amount ofoxygen (02) in the fetal circulation
3. Inferior vena cava blood drains into the rightatrium: most blood is directly shunted into the leftatrium through the foramen ovale.
4. Superior vena cava blood: most blood is directed intothe right ventricle and from there into the pulmo-nary artery.
5. Pulmonary artery blood: most blood is shuntedthrough a patent ductus arteriosus (kept open by pros-taglandin E2) into the aorta.
6. Changes at birth: ductus arteriosus closes (becomes lig-amentum arteriosum), gas exchange occurs in thelungs, and the foramen ovale closes.
B. Congenital heart disease1. 02 saturation (Sa0 2) in shunts
a. Left-sided to right-sided heart shunts: increasedSa02 from 75% to about 80% in affected chambersand vessels
b. Right-sided to left-sided heart shunts: decreasedSa02 from 95% to about 80% in affected cham-bers and vessels
2. Left-sided to right-sided heart shuntsa. Volume overload occurs in the right side of the
heart; complications include:(1) Pulmonary hypertension(2) RVH due to increased afterload imposed on
the ventricle as a result of pulmonaryhypertension
(3) LVH due to volume overload of blood origi-nating from the right side of the heart
b. Reversal of the shunt occurs when RVH overridesleft ventricular pressure; cyanosis (Eisen-menger's syndrome) develops.
c. VSD (Figure 10-3)(1) Defect in the membranous septum; associ-
ated with cri du chat syndrome, trisomy 13,and trisomy 18
(2) Increased Sa0 2 in right ventricle and pulmo-nary artery
(3) Most spontaneously close.
Most commoncongenital heartdisease in children:VSD
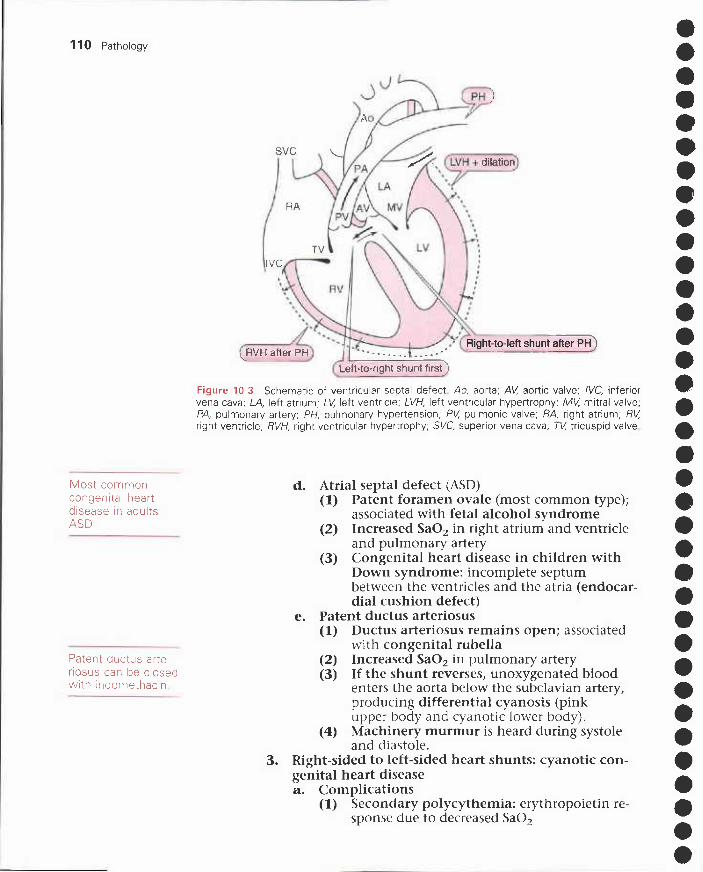
110 Pathology
A
SVCLVH + dilation
RA
TVIVC
- .......
Left-to-right shunt first)
Figure 10-3 Schematic of ventricular septal defect Ao, aorta; AV aortic valve; IVC, inferiorvena cava; LA, left atrium; LV, left ventricle; LVH, left ventricular hypertrophy; MV mitral valve;PA, pulmonary artery; PH, pulmonary hypertension; PV pulmonic valve; RA, right atrium; RVright ventricle; RVH, right ventricular hypertrophy; SVC, superior vena cava; TV, tricuspid valve
`RVH after P/-1Right-to-left shunt after PH)
d. Atrial septal defect (ASD)(1) Patent foramen ovate (most common type);
associated with fetal alcohol syndrome(2) Increased Sa02 in right atrium and ventricle
and pulmonary artery(3) Congenital heart disease in children with
Down syndrome: incomplete septumbetween the ventricles and the atria (endocar-dial cushion defect)
e. Patent ductus arteriosus(1) Ductus arteriosus remains open; associated
with congenital rubella(2) Increased Sa02 in pulmonary artery(3) If the shunt reverses, unoxygenated blood
enters the aorta below the subclavian artery,producing differential cyanosis (pinkupper body and cyanotic lower body).
(4) Machinery murmur is heard during systoleand diastole.
3. Right-sided to left-sided heart shunts: cyanotic con-genital heart diseasea. Complications
(1) Secondary polycythemia: erythropoietin re-sponse due to decreased Sa02
Most commoncongenital heartdisease in adults:ASD
Patent ductus arte-riosus can be closedwith indomethacin
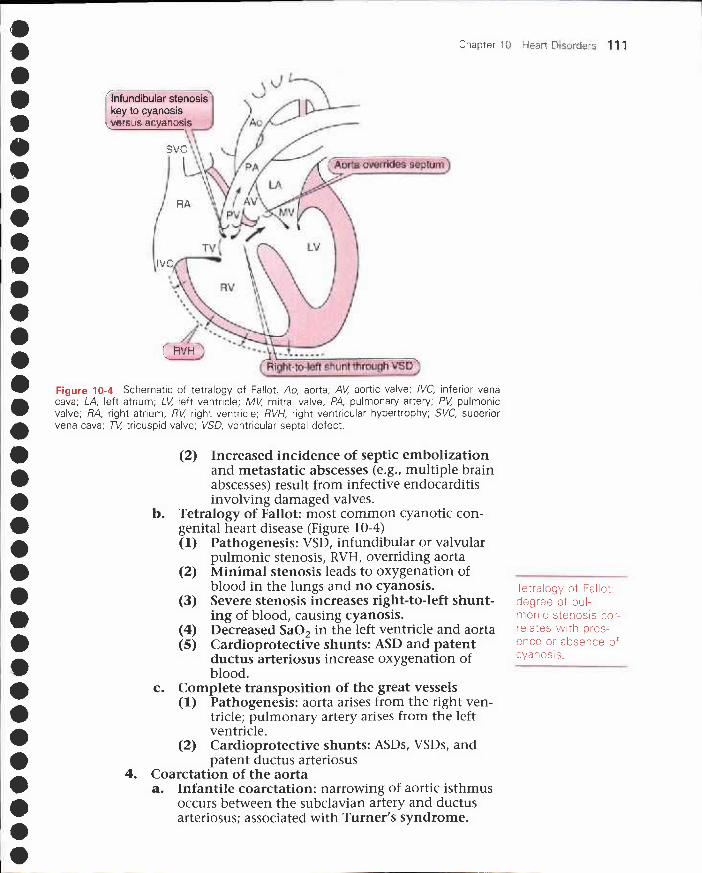
Infundibular stenosiskey to cyanosisversus acyanosis
Chapter 10 Heart Disorders 111
Figure 10 -4 Schematic of tetralogy of Fallot. Ao, aorta; AV aortic valve; IVC, inferior venacava; LA, left atrium; LV, left ventricle; MV mitral valve; PA, pulmonary artery; PV pulmonicvalve; RA, right atrium; RV right ventricle; RVH, right ventricular hypertrophy; SVC, superiorvena cava; TV, tricuspid valve; VSD, ventricular septal defect
(2) Increased incidence of septic embolizationand metastatic abscesses (e.g., multiple brainabscesses) result from infective endocarditisinvolving damaged valves.
b. Tetralogy of Fallot: most common cyanotic con-genital heart disease (Figure 10-4)(1) Pathogenesis: VSD, infundibular or valvular
pulmonic stenosis, RVH, overriding aorta(2) Minimal stenosis leads to oxygenation of
blood in the lungs and no cyanosis.(3) Severe stenosis increases right-to-left shunt-
ing of blood, causing cyanosis.(4) Decreased Sa02 in the left ventricle and aorta(5) Cardioprotective shunts: ASD and patent
ductus arteriosus increase oxygenation ofblood.
c. Complete transposition of the great vessels(1) Pathogenesis: aorta arises from the right ven-
tricle; pulmonary artery arises from the leftventricle.
(2) Cardioprotective shunts: ASDs, VSDs, andpatent ductus arteriosus
4. Coarctation of the aortaa. Infantile coarctation: narrowing of aortic isthmus
occurs between the subclavian artery and ductusarteriosus; associated with Turner's syndrome.
•••••••••••••••••••••••••••••••••••
Tetralogy of Fallot:degree of pul-monic stenosis cor-relates with pres-ence or absence ofcyanosis.

1 1 2 Pathology
b. Adult coarctation(1) Pathogenesis: constriction is distal to the lig-
amentum arteriosum; blood flow is increasedproximal to the constriction and decreaseddistally.
(2) Proximal to the constriction: increasedupper extremity blood pressure; dilation ofaorta and aortic valve ring (regurgitation);increased cerebral blood flow (danger ofaneurysms)
(3) Distal to the constriction: decreased bloodpressure in lower extremity; decreased renalblood flow (activation of renin-angiotensin-aldosterone system, causing hypertension)
(4) Collateral circulation: intercostal arteriesbypass obstruction; rib notching on inferiorpart of rib
V. Acquired Valvular Heart DiseaseA. Rheumatic fever
1. Acute, immune-mediated multisystem disease thatfollows group A streptococcal pharyngitis after an in-terval of a few weeks
2. Epidemiologya. Occurs at 5-15 years of ageb. Develops 1-5 weeks after group A streptococcal
pharyngitis3. Pathogenesis
a. Antibodies develop against group A streptococ-cal M proteins that cross-react with similar pro-teins in human tissue
b. Type II hypersensitivity reaction4. Carditis: inflammation of all layers of the heart
a. Fibrinous pericarditis: precordial chest pain withfriction rub
b. Myocarditis(1) Most common cause of death in acute
disease(2) Aschoff bodies are present: central area of fi-
brinoid necrosis surrounded by Anitschkow'scells (reactive histiocytes)
c. Endocarditis: usually involves the mitral valve(then aortic valve)(1) Sterile, verrucoid-appearing vegetations
develop along the line of closure of thevalve; embolism is uncommon (Figure 10-5).
(2) Mitral valve regurgitation and/or aorticvalve regurgitation: may result in CHF
(3) Recurrent infection of the mitral and aorticvalves leads to mitral stenosis or aorticstenosis.
Rheumatic fever:mitral regurgitationin acute attack;mitral stenosis inchronic disease

Chapter 10 Heart Disorders 113
Figure 10-5 Acute rheumaticfever Uniform, verrucoid-appearing sterile vegetationsappear along the line of closureof the mitral valve.
5. Associated symptomsa. Migratory polyarthritis: occurs in large joints
(knees) and small joints (wrists); no permanentjoint damage
b. Subcutaneous nodules: occur on extensor surfacesc. Erythema marginatum: coalescing circular rings of
erythema that develop over the trunk andextremities
d. Sydenham's chorea: reversible rapid, involuntarymovements affecting all muscles
6. Diagnosis of acute rheumatic fever: revised Jones cri-teria have been established using anti-streptolysin 0(ASO) titers and other laboratory tests.
B. Mitral stenosis1. Narrowing of the mitral valve orifice2. Etiology: most often caused by recurrent attacks of
rheumatic fever occurring over many years3. Pathophysiology: volume overload occurs in the
left atrium and lungs, leading to biventricular failure,pulmonary venous hypertension, and RVH(Figure 10-6).
4. Clinical findingsa. Heart murmur: opening snap in diastole, followed
by a mid-diastolic rumbling murmurb. Dyspnea and hemoptysis with rust-colored
sputum due to pulmonary congestion and hemor-rhage with production of heart failure cells
c. Atrial fibrillation(1) Due to left atrial dilation and hypertrophy(2) Intra-atrial thrombus develops due to stasis;
danger of systemic embolization.C. Mitral regurgitation
1. Retrograde blood flow into the left atrium duringsystole due to an incompetent mitral valve or dilatedmitral valve ring
2. Etiology: mitral valve prolapse; infective endocarditisand rupture or dysfunction of the papillary muscle
Most commoncause of mitral re-gurgitation: mitralvalve prolapse
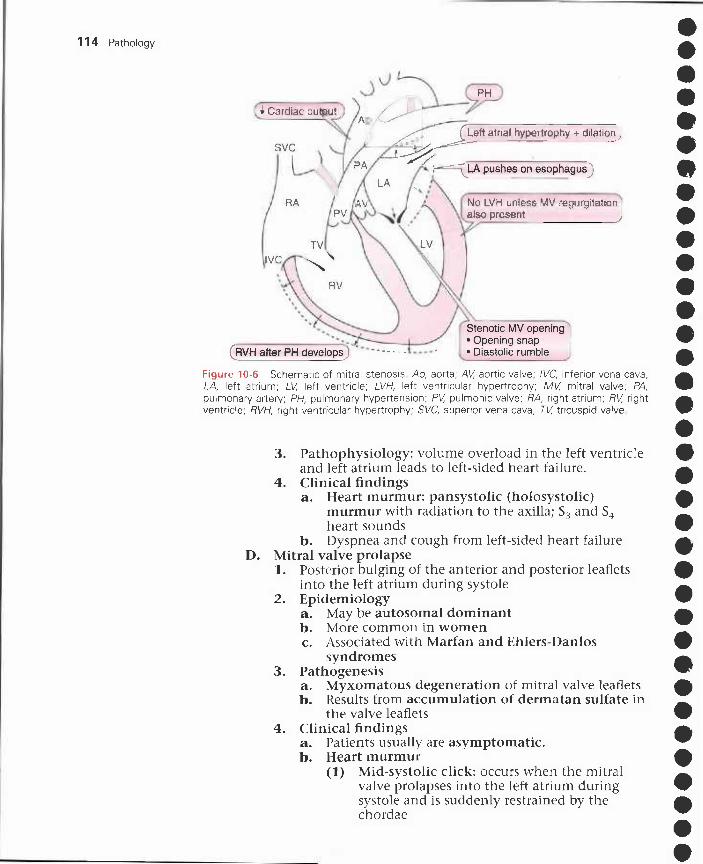
PH
Left atrial hypertrophy + dilation I
'r----CLA pushes on esophagus
1 Cardiac outputAo
SVC
PA
LA
jNo LVH unless MV regurgitationalso present
RA AVPV
LVTVIVC
RV
Stenotic MV openine• Opening snap• Diastolic rumble(RVH after PH develops)
114 Pathology
Figure 10 -6 Schematic of mitral stenosis Ao, aorta; AV aortic valve; IVC, inferior vena cava;LA, left atrium; LV, left ventricle; LVH, left ventricular hypertrophy; MV mitral valve; PA,pulmonary artery; PH, pulmonary hypertension; PV pulmonic valve; RA, right atrium; RV rightventricle; RVH, right ventricular hypertrophy; SVC, superior vena cava; TV, tricuspid valve
3. Pathophysiology: volume overload in the left ventricleand left atrium leads to left-sided heart failure.
4. Clinical findingsa. Heart murmur: pansystolic (holosystolic)
murmur with radiation to the axilla; S 3 and S4
heart soundsb. Dyspnea and cough from left-sided heart failure
D. Mitral valve prolapse1. Posterior bulging of the anterior and posterior leaflets
into the left atrium during systole2. Epidemiology
a. May be autosomal dominantb. More common in womenc. Associated with Marfan and Ehlers-Danlos
syndromes3. Pathogenesis
a. Myxomatous degeneration of mitral valve leafletsb. Results from accumulation of dermatan sulfate in
the valve leaflets4. Clinical findings
a. Patients usually are asymptomatic.b. Heart murmur
(1) Mid-systolic click: occurs when the mitralvalve prolapses into the left atrium duringsystole and is suddenly restrained by thechordae

Chapter 10 Heart Disorders 115
(2) Mid to late systolic murmur follows theclick as a result of mitral valve regurgitation.
(3) Click and murmur move closer to S i heartsound with a decrease in left ventricularpreload; causes of decreased preload include:(a) Anxiety: increased heart rate decreases
diastolic filling of left ventricle.(b) Standing: decreases venous return to the
right side of the heart(4) Click and murmur move closer to S2 heart
sound with an increase in left ventricularpreload; causes of increased preload include:(a) Reclining: increases venous return to the
right side of the heart(b) Squatting or sustained hand grip: in-
creases systemic vascular resistance,which impedes emptying of the leftventricle
c. Symptoms: palpitations, chest pain, fatigueE. Aortic stenosis
1. Obstruction to left ventricular outflow during systole2. Etiology: most often due to age-related sclerosis and
dystrophic calcification of a previously normal aorticvalve or to a congenital bicuspid aortic valve
3. Pathophysiology: reduction in the aortic valveorifice area decreases cardiac output and producesconcentric LVH.
4. Clinical findingsa. Heart murmur: crescendo-decrescendo ejection
murmur during systole; murmur radiates intocarotid arteries.
b. Angina with exercise: decreased cardiac outputleads to less filling of the coronary arteries duringdiastole; hypertrophied heart receives less blood.
c. Hemolytic anemia with schistocytes (seeChapter 11)
F. Aortic regurgitation1. Retrograde blood flow into the left ventricle due to an
incompetent valve or dilated valve ring2. Etiology: long-standing essential hypertension,
chronic rheumatic fever, infective endocarditis, aorticdissection
3. Pathophysiology: volume overload of the left ventricleincreases cardiac output and increases pulse pressure.
4. Clinical findingsa. Heart murmur: high-pitched "blowing" early
diastolic murmur is heard immediately after an S2heart sound.
b. Hyperdynamic circulation with bounding arterialpulses
•••••••••••••••••••••••••••••••••••
Most commonvalvular lesioncausing hemolyticanemia with schisto-cytes: aorticstenosis
Aortic regurgitationproduces bound-ing arterial pulses

116 Pathology
Figure 10 -7 Acute bacterial en-docarditis. Large, friable, and ir-regular vegetation (arrow) ispresent on the margin of themitral valve. Smaller vegetationsare present along the line ofclosure of the valve.
G. Tricuspid regurgitation1. Retrograde blood flow into the right atrium during
systole leads to right ventricular overload and right-sided heart failure.
2. Etiology: stretching of the tricuspid valve ring inright-sided heart failure, infective endocarditis in in-travenous drug abuse, carcinoid heart disease
3. Pathophysiology: blood regurgitates into the rightatrium and jugular venous system during systole.
4. Clinical findingsa. Heart murmur: pansystolic murmur, increasing
in intensity with inspirationb. Pulsating liver: blood regurgitates into the venous
system with systole.H. Carcinoid heart disease
1. Increased serotonin from liver metastasis of a carcinoidtumor causes valve fibrosis.
2. Results in tricuspid valve regurgitation and pulmonicvalve stenosis
I. Infective endocarditis1. Etiology
a. Streptococcus viridans is the most common cause,followed by Staphylococcus aureus, which is themost common cause in intravenous drug abuse.
b. Staphylococcus epidermidis is associated with en-docarditis involving prosthetic devices.
2. Pathogenesisa. Streptococcus viridans infects previously damaged
valves.b. Staphylococcus aureus infects normal or previously
damaged valves.3. Pathology
a. Valves involved: mitral valve (most common),aortic valve; tricuspid and aortic valves mostoften involved in intravenous drug abuse
b. Bulky vegetations embolize, producing abscessesand infarctions in distant organ sites (Figure 10-7).
c. Valve destruction leads to regurgitation murmurs.
Tricuspid valve re-gurgitation in in-travenous drugabusers is due to in-fective endocarditis.

•• Chapter 10 Heart Disorders 117
4. Clinical findingsa. Immunocomplex vasculitis: splinter hemorrhages
in nail beds, glomerulonephritisb. Fever, splenomegaly, positive blood cultures
J. Libman-Sacks endocarditis1. Associated with systemic lupus erythematosus (SLE)2. Sterile vegetations are located over the mitral valve
surface.K. Nonbacterial thrombotic endocarditis (marantic
endocarditis)1. Paraneoplastic syndrome (see Chapter 8)2. Sterile vegetations on the mitral valve are associated
with mucin-producing tumors of the colon andpancreas.
VI. Myocardial and Pericardial DisordersA. Myocarditis
1. Etiologya. Microbial pathogens: coxsackievirus, Trypano-
soma cruzi (Chagas' disease; leishmanial formsinfect cardiac muscle)
b. Acute rheumatic fever, drugs (e.g., doxorubicin),SLE
2. Pathology: endocardial biopsy is warranted if infectionis suspected; a lymphocytic infiltrate is highly pre-dictive of coxsackievirus.
3. Clinical findings: fever, chest pain, CHF, elevatedCK-MB and troponins
B. Pericarditis1. Etiology
a. Similar to myocarditisb. Coxsackievirus most common overall cause
2. Pathologya. Fibrinous type of pericardial exudate; often accom-
panied by an effusionb. Dense scar tissue with dystrophic calcification may
cause constrictive pericarditis.3. Clinical findings
a. Precordial chest pain: pain is relieved when sittingup and leaning forward and increases withinspiration.
b. Pericardial friction rub: scratchy, three-component rub (systole, early and late diastole)
c. Pericardial effusion(1) Muffled heart sounds: fluid surrounds the
heart; all chamber pressures are uniformlyincreased.
(2) Hypotension associated with pulsus para-doxus: drop in systolic blood pressure > 10mm Hg during inspiration
•••••••••••••••••••••••••••••••
Most commoncause of myocarditisand pericarditis:coxsackievirus
•••

118 Pathology
(3) Neck vein distention on inspiration: bloodis unable to enter the right atrium and refluxesinto the jugular vein (Kussmaul's sign).
d. Constrictive pericarditis: incomplete filling of thecardiac chambers due to thickening of the pari-etal pericardium(1) Etiology: tuberculosis is the most common
cause worldwide.(2) Pathophysiology: incomplete filling of all
chambers
VII. CardiomyopathyA. Congestive (dilated) cardiomyopathy
1. Most common cardiomyopathy; characterized by anenlarged heart with dilation of atria and ventricles
2. Etiology: idiopathic (most common), drugs (e.g., doxo-rubicin, cocaine), postpartum state, thiamine defi-ciency (alcoholism), hypothyroidism
3. Pathophysiology: decreased contractility with a lowEF 25%)
4. Clinical findingsa. Massive cardiac enlargement: all chambers are
dilated; echocardiography shows poor contractility.b. Biventricular CHF with bundle branch blocks and
atrial and ventricular arrhythmiasB. Hypertrophic cardiomyopathy
1. Hypertrophy of the myocardium with disproportion-ately greater thickening of the interventricular septumthan of the free left ventricular wall
2. Epidemiology: sudden death in young individuals;familial form (autosomal dominant) in young indi-viduals (50% of cases)
3. Pathophysiologya. Obstruction of blood flow is below the aortic
valve: the anterior leaflet of the mitral valve isdrawn against the asymmetrically hypertrophiedseptum as blood exits the left ventricle(Figure 10-8).
b. Aberrant myofibers and conduction system inthe interventricular septum; conduction distur-bances are responsible for sudden death.
c. Decreased diastolic filling: muscle thickening re-stricts filling.
4. Clinical findingsa. Systolic ejection type murmurb. Murmur intensity increases (obstruction worsens)
with decreased preload (e.g., standing up) and useof inotropic drugs (e.g., digitalis).
c. Murmur intensity decreases (obstruction lessens)with increased preload (e.g., reclining) and drugsdecreasing cardiac contractility (e.g., (3-blockers).
Young women withpericarditis andeffusion most likelyhave SLE
Most commoncause of suddendeath in young indi-viduals: hypertro-phic cardiomyopathy

Anterior MV leafletdrawn against septum,
Chapter 10 Heart Disorders 119•••••••••
diac output• Anterior MV leaflet
obstructs blood flow
•Asymmetric hypertrophy) • Asymmetric hypertrophyof se turn of septum
•
•
•
• C. Restrictive cardiomyopathy• 1. Characterized by decreased ventricular compliance,
usually secondary to infiltrative disease of the• myocardium
•2. Etiology
a. Tropical endomyocardial fibrosis: most commoncause worldwide
•b. Pompe's glycogenosis, hemochromatosis, amyloid-
osis, endocardial fibroelastosis in a child (thick
•fibroelastic tissue in the endocardium), sarcoidosis
3. Clinical findings: arrhythmias (conduction defects),• CHF
• VIII. Tumors of the heart
• A. Myxoma1. Benign primary mesenchymal tumor; most common
• primary adult tumor
•2. Pathology
a. Approximately 80% arise from the left atriumb. Sessile or pedunculated
•c. "Ball-valve" effect blocks the mitral valve orifice
and blocks diastolic filling of the ventricle, simu-• lating mitral valve stenosis.
Figure 10-8 Schematic of hypertrophic cardiomyopathy. Ao, aorta; AV, aortic valve; IVC,inferior vena cava; IVS, interventricular septum; LA, left atrium; LV, left ventricle; LVH, leftventricular hypertrophy; MV mitral valve; PA, pulmonary artery; PV, pulmonic valve; RA, rightatrium; RV, right ventricle; SVC, superior vena cava; TV, tricuspid valve.
••

120 Pathology
3. Clinical findingsa. Nonspecific: fever, fatigue, malaise, anemiab. Diagnosis: transesophageal ultrasound (most useful
study for viewing the left atrium)r. Complications: embolization; syncopal episodes
(block mitral valve orifice)B. Rhabdomyoma
1. Benign tumor arising from cardiac muscle2. Most common primary tumor of the heart in infants
and children; major association with tuberous sclero-sis (see Chapter 25)
••••••••••••••
Myxomas occurin adults; rhab-domyomas occurin children
•••••••••••••••••••••

Red Blood CellDisorders
I. Erythropoiesis• Erythropoiesis is the production of red blood cells (RBCs) in the
bone marrow and is dependent on the release of erythropoietinfrom the kidneys.
A. Erythropoiesis and erythropoietin (EPO)1. Stimuli for EPO release: hypoxemia, severe anemia,
left-shift of oxygen (02)-binding curve, high altitude EPO is synthesized
2. Increased 02 content suppresses EPO release (e.g.,polycythemia vera).
in peritubularcapillaries.
3. EPO accelerates erythropoiesis in the bone marrow bystimulating the erythroid stem cell to divide.
EPO increases the 0 2-carrying capacity of bloodby increasing the total number of RBCs. Epoetin alfa,a form of EPO produced by recombinant DNA tech-nology, is frequently abused by athletes to increasetheir energy level. It also is used in the treatment ofanemia associated with renal failure, chronic disease,and chemotherapy.
4. Markers of erythropoiesis: peripheral bloodreticulocytesa. Reticulocytes are newly released erythrocytes identi-
fied with supravital stains that detect thread-likeRNA filaments in the cytoplasm (Figure 11-1).
b. They require 24 hours to become mature RBCs.B. Reticulocyte count: marker of effective erythropoiesis
(bone marrow response to anemia)1. Usually reported as a percentage: normal = 0.5-1.5%2. Initial percentage is corrected for the degree of
anemia.
Reticulocyte countis a measure ofeffective erythropoi-esis and is cor-rected for thedegree of anemia.
121

122 Pathology
Figure 11-1 Peripheral bloodreticulocytes with supra yital stain(new methylene blue). Red bloodcells with thread-like material inthe cytosol represent residualRNA filaments and protein.
a. Corrected reticulocyte count = (actual Hct/45) xreticulocyte count, where 45 represents thenormal hematocrit (Hct)
b. Example: If the Hct is 15%, and the reticulocytecount is 9%, the corrected reticulocyte count is 3%(15/45 x 9% = 3%).
3. Corrected reticulocyte count > 3%: good bone marrowresponse to anemia (effective erythropoiesis), such asin autoimmune hemolytic anemia
4. Corrected reticulocyte count < 2%: poor bone marrowresponse to anemia (ineffective erythropoiesis), suchas in iron deficiency
II. Extramedullary HematopoiesisA. RBC, white blood cell (WBC), and platelet production
(hematopoiesis) occurs outside the bone marrow.1. Common sites include the liver and spleen.2. Extramedullary hematopoiesis causes hepatomegaly and
splenomegaly.B. Pathogenesis
1. Intrinsic bone marrow disease (e.g., myelofibrosis)2. Accelerated erythropoiesis (e.g., severe hemolysis in
sickle cell disease)a. Expands the bone marrow cavityb. Produces frontal bossing of the skull and "hair-on-
end" appearance on radiographs of the skull
•••••••
00
0•••••••••••
HI. Complete Blood Cell Count (CBC) and Other StudiesA. Components of a CBC: hemoglobin (lib), Hct, RBC count,
RBC indices, RBC distribution width (RDW), WBC countwith a differential count, platelet count, evaluation of theperipheral blood morphology
B. Hb, Hct, and RBC counts1. Factors affecting the normal ranges of Hb, Hct, and
RBC countsa. Age: newborns have higher normal ranges than do
infants and children, because HIDE (2c(/27 globinchains) shifts the 02-binding curve to the left.
•••••••

Microcytic anemias (MCV < 80 p.m3)Iron deficiencyAnemia of chronic diseaseThalassemiaSideroblastic anemia
Macrocytic anemias (MCV > 100 j..i.rn3)Folate deficiencyVitamin [3 12 (cobalamin) deficiency
Normocytic anemias (MCV 80-1001m3)
Corrected reticulocyte count < 2% Corrected reticulocyte count > 3% Corrected reticulocyte count > 3%
Acute blood lossEarly-stage iron deficiencyEarly-stage anemia of chronicdisease
Aplastic anemiaRenal disease
Intrinsic RBC defectMembrane defectsCongenital spherocytosisHereditary elliptocytosisParoxysmal nocturnal
hemoglobinuriaAbnormal hemoglobins
Sickle cell diseaseDeficient enzymesG6PD deficiencyPyruvate kinase deficiency
Extrinsic RBC defectAutoimmune hemolytic anemiaMicroangiopathic and
macroangiopathic hemolyticanemia
Malaria
I
Chapter 11 Red Blood Cell Disorders 123
Figure 11-2 Classification of anemia using mean corpuscular volume (MCV) An intrinsic red blood cell (RBC) defectindicates a structural or biochemical flaw in the RBCs. An extrinsic RBC defect indicates that the RBCs are structurallynormal, but that other factors cause the anemia. G6PD, glucose-6-phosphate dehydrogenase,
b. Sex: men have higher reference intervals than dowomen, because of increased testosterone levels(stimulates erythropoiesis) and lack of menses.
c. Pregnancy: pregnant women have lower referenceintervals than do nonpregnant women, because of agreater increase in plasma volume than in RBCmass.
2. Anemiaa. Decrease in Hb, Hct, or RBC concentrationb. Sign of an underlying disease rather than of a spe-
cific diagnosisc. Clinical findings are nonspecific: fatigue, dyspnea
with exertion, anorexia, insomnia, inability toconcentrate, dizziness
C. RBC indices1. Mean corpuscular volume (MCV): average volume of
RBCsa. Used to determine type of anemia: microcytic
(< 80 pin3), normocytic (80-100 mm 3 ), macrocytic(> 100 µm 3 ) (Figure 11-2)
b. Dimorphic RBC population: mixed microcytic andmacrocytic RBCs produce a normal MCV.
In anemia, satu-ration and arterialPo, are normal.
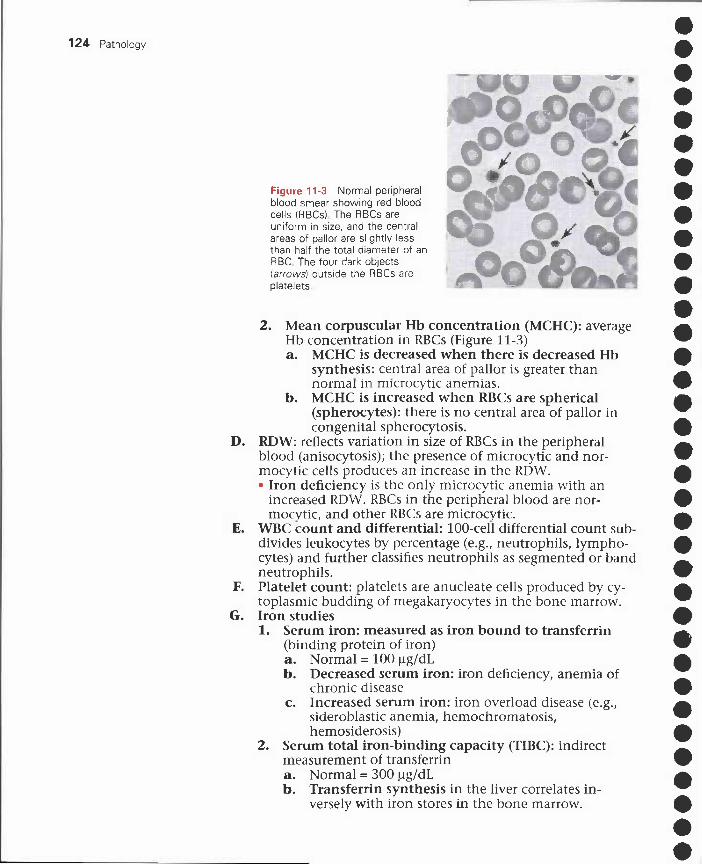
124 Pathology
Figure 11-3 Normal peripheralblood smear showing red bloodcells (RBCs) The RBCs areuniform in size, and the centralareas of pallor are slightly lessthan half the total diameter of anRBC The four dark objects(arrows) outside the RBCs areplatelets
2. Mean corpuscular Hb concentration (MCHC): averageHb concentration in RBCs (Figure 11-3)a. MCHC is decreased when there is decreased Hb
synthesis: central area of pallor is greater thannormal in microcytic anemias.
b. MCHC is increased when RBCs are spherical(spherocytes): there is no central area of pallor incongenital spherocytosis.
D. RDW: reflects variation in size of RBCs in the peripheralblood (anisocytosis); the presence of microcytic and nor-mocytic cells produces an increase in the RDW.• Iron deficiency is the only microcytic anemia with an
increased RDW. RBCs in the peripheral blood are nor-mocytic, and other RBCs are microcytic.
E. WBC count and differential: 100-cell differential count sub-divides leukocytes by percentage (e.g., neutrophils, lympho-cytes) and further classifies neutrophils as segmented or bandneutrophils.
F. Platelet count: platelets are anucleate cells produced by cy-toplasmic budding of megakaryocytes in the bone marrow.
G. Iron studies1. Serum iron: measured as iron bound to transferrin
(binding protein of iron)a. Normal = 1001.1g/dLb. Decreased serum iron: iron deficiency, anemia of
chronic diseasec. Increased serum iron: iron overload disease (e.g.,
sideroblastic anemia, hemochromatosis,hemosiderosis)
2. Serum total iron-binding capacity (TIBC): indirectmeasurement of transferrina. Normal = 300 tg/dLb. Transferrin synthesis in the liver correlates in-
versely with iron stores in the bone marrow.

Heme
Iron Ferrochetalase
Succinyl-CoA
Glycine + vitamin B6ALA synthase
Decreased in iron deficiencyI. and anemia of chronic disease
Decreased ina- and J3-thalassemia
Iron Globin chains (a, 13, 8, y)Hemoglobin
ALA Protopo phyrin IXMitochondria
ALA dehydrase Cytosol Defect in heme synthesisin sideroblastic anemias
Porphobilinogen
•
•
Chapter 11 Red Blood Cell Disorders 125
•••••••••
Figure 11-4 Pathophyslology of microcytic anemias. 8-ALA, aminolevulinic acid.••
• (1) Decreased iron stores in the bone marrow
•cause increased liver synthesis of transferrin:serum TIBC is increased (e.g., iron deficiency).
• (2) Increased iron stores in the bone marrowcause decreased liver synthesis of transferrin:
• serum TIBC is decreased (e.g., anemia ofchronic disease, iron overload diseases).
3. Percent saturation of iron: percentage of transferrin oc-• cupied by iron
•a. Normal = 100/300 = 33%b. Decreased iron saturation: iron deficiency, anemia
• of chronic diseasec. Increased iron saturation: iron overload diseases
• 4. Serum ferritin: primary iron storage protein
Ia. Decreased serum ferritin: iron deficiencyb. Increased serum ferritin: anemia of chronic
• disease, iron overload diseases
• IV. Microcytic Anemias
•A. Pathogenesis: defects in Hb synthesis (Figure 11-4)
1. Defects in heme (iron + protoporphyrin) synthesis
• (e.g., iron deficiency, anemia of chronic disease, sidero-
•blastic anemias)
2. Defects in globin chain synthesis (e.g., a-thalassemia,
• (3-thalassemia)B. Iron-deficiency anemia
• 1. Most common anemia
•2. Bleeding is the most common cause.
a. Newborns and children: bleeding Meckel's
• diverticulumb. Males < 50 years of age: bleeding peptic ulcer (duo-
• denal, gastric)
•c. Females < 50 years of age: menorrhagia
Iron deficiency ismost commonlycaused by bleeding.
•
126 Pathology
Figure 11-5 Peripheral bloodsmear in iron deficiency anemiaThe enlarged central area ofpallor in the red blood cell(arrow) indicates a decrease inhemoglobin synthesis, whichis characteristic of the microcyticanemias The mean corpuscularhemoglobin concentration isdecreased
d. Males and females > 50 years of age: gastrointesti-nal bleeding (polyp, colorectal cancer)
3. Clinical and laboratory findingsa. Glossitis, gastritis, spooning of the nails
(koilonychia)
Plummer-Vinson syndrome is due to severeiron deficiency. Characteristic findings includeformation of a partially obstructing esophagealweb, gastritis, achlorhydria (absent acid), glossi-tis, and spoon nails.
b. Laboratory findings(1) Iron studies: decreased serum iron, % satura-
tion, and ferritin; increased TIBC
The stages of iron deficiency, in se-quence, are absent iron stores; decreasedserum ferritin, serum iron, and % satura-tion; increased TIBC; normocytic anemia;and microcytic anemia.
(2) Thrombocytosis (common in chronic iron defi-ciency), increased RDW, decreased MCHC(Figure 11-5)
C. Anemia of chronic disease1. Etiology: chronic inflammation (e.g., rheumatoid arthri-
tis, tuberculosis), alcoholism, cancer2. Pathogenesis: iron is trapped in bone marrow macro-
phages and is unable to be used to synthesize Hb.3. Laboratory findings
a. Hb: rarely < 9 g/dLb. Iron studies: decreased serum iron, TIBC, and % sat-
uration; increased serum ferritinD. a -Thalassemia
1. Autosomal recessive disorder2. Usually seen in Southeast Asians and in African
Americans
Anemia of chronicdisease: TIBC,T serum ferritin

Chapter 11 Red Blood Cell Disorders 127
3. Pathogenesis: decrease in a-globin chain synthesis onchromosome 16 due to gene deletionsa. One-gene deletion: silent carrier; no anemiab. Two-gene deletions: a-thalassemia trait
(1) Mild anemia: increased RBC count (unknowncause)
(2) Hb electrophoresis: normal; decrease ina-globin chain synthesis proportionately de- cc-Thalassemia traitcreases the synthesis of HbA (2a/2(3), HbA2 normal Hb
(2a/26), and HbF (2a/2y). electrophoresis
c. Three-gene deletions: HbH disease (four (3-chains)(1) Severe hemolytic anemia: due to the presence
of (3-chain inclusions(2) Hb electrophoresis: detects HbH
d. Four-gene deletions: hemoglobin Bart's disease(four y-chains)(1) Incompatible with life; increased incidence of
spontaneous abortion(2) Hb electrophoresis: detects Bart's Hb
E. 0-Thalassemia1. Autosomal recessive disorder2. Usually seen in African Americans and individuals of
Mediterranean descent3. Pathogenesis
a. Decrease in 13-globin chain synthesis on chromo-some 11(1) Mild anemia: most often due to DNA splicing
defects(2) Severe anemia: due to nonsense mutation
with formation of a stop codonb. Normal synthesis of ei-, 8-, and y-globin chains:
some p-globin chain synthesis is designated 0+;no p-globin chain synthesis is designated 13°.
c. 13-Thalassemia minor (0/(3 +) 3-Thalassemia(1) Mild anemia minor: T HbA2,
(a) Mild protective effect against Plasmodium T HbF
falciparum malaria(b) Teardrop cells and target cells in the pe-
ripheral blood(c) Increased RBC count
(2) Hb electrophoresis: decreased HbA (2a/213),increased HbA2 (2a/28) and HbF (20/12y)
d. 13-Thalassemia major (Cooley's anemia; (r/fror 13-713°)(1) Severe hemolytic anemia: increased transfu-
sion requirement, danger of iron overload(hemosiderosis)
(2) Extramedullary hematopoiesisF. Sideroblastic anemias
• Group of anemias with a defect in heme synthesis in themitochondria

128 Pathology
1. Called sideroblastic because iron accumulates in themitochondria of nucleated RBCs in the bone marrowand forms ringed sideroblasts
2. Etiologya. Alcoholism
(1) Most common cause of sideroblastic anemia(2) Mitochondrial poison that damages heme
synthesis in the mitochondriab. Pyridoxine (vitamin B6) deficiency
(1) Vitamin B6 is a cofactor for 8-aminolevulinicacid (8-ALA) synthase, which catalyzes therate-limiting reaction in heme synthesis.
(2) Most common cause of deficiency is isoniazid,which is used in the treatment of tuberculosis.
c. Lead poisoning(1) Causes
(a) Pica (abnormal craving) for lead-basedpaint
(b) Working with or living near a source oflead (e.g., automobile or battery factory)
(c) Pottery painting (use of lead-based paints)(2) Pathogenesis: lead denatures enzymes in heme
synthesis.(a) Ferrochelatase: is unable to combine iron
with protoporphyrin to form heme(b) ALA dehydrase: cannot convert 8-ALA to
porphobilinogen(3) Clinical findings
(a) Abdominal colic with diarrhea(b) Encephalopathy in children: 8-ALA
(proximal to ALA dehydrase block)damages neurons, increases vessel perme-ability (cerebral edema), and causesdemyelination.
(c) Growth retardation in children: lead de-posits in the epiphysis of growing bone;radiograph shows increased density in theepiphyses.
(d) Peripheral neuropathy in adults: foot-drop (peroneal nerve palsy)
(4) Laboratory findings(a) Increased lead levels in blood and urine(b) Coarse basophilic stippling of RBCs: per-
sistence of ribosomes due to denaturationof ribonuclease (Figure 11-6)
3. Laboratory findings in the sideroblastic anemiasa. Iron overload: increased serum iron, % saturation,
and ferritin; decreased TIBCb. Ringed sideroblasts in the bone marrow
(Figure 11-7)G. Microcytic anemias: laboratory findings are listed in
Table 11-1.
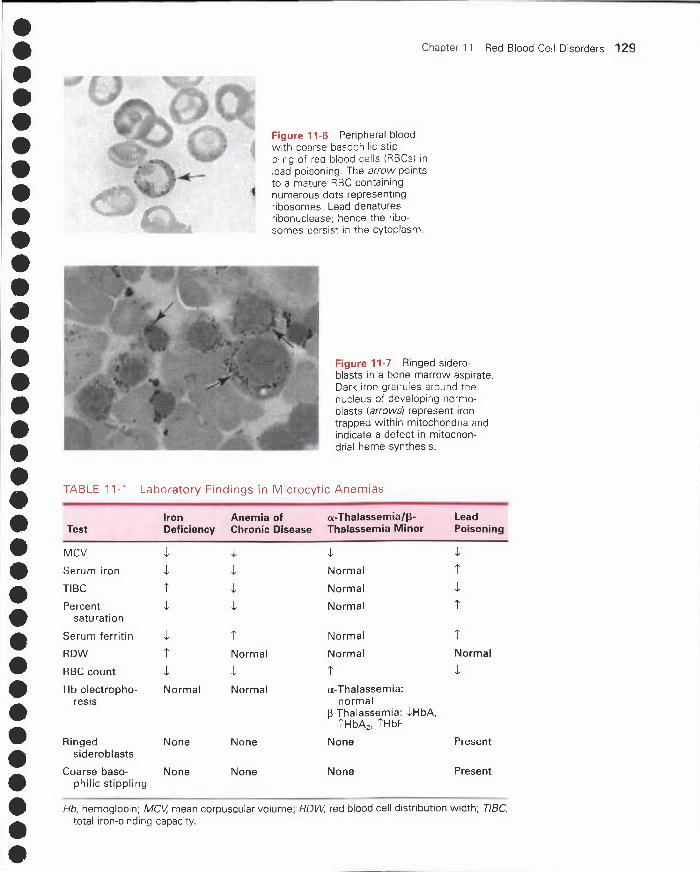
4
Chapter 11 Red Blood Cell Disorders 129
Figure 11 -6 Peripheral bloodwith coarse basophilic stip-pling of red blood cells (RBCs) inlead poisoning The arrow pointsto a mature RBC containingnumerous dots representingribosomes Lead denaturesribonuclease; hence the ribo-somes persist in the cytoplasm
Figure 11 -7 Ringed sidero-blasts in a bone marrow aspirate.Dark iron granules around thenucleus of developing normo-blasts (arrows) represent irontrapped within mitochondria andindicate a defect in mitochon-dria! heme synthesis.
TABLE 11-1 Laboratory Findings in Microcytic Anemias
TestIronDeficiency
Anemia ofChronic Disease
et-Thalassemia/13-Thalassemia Minor
LeadPoisoning
MCV
Serum iron1
14,
1' Normal
1
T
TIBC 1 Normal 1
Percentsaturation
1 Normal iSerum ferritin 4. NormalRDW Normal Normal Normal
RBC count 4. 1. .1.
Hb electropho- Normal Normal a-Thalassemia:resis normal
13-Thalassemia: 4.HbA,THbA2, THbF
Ringedsideroblasts
None None None Present
Coarse baso-philic stippling
None None None Present
Hb, hemoglobin; MCV, mean corpuscular volume RDW, red blood cell distribution width; TIBC,total iron-binding capacity.

130 Pathology
V. Macrocytic AnemiasA. Major causes of macrocytic anemia are folate deficiency
and vitamin B 12 deficiency.B. Folate deficiency
1. Pathogenesisa. Defect in DNA synthesis: delayed nuclear matura-
tion results in large nucleated cells throughout thebody.
b. Ineffective erythropoiesis(1) Enlarged hematopoietic cell precursors (mega-
loblastic) outside the bone marrow sinusoidsare destroyed by macrophages or undergoapoptosis.
(2) Pancytopenia in peripheral blood: anemia,neutropenia, and thrombocytopenia
2. Folate metabolisma. Present in the polyglutamate form in food (fruits,
vegetables, and grains)b. Converted to monoglutamates by intestinal
conjugasec. Monoglutamate is reabsorbed in the jejunum.d. Reabsorbed folate circulates in blood as methyl-
tetrahydrofolate: only 3-4 months' supply of folatein the liver.
Most commoncause of a macro-cytic anemia. folatedeficiency
Vitamin B 12 removes the methyl group frommethyltetrahydrofolate and transfers themethyl group to homocysteine, which is con-verted to methionine. Deficiency of either fo-late or vitamin B12 causes an increase in serumhomocysteine levels, which increases the riskof vessel thrombosis (see Chapter 9).
3. Etiologya. Dietary deficiency (most common cause)
(1) Alcoholism: inadequate folate in alcohol prod-ucts (except beer); direct inhibition of jejunalreabsorption
(2) Pregnant women not taking prenatal vita-mins; vitamins normally contain folate andiron, both of which are used by the fetus.
(3) Elderly individuals with poor dietb. Drugs
(1) Phenytoin: inhibits intestinal conjugase (pre-vents folate reabsorption)
(2) Alcohol and oral contraceptives: prevent re-absorption of monoglutamate in the jejunum
(3) Methotrexate and trimethoprim: inhibit di-hydrofolate reductase
Dihydrofolate re-ductase is inhibitedby methotrexate andtrimethoprim.

Chapter 11 Red Blood Cell Disorders 131
Dihydrofolate reductase reduces dihydro-folate to tetrahydrofolate, which is re-quired for DNA synthesis. Drugs that blockthe enzyme cause macrocytic anemia. Leu-covorin (folinic acid), a fully activatedform of folic acid, prevents these macro-cytic anemias.
c. Malabsorption: disorders affecting the jejunum(e.g., celiac disease) prevent folate reabsorption
4. Clinical finding: glossitis (smooth, sore tongue; atrophyof papillae)
5. Laboratory findingsa. Peripheral blood findings: pancytopenia, oval-
shaped macrocytes, hypersegmented neutrophils(� 5 nuclear lobes) (Figure 11-8)
b. Decreased serum folate and RBC folate (best screen-ing test)
c. Increased serum homocysteineC. Vitamin B 12 deficiency
1. Vitamin B 12 is present in meat, eggs, and dairyproducts.
2. Pathogenesis: same as for folate deficiency (seesection V B)
3. Vitamin B 1 , metabolisma. Vitamin B 12 is bound to R factor in saliva; R factor
prevents acid destruction of vitamin B1,.b. Parietal cells in the body and fundus of the
stomach synthesize intrinsic factor (IF) and alsoproduce acid for the digestive process.
c. Pancreatic enzymes cleave off R factor, allowingvitamin B 12 to bind to IF.
d. Vitamin B 12-IF complex is reabsorbed in the termi-nal ileum.
e. After reabsorption, vitamin B 12 is delivered to meta-bolically active cells or stored in the liver (6- to9-year supply).
••••••••••S••••••
•••••••••••••••
Figure 11-8 Peripheral blood inmegaloblastic anemia showingthe hypersegmented neutro-phil (arrow) with nine lobes Neu-trophils normally have less thanfive nuclear segments Hyper-segmented neutrophils are excel-lent markers of folate andvitamin [3 12 deficiency The en-larged, egg-shaped red bloodcells (macro-ovalocytes) charac-teristic of macrocytic anemiasare associated with problems inDNA synthesis.

132 Pathology
TABLE 11-2 Clinical and Laboratory Findings in Vitamin B 12 and FolateDeficiencies
Laboratory/Clinical FindingPerniciousAnemia
Other Vitamin B„Deficiencies
FolateDeficiency
Achlorhydria Present Absent AbsentAutoantibodies Present Absent AbsentChronic atrophic gastritis Present Absent AbsentGastric carcinoma risk T None NoneHypersegmented neutrophils Present Present PresentMean corpuscular volume T T TNeurologic disease Present Present NonePancytopenia Present Present PresentPlasma homocysteine T T ISerum gastrin level T Normal NormalUrine methylmalonic acid I T Normal
4. Etiologya. Pure vegan diet: diet lacking meat, eggs, and dairy
productsb. Pernicious anemia
(1) Autoimmune destruction of parietal cells: an-tibodies are directed against parietal cells.
(2) Consequences: absence of IF; achlorhydria (noacid is produced); type A chronic atrophicgastritis
c. Chronic pancreatitis: no enzymes to cleave off Rfactor
d. Fish tapeworm (Diphyllobothrium latum) infestation,bacterial overgrowth (destroy vitamin B12-IFcomplex), terminal ileal disease (e.g., Crohn'sdisease)
5. Clinical findings (Table 11-2)a. Similar to folate deficiencyb. Findings unique to pernicious anemia
(1) Maldigestion of food: due to chronic gastritisand achlorhydria
(2) Increased incidence of gastric cancerc. Subacute combined degeneration of the spinal
cord: demyelination of posterior columns and lateralcorticospinal tract(1) Posterior column signs: unsteady gait, lack of
proprioception (joint sense), loss of vibratorysensation
(2) Lateral corticospinal tract signs: spasticity,positive Babinski's sign
Vitamin B 12 defi-ciency producessubacute combineddegeneration inthe spinal cord.

Chapter 11 Red Blood Cell Disorders 133
Vitamin B12 is involved in propionatemetabolism. It serves as a cofactor in thereaction that converts methylmalonyl CoAto succinyl CoA. Vitamin B 12 deficiencyleads to an accumulation of methylma-Ionic acid and propionic acid, which pro-duce demyelination in the spinal cord.Folate is not involved in propionate metab-olism, and folate deficiency does not leadto neurologic abnormalities.
6. Laboratory findings (see Table 11-2)a. Decreased serum vitamin B12b. Increased serum homocysteine and urine methyl-
malonic acidc. Peripheral blood findings: similar to folate
deficiencyd. Hypergastrinemia: present only in pernicious
anemia due to loss of negative feedback of gastricacid
e. Schilling test(1) Identifies the cause of vitamin B 12 deficiency(2) Example: reabsorption of vitamin B 12 after ad-
ministration of IF confirms the diagnosis ofpernicious anemia.
VI. Normocytic Anemias with Corrected Reticulocyte Count < 2%A. Acute blood loss
1. Loss of whole blood causes signs of volume depletionwithout an initial change in Hb and Hct.a. The bone marrow requires at least 5-7 days before
reticulocytosis occurs.b. Beyond 7 days, loss of blood is associated with
reticulocytosis.2. Plasma is replaced first, and the plasma replacement
reveals the RBC deficit; infusion of 0.9% normal salineimmediately indicates the deficit.
3. Major causes of blood loss: bleeding peptic ulcer(most common), trauma (e.g., ruptured spleen), divertic-ulosis, angiodysplasia
B. Aplastic anemia1. Pathogenesis: suppression or destruction of the trilin-
eage myeloid stem cells2. Etiology: most cases are idiopathic.
a. Drugs (e.g., chloramphenicol, chemotherapy agents)b. Infection (e.g., parvovirus B19, which also produces
pure RBC aplasia)c. Chemicals (e.g., benzene), irradiation
3. Clinical findings: fever (infection associated with neu-tropenia), bleeding (thrombocytopenia), fatigue(anemia)
Hb and Hct: normalin acute blood loss

••••••••••••••••••••••••S•••••••••••
134 Pathology
4. Laboratory findings: pancytopenia, hypocellular bonemarrow
VII. Normocytic Anemias with Corrected Reticulocyte Count > 3%Memo' ytic Anemias)A. Pathogenesis (Table 11-3)
1. Intrinsic defect: defect in RBCs causes anemia (e.g.,membrane defect, abnormal Hb, enzyme deficiency).
2. Extrinsic defect: factor outside the RBC causes hemoly-sis (e.g., calcified and stenotic aortic valve, mechanicalheart valve)
3. Extravascular and intravascular hemolysisa. Extravascular hemolysis: fixed macrophages (e.g.,
spleen, liver) phagocytose RBCs coated by IgG orC3b (e.g., autoimmune hemolytic anemia) or abnor-mally shaped RBCs (e.g., spherocytes).(1) Increase in serum unconjugated bilirubin:
end-product of Hb degradation bymacrophages
(2) May cause jaundiceb. Intravascular hemolysis: hemolysis occurs within
the circulation; causes:(1) Structural defects, such as deficiency of
glucose-6-phosphate dehydrogenase (G6PD)(2) Destruction by complement, such as IgM-
mediated hemolysis(3) Mechanical damage, such as calcific aortic
stenosisc. Consequences of chronic intravascular hemolysis
(1) Increased plasma and urine Hb: chronic he-moglobinuria may result in iron deficiency.
(2) Decreased serum haptoglobin: haptoglobincombines with Hb to form a complex that isphagocytosed and degraded by macrophages.
B. Congenital spherocytosis1. Pathogenesis: autosomal dominant disorder
a. Intrinsic defect with extravascular hemolysisb. Defect in spectrin in RBC membrane that results
in the loss of membrane fragments and formation ofspherocytes
c. Defects in membrane proteins other than spectrinproduce hereditary elliptocytosis and a benign,mild hemolytic anemia.
2. Clinical findingsa. Jaundiceb. Increased incidence of calcium bilirubinate
gallstonesc. Splenomegaly: splenectomy prevents hemolysisd. Self-limited aplastic crisis: most often due to par-
vovirus B19 infection
Congenital sphero-cytosis: defect inspectrin; increasedosmotic fragility
Intravascular hemo-lysis: L serumhaptoglobin;hemoglobinuria
Extravascular he-molysis: macro-phage phagocytosisof RBCs; T uncon-jugated bilirubin
Chapter 11 Red Blood Cell Disorders 135
TABLE 11-3 Summary of Normocytic Anemias
Anemia Pathogenesis Comments
•••••••••••••••••••••••••••••••••••
Loss of whole blood
Decreased iron stores
Iron trapped in macrophages
Suppression of trilineage myeloidstem cells
Deficiency of EPO
Autosomal dominant with defect inspectrin in RBC membrane
Extravascular hemolysis
Loss of anchor for DAF in myeloidstem cell
Complement destruction of hemato-poietic cells
Intravascular hemolysis
Autosomal recessiveSubstitution of valine for glutamic
acid at 6th position of p-globinchain
Extravascular hemolysis
X-linked recessiveDeficiency of glutathione with
oxidant damage to RBCsIntravascular hemolysis
Autosomal recessiveDecreased ATP synthesisExtravascular hemolysis
IgG with or without C3b coats RBCsMacrophage removal of cellsExtravascular hemolysis
IgM with C3bHemolysis due to complement
destruction of RBCsIntravascular hemolysis
Mechanical destruction of RBCs withformation of schistocytes
Intravascular hemolysisTransmitted by female Anopheles
mosquitoIntravascular hemolysis
Initial Hb and Hct normalSigns of volume depletion
Normocytic anemia before microcyticAll iron studies abnormal
Normocytic anemia before microcyticAll iron studies abnormal
PancytopeniaHypocellular marrow
Treat with recombinant EPO
Increased osmotic fragilityTreat with splenectomy
PancytopeniaScreen with sugar water testConfirm with acidified serum test
HbAS: HbA 55-60%, HbS 40-45%HbSS: HbS 90-95%, HbF 5-10%,
no HbA
Heinz body preparation: screenduring active hemolysis
Enzyme assay: confirming test whenno hemolysis is present
Increased 2,3-BPG (proximal toenzyme block)
Dehydrated RBCs with "thorny"projections
Positive direct Coombs' testAssociation with SLE, drugs (penicil-
lin, methyldopa)Association with Mycoplasma pneu-
moniae, chronic lymphocyticleukemia, drugs (quinidine)
Chronic hemoglobinuria results iniron deficiency
Intravascular rupture of RBCs corre-sponds with fever
Corrected reticulocyte count < 2%
Acute blood loss
Early-stage iron deficiency
Early-stage anemia of chronicdisease
Aplastic anemia
Renal disease
Corrected reticulocyte count > 3%
Congenital spherocytosis
Paroxysmal nocturnalhemoglobinuria
Sickle cell anemia
G6PD deficiency
Pyruvate kinase deficiency
Warm autoimmune hemolyticanemia
Cold autoimmune hemolyticanemia
Microangiopathic andmacroangiopathic hemolyticanemia
Malaria
ATP adenosine triphosphate; BPG, bisphosphoglycerate; DAF, decay-accelerating factor; EPO, erythropoietin; G6PD,glucose-6-phosphate dehydrogenase; Hb, hemoglobin; HbAS, sickle cell trait; HbSS, sickle cell disease; Hct, hematocrit;RBC, red blood cell; SLE, systemic lupus erythematosus

136 Pathology
Figure 11-9 Peripheral bloodwith spherocytes in congeni-tal spherocytosis. Numerous,round, dense red blood cells(RBCs) without central areas ofpallor represent spherocytes(arrows), or RBCs that have lostpieces of their cell membraneThe mean corpuscular hemoglo-bin concentration is increased
3. Laboratory findingsa. Normocytic anemia with reticulocytosis: increased
MCHC due to increased spherocytes (Figure 11-9)b. Increased RBC osmotic fragility: decreased surface
area-to-volume ratio causes spherocytes to rupturein mildly hypotonic salt solutions.
C. Paroxysmal nocturnal hemoglobinuria1. Pathogenesis: intrinsic defect with intravascular
hemolysisa. Acquired membrane defect in myeloid stem cells:
gene mutation causes a loss of the anchor for decay-accelerating factor.
Decay-accelerating factor normally neutralizescomplement attached to RBCs, neutrophils, andplatelets when there is a mild respiratory acidosis,such as occurs during sleep.
b. Intravascular complement destruction of RBCs,neutrophils, and platelets
2. Clinical findingsa. Episodic hemoglobinuria: causes iron deficiencyb. Increased risk of acute leukemia
3. Laboratory findingsa. Positive sugar water test: screening testb. Positive acidified serum test: confirmatory testc. Peripheral blood findings: pancytopenia,
reticulocytosisD. Sickle cell anemia
1. Epidemiologya. Autosomal recessive disorder
(1) Heterozygotes have sickle cell trait (HbAS).(2) Homozygotes have sickle cell disease (HbSS).
b. Most common hemoglobinopathy in AfricanAmericans: prevalence of HbSS is 1/600; carrier rateis about 1/12.
Child of male andfemale with HbAS25% normal, 50%HbAS, 25% HbSS

Chapter 11 Red Blood Cell Disorders 137
2. Pathogenesis: intrinsic defect with predominantly extra-vascular hemolysisa. Missense mutation: substitution of valine for glu-
tamic acid at sixth position of 13-globin chainb. Factors increasing deoxyhemoglobin cause HbS
molecules to polymerize and form sickle cells.Factors enhancing sickling include:(1) HbS > 60%: most important factor(2) Reduced 02 tension: basis for inducing sick-
ling in screening tests; increase in 02 revertssome sickled cells to normal.
(3) Volume depletion: concentrates HbS(4) Acidosis: increases 0 2 release, causing increased
deoxyhemoglobinc. HbF prevents sickling.
(1) Increased HbF at birth prevents sickling inHbSS.
(2) Hydroxyurea increases synthesis of HbF andreduces sickle cell crises.
3. Clinical findings: associated with chronic hemolyticanemia and painful vaso-occlusive crises in whichsickle cells block the microcirculation, causing ischemicdamagea. Dactylitis in infants: swelling of hands and feet due
to bone infarctsb. Acute chest syndrome: chest pain, lung infiltrates,
hypoxemia; most common cause of death inadults
c. Stroke: blockage of CNS vessels; common cause ofdeath
d. Aplastic crises(1) Erythropoiesis temporally shuts down; periph-
eral blood reticulocytes disappear.(2) Associated with parvovirus B19
e. Complications related to ischemic damage(1) Aseptic necrosis of femoral head(2) Autosplenectomy
(a) Spleen is enlarged but dysfunctional by2 years of age.
(b) Spleen is fibrosed and diminished in sizein young adults.
f. Increased susceptibility to infection(1) Children are at risk for Streptococcus pneumo-
niae sepsis.(2) Increased incidence of osteomyelitis due to
Salmonella speciesg. Calcium bilirubinate gallstones: jet black stones;
due to increased bilirubin in bile from chronichemolysis
Osteomyelitis inHbSS: due toSalmonella
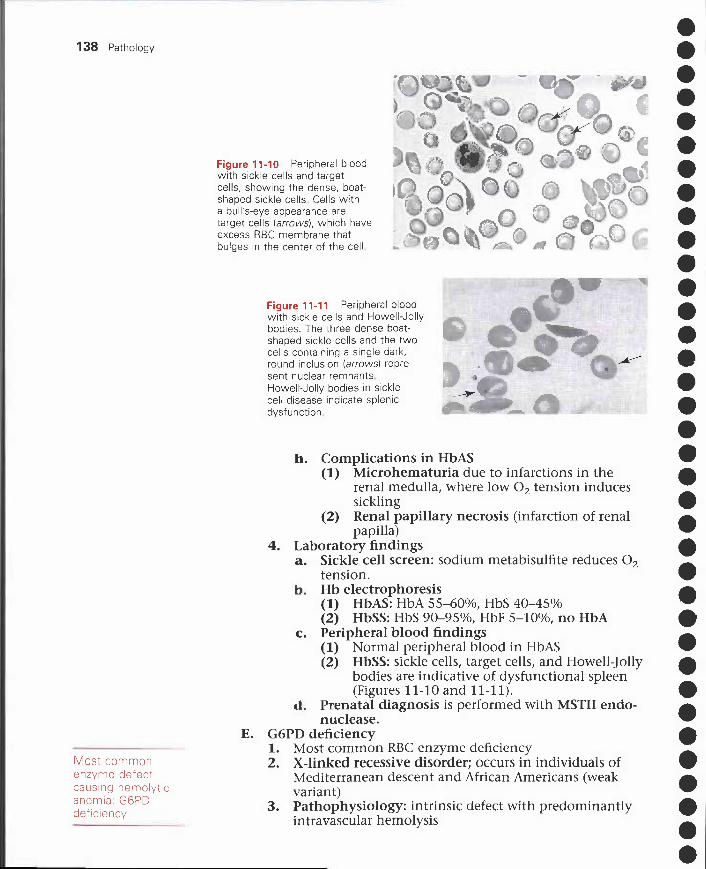
138 Pathology
Figure 11-10 Peripheral bloodwith sickle cells and targetcells, showing the dense, boat-shaped sickle cells Cells witha bull's-eye appearance aretarget cells (arrows), which haveexcess RBC membrane thatbulges in the center of the cell
rOtk),!E4 115411 -
0464,z D(13 QC) ''cia'0ro c (j4c
o,wti 0 0.29, cro\ 0 0 0
no 0 ovV°Apo
Figure 11-11 Peripheral bloodwith sickle cells and Howell-Jollybodies. The three dense boat-shaped sickle cells and the twocells containing a single dark,round inclusion (arrows) repre-sent nuclear remnantsHowell-Jolly bodies in sicklecell disease indicate splenicdysfunction
spfi,t) w
fe4120
h. Complications in HbAS(1) Microhematuria due to infarctions in the
renal medulla, where low 0 2 tension inducessickling
(2) Renal papillary necrosis (infarction of renalpapilla)
4. Laboratory findingsa. Sickle cell screen: sodium metabisulfite reduces 02
tension.b. Hb electrophoresis
(1) HbAS: HbA 55-60%, HbS 40-45%(2) HbSS: HbS 90-95%, HbF 5-10%, no HbA
c. Peripheral blood findings(1) Normal peripheral blood in HbAS(2) HbSS: sickle cells, target cells, and Howell-Jolly
bodies are indicative of dysfunctional spleen(Figures 11-10 and 11-11).
d. Prenatal diagnosis is performed with MSTII endo-nuclease.
E. G6PD deficiency1. Most common RBC enzyme deficiency2. X-linked recessive disorder; occurs in individuals of
Mediterranean descent and African Americans (weakvariant)
3. Pathophysiology: intrinsic defect with predominantlyintravascular hemolysis
Most commonenzyme defectcausing hemolyticanemia: G6PDdeficiency

•
•Chapter 11 Red Blood Cell Disorders 139
• a. Decreased production of glutathione in the
• pentose phosphate shunt(1) Glutathione is necessary to neutralize perox-
• ide, an oxidant produced in RBC metabolism.
•(2) Peroxide denatures Hb (forms Heinz bodies)
and damages the RBC membrane.
• b. Oxidant stresses inducing hemolysis(1) Infection (most common)
• (2) Drugs (e.g., primaquine, dapsone,sulfonamides)
(3) Fava beans (mainly in the Mediterraneanvariant, where G6PD levels in RBCs are lowest)
•4. Clinical findings: sudden onset of back pain with
hemoglobinuria
•5. Laboratory findings
a. Heinz body preparation: supravital stain; screening
• test during active hemolysis
•b. RBC enzyme analysis: confirmatory test when he-
molysis has subsided
• c. Peripheral blood findings: bite cells (macrophageremoval of RBC membrane), reticulocytosis
• F. Pyruvate kinase deficiency
•1. Pathogenesis: intrinsic defect with extravascular
hemolysis
• a. Autosomal recessive disorder
•b. RBCs lack ATP and are unable to maintain mem-
brane pumps (RBCs become dehydrated).
• 2. Clinical findingsa. Mild hemolytic anemia and jaundice beginning at
• birth
IIb. Increase in 2,3-bisphosphoglycerate proximal to
enzyme block shifts the 02-binding curve to the
• right, causing increased release of 02.3. Laboratory findings
• a. Reticulocytosis, RBCs with "thorny" projectionsb. RBC enzyme assay is the confirmatory test.
G. Autoimmune hemolytic anemia1. Pathogenesis
•a. Warm antibody type (most common): IgG with
or without C3b coats RBCs; splenic/liver macro-• phages phagocytose cells; extrinsic defect with extra-
vascular hemolysis.
• (1) Systemic lupus erythematosus: most commoncause of autoimmune hemolytic anemia
(2) Drugs
• (a) IgG antibody attaches to penicillin onRBCs (type II hypersensitivity reaction).
• (b) IgG antibody attaches to Rh antigens
•altered by methyldopa (type II hyper-sensitivity reaction).
•
0•

0 • •0
04 0*0.0;Figure 11 - 12 Peripheral bloodwith schistocytes. The frag-mented red blood cells (RBCs)with absence of central pallor,schistocytes, are produced whenRBCs are mechanically injuredby calcium deposits in an aorticvalve, platelet thrombi, or fibrinclots in the microvasculature.
140 Pathology
b. Cold antibody type: IgM with C3b coats RBCs;IgM activates complement system, resulting in intra-vascular hemolysis.(1) Drugs (e.g., quinidine) form immunocomplex
with IgM and attach to the RBC membrane,causing complement-mediated intravascularhemolysis (type III hypersensitivity reaction).
(2) Infection (e.g., Mycoplasma pneumoniae pneu-monia), chronic lymphocytic leukemia
2. Clinical findings: hepatosplenomegaly, generalizedlymphadenopathy, jaundice (only warm type)
3. Laboratory findingsa. Positive direct Coombs' test: detects IgG, IgM, and
C3b attached to RBCsb. Indirect Coombs' test: less sensitive (detects anti-
bodies in serum)c. Peripheral blood findings: spherocytes (macro-
phage removal of RBC membrane), reticulocytosisH. Microangiopathic and macroangiopathic hemolytic
anemias1. Pathogenesis: extrinsic defect with intravascular
hemolysisa. Microangiopathic: intravascular damage to RBCs
with production of fragmented RBCs (schistocytes)(1) Platelet thrombi (e.g., hemolytic uremic
syndrome)(2) Intravascular fibrin clots (e.g., disseminated
intravascular coagulation)b. Macroangiopathic
(1) Intravascular damage from calcific aortic ste-nosis or prosthetic heart valves
(2) May cause iron deficiency due to loss of Hb inthe urine
2. Laboratory findings: schistocytes (Figure 11-12)
Most commoncause of macroan-giopathic hemo-lytic anemia: calcificaortic stenosis
Autoimmune he-molytic anemias:positive directCoombs' test
••••••••••••••••••••••••••••••••••••

••••••••••
Chapter 11 Red Blood Cell Disorders 141
Figure 11-13 Plasmodium fal-ciparum ring forms in redblood cells (RBCs) This RBC hastwo ring forms Multiple infes-tation of an RBC is characteristicof P falciparum malaria.
• I. Malaria
•1. Pathogenesis
a. Female Anopheles mosquito transmits plasmodium
•to human; intraerythrocytic parasite causes intra-vascular hemolysis and fever spikes.
• b. Extrinsic defect with intravascular hemolysis:minor component of extravascular hemolysis
2. Clinical findings: fever and splenomegaly
• a. Plasmodium vivax: most common type; tertian
•fever pattern (every 48 hours)
b. P. falciparum: most lethal type; quotidian fever
•pattern (daily spikes with no pattern)
c. P. malariae: association with nephrotic syndrome;
• quartan fever pattern (every 72 hours)
•3. Laboratory findings: thin and thick smears identify or-
ganisms in RBCs; P. falciparum malaria only has ring
•forms and gametocytes (banana-shaped) (Figure 11-13).
•••••••••••••••

12IIPMPIllkhite Blood Cel
Disorders
1. Benign Qualitative White Blood Cell DisordersA. Pathogenesis
1. Defects in leukocyte structure (e.g., microtubules) orfunction (e.g., adherence, chemotaxis, phagocytosis,killing)
2. Examples: Chediak-Higashi syndrome, chronic granu-lomatous disease of childhood, myeloperoxidase defi-ciency, congenital leukocyte adhesion molecule defect
B. Clinical findings1. Unusual pathogens (e.g., coagulase-negative
Staphylococcus)2. Frequent infections and growth failure in children3. Lack of an inflammatory response (e.g., production of
"cold" abscesses)
Job's syndrome is an autosomal recessive disorder ofneutrophils, characterized by abnormal chemotaxisleading to "cold" soft tissue abscesses due to Staphy-lococcus aureus. Patients have red hair, a leonine face,chronic eczema, and increased IgE (hyperimmune Esyndrome).
C. Unusual benign leukocyte reactions1. Leukemoid reaction
a. Absolute leukocyte count: percentage of the specificwhite blood cell (WBC) x total WBC count; usually> 50,000/m1,
b. Pathophysiology(1) Exaggerated response to infection(2) May involve neutrophils, lymphocytes, or
eosinophils142

Chapter 12 White Blood Cell Disorders 143
c. Etiology: perforating appendicitis (neutrophils),whooping cough (lymphocytes), cutaneous larvamigrans (eosinophils)
2. Leukoerythroblastic reactiona. Pathogenesis
(1) Bone marrow infiltrative disease (e.g., fibrosis,metastatic breast cancer) or multiple bonefractures
(2) Immature bone marrow cells enter the periph-eral blood.
b. Peripheral blood findings: myeloblasts, progranulo-cytes, nucleated red blood cells (RBCs) intermixedwith normal peripheral blood WBCs
II. Benign Quantitative WBC DisordersA. Disorders involving neutrophils
1. Neutrophilic leukocytosis: absolute neutrophil count> 7000/4a. Pathogenesis
(1) Increased bone marrow production or release ofneutrophils
(2) Decreased adhesion to endothelial cells (e.g.,due to corticosteroid use)
b. Etiology(1) Infection (e.g., acute appendicitis)(2) Sterile inflammation with necrosis (e.g., acute
myocardial infarction)(3) Drugs (e.g., corticosteroids)
2. Neutropenia: absolute neutrophil count < 15004LLa. Pathogenesis
(1) Decreased production(2) Autoimmune destruction(3) Activation of neutrophil adhesion molecules
(e.g., endotoxins)b. Etiology
(1) Aplastic anemia(2) Autoimmune destruction (e.g., systemic lupus
erythematosus, SLE; chemotherapy)(3) Septic shock
B. Disorders involving eosinophils1. Eosinophilia: absolute eosinophil count > 700/4
a. Pathogenesis: release of eosinophil chemotacticfactor from mast cells
b. Etiology(1) Type I hypersensitivity reaction (e.g., bron-
chial asthma, reaction to penicillin)(2) Invasive helminthic infection (e.g., strongy-
loidiasis); pinworms do not cause eosinophilia(noninvasive).
(3) Polyarteritis nodosa, Addison's disease (cortisoldeficiency)
Leukoerythroblasticreaction in awoman > 50 yearsof age is usually dueto metastatic breastcancer.

144 Pathology
2. Eosinopenia: corticosteroids sequester eosinophils inlymph nodes.
C. Disorders involving basophils1. Basophilia: absolute basophil count > 110/uL2. Basophilia usually occurs in neoplastic myeloprolifera-
tive diseases (see section III).D. Disorders involving lymphocytes
1. Lymphocytosis: absolute lymphocyte count > 4000/4 inadults or > 8000/4 in childrena. Pathogenesis
(1) Increased production(2) Decreased entry into lymph nodes (e.g.,
lymphocytosis-promoting factor produced byBordetella pertussis)
b. Etiology(1) Viral (e.g., mononucleosis) or bacterial (e.g.,
whooping cough)(2) Drugs (e.g., phenytoin)
2. Atypical lymphocytosisa. Pathogenesis: antigenically stimulated lymphocytes,
which have prominent nucleoli and abundant bluecytoplasm
b. Etiology(1) Infection (e.g., mononucleosis, viral hepatitis,
cytomegalovirus, toxoplasmosis)(2) Drugs (e.g., phenytoin)
3. Infectious mononucleosisa. Pathogenesis: Epstein-Barr virus (EBV) infection
(1) Primarily transmitted by kissing: EBV initiallyreplicates in the salivary glands and thendisseminates.
(2) EBV attaches to CD21 receptors on B cells,causing B-cell proliferation and increased syn-thesis of antibodies.
b. Clinical findings(1) Extreme fatigue, tonsillitis, hepatosplenomeg-
aly, generalized lymphadenopathy(2) Rash develops if treated with ampicillin.
c. Laboratory findings(1) Atypical lymphocytosis
(a) Usually > 20% of the total WBC count(b) Atypical lymphocytes are antigenically
stimulated T cells (Figure 12-1).(2) Positive heterophil antibody test: detects IgM
antibodies against horse (most common),sheep, and bovine RBCs
(3) Positive antiviral capsid antigen test: mostsensitive test
(4) Increased serum transaminases from hepati-tis: jaundice is rare.
4. Lymphopenia: absolute lymphocyte count < 1500/4 inadults or < 3000/4 in children
B cells have CD21receptor sitesfor EBV.
••••••••••••••••••••••••••••••••••••

Chapter 12 White Blood Cell Disorders 145••••0•••
Figure 12 -1 Peripheral blood
• with atypical lymphocyte The
and coarse nuclear chromatin.cell shows prominent nucleoli
The cytoplasm is abundant and
• is indented by adjacent red blood
•
cells
•••
a. Pathogenesis: decreased production, decreasedrelease from lymph nodes, increased destruction(autoimmune)
b. Etiology
• (1) HIV: lysis of CD4 helper T cells(2) Immunodeficiency: DiGeorge syndrome (T-cell
deficiency), severe combined immunodeficiency(SCID)
(3) Other causes: autoimmune destruction (e.g.,
• SLE), corticosteroids (sequester lymphocytes inlymph nodes)• E. Disorders involving monocytes
•1. Monocytosis: absolute monocyte count > 800/42. Pathogenesis: response to chronic inflammation
• 3. Etiology
•a. Chronic infection (e.g., tuberculosis)b. Chronic inflammation (e.g., rheumatoid arthritis)
• c. Malignancy (e.g., carcinoma, malignant lymphoma)
• III. Neoplastic Myeloid Disorders
•A. Chronic myeloproliferative disorders
1. Pathogenesis: unregulated proliferation of neoplastic
• stem cells involving one or more bone marrow cell lines
•a. Splenomegalyb. Propensity for reactive bone marrow fibrosis
•("spent phase") and transformation to acuteleukemia
• 2. Polycythemia: increased hemoglobin (Hb), hematocrit(Hct), and RBC count (Table 12-1)a. RBC count versus RBC mass
• (1) RBC count: number of RBCs per ttL of blood
Corticosteroidsproduce neutrophilicleukocytos[s,eosinopenia,and iymphopenia.
••

146 Pathology
TABLE 12-1 Laboratory Findings in Polycythemias
Polycythemia RBC MassPlasmaVolume Sa02 EPO
Polycythemia vera T T Normal J
Appropriate polycythemia (e.g., T Normal .1. TCOPD, cyanotic congenital heartdisease)
Inappropriate polycythemia: ectopic T Normal Normal 1EPO (e.g., renal disease)
Relative polycythemia (e.g., volumedepletion)
Normal .1• Normal Normal
COPD, chronic obstructive pulmonary disease; EPO, erythropoietin; RBC, red blood cell; Sa02,oxygen saturation.
(2) RBC mass: total number of RBCs in the body inmL/kg
b. Relative polycythemia(1) Increased RBC count is due to a decrease in
plasma volume (e.g., volume depletion fromsweating).
(2) RBC mass is normal.c. Absolute polycythemia
(1) Increase in bone marrow RBC production: in-creases RBC count and RBC mass
(2) It is an appropriate absolute polycythemia ifthere is a hypoxic stimulus for erythropoietin(EPO) release. For example:(a) Primary lung disease (e.g., chronic ob-
structive lung disease)(b) Cyanotic congenital heart disease (e.g.,
tetralogy of Fallot)(c) Living at high altitude
(3) It is an inappropriate absolute polycythemiaif there is no hypoxic stimulus for EPO release.For example:(a) Polycythemia vera(b) Ectopic secretion of EPO: renal disorders
(cysts, cancer); hepatocellular carcinoma3. Polycythemia vera (see Table 12-1)
a. Pathogenesis(1) Clonal expansion of the trilineage myeloid
stem cell(2) Increase in RBCs, granulocytes (neutrophils,
eosinophils, basophils), and plateletsb. Clinical findings
(1) Splenomegaly(2) Thrombotic events: due to hyperviscosity (e.g.,
hepatic vein thrombosis, bowel infarction)

Chapter 12 White Blood Cell Disorders 147
(3) Signs of increased histamine (released frommast cells), such as ruddy face, pruritus afterbathing, and peptic ulcer disease (histaminestimulates production of gastric acid)
(4) Gout: increased breakdown of nucleated cellswith release of purines (converted to uric acid)
c. Laboratory findings(1) Hct increased: > 52% in men, > 48% in women(2) Increased RBC mass and plasma volume: only
type of polycythemia with an increase inplasma volume
(3) Absolute leukocytosis (leukocytes > 12,000/4)and thrombocytosis (platelets > 400,000/4)
(4) Decreased EPO: increased oxygen content in-hibits EPO release; only type of polycythemiawith decreased EPO
(5) Normal arterial blood gases, hypercellular bonemarrow with fibrosis in later stages
4. Chronic myelogenous leukemia (CML)a. Epidemiology
(1) Usually occurs at 40-60 years of age(2) Risk factors: exposure to ionizing radiation and
benzeneb. Pathogenesis
(1) Clonal expansion of pluripotential stem cell:capacity to differentiate into a lymphoid ortrilineage myeloid stem cell
(2) t9;22 translocation of ABL proto-oncogene,which fuses with the break cluster region (BCR)on chromosome 22 (BCR-ABL fusion gene)
c. Clinical findings(1) Hepatosplenomegaly and generalized lymph-
adenopathy: metastatic CML(2) Blast crisis: usually occurs after about 3 years;
increase in numbers of myeloblasts orlymphoblasts
d. Laboratory findings(1) Peripheral WBC count: 50,000-200,000
cells/µL (Figure 12-2)(2) Normocytic to macrocytic anemia: macrocytic
if folate is depleted in the production of leuke-mic cells
(3) Platelet count: thrombocytosis (40-50%),thrombocytopenia
(4) Bone marrow findings: hypercellular; fibrosisis common.
(5) Positive Philadelphia chromosome and BCR-ABL fusion gene
(6) Decreased leukocyte alkaline phosphatase(LAP)(a) LAP absent in neoplastic granulocytes
Polycythemia vera:only type of poly-cythemia withT plasma volumeand EPO
Philadelphia chro-mosome = chromo-some 22

148 Pathology
0 ovip 1 0 :act'''.o a w dip0 0 Orr i0 , Vko fie00 • 0 •.-*, op oil%0- c e,0 00,, ,itti Apoio gip0 0 wp ac, 0 M C- 09• 04 co 0 0- 4tils'n IA() A-`
..00- " C3 W.A
Figure 12-2 Peripheral blood in chronic myelogenous leukemia. Marked leukocytosis showsneutrophils at different stages of development (segmented and band neutrophils, metamyelo-cytes and myelocytes). The cell in the center (arrow) depicts a basophil with dark granules inthe cytosol and overlying the nucleus. Basophilia is prominent in chronic myeloproliferativediseases.
(13) Excellent test to differentiate chronic my-elogenous leukemia from leukemoid reac-tion (increased LAP)
5. Myelofibrosis and myeloid metaplasiaa. Pathogenesis
(1) Bone marrow fibrosis occurs earlier than inother types of myeloproliferative disease.
(2) Neoplastic cells are produced in the spleen andother sites (extramedullary hematopoiesis).
b. Clinical findings: splenomegaly with portal hyper-tension, splenic infarcts with left-sided pleuraleffusions
c. Laboratory findings(1) Bone marrow fibrosis due to stimulation of
fibroblasts(2) Peripheral WBC count: 10,000-50,000 cells/4(3) Normocytic anemia: teardrop cells (damaged
RBCs); leukoerythroblastic reaction(4) Platelets: thrombocytosis in 50% of cases
6. Essential thrombocythemiaa. Pathogenesis: stem cell disorder with proliferation
of megakaryocytes; platelets increased butnonfunctional
b. Clinical findings: bleeding (usually gastrointestinalwith concomitant iron deficiency), splenomegaly
c. Laboratory findings: thrombocytosis (platelets> 600,000/gL), mild leukocytosis, abnormal plateletmorphology
B. Myelodysplastic syndrome1. Pathogenesis
a. Stem cell disorder usually seen in men 50-80 yearsof age

Chapter 12 White Blood Cell Disorders 149
b. Frequently progresses to acute myelogenous leuke-mia (AML): "preleukemia"
c. Chromosomal abnormalities in 50% of cases(5q-, trisomy 8)
2. Laboratory findingsa. Severe pancytopenia
(1) Normocytic to macrocytic anemia: dimorphicRBC population (microcytic and macrocytic)
(2) Leukoerythroblastic reaction: myeloblasts<5%
b. Bone marrow findings: ringed sideroblasts (nucle-ated RBCs with excess iron), myeloblasts < 30% (if> 30%, disease is progressing to AML)
C. Leukemias (acute and chronic)1. Leukemias are malignant diseases of bone marrow stem
cells that may involve all cell lines.2. Epidemiology
a. Risk factors(1) Chromosomal abnormalities (e.g., Down syn-
drome, chromosome instability syndromes)(2) Ionizing radiation(3) Chemicals (e.g., benzene)(4) Alkylating agents (particularly busulfan)
b. Age ranges for common leukemias(1) Newborn to 14 years: acute lymphoblastic leu-
kemia (ALL)(2) 15-39 years: AML(3) 40-60 years: AML (- 60%) and CML (- 40%) Most common type(4) > 60 years: chronic lymphocytic leukemia (CLL) of leukemia: CLL
3. Pathogenesisa. Block in stem cell differentiation: monoclonal pro-
liferation of neoplastic leukocytes behind the blockb. Leukemic cells ultimately replace the bone marrow,
enter the peripheral blood, and infiltrate tissuesthroughout the body.
4. Clinical findings associated with acute leukemiaa. Abrupt onset of signs and symptomsb. Fever (infection), bleeding (thrombocytopenia),
fatigue (anemia)c. Metastatic disease: hepatosplenomegaly, generalized
lymphadenopathy, central nervous system (CNS)involvement (especially in ALL)
d. Bone pain and tenderness: from bone marrow ex-pansion by leukemic cells
5. Laboratory findings associated with acute leukemiaa. Peripheral WBC count: < 10,000/4 (normal) to
> 100,000/4; > 30% blasts are present (e.g., myelo-blasts, lymphoblasts).
b. Normocytic to macrocytic anemia: macrocytic iffolate is depleted in production of leukemic cells

150 Pathology
Figure 12-3 Peripheral bloodwith promyelocyte filled withAuer rods in acute promyelocyticleukemia The promyelocyte hasnumerous splinter-shaped in-clusions in the cytoplasm (arrow)representing Auer rods
c. Platelets: thrombocytopenia (usually < 100,000/viL)d. Bone marrow findings: hypercellular with > 30%
blasts (usually > 60%)6. Clinical findings associated with chronic leukemia
a. Insidious onsetb. Hepatosplenomegaly and generalized lymph-
adenopathy7. Laboratory findings associated with chronic leukemia
a. Peripheral WBC count(1) Similar to that of acute leukemia(2) Blast cells usually < 10%(3) Evidence of maturation of cells
b. Normocytic to macrocytic anemia: macrocytic iffolate is depleted in production of leukemic cells
c. Platelets: thrombocytopenia (usually < 100,000/4)d. Bone marrow findings: hypercellular with < 10%
blasts8. AML
a. Cytogenetic abnormalities are common: t(15;17)in acute promyelocytic leukemia
Most important testfor diagnosing leu-kemia: bone marrowexamination
Acute promyelocytic leukemia is associatedwith an abnormality in retinoic acid metabolism.High doses of vitamin A may induce remissionby maturing the cells.
b. Clinical findings(1) Disseminated intravascular coagulation (DIG)
is invariable in acute promyelocytic leukemia.(2) Gum infiltration is common in acute mono-
cytic leukemia.c. Auer rods: splinter-shaped to rod-shaped structures
in the cytosol of blast cells (fused azurophilicgranules) seen only in AML (Figure 12-3)
AML: Auer rods inthe cytoplasm ofmyeloblasts

Chapter 12 White Blood Cell Disorders 151
IV. Lymphoid LeukemiasA. ALL
1. Epidemiologya. Most common leukemia in children (newborn to
14 years of age)b. Subtypes include early pre-B-cell ALL (80%), and
pre- ii - , Ii-, and -cell ALL.2. Pathogenesis: clonal lymphoid stem cell disease
a. Early pre-B-ceil ALL: positive marker studies forcommon ALL antigen (CALLA, 0) and terminaldeoxvnucleotidvl transferase (IdT)
b. T-cell ALL: CD10- and TdT+3. Clinical findings
a. Metastatic sites similar to those of AMLb. B-cell types: commonly metastasize to the CNS and
testiclesc. T-cell type: presents as anterior mediastinal mass
or acute leukemia4. Laboratory findings
a. Peripheral WBC count: 10,000-100,000/pL, > 30%I ymphoblasts in peripheral blood
b. Normocytic anemia with thrombocytopeniac. Bone marrow findings: bone marrow often totally
replaced by lymphoblasts5. Prognosis: early pre-B-cell ALL has a 5-year survival rate
of more than 85%.B. Chronic lymphocytic leukemia (CLL)
1. Epidemiology: occurs in individuals > 60 years of age2. Pathogenesis: neoplastic disorder of virgin B cells (B cells
that cannot differentiate into plasma cells)3. Clinical findings
a. Generalized lymphadenopathyb. Metastatic sites similar to those of AMLc. Increased incidence of autoimmune hemolytic
anemia: warm and cold types4. Laboratory findings
a. Peripheral WBC count: 15,000-200,000/µL, lym-phoblasts < 10%, neutropenia, numerous "smudge"cells (fragile leukemic cells) (Figure 12-4)
b. Normocytic anemia (50% of cases) and thrombocy-topenia (40% of cases)
c. Bone marrow findings: usually replaced by neoplas-tic B cells
d. Hypogammaglobulinemia is common.C. Adult T-cell leukemia
1. Malignant lymphoma associated with human T-cell leu-kemia virus (HTLV-1)
2. Pathogenesis: activation of TAX gene, which inhibits theTP53 (p53) suppressor gene, leading to monoclonal pro-liferation of neoplastic CD4 helper T cells
Most commonacute leukemia inchildren: ALL
Most commoncause of generalizedlymphadenopathyin individuals> 60 years ofage: CLL

152 Pathology
' C)- a Onl
Pe V '0 v0 4 0 GP 46 ---) CC)DC1(0 ,c913,P cpou IV IA• w-g 0 0 C7661i Q c='‘''0
Figure 12 -4 Peripheral blood in chronic lymphocytic leukemia There are an increasednumber of lymphocytes with dense nuclear chromatin and scant cytoplasmic borders Thelymphocytes are extremely fragile and produce characteristic "smudge" cells (arrows) duringpreparation of a slide
3. Clinical findingsa. Hepatosplenomegaly and generalized
lymphadenopathyb. Skin infiltration: common finding in all T-cell
malignanciesc. Lytic bone lesions
(1) Due to lymphoblast release of osteoclast-activating factor
(2) Associated with hypercalcemia4. Laboratory findings
a. Peripheral WBC count: 10,000-50,000/p L, > 30%lymphoblasts (CD4+, TdT-)
b. Normocytic anemia and thrombocytopeniac. Bone marrow findings: replaced by CD4
lymphoblastsD. Hairy cell leukemia
1. Type of B-cell leukemia; most common in middle-aged men
2. Clinical findingsa. Splenomegaly: 90% of casesb. Absence of lymphadenopathy: only leukemia
without lymphadenopathy
c. Hepatomegaly (20% of cases), autoimmune vasculitisand arthritis
d. Excellent response to 2-chlorodeoxyadenosine(purine nucleoside)
3. Laboratory findingsa. Pancytopenia; cells have hair-like projections; bone
marrow packed with leukemic cellsb. Positive tartrate-resistant acid phosphatase stain
(TRAP)

411141‘10Lymphoid TissueDisorders
I. LymphadenopathyA. Locations of lymphoid tissue: regional lymph nodes,
tonsils and adenoids (Waldeyer's ring), Peyer's patches andappendix, thymus, white pulp of spleen
B. Specific cell locations in lymphoid tissue1. B cells: germinal follicles in lymph nodes and extra-
nodal tissue (e.g., appendix), peripheral areas of whitepulp of spleen
2. T cells: paracortex in lymph nodes and extranodaltissue, periarteriolar sheath in spleen, thymus
3. Histiocytes: sinuses in lymph nodes, red pulp of spleenC. Sites of pathologic processes in lymph nodes (Figure 13-1)D. Epidemiology
1. Patients younger than 30 years of age: in about 80%of cases, nodal enlargement usually indicates benigndisease, such as reactive lymphadenitis due to infection.
2. Patients older than 30 years of age: in about 60% ofcases, nodal enlargement usually indicates malig-nant disease, such as non-Hodgkin's lymphoma (NHL)and metastasis.
E. Clinical findings1. Painful mobile nodes: imply inflammation (e.g.,
infection)a. Localized: drain sites of infection (e.g., tonsillitis);
most common sites are the anterior cervical nodesand the inguinal nodes.
b. Generalized: systemic disease (e.g., mononucleosis;systemic lupus erythematosus, SLE)
2. Painless nodes: nodes are indurated and often fixed tosurrounding tissue.a. Localized: nodes draining a primary cancer site
(e.g., axillary nodes in breast cancer); Hodgkin'slymphoma
Painless nodesusually containmetastatic cancer.
153

154 Pathology
(
Germinal follicle: reactivelymphadenitis, follicular B-celllymphoma
Sinus: metastasis, sinushistiocytosis, Langerhans cellhistiocytosis, histiocytic lymphoma'
Paracortex: reactive hyperplasia,T-cell lymphomas
Figure 13-1 Sites of pathologic processes in lymph nodes. Some lymphoid disorders initiallylocalize in the germinal follicles, where B cells are located; others localize in the paracortex,where T cells are located Mixed B- and T-cell reactions also may occur Histiocytic disordersinvolve the sinuses.
b. Generalized: metastasis in leukemia; follicularB-cell lymphoma
3. Key nodal groups involved in primary or metastaticcancera. Submental: metastatic squamous carcinoma in the
floor of the mouthb. Cervical: metastatic head and neck tumors (e.g.,
larynx, thyroid, nasopharynx); Hodgkin'slymphoma
c. Left-sided supraclavicular (Virchow's nodes):metastatic abdominal cancers (e.g., stomach,pancreas)
d. Axillary: metastatic breast cancere. Hilar: metastatic lung cancerf. Mediastinal: Hodgkin's lymphomag. Para-aortic: metastatic testicular cancer, Burkitt's
lymphoma
II. Reactive Lymphadenitis- Reactive lymphadenitis is due to hyperplasia of B cells, T cells,
or histiocytes in response to antigenic stimulation of thelymph node.
A. Follicular hyperplasia1. B-cell antigenic response: germinal follicles are sharply
demarcated from the paracortex, and cells are in differ-ent developmental stages.
2. Examples: early stages of HIV, SLEB. Paracortical hyperplasia: T-cell antigenic response; causes
include:1. Dermatopathic lymphadenitis: nodes draining
chronic dermatitis (e.g., psoriasis, eczema) with macro-phage phagocytosis of melanin pigment
2. Phenytoin, viral infections
Most commoncause of reactivelymph adenitisiinfection

Chapter 13 Lymphoid Tissue Disorders 155
C. Mixed B- and T-cell hyperplasia1. Cat-scratch disease is caused by Bartonella henselae.2. Granulomatous microabscesses are present in regional
lymph nodes.D. Sinus histiocytosis
1. Benign histiocytic response in lymph nodes draining atumor
2. Favorable prognostic sign in breast cancer
III. Non-Hodgkin's LymphomaA. Epidemiology
1. Accounts for about 60% of lymphomas in adults;more than 80% are of B-cell origin and derive from thegerminal follicle.
2. Accounts for about 60% of lymphomas in children;usually T-cell lymphoblastic lymphoma or Burkitt'slymphoma
3. Risk factorsa. Viruses: such as Epstein-Barr virus (EBV) in Bur-
kitt's lymphoma or central nervous system (CNS)lymphoma in HIV
b. Helicobacter pylori: lymphoma from mucosa-associated lymphoid tissue (MALT) in the stomach
c. Autoimmune disease: SjOgren's syndrome (sali-vary gland and gastrointestinal lymphomas)
d. AIDS, immunosuppressive therapy, irradiationB. Pathogenesis
1. Mutation produces a blockage at a specific stage indevelopment of B or T cells.
2. Neoplastic cells accumulate proximal to the block.C. B-cell lymphomas (Table 13-1)D. T-cell lymphomas
1. Precursor T-cell lymphoblastic lymphoma andleukemiaa. Precursor T-cell lymphoma: 40% of childhood
lymphomas(1) Primarily involves the anterior mediastinum
and cervical nodes(2) Bone marrow and CNS involvement are
common.b. Precursor T-cell lymphoblastic leukemia: leuke-
mic variant of precursor T-cell lymphoma2. Mycosis fungoides and Sezary syndrome
a. Both lymphomas involve neoplastic peripheralCD4 helper T cells.
b. They usually affect adults 40--60 years of age.c. Mycosis fungoides
(1) Begins in skin (rash to plaque to nodularmasses) and progresses to lymph nodes, lung,liver, and spleen
•S
•••••••••0•••••0••••••••••••••••••
Most commonB-cell childhood lym-phoma: Burkitt'slymphoma

156 Pathology
(2) Pautrier's microabscesses: nests of neoplasticcells in the epidermis
d. Sezary syndrome: mycosis fungoides with a leuke-mic phase
IV. Hodgkin's LymphomaA. Epidemiology and etiology
1. Accounts for about 40% of adult lymphomas2. Slightly more common in men (except for nodular scle-
rosing type)3. Bimodal age distribution: first large peak in third
decade, second smaller peak in individuals older than45-50 years of age
4. Associated factors: defects in cutaneous anergy tocommon antigens; EBV (mixed-cellularity type)
B. Classification (Table 13-2)1. Lymphocyte predominant2. Nodular sclerosing3. Mixed cellularity4. Lymphocyte depletion (not discussed in this text)
C. Reed-Sternberg (RS) cell: neoplastic cell of Hodgkin'slymphoma1. Generally a transformed germinal center B cell
(CD15+, CD30+)
TABLE 13-1 Common Types of B-cell Non-Hodgkin's Lymphoma
Type EpidemiologyDescription/Immunophenotype Clinical Findings
Burkitt's lymphoma 30% of children withnon-Hodgkin'slymphoma (NHL)
EBV relationship with translo-cation t(8;14)
American variant: GI tract,retroperitoneum
African variant: jawBone marrow involvement
and leukemic phasecommon
Diffuse large B-cell 50% of adults with Derives from germinal center Localized disease with extra-lymphoma NHL; elderly
and childhoodpopulations
nodal involvement: GItract, brain (EBV associa-tion with AIDS)
Extranodal marginalzone lymphoma
Derives from MALT Stomach (Helicobacter pylori)
Follicular lymphoma 40% of adults with Derives from germinal center Generalized lymphadenop-NHL; elderly Translocation t[14;18]) with athypatients overexpression of BCL2
anti-apoptosis geneBone marrow involvement
Small lymphocytic Patients usually > 60 Neoplasm of small, mature B Generalized lymph-lymphoma (SLL) years of age lymphocytes adenopathy
SLL if confined to lymphnodes
CLL if leukemic phase ispresent
CLL, chronic lymphocytic leukemia; EBV, Epstein-Barr virus; GI, gastrointestinal; MALT, mucosa-associated lymphoidtissue.

Chapter 13 Lymphoid Tissue Disorders 157
2. Classic RS cell: two mirror-image nuclei, each with aneosinophilic nucleolus surrounded by a clear halo("owl's eyes") (Figure 13-2)
3. L and H variant: large, pale-staining, multilobed cell("popcorn cell") seen in lymphocyte predominanttype
4. Lacunar cell variant: pale cell with multilobed nucleuscontaining many small nucleoli; seen in nodular scle-rosing type
D. Pathologic findings1. Diagnosis depends on the presence of classic RS cells or
RS variant cells in a background of reactive cells (e.g.,eosinophils, plasma cells).
2. Localized groups of nodes with contiguous spread (e.g.,cervical or supraclavicular nodes); cut section hasbulging "fish-flesh" appearance.
E. Clinical findings1. General: fever, unexplained weight loss, pruritus, night
sweats
TABLE 13-2 Some Types of Hodgkin's Lymphoma
Type Epidemiology Clinical Findings
Lymphocyte 5% of casespredominant Occurs mainly in males
Mixed cellularity 30% of casesMen > 50 years of ageStrong Epstein-Barr virus
association
Nodular sclerosing 60% of casesOccurs mainly in females
Asymptomatic young male withcervical or supraclavicularnodes
Difficult to find classic RS cellsL and H variants present
RS cells numerousTEosinophils, plasma cells,
histiocytes
Usually involves anterior medias-tinal nodes and either cervicalor supraclavicular nodes
RS cells infrequentLacunar cells presentCollagen separates nodular areas
RS, Reed-Sternberg
Figure 13-2 Classic Reed-Sternberg (RS) cell The large,multilobed cell with prominentnucleoli is surrounded by a haloof clear nucleoplasm ClassicRS cells are more easily found inmixed-cellularity Hodgkin's lym-phoma than in lymphocyte-predominant and nodular-sclerosing Hodgkin's lymphoma

158 Pathology
2. Hematologic: normocytic anemia; painless enlarge-ment of single groups of lymph nodes
F. Prognosis1. Clinical stage is more important than type of Hodg-
kin's disease in determining prognosis.2. Most patients in stages I and II have lymphadenopathy
above the diaphragm, usually involving the supra-clavicular and anterior mediastinal nodes.
3. Increased risk of secondary malignancies (acute my-elogenous leukemia or NHL) is a complication of radia-tion therapy and alkylating agents (most oftenresponsible).
V. Plasma Cell DyscrasiasA. Plasma cell dyscrasias are monoclonal B-cell disorders with
an increase in a single immunoglobulin and correspondinglight chain.1. Immunoglobulin is detected as a monoclonal spike
(M component) on serum protein electrophoresis.2. Clinical significance of M components
a. Characteristic of plasma cell dyscrasias (e.g., mul-tiple myeloma)(1) Most commonly due to an increase in IgG(2) Other plasma cell clones are suppressed.
b. Bence Jones protein (x or X light chains excretedin urine) is associated with B-cell (plasma cell)malignancy.
c. Immunoelectrophoresis identifies the immuno-globulin and light chain in serum and light chainsin urine.
B. Multiple myeloma1. M spike in 80-90% of patients: usually IgG x followed
by IgA and pure light chain myeloma; urine is posi-tive for Bence Jones protein in 60-80% of cases.
2. Epidemiologya. Rare in individuals younger than 40 years of ageb. Increased risk when exposed to radiation
3. Microscopic findingsa. Sheets of malignant plasma cells in bone marrow
aspirateb. Plasma cells account for more than 10% of WBCs.
4. Skeletal findingsa. Bone pain is due to lytic ("punched out") lesions
(vertebra most common site; also seen in ribs,skull, and pelvis) and pathologic fractures(Figure 13-3).
b. Hypercalcemia, diffuse bone demineralization5. Renal findings: renal failure; myeloma kidney due to:
a. Proteinaceous tubular casts composed of BenceJones protein, which damages tubular epitheliumand causes an intratubular multinucleated giantcell reaction
Most commonprimary malignantplasma cell dyscra-sia: multiplemyeloma
••••••••••
Chapter 13 Lymphoid Tissue Disorders 159
Figure 13-3 Radiograph of askull showing multiple "punchedout" I ytic lesions in multiplemyeloma. When these lesionsare located in other areas, suchas the ribs, pathologic frac-tures frequently occur
•
• TABLE 13-3 Some Plasma Cell Dyscrasias
•
•
•
••••••••
MGUS
Lymphoplasmacyticlymphoma(WaldenstrOm'smacroglobulin-emia)
Dyscrasia
MGUS, monoclonal gammopathy of undetermined significance
Characteristics
Occurs in elderly patientsSmall IgG M spikePlasma cells < 3% in bone
marrowBence Jones protein absent
Common in elderly menM spike with IgMNeoplastic lymphoplasmacy-
toid B cellsBence Jones proteins presentGeneralized lymphadenopathyAnemiaBone marrow involvedHyperviscosity syndrome due
to increased IgM: retinalhemorrhages, strokes
Prognosis
Myeloma, macroglobulinemia,or primary amyloidosisdevelops in 20-24% of cases
Median survival 5 years
•••••••••••••
b. Nephrocalcinosis: metastatic calcification of thetubular cell basement membranes
c. Primary amyloidosis: light chains are converted toamyloid; nephrotic syndrome.
6. Hematologic findings: normocytic anemia with rou-leaux, increased erythrocyte sedimentation rate
7. Prognosis: median survival is 6 months withouttreatment.
C. Other plasma cell dyscrasias (Table 13-3)
VI. Langerhans' Cell Histiocvtoses• Langerhans' histiocytes: CD1+; on electron microscopy, have
Birbeck granules (tennis racket appearance)A. Letterer-Siwe disease: malignant histiocytosis
1. Occurs in infants and in children younger than 2 yearsof age
Most commoncause of death inmultiple myeloma:recurrent infection

160 Pathology
2. Clinical findings: diffuse eczematous rash; multipleorgan involvement; cystic defects in the skull, pelvis,and long bones
3. Prognosis: rapidly fatalB. Hand -Schuller-Christian disease: malignant histiocytosis
1. Mainly affects children2. Clinical findings
a. General: fever, localized rash on scalp and in earcanals
b. Classic triad caused by infiltrative disease: cysticskull defects, diabetes insipidus due to invasionof posterior pituitary, exophthalmos from infiltra-tion of the orbit
3. Prognosis: somewhat better than for Letterer-Siwedisease
C. Eosinophilic granulomas: benign histiocytosis1. Unifocal lytic lesions in bone (skull, ribs, and femur)
occur in adolescents and young adults.2. Bone pain and pathologic fractures are common.
VII. Mast Cell DisordersA. Urticaria pigmentosa: localized
1. Multiple oval, red-brown, nonscaling macules orpapules
2. Scratching results in erythematous swelling of thelesions and pruritus due to mast cell release ofhistamine.
3. Lesions remain hyperpigmented after regression.4. Skin biopsy: mast cells have metachromatic granules
that stain positive with toluidine blue and Giemsa stain.B. Mastocytosis: systemic
1. Multiple organ involvement by mast cells2. Mast cells are often present in peripheral blood.
VIII. Disorders of the SpleenA. Splenomegaly; causes include:
1. Autoimmune disease: SLE, rheumatoid arthritis2. Infectious disease: mononucleosis, visceral
leishmaniasis3. Infiltrative disease: lysosomal storage disease,
leukemias4. Extramedullary hematopoiesis: myeloproliferative
disease (e.g., myelofibrosis, myeloid metaplasia)5. Vascular congestion: portal hypertension in cirrhosis
B. Hypersplenism1. Red and white blood cells and platelets, either singly or
in combination, are sequestered and destroyed.2. The most common cause is portal hypertension.
Macrophages havea fibrillary appear-ance in Gaucher'sdisease; soapbubble appearancein Niemann-Pickdisease

Chapter 13 Lymphoid Tissue Disorders 161
3. Clinical findingsa. Splenomegalyb. Peripheral blood cytopenias: anemia, thrombocy-
topenia, neutropenia alone or in combinationC. Splenic dysfunction
1. Howell Jolly bodies (nuclear remnants) seen in redblood cells
2. Predisposition to infections: septicemia, peritonitisa. Mechanism: concentration of IgM drops, decreas-
ing complement system activation and reducingC3b, an opsonizing agent.
b. Pathogens involved: Streptococcus pneumoniae fol-lowed by Haemophilus influenzae
c. Immunization helps prevent infectiouscomplications.

14Coagulation Disorders
AO'w
I. Normal Hemostasis• Proper blood clotting and prevention of blood loss requires the
interaction of blood vessels, platelets, coagulation factors, andfibrinolytic agents.
A. Anticoagulants normally found in small blood vessels(capillaries, venules, arterioles)1. Heparin: enhances antithrombin III activity, thus neu-
tralizing serine protease coagulation factors, such as XII,XI, IX, X, II (prothrombin), and thrombin
2. Prostaglandin 12 (prostacyclin): vasodilator that inhib-its platelet aggregation; synthesized by endothelial cells
3. Proteins C and S: vitamin K-dependent factors that inac-tivate factors V and VIII and enhance fibrinolysis
4. Tissue plasminogen activator (tPA): activates plasmin-ogen to release plasmin, which degrades coagulationfactors and lyses fibrin clots
B. Procoagulants released in small-vessel injury1. Thromboxane A2 (TXA2)
a. Synthesis: converted from prostaglandin H 2 bythromboxane synthase in platelets
b. Functions: vasoconstriction, enhanced plateletaggregation
2. Von Willebrand's factor (vWF)a. Synthesis: by endothelial cells and megakaryocytesb. Functions
(1) Binds platelets to collagen, thus contributingto platelet adhesion
(2) Complexes with factor VIII coagulant(VIII:C) in the circulation, thereby stabilizing it
Factor VIII:C is synthesized in the liver.When VIII:C is activated by thrombin, itdissociates from the VIII:vWF complex andfunctions as a coagulant.
A decrease in vWFsecondarily de-creases procoagu-lant factor (VIII:C)activity
162

Intrinsic system
HMWK, Activates plasminogencollagen //
XII Xlla
Activates kininogen systemXI Xla
IX )0 IXa
VIII + IXa + PF3 + Ca2+
<Xa
Chapter 14 Coagulation Disorders 163
Extrinsic system
Tissue thromboplastinVII
Vila
1
Final common pathway:!_actor X to fibrin clot X
V + Xa + PF3 + Ca2+
"Prothrombin complex"
Prothrombin (II)
0 Thrombin (enzyme)
Fibrinogen (I)
0 Fibrin monomer + fibrinopeptides A + B
Fibrin monomers aggregate Soluble fibrin 7-0- Cross-linked insoluble fibrin
XIII XlllaThrombin
Figure 14-1 Coagulation cascade Both the extrinsic and intrinsic coagulation systems usethe final common pathway for the formation of a fibrin clot HMWK, high-molecular-weightkininogen; PF3, platelet factor 3
3. Tissue thromboplastin (factor III): exposed on damagedtissue and activates factor VII in the extrinsic coagula-tion system
4. Extrinsic and intrinsic coagulationC. Platelets
1. Derivation: cytoplasmic fragmentation of megakaryo-cytes (- 1000-3000 platelets per megakaryocyte)
2. Locations: peripheral blood (life, - 9-10 days); spleen(approximately one third of total platelet pool)
3. Membrane componentsa. Glycoprotein receptor for vWF: GpIbb. Glycoprotein receptor for fibrinogen: GpIIb:IIIac. Platelet membrane with platelet factor 3 (PF3):
phospholipid substrate required for the clottingsequence
4. Structurea. Contractile elements (e.g., thrombosthenin) aid in
clot retraction.b. Dense bodies contain adenosine diphosphate (ADP),
an aggregating agent, and calcium (Ca2±), a bindingagent for vitamin K-dependent factors.
c. a-Granules contain vWF and PF4 (heparin-neutralizing factor).
D. Coagulation cascade (Figure 14-1)1. Extrinsic system: factor VII
••••••••••
•••••••••••••••••••••••••

164 Pathology
a. Tissue thromboplastin released from injured tissueactivates factor VII, resulting in the formation offactor VIIa.
b. In the final common pathway, factor VIIa activatesfactor X.
2. Intrinsic system: factors XII, XI, IX, and VIIIa. Exposed subendothelial collagen and high-
molecular-weight kininogen activate factor XII(Hageman factor) to form factor XIIa.
b. Factor XIIa activates three substances.(1) Factor XI to form XIa(2) Plasminogen to form plasmin(3) Kininogen system to produce kallikrein and
bradykininc. XIa activates factor IX to form a four-component
complex (factor IXa, factor VIII, PF3, Ca2+). In thefinal common pathway, this complex activatesfactor X.
3. Final common pathway: factors X, V, prothrombin, andI (fibrinogen)a. Prothrombin complex: four-component system
(factor Xa, factor V, PF 3, Ca2+) cleaves prothrombinto the enzyme thrombin.
b. Thrombin(1) Acts on fibrinogen to produce soluble fibrin
monomers and fibrinopeptides A and B(2) Activates fibrin-stabilizing factor XIII(3) Activated fibrin-stabilizing factor XIIIa con-
verts soluble fibrin monomers to insolublefibrin by enhancing cross-linking between pro-teins to strengthen the fibrin clot.
4. Coagulation factors consumed in a clot: fibrinogen,prothrombin, factors V and VIII
•••••••••••••••••••••••A fibrin clot forms when blood is drawn into a clottube (no added anticoagulant). When the tube isspun in a centrifuge, the supernatant is serum. Unlikeplasma, it lacks fibrinogen, prothrombin, and factorsV and VIII.
5. Vitamin K-dependent factorsa. These factors are synthesized in the liver as non-
functional precursor proteins.b. Epoxide reductase activates vitamin K in the liver to
become vitamin K1.c. The factors are functional because of y-carboxylation
by vitamin K 1 , which binds them to Ca2+ and PF3in the clotting cascade.
•••••••••••••

•• Chapter 14 Coagulation Disorders 165
•• Vascular phase t Vessel injury with activation of coagulation cascade
•••
Platelet adhesion to vWF
Defective in vWD, Bernard-Soulier disease
Platelet release of aggregating agents
Platelet synthesis of TXA2 Inhibited by aspirin)
Temporary platelet plug --
1Coagulation phase Formation of stable platelet plug
1 Dissolution of platelet plugi Fibrinolytic phase 4'
Reestablishment of blood flow
Patients with clotting disorders who must takeanticoagulants are initially placed on both hepa-rin and warfarin. Heparin has an immediateanticoagulant effect, and warfarin inhibitsepoxide reductase and prevents any furthery-carboxylation of vitamin K-dependent factors.Because previously y-carboxylated factors are stillpresent, complete anticoagulation does not im-mediately occur. Prothrombin has the longesthalf-life of all the vitamin K-dependent factors.Therefore, complete anticoagulation takes atleast 3-4 days, until all functional prothrombinhas been depleted.
E. Fibrinolytic system1. Activation
a. tPA activates plasminogen, forming the enzymeplasmin.
b. Factor XIIa also activates plasminogen.2. Functions of plasmin: cleaves insoluble fibrin mono-
mers and fibrinogen into fibrin and fibrinogen deg-radation products (FDPs)
F. Small-vessel hemostasis response to injury (Figure 14-2)1. Vascular phase
a. Vasoconstriction occurs immediately after injury.b. Factor VII is activated by tissue thromboplastin;
exposed collagen activates factor XII.2. Platelet phase
a. Adhesion: GpIb receptors on the platelets adhere toexposed vWF in damaged endothelial cells.
Platelet phase
•••••••••••0
S
••••••••••S
Defective in Glanzmann's disease
Defective in factor deficiencies
Figure 14-2 Small-vessel hemostasis response to injury TXA2, thromboxane A2 ; vWD, von Willebrand's disease; vWF,von Willebrand's factor.

166 Pathology
TABLE 14-1 Laboratory Findings in Some Hemostatic Disorders
Disorder Platelet Count Bleeding Time PT PTT
Thrombocytopenia
Von Willebrand's disease
Hemophilia A
DIC
1
Normal
Normal
I,
T
T
Normal
T
Normal
Normal
Normal
T
Normal
1'
T
T
DIC, disseminated intravascular coagulation; PT, prothrombin time; PTT, partial thromboplastintime.
b. Release reaction: release of ADP causes platelet ag-gregation in the lumens of injured vessels.
c. Synthesis and release of TXA2 : vessels constrict, re-ducing blood flow; this action further enhancesplatelet aggregation.
d. Temporary platelet plug stops bleeding: aggregatedplatelets with fibrinogen attached to their Gpllb-Ma receptors form a plug.
3. Coagulation phase: thrombin produced by activation ofthe coagulation cascade converts fibrinogen into insol-uble fibrin monomers to form a stable plug.
4. Fibrinolytic phase: plasmin cleaves the insoluble fibrinmonomers that hold the platelet plug together and re-establishes blood flow.
II. Laboratory Findings Associated With Hemostasis (Table 14-1)A. Blood studies of platelets and platelet function
1. Platelet count2. Bleeding time
a. Detects platelet abnormalities that occur before theformation of the temporary platelet plug
b. May be prolonged in several disorders (Table 14-2)3. vWF tests
a. Ristocetin cofactor assay: evaluates vWF function(1) Deficient vWF in classic von Willebrand's
disease(2) Absent GpIb receptor for vWF in Bernard-
Soulier syndromeb. vWF antigen assay: measures the quantity of vWF
(regardless of function); decreased vWF antigen inclassic von Willebrand's disease
c. Agar gel electrophoresis: evaluates the size distribu-tion of circulating vWF multimers and helps dif-ferentiate subtypes of von Willebrand's disease
B. Laboratory studies used to evaluate coagulation factors1. Prothrombin time (PT)
a. International normalized ratio standardizes the PT,so that the results are the same regardless of the re-agents used.
Most commoncause of prolongedbleeding time:aspirin
PT is prolonged onlywhen the level ofa clotting factoris 30-40% ofnormal.

Chapter 14 Coagulation Disorders 167
TABLE 14-2 Causes of Prolonged Bleeding Time
Cause
Nature of Defect
Comments
Aspirin or NSAIDs Platelet aggregation defect Normal platelet count
Inhibition of platelet cyclooxygenase,which ultimately inhibits synthesis ofthromboxane A2
Bernard-Soulier syndrome Platelet adhesion defect
Thrombocytopenia, giant plateletsAutosomal recessive disease with
Lifelong bleeding problem
absent Gplb platelet receptors for vWF
Glanzmann's disease Platelet aggregation defect
Lifelong bleeding problemAutosomal recessive disease with
absent GpIlb-Illa fibrinogen receptors;absent thrombosthenin
Renal failure Platelet aggregation defect
Reversed with dialysis and desmo-Inhibition of platelet phospholipid by
pressin acetatetoxic products
Scurvy Vascular defect May cause hemarthroses
Caused by vitamin C deficiency; defec-tive collagen resulting from poorcross-linking
Thrombocytopenia Decreased platelet number
Prolonged bleeding time when plateletcount < 90,000 cells/4
Von Willebrand's disease
Platelet adhesion defect
Other factor VIII coagulation defectsAutosomal dominant disorder with
absent or defective vWF
NSAID, nonsteroidal ant-inflammatory drug; vWF, von Willebrand's factor
b. Uses(1) Evaluates the extrinsic system in clot forma-
tion: factors VII, X, V, prothrombin, and fibrin-ogen (see Figure 14-1)
(2) Monitors patients taking warfarin derivatives(3) Evaluates liver function
2. Partial thromboplastin time (PIT)a. Evaluates the intrinsic system in clot formation:
factors XII, XI, IX, VIII, X, V, prothrombin, and fi-brinogen (see Figure 14-1)
b. Monitors patients taking heparin
Anticoagulation with heparin or warfarin causesboth the PT and PTT to become prolonged,because both drugs inhibit factors in the finalcommon pathway. Studies show that the PTmonitors warfarin more accurately, whereas thePTT monitors heparin more accurately.
••••••••••••••••••••••••••••••••••••

168 Pathology
Figure 14-3 Petechiae in idio-pathic thrombocytopenic purpurashowing pinpoint hemorrhages,a sign of platelet dysfunction,in the skin over the thorax andshoulders. When touched, pete-chiae do not blanch withpressure
C. Studies used to evaluate fibrinolysis1. Tests
a. FDP assay: detects all products of plasmin cleavageof fibrinogen or insoluble fibrin clots
b. D-dimer assay: detects only cross-linked insolublefibrin monomers in a fibrin clot
2. Usesa. Screening for disseminated intravascular coagulation
(DIC) and pulmonary embolib. Evaluating response to fibrinolytic therapy for de-
grading venous and arterial clots
III. Platelet DisordersA. Clinical findings associated with platelet dysfunction
1. Epistaxis (nosebleeds): most common symptom of plate-let dysfunction
2. Petechiae and multiple small ecchymoses (purpura)a. Petechiae: pinpoint areas of hemorrhage
(Figure 14-3)b. Ecchymoses (purpura): larger areas of hemorrhage
(diameter, 2 cm)
Palpable (raised) purpura is a sign of a small-vessel vasculitis, not a platelet disorder. Senilepurpura is a normal finding in elderly patientsand results from impaired collagen productionand capillary fragility. Nonpalpable purpura de-velops in areas of trauma (e.g., back of hands,shins) (Figure 14-4).
3. Bleeding from superficial cuts or abrasions: no tempo-rary platelet plug is present to stop bleeding.
4. Easy bruisability, bleeding from mucosal membranes;central nervous system, gastrointestinal, and genitouri-nary bleeding occur in cases of very severe thrombo-cytopenia.
B. Quantitative platelet disorders1. Thrombocytopenia: decreased number of platelets
a. Pathogenesis
Chronic autoim-mune thrombocyto-penic purpura ismost common inwomen with SLE.

Chapter 14 Coagulation Disorders 169
Figure 14-4 Senile purpurashowing the large, irregular areasof hemorrhage on the back ofboth hands This benign condi-tion primarily occurs in bodyareas that are frequently trauma-tized
(1) Decreased production (e.g., aplastic anemia,leukemia)
(2) Immunologic destruction (e.g., autoimmunerombocyto pem a I
(3) Increased consumption (e.g., thromboticthromhocytopenic purpura, DIC)
(4) Sequestration in the spleen (e.g., hypersplen-ism in portal hypertension)
b. Types (Table 14-3)2. Thrombocytosis: increased platelet count
a. Reactive thrombocytosis (e.g., chronic iron defi-ciency, infections, splenectomy, malignancy)
b. Primary thrombocytosis (e.g., essential throm-bocythemia, polycythemia Vera)
C. Qualitative platelet disorders: acquired (e.g., aspirin use) orhereditary (e.g., Glanzmann's disease) (see Table 14-2)
IV. Coagulation DisordersA. Pathogenesis
1. Decreased production (e.g., hemophilia A, cirrhosis)2. Pathologic inhibition (e.g., acquired circulating anti-
bodies, or inhibitors, of coagulation factors)3. Excessive consumption of coagulation factors in fibrin
clots (e.g., DIC)B. Clinical findings
1. Delayed bleeding (e.g., tooth extraction, appendectomy)a. A temporary platelet plug is the only mechanical
block preventing bleeding; this plug is easily dis-lodged, causing delayed bleeding.
h. Lack of thrombin prevents formation of a stableplatelet plug.
2. Menorrhagia3. Gastrointestinal and genitourinary bleeding4. Hemarthroses and retroperitoneal bleeding: only in
severe factor deficiencies (factor levels < 5%)
Most commoncause of thrombocy-topenia in children:idiopathic throm-bocytopenic purpura

IgG antibodies directed against GpIlb:Illareceptors (type II hypersensitivityreaction)
Macrophages phagocytose platelets
IgG antibodies directed against Gpllb:lllareceptors (type II hypersensitivityreaction)
Type I variant: nonimmune; developsearly; transient
Type II variant: macrophage removal ofplatelets surfaced by IgG antibodydirected against heparin attached toPF 4 (type II hypersensitivity)
Similar to ITP
Deficiency in vWF-cleaving metallopro-tease in endothelial cells
Increase in circulating multimers of vWFincreases platelet adhesion to areas ofendothelial injury at arteriole-capillaryjunctions
Platelets are consumed because ofproduction of platelet thrombi in areasof injury (not DIC)
Endothelial damage at arteriole-capillaryjunction caused by Shiga-like toxin of0157:H7 serotype of Escherichia coli;organisms proliferate in undercookedbeef
Causes thrombocytopenia in children;abrupt onset after upper respiratorytract infection
Responds well to corticosteroids
Most common in women with SLE;insidious onset
Splenomegaly
Occurs 5-14 days after heparin treat-ment
Must discontinue heparin
Most common hematologic abnormalityin HIV (not AIDS-defining condition)
Occurs in women; usually acquiredClinical pentad: fever, thrombocyto-
penia, renal failure, microangiopathichemolytic anemia with schistocytes(damage by platelet thrombi), CNSdeficits
Occurs in childrenClinical findings similar to TM; CNS
findings are less frequent
PF4, platelet factor 4; SLE, systemic lupus
Factor VIII:C activityof < 1`)/0 leads tosevere hemophilia
170 Pathology
TABLE 14-3 Types of Thrombocytopenia
Type
Pathogenesis
Comments
Idiopathic thrombocyto-penic purpura (ITP)
Chronic autoimmunethrombocytopenicpurpura
Heparin-induced throm-bocytopenia
HIV thrombocytopenia
Thrombotic thrombocy-topenic purpura (TTP)
Hemolytic uremicsyndrome
••••••••••••••••••••••••••••••••••••
CNS, central nervous system; DIC, disseminated intravascular coagulation;erythematosus; vWF, von Willebrand's factor.
C. Hemophilia A1. Epidemiology
a. X-linked recessive trait: females are usuallyasymptomatic carriers who transmit the X chromo-some to 50% of their male offspring and to 50% oftheir female offspring.
b. Female carriers with symptomatic disease: causedby inactivation of more maternal than paternal Xchromosomes; females are homozygous for the ab-normal X chromosome.
c. No family history of hemophilia: disease mostlikely caused by a mutation (30% of cases)
2. Pathogenesis: decreased synthesis of factor VIII:coagu-lant (VIII:C); disease severity correlates with factor VIII:Cactivity.

Chapter 14 Coagulation Disorders 171
3. Clinical findingsa. Bleeding problems related to circumcision or um-
bilical cord separation (factor VIII:C levels < 30%)b. Severe disease: spontaneous hemarthroses
Hemophilia B (Christmas disease) is an X-linkedrecessive disorder that involves a deficiency offactor IX. It is clinically indistinguishable fromhemophilia A.
4. Laboratory findings in hemophilia Aa. Prolonged PTT, with normal PTb. Decreased factor VIII:C activityc. Decreased factor VIII antigen (VIII:Ag): uses anti-
bodies directed against the protein component offactor VIII
d. Normal bleeding time: vWF is present.e. Detection of female carriers: DNA techniques are
most sensitive.5. Treatment
a. Mild cases: desmopressin acetate, which increasesVIII:C and vWF synthesis
b. More severe cases: recombinant factor VIII, whichcarries no risk of HIV
D. Classic von Willebrand's disease: autosomal dominant coag-ulation disorder1. Pathogenesis: combination of platelet and coagulation
factor dysfunction; decreased vWF and factor VIII:Cactivity
2. Clinical findingsa. Increased bruising, bleeding from mucous mem-
branes, menorrhagiab. Associated angiodysplasia of the right colon (see
Chapter 17)3. Laboratory findings
a. Prolonged PTT, with normal PTb. Prolonged bleeding time caused by a platelet adhe-
sion defectc. Abnormal ristocetin cofactor assay: decreased func-
tioning of vWFd. Decreased vWF antigen: decreased quantity of vWFe. Decreased VIII:Ag and VIII:C activity
4. Treatment: desmopressin acetate (increases vWF andVIII:C synthesis); oral contraceptives (action of estrogenis similar to that of desmopressin)
E. Disseminated intravascular coagulation (DIC)1. Pathogenesis
a. Activation of the coagulation system results fromexposure to tissue thromboplastin and/or endothelialcell injury.
Most common he-reditary coagula-tion disorder: vonWillebrand's disease
Treatment of choicefor mild von Wide-brand's disease andhemophilia A: des-mopressin acetate

172 Pathology
b. Obstruction of blood flow and consumption of co-agulation factors (fibrinogen, prothrombin, factorsV and VIII) and platelets result from the develop-ment of fibrin clots (thrombi) in the microcircula-tion.
c. Activation of fibrinolytic system involves second-ary fibrinolysis caused by activation of plasmino-gen directed by factor XII.
d. Consumption of coagulation factors causesbleeding.
2. Etiologya. Sepsis (most common cause): most often due to
Escherichia coli; also common in meningococcemiadue to Neisseria meningitidis
b. Disseminated malignancy (e.g., acute promyelo-cytic leukemia, pancreatic cancer)
c. Trauma with crush injuries, rattlesnakeenvenomation
3. Clinical findings: shock; diffuse, slow bleeding frombreaks in the skin and mucous membranes
4. Laboratory findingsa. Coagulation tests: prolonged PT and PTT; decreased
fibrinogenb. Platelet tests: thrombocytopenia, prolonged bleed-
ing timec. Fibrinolysis tests: presence of FDPs and D-dimers
(most sensitive overall test for DIC)d. Normocytic anemia with schistocytes and reticu-
locytosis: fibrin clots damage red blood cells, produc-ing schistocytes (microangiopathic hemolyticanemia)
5. Treatment: treat underlying disease (most important)
V. Thrombotic DisordersA. Acquired thrombosis
1. Associated conditions: multiple abortions (thrombosisof placental bed vessels), strokes, thromboembolism
2. Etiologya. Postoperative state: stasis of blood flowb. Malignancy: increase in coagulation factors, throm-
bocytosis, release of procoagulants from tumorsc. Folate deficiency: increased plasma homocysteine
levels (see Chapter 11)d. Oral contraceptives: increased synthesis of coagula-
tion factors and decreased antithrombin III causedby estrogen
e. Hyperviscosity: polycythemia vera, WaldenstrOm'smacroglobulinemia
B. Hereditary thrombosis: autosomal dominant disorders
DIC is a consump-tion coagulopathy.

•• Chapter 14 Coagulation Disorders 173
1. Clinical findingsa. Deep venous thrombosis and pulmonary emboli at
an early ageb. Venous thrombosis at unusual sites (e.g., hepatic
vein)2. Factor V Leiden mutant: proteins C and S cannot
degrade factor V Leiden.3. Antithrombin III deficiency
a. Functions of antithrombin III: activity is enhancedby heparin; neutralizes serine proteases (e.g., factorsXII, XI, IX, X, prothrombin, thrombin)
b. Clinical finding: no lengthening of the PTT withheparin therapy at a standard dose
4. Protein C and S deficiency: inability to inactivate factorsV and VIII
•••, •i o1 •, •••••••
Most common he-reditary cause ofthrombosis: factor VLeiden
•••••••••••••••••••••

1544614DBlood Transfusion
04
Disorders
I. ABO Blood GroupsA. Group 0
1. Most common blood group: no blood group antigensare present on the red blood cell (RBC) membrane.
2. Natural antibodies in serum: anti-A IgM, anti-B IgM;some individuals also have anti-AB IgG.
3. Increased incidence of duodenal ulcersB. Group A
1. Anti-B IgM antibodies2. Increased incidence of gastric carcinoma
C. Group B: anti-A IgM antibodiesD. Group AB
1. Least common blood group2. No natural antibodies
E. Newborns do not have natural antibodies at birth; anyantibodies in a newborn's serum are IgG antibodies of ma-ternal origin.
F. Elderly individuals often lose their natural blood group anti-bodies and therefore may not have an immune reaction ifthey are transfused with blood of the wrong ABO group.
II. Rh and Non-Rh Antigen SystemsA. Rh antigen system
1. The Rh antigen system has three adjoining gene loci: onelocus codes for D antigen, another locus codes for Cand/or c antigen, and the remaining locus codes for Eand/or e antigen.
2. An individual who is Rh positive is D antigen positive.a. About 85% of the population is D antigen positive.b. Those lacking D antigen are considered Rh negative.
B. Clinically important non-Rh antigens1. Duffy (Fy) antigens: Fy antigens are the binding site for
infestation of RBCs by Plasmodium vivax.
Newborns do nothave natural bloodgroup antibodies;the elderly may losethem
Many AfricanAmericans lack theFy antigen, whichoffers protectionagainst contractingP vivax malaria.
174

Chapter 15 Blood Transfusion Disorders 175
2. I antigen system: cold-reacting IgM antibodies (cold ag-glutinins) against i antigen or I antigen may result incold autoimmune hemolytic anemia.
III. Blood Transfusion TherapyA. Tests performed on donor blood
1. Group (ABO) and type (Rh)2. Antibody screen (indirect Coombs' test): detects atypi-
cal antibodies (e.g., anti-D, anti-Kell)3. Screening for infectious diseases: serologic test for syph-
ilis, hepatitis B surface antigen for hepatitis B, hepatitisC antibodies, HIV-1 and -2 antibodies, HTLV-1 anti-bodies, West Nile virus
A risk remains for transmitting infection when trans-fusing blood, because antibodies develop at differentrates against different infections. The risk of contract-ing disease per unit of blood is 1:3300 for hepatitis C,1:200,000 for hepatitis B, and 1:676,000 for HIV. Themost common infectious agent transmitted by bloodtransfusion is cytomegalovirus, which lives in donorlymphocytes.
B. Patient cross-match1. Components of a standard cross-match: ABO group
and Rh type; antibody screen for atypical antibodies;direct Coombs' test (to identify IgG antibodies on RBCs)
2. Major cross-matcha. Serum is mixed with a sample of RBCs from the
donor unit.b. Purpose of cross-match: to detect atypical antibod-
ies in the recipient's serum that are directed againstforeign antigens on donor RBCs
c. A compatible cross-match has no RBC agglutinationor hemolysis; it does not guarantee that the recipi-ent will not develop atypical antibodies, a transfu-sion reaction, or an infection.
Atypical antibodies develop when an individualis exposed to an Rh or non-Rh antigen (e.g., Kellantigen) they lack. They may produce a hemo-lytic transfusion reaction if blood containing theforeign antigen is infused into the patient.
3. Group 0 packed RBCs can be transfused into anypatient, regardless of blood group.a. Individuals that are blood group 0 are universal
donors, because there are no blood group antigens

176 Pathology
on the surface of blood cells against which thenatural antibodies in transfused blood might react.
b. Individuals that are blood group 0 can receiveonly 0 blood, because they have anti-A IgM andanti-B IgM antibodies, which can destroy transfusedA, B, or AB RBCs.
4. Individuals that are blood group AB are universal re-cipients, because they have no natural antibodies todestroy transfused A, B, or AB blood cells.
Before blood is transfused into newborns or individu-als with immunodeficiencies, the blood must be irra-diated to kill donor leukocytes. This prevents therecipient from developing a graft-versus-host reac-tion or cytomegalovirus infection.
C. Transfusion reactions1. Allergic reaction: most common transfusion reaction
a. Pathogenesis: type I IgE-mediated hypersensitivityreaction against proteins in the donor blood
b. Clinical findings: urticaria, fever, wheezing, and thepotential for anaphylactic shock; mild cases aretreated with antihistamines.
2. Febrile reactiona. Pathogenesis: type II cytotoxic antibody hypersen-
sitivity reaction; recipient has anti-HLA antibod-ies directed against foreign HLA antigens ondonor leukocytes (no HLA antigens on RBCs).
b. Clinical findings: fever, chills, headache, and flush-ing; treated with antipyretics
3. Acute hemolytic transfusion reaction: may be intravas-cular or extravascular hemolysisa. Intravascular hemolysis: IgM-mediated hemolysis
of donor RBCs(1) Patient is transfused with an incompatible
blood group.(2) Example: patient with blood group B receives
group A blood.b. Extravascular hemolysis
(1) An atypical antibody in a recipient reacts with aforeign antigen on the donor RBCs, resulting inmacrophage phagocytosis of the RBCs coatedby the atypical antibody.
(2) Jaundice occurs because unconjugated biliru-bin is the end-product of macrophage degrada-tion of hemoglobin.
Patients who were infused with blood20-30 years earlier may have been exposedto a foreign blood group antigen and devel-
Blood group 0 isthe universal donor;blood group AB isthe universalrecipient.

Chapter 15 Blood Transfusion Disorders 177
oped atypical antibodies that are no longercirculating. Reexposure to the foreign anti-gen causes memory B cells to transforminto plasma cells and produce the antibody,resulting in an extravascular hemolytic ane-mia. A delayed hemolytic transfusion re-action may occur 3-10 days after the trans-fusion.
c. Clinical findings: hypotension, fever, disseminatedintravascular coagulation, oliguria (renal failure)
d. Laboratory findings in recipients(1) Positive direct Coombs' test (antibodies and/or
complement coating donor RBCs) and positiveindirect Coombs' test (atypical antibodies arepresent in serum)
(2) No increase in hemoglobin over pretransfu-sion levels
(3) Hemoglobinuria (sign of intravascular hemoly-sis), unconjugated hyperbilirubinemia (signof extravascular hemolysis)
IV. Hemolytic Disease of the Newborn (HDN)• HDN occurs from transplacental passage of maternal IgG anti-
bodies (e.g., anti-D antibodies, anti-AB antibodies in blood group0 mothers), resulting in an extravascular hemolytic anemia inthe fetus.
A. ABO HDN1. Most common HDN: 20-25% of all pregnancies; may
occur in any pregnancy2. Pathogenesis
a. Mother is blood group 0 and fetus is either bloodgroup A or B: some individuals with blood group0 have naturally occurring anti-AB IgG antibodies.
b. Anti-AB IgG antibodies cross the placenta andattach to fetal A or B RBCs; fetal splenic macrophagesphagocytose and destroy the RBCs, causing anemia.
c. Unconjugated bilirubin from extravascular hemoly-sis in the fetus is conjugated in the mother's liver.
3. Clinical and laboratory findingsa. Jaundice develops within the first 24 hours after
birth.(1) Jaundice develops because the conjugating
enzymes in the fetal liver are unable to processthe excess bilirubin load.
(2) Risk of kernicterus is minimal: kernicterus isdeposition of lipid-soluble unconjugated biliru-bin in the basal ganglia; it causes severe braindamage.
b. Mild normocytic anemia, or no anemia
Most commoncause of jaundice infirst 24 hours afterbirth: ABO HDN

178 Pathology
c. Positive direct Coombs' test on fetal cord bloodRBCs from anti-AB IgG antibodies coating fetal A orB RBCs
d. Spherocytes in cord blood peripheral smear: mac-rophages remove a portion of the RBC membrane,causing the cells to become spherocytes.
B. Rh HDN1. Pathogenesis
a. Mother is Rh (D antigen) negative and fetus is Rhpositive.
b. Mother may be exposed to fetal Rh-positive blood(fetomaternal bleed) during the last trimester orduring childbirth.
c. Anti-D IgG antibodies develop in the mother onexposure to fetal Rh-positive cells after delivery;hence, the first Rh-incompatible pregnancy does notaffect the fetus.
d. Subsequent Rh-incompatible pregnancies result inextravascular hemolytic anemia in the fetus.(1) Anti-D IgG antibodies cross the placenta and
attach to fetal Rh-positive RBCs; fetal macro-phages phagocytose RBCs, causing severeanemia.
(2) Fetus may develop cardiac failure in utero,leading to hydrops fetalis and death.(a) Hydrops fetalis is biventricular congestive
heart failure with ascites and edema.(b) Extramedullary hematopoiesis in the
spleen and liver is commonly present inthe fetus.
e. Rh-negative mothers who are negative for anti-Dantibodies receive anti-D globulin (Rh immuno-globulin) during week 28 of pregnancy.(1) Anti-D globulin does not cross the placenta.(2) Anti-D globulin protects the mother from sensi-
tization to fetal Rh-positive cells that may enterher circulation during the last trimester.
f. Additional anti-D globulin is given to the motherafter delivery if the infant is Rh positive.
Anti-D globulin masks the antigenic sites on thefetal RBCs, so that the mother does not developan antibody response against the D antigen. Ifthe mother develops anti-D antibodies, there isno indication for giving anti-D globulin eitherduring or after delivery, because its main purposeis to prevent sensitization.
2. Clinical and laboratory findingsa. Anemia is severe in Rh HDN.b. Jaundice develops shortly after birth.

Chapter 15 Blood Transfusion Disorders 179
(1) Level of unconjugated bilirubin is much higherthan in ABO HDN: much of the unconjugatedbilirubin is not bound by albumin and cir-culates freely in the blood.
(2) Increased risk of kernicterusc. Positive direct and indirect Coombs' tests on fetal
cord bloodd. Exchange transfusions usually are required: the in-
fant's blood is removed and replaced with freshblood.
ABO incompatibility protects the motheragainst Rh sensitization. For example, if themother is 0 negative and the fetus is A positive,any A-positive fetal RBCs entering the mother'scirculation are immediately destroyed by mater-nal anti-A IgM antibodies, thereby preventingsensitization.
C. Treatment of jaundice in HDN1. Unconjugated bilirubin in the skin absorbs light energy
from blue fluorescent light.2. Photoisomerization converts unconjugated bilirubin to
a nontoxic, unconjugated water-soluble dipyrrole(lumirubin) that is excreted in either bile or urine.

161116. 4,4*-
Respiratory Disorders 7
I. Upper Airway DisordersA. Choanal atresia
1. Unilateral or bilateral bony septum between the noseand the pharynx prevents newborn from breathingthrough the nose.
2. Newborn turns cyanotic when breastfeeding; cryingcauses infant to "pinken up" again.
B. Types of nasal polyps1. Allergic: most common type of polyp
a. Most often seen in adults with a history of IgE-mediated allergies
b. Nasal smears show numerous eosinophils.2. Associated with nonsteroidal anti-inflammatory
drugs (NSAIDs)a. Polyps most often occur in women with various
pain syndromes (e.g., headache).b. Clinical triad: NSAIDs may lead to asthma and
the formation 01 nasal polyps.(1) NSAIDs block cyclooxygenase, opening the
lipoxygenase pathway.(2) Leukotrienes C, D, and E4 are produced and
serve as potent bronchoconstrictors.3. Associated with cystic fibrosis (CF): nasal pol yps in a
child warrant a sweat test to rule out CF.C. Obstructive sleep apnea
• Apnea is the interval of breath cessation associated withexcessive snoring.
1. Pathogenesis: airway obstruction causes carbon dioxide(CO?) retention, leading to hypoxemia.
2. Etiology: obesity, pharyngeal muscle collapse on inspi-ration, tonsillar hypertrophy, nasal septum deviation
3. Clinical findings: excessive snoring with episodes ofapnea, daytime somnolence, headache, irritability
NSAIDs —> asthma-> nasal polyps
Polysomnographydocuments periodsof apnea duringsleep.
180

Chapter 16 Respiratory Disorders 181
4. Complicationsa. Cor pulmonale: pulmonary hypertension (due to
vasoconstrictive effects of chronic hypoxemia andrespiratory acidosis) and right ventricularhypertrophy
b. Secondary polycythemia: due to hypoxemic stim-ulus for erythropoietin release
D. Sinusitis1. Sites: maxillary sinus is most often involved in adults,
ethmoid sinus in children.2. Pathogenesis
a. Blockage of drainage into the nasal cavity (e.g.,viral upper respiratory infection, deviated nasalseptum)
b. Pathogens causing sinusitis: Streptococcus pneumo-niae, rhinoviruses, anaerobes (chronic sinusitis),systemic fungi (e.g., Mucor, especially in diabetesmellitus)
3. Clinical and laboratory findings: fever, nasal conges-tion, pain over sinuses; computed tomography is themost sensitive test.
E. Nasopharyngeal carcinoma1. Epidemiology: most common malignant tumor of the
nasopharynx; occurs more often in males2. Pathogenesis: causal relationship with Epstein-Barr
virus3. Clinical and pathologic findings
a. Keratinizing or nonkeratinizing squamous cellcancer or undifferentiated cancer
b. Metastasizes to cervical lymph nodes: nasophar-ynx should be biopsied to determine if there iscervical lymph node metastasis when no primarycancer is found.
F. Laryngeal carcinoma1. More common in men2. Risk factors: cigarette smoking, alcohol (synergistic
effect with smoking), asbestos exposure, squamous pap-illomas (human papilloma virus association)
3. Most are keratinizing squamous cell carcinomaslocated on the vocal cords.
4. Clinical findings: persistent hoarseness, palpable cer-vical lymph nodes (due to metastasis)
II. Atelectasis• Atelectasis is loss of lung volume due to inadequate expansion
of the air spaces (collapse).A. Resorption atelectasis
1. Pathogenesisa. Airway obstruction prevents air from reaching the
alveoli.
••••S
•So•••••••••••••••••••••••••••
Most commoncause of sinusitis:S. pneumoniae
Most commoncause of squamouscell carcinomas inthe larynx andoropharynx:smoking

182 Pathology
b. Causes of obstruction: mucous or mucopurulentplug, aspiration of foreign material, broncho-genic carcinoma
c. Cause of alveolar collapse: lack of air and distal re-sorption of preexisting air; may involve a com-plete lung, an entire lobe, or segments of a lobe
2. Clinical findingsa. Fever and dyspneab. Dullness to percussion and absent vocal vibra-
tory sensation (tactile fremitus) over the area ofconsolidation
Resorption atelectasis due to mucous plugsblocking the terminal bronchioles is commonin the postoperative period. During this time, apatient's breathing may be restricted because ofpain and fear of coughing. The patient mayneed breathing treatments to help lyse themucous plugs to prevent atelectasis, which of-ten develops into pneumonia if untreated.
B. Compression atelectasis• Air or fluid in the pleural cavity under increased pressure
(e.g., tension pneumothorax, effusion) collapses smallairways under the pleura.
C. Atelectasis due to loss of surfactantRespiratory distress syndrome (RDS) in newborns
1. Causes of decreased surfactant in the fetal lungsa. Prematurityb. Maternal diabetes: fetal hyperglycemia increases
insulin release, which inhibits surfactant synthesis.c. Cesarean section: no stress-induced increase in
cortisol from a vaginal delivery
Women who must deliver their infants prema-turely receive glucocorticoids to increase fetalsurfactant synthesis, hence reducing the poten-tial for RDS developing in the newborn. Sur-factant (lecithin) decreases surface tension,which keeps the small airways open duringexpiration.
RDS is decreasedsynthesis of surfac-tant resulting incollapse of alveoli.
Most commoncause of fever24-36 hours aftersurgery: resorptionatelectasis
••••••••••••••••••••••••••••
2. Widespread atelectasis: causes massive intrapulmo-nary shunting, because pulmonary capillary blood isnot being oxygenated
3. Gross findings: purple-red lungs4. Microscopic findings: collapsed alveoli lined by
hyaline membranes derived from necrotic cellulardebris and proteins leaking from damaged pulmonaryvessels (Figure 16-1)
•••••••

Chapter 16 Respiratory Disorders 183
Figure 16-1 Neonatal respira-tory distress syndrome Some ofthe dilated respiratory bronchi-oles and alveolar ducts are linedwith a fibrin-rich membrane(hyaline membrane; arrow). Thesubjacent alveoli are collapsed.
5. Clinical findings: respiratory difficulty within a fewhours after birth, cyanosis, grunting with nasal flaring
6. Complicationsa. Superoxide free radical (02i) damage from
oxygen (02) therapy: may result in blindness andpermanent damage to small airways (broncho-pulmonary dysplasia)
b. Intraventricular hemorrhage, patent ductus arterio-sis with machinery murmur (due to persistenthypoxemia), necrotizing enterocolitis
III. Respiratory InfectionsA. Pneumonia
1. Classified as community acquired (typical or atypical)or nosocomial (hospital acquired)
2. Typical community-acquired pneumonia (Table 16-1)a. Majority of cases caused by bacterial pathogens;
most often due to S. pneumoniae (Figure 16-2)b. Risk factors for S. pneumoniae (pneumococcus)
pneumonia include splenic dysfunction andimmunodeficiency.
c. Bronchopneumonia: begins as acute bronchitisand spreads locally into the lungs(1) Usually involves the lower lobes or right
middle lobe(2) Patchy yellow areas of consolidation; micro-
abscesses are present in the areas ofconsolidation.
d. Lobar pneumonia: complete or almost completeconsolidation of a lobe of the lung
e. Complications of bronchopneumonia and lobarpneumonia: lung abscesses, empyema (pus in thepleural cavity), excessive scar tissue, sepsis
f. Clinical findings(1) Sudden onset of symptoms: high fever with
productive cough
Most commoncause of typicalcommunity-acquiredpneumonia: S.pneumoniae

184 Pathology
TABLE 16-1 Pathogens That Cause Bacterial Respiratory Infections
Reaction onPathogen Gram Stain
Comments
Haemophilus Gram-negative rod Bronchitis, bronchopneumonia, sinusitis,influenzae acute epiglottitis (inspiratory stridor)
Klebsiella Gram-negative rod Typical pneumonia in individuals with alco-pneumoniae holism and elderly patients in nursing
homesBlood-tinged, thick, mucoid sputum
Legionella Gram-negative rod Water-loving bacteria (water coolers, waterpneumophila mists on vegetables in grocery stores)
High fever, dry cough, malaise, and flu-likesymptoms
Mycobacterium Gram-resistant Primary TB involves upper part of lowertuberculosis lobe or lower part of upper lobe
Reactivation TB involves upper lobes andcavitates
Pseudomonas Gram-negative rod Water-loving bacteria transmitted byaeruginosa respirators
Green-colored sputum (pyocyanin)Common cause of pneumonia and death in
cystic fibrosis
Staphylococcus Gram-positive coccus Pneumonia often superimposed on influ-aureus enza pneumonia
Lung pathogen in cystic fibrosisProduces pleural tension pneumatocysts
(danger of pneumothorax)
Streptococcus Gram-positive Common cause of community-acquiredpneumon,ae diplococcus pneumonia
Risk factors: older age, splenic dysfunction,AIDS
TB, tuberculosis
Figure 16-2 Gram stain of .1:Streptococcus pneumoniae The ri A(sputum stain shows numerous ; 3y..: -%-
.': kv, .. ,:..;lancet-shaped diplococci with the .■• ,... 1,... ...... "_ztapered ends pointing to each l.cother. A few neutrophils contain .- ..r.
..91 1. 1phagocytosed bacteria

Chapter 16 Respiratory Disorders 185
TABLE 16-2 Pathogens That Cause Chlamydial, Rickettsial, Mycoplasmal,and Viral Respiratory Infections
Pathogen Comments
Chlamydiapneumoniae
C. psittaci
C. trachomatis
Coxiella burnetii(Q fever)
Mycoplasmapneumoniae
Cytomegalovirus
Influenza virus
Parainfluenza virus
Respiratorysyncytial virus
Rhinovirus
Rubeola
Common cause of atypical pneumonia
Causes ornithosis, a zoonosis transmitted by avian intermediary(e.g., parrot)
Atypical pneumonia
Common cause of pneumonia in newbornsAfebrile, staccato cough (choppy cough), conjunctivitis, wheezing
(bronchiolitis)
Rickettsia transmitted without vectorContracted by dairy farmers and veterinarians associated with
birthing process of sheep, cattle, and goats, and handlers ofmilk or feces from these animals
Cause of atypical pneumoniaCommon among adolescents and military recruitsComplications: bullous myringitis (vesicles on membranes),
erythema multiforme, intravascular hemolytic anemia (coldagglutinins in blood)
Pneumonia in immunocompromised hostsEnlarged alveolar macrophages, pneumocytes, and endothelial
cells contain basophilic intranuclear inclusions surroundedby halo
Common cause of death in elderly patients with underlying renal,cardiac, or lung disorder
Cause of croup (laryngotracheobronchitis) in infantsInspiratory stridor due to tracheal obstruction
Common viral cause of pneumonia and bronchiolitis (wheezing)in children
Cause of common cold
Pneumonia is most common cause of deathWarthin-Finkeldey multinucleated giant cells are characteristic
finding
(2) Signs of consolidation (alveolar infiltrate):dullness to percussion; increased vocal tactilefremitus
(3) Chest radiograph: patchy infiltrates (bron-chopneumonia) or lobar consolidation
g. Laboratory findings: positive Gram stain, neutro-philic leukocytosis
3. Atypical community-acquired pneumonia(Table 16-2)a. Epidemiology: usually involves school-age chil-
dren and young adults; occurs in crowded condi-tions (schools, prisons, military barracks)
b. Etiology: most often caused by Mycoplasma pneu-moniae; additional pathogens include Chlamydiapneumoniae, C. trachomatis, and viruses (respiratorysyncytial, influenza, adenovirus).
Most commoncause of atypicalcommunity-acquiredpneumonia: M.pneumoniae

186 Pathology
Figure 16-3 CytomegalovirusThe enlarged nuclei of manyof the type I pneumocytescontain large inclusions (baso-philic staining with H&E stain)surrounded by a clear halo
Figure 16-4 Pneumocystiscarinii pneumonia. This silver-impregnated cytologic smearprepared from bronchial wash-ings in an HIV-positive patientcontains numerous P cariniicysts Some cysts look likecrushed ping-pong balls
4
c. Patchy interstitial pneumonia: mononuclear infil-trate; alveolar spaces usually free of exudate (nosigns of consolidation)
d. Clinical findings(1) Insidious onset with low-grade fever(2) Flu-like symptoms: pharyngitis, laryngitis,
mildly productive cough(3) No signs of consolidation
4. Nosocomial pneumoniaa. Associated with severe underlying disease, antibi-
otic therapy, immunosuppression, respirators, andindwelling venous catheters
b. Due to gram-negative bacteria (e.g., Escherichiacolt, Pseudomonas aeruginosa) and gram-positivebacteria (e.g., Staphylococcus aureus)
5. Pneumonia in immunocompromised hostsa. Usually occurs in AIDS and in bone marrow
transplantationb. Common opportunistic infections
(1) Cytomegalovirus (Figure 16-3; see Table 16-2)(2) Pneumocystis carinii (Figure 16-4; Table 16-3)
P aeruginosa pneu-monia is mostcommonly trans-mitted in hospitalsvia respirators.

Chapter 16 Respiratory Disorders 187
B. Tuberculosis (TB)1. Epidemiology
a. Inhalation of the bacterium (droplet infection)b. Screening: purified protein derivative (PPD) intra-
dermal skin test; does not distinguish active frominactive disease
2. Etiology: caused by Mycobacterium tuberculosis; strictaerobe; acid-fast due to mycolic acid in the cell wall
3. Primary TBa. Initially localizes in a subpleural location: upper
part of the lower lobes or lower part of the upperlobes(1) Ghon's focus: area of caseous necrosis in sub-
pleural location(2) Ghon's complex: spread of infection to hilar
lymph nodes with caseous necrosis in nodesas well
b. Usually resolves: calcified granuloma or area ofscar tissue may be a nidus for reactivation TB.
•••••••••••••••••••
•••••••S••••••
TABLE 16-3 Pathogens That Cause Systemic Fungal Respiratory Infections
Pathogen Comments
Aspergillus fumigatus
Aspergilloma (fungus ball in abandoned cavities)Allergic bronchopulmonary disease (IgE levels increased;
eosinophilia)Vessel invasion produces hemorrhagic infarcts
Blastomyces Occurs in sites similar to histoplasmosis; more commondermatitidis in men
Produces skin and lung diseaseSkin lesions simulate squamous cell carcinoma
Candida albicans
Lung disease contracted from sepsis secondary to infections
from indwelling venous cathetersVessel invasion produces hemorrhagic infarcts
Coccidioides immitis
Contracted by inhaling arthrospores in dust in arid desert
areas in southwestern United States ("valley fever");increased after earthquakes (increased dust)
Flu-like symptoms, erythema nodosum (painful nodules onlower legs)
Cryptococcus Opportunistic fungal infectionneoformans Found in pigeon excreta
Histoplasma Endemic in Ohio and central Mississippi river valleyscapsulatum Associated with excreta of bats (increased incidence in
spelunkers), starlings, and chickens (common amongchicken farmers)
Mucor species
Common in individuals with diabetes mellitus and immuno-suppressed patients
Vessel invasion produces hemorrhagic infarcts
Pneumocystis carinii
Primarily an opportunistic infection; most common initialAIDS-defining infection
Occurs when CD4 helper T cell count < 200/4
Most common in-fectious disease thatcauses deathworldwide: TB

,...:400-■ E0..!: vir •.telmr,tpilii&11-4 SIP ..#' a.46114.71-r_t4eli.;*
‘#
' 6...
. • • 41"
' N. '* •,, I • afr ' . • i.‘
c, !■ t ' f, • z lef "'• ii ■•g • &
' e -sf 4., "'slffer, f- • " . ki.„,T.WI
, 45 14i414/80 t. 6 • • .1.-•li" sa; ii• .. &,
mtatufift;..-- :74.14: ill-.4, Igi - :
,_ grat,41, 0014 .,-..''...?131+ ! 10
188 Pathology
Figure 16-5 Histoplasma capsulatum in a section of adrenal gland from a patient withdisseminated histoplasmosis. Numerous macrophages are filled with small yeast forms (smalldots surrounded by a clear space) of H. capsulatum.
4. Secondary (reactivation) TBa. Reactivation of a previous primary site is the
most common cause.b. Cavitary lesion usually involves one or both
apices in the upper lobes.c. Clinical findings: fever, drenching night sweats,
weight loss, hemoptysisd. Complications
(1) Miliary spread in lungs: invasion into thebronchus or lymphatics
(2) Systemic miliary spread: invasion of pulmo-nary vein; kidneys most common extrapul-monary site
(3) Massive hemoptysis, bronchiectasis, scarcarcinoma
C. Systemic fungal infections (see Table 16-3)a. Usually produce a granulomatous inflammatory
reaction with or without caseationb. Histoplasma capsulaturn is the most common
systemic fungal infection (Figure 16-5).c. Coccidioides immitis is the most common systemic
fungal infection in the desert regions of the south-western United States.
D. Lung abscess1. Etiology
a. Aspiration of oropharyngeal material (e.g.,carious teeth, infected sinus or tonsillar material)
b. Complication of bacterial pneumonia (e.g., Kleb-siella pneumoniae, P. aeruginosa, Staphylococcusaureus, Streptococcus pyogenes)
2. Gross findings: vary in size and location; those due toaspiration are primarily on the right side of the lung(Box 16-1).
Most commoncause of TB inAIDS: M.avium-in tracellulare

-- Right middlelobe
Right lung
••••
Chapter 16 Respiratory Disorders 189
BOX 16-1 Aspiration Sites in the Lungs
• Foreign material localizes to different portions of the lung, depending onthe position of the patient. In the standing or sitting position, material lo-
• calizes in the posterobasal segment of the right lower lobe; in the supineposition, the superior segment of the right lower lobe; and in the right-sided position, the right middle lobe or the posterior segment of the right
•upper lobe. The most common aspiration site is the superior segment ofthe right lower lobe.
•••
Posterior segmentof right upper lobe
•
•
•Posterobasalsegment of
• right lower lobe --
•I
ID.3. Clinical findings
a. Spiking fever with productive cough: foul-smelling sputum in aspiration types is due to the
• presence of anaerobes (e.g., Fusobacterium nu-
•
b .cleatum, Prevotella melanogenicus).Chest radiograph: cavitation with air-fluid level• IV. Vascular Lung Lesions
A. Pulmonary thromboembolism
• 1. Epidemiology and pathogenesisa. Source: 95% originate in deep veins of the lower
• extremity above the popliteal vein (e.g., femoralvein).
• b. Risk factors: postoperative state, trauma, oral con-• traceptives, congestive heart failure, prolonged
c. Size of the embolus determines the caliber of thesitting
•pulmonary artery vessel that is occluded.(1) Large emboli occlude major vessels (saddle
• embolus).(2) Small emboli occlude medium- and small-
sized pulmonary arteries.
• d. Hypoxemia is a potential consequence of pulmo-nary artery occlusion.
•
Superior segmentof right lower lobe —
Bronchial arteryprotects against pul-monary infarction.
•

190 Pathology
Figure 16-6 Hemorrhagic pulmonary infarction The dark, triangle-shaped area of hemor-rhagic infarction extends to the periphery of the lung. The base of the triangle rests on thepleural surface The apex of the triangle normally points to the occluded pulmonary vessel (thevessel is not visible in this specimen)
In a patient with normal bronchial artery bloodflow and ventilation, a pulmonary embolusresults in a hemorrhagic infarct in less than10% of cases. However, if the patient has com-promised cardiac function (e.g., heart failure),decreased bronchial artery blood flow (e.g., de-creased cardiac output), or a previously under-ventilated lung (e.g., obstructive lung disease),then occlusion of the pulmonary vessel likelywill result in a hemorrhagic infarct, whichsignificantly increases morbidity and mortality.
2. Gross and microscopic findings of pulmonaryinfarctiona. Red-blue colored wedge-shaped area that extends
to the pleural surface: pleural surface often has afibrinous exudate (Figure 16-6).
b. Majority of infarcts are in the lower lobes: perfu-sion is greater than ventilation in the lower lobes.
c. Coagulation necrosis with alveolar spaces filledwith blood and alveolar macrophages
3. Clinical findings of pulmonary infarction: suddenonset of dyspnea and tachypnea
4. Laboratory findings of pulmonary infarction: hypox-emia, respiratory alkalosis
B. Pulmonary hypertension1. Primary pulmonary hypertension: more common in
women; due to endothelial cell dysfunction2. Hypoxemia and acidosis: potent stimuli for pulmo-
nary artery vasoconstriction, causing smooth musclehyperplasia and hypertrophy

Chapter 16 Respiratory Disorders 191
3. Etiology of secondary pulmonary hypertensiona. Chronic hypoxemia: living at high altitude,
chronic lung diseaseb. Chronic respiratory acidosis (e.g., chronic
bronchitis)c. Loss of pulmonary vasculature (decrease in cross-
sectional area of pulmonary vasculature): placesincreased demand on remaining vessels (e.g.,chronic lung disease)
d. Left-to-right cardiac shunts: volume overloadspulmonary arteries.
e. Mitral stenosis: backup of blood into pulmonaryveins, leading to pulmonary venous hypertension
4. Gross and microscopic findingsa. Atherosclerosis of main elastic pulmonary arteriesb. Medium- and small-sized pulmonary vessels: pro-
liferation of myointimal cells and smooth musclecells
5. Clinical and laboratory findings: progressive dyspneaand chest pain with exertion; cor pulmonale
V. Restrictive Lung Diseases• Restrictive lung disease includes interstitial lung diseases char-
acterized by decreased compliance (stiff lungs), leading to de-creased total lung capacity.
A. Pathogenesis and general features1. Decreased lung compliance
a. Decreased expansion of the lung parenchymaduring inspiration: results in dyspnea
b. Restriction of movement of the chest wall (e.g., ky-phoscoliosis) or lung parenchyma (increasedfibrosis or fluid in the interstitium)
c. Damage to the interstitium affects types I and IIalveolar cells and endothelial cells: leads to afunctional loss of alveolar and capillary units
2. Increased lung elasticity: recoil of the lung on expira-tion is increased.
3. Etiologya. Acute restrictive lung disease: acute respiratory
distress syndrome (ARDS)b. Chronic restrictive lung diseases: pneumoconio-
ses (e.g., coal worker's pneumoconiosis, silicosis, as-bestosis, berylliosis), sarcoidosis, hypersensitivitypneumonitis, collagen vascular diseases
4. General clinical findings in restrictive lung diseasesa. Exertional dyspnea and dry coughb. Respiratory alkalosis (arterial Pco 2 < 33 mm Hg)
and hypoxemiac. All lung volumes and lung capacities are de-
creased; increased forced expiratory volume in 1second (FEV 1 )/forced vital capacity (FVC) ratio
••••••••••••••••••••••••••••••••••
Cor pulmonale: pul-monary hyperten-sion + rightventricular hypertro-phy, causing right-sided heart failure
Restrictive lungdisease: compli-ance, T elasticity; in-terstitial fibrosis
Restrictive lungdisease: T FEVi/FVC
••

192 Pathology
d. Chest radiograph: diffuse bilateral reticular infil-trates ("ground glass" appearance)
B. ARDS• Type of noncardiogenic pulmonary edema resulting from
acute damage to the alveoli1. Pathogenesis
a. Damage due to neutrophils and alveolarmacrophages(1) Pulmonary capillaries leak fluid into the
alveoli, producing edema and hyalinemembranes.
(2) Damage to type H pneumocytes produceswidespread atelectasis.
b. Clinical disorders commonly associated withARDS: sepsis, gastric aspiration, severe trauma withshock, pneumonia
2. Gross findings: lungs are dark red and heavy.3. Microscopic findings
a. Early findings: diffuse alveolar damage and ne-crosis, alveolar edema, collapsed alveoli; somealveoli are lined by hyaline membranes.
b. Late findings: progressive interstitial fibrosisinterspersed with dilated alveoli, resembling ahoneycomb
4. Clinical findings: dyspnea with severe hypoxemia andrespiratory acidosis develops within 72 hours of theinitial insult; the mortality rate approaches 100%.
C. Pneumoconioses1. Inhalation of mineral dust (coal dust, silica, asbestos,
beryllium) into the lungs leads to interstitial fibrosis.2. Particle size determines site of lung deposition: 1- to
5-1.im particles reach the bifurcation of respiratorybronchioles and alveolar ducts; particles < 0.5 .rmreach the alveoli and are phagocytosed by alveolarmacrophages.
3. Coal worker's pneumoconiosisa. Source of coal dust (anthracotic pigment): coal
mines, large urban centers, cigarette smokeb. Pulmonary anthracosis: innocuous disease with
anthracotic pigment in the interstitial tissue,hilar nodes, and alveolar macrophages ("dustcells")
c. Simple type: fibrotic opacities < 1 cm in upperlobes and upper portions of lower lobes; centrilob-ular emphysema sometimes develops.
d. Complicated type (progressive massive fibrosis):fibrotic opacities > 1-2 cm; necrotic centers; crip-pling lung disease ("black lung" disease)
e. Complications: cor pulmonale, Caplan's syn-drome (coal worker's pneumoconiosis plus largecavitating rheumatoid nodules in the lungs)
Most commoncause of ARDS:sepsis
Coal worker'spneumoconiosis:no increased inci-dence of canceror TB

Chapter 16 Respiratory Disorders 193
Figure 16 -7 Asbestos bodyThe straight, beaded asbestosbody (ferruginous body) rep-resents an asbestos fiber coatedby iron and protein.
4. Silicosisa. Sources of silica (quartz): foundries, quarriesb. Silica is highly fibrogenic.c. Chronic exposure: nodular opacities, calcification
of hilar nodesd. Associations: cor pulmonale, Caplan's syndrome
5. Asbestosisa. Sources: pipe-fitting in shipyards, roofing (> 20
years), demolition of buildings containing asbestosb. Asbestos (ferruginous) bodies (Figure 16-7)
(1) Asbestos fibers are coated by iron andprotein.
(2) Do not enter the pleurac. Asbestos-related disease
(1) Benign pleural plaque: often calcified and in-volves visceral and parietal pleura and domeof the diaphragm
(2) Pleural effusions(3) Diffuse interstitial fibrosis: involves respira-
tory bronchioles, alveolar ducts, and alveoli(4) Primary bronchogenic carcinoma: most
common cause of cancer-related deathswith or without smoking tobacco; markedlyenhanced with smoking tobacco
(5) Malignant mesothelioma of pleura andperitoneum: no causal relationship withsmoking tobacco; tumor encases and locallyinvades subpleural lung tissue; requires25-40 years to develop.
d. Associations: cor pulmonale, Caplan's syndrome6. Beryl liosis
a. Source: inhalation of dust or oxides of metallicberyllium in the nuclear and aerospace industries
b. Diffuse interstitial fibrosis with noncaseatinggranulomas
c. Increased incidence of cor pulmonale andprimary lung cancer
Silicosis: increasedrisk of primarylung cancer and TB
Asbestosis: benignpleural plagues mostcommon lesion

194 Pathology
D. Sarcoidosis1. Epidemiology: most common noninfectious granu-
lomatous disease of the lungs; more common inAfrican Americans and nonsmokers
2. Pathogenesis: an unknown antigen interacts with CD4helper T cells, leading to formation of noncaseatinggranulomas.
3. Clinical findingsa. Lung disease
(1) Noncaseating granulomas in the lung intersti-tium and hilar lymph nodes
(2) Hilar nodes contain multinucleated giantcells with laminated calcium concretions(Schaumann's bodies) and stellate inclusions(asteroid bodies).
(3) Diffuse interstitial fibrosis(4) Dyspnea is the most common symptom.
b. Skin lesions: nodular lesions containinggranulomas
c. Inflammation of the uveal tract: causes blurryvision
d. Hepatomegaly: sarcoidosis is the most commoncause of noninfectious granulomatous hepatitis.
4. Laboratory findingsa. Hypercalcemia: occurs in 5% of cases; due to syn-
thesis of la-hydroxylase by macrophages in granu-lomas (increases vitamin D)
b. Polyclonal gammopathy, cutaneous anergy tocommon skin antigens (e.g., Candida)
5. Chest radiograph: enlarged hilar lymph nodes ("potatonodes"), reticulonodular densities throughout the lungparenchyma
6. Prognosis: majority of patients recover with corticoste-roid therapy.
E. Hypersensitivity pneumonitis1. Extrinsic allergic alveolitis associated with exposure to
a known inhaled antigen2. Farmer's lung: exposure to a thermophilic bacterium in
moldy hay3. Silo filler's disease: inhalation of nitrogen dioxide
fumes from fermenting corn in a closed space4. Byssinosis: dyspnea due to contact with cotton, linen,
and hemp products in a textile factoryF. Drugs producing restrictive lung disease (e.g., amio-
darone, bleomycin, methotrexate, nitrofurantoin)G. Collagen vascular diseases producing restrictive lung
disease (e.g., systemic sclerosis, systemic lupus erythemato-sus, rheumatoid arthritis)
VI. Obstructive Lung Diseases• Obstructive lung diseases are characterized by obstruction to
airflow out of the lungs.

Chapter 16 Respiratory Disorders 195
A. Asthma1. Episodic, reversible, hyperreactive airway disease that
mainly targets the bronchi and terminal bronchioles2. Pathogenesis: exaggerated bronchoconstrictor re-
sponse of the terminal (nonrespiratory) bronchioles inassociation with bronchial inflammation
3. Extrinsic asthmaa. Type I hypersensitivity reaction with exposure to
extrinsic allergens: typically develops in childrenwith a family history of atopic allergy
b. Mast cell release of mediators (e.g., histamine,prostaglandins, leukotrienes C, D, and E4): potentbronchoconstrictors that increase vessel permeabil-ity, leading to mucosal swelling
c. Recruitment of inflammatory cells: eosinophilsrelease major basic protein and eosinophil cationicprotein that damage epithelial cells.
d. Changes in bronchial epithelium: inflammation,hyperplasia of submucosal glands and smoothmuscle cells
e. Changes in terminal bronchioles: mucous plugsblock the lumen; goblet cell metaplasia
f. Clinical findings: episodic expiratory wheezing,nocturnal cough, increased anteroposterior di-ameter due to air trapping
g- Laboratory findings: respiratory alkalosis may pro-gress to respiratory acidosis.
4. Intrinsic asthmaa. Nonatopic asthma: typically develops later in lifeb. Examples: NSAID sensitivity, stress, exercise, envi-
ronmental pollutants (e.g., ozone), smoke
Asthma may be caused by ozone (0 3), an airpollutant that derives from interactions of 0,with nitrogen and sulfur oxides and hydrocar-bons. It forms highly reactive free radicals inthe airways that cause inflammation and irrita-tion, often precipitating asthma.
B. Emphysema1. Permanent enlargement of all or part of the respiratory
unit (respiratory bronchioles, alveolar ducts, alveoli)2. Cigarette smoking is the most common cause of cen-
triacinar (centrilobular) emphysema and panacinaremphysema.
3. Pathogenesisa. Increased compliance and decreased elasticity:
imbalance between elastase in neutrophils and anti-elastases (ocrantitrypsin)
b. Cigarette smoke is chemotactic to neutrophils andmacrophages that contain elastase.
Most commonchronic respiratorydisease in chil-dren: asthma
Emphysema: de-struction of elastictissue in respiratoryunit

Terminalbronchiole
Alveolarduct
Alveoli
Distendedrespiratorybronchiole
A Centriacinar emphysema B Panacinar emphysema
196 Pathology
Respiratorybronchiole
Normal
Distendedalveolar ductand alveoli
C Paraseptal emphysema
Figure 16-8 Types of emphysema. he senematic shows a normal distal airway, including aterminal bronchiole leading into the respiratory unit consisting of a respiratory bronchiole,alveolar duct, and alveoli. Centriacinar emphysema {A) is characterized by trapping of air inthe respiratory bronchioles. Panacinar emphysema {B) is characterized by trapping of air in theentire respiratory unit. Paraseptal emphysema IC) is associated with trapping of air in thealveolar duct and alveoli.
c. Free radicals in cigarette smoke inactivate arantitrypsin.
4. Centriacinar (centrilobular) emphysemaa. Most common type of emphysema: strong associ-
ation with cigarette smoking; more common andmore severe in men
b. Respiratory bronchioles are the primary site ofelastic tissue destruction.
c. Usually involves the apical segments of the upperlobes
d. Respiratory bronchioles distend and trap air: in-creases residual volume and total lung capacity(Figure 16-8, A)
5. Panacinar emphysemaa. Associated with hereditary arantitrypsin defi-
ciency: autosomal recessive disorder; emphy-sema develops at an early age.
b. MM phenotype is normal; ZZ phenotype is associ-ated with very low serum levels of arantitrypsin.

Chapter 16 Respiratory Disorders 197
c. Absent a l -globulin peak in serum proteinelectrophoresis
d. Entire respiratory unit is uniformly distended:mainly affects the lower lobes (Figure 16-8, B)
6. Paraseptal emphysemaa. Mainly targets the alveolar ducts and alveoli:
generally affects the upper half of the lungs(Figure 16-8, C)
b. Increased incidence of spontaneous pneumotho-rax: due to rupture of subpleural blebs
7. Clinical findingsa. Progressive dyspnea and hyperventilation: pa-
tients (sometimes called "pink puffers") have towork hard at breathing.
b. Scant sputum production; breath sounds dimin-ished due to lung hyperinflation
c. Increased total lung capacity: due to increase inresidual volume from air trapped in the respiratoryunit; decreased FEV1/FVC
d. Normal to decreased arterial Pco 2 (respiratory al-kalosis); hypoxemia Emphysema:
e. Cor pulmonale may develop late in the disease. L FEV,/FVC
f. Chest radiograph: hyperlucent lung fields, in-creased anteroposterior diameter, vertically ori-ented heart, depressed diaphragm
C. Chronic bronchitis1. Clinical diagnosis: defined as a productive cough for at
least 3 months for 2 consecutive years2. Etiology: strong association with cigarette smoking,
urban residence3. Pathogenesis
a. Hypersecretion of mucus in bronchi and obstruc-tion to airflow in the terminal bronchioles: airflowobstruction in chronic bronchitis is proximal to theobstruction seen in emphysema.
b. Changes in bronchi: inflammation, enlarged sub-mucosal glands, squamous cell metaplasia
c. Changes in terminal bronchioles: inflammation,mucous plugs, goblet cell metaplasia
4. Clinical findingsa. Productive coughb. Cyanosis from CO 2 retention (patient sometimes
called "blue bloater"); wheezing and sibilantrhonchi are commonly heard.
c. Chronic respiratory acidosis: arterial Pco, > 45mm Hg and HCO3- > 30 mEq/L; hypoxemia
d. Cor pulmonale is commonly present.c. Chest radiograph: increased bronchial markings
D. Bronchiectasis1. Permanent dilation of the bronchi and bronchioles due
to destruction of cartilage and elastic tissue bychronic necrotizing infections
198 Pathology
TABLE 16-4 Tumors and Tumor-like Disorders of the Lung
Type of Tumor Locationor Disorder in Lung Comments
Peripheral More common in womenAssociated with cigarette smokingTumors may develop in scars or spread along
alveolar walls and mimic lobar pneumonia(bronchioloalveolar)
Central More common in menStrong association with cigarette smokingTend to cavitateMay ectopically secrete PTH-related protein
Central More common in menStrong association with cigarette smokingArise from neuroendocrine cells (Kulchitsky
cells)Rapidly growing cancer that metastasizes earlyMay ectopically secrete ADH or ACTH
Peripheral Undifferentiated cancer that metastasizes early
Central No association with cigarette smokingLow-grade cancer of neuroendocrine originCarcinoid syndrome is rare (does not require
liver metastasis)
Multifocal More common than primary cancerUsually presents with dyspnea
Central Non-neoplastic proliferation of cartilage andadipose tissue
Appears as solitary "coin" lesion on chestradiograph
Adenocarcinoma
Squamous cellcarcinoma
Small cell (oat cell)carcinoma
Large cell carcinoma
Bronchial carcinoid
Carcinoma metastasicto the lung
Bronchial hamartoma
ACTH, adrenocorticotropic hormone; ADH, antidiuretic hormone; PTH, parathyroid hormone
2. Etiology: CF, TB, bronchial obstruction, immotile ciliasyndrome (absent dynein arm)
3. Gross findings: most commonly occurs in the lowerlobes; bronchi and bronchioles are filled with pus andextend to the lung periphery.
4. Clinical findings: copious amounts of sputum,hemoptysis
5. Chest radiograph: bronchial markings extend to thelung periphery.
VII. NeoplasmsA. Epidemiology
1. Rate of increase in lung cancer is declining in menbut increasing in women: peak incidence is 55-65years of age.
2. Etiologya. Cigarette smoking (polycyclic hydrocarbons) is the
most common cause (lower risk with pipe and cigarsmoking).
b. Radon gas (uranium mining), asbestos, silica, beryl-lium, arsenic
Most commoncause of bronchiec-tasis: CF
Most commoncause of cancerdeaths in both menand women: primarylung cancer
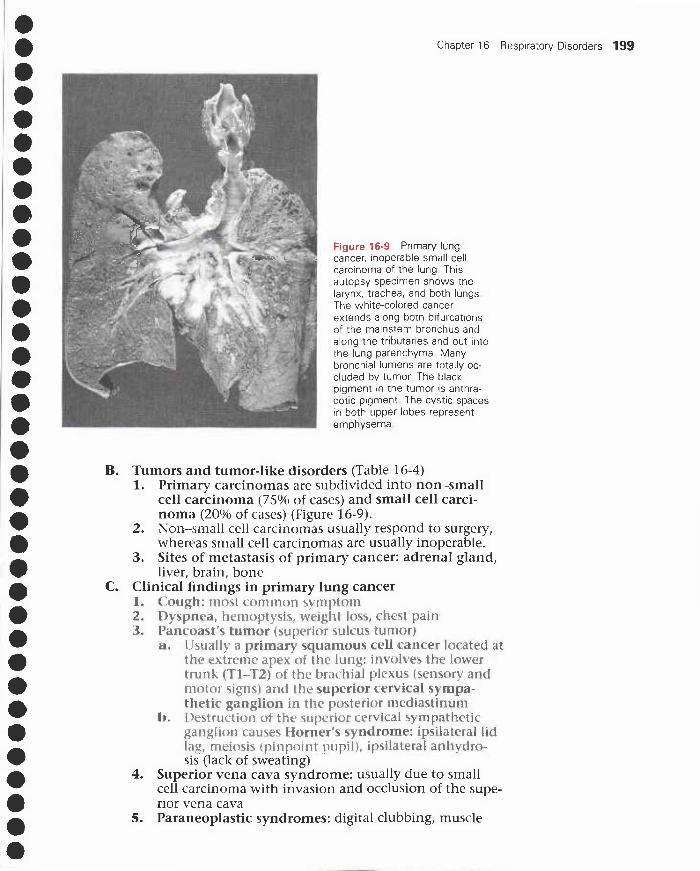
Chapter 16 Respiratory Disorders 199
Figure 16-9 Primary lungcancer, inoperable small cellcarcinoma of the lung Thisautopsy specimen shows thelarynx, trachea, and both lungsThe white-colored cancerextends along both bifurcationsof the mainstem bronchus andalong the tributaries and out intothe lung parenchyma Manybronchial lumens are totally oc-cluded by tumor The blackpigment in the tumor is anthra-cotic pigment The cystic spacesin both upper lobes representemphysema
B. Tumors and tumor-like disorders (Table 16-4)1. Primary carcinomas are subdivided into non-small
cell carcinoma (75% of cases) and small cell carci-noma (20% of cases) (Figure 16-9).
2. Non-small cell carcinomas usually respond to surgery,whereas small cell carcinomas are usually inoperable.
3. Sites of metastasis of primary cancer: adrenal gland,liver, brain, bone
C. Clinical findings in primary lung cancer1. Cough: most common symptom2. Dyspnea, hemoptysis, weight loss, chest pain3. Pancoast's tumor (superior sulcus tumor)
a. Usually a primary squamous cell cancer located atthe extreme apex of the lung: involves the lowertrunk (T1-T2) of the brachial plexus (sensory andmotor signs) and the superior cervical sympa-thetic ganglion in the posterior mediastinum
b. Destruction of the superior cervical sympatheticganglion causes Horner's syndrome: ipsilateral lidlag, meiosis (pinpoint pupil), ipsilateral anhydro-sis (lack of sweating)
4. Superior vena cava syndrome: usually due to smallcell carcinoma with invasion and occlusion of the supe-rior vena cava
5. Paraneoplastic syndromes: digital clubbing, muscle

200 Pathology
weakness (Eaton-Lambert syndrome), ectopic hormonesecretion
VIII. Disorders of the PleuraA. Movement of pleural fluid
1. Depends on the balance between hydrostatic andoncotic pressures in the capillaries in the parietal andvisceral pleura
2. Fluid normally moves from the parietal pleura into thepleural space and into the lungs.
B. Etiology of pleural effusion1. Increased hydrostatic pressure in the visceral pleura
(e.g., congestive heart failure)2. Decreased oncotic pressure (e.g., nephrotic syndrome)3. Obstruction of lymphatic drainage from the visceral
pleura (e.g., lung cancer)4. Increased vessel permeability of visceral pleural cap-
illaries (e.g., pneumonia, pulmonary infarction)C. Types of pleural effusion
1. Transudates: ultrafiltrates of plasma involving distur-bances in Starling's forces
2. Exudates: protein- and cell-rich fluids; increase in vesselpermeability
D. Spontaneous pneumothorax1. Etiology: idiopathic, paraseptal emphysema, Marfan
syndrome, rising to the surface when deep-sea diving2. Pathogenesis
a. Rupture of a subpleural or intrapleural bleb pro-duces a hole in the pleura.
b. Pleural cavity pressure is the same as the atmo-spheric pressure, causing a portion of the lungor the entire lung to collapse.
3. Clinical findingsa. Sudden onset of dyspnea with pleuritic type of
chest painb. Tympanitic percussion note and absent breath
sounds: the trachea deviates to the side of thecollapse.
E. Tension pneumothorax1. Etiology: penetrating trauma to the lungs (e.g., knife
wound), rupture of tension pneumatocysts2. Pathogenesis
a. Flap-like pleural tear allows air into the pleuralcavity but prevents its exit.
b. Increased intrapleural pressure produces compres-sion atelectasis.
3. Clinical findingsa. Sudden onset of severe dyspneab. Tympanitic percussion note and absent breath
sounds: the trachea and mediastinal structuresdeviate to the contralateral side, compromisingvenous return to the heart.
Most commoncause of pleural ef-fusion: conges-tive heart failure
Tension pneumo-thorax: mediastinalshift compromisesvenous return to theheart

17a-NOWA
1"1111111 GastrointestinalDisorders
I. Disorders of the Oral Cavity: Mouth and JawA. Cleft lip
1. Most common congenital disorder of oral cavity;occurs most often in males
2. Multifactorial inheritance; associated with cleft palatein 50% of cases
3. Results from the failure of facial processes to fuseB. Inflammatory disorders
1. Infectious conditions (Table 17-1)2. Aphthous ulcers (canker sores or stress ulcers)
a. May be of autoimmune originb. Painful ulcers of the soft palate, tongue, and buccal
mucosa; covered by a "shaggy" gray membrane3. Glossitis (inflammation of tongue): caused by chronic
iron deficiency, vitamin B 12 or folate deficiency, scurvy,or scarlet fever
C. Leukoplakia (white patch) and erythroplakia (red patch)1. Located on the vermilion border of the lower lip
(most common), buccal mucosa, palate, and the floor ofthe mouth; lesions cannot be scraped off.
2. Caused by chronic irritation (e.g., dentures; mostcommon); tobacco use; alcohol abuse; and human pap-illoma virus (HPV)
3. Result from squamous hyperplasia of the epidermis4. Risk of squamous dysplasia or squamous cell cancer
associated with erythroplakia in 80-90% of cases andwith leukoplakia in 2-6% of cases (Figure 17-1)
D. Malignant tumors of the mouth1. Squamous cell carcinoma (> 95%)
a. Occurs most often in men older than 50 years ofage; most tumors are well-differentiated.
Biopsy leukoeryth-roplakia to ruleout squamous dys-plasia or cancer
201
BacterialCervicofacial
actinomycosis
Diphtheria
Peritonsillarabscess
ViralExudative
tonsillitis
Hairy leukoplakia
Herpes labialis
Mumps
Other AgentsCongenital
syphilis
Oral thrush
EBV, Epstein-Barr virus
Presents as draining sinus tract from facial and/or cervicalarea; "sulfur granules" in pus
Complication due to extraction of abscessed tooth
Toxin produces "shaggy" gray pseudomembrane in poste-rior pharynx and upper airways
Uvula deviates to contralateral sideComplication due to tonsillitis
Associated with tonsillitis; potential for acute rheumaticfever and glomerulonephritis
Acute bacterial inflammation of major salivary glandUsually secondary to calculus, which obstructs duct in
postoperative patients
Culture is necessary to differentiate bacterial versus viralinfection
Glossitis associated with bilateral white, hairy excrescenceson lateral border of tongue
Pre-AIDS defining lesion
Recurrent infection; reactivated by stress, sunlight, andmenses
Remains dormant in cranial sensory ganglia
Bilateral parotitis (70%) with elevation of serum amylaseComplications include meningoencephalitis, unilateral
orchitis in adolescents, oophoritis, and pancreatitis
Abnormalities involving incisors (tapered like a peg) andmolar teeth (resemble mulberries)
May occur in neonates, immunocompromised patients(common pre-AIDS defining lesion), diabetes mellitus,and following antibiotic therapy
202 Pathology
TABLE 17-1 Infections of the Oral Cavity
Infection Pathogen Features
Actinomyces israelii
Viruses: most cases
Corynebacteriumdiphtheriae
Streptococcus pyogenes
EBV
Herpes simplex virustype 1
Paramyxovirus
Treponema pallidum(spirochete)
Candida albicans (yeast)
•••••••••••••••••••••••••
Pharyngitis S. pyogenes
Sialadenitis Staphylococcus aureus
b. Risk factors: tobacco use (most common), alcoholabuse (synergistic with smoking), HPV, chronic ir-ritation from dentures
c. Sites: lower lip (vermilion border), floor of themouth, lateral tongue
2. Basal cell carcinoma: most common cancer of theupper lip; associated with exposure to ultraviolet B light
II. Salivary Gland Tumors• Most tumors in the parotid glands are benign; tumors of the
minor salivary glands are more likely to be malignant.A. Pleomorphic adenoma (mixed tumor): usually benign
1. Most common benign tumor of salivary glands; occursmost often in women
Most common siteof tumors of the sal-ivary glands: parotidgland
••••••••••

••
Chapter 17 Gastrointestinal Disorders 203
•••••••••••
Figure 17 -1 Leukoplakia of thetongue with invasive squa-mous cell carcinoma. Discreteraised white patches are evidenton both sides of the tongue.The diffuse white discolorationon the left side represents an ex-tensive area of invasive squa-mous cell carcinoma.
111 2. Painless, movable mass at the angle of the jaw; corn-
•posed of epithelial cells intermixed with myxoma-tous and cartilaginous stroma
• 3. Tends to recur after removal4. May become malignant; facial nerve involvement is a
• sign of malignancy.
•B. Warthin's tumor (papillary cystadenoma lymphomato-
sum): benign
• 1. Occurs most often in men2. Heterotopic salivary gland tissue trapped in a lymph
• node; cystic glandular structures are located withinbenign lymphoid tissue.
C. Mucoepidermoid carcinoma: malignant
• 1. Most common malignant tumor of the salivary glands;most commonly located in the parotid gland
2. Mixture of neoplastic squamous and mucus-secreting
• cells
• HI. Disorders of the Esophagus
•A. Signs and symptoms of esophageal disease
1. Heartburn (e.g., gastroesophageal reflux disease, GERD)2. Dysphagia for solids: symptom of an obstructive
lesion (e.g., cancer, esophageal web)3. Dysphagia for solids and liquids: symptom of a motif-
• ity disordera. Upper esophageal dysphagia: striated muscle
• weakness, as occurs in dermatomyositis and myas-
•thenia gravis
b. Lower esophageal dysphagia: smooth muscle dys-
•motility, as occurs in systemic sclerosis; CRESTsyndrome (calcinosis, Raynaud's phenomenon,•
•

204 Pathology
Trachea — — Proximal esophagus:ends blindly
Distal esophagus:arises from trachea
Stomachit
Danger of chemical pneumonia Distended with air
Figure 17-2 Tracheoesophageal fistula The proximal esophagus ends blindly, and the distalesophagus arises from the trachea The stomach is distended with air due to communicationof the esophagus with the trachea
esophageal dysfunction, sclerodactyly, telangiec-tasia); achalasia
B. Anatomic disorders1. Tracheoesophageal fistula
a. Morphology: proximal esophagus ends blindlyand distal esophagus arises from the trachea(Figure 17-2).
b. Maternal polyhydramnios (excess amniotic fluid):small intestine is unable to resorb swallowed amni-otic fluid.
c. Abdominal distention: air in stomachd. VATER syndrome
2. Esophageal web (Plummer-Vinson syndrome): causedby chronic iron deficiency; usually occurs in womenin their 30s and 40s
3. Esophageal diverticulaa. True diverticulum: outpouching of the esophageal
wall lined by mucosa, submucosa, muscularispropria, and adventitia
b. False (pulsion) diverticulum(1) Increased pressure pushes the mucosa, submu-
cosa, and part of the muscularis propriathrough an area of weakness in the underlyingmuscle wall.
(2) Zenker's diverticulum: pulsion diverticulumof the upper esophagus causing episodic noc-turnal regurgitation of food
4. Hiatal herniaa. Sliding hernia (most common): herniation of the
stomach through a widened diaphragmatic hiatus;occurs when the stomach protrudes above thediaphragm
b. Clinical findings: heartburn and nocturnal epigas-tric distress from acid reflux, hematemesis (vomitingblood), ulceration, stricture formation
5. Esophageal lacerations and rupturea. Most often caused by retching in alcoholics
Most commonesophageal divertic-ulum: Zenker'sdiverticulum
VATER syndrome:vertebral abnormali-ties, anal atresia,tracheoesophagealfistula, renaldisease, and absentradius
•••••••••••••••••••••••
•••••••••••

Chapter 17 Gastrointestinal Disorders 205
Figure 17-3 Esophagealvarices, showing many linear-oriented dilated and tortuousveins in the submucosa of thedistal esophagus.
b. Mallory-Weiss syndrome: mucosal tear in the prox-imal stomach and distal esophagus resulting inhematemesis
c. Boerhaave's syndrome: rupture of the distal esoph-agus resulting in pleural effusion and air in themediastinum
6. Esophageal varicesa. Dilated subepithelial and submucosal left gastric
coronary vein; coronary vein drains into theportal vein; empties blood from the distal esophagusand proximal stomach (Figure 17-3)
b. Caused by portal hypertension due to cirrhosis ofthe liver
C. Achalasia1. Dilation of the esophagus proximal to the lower esoph-
ageal sphincter (LES) accompanied by absent peristalsis2. Pathogenesis
a. Primary achalasia: incomplete relaxation of theLES; lack of ganglion cells in myenteric plexusresults in the loss of vasointestinal peptide, whichnormally relaxes the LES.
b. Acquired achalasia: Chagas' disease with destruc-tion of ganglion cells by leishmanial forms
3. Clinical findings: nocturnal regurgitation of un-digested food
D. Esophagitis1. Infectious esophagitis
a. Usually a complication of AIDSb. Caused by herpes simplex virus, cytomegalovirus,
or Candida
Most commoncause of death incirrhosis: rupturedesophageal varices
Most commonneuromuscular dis-order of the esopha-gus: achalasia

206 Pathology
Figure 17 -4 Barrett's esopha-gus, showing an extensivearea of glandular metaplasia withnumerous goblet cells A smallsection of squamous epitheliumremains on the right
2. Corrosive esophagitisa. Ingestion of strong alkali (e.g., ammonia, lye) or
acid (e.g., sulfuric acid)b. Causes strictures and possibly squamous cell carci-
nomaE. Reflux disease
1. GERD: reflux of acid and bile into the distal esophagusa. Pathogenesis: defective LES; ineffective esophageal
clearance of reflux materialb. Risk factors: smoking tobacco, alcohol, caffeine,
fatty foods, hiatal herniac. Clinical findings
(1) Most common cause of noncardiac chestpain, nocturnal cough, and nocturnalasthma
(2) Often manifested by heartburn and acid injuryto tooth enamel
d. Complication: Barrett's esophagus2. Barrett's esophagus: glandular metaplasia in distal
esophagusa. Columnar epithelium contains gastric-type cells
(mucous cells) and small intestine-type cells(goblet cells) (Figure 17-4)
b. Complications: ulceration with stricture formation,adenocarcinoma, glandular dysplasia
F. Esophageal tumors1. Leiomyoma: most common benign tumor of esophagus;
usually asymptomatic2. Adenocarcinoma: most common predisposing cause is
Barrett's esophagus.3. Squamous cell carcinoma
a. Most common primary cancer of esophagus indeveloping countries: occurs most often in blacksand in men worldwide
b. Risk factors: tobacco smoking, alcohol abuse,lye strictures, achalasia, Plummer-Vinsonsyndrome, HPV
c. Location: midesophagus (50%), loweresophagus (30%)
Most commoncause of heartburn:GERD
Most commonprimary cancer ofthe esophagus:adenocarcinoma
Most common riskfactor for squamouscell carcinoma ofthe esophagus:tobacco smoking

Chapter 17 Gastrointestinal Disorders 207
d. Clinical findings: dysphagia for solids, weight losse. Metastasis: spreads to local nodes first, then to liver
and lungs; 5-year survival rate less than 15%
IV. Stomach DisordersA. Signs and symptoms of stomach disease
1. Hematemesis2. Melena: dark, tarry stools signify a bleed proximal to the
duodenojejunal junction.B. Pyloric stenosis
1. Congenital pyloric obstruction: occurs in 1:500 livebirths; more common in boys; multifactorial inheritance
2. Pathogenesis: progressive hypertrophy of the circularmuscles in the pyloric sphincter occurs over the ensuing2-4 weeks
3. Clinical findings: projectile vomiting of non—bile-stained fluid 2-4 weeks after birth
4. Acquired pyloric obstruction: complication of chronicduodenal ulcer disease
C. Gastritis1. Acute hemorrhagic (erosive) gastritis
a. Etiology: bleeding results from erosions (breach inthe epithelium of the mucosa) and is caused by:(1) Nonsteroidal anti-inflammatory drugs
(NSAIDs)(2) Smoking tobacco, alcohol, burns, central
nervous system injury, uremia, consumption ofAnisakis (worm in raw fish)
b. Clinical findings: hematemesis, melena, irondeficiency
2. Chronic atrophic gastritisa. Type A: involves the body and fundus of the
stomach(1) Most commonly caused by autoimmune
destruction of parietal cells (e.g., perniciousanemia)
(2) Complications: achlorhydria with hypergas-trinemia, macrocytic anemia, increased risk ofadenocarcinoma
b. Type B: involves the antrum and pylorus of thestomach(1) Commonly caused by Helicobacter pylori:
produces urease and cytotoxins; colonizes themucous layer that lines the mucosa
(2) Microscopic findings: chronic inflammatoryinfiltrate in the lamina propria consistingmainly of lymphocytes with prominent germi-nal follicles; the intestinal metaplasia (gobletcells, Paneth cells) is a precursor of adenocar-cinoma.
(3) Tests used to identify H. pylori: tests thatdetect urease; serologic tests
Most commoncause of gastritis.NSAIDs
Most commoncause of type Bchronic atrophic gas-tritis: H. pylori

208 Pathology
TABLE 17-2 Comparison of Gastric Ulcers and Duodenal Ulcers
Feature
Gastric Ulcers
Duodenal Ulcers
Percent of ulcer cases 25%
75%
Epidemiology Male:female ratio: 1:1; risk
Male:female ratio: 2:1; risk
increased with blood
increased with blood groupgroup A
0 and MEN ISmoking does not cause
Increased in cirrhosis, COPD,
PUD but delays healing renal failure, hyperparathy-roidism
Helicobacter pylori - 80% of cases
90-95% of cases
Pathogenesis Defective mucosal barrier
Defective mucosal barrier duedue to H. pylori
to H. pyloriMucosal ischemia (reduced
Increased acid production
PGE), bile reflux, delayed
(increased parietal cell mass)gastric emptying
Location Single ulcer on lesser
Single ulcer on anteriorcurvature of antrum; portion of first part ofsame location for cancer
duodenum followed bysingle ulcer on posteriorportion (danger of perfora-tion into pancreas)
Complications Bleeding (most commonly
Bleeding (most commonly in
in left gastric artery)
gastroduodenal artery)Perforation
Perforation (air underdiaphragm, pain radiates toleft shoulder)
Gastric outlet obstruction;pancreatitis
Clinical findings Burning epigastric pain
Burning epigastric pain 1-3
soon after eating
hours after eating
COPD, chronic obstructive pulmonary disease; MEN, multiple endocrine neoplasia; PGE,prostaglandin E
(4) Complications: cancers associated with H.pylori (e.g., gastric adenocarcinoma, low-gradeB-cell malignant lymphoma)
c. Menetrier's disease: hypertrophic gastropathy(1) Characterized by giant rugal folds caused by
hyperplasia of mucus-secreting cells and hypo-proteinemia
(2) Results in atrophy of parietal cells (achlor-hydria) and risk of adenocarcinoma
D. Peptic ulcer disease (PUD) (Table 17-2)1. Ulcers are breaches in the mucosa that extend into the
submucosa or deeper.2. Peptic ulcers occur in the stomach or in the first part of
the duodenum.3. All peptic ulcers (gastric or duodenal) are identical in
appearance.a. Gross: clean, sharply demarcated lesions that are
slightly elevated at the edges
Most commoncause of gastric andduodenal ulcers:H pylori
•••••••••••••••••••••••••••••••••••

•••••••••••••••••••••••••••:••••••
Chapter 17 Gastrointestinal Disorders 209
b. Microscopic: four layers: necrotic debris; inflam-mation, with neutrophils predominating; granula-tion (repair) tissue; fibrotic tissue
4. Zollinger-Ellison syndrome: malignant islet cellgastrin-secreting tumora. Located in pancreatic islet cells; associated with
multiple endocrine neoplasia type I (20-30% ofcases)
b. Clinical findings: epigastric pain with weight loss,peptic ulceration (single or multiple), diarrhea
c. Laboratory findings(1) Increased basal and maximal output of acid(2) Serum gastrin > 1000 pg/mL
E. Gastric polyps and tumors1. Gastric polyps: develop in the setting of chronic gastri-
tis and achlorhydriaa. Hyperplastic polyps (most common): hamartomas
with no malignant potentialb. Adenomatous polyps: neoplastic and usually
sessile; potential for malignant transformation ifpolyps > 2 cm
2. Leiomyoma: stomach is most common gastrointesti-nal (GI) site; may ulcerate or bleed; no malignantpotential
3. Gastric adenocarcinoma: intestinal typea. Epidemiology: decreased incidence in the United
States; increased in Japan; occurs most often inmen
b. Risk factors: H. pylori, nitrosamines, smoked foods,diet lacking fruits and vegetables (decreased anti-oxidants), type A gastritis, Menetrier's disease, ade-nomatous polyps
c. Precursor lesion: intestinal metaplasiad. Locations: lesser curvature of pylorus and antrum
(50-60% of cases), cardia (25% of cases), body andfundus
e. Gross and microscopic findings: polypoid, ulcer-ated appearance(1) Early lesions are confined to the mucosa and
submucosa.(2) Advanced lesions extend into the muscle wall.
f. Clinical findings(1) Weight loss, epigastric pain(2) Paraneoplastic lesions (acanthosis nigricans,
multiple outcroppings of seborrheic keratoses)g. Common metastatic sites: lymph nodes (especially
left supraclavicular node), liver, lungs, ovariesh. Prognosis: overall 5-year survival rate about 20%
4. Gastric adenocarcinoma: diffuse type (less common)a. No association with chronic gastritis or H. pylorib. Characterized by diffuse infiltration of the
stomach wall (linitis plastica) and no peristalsis;
Most commoncause of hemateme-sis and melena:PUD
Most commoncause of gastric ade-nocarcinoma:H. pylori
•

•••••••••••••••••••••••••••:•••••••
210 Pathology
composed of "signet-ring" cells (mucin-filled neo-plastic cells with the nucleus pushed to theperiphery)
c. Hematogenous spread, causing bilateral Kruken-berg tumors in ovaries
5. Primary gastric malignant lymphomaa. Stomach is the most common site of extranodal
malignant lymphomas.b. Low-grade B-cell lymphoma: MALToma;
H. pylon-relatedc. High-grade lymphomas: B- or T-cell lymphomas
V. Disorders of the Small and Large BowelsA. Signs and symptoms of bowel disease
1. Small bowel diseasea. Colicky pain, constipation (gas but no defecation),
or obstipation (no gas or defecation, such asobstruction)
b. Hematemesis and melena (e.g., duodenal ulcer)c. Diarrhea (e.g., infection; malabsorption; infarction,
if bloody)d. Anemia (e.g., malabsorption of iron, folate, or
vitamin B12)2. Large bowel disease
a. Diarrhea (e.g., infection; infarction, if bloody)b. Pain (e.g., inflammatory bowel disease, appendicitis)c. Iron deficiency (e.g., polyps or colorectal cancer )d. Hematochezia (loss of blood per rectum; caused by
sigmoid diverticulosis or angiodysplasia)B. Congenital disorders
1. Duodenal atresia: distal to the entry to the commonbile ducta. Associated with Down syndromeb. Clinical findings: vomiting bile-stained fluid at
birth, "double bubble sign" (air in stomach and inproximal duodenum), maternal polyhydramnios
2. Hirschsprung's disease (congenital megacolon)a. Most often involves distal sigmoid colon and
rectum; proximal bowel is dilated but capable ofperistalsis.
b. Occurs most often in males; associated with Downsyndrome
c. Caused by lack of ganglion cells in Meissner's sub-mucosal plexus and Auerbach's myenteric plexus;acquired form is caused by Chagas' disease.
d. Clinical findings: difficulty expelling meconiumat birth, alternating signs of obstruction withdiarrhea
e. Complications: enterocolitis of dilated bowel(danger of perforation); may involve the ureters.
MALToma:rnucosa-associatedlymphoid tissuelymphoma

Chapter 17 Gastrointestinal Disorders 211
Figure 17-5 Hemorrhagic in-farction of small bowel, showingthe diffuse dark discolorationof the small bowel The arrowshows a thrombosed arteryattached to the aorta
3. Meckel's diverticuluma. Vitelline (omphalomesenteric) duct remnant: true
diverticulumb. Newborn: feces leaking out of umbilical area if vitel-
line duct connection persistsc. Complications: bleeding (most common), inflam-
mation (simulates acute appendicitis)C. Vascular disorders
1. Bowel infarctiona. Ischemic damage is more likely to occur in the
small bowel than in the large bowel. The smallbowel is supplied by the superior mesenteric artery.
b. Types(1) Transmural infarction (all visceral layers)
involves occlusion of a major mesenteric vessel(embolus, thrombus), usually the superiormesenteric artery (most common) or vein(Figure 17-5).
(2) Mural (mucosa and submucosa) and mucosalinfarctions (extends to muscularis mucosae)usually occur in states of hypoperfusion(e.g., shock).
c. Clinical findings: sudden onset of diffuse abdomi-nal pain, bowel distention, bloody diarrhea; ileus(absent bowel sounds)
d. Laboratory findings: neutrophilic leukocytosis,increased amylase (derives from infarcted bowel)
e. Radiographic findings: "thumbprint sign" due toedema in the bowel wall; bowel distention with air-fluid levels
2. Ischemic colitisa. Occurs in the splenic flexure (area of overlap
between superior and inferior mesenteric arteries)
••••••••••••••••••••••••••••••••••••
Meckel's diverticu-lum rule of 2s: 2"long, 2' from ileoce-cal valve, 2% ofpopulation, 2%symptomatic
Most commoncause of iron defi-ciency in newbornsand young chil-dren: Meckel'sdiverticulum
Most common ar-rhythmia predispos-ing to embolicsmall bowel infarc-tion: atrial fibrillation

TABLE 17-3 Types of Diarrhea
Invasive Pathogens invadeenterocytes
Diarrhea with bloodand leukocytes
Secretory Loss of isotonic fluidNo inflammatory
changes present inbowel mucosa
Osmotic Osmotically activesubstance isdrawing hypotonicsalt solution out ofbowel
Shigella spp., Campy-lobacter jejuni,Entamoebahistolytica
LaxativesVibrio cholerae;
ETEC; foodpoisoning due toBacillus cereus andClostridium perfrin-gens
Disaccharidase defi-ciency
Ingestion of poorlyabsorbable solutes(e.g., magnesiumsulfate laxatives)
Fecal smear forleukocytes: positive
Stool culture; ovaand parasites
Fecal smear forleukocytes: nega-tive in most cases
Fecal smear forleukocytes:negative
••••••••••••••
Type Characteristics
Causes
Screening Tests
212 Pathology
Diarrhea: > 250 gstool/day; acute: < 3weeks; chronic:> 3 weeks
ETEC, enterotoxigenic Escherichia cofi
b. Clinical findings: severe pain in the splenic flexureshortly after eating (mesenteric angina), weightloss, bloody diarrhea
c. Repair of the infarction site may result in fibrosis,leading to ischemic strictures and signs ofobstruction.
3. Angiodysplasia: dilation of mucosal and submucosalvenules in the cecum and right colona. Most often occurs in elderly individualsb. Pathogenesis: increased stress on the bowel wall
stretches the venules.c. Clinical findings: hematochezia (passage of bright
red bloody stools)4. Hemorrhoids
a. Internal: dilated superior hemorrhoidal veins inmucosa and submucosa(1) Etiology: straining when attempting to defe-
cate (most common), pregnancy, portalhypertension
(2) Clinical findings: often prolapsed from therectum; bleeding
b. External: dilated inferior hemorrhoidal veins;painful thrombosis
D. DiarrheaTypes and causes (Tables 17-3 and 17-4)
Lactase deficiency is a common genetic defect inNative Americans, Asian Americans, and AfricanAmericans. Anaerobic bacteria in the colon degrade
••••••••••••••••••••••

Chapter 17 Gastrointestinal Disorders 213
TABLE 17-4 Microbial Pathogens That Cause Diarrhea
Pathogen Comments
VirusesCytomegalovirus Common cause of diarrhea in AIDSNorwalk Most common cause of adult gastroenteritisRotavirus Most common cause of childhood diarrhea; occurs in winter
BacteriaBacillus cereus Food poisoning with preformed toxin; associated with fried rice or tacosCampylobacter jejuni Produces dysentery with crypt abscesses and ulcers resembling ulcerative colitisClostridium botulinum
(adult) Food poisoning with preformed toxin (blocks release of acetylcholine)Causes paralysis and mydriasis
(infant) Food poisoning contracted by eating spores in honeyClostridium difficile Associated with pseudomembranous colitis; antibiotics (e.g., ampicillin) cause
overgrowth of toxin-producing C. difficile in colon; pseudomembrane coverscolon mucosa (creamy to greenish flat plaques)
Toxin assay of stool is best confirmatory testEscherichia coli ETEC: produces heat-stable toxin that stimulates guanylate cyclase, causing
secretory diarrhea (traveler's diarrhea)STEC (0157:H7 serotype): contracted by eating undercooked beef; may produce
hemorrhagic colitisMycobacterium tuberculosis Organisms swallowed from primary focus in lung; invade Peyer's patches;
circumferential spread in lymphatics leads to stricture formationSalmonella species Pathogenic Salmonella: S. typhi, S. paratyphi, S. enteritidis (food poisoning)
Typhoid fever caused by S. typhi:Week 1: invades Peyer's patches and produces sepsis (blood culture best for
diagnosis)Week 2: diarrhea (positive stool culture); classic triad of bradycardia, neutro-
penia, splenomegalyChronic carrier state due to gallbladder disease
Shigella species Mucosal ulceration, pseudomembranous inflammation, dysenteryStaphylococcus aureus Food poisoning with preformed toxin that occurs in 1-6 hours after eating
Culture food, not stoolVibrio cholerae Enterotoxin stimulates adenylate cyclase in small bowelYersinia enterocolitica Dysentery, mesenteric lymphadenitis (granulomatous microabscesses)
ProtozoaBalantidium coliCryptosporidium parvum
Entamoeba histolytica
Giardia lamblia
Helminths
Ascaris lumbricoidesDiphyllobothrium latumEnterobius vermicularisNecator americanusStrongyloides stercoralis
Trichuris trichiura
Produces colonic ulcers with bloody diarrheaMost common cause of diarrhea in AIDSContracted by ingesting oocysts (acid-fast positive; attach to brush border)Detected with string test (organisms attach to string that is swallowed)Produces dysentery with flask-shaped ulcers in cecumTrophozoites phagocytose red blood cellsMost common protozoal cause of diarrhea in United StatesProduces acute and chronic diarrhea with malabsorptionDetected with string test or examination of stool
Bowel obstruction in adult phaseAdult worms produce vitamin B 12 deficiencyEggs deposited in anus cause anal pruritusAdults attach to villi, resulting in blood loss and iron deficiencyProduces abdominal pain and diarrheaPositive string testAbdominal pain, diarrhea; rectal prolapse in children
ETEC, enterotoxigenic Escherichia coif; STEC, Shiga toxin E coli

214 Pathology
undigested lactose to lactic acid (acid pH stool) andhydrogen gas, leading to abdominal distention withexplosive diarrhea. Treatment is avoidance of dairyproducts.
E. Malabsorption• Increased fecal excretion of fat with concurrent deficien-
cies of vitamins (particularly fat-soluble vitamins), miner-als, carbohydrates, and proteins
A qualitative or quantitative fecal test for fat is thegold standard screening test for malabsorption. Serumbeta carotene levels are decreased because of the de-creased reabsorption of fat-soluble vitamins.
1. Pathogenesisa. Pancreatic insufficiency
(I) Maldigestion of tats caused by diminishedlipase and/or colipase activity; ma [digestion ofproteins caused by diminished trypsin
(2) Carbohydrate digestion unaffected (amylasein salivary glands and presence of brush borderenzymes)
(3) Etiology: chronic pancreatitis (e.g., alcoholabuse, cystic fibrosis)
b. Bile salt deficiency(1) Defective micellarization of fats for easy ab-
sorption by villi(2) Etiology: cirrhosis (decreased synthesis), chole-
stasis (decreased bile transport), bacterial over-growth (destruction of bile salts), terminalileal disease (decreased recycling)
c. Small bowel disease (Table 17-5)(1) Inability to reabsorb micelles due to loss of
villous surface (e.g., celiac disease, Crohn'sdisease, Whipple's disease)
(2) Lymphatic obstruction (e.g., Whipple'sdisease, malignant lymphoma)
(3) Abetalipoproteinemia: loss of apolipoproteinB-48 prevents movement of chylomicrons intolymphatics; lipids accumulate in intestinalcells.
(4) D-Xylose screening test: inability to absorborally administered xylose into the blood orurine indicates small bowel disease.
2. Clinical findingsa. Weight loss: due to calorie lossb. Steatorrhea: excessive, bulky, greasy stools that floatc. Deficiencies of fat-soluble vitamins (A, D, E, K) and
water-soluble vitamins (see Chapter 7)

Chapter 17 Gastrointestinal Disorders 215
TABLE 17-5 Small Bowel Disorders That Cause Malabsorption
Disorder
Characteristics Clinical Findings
Celiac disease
Whipple'sdisease
Autoimmune disease producingantibodies against gliadin frac-tion in gluten
Associated with HLA-DQ2 andHLA-DQ8
Usually begins in infancy; mostcommon in females
Blunting of villi in duodenumand jejunum
Most common in malesCaused by Tropheryma whippelii
bacilliBlunting of villi; foamy
PAS-positive macrophages inlamina propria
Associated with dermatitisherpetiformis (autoimmunevesicular skin disease)
May produce T-cell lymphomaof stomach and/or smallintestine
Restrict or eliminate gluten fromdiet
Fever, recurrent polyarthritis,generalized lymphadenopathy,increased skin pigmentation
HLA, human leukocyte antigen; PAS, periodic acid-Schiff.
d. Combined anemias (e.g., macrocytic anemia fromfolate or vitamin B 12 deficiency combined withiron deficiency)
e. Ascites and pitting edema caused by hypo-proteinemia
F. Inflammatory bowel disease (Table 17-6)1. Ulcerative colitis
a. Chronic, relapsing ulceroinflammatory diseaseb. Ulcerations involve the mucosa and submucosa of
the rectum and colon (Figure 17-6).2. Crohn's disease
a. Chronic, granulomatous, ulceroconstrictivedisease
b. Transmural inflammation is discontinuousthroughout entire GI tract (Figure 17-7).
G. Diverticulosis1. Meckel's diverticulum2. Small bowel diverticulosis: pulsion diverticula
a. Most often occurs in duodenum; wide-moutheddiverticula suggest systemic sclerosis.
b. Complications: diverticulitis (danger of perfora-tion), bacterial overgrowth (produces bile salt defi-ciency and vitamin B 12 deficiency)
3. Colonic diverticulosis: pulsion diverticulaa. Most commonly occurs in sigmoid colonb. Caused by a low-fiber diet, resulting in increased
constipationc. Pathogenesis: area of weakness is site where the
vessels penetrate the muscular propria; diverticulumis juxtaposed to a blood vessel (Figure 17-8).
Most common in-flammatorybowel disease:ulcerative colitis
216 Pathology
TABLE 17-6 Comparison of Ulcerative Colitis and Crohn's Disease
Feature
Ulcerative Colitis
Crohn's Disease
Epidemiology
Extent
Location
Gross features
Microscopic features
Clinical findings
Radiography
Complications
More common in whitesthan blacks
No sex predilectionAffects young adults
Mucosal and submucosalMainly rectum; extends
continuously into leftcolon; may involve entirecolon
Does not involve otherareas of GI tract
Inflammatory pseudopolypsAreas of friable, bloody
residual mucosa; ulcera-tion and hemorrhage
Ulcers and crypt abscessescontaining neutrophils
Dysplasia or cancer may bepresent
Recurrent left-sided abdom-inal cramping withbloody diarrhea andmucus
"Lead pipe" appearance inchronic disease
Toxic megacolon (hypotonicand distended bowel)
Primary sclerosing cholan-gitis (fibrosis aroundcommon bile duct leadingto jaundice); HLA-B27–positive spondylo-arthropathy
Adenocarcinoma (greatestrisks are pancolitis, earlyonset, duration of disease> 10 years)
More common in whitesthan blacks, in Jews thannon-Jews
More common in womenAffects young adults
Transmural
Terminal ileum alone (30%),ileum and colon (50%),colon alone (20%)
Involves other areas of GItract (mouth to anus)
Thick bowel wall andnarrow lumen (leads toobstruction)
Aphthous ulcers (early sign)Skip lesions, strictures,
fistulas; deep linear ulcerswith cobblestone pattern
Fat creeping around serosa
Noncaseating granulomas(60%), lymphoid aggre-gates
Dysplasia or cancer lesslikely
Recurrent right lower quad-rant colicky pain (obstruc-tion) with diarrhea
Bleeding occurs only withcolon or anal involvement
"String" sign in terminalileum from lumina!narrowing by inflamma-tion, fistulas
Fistulas, obstructionCalcium oxalate renal
calculi (increased reab-sorption of oxalatethrough inflamedmucosa)
Malabsorption due to bilesalt deficiency
Macrocytic anemia due tovitamin B 12 deficiency
GI, gastrointestinal

Chapter 17 Gastrointestinal Disorders 217
Figure 17 -6 Ulcerative colitis.The colon shows diffuse ul-ceration of the mucosal surfaceand residual islands of in-flamed mucosa (pseudopolyps)
Figure 17 -7 Crohn's disease,showing a resection of the termi-nal ileum with attached cecumand appendix; the appendix is tothe left The thickened terminalileal wall causes the narrow-ing (arrow) at the junction of theileum and the cecum The ilea!mucosa has a cobblestoneappearance due to linear ulcer-ations (aphthous ulcers) that cutinto the underlying submucosa
Figure 17 -8 Sigmoid diverticu-losis viewed on barium enemaMultiple diverticula sacs areoutlined by the double-contrastbarium technique

d. Complications(1) Diverticulitis: fecalith in diverticulum sac
causes ulceration and ischemia, possible per-foration (peritonitis and air under diaphragm),and abscess.
(2) Hematochezia most commonly caused by di-verticulosis; formation of fistulas (connectionbetween hollow structures)
H. Small bowel obstruction1. Signs: colicky pain; radiograph shows bowel distention
and air-fluid levels with a stepladder appearance2. Etiology
a. Adhesions from previous surgeryb. Hernia: small bowel trapped in indirect hernia sac;
second most common cause of obstruction in adultsc. Intussusception
(1) Occurs most often in children(2) Terminal ileum telescopes into the cecum,
causing a combination of obstruction andischemia as well as colicky pain with bloodydiarrhea.
d. Meconium ileus: complication in newborns withcystic fibrosis
e. Volvulus: bowel twists around mesenteric root, re-sulting in obstruction and strangulation.
f. Duodenal atresia, Hirschsprung's diseaseI. Small and large bowel polyps
1. Non-neoplastic (hamartomatous) polypsa. Hyperplastic polyps: most common polyp; no asso-
ciated polyposis syndromes; histologically have a"sawtooth" appearance
b. Juvenile (retention) polyps: most common polypin children; the most common location is therectum, where polyps may prolapse and bleed.
c. Peutz-Jeghers polyps: autosomal dominant; mostoften occur in the small bowel; mucosal pigmen-tation of buccal mucosa and lips
2. Neoplastic polyps: adenomasa. Premalignant dysplastic colonic polyps; more
common with increasing ageb. Tubular adenoma (60% of cases)
(1) Occur most often in the sigmoid colon(2) Stalked polyp; complex branching of glands
(adenomatous change) (Figure 17-9)c. Tubulovillous adenoma: usually stalked polyp;
combination of adenomatous and villous architec-ture (similar to small bowel villi)
d. Villous adenoma: large and sessile with mainlyvillous architecture; rectosigmoid location; secretesprotein and potassium-rich mucus
e. Risk factors for malignancy in adenomas: size> 2 cm, multiple polyps, villous architecture
218 Pathology
Most commoncomplication ofdiverticulosis:diverticulitis
Most commoncause of smallbowel obstruction:adhesions
Most common siteof GI polyps:sigmoid colon
••••••••••••••••••••••••••••••••••••

Chapter 17 Gastrointestinal Disorders 219
Figure 17 -9 Tubular adenoma. The head of the stalked polyp has a lobulated, raspberry-likeappearance. The arrow points to the stalk.
3. Familial polyposis syndromesa. Autosomal dominant disorders; inactivation of the
adenomatous polyposis coli (APC) suppressor geneon chromosome 5
b. Polyps develop between 10 and 20 years of age; ma-lignant transformation occurs between 35 and40 years of age (treatment is prophylacticcolectomy).
c. Gardner's syndrome: autosomal dominant poly-posis; characterized by colonic polyps, benign osteo-mas, and benign soft tissue tumors
d. Turcot's syndrome: autosomal recessive polypo-sis; characterized by colonic polyps and malig-nant brain tumors (e.g., astrocytoma)
J. Small bowel cancer1. Carcinoid tumor
a. Neuroendocrine tumor contains neurosecretorygranules.
b. Metastasis correlates with size (> 2 cm) and depthof invasion (> 50% of bowel thickness).
c. Most common location is the tip of the appendix(usually too small to metastasize to liver); appearsas a bright yellow tumor
d. Carcinoid syndrome(1) Develops when carcinoid tumor metastasizes
to liver(2) Clinical findings: caused by serotonin, which
produces cutaneous flushing, diarrhea, tricus-pid regurgitation, and pulmonic stenosis
2. Malignant lymphoma: most often occurs in Peyer'spatches of terminal ileum; usually B-cell origin
3. Primary adenocarcinoma: most often located in theduodenum
K. Colorectal carcinoma1. Common cause of death due to cancer in both men
and women
Most commonprimary site of me-tastasis causingcarcinoid syndrome:terminal ileum
Carcinoid syn-drome: T urine5-hydroxyindoleaceticacid from serotonindegradation

220 Pathology
Figure 17-10 Adenocarcinomaof the sigmoid colon Resec-tion of the rectosigmoid showsan annular and ulcerating growth,causing a stricture.
2. Risk factorsa. Age older than 50 yearsb. Diet: low-fiber, high saturated fatc. Cigarette smoking, familial polyposis syndrome,
family history, ulcerative colitis3. Pathogenesis
a. Adenoma-carcinoma sequence: sequential loss ofgenes (APC, K-RAS, 18q21 deletion, TP53)
b. Inactivation of DNA mismatch genes, which nor-mally correct mismatches of nucleotide bases
Hereditary (autosomal dominant) nonpoly-posis syndrome (Lynch syndrome) is character-ized by inactivation of DNA mismatch genes.Type I is associated with right-sided colorectalcarcinoma (70%). Type II is associated withcolorectal carcinoma plus adenocarcinomasinvolving other tissues (e.g., endometrium,pancreas).
4. Location: rectosigmoid (50% of cases), ascendingcolon, descending colon, transverse colon and cecum
5. Screening tests: fecal occult blood test, flexible sigmoid-oscopy, colonoscopy (gold standard), barium enema
6. Clinical findingsa. Rectosigmoid location: obstruction; annular,
"napkin-ring" appearance; change in bowel habits(constipation and/or diarrhea) (Figure 17-10)
b. Ascending colon: bleeding, polypoid tumors, irondeficiency
7. Sites of metastasis: liver, lungs, bone, brain8. Prognosis: most important factor is the stage of cancer;
5-year survival rate about 35% overall; use carcinoem-bryonic antigen to track patients for recurrences.

Chapter 17 Gastrointestinal Disorders 221
VI. Anorectal DisordersA. Rectal prolapse
1. Intussusception of rectum through the anus becauseof weak rectal support
2. Etiology: in children younger than 2 years of age, severecoughing, cystic fibrosis; in adults, straining at stool
B. Pilonidal cyst and abscess: painful sacrococcygeal masswith purulent drainage caused by excess hair in a deepgluteal fold becoming traumatically buried in a sinus
C. Anal carcinoma1. Basaloid (epidermoid or cloacogenic) carcinoma:
most common type; occurs most often in women;located in the transitional zone above the dentate line
2. Squamous cell carcinoma: located in the anal canal;majority occur in homosexual men (associated withHPV)
VII. Acute AppendicitisA. Pathogenesis
1. Children: lymphoid hyperplasia (60% of cases) oftensecondary to a viral infection (e.g., adenovirus, measles)
2. Adultsa. Obstruction of the proximal lumen by a fecalith
causes increased intraluminal pressure, leading tomucosal injury and bacterial invasion (e.g., Esche-richia coli, Bacteroides fragilis).
b. May also be caused by sunflower seeds or pinworminfection
B. Clinical findings1. Initial colicky periumbilical pain; nausea, vomiting,
and fever2. Pain moves to right lower quadrant: rebound tender-
ness at McBurney's point3. Laboratory findings: neutrophilic leukocytosis; protein-
uria, hematuria, and pyuriaC. Periappendiceal abscess with or without perforation: most
common complication; subphrenic abscess may develop(usually result of B. fragilis).
D. Disorders mimicking appendicitis: viral gastroenteritis,ruptured follicular cyst, ruptured ectopic pregnancy, mesen-teric lymphadenitis, Meckel's diverticulitis

18dist 1_Hepatobiliary andPancreatic Disorders
I. Laboratory Evaluation in Liver Cell InjuryA. Liver function tests (Table 18-1)B. Results of liver cell injury
1. Damaged hepatocyte integrity; serum transaminases(alanine transaminase, ALT; aspartate transaminase,AST) are elevated.
2. Disrupted bilirubin metabolism; biliary excretion isaffected (Figure 18-1).a. Unconjugated bilirubin is the end product of
heme degradation by macrophages in the spleen;unconjugated bilirubin is lipid soluble.
b. Unconjugated bilirubin combines with albuminin the blood, is taken up by hepatocytes, and isthen conjugated by uridine glucuronosyltrans-ferases.
c. Conjugated bilirubin is secreted into the intrahe-patic bile ducts and eventually enters the small in-testine; conjugated bilirubin is water soluble.
d. Intestinal bacteria convert conjugated bilirubin tourobilinogen, which is oxidized to urobilin;stool and urine derive their color from urobilin.
e. A small amount of urobilinogen is recycled to theliver and kidneys; some is normally present inthe urine.
3. Reduced hepatocyte function; protein synthesis (e.g.,albumin, coagulation factors); cell metabolism (e.g.,urea cycle for disposal of ammonia)
C. Jaundice: hyperbilirubinemia1. Characterized by yellowing of the sclerae, skin, or
mucous membranes2. Determination of type and cause is based on the per-
centage of conjugated bilirubin (Table 18-2).
% conjugatedbilirubin = con-jugated bilirubin÷ total bilirubin
222

Chapter 18 Hepatobiliary and Pancreatic Disorders 223
TABLE 18-1 Liver Function Tests
Test Significance
Hepatocyte function
Serum albumin
ALT > AST: viral hepatitisAST > ALT: alcoholic hepatitis
Induction of cytochrome P-450 system (e.g.,alcohol) increases GGT
Normal GGT and increased ALP: source ofALP other than liver
Increased GGT and ALP: liver cholestasis
Mainly unconjugated hyperbilirubinemia (e.g.,congenital spherocytosis)
Mixed hyperbilirubinemia (e.g., viral hepatitis)Mainly conjugated hyperbilirubinemia (e.g.,
gallstone in common bile duct)Bilirubinuria: viral hepatitis, obstruction of bile
ducts (e.g., gallstone in common bile duct)Increased urine urobilinogen: extravascular
hemolytic anemias (e.g., congenital sphero-cytosis), viral hepatitis
Absent urine urobilinogen: bile duct obstruc-tion (e.g., gallstone in common bile duct)
Albumin is synthesized by the liverHypoalbuminemia: severe liver disease (e.g.,
cirrhosis)Majority of coagulation factors are synthesized
in liverProlonged PT: severe liver disease
Urea cycle occurs in liverDecreased serum BUN: cirrhosis
Ammonia is metabolized in the urea cycleIncreased serum ammonia: cirrhosis, Reye's
syndrome
Liver cell necrosis
Serum alanine transaminase (ALT)Serum aspartate transaminase (AST)
Cholestasis
Serum y-glutamyltransferase (GGT)Serum alkaline phosphatase (ALP)
Biliruhin excretion
Conjugated bilirubin'
<20%
20-50%> 50%
Urine bilirubin
Urine urobilinogen
Prothrombin time (PT)
Blood urea nitrogen (BUN)
Serum ammonia
"Conjugated bilirubin % = conjugated bilirubin total bilirubin.
II. Viral Hepatitis• Most infections that cause liver cell necrosis are due to viruses.A. Pathology
1. Microscopic findings in acute viral hepatitisa. Lymphocytic infiltrate with destruction of
hepatocytesb. Apoptosis of hepatocytes (Councilman's bodies)
2. Microscopic findings in chronic viral hepatitisa. Inflammation disrupts the normal demarcation
between the portal triad and hepatocytes locatedaround the portal triad (piecemeal necrosis).
Most commoncause of massivehepatic necrosis:viral hepatitis

224 Pathology
Senescent RBC Globin chain splits off
Macrophage - Heme
Iron
Unbound unconjugated bilirubin (lipid soluble)
Blood -[ Unconjugated bilirubin (bilirubin + albumin)
UGT
Liver -Conjugated bilirubin (water soluble)
Secreted into bile
Bowel -80%
Urobilinogen Urobilin (color of stool)20%
Enterohepaticcirculation
Liver (90%) Kidneys (10%) —)-Urobilin (color of urine)
Figure 18-1 Bilirubin metabolism. RBC, red blood cell; UGT, uridine glucuronosyltransferase
b. Fibrosis connects a portal triad to another portaltriad or the portal triad to the central vein.
B. Phases of acute viral hepatitis1. Prodrome: fever, hepatomegaly; steadily increasing
serum transaminases, which peak just before jaundiceoccurs; atypical lymphocytosis
2. Icteric: severity of jaundice varies depending on thetype of hepatitis; bilirubinuria, increased urineurobilinogen
3. Recovery: jaundice resolves.C. Transmission and clinical features (Table 18-3); acute hep-
atitis becomes chronic when symptoms last more than 6months.
D. Serology1. Hepatitis A virus (HAV)
a. IgM anti-HAV indicates active infection.b. IgG anti-HAV indicates recovery from infection or
vaccination (protective antibody).2. Hepatitis B virus (HBV) (Figure 18-2; Table 18-4)
a. Hepatitis B surface antigen (HBsAg)(1) Appears within 2-8 weeks; first marker of
infection(2) Persists up to 4 months in acute HBV and
more than 6 months in chronic HBVb. Hepatitis B e antigen (HBeAg) and HBV DNA
(1) Infective particles; appear after HBsAg anddisappear before HBsAg
Most common typeof hepatitis associ-ated with accidentalneedlestick and in-travenous drug use:HBV
Chapter 18 Hepatobiliary and Pancreatic Disorders 225
TABLE 18-2 Causes of Jaundice
Type of Urine UrineHyperbilirubinemia Bilirubin Urobilinogen Examples of Disorders
UnconjugatedConjugated bilirubin
< 20%
Increased produc- Absent 1. Extravascular hemolytic anemias:tion of unconju- congenital spherocytosis, Rh andgated bilirubin ABO hemolytic disease of newborn
Destruction of RBCs in bone marrow:severe j3-thalassemia, deficienciesof vitamin B12 and folate
Decreased conjuga- Absent Normal Gilbert syndrome: genetic defect intion of unconju- uptake of bilirubin; jaundice occursgated bilirubin with fasting
Crigler-Najjar syndrome: geneticdisorder caused by decrease inconjugating enzymes
Physiologic jaundice of newborn:begins on day 3 of life (caused bydestruction of fetal RBCs)
MixedConjugated bilirubin T T Viral hepatitis: also a defect in uptake
20-50% of bilirubin
Obstructive
Conjugated bilirubin T> 50%
Absent Decreased intrahepatic bile flowDrug-induced (e.g., oral contra-
ceptives)Primary biliary cirrhosisDubin-Johnson syndrome: genetic
defect in secretion into intrahe-patic bile ducts; black pigment inhepatocytes
Rotor's syndrome: similar toDubin-Johnson syndromebut without black pigment inhepatocytes
Decreased extrahepatic bile flowGallstone in common bile ductCarcinoma of head of pancreas
RBCs, red blood cells.
(2) Presence for more than 6 months in conjunc-tion with HBsAg indicates an infectivechronic carrier.
c. IgM anti-HBV core antibody (IgM anti-HBc)(1) Nonprotective antibody; remains positive in
acute and chronic infections.(2) Persists during the "window phase," or "se-
rologic gap," when HBsAg, HBV DNA, andHBeAg are absent
(3) Converts to IgG anti-HBc when there is noviral replication
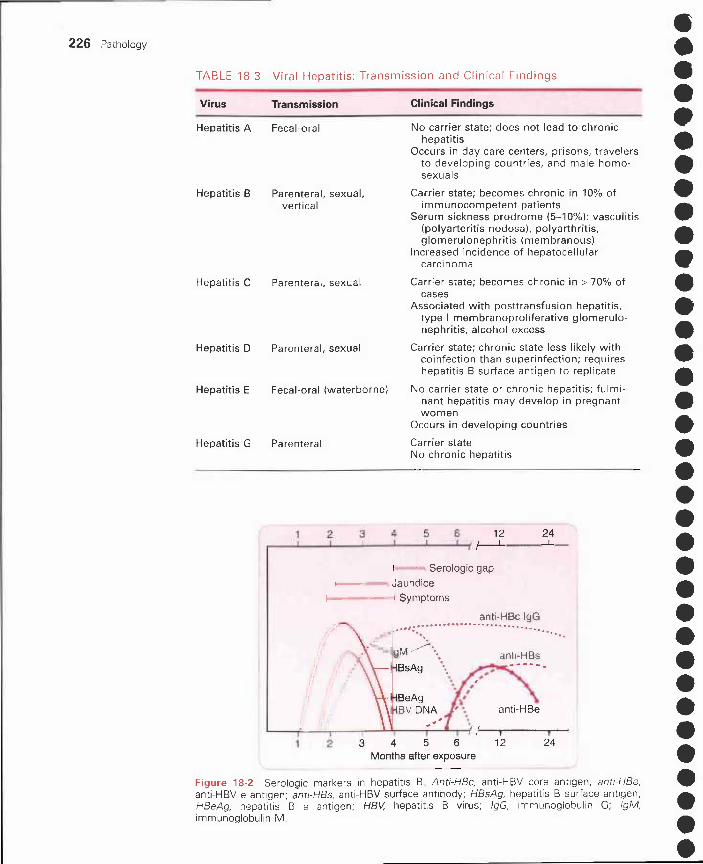
.............. anti-H Bc Ig G•..
anti-HBsHBsAg.....
226 Pathology
TABLE 18-3 Viral Hepatitis: Transmission and Clinical Findings
Virus Transmission Clinical Findings
Hepatitis A Fecal -oral
Hepatitis B Parenteral, sexual,vertical
No carrier state; does not lead to chronichepatitis
Occurs in day care centers, prisons, travelersto developing countries, and male homo-sexuals
Carrier state; becomes chronic in 10% ofimmunocompetent patients
Serum sickness prodrome (5-10%): vasculitis(polyarteritis nodosa), polyarthritis,glomerulonephritis (membranous)
Increased incidence of hepatocellularcarcinoma
Hepatitis C Parenteral, sexual Carrier state; becomes chronic in > 70% ofcases
Associated with posttransfusion hepatitis,type I membranoproliferative glomerulo-nephritis, alcohol excess
Hepatitis D Parenteral, sexual Carrier state; chronic state less likely withcoinfection than superinfection; requireshepatitis B surface antigen to replicate
Hepatitis E Fecal-oral (waterborne) No carrier state or chronic hepatitis; fulmi-nant hepatitis may develop in pregnantwomen
Occurs in developing countries
Hepatitis G Parenteral Carrier stateNo chronic hepatitis
12
24 /
Serologic gap
JaundiceSymptoms
HBeAgHBV DNA anti-HBe
,••
3 4 5 6
112 214Months after exposure
Figure 18-2 Serologic markers in hepatitis B Anti-HBc, anti-HBV core antigen; anti-HBe,
anti-HBV e antigen; anti-Has, anti-HBV surface antibody; HBsAg, hepatitis B surface antigen;HBeAg, hepatitis B e antigen; HBV, hepatitis B virus; IgG, immunoglobulin G; /gM,immunoglobulin M

Chapter 18 Hepatobiliary and Pancreatic Disorders 227
TABLE 18-4 Serologic Studies in Hepatitis B
HBeAg, IgM IgGHBsAg HBV DNA Anti-HBc Anti-HBc Anti-HBs Interpretation
+ - Earliest phase of acute HBV
+ + + - Acute or chronic HBV
- + - - Window phase (serologic gap)
- - + + Recovery from HBV
- - - + Immunization against HBV
HBV, hepatitis B virus.
d. Anti-HBV surface antibody (anti-HBs): protectiveantibody; only marker of immunization afterHBV vaccination
e. Chronic HBV: persistence of HBsAg more than 6months (1gM anti-HBc remains positive)(1) "Healthy" chronic carriers: presence of
HBsAg and absence of infective particles (HBVDNA, HBeAg)
(2) Infective chronic carriers: presence of bothHBsAg and infective particles (HBV DNA,HBeAg); increased risk of cirrhosis and hepa-tocellular carcinoma
3. Hepatitis C virus (HCV): presence of IgG or IgM anti-HCV indicates infection.
4. Hepatitis D virus (HDV): presence of IgG or IgM anti-HDV indicates active infection.
5. Hepatitis E virus (HEV): presence of 1gM anti-HEV in-dicates active infection; IgG anti-HEV indicates recovery(protective antibody).
E. Vaccination against HBV decreases the risk of HBV andHDV and decreases the risk of hepatocellular carcinoma.
III. Other Inflammatory Hepatic DisordersA. Autoimmune hepatitis
1. Occurs most often in young women2. Clinical findings: fever, jaundice, hepatosplenomegaly3. Laboratory findings: positive serum antinuclear anti-
body, anti—smooth muscle antibodiesB. Neonatal hepatitis
1. Idiopathic or associated with infections (e.g., cyto-megalovirus) or inborn errors of metabolism (e.g.,galactosemia)
2. Characterized by the presence of multinucleated giantcells
C. Reye's syndrome1. Usually develops in children younger than 4 years of
age; often follows chickenpox or influenza

228 Pathology
TABLE 18-5 Some Infectious Diseases That Affect the Liver
Disease Causal Pathogen(s)
Characteristics
Entamoeba histolytica
Escherichia coli in adults,Streptococcus pneumoniaein children
Mycobacterium tuberculosis,Histoplasma capsulatum
Echinococcus granulosus(sheepherder's disease)
Schistosoma mansoniEggs incite a fibrotic response
in the portal vein ("pipestemcirrhosis")
Amebiasis; abscess usuallyinvolves right lobe
Develops in ascites (e.g.,cirrhosis, nephroticsyndrome)
Sign of miliary spread
Single or multiple cystscontaining larval forms
Complications of cirrhosis:portal hypertension, ascites,and esophageal varices
Liver abscess
Spontaneousperitonitis
Granulomatoushepatitis
Echinococcosis
Schistosomiasis
2. Pathogenesis: mitochondrial damage (may be causedby viral infection or salicylates), with disruption ofthe urea cycle (increase in serum ammonia) and defec-tive 13-oxidation of fatty acids
3. Pathology: microvesicular type of fatty liver (smallcytoplasmic globules without nucleus displacement)
4. Clinical findings: encephalopathy (cerebral edema,coma, convulsions); hepatomegaly
5. Laboratory findings: elevated serum transaminases(ALT > AST), normal to slightly elevated total bilirubin
D. Other infectious diseases affecting the liver (Table 18-5)
IV. Circulatory Disorders of the LiverA. Prehepatic obstruction to blood flow
1. Hepatic artery thrombosis with infarctiona. Liver infarction is uncommon because of a dual
blood supply (e.g., tributaries of the hepatic arteryand portal vein empty their blood into thesinusoids).
b. Etiology: liver transplantation, vasculitis due topolyarteritis nervosa
2. Portal vein thrombosisa. Etiology: pylephlebitis (inflammation of the
portal vein), polycythemia vera, hepatocellular car-cinoma (tumor invasion of the portal vein)
b. Clinical findings: portal hypertension, ascites,splenomegaly; no hepatomegaly
B. Intrahepatic obstruction to blood flow1. Etiology: passive congestion with or without centrilob-
ular hemorrhagic necrosisa. Passive congestion: backup of systemic venous
blood into the central veins and sinusoids; usuallysecondary to right-sided heart failure
Most commoncause of pylephlebi-tis: acuteappendicitis

Chapter 18 Hepatobiliary and Pancreatic Disorders 229
Figure 18 -3 Centrilobular hem-orrhagic necrosis ("nutmeg"liver) The liver has a mottled cutsurface Dark areas representcongested central veins andsinusoids
b. Centrilobular hemorrhagic necrosis: results froma combination of left- and right-sided heartfailure(1) Left-sided heart failure: produces hypoperfu-
sion of the liver leading to ischemic necrosisof hepatocytes around the central vein
(2) Right-sided heart failure: causes congestionof the central veins and hemorrhage into thesurrounding parenchyma
2. Gross findings: hepatomegaly with a mottled red ap-pearance ("nutmeg liver") (Figure 18-3)
3. Microscopic findings: congestion of the central veinsalong with hemorrhage into the subjacent liver pa-renchyma; necrosis of hepatocytes around the centralvein
4. Clinical and laboratory findingsa. Enlarged painful liver, jaundiceb. Elevated transaminases caused by ischemic necrosisc. May progress to cardiac cirrhosis (fibrosis around
central veins)C. Posthepatic obstruction to blood flow: hepatic vein
thrombosis (Budd-Chiari syndrome)1. Etiology: polycythemia vera, oral contraceptive use,
hepatocellular carcinoma invading the hepatic vein2. Clinical findings: enlarged painful liver, ascites, portal
hypertension, splenomegaly3. Laboratory findings: elevated transaminases, pro-
longed prothrombin time
s igns of hepaticvein thrombosis: he-patomegaly, ascites,portal hypertension

230 Pathology
TABLE 18-6 Drug- and Chemical-induced Liver Diseases
Disease
Cause
Tumors
Angiosarcoma
Cholangiocarcinoma
Hepatocellular carcinoma
Liver cell adenoma
Other liver diseases
Acute hepatitis
Cholestasis
Fatty change
Fibrosis
Vinyl chloride, arsenic, thorium dioxide (radioactivecontrast material)
Thorium dioxide
Vinyl chloride, aflatoxin (due to Aspergillus mold)
Oral contraceptives
Isoniazid (caused by toxic metabolite), halothane, acet-aminophen, methyldopa
Oral contraceptives (estrogen interferes with intrahepaticbile secretion), anabolic steroids, erythromycin estolate
Amiodarone (resembles alcoholic hepatitis; Mallory'sbodies and progression to cirrhosis), methotrexate
Methotrexate, retinoic acid, amiodarone
V. Alcohol-related and Drug- and Chemical-induced LiverDisordersA. Alcohol-related disorders
1. Fatty change; most common alcohol-related liverdisordera. Substrates of alcohol metabolism are used to syn-
thesize liver triacylglycerol; reversible withalcohol abstinence
b. Clinical findings: tender hepatomegaly withoutfever
c. Laboratory findings: AST > ALT, increasedy-glutamyltransferase (GGT)
2. Alcoholic hepatitisa. Pathogenesis: caused by acetaldehyde damage to
hepatocytes and stimulation of collagen synthe-sis around the central vein (perivenular fibrosis)
b. Microscopic findings: fatty change, neutrophil in-filtration, Mallory's bodies (damaged cytokeratinintermediate filaments in hepatocytes), perivenularfibrosis
c. Clinical findings: fever, painful hepatomegaly,ascites, hepatic encephalopathy
3. Alcoholic cirrhosis (see section VII)B. Chemical- and drug-induced liver disease (Table 18-6)
Vl. Cholestatic (Obstructive) Liver DiseaseA. Types of cholestatic liver disease
1. Intrahepatic cholestasis: blockage of intrahepatic bileductsa. Etiology: drugs (e.g., oral contraceptives), neonatal
hepatitis
Alcohol-related dis-orders: fatty change,alcoholic hepatitis,cirrhosis

Chapter 18 Hepatobiliary and Pancreatic Disorders 231
b. Pregnancy-induced cholestasis (benign intrahe-patic cholestasis of pregnancy)(1) Caused by estrogen inhibition of bile secretion(2) Presents no danger to mother or fetus
2. Extrahepatic cholestasis: blockage of common bile ducta. Etiology: gallstone in the common bile duct,
primary sclerosing cholangitis, pancreatic carci-noma
b. Extrahepatic biliary atresia(1) Cause of jaundice in newborns; inflamma-
tory destruction of all or part of the extrahe-patic bile ducts; bile duct proliferation inthe portal triads
(2) Accounts for most liver transplantations inchildren
B. Findings associated with cholestasis1. Gross findings: enlarged liver with green discoloration2. Clinical findings
a. Jaundice with pruritus: due to bile salts deposited inthe skin
b. Malabsorption (bile salts do not enter the smallintestine)
c. Cholesterol deposits in the skin (from cholesterolin bile)
d. Light-colored stools (lack of urobilin)3. Laboratory findings
a. Percent conjugated bilirubin > 50%, bilirubinuriab. No urine urobilinogenc. Increased serum alkaline phosphatase and GGT
C. Primary sclerosing cholangitis1. Obliterative fibrosis of intrahepatic and extrahepatic
bile ducts: more common in males; associated withulcerative colitis
2. Clinical findings: jaundice, cirrhosis; increased inci-dence of cholangiocarcinoma
VII. CirrhosisA. Pathology
1. Irreversible diffuse fibrosis2. Regenerative nodules: hepatocyte reaction to injury
a. Lack normal liver architecture; surrounded bybands of fibrosis (Figure 18-4)
b. Compress sinusoids and central veins, producingintrasinusoidal hypertension with an overallreduction in functional sinusoids
c. Cirrhosis is sometimes classified by nodule size: mi-cronodular, macronodular, or mixed.
B. Etiology1. Alcoholic liver disease2. Infection (postnecrotic cirrhosis caused by HBV or
HCV)3. Autoimmune disease (biliary cirrhosis)
Extrahepatic biliaryatresia: majorreason for livertransplantation inchildren
Most common typeof cirrhosis:alcoholic cirrhosis

232 Pathology
Figure 18-4 Alcoholic cirrhosis,showing diffuse micronodularsurface of the liver. The regener-ative nodules are surroundedby fibrosis
4. Metabolic disease (hemochromatosis, Wilson's disease,al -antitrypsin deficiency)
C. Conditions associated with cirrhosis1. Hepatic failure: end point of progressive damage to the
livera. Most often caused by chronic liver diseaseb. Clinical findings
(1) Multiple coagulation defects: caused by in-ability of hepatocytes to synthesize coagula-tion factors; hemorrhagic diathesis
(2) Hypoalbuminemia: caused by decreased syn-thesis of albumin; dependent pitting edemaand ascites
(3) Hepatic encephalopathy: caused by increasedsynthesis of false neurotransmitters (e.g.,GABA) and increased serum ammonia; alter-ations in mental status, asterixis (flappingtremor), coma
2. Portal hypertensiona. Caused by resistance to intrahepatic blood flow
(due to intrasinusoidal hypertension) and anasto-moses between portal vein tributaries and the arte-rial system
b. Complications: ascites, congestive splenomegaly,esophageal varices, hemorrhoids, periumbilicalvenous collaterals (caput medusae)
3. Ascitesa. Pathogenesis
(1) Portal hypertension: increases portal vein hy-drostatic pressure, forcing fluid (transudate)into the peritoneal cavity
(2) Hypoalbuminemia: decreases oncotic pres-sure, causing loss of fluid (transudate) into theperitoneal cavity
(3) Secondary hyperaldosteronism: caused bydecreased cardiac output, leading to activationof the renin-angiotensin-aldosterone system(retention of sodium and water)
Most commoncause of esophagealvarices: portalhypertension

Chapter 18 Hepatobiliary and Pancreatic Disorders 233
b. Clinical findings: abdominal distention with afluid wave
4. Hepatorenal syndrome: renal failure without intrinsicrenal parenchymal diseasea. Decreased renal blood flowb. Preservation of renal tubular function
5. Hyperestrinism in malesa. Pathogenesis: liver is unable to degrade hormones
such as estrogens and 17-ketosteroids (con-verted to estrogen).
b. Clinical findings: gynecomastia, spider angiomas,female distribution of hair
D. Postnecrotic cirrhosis1. Most often caused by chronic hepatitis due to HBV
and HCV2. Increased incidence of hepatocellular carcinoma
E. Primary biliary cirrhosis: autoimmune disorder1. Occurs more often in women 40-50 years of age2. Pathogenesis: granulomatous destruction of bile
ducts in the portal triads; progresses from a chronicinflammatory reaction to cirrhosis
3. Clinical findings: pruritus, hepatomegaly, jaundice(occurs later in the disease when most of the bile ductshave been destroyed), portal hypertension
4. Laboratory findings: antimitochondrial antibodies(> 90% of cases), increased IgM
F. Hereditary hemochromatosis: autosomal recessive disorder1. Occurs more often in men older than 40 years of age;
in women, symptoms develop after menopause.2. Pathogenesis
a. Increased reabsorption of iron in the small intestineb. Mutations involving hereditary hemochromatosis
gene (1:10 carrier rate)3. Iron deposits in multiple organs: liver, pancreas,
heart, joints, skin; iron produces hydroxyl free radicals,which damage tissue and cause fibrosis.
Hemosiderosis (secondary hemochromatosis) iscaused by multiple blood transfusions (e.g., sicklecell anemia, thalassemia major); alcohol abuse (al-cohol increases iron reabsorption); and well water(iron pipes). Iron deposits are more prevalent inmacrophages than in parenchymal tissue.
4. Clinical and laboratory findingsa. Cirrhosis: iron deposits in hepatocytes; increased
risk of hepatocellular carcinomab. "Bronze" diabetes: diabetes mellitus (caused by
destruction of (3-islet cells), hyperpigmentation(caused by iron deposits and increased melaninproduction)
Most commonpathologic cause ofgynecomastia:cirrhosis
Signs of hemochro-matosis: "bronze"diabetes and Tserum ferritin

234 Pathology
c. Malabsorption (destruction of exocrine pancreas),restrictive cardiomyopathy, degenerative jointdisease
d. Increased serum iron, percent iron saturation, andferritin; decreased total iron binding capacity
G. Wilson's disease (hepatolenticular degeneration): autoso-mal recessive disorder of copper metabolism1. Rarely develops in children younger than 6 years of age2. Pathogenesis
a. Defective hepatocyte transport system for coppersecretion into bile and decreased synthesis of ce-ruloplasmin (binding protein of copper in blood)
b. Unbound copper accumulates in blood and is de-posited in other tissues, causing a toxic effect.
3. Clinical and laboratory findingsa. Liver disease: acute hepatitis progresses to
cirrhosis.b. Kayser-Fleischer ring: free copper deposits in the
cornea (Descemet's membrane)c. Central nervous system disease: copper deposits
in the putamen, producing a movement disor-der resembling parkinsonism, and subthalamicnucleus, producing hemiballismus
d. Decreased total serum copper (because of decreasedceruloplasmin), increased serum and urine freecopper
H. a l -Antitrypsin deficiency: autosomal recessive disorder1. Pathogenesis
a. Alleles are inherited codominantly; each alleleexpresses itself.
b. Normal genotype is PiMM; most common abnor-mal allele is Z.
c. PiZZ variant has decreased arantitrypsin levelsin serum(1) Hepatocytes produce a mutant protein that
cannot be secreted into the blood.(2) Accumulation of arantitrypsin (visible with
special stains) in hepatocytes causes liverdamage.
2. Clinical findings: neonatal cholestasis, cirrhosis, hepa-tocellular carcinoma
VIII. Liver TumorsA. Benign tumors
1. Cavernous hemangioma: most common benigntumor; rarely causes intraperitoneal hemorrhage
2. Liver cell adenoma: benign tumor of hepatocytesa. Usually occurs in women of childbearing age; as-
sociated with use of oral contraceptivesb. Highly vascular tumor; tends to rupture during
pregnancy; does not become malignant
Wilson's diseasecauses chronic liverdisease and move-ment disorders.
Most commoncause of cirrhosisand hepatocellu-lar carcinoma in chil-dren: a,-antitrypsindeficiency

Chapter 18 Hepatobiliary and Pancreatic Disorders 235
Figure 18 -5 Hepatocellular car-cinoma Multiple large, hemor-rhagic tumor masses are presentin the liver. There is also diffuseinfiltration of tumor blending inwith the remaining liver
B. Malignant tumors1. Metastasis: primary cancers of the lung (most
common), gastrointestinal tract and breast; multiplenodular masses
2. Hepatocellular carcinoma: most common primaryliver cancera. Epidemiology: more common in men; peaks about
60 years of ageb. Pathogenesis: usually associated with preexisting
cirrhosisc. Etiology
(1) Chronic HBV (Africa, most of Asia); chronicHCV (western countries, Japan)
(2) Aflatoxins (from Aspergillus mold in grains andpeanuts), hereditary hemochromatosis, alco-holic cirrhosis, primary biliary cirrhosis,arantitrypsin deficiency
d. Gross findings: focal, multifocal, or diffusely in-filtrating cancer with or without preexisting cir-rhosis; evidence of portal and hepatic veininvasion (Figure 18-5)
e. Microscopic findings: presence of bile in neoplas-tic cells
f. Clinical findings: fever (liver cell necrosis), rapidenlargement of the liver, increased ascites,blood in ascitic fluid
g. Laboratory findings: increase in a-fetoprotein,production of ectopic hormones (erythropoietin,insulin-like factor)
h. Metastases: lung most common site3. Angiosarcoma: associated with exposure to vinyl chlo-
ride, arsenic, or thorium dioxide
IX. Gallbladder and Biliary Tract DisordersA. Cholelithiasis
1. Gallstones composed of cholesterol: 80% of stonesa. Risk factors: women older than 40 years of age,
obesity, rapid weight loss, use of oral contracep-
Most cases of he-patocellular car-cinoma are causedby postnecroticcirrhosis due to HBVor HCV.
Risk factors forgallstones: female,fat, 40

236 Pathology
tives, Native American ethnicity (e.g., Pima,Navajo)
b. Pathogenesis: supersaturation of bile with choles-terol; decreased bile salts or lecithin in bile(both normally solubilize cholesterol)
c. Complications: cholecystitis, common bile ductobstruction (choledocholithiasis), ascendingcholangitis (causes multiple liver abscesses), gall-bladder cancer, acute pancreatitis
2. Black pigment stones: composed of calcium bilirubinsalts; sign of chronic extravascular hemolytic anemia(e.g., congenital spherocytosis)
B. Acute cholecystitis1. Pathogenesis: stone impacted in the cystic duct; in-
creased intraluminal pressure with ischemia to bowelwall, leading to mucosal injury and infection (usuallydue to Escherichia coli)
2. Clinical and laboratory findingsa. Nausea, vomiting, and fever: usually 15-30 minutes
postprandialb. Midepigastric colicky pain with shift to the right
upper quadrant; pain may radiate to the rightscapula.
c. Jaundice suggests stone in the common bile duct.d. Neutrophilic leukocytosis
3. Ultrasound is the gold standard to identify gallstones;radionuclide scan identifies stone in the cystic duct.
C. Chronic cholecystitis1. May or may not occur after repeated attacks of acute
cholecystitis2. Supersaturation of bile usually causes chronic inflam-
mation; gallstones are present in most cases.D. Adenocarcinoma of the gallbladder
1. Most often occurs in elderly women2. Pathogenesis: cholelithiasis (95% of cases); porcelain
gallbladder (gallbladder with dystrophic calcification)E. Cholangiocarcinoma
1. Most common malignancy of intrahepatic and extra-hepatic bile ducts: most are adenocarcinomas.
2. Etiology: primary sclerosing cholangitis (mostcommon cause in the United States), Opisthorchis sinen-sis (Chinese liver fluke), thorium dioxide
3. Clinical findings: obstructive jaundice, palpable gall-bladder (Courvoisier's sign)
X. Cystic FibrosisA. Epidemiology: autosomal recessive; 1:25 carrier rateB. Pathogenesis
1. Most common mutation is a three—nucleotide base de-letion on chromosome 7.
Most commonprimary cancerof biliary tract:gallbladderadenocarcinoma
Most common lethalgenetic disease inwhite children: CF

Chapter 18 Hepatobiliary and Pancreatic Disorders 237
a. Produces a defective cystic fibrosis transmem-brane regulator (CFTR), causing a defect in chlo-ride ion (cr) transport.
b. Mutant CFTR degrades in the Golgi apparatusbefore reaching the cell membrane.
2. Sodium (Nat) and a- reabsorption in the sweatglands is impaired (basis of the sweat test).
3. Increased Na+ reabsorption and decreased CI- secre-tion into duct lumens (e.g., terminal bronchioles, pan-creatic ducts); causes obstruction of ducts by secretingthick, dehydrated mucus
C. Clinical findings1. Recurrent respiratory infections leading to respira-
tory failure and deatha. Infections: bronchitis and pneumonia; pathogens
include Burkholderia cepacia, Pseudomonas aerugi-nosa, and Staphylococcus aureus.
b. Bronchiectasis caused by infection and obstruction2. Chronic pancreatic insufficiency
a. Exocrine deficiency: due to gland atrophy fromduct obstruction; causes malabsorption
b. Endocrine deficiency: type 1 diabetes mellitusdue to fibrosis of pancreatic stroma
3. Men have atresia of vas deferens (95% are infertile).4. Women have thick cervical mucus; are able to become
pregnant5. Meconium ileus in newborn, nasal polyps in children,
secondary biliary cirrhosis (due to chronic extrahe-patic bile duct obstruction)
XI. Pancreatic DisordersA. Acute pancreatitis
1. Pathogenesis: proenzymes in acinar cells are activatedby trypsin, leading to damage of the acinar cells, en-zymatic fat necrosis, and destruction of elastic tissue inblood vessels (hemorrhage).
2. Etiology: alcohol abuse; obstruction of the distal endof the common bile duct by gallstones, duodenalulcer, infection (e.g., mumps, cytomegalovirus in AIDS);hypercalcemia; hypertriglyceridemia
3. Clinical findingsa. Sudden onset of fever, nausea, vomiting, and
dyspneab. Midepigastric pain with radiation to the back
due to the retroperitoneal location of the pancreasc. Shock, flank hemorrhage (Turner's sign), perium-
bilical hemorrhage (Cullen's sign), tetany (hypocal-cemia from enzymatic fat necrosis)
Most commoncause of death inCF: respiratory infec-tion due to Paeruginosa
Most commoncauses of acute pan-creatitis: alcoholabuse and obstruc-tion of bile ductby gallstones

238 Pathology
4. Laboratory findingsa. Increased serum amylase: occurs within 2-12
hours; returns to normal within 2-3 days (increasedrenal clearance); amylase lacks specificity (presentin small bowel and salivary glands).
b. Increased serum lipase: more specific for pancre-atitis; similar time frame as for amylase, exceptserum levels return to normal within 3-5 days
c. Neutrophilic leukocytosis, hypocalcemia, hyper-glycemia
5. Radiographic findings: sentinel loop (localized ileus)near duodenum or transverse colon (cutoff sign),left-sided pleural effusion (contains amylase)
6. Complicationsa. Pseudocyst: collection of fluid and necrotic debris
around the pancreas; abdominal mass with in-creased serum amylase persisting longer than10 days
b. Acute respiratory distress syndrome: destructionof surfactant by increased circulating lecithinase
B. Chronic pancreatitis1. Pathogenesis: usually caused by repeated attacks of
pancreatitis, leading to loss of pancreatic parenchymaand replacement by fibrous tissue
2. Etiology: alcohol abuse, CF, protein-caloriemalnutrition
3. Clinical findings: severe pain radiating to the back,malabsorption, diabetes mellitus, pancreatic pseudocyst
4. Laboratory and radiographic findings: increasedserum amylase and lipase; pancreatic calcifications
C. Carcinoma of the pancreas (adenocarcinoma)1. Most commonly caused by smoking tobacco; associ-
ated with mutations of K-RAS and suppressor genesCDKN2A and TP53
2. Location: most lesions occur in the pancreatic head;the remainder occur in the body and tail of thepancreas.
3. Clinical and laboratory findingsa. Epigastric pain with weight loss, superficial migra-
tory thrombophlebitisb. Jaundice, light-colored stools, palpable gallbladder
(Courvoisier's sign); signs of carcinoma of head ofpancreas with common bile duct obstruction
c. Increased CA 19-9
Most commoncause of chronicpancreatitis: alcoholabuse

Sok
9,1
Kidney Disorders
•••••••••••S
• I. Laboratory Studies Used to Assess Renal Function
•A. Serum blood urea nitrogen (BUN)
1. End product of amino acid and pyrimidine metabo-
• lism: produced via the urea cycle in the liver, filteredin the kidneys, and reabsorbed in the proximal tubule
111 2. Causes of increased serum BUN
•a. Hypoperfusion of the kidneys: decreased glomer-
ular filtration rate (GFR) (e.g., heart failure, shock,i1 glomerulonephritis)
—b. Renal failure: acute or chronic
3. Causes of decreased serum BUN0 a. Chronic liver disease: dysfunctional urea cycle
b. Pregnancy: dilutional effect from increased plasma• volume; increased renal clearance
OB. Serum creatinine
1. Metabolic end product of creatine in muscle: filteredin the kidneys but not reabsorbed or secreted
O2. Causes of increased serum creatinine: hypoperfusion
of the kidneys (decreased GFR), renal failure (acute
Oor chronic)
3. Cause of decreased serum creatinine: normal• pregnancy
• C. Serum BUN:creatinine ratio (- 10:1)1. Azotemia is an increase in serum BUN and an in-
crease in creatinine.2. The serum BUN:creatinine ratio is useful in determining
O the cause of azotemia.
Oa. Prerenal azotemia: BUN:creatinine ratio > 15:1
(1) Implies renal hypoperfusion (decreased GFR)(2) Increased proximal tubule reabsorption of
Ourea causes a disproportionate increase inserum BUN when compared with serumcreatinine (BUN = 80 mg/dL; creatinine =4 mg/dL; BUN:creatinine = 80:4 = 20:1).• 239
•

240 Pathology
(3) Examples: dehydration (e.g., diarrhea, vomit-ing, diuretics), congestive heart failure, shock
b. Renal azotemia (uremia): BUN:creatinine ratio< 15:1(1) Increase in serum BUN and creatinine associ-
ated with intrinsic renal failure(2) Filtration and clearance of urea and creatinine
are equally affected because of parenchymaldamage; both BUN and creatinine in-crease proportionately (BUN = 80 mg/dL;creatinine = 8 mg/dL; BUN:creatinine =80:8 = 10:1).
(3) Examples: acute tubular necrosis, chronicrenal failure
c. Postrenal azotemia: BUN:creatinine ratio > 15:1(1) Caused by urinary tract obstruction below
the kidneys(2) Back-diffusion of urea into the blood; chronic
obstruction results in parenchymal damageand renal azotemia.
(3) Example: prostatic hyperplasia with obstruc-tion of urethra
D. Creatinine clearance: measure of GFR1. Causes of decreased creatinine clearance: renal
failure, increasing age2. Causes of increased creatinine clearance
a. Normal pregnancy: increase in plasma volume in-creases GFR.
b. Diabetic nephropathy: hyperfiltration is an earlyfeature of diabetic nephropathy.
E. Urinalysis (Table 19-1)
II. Congenital Anomalies and Cystic Diseases of the KidneysA. Horseshoe kidney
1. Two kidneys joined at their lower poles2. Increased incidence in Turner's syndrome3. Increased risk of infection and renal calculi
B. Cystic diseases of the kidney1. Renal dysplasia
a. Abnormal development of one or both kidneys;persistence of abnormal structures (e.g., cartilage,undifferentiated mesenchyme) in the kidneys
b. Clinical finding: unilateral flank mass2. Autosomal recessive (juvenile) polycystic kidney
diseasea. Bilateral cystic disease; cysts are present in the
cortex and medulla.b. Associated with maternal oligohydramnios: de-
creased amount of amniotic fluid resulting fromdysfunctional kidneys
c. Clinical findings(1) Enlarged kidneys at birth
Most common riskfactor for renalazotemia: prerenalazotemia
Most common con-genital kidney dis-order: horseshoekidney
Most commoncystic disease inchildren: renaldysplasia

Chapter 19 Kidney Disorders 241
TABLE 19-1 Laboratory Tests Used in Urinalysis
Test
Result
Cause
Sediment
Cells
Casts
Crystals
Dark yellow
Red or pinkFixed (e.g., 1.010)
Inability to concentrate urine
Acid or alkaline pHProteinuria
Increased serum glucose +glucosuria
Normal serum glucose +glucosuria
KetonuriaBilirubinuriaAbsentIncreased
HemoglobinuriaHematuriaMyoglobinuriaIncreased
Presence of esterase in neutro-phils (pyuria)
Sterile pyuria (neutrophilspresent but negative stan-dard urine culture)
BacteriaRed blood cells (hematuria)
Neutrophils (pyuria)Oval fat bodies
HyalineRed blood cell
White blood cell
Renal tubularFattyWaxy (refractile, acellular)BroadCalcium oxalate
Uric acid
Cystine
Concentrated urine (e.g., dehydration), bilirubi-nuria, increased urobilinogen
Hematuria, hemoglobinuria, myoglobinuriaLack of concentration and dilution (e.g., chronic
renal failure)First sign of intrinsic renal disease
Determined by diet and acid-base statusFirst metabolic abnormality in intrinsic renal
diseaseDiabetes mellitus
Normal pregnancy, benign glucosuria
Diabetic ketoacidosis, starvation, ketogenic dietsViral hepatitis, obstructive jaundiceObstructive jaundiceViral hepatitis, extravascular hemolytic anemia
(e.g., spherocytosis)Intravascular hemolytic anemiaRenal calculiCrush injuriesProduced by nitrate-reducing uropathogens (e.g.,
Escherichia coil)Infections (e.g., urethritis, cystitis, pyelonephritis)
Chlamydia trachomatis urethritis, tuberculosis
Usually sign of urinary tract infectionRenal stone; cancer (bladder, renal); glomerulo-
nephritisSame as aboveRenal tubular cells with lipid (nephrotic
syndrome)No significance in absence of proteinuriaNephritic type of glomerulonephritis (e.g., post-
streptococcal glomerulonephritis)Acute pyelonephritis, acute tubulointerstitial
nephritis (drugs)Acute tubular necrosisNephrotic syndrome (e.g., lipoid nephrosis)Chronic renal failureChronic renal failurePure vegan diet, ethylene glycol poisoning,
calcium oxalate calculiHyperuricemia associated with gout or massive
destruction of cells postchemotherapyCystinuria, hexagonal cystine crystals
General ExaminationColor
Specific gravity
Chemical Dipsticks
pHProtein
Glucose
KetonesBilirubinUrobilinogen (trace
amount normal)
Blood
Nitrites
Leukocyte esterase

242 Pathology
Figure 19-1 Adult polycystickidney disease. There is com-plete effacement of normalkidney architecture by cystswithin the cortex and medullaof both kidneys
(2) Potter's facies in newborns: deformationcaused by oligohydramnios; low-set ears,"parrot beak" nose, and lung hypoplasia
(3) Cysts also occur in the liver; danger of con-genital hepatic fibrosis, leading to portalhypertension
3. Hereditary adult polycystic kidney diseasea. Autosomal dominant disease with high
penetrance(1) Cysts occur by 20-25 years of age; accounts
for approximately 10% of chronic renal failurein adults
(2) Associated risk of renal cell carcinomab. Clinical findings
(1) Hypertension causing intracranial berry an-eurysms and the potential for subarachnoidhemorrhage; mitral valve prolapse
(2) Bilaterally palpable kidneys (Figure 19-1),chronic renal failure by 70 years of age,hematuria
(3) Sigmoid diverticulosis, cysts in liver andpancreas
4. Medullary sponge kidneya. Striations in the papillary ducts of the medulla and
"Swiss cheese" appearance most often seen on in-travenous pyelogram
b. Clinical findings: recurrent urinary tract infec-tions, hematuria, renal calculi
5. Simple retention cyst: derived from tubular obstruc-tion; may be confused with renal cell carcinoma
III. Glomerular Disorders• Glomerular disease associated with inflammation is termed
glomerulonephritis.A. Terminology (Table 19-2)B. Mechanisms of glomerular disease
Most commoncomplication of adultpolycystic kidneydisease:hypertension
Most common adultrenal cyst: simpleretention cyst

Chapter 19 Kidney Disorders 243
TABLE 19-2 Nomenclature and Description of Glomerular Disorders
Term Description
Only a few glomeruli are abnormalAll glomeruli are abnormal> 100 nuclei in affected glomeruliThick GBM, no proliferative changeThick GBM, hypercellular glomeruli
Fibrosis involving only segment of involvedglomerulus
Proliferation of parietal epithelial cellsaround glomerulus
Involves only glomeruli and no other targetorgans (e.g., minimal change disease)
Involves glomeruli and other target organs(e.g., systemic lupus erythematosus)
Focal glomerulonephritisDiffuse glomerulonephritisProliferative glomerulonephritisMembranous glomerulonephritisMembranoproliferative
glomerulonephritisFocal segmental glomerulosclerosis
Crescentic glomerulonephritis
Primary glomerular disease
Secondary glomerular disease
GBM, glomerular basement membrane.
1. Immunocomplexes in glomeruli (type III hypersensi-tivity): most common causea. Complexes activate complement; the C5a pro-
duced is chemotactic to neutrophils, damaging theglomeruli.
b. Example: DNA–anti-DNA complexes in systemiclupus erythematosus (SLE)
2. Antibodies directed against glomerular basementmembrane (GBM) antigens (e.g., Goodpasture's syn-drome, type II hypersensitivity)
3. Defects in T cell–mediated immunity: cytokinesdamage visceral epithelial cells or result in loss of thenormal negative charge of the GBM.
C. Evaluation of glomerular disease using renal biopsy1. H&E stain: helps determine the type of glomerular
disease2. Immunofluorescence stain: identifies the deposition
patterns of various proteins (e.g., IgG, complement)a. Linear pattern: characteristic finding in anti-GBM
disease; antibodies form a line along the GBM(e.g., Goodpasture's syndrome) (Figure 19-2)
b. Granular ("lumpy-bumpy") pattern: usually indi-cates immunocomplex deposition in the glo-merulus (Figure 19-3)
3. Electron microscopya. Detects submicroscopic defects in the glomerulus
(e.g., fusion of podocytes, damage to visceral epi-thelial cells)
b. Detects electron-dense immunocomplex depositsin subendothelial, intramembranous, subepithe-lial, or mesangial sites
D. Nephritic syndrome: clinical subtype of glomerular disease(Table 19-3)1. Clinical and laboratory findings

244 Pathology
Figure 19-2 Goodpasture's syn-drome, linear immunofluores-cence. The uninterrupted smoothimmunofluorescence along theglomerular basement membraneis caused by deposition of IgGantibodies directed againstthe membrane.
Figure 19-3 Acute postinfec-tious glomerulonephritis, granularimmunofluorescence. Granularirregular deposits in the capillar-ies are caused by immuno-complex deposition.
a. Key finding: red blood cell (RBC) casts; whiteblood cell (WBC) casts may be present.
b. Hypertension: caused by sodium retention (alsoperiorbital puffiness)
c. Oliguria (< 400 mL urine/day): caused by de-creased GFR from inflamed glomeruli
d. Hematuria with dysmorphic RBCs: abnormal cellshapes are caused by glomerular damage.
e. Neutrophils in the sediment: particularly in im-munocomplex types
f. Mild to moderate proteinuria: < 2 g/dayg. Azotemia
2. IgA glomerulonephritis (Berger's disease)a. Incidence of nephritic or nephrotic presentation
occurs in children and adults at the same rate.b. Increased mucosal synthesis and decreased clear-
ance of IgA: immunocomplexes contain IgA; acti-vation of alternative complement pathway
c. May have features in common with Henoch-Schtinlein purpura
Most common glo-merulonephritis: IgAglomerulonephritis

Chapter 19 Kidney Disorders 245
TABLE 19-3 Glomerular Diseases
Disease
Morphologic Characteristics
Mesangial IgA immunocomplex deposits, granularimmunofluorescence
Subepithelial immunocomplex deposits, granularimmunofluorescence
Subendothelial immunocomplex deposits, wirelooping, granular immunofluorescence
May have crescent formationGoodpasture's syndrome: anti-GBM antibodies,
linear immunofluorescenceSplit GBM, lipid in visceral epithelial cells (foam
cells), negative immunofluorescenceSensorineural hearing loss, ocular abnormalities
Fusion of podocytes, negative immunofluorescence
Fusion of podocytes, negative immunofluorescence
Fusion of podocytes, granular immunofluorescence,subepithelial deposits ("spike and dome" pattern)
Fusion of podocytes, granular immunofluorescence,subendothelial deposits, tram tracks
Fusion of podocytes, granular immunofluorescence,intramembranous deposits, tram tracks, C3nephritic factor
Fusion of podocytes, negative immunofluorescence,afferent and efferent hyaline arteriolosclerosis,nodular mesangial deposits
Fusion of podocytes, negative immunofluorescence,amyloid positive for Congo red stain, apple-greenbirefringence when polarized
NephriticIgA glomerulonephritis
(Berger's disease)Poststreptococcal
glomerulonephritisDiffuse proliferative
glomerulonephritis (SLE)Rapidly progressive
glomerulonephritis
Alport's syndrome (hereditarynephritis)
NephroticMinimal change disease
(lipoid nephrosis)Focal segmental
glomerulosclerosisDiffuse membranous
glomerulonephritisType I MPGN
Type II MPGN
Diabetic nephropathy
Renal amyloidosis
GBM, glomerular basement membrane; MPGN, membranoproliferative glomerulonephritis;SLE, systemic lupus erythematosus
d. Granular immunofluorescence in the mesangiume. Clinical and laboratory findings
(1) Episodic bouts of hematuria, usually follow-ing an upper respiratory infection
(2) Slow progression to chronic renal failure(3) Increased serum IgA
3. Acute poststreptococcal glomerulonephritisa. Usually follows group A streptococcal infection of
the skin or pharynx (e.g., scarlet fever); immuno-complexes activate the alternative complementsystem.
b. Microscopic findings: diffuse proliferativepattern with neutrophil infiltration, granular im-munofluorescence, subepithelial deposits(Figure 19-4)
c. Clinical and laboratory findings(1) Hematuria with smoky-colored urine (1-3
weeks after group A streptococcal infection)
Most common typeof postinfectiousglomerulonephritis:acute poststrep-tococcal glomerulo-nephritis
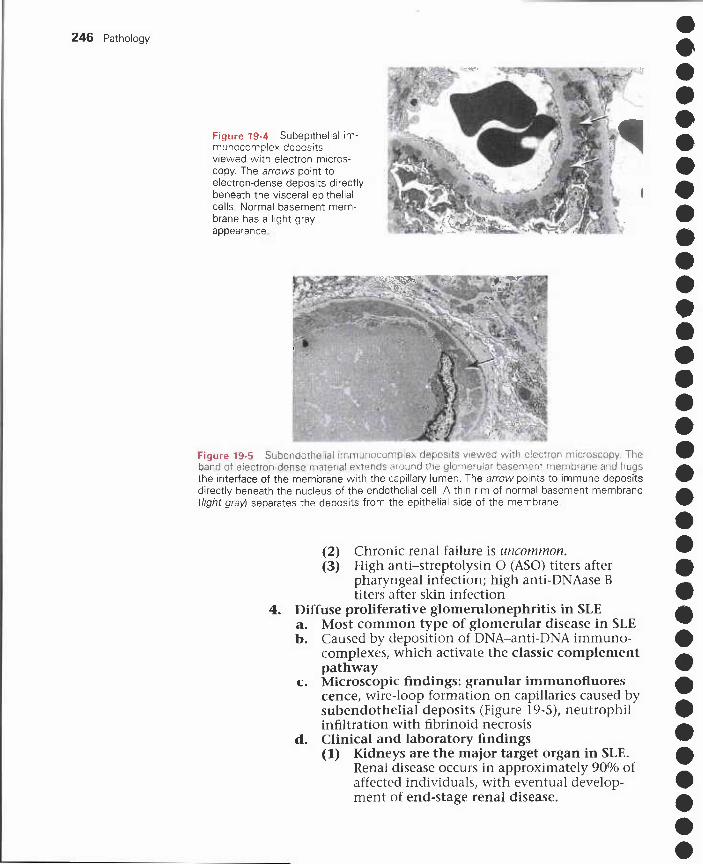
246 Pathology
Figure 19-4 Subepithelial im-munocomplex depositsviewed with electron micros-copy. The arrows point toelectron-dense deposits directlybeneath the visceral epithelialcells Normal basement mem-brane has a light grayappearance
Figure 19-5 Subendothelial irnmunocomplex deposits viewed with electron microscopy. Theband of electron-dense material extends around the glornerular basement membrane and hugsthe interface of the membrane with the capillary lumen. The arrow points to immune depositsdirectly beneath the nucleus of the endothelial cell A thin rim of normal basement membrane(light gray) separates the deposits from the epithelial side of the membrane.
(2) Chronic renal failure is uncommon.(3) High anti—streptolysin 0 (ASO) titers after
pharyngeal infection; high anti-DNAase Btiters after skin infection
4. Diffuse proliferative glomerulonephritis in SLEa. Most common type of glomerular disease in SLEb. Caused by deposition of DNA—anti-DNA immuno-
complexes, which activate the classic complementpathway
c. Microscopic findings: granular immunofluores-cence, wire-loop formation on capillaries caused bysubendothelial deposits (Figure 19-5), neutrophilinfiltration with fibrinoid necrosis
d. Clinical and laboratory findings(1) Kidneys are the major target organ in SLE.
Renal disease occurs in approximately 90% ofaffected individuals, with eventual develop-ment of end-stage renal disease.

Chapter 19 Kidney Disorders 247
Figure 19 -6 Crescentic glomer-ulonephritis. The arrows pointto a proliferation of parietal epi-thelial cells in Bowman'scapsule, occupying approxi-mately 50% of the entire urinaryspace The cells encase andeventually compress the glomer-ular tuft
(2) Serum antinuclear antibody (ANA) test:usually a rim pattern indicating presence ofanti—double-stranded DNA antibodies
5. Rapidly progressive glomerulonephritisa. Rapid loss of renal function progressing to acute
renal failure over days to weeks; poor prognosisb. May or may not be associated with crescent forma-
tion (crescentic glomerulonephritis; Figure 19-6)c. Clinical associations: Goodpasture's syndrome,
microscopic polyarteritis, Wegener's granulo-matosis
6. Goodpasture's syndromea. Occurs most often in young malesb. Caused by anti-GBM antibodies, which are di-
rected against type IV collagen in both the glomer-ular and pulmonary capillaries
c. Crescentic glomerulonephritis; linear immunoflu-orescence; no electron-dense deposits
7. Alport's syndromea. X-linked dominant disease (85%): defect in the
synthesis of type IV collagen in the GBMb. Microscopic findings: negative immunofluores-
cence; split GBM; lipid accumulation in visceralepithelial cells, producing foam cells
c. Clinical findings: sensorineural hearing loss,ocular abnormalities
E. Nephrotic syndrome: clinical subtype of glomerular disease(see Table 19-3)1. Clinical and laboratory findings
a. Proteinuria (gold standard for diagnosis): > 3.5g/day
b. Key finding: fatty casts with Maltese crossesc. Generalized pitting edema and ascites: caused by
hypoalbuminemia; risk of spontaneous peritoni-tis caused by Streptococcus pneumoniae
d. Hypertension: caused by sodium retention
Goodpasture's syn-drome begins withhemoptysis andends with renalfailure.

248 Pathology
Figure 19-7 Fusion of thepodocytes in minimal changedisease. The arrows show fusionof the podocytes, which shouldbe separated by slit pores
e. Hypercholesterolemia: caused by hypoalbumin-emia, leading to increased synthesis of choles-terol by the liver
f. Hypogammaglobulinemia: caused by loss ofy-globulins in the urine
2. Minimal change disease (lipoid nephrosis)a. More common in girls; occurs in 20-25% of adultsb. T-cell immune reaction against visceral epithe-
lial cells causes a loss in the negative charge of theGBM (polyanion loss), resulting in loss ofalbumin.
c. Secondary causes: nonsteroidal anti-inflammatorydrugs (NSAIDs), Hodgkin's disease, bee stings
d. Microscopic findings: structurally normal glomer-uli; positive fat stains in the glomerulus andtubules (become oval fat bodies); negative immu-nofluorescence; fusion of podocytes (Figure 19-7)
e. Clinical and laboratory findings(1) Often preceded by a respiratory infection or
routine immunization(2) Normal blood pressure (unlike other causes
of nephrotic syndrome)3. Focal segmental glomerulosclerosis
a. Occurs as primary or secondary disease; second-ary causes include HIV infection and intrave-nous heroin abuse.
b. Microscopic findings: normal immunofluores-cence; fusion of podocytes with detachment of vis-ceral epithelial cells
c. Clinical and laboratory findings(1) Most common initial presentations: protein-
uria, microscopic hematuria
Most commoncause of nephroticsyndrome in chil-dren: minimalchange disease
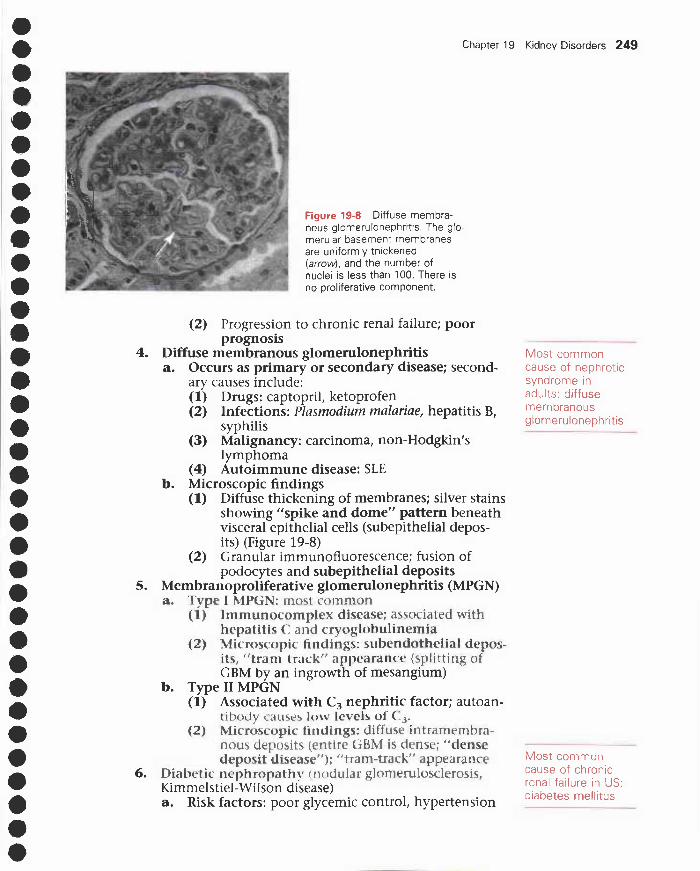
Chapter 19 Kidney Disorders 249
Figure 19-8 Diffuse membra-nous glomerulonephritis. The glo-merular basement membranesare uniformly thickened(arrow), and the number ofnuclei is less than 100. There isno proliferative component.
(2) Progression to chronic renal failure; poorprognosis
4. Diffuse membranous glomerulonephritisa. Occurs as primary or secondary disease; second-
ary causes include:(1) Drugs: captopril, ketoprofen(2) Infections: Plasmodium malariae, hepatitis B,
syphilis(3) Malignancy: carcinoma, non-Hodgkin's
lymphoma(4) Autoimmune disease: SLE
b. Microscopic findings(1) Diffuse thickening of membranes; silver stains
showing "spike and dome" pattern beneathvisceral epithelial cells (subepithelial depos-its) (Figure 19-8)
(2) Granular immunofluorescence; fusion ofpodocytes and subepithelial deposits
5. Membranoproliferative glomerulonephritis (MPGN)a. Type I MPGN: most common
(1) Immunocomplex disease; associated withhepatitis C and cryoglobulinemia
(2) Microscopic findings: subendothelial depos-its, "tram-track" appearance (splitting ofGBM by an ingrowth of mesangium)
b. Type II MPGN(1) Associated with C3 nephritic factor; autoan-
tibody causes low levels of C3.(2) Microscopic findings: diffuse intramembra-
nous deposits (entire GBM is dense; "densedeposit disease"); "tram-track" appearance
6. Diabetic nephropathy (nodular glomerulosclerosis,Kimmelstiel-Wilson disease)a. Risk factors: poor glycemic control, hypertension
Most commoncause of nephroticsyndrome inadults: diffusemembranousglomerulonephritis
Most commoncause of chronicrenal failure in US:diabetes mellitus

250 Pathology
Figure 19-9 Diabetic glomerulo-sclerosis The broken arrowpoints to an afferent or efferentarteriole that has hyaline arte-riolosclerosis, with an increase inproteinaceous material in thewall of the vessel. The solidarrow shows a mesangial nodulecontaining type IV collagen andtrapped protein
b. Pathogenesis(1) Nonenzymatic glycosylation of the GBM
membranes and tubule basement membranes,increasing vessel and tubular cell permeabil-ity to proteins
(2) Nonenzymatic glycosylation of the afferentand efferent arterioles, causing hyaline arte-riolosclerosis of the efferent arterioles beforethe afferent arterioles
(3) Hyperfiltration damage to the mesangiumcaused by the selective hyaline arteriolosclero-sis of efferent arterioles, increasing GFR, whichdamages mesangial cells
(4) Increased deposition of type IV collagen inthe GBM and mesangium
c. Microscopic findings(1) Afferent and efferent hyaline arterioloscle-
rosis: when the afferent arteriole becomes hy-alinized, GFR decreases (Figure 19-9).
(2) Nodular masses in the mesangial matrix:caused by increased type IV collagen andtrapped proteins
d. Clinical and laboratory findings(1) Microalbuminuria
Angiotensin-converting enzyme (ACE)inhibitors are prescribed when microal-buminuria is first detected. ACE inhibitorsslow the progression of diabetic nephrop-athy by decreasing pressure in the glomer-ular capillaries.
(2) Other renal problems associated with diabe-tes mellitus: renal papillary necrosis, acuteand chronic pyelonephritis
Initial laboratoryfinding in diabeticnephropathy:microalbuminuria

Chapter 19 Kidney Disorders 251
7. Nephropathy associated with amyloidosis: associatedwith both primary and secondary amyloidosis (seeChapter 3)
F. Chronic glomerulonephritis1. Causes (descending order of incidence): rapidly progres-
sive glomerulonephritis, focal segmental glomerulo-sclerosis, type 1 MPGN, IgA glomerulonephritis
2. Gross and microscopic findings: "shrunken" kidneys,glomerular sclerosis, tubular atrophy
IV. Disorders Affecting Tubules and InterstitiumA. Acute tubular necrosis: mainly involves the tubules
1. Ischemic typea. Prerenal azotemia secondary to hypovolemia
(most common cause)b. Ischemia damages endothelial cells by causing
vasoconstriction of afferent arterioles and decreas-ing GFR.
c. Ischemia damages tubule cells.(1) Causes detachment of tubular cells, produc-
ing renal tubular cell casts(2) Casts obstruct the lumen, causing an in-
crease in intratubular pressure, which de-creases GFR and pushes fluid into the intersti-tium, resulting in oliguria.
d. Sites of tubular damage(1) Straight segment of the proximal tubule and
medullary segment of the thick ascendinglimb of the loop of Henle
(2) Tubular basement membrane: disruptionprevents tubular cell regeneration.
2. Nephrotoxic typea. Etiology: aminoglycosides (most common
cause), radiocontrast agentsb. Microscopic findings: nephrotoxic agents mainly
damage the proximal convoluted tubules;intact basement membrane
3. Clinical and laboratory findings (both types): suddenonset of oliguria associated with pigmented renaltubular cell casts
B. Tubulointerstitial nephritis: involves the tubules andinterstitium1. Etiology: acute pyelonephritis (most common cause),
drugs, infections (e.g., legionnaires' disease), SLE, leadpoisoning, urate nephropathy, multiple myeloma
2. Acute pyelonephritisa. Vesicoureteral reflux with ascending infection is
the most common mechanism for infections ofthe lower urinary tract (e.g., urethritis, cystitis)and the upper urinary tract (e.g., acute pyelo-nephritis) in females.
Most commoncause of acute renalfailure: acute tubularnecrosis

252 Pathology
(1) In vesicoureteral reflux, the intravesicalportion of the ureter is oriented horizon-tally, which predisposes to reflux duringmicturition.
(2) Infection then ascends into the renal pelvisand from there into the renal parenchyma, re-sulting in acute pyelonephritis.
Colonization by Escherichia coli may occurin the distal urethra and vaginal introitus.In urethritis, bacteria ascend into the ure-thra, and in cystitis, they ascend into thebladder. Urinary tract infections are morecommon in females, because they have ashort urethra. • Imm■
b. Risk factors: urinary tract obstruction, diabetesmellitus, pregnancy, sickle cell trait and sickle celldisease
c. Gross and microscopic findings: grayish whiteareas of abscess formation; multifocal areas of mi-croabscess formation in the tubular lumens(source of the WBC casts) and interstitium
d. Clinical findings: spiking fever, flank pain, urinaryfrequency, dysuria (painful urination)
e. Laboratory findings: pyuria, bacteriuria, hematu-ria, WBC casts
f. Complications: chronic pyelonephritis, renal pap-illary necrosis, septicemia with endotoxic shock
3. Chronic pyelonephritisa. Pathogenesis: vesicoureteral reflux in young girls;
lower urinary tract obstruction (e.g., prostatichyperplasia, renal calculi)
b. Reflux type of pyelonephritis: cortical scarsoverlie a blunt calyx that has been destroyed byinflammation and replaced by scar tissue (visible onintravenous pyelography).
c. Obstructive type of pyelonephritis: uniform dila-tion of the calyces and diffuse thinning of corti-cal tissue caused by hydronephrosis
d. Microscopic findings: tubular atrophy (tubulescontain eosinophilic material resembling thyroidtissue), secondary scarring of the glomeruli
4. Acute drug-induced tubulointerstitial nephritisa. Pathogenesis: combination of type I and type IV
hypersensitivity reactionsb. Common drug associations: penicillin (particu-
larly methicillin), rifampin, sulfonamides, NSAIDs,diuretics

Chapter 19 Kidney Disorders 253
c. Clinical findings: abrupt onset of oliguria, fever,and rash; withdrawal of the drug causes reversalof the disease.
d. Laboratory findings: BUN:creatinine ratio < 15:1;eosinophilia; WBC casts
5. Analgesic nephropathy: common cause of chronicdrug-induced tubulointerstitial nephritisa. Pathogenesis: combination of acetaminophen
(a metabolite of phenacetin) and aspirin in apatient (usually a female) with chronic pain (e.g.,headache)(1) Free radicals from acetaminophen damage
the renal tubules in the renal medulla.(2) Aspirin inhibits renal synthesis of prosta-
glandin E2 (vasodilator), causing ischemicdamage.
b. Complications(1) Renal papillary necrosis: sloughing of the
renal papillae with gross hematuria, protein-uria, and colicky flank pain• Diabetes mellitus, sickle cell trait or sickle
cell disease, acute pyelonephritis, and ob-structive uropathy may also cause renal pap-illary necrosis.
(2) Hypertension, renal pelvic and bladder transi-tional cell carcinomas, chronic renal failure
6. Urate nephropathya. Deposition of urate crystals in the tubules and
interstitiumb. Etiology: massive release of purines (precursor of
uric acid) resulting from aggressive treatment of dis-seminated cancer (e.g., leukemia); lead poisoning;gout
Patients with disseminated cancers should begiven allopurinol, a xanthine oxidase inhibi-tor, before being treated with chemotherapy.Allopurinol prevents urate nephropathy (tu-mor lysis syndrome) and acute renal failure.
7. Chronic lead poisoninga. Lead decreases secretion of uric acid (urate ne-
phropathy); its direct toxic effect on the kidneysalso causes tubulointerstitial nephritis and neph-rotoxic acute tubular necrosis.
b. Acid-fast inclusions are evident in the nuclei ofthe proximal tubule cells.
8. Multiple myeloma: associated mechanisms of renaldisease include:a. Bence Jones proteinuria: production of tubular
casts obstructs the lumen and incites a foreign

254 Pathology
body giant cell reaction; light chains are toxic torenal tubular epithelium.
b. Hypercalcemia secondary to lytic lesions inbone: resulting nephrocalcinosis (metastatic calci-fication of renal tubular cell basement mem-branes) leads to tubular dysfunction and renalfailure.
c. Primary amyloidosis: light chains are converted toamyloid.
V. Chronic Renal Failure• Progressive irreversible azotemia develops over months to
years; symptoms and signs occur when GFR is less than 10-15mL/min.
A. Causes (in descending order of importance): diabetic melli-tus, hypertension, glomerulonephritis
B. Clinical findings1. Hematologic: normocytic anemia (decreased erythro-
poietin), qualitative platelet defects (prolonged bleedingtime)
2. Renal osteodystrophya. Osteitis fibrosa cystica
(1) Caused by secondary hyperparathyroidismfrom hypocalcemia related to hypovitamin-osis D: increased resorption of bone
(2) Cystic lesions in bone: commonly occur inthe jaw; hemorrhage into cysts causes browndiscoloration.
b. Osteomalacia: hypocalcemia caused by hypovita-minosis D leads to decreased mineralization ofbone.
c. Osteoporosis: excess hydrogen ions from metabolicacidosis are buffered by bone with a subsequentloss of bone mass.
3. Cardiovasculara. Hypertension due to sodium retention, hemor-
rhagic fibrinous pericarditisb. Congestive heart failure, accelerated atherosclerosis
(high triacylglycerol levels due to decreasedmetabolism)
4. Hemorrhagic gastritis, uremic frost (deposit of urea crys-tals on skin)
C. Laboratory findings1. Electrolytes
a. Hyponatremia, hyperkalemia, increased anion gapmetabolic acidosis
b. Hypocalcemia: hypovitaminosis D (decreased syn-thesis of 1 a-hydroxylase), hyperphosphatemia(forces calcium into bone and soft tissue)
2. Urinalysis: fixed specific gravity; waxy and broad casts3. Azotemia with BUN:creatinine ratio < 15:1

Chapter 19 Kidney Disorders 255
VI. Vascular DisordersA. Benign nephrosclerosis
1. Pathogenesis: hyaline arteriolosclerosis of small arter-ies and arterioles in the renal cortex, causing tubularatrophy, interstitial fibrosis, glomerular sclerosis, andsmall kidneys
2. Cortex of kidney has a finely granular surface.3. Laboratory findings: mild proteinuria, hematuria (no
RBC casts), renal azotemiaB. Malignant hypertension
1. Etiology: preexisting benign nephrosclerosis (mostcommon cause), hemolytic uremic syndrome, systemicsclerosis
2. Vascular damage to arterioles and small arteriesa. Fibrinoid necrosis and necrotizing arteriolitis
and glomerulitis: rupture of small vessels producespinpoint hemorrhages on the cortical surface("flea bitten" kidneys).
b. Hyperplastic arteriolosclerosis: "onion skin"lesions
3. Clinical findingsa. Rapid increase in blood pressure 210/120
mm Hgb. Hypertensive encephalopathy
(1) Cerebral edema with papilledema: loss ofmargin of normal optic nerve disk
(2) Retinopathy (flame hemorrhages, exudates);potential for intracerebral hemorrhage (stroke)
c. Acute renal failure4. Laboratory findings: azotemia with BUN:creatinine
ratio < 15:1; hematuria with RBC casts; proteinuriaC. Renal infarction
1. Etiology: embolization from thrombi in the left sideof the heart (most common cause), atheroembolicrenal disease, vasculitis (e.g., classic polyarteritisnodosa)
2. Gross and microscopic appearance: pale infarctions ofthe cortex, representing coagulation necrosis
3. Clinical findings: sudden onset of flank pain andhematuria
D. Sickle cell nephropathy1. Occurs with sickle cell trait or sickle cell disease2. Clinical findings: asymptomatic hematuria (renal in-
farctions in medulla), inability to concentrate urine,renal papillary necrosis, pyelonephritis
E. Diffuse cortical necrosis1. Complication of an obstetric emergency (e.g., pre-
eclampsia, abruptio placentae)2. Caused by disseminated intravascular coagulation
limited to the renal cortex
Most common renaldisease in essen-tial hypertension:benign nephro-sclerosis

256 Pathology
3. Clinical findings: pregnant women with sudden onsetof anuria (absence of urine) followed by acute renalfailure
VII. Obstructive DisordersA. Sites of obstruction
1. Upper urinary tract: ureter (most common), renaltubule, renal pelvis
2. Lower urinary tract: prostate (most common),urethra, bladder
B. Hydronephrosis1. Etiology: ureteral calculus (most common cause),
retroperitoneal fibrosis, cervical cancer2. Gross findings: dilated ureter and renal pelvis with
compression atrophy of the renal medulla and cortex3. Laboratory finding: postrenal azotemia
C. Urolithiasis (renal calculi)1. Risk factors
a. Hypercalciuria in the absence of hypercalcemia:caused by increased gastrointestinal reabsorptionof calcium
b. Decreased urine volume (concentrates urine),reduced urine citrate (citrate normally chelatescalcium), primary hyperparathyroidism
2. Types of renal calculia. Calcium oxalate: most common in adults; in-
creased incidence in pure vegansb. Calcium phosphate: most common in children;
associated with excess ingestion of dairy productsc. Magnesium ammonium phosphate: "staghorn
calculus"; associated with urease producers (e.g.,Proteus) and characterized by alkaline urine thatsmells like ammonia
d. Uric acid associated with hyperuricemia, cystineassociated with cystinuria
3. Clinical and laboratory findingsa. Sudden onset of ipsilateral colicky pain in the
flank radiating to the groinb. Hematuria (gross and microscopic)c. Visible on routine radiograph, because the majority
of calculi contain calcium
VIII. Tumors of the KidneyA. Angiomyolipoma: benign hamartoma composed of blood
vessels, smooth muscle, and adipose tissue; associatedwith tuberous sclerosis
B. Renal cell carcinoma (or Grawitz' tumor, clear cell carci-noma, hypernephroma)1. Epidemiology: more common in men in the sixth or
seventh decades of lifea. Types: sporadic (most common) and hereditary
Most commoncomplication ofupper urinary tractobstruction:hydronephrosis
Most commonmetabolic abnormal-ity in urolithiasis:hypercalciuria

Chapter 19 Kidney Disorders 257
Figure 19-10 Renal adenocarci-noma, The large u pper polemass with multifocal areas ofhemorrhage extends into therenal pelvis.
b. Risk factors: smoking tobacco (most common),von Hippel-Lindau disease
2. Pathogenesis: cancer derives from proximal tubulecells.
3. Gross and microscopic findingsa. Upper pole mass with cysts and hemorrhage:
usually > 3 cm; bright yellow tumor (Figure 19-10)b. Composed of clear cells that contain lipid and
glycogenc. Tendency for renal vein invasion: tumor may
ascend the inferior vena cava and extend to theright side of the heart.
4. Clinical and laboratory findingsa. Triad of hematuria, flank mass, and costoverte-
bral angle painb. Ectopic secretion of hormones: erythropoietin
(secondary polycythemia), parathyroid hormone-related peptide (hypercalcemia)
5. Metastasesa. Sites: lungs (most common; hemorrhagic with
metastases); bone (lytic lesions); skin (hemorrhagicnodules)
b. Late metastases may occur 10-20 years after thetumor has been removed.
6. Prognosis: average 5-year survival rate is 45%.C. Cancers of the renal pelvis
1. Transitional cell carcinomaa. May also occur in renal calyces, ureter, or bladder;
approximately 50% of affected individuals havesimilar tumors elsewhere in the urinary tract.
b. Risk factors: smoking tobacco, phenacetin abuse,aromatic amines (aniline dyes), cyclophosphamide
2. Squamous cell carcinoma: risk factors include renalcalculi and chronic infection.
Most commoncause of renal cellcarcinoma and tran-sitional cell carci-noma: tobaccosmoking

258 Pathology
D. Wilms' tumor1. Occurs in children 2-5 years of age2. Types: sporadic and genetic (autosomal dominant trait
on chromosome 11)3. Clinical associations
a. WAGR syndrome: Wilms' tumor, aniridia (absentiris), genital abnormalities, retardation
b. Beckwith-Wiedemann syndrome: Wilms' tumor,enlarged body organs (e.g., kidneys), hemihypertro-phy of extremities
4. Large, necrotic, grayish-tan colored tumor: containsabortive glomeruli and tubules, primitive cells, andrhabdomyoblasts
5. Clinical findings: unilateral palpable mass and hy-pertension caused by renin secretion
6. Prognosis: frequently metastasizes to the lungs; goodprognosis
Most commonprimary renal tumorin children: Wilms'tumor

20sqlol
Lower Urinary Tractand Male ReproductiveDisorders
1. Disorders of the Urethra and BladderA. Congenital anomalies
1. Persistent urachal sinus: urine draining from a sinusopening in the umbilicus in newborns; risk factor foradenocarcinoma of the bladder
2. Exstrophy: absence of the anterior part of the bladderand abdominal wall; risk factor for adenocarcinoma ofthe bladder
B. Diverticula: most common in elderly men with urethral ob-struction caused by prostatic hyperplasia
C. Acute cystitis1. Etiology
a. Pathogens(1) Escherichia coli (80-90% of cases), Staphylococ-
cus saprophyticus (10-20% of cases in young,sexually active females); adenovirus (hemor-rhagic cystitis)
(2) Most E. coli infections are caused by ascendinginfection from the urethra.
b. Cyclophosphamide (hemorrhagic cystitis)2. Clinical findings: afebrile, dysuria with increased fre-
quency of urination, suprapubic pain3. Laboratory findings: pyuria, hematuria, and bacteriuria;
positive dipstick reactions for nitrite and leukocyte es-terase; absence of white blood cell casts
D. Sexually transmitted urethritis1. Termed acute urethral syndrome in women and non-
specific urethritis in men2. Pathogens: Chlamydia trachomatis, Mycoplasma
horninis, Neisseria gonorrhoeae, Ureaplasma urealyticum
Bladder diverticulaare mostcommon in menwith prostatichyperplasia
Acute cystitis ismore common infemales because ofshort urethra
Most commoncause of sexuallytransmitted urethri-tis: C. trachomatis
259

260 Pathology
3. Clinical findings: urethral discharge (positive Gramstain with Neisseria), dysuria, increased urinary frequency
E. "Sterile" pyuria1. Pathogens: C. trachomatis, M. hominis, Mycobacterium tu-
berculosis (e.g., renal tuberculosis), U. urealyticum2. Neutrophils are present in the urine, but routine cul-
tures are negative.F. Bladder tumors
1. Transitional cell carcinomaa. Epidemiology
(1) Transitional cell carcinoma occurs more oftenin men older than 50 years of age.
(2) Risk factors are smoking tobacco, aniline dyes,and cyclophosphamide.
b. Pathology(1) Most common sites are lateral or posterior walls
at the base of the bladder; multifocal tumorsand recurrences are typical.
(2) Sites of metastases are the regional lymphnodes, liver, lungs, and bones.
(3) Low-grade cancers are papillary and usuallyare not invasive; high-grade cancers are papil-lary or flat and usually are invasive.
c. Clinical finding: painless hematuria (gross ormicroscopic)
2. Squamous cell carcinoma: risk factors include Schisto-soma haematobium infection in the venous plexus of thebladder and chronic bladder irritation.
3. Embryonal rhabdomyosarcomaa. Occurs in boys younger than 4 years of ageb. Sarcoma is derived from striated muscle; a grape-like
mass protrudes from the urethral orifice.c. Poor prognosis
II. Disorders of the PenisA. Congenital anomalies
1. Hypospadias: abnormal opening on the ventral surfaceof the penis caused by faulty closure of the urethralfolds
2. Epispadias: abnormal opening on the dorsal surface ofthe penis caused by a defect in the genital tubercle
B. Phimosis: orifice of the prepuce that is too small to retractover the head of the penis
C. Peyronie's disease: fibromatosis causing painful contracturesof the penis leading to lateral curvature of the penis; maycause infertility
D. Priapism: persistent and painful erection; caused by sicklecell disease and penile trauma
E. Cancers of the penis1. Carcinoma in situ: associated with human papilloma-
virus (HPV) type 16; precursor of invasive squamouscancer
Most commonprimary bladdercancer: transitionalcell carcinoma
Phimosis is com-monly associatedwith infection.

Chapter 20 Lower Urinary Tract and Male Reproductive Disorders 261
a. Bowen's disease: leukoplakic lesion involving theshaft of the penis and/or scrotum
b. Erythroplasia of Queyrat: shiny red lesions on themucosal surface of the glans and prepuce
2. Squamous cell carcinomaa. Usually affects uncircumcised men 40-70 years
of ageb. Risk factors: I-I PV types 16 and 18, smoking tobacco,
smegma (material that collects under the prepuce)c. Most common site is the glans or mucosal surface of
prepuce.d. Sites of metastasis are the inguinal and iliac nodes.
III. Disorders of the Scrotum, Testis, and EpididymisA. Hydrocele: accumulation of clear fluid in the scrotum
1. Caused by a persistent tunica vaginalis2. Most common cause of scrotal enlargement in boys3. Other fluids that may accumulate in the tunica vagi-
nalis: blood (hematocele), sperm (spermatocele)B. Varicocele: dilation of the veins of the pampiniform plexus
of the spermatic cord1. "Bag of worms" appearance2. Caused by venous drainage of the left testis (90% of
cases) to the left renal vein, leading to increased retro-grade venous pressure; blockage of the left renal vein,such as occurs in renal cell carcinoma, may also producea varicocele.
3. Common cause of infertility; heat from a varicocele de-creases spermatogenesis.
C. Cryptorchidism: incomplete descent of one or both testesinto the scrotal sac1. Most common location of the cryptorchid testis is the
inguinal canal, where a mass is palpable.2. Most cases are unilateral.3. Histologic changes (e.g., atrophy, neoplasia) may occur
in both the cryptorchid testis and the contralateral de-scended testis.
4. Complications include potential infertility andseminoma.
D. Orchitis: inflammation of testis1. May be caused by mumps (usually unilateral), syphilis,
or AIDS2. Spreads from acute epididymitis
E. Epididymitis1. Causal pathogens
a. Patients younger than 35 years of age: C. trachoma-tis, N. gonorrhoeae
b. Patients older than 35 years of age: E. coli, Pseudo-monas aeruginosa
2. Tuberculous epididymitis begins in the epididymis andspreads to the seminal vesicles, prostate, and testicles.
Most commoncause of squamouscell carcinoma ofpenis: nocircumcision
Varicoceles mostcommonly occur inthe left scrotal sac
Males with unilat-eral cryptorchidismare at risk forseminomas in boththe descendedand undescendedtestes.

262 Pathology
3. Clinical findings: scrotal pain and swelling, epididymaltenderness, Prehn's sign
F. Torsion of the testicle1. Twisting of the spermatic cord leads to ischemic pain
and potential infarction.2. Predisposing causes include violent movement or physi-
cal trauma (most common) and cryptorchidism.3. Clinical findings include sudden onset of testicular pain,
absent cremasteric reflex, and a testicle in the inguinalcanal.
G. Testicular cancer (Table 20-1)1. Epidemiology: most common cancer in men 15-35
years of age; more common in whites than in blacks2. Classification
a. Most testicular cancers (95%) derive from germ cells.b. Tumors may contain one cell type (40% of cases) or
mixtures of cell types (60% of cases).3. Risk factors: cryptorchidism (most common), testicular
feminization, Klinefelter's syndrome4. Presenting symptom: unilateral painless enlargement
of the testis that does not transilluminate5. Tumor markers: a-fetoprotein, human chorionic
gonadotropin
IV. Prostate DisordersA. Clinical anatomy
1. Dihydrotestosterone (DHT) is the only hormone respon-sible for prostate development in the fetus.
2. Prostate encircles the neck of the bladder and the urethra.3. Prostate is divided into central and peripheral zones.
a. Prostate hyperplasia develops centrally, around theurethra.
b. Prostate cancer develops peripherally; tumors maybe palpable on digital examination.
B. Prostatitis1. Causes of acute prostatitis
a. Most common: E. colib. C. trachomatis, Klebsiella pneumoniae, P. aeruginosa,
U. urealyticum2. Causes of chronic prostatitis
a. Previous acute prostatitis: infection with C. tra-chomatis, U. urealyticum
b. Abacterial prostatitis (e.g., trauma from cycling)c. Granulomatous prostatitis: nonspecific response to
prostatic secretions or systemic disease (e.g., sarcoido-sis, tuberculosis)
3. Clinical findingsa. Fever (acute prostatitis), low back pain, perineal
pain, dysuria and hematuriab. Pain on digital rectal examination; increased
prostate-specific antigen (PSA)
Prehn's sign: L inpain with scrotalelevation
Most commongerm cell tumor:seminoma
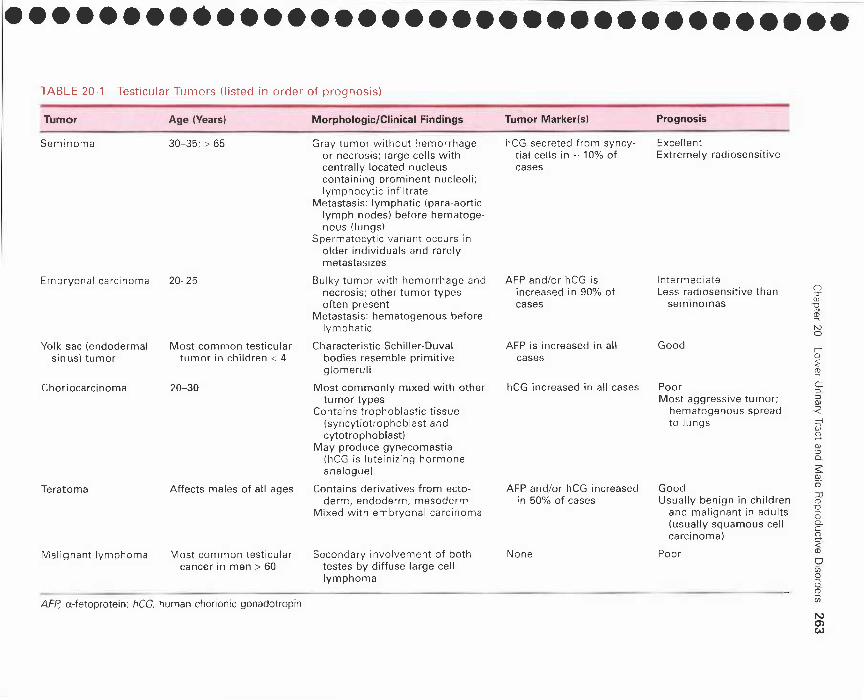
• • • • • • • • ••• • • VI • • • • • • ••• IP • • ••••
TABLE 20-1 Testicular Tumors (listed in order of prognosis)
Tumor
Age (Years)
Morphologic/Clinical Findings
Tumor Marker(s)
Prognosis
Seminoma 30-35; > 65 Gray tumor without hemorrhageor necrosis; large cells withcentrally located nucleuscontaining prominent nucleoli;lymphocytic infiltrate
Metastasis: lymphatic (para-aorticlymph nodes) before hematoge-nous (lungs)
Spermatocytic variant occurs inolder individuals and rarelymetastasizes
hCG secreted from syncy- Excellenttial cells in - 10% of Extremely radiosensitivecases
Embryonal carcinoma 20-25 Bulky tumor with hemorrhage and AFP and/or hCG isnecrosis; other tumor types increased in 90% ofoften present cases
Metastasis: hematogenous beforelymphatic
Yolk sac (endodermal Most common testicular Characteristic Schiller-Duval AFP is increased in allsinus) tumor tumor in children < 4 bodies resemble primitive cases
glomeruli
Choriocarcinoma 20-30 Most commonly mixed with othertumor types
Contains trophoblastic tissue(syncytiotrophoblast andcytotrophoblast)
May produce gynecomastia(hCG is luteinizing hormoneanalogue)
Teratoma Affects males of all ages Contains derivatives from ecto-derm, endoderm, mesoderm
Mixed with embryonal carcinoma
IntermediateLess radiosensitive than
seminomas
Good
hCG increased in all cases PoorMost aggressive tumor;
hematogenous spreadto lungs
AFP and/or hCG increased Goodin 50% of cases Usually benign in children
and malignant in adults(usually squamous cellcarcinoma)
Malignant lymphoma Most common testicular
Secondary involvement of both
None
Poorcancer in men > 60
testes by diffuse large celllymphoma
AFP, cc-fetoprotein; hCG, human chorionic gonadotropin
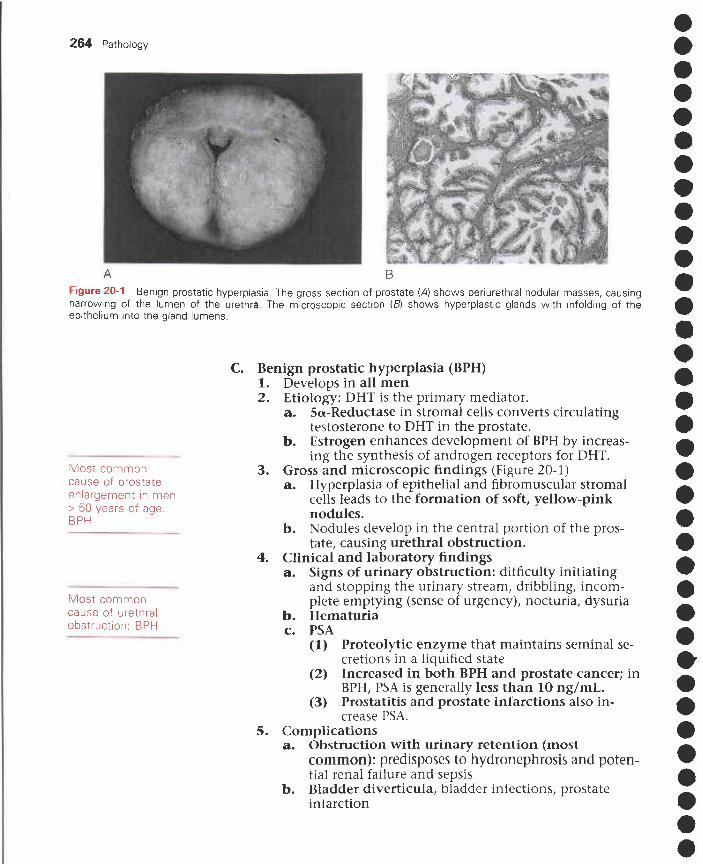
264 Pathology
A
BFigure 20-1 Benign prostatic hyperplasia The gross section of prostate (A) shows periurethral nodular masses, causingnarrowing of the lumen of the urethra The microscopic section (B) shows hyperplastic glands with infolding of theepithelium into the gland lumens.
C. Benign prostatic hyperplasia (BPH)1. Develops in all men2. Etiology: DHT is the primary mediator.
a. 5a-Reductase in stromal cells converts circulatingtestosterone to DHT in the prostate.
b. Estrogen enhances development of BPH by increas-ing the synthesis of androgen receptors for DHT.
3. Gross and microscopic findings (Figure 20-1)a. Hyperplasia of epithelial and fibromuscular stromal
cells leads to the formation of soft, yellow-pinknodules.
b. Nodules develop in the central portion of the pros-tate, causing urethral obstruction.
4. Clinical and laboratory findingsa. Signs of urinary obstruction: difficulty initiating
and stopping the urinary stream, dribbling, incom-plete emptying (sense of urgency), nocturia, dysuria
b. Hematuriac. PSA
(1) Proteolytic enzyme that maintains seminal se-cretions in a liquified state
(2) Increased in both BPH and prostate cancer; inBPH, PSA is generally less than 10 ng/mL.
(3) Prostatitis and prostate infarctions also in-crease PSA.
5. Complicationsa. Obstruction with urinary retention (most
common): predisposes to hydronephrosis and poten-tial renal failure and sepsis
b. Bladder diverticula, bladder infections, prostateinfarction
Most commoncause of prostateenlargement in men> 50 years of age:BPH
Most commoncause of urethralobstruction: BPH
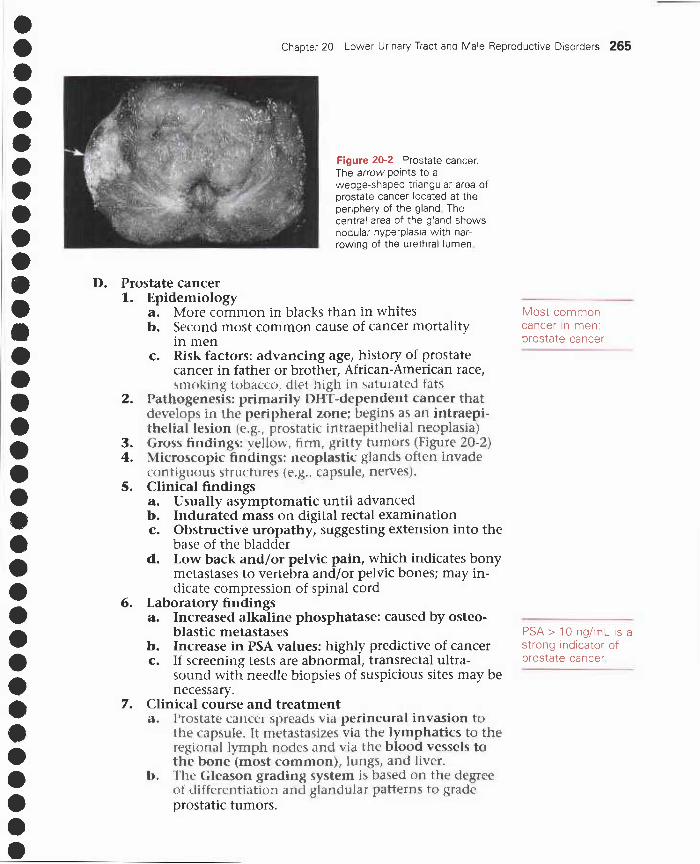
Chapter 20 Lower Urinary Tract and Male Reproductive Disorders 265
Figure 20-2 Prostate cancer.The arrow points to awedge-shaped triangular area ofprostate cancer located at theperiphery of the gland Thecentral area of the gland showsnodular hyperplasia with nar-rowing of the urethral lumen
D. Prostate cancer1. Epidemiology
a. More common in blacks than in whitesb. Second most common cause of cancer mortality
in menc. Risk factors: advancing age, history of prostate
cancer in father or brother, African-American race,smoking tobacco, diet high in saturated fats
2. Pathogenesis: primarily DHT-dependent cancer thatdevelops in the peripheral zone; begins as an intraepi-thelial lesion (e.g., prostatic intraepithelial neoplasia)
3. Gross findings: yellow, firm, gritty tumors (Figure 20-2)4. Microscopic findings: neoplastic glands often invade
contiguous structures (e.g., capsule, nerves).5. Clinical findings
a. Usually asymptomatic until advancedb. Indurated mass on digital rectal examinationc. Obstructive uropathy, suggesting extension into the
base of the bladderd. Low back and/or pelvic pain, which indicates bony
metastases to vertebra and/or pelvic bones; may in-dicate compression of spinal cord
6. Laboratory findingsa. Increased alkaline phosphatase: caused by osteo-
blastic metastasesb. Increase in PSA values: highly predictive of cancerc. If screening tests are abnormal, transrectal ultra-
sound with needle biopsies of suspicious sites may benecessary.
7. Clinical course and treatmenta. Prostate cancer spreads via perineural invasion to
the capsule. It metastasizes via the lymphatics to theregional lymph nodes and via the blood vessels tothe bone (most common), lungs, and liver.
b. The Gleason grading system is based on the degreeof differentiation and glandular patterns to gradeprostatic tumors.
Most commoncancer in men:prostate cancer
PSA > 10 ng/mL is astrong indicator ofprostate cancer

266 Pathology
c. Treatment is radical prostatectomy or radiotherapy.Hormone therapy blocks the effect of androgens ontumor growth.
V. Male Hypogonadism and Erectile DysfunctionA. Hypogonadism: decreased production of testosterone or re-
sistance to testosterone1. May result from Klinefelter's syndrome or testicular
feminization (see Chapter 5)2. Clinical presentation: erectile dysfunction (failure to
sustain an erection), loss of male secondary sex character-istics, infertility
3. Primary hypogonadism: Leydig cell dysfunctiona. Etiology: alcohol abuse, orchitis, radiationb. Laboratory findings: decreased serum testosterone
and increased luteinizing hormone (LH), de-creased sperm count (testosterone stimulatesspermatogenesis)
4. Secondary hypogonadism: hypothalamic or pituitarydysfunctiona. Hypothalamic dysfunction: Kallmann's syndrome
(e.g., absent gonadotropin-releasing hormone, inabil-ity to taste and smell)
b. Pituitary dysfunction: hypopituitarism caused bynonfunctioning pituitary adenoma
c. Laboratory findings: decreased LH and decreasedtestosterone
B. Erectile dysfunction; causes:1. Psychogenic problems: nighttime erections (nocturnal
penile tumescence) still occur.2. Decreased testosterone (decreased libido)3. Vascular insufficiency (e.g., Leriche syndrome, or aorto-
iliac atherosclerosis)4. Neurologic disease (autonomic neuropathy in multiple
sclerosis or diabetes mellitus)5. Drugs that block androgen receptors (e.g., alcohol, leu-
prolide, methyldopa, psychotropics, spironolactone)
Sildenafil is the most common drug used for thetreatment of erectile dysfunction. It inhibits thebreakdown of cyclic guanosine monophosphate(cGMP) by type 5 phosphodiesterase. This actionincreases levels of cGMP, which causes vasodilationin the corpus cavernosum and the penis. Yohimbe (aplant product) also causes vasodilation of vessels.
6. Endocrine disorders (e.g., diabetes mellitus, prolacti-noma)
Most commoncause of erectiledysfunction in men> 50 years of age:vascular insuffi-ciency

••••••••• Female Reproductive• Disorders and Breast•• Disorders••••
I. Sexually Transmitted Diseases (STDs) and Other GenitalInfections
• A. Sexually transmitted diseases1. Herpes simplex virus type 2 (HSV-2) infections
• a. Recurrent vesicles that ulcerate are located on thevulva, cervix, and perianal area.
b. Tzanck preparation: stained scrapings removed• from the base of an ulcer show multinucleated
•squamous cells with eosinophilic intranuclearinclusions.
•2. Human papillomavirus (HPV) infections
a. HPV types 6 and 11 are associated with condy-• loma acuminata (venereal warts); these fern - like
•or flat lesions occur in the genital area (e.g.,vulva, cervix, perianal area).
• b. HPV types 16 and 18 are associated with dysplasiaand squamous cell carcinoma.
• c. HPV produces koilocytic change in squamous
•epithelium; cells with wrinkled pyknotic nuclei aresurrounded by a dear halo (Figure 21-1).
3. Chlamydia: caused by Chlamydia trachomatis, a
•gram-negative intracellular parasite; often coexists with Neisseria gonorrhoeae Most common STD
•a. Primary infection sites in males and
(1) Males: urethra (nonspecific urethritis), females: chlamydia
• epididymis, anus (proctitis)• (2) Females: Bartholin's glands, urethra (acute
urethral syndrome), cervix (cervicitis), fallo-• pian tubes and ovaries (pelvic inflamma-
tory disease; PID), anus• 267•

Figure 21-1 Koilocytosiscaused by human papilloma-virus The squamous cells havewrinkled pyknotic nuclei sur-rounded by a clear halo (arrows)
268 Pathology
Chancroid: painfululcers
(3) Newborns: eyes (ophthalmia neonatorum;from passage through the birth canal)
b. Produces red inclusions (elementary bodies) inphagocytic vacuoles in infected metaplastic squa-mous cells
c. Symptoms: dysuria, vaginal or penile discharge,pelvic pain
d. Lymphogranuloma venereum: caused by a sub-species of C. trachomatis(1) Produces papules in the genital region; no
ulceration(2) Inguinal lymphadenitis with granulomatous
microabscesses and draining sinuses, pro-ducing lymphatic scarring (lymphedemaof vulva); can lead to rectal strictures
4. Gonorrhea: caused by N. gonorrhoeae, gram-negativediplococcusa. Infection of glandular or transitional epitheliumb. Primary infection sites: same as for chlamydia;
jointsc. Symptoms: same as for chlamydia; joint pain
(septic arthritis)d. Complications of untreated disease: ectopic preg-
nancy, male sterility, disseminated gonococce-mia (C5—C9 deficiency)
5. Chancroid: caused by Haemophilus ducreyi, gram-negative roda. Produces painful genital and perianal ulcersb. Suppurative inguinal nodes
6. Granuloma inguinale: caused by Calymmatobacte-Hum granulomatis, gram-negative coccobacillusa. Macrophages (Donovan bodies) phagocytose bac-
terium.b. Creeping, raised sore heals by scarring; no lymph-
adenopathy.7. Syphilis: caused by Treponema pallidum, gram-
negative spirochete

S•
••
Chapter 21 Female Reproductive Disorders and Breast Disorders 269
• a. Causes endarteritis obliterans (vasculitis involving
•arterioles); characteristic plasma cell infiltrate painlessSyphilis:
b. Primary syphilis: solitary, painless chancre (e.g., ulcers• on labia); resolves within 3-6 weeks
c. Secondary syphilis: diffuse maculopapular rash• (trunk, palms, soles); generalized lymphadenop-• athy; condyloma lata (flat lesions occur in the
same area as condyloma acuminata)
• d. Tertiary syphilis: central nervous system disease;
•syphilitic aortitis; locally destructive disease withgummas
•e. Congenital syphilis (see Table 5-5)
8. Trichomoniasis: caused by Trichomonas vaginalis,• flagellated protozoan
Oa. Causes vaginitis, cervicitis, and urethritisb. Strawberry-colored cervix and inflamed vaginal
• mucosa; greenish, frothy dischargeB. Other genital infections
e 1. Bacterial vaginosis: caused by Gardnerella vaginalis,
•gram-negative roda. Malodorous vaginal discharge
• b. Occurs when the vaginal pH > 5
•c. Organisms adhere to squamous cells; clue cells
are found. Clue cells are char-• 2. Candidal vaginosis: caused by Candida albicans, yeast acteristic of bacterial
with pseudohyphae vaginosis
• a. Predisposing conditions: diabetes mellitus, sys-
•temic antibiotics, pregnancy, oral contraceptives
b. Pruritic vaginitis with a white discharge; in-• flamed mucosa
3. Toxic shock syndrome: due to a toxin-producing• strain of Staphylococcus aureus
• a. Associated with use of highly absorbent tamponsb. Clinical findings: high fever, mental confusion, di-
arrhea, hypotension, erythematous rash; rash
•appears during or soon after menses anddesquamates.
• II. Disorders of the Vulva• A. Non-neoplastic dermatoses
•1. Lichen sclerosis
a. Thinning of epidermis, parchment-like appearance
• of skinb. Usually occurs in postmenopausal women;
e minimal risk of squamous cell carcinoma
•2. Lichen simplex chronicus
a. White plaque-like lesion (leukoplakia) caused by
• epithelial hyperplasia
• B. Tumbo.rs No risk of squamous cell carcinoma
•1. Vulvar intraepithelial neoplasia (VIN)
a. Mild to severe dysplasia; carcinoma in situ

270 Pathology
Figure 21 -2 ExtramammaryPaget's disease Large, malig-nant Paget's cells (nucleatedcells surrounded by a clear halo)are disposed singly and inclusters within the epidermis(arrows) The concentration ofcells is greatest along the basalcell layer: all layers of the epider-mis are involved
b. Strongly associated with HPV type 16; precursorof squamous cell carcinoma
2. Squamous cell carcinomaa. Risk factors: HPV type 16, VIN, smoking tobacco,
immunodeficiency (e.g., AIDS)b. Metastasis: first to inguinal nodes
3. Extramammary Paget's diseasea. Red, crusted vulvar lesion: often confused with
eczemab. Intraepithelial adenocarcinoma: malignant
Paget's cells have clear halos because of secretion ofmucopolysaccharides (Figure 21-2).
4. Malignant melanoma: melanoma cells are histologi-cally similar to Paget's cells.
III. Disorders of the VaginaA. Gartner's duct cyst: remnant of wolffian (mesonephric)
duct; benign cyst on the lateral wall of vaginaB. Cystocele: relaxation of pelvic support causing uterine
descent, with subsequent bladder protrusion into the vaginaC. Tumors
1. Sarcoma botryoides (also known as embryonal rhab-domyosarcoma): necrotic, grape-like mass protrud-ing from the vagina in girls younger than 5 years of age
2. Clear cell adenocarcinoma of the vaginaa. Occurs in daughters of women who received dieth-
ylstilbestrol (DES) during pregnancy; DES inhib-its mullerian differentiation.
b. Low risk (1:1000)c. May also involve the cervixd. Vaginal adenosis: benign lesion; precursor of
clear cell adenocarcinoma3. Vaginal squamous cell carcinoma
a. Associated with HPV type 16b. Usually an extension from a cervical squamous
cell cancer rather than a primary cancer
Most commoncancer of the vulva,vagina, and cervix:squamous cellcarcinoma

••
Chapter 21 Female Reproductive Disorders and Breast Disorders 271
IV. Disorders of the Cervix
•A. Cervicitis
1. Acute cervicitis: normally occurs in young women
•a. Squamous epithelium of the exocervix is derived
from the metaplastic conversion of acutely in-• flamed mucus-secreting columnar cells that migrate
•from the endocervix to the surface of the exocer-vix (transformation zone).
• b. Gland orifices covered by metaplastic squamous ep-ithelium obstruct the outflow of mucus, resulting
• in nabothian cysts.
•2. Pathologic acute cervicitis: commonly occurs with in-
fections associated with C. albicans, C. trachomatis,• HSV-2, N. gonorrhoeae, and T. vaginalis
•B. Cervical polyp: non-neoplastic polyp
1. Protrudes from the cervical os, producing postcoital
•bleeding
2. Cervical polyps are not precancerous.• C. Cervical intraepithelial neoplasia (CIN)
•1. Associated with HPV in most cases (high risk: types
16, 18; low risk: types 6, 11)• 2. Risk factors: early age of sexual intercourse, multiple
partners, high-risk male partners (e.g., intravenous drug• abusers), HPV types 16 and 18, smoking tobacco, oral
contraceptives, immunodeficiency3. CIN types
• a. I: mild dysplasia, involving lower one third of theepithelium
b.b. II: moderate dysplasia, involving lower two thirds
• of the epitheliumc. III: severe dysplasia to carcinoma in situ, involv-
e ing full thickness of the epithelium
•d. Progression from CIN Ito CIN III and invasive
cancer (average age — 45 years) occurs over a period• of approximately 10 years.
D. Cervical cancer: least common gynecologic cancer because• of detection of CIN with cervical Papanicolaou (Pap) smears
•and subsequent removal of the dysplastic epithelium
•••••••••S
Cells to be sampled for a cervical Pap smear are takenfrom the lateral wall of the vagina, exocervix, andtransformation zone (site where cervical dysplasia be-gins). Cell types that are evaluated in a Pap smearinclude superficial, intermediate, and parabasal cells.Superficial cells are estrogen-stimulated, and intermedi-ate cells are progesterone-stimulated. Parabasal cellsindicate a lack of stimulation by hormones. Nonpreg-nant adult women normally have 70% superficial squa-mous cells and 30% intermediate squamous cells; noparabasal cells should be seen.
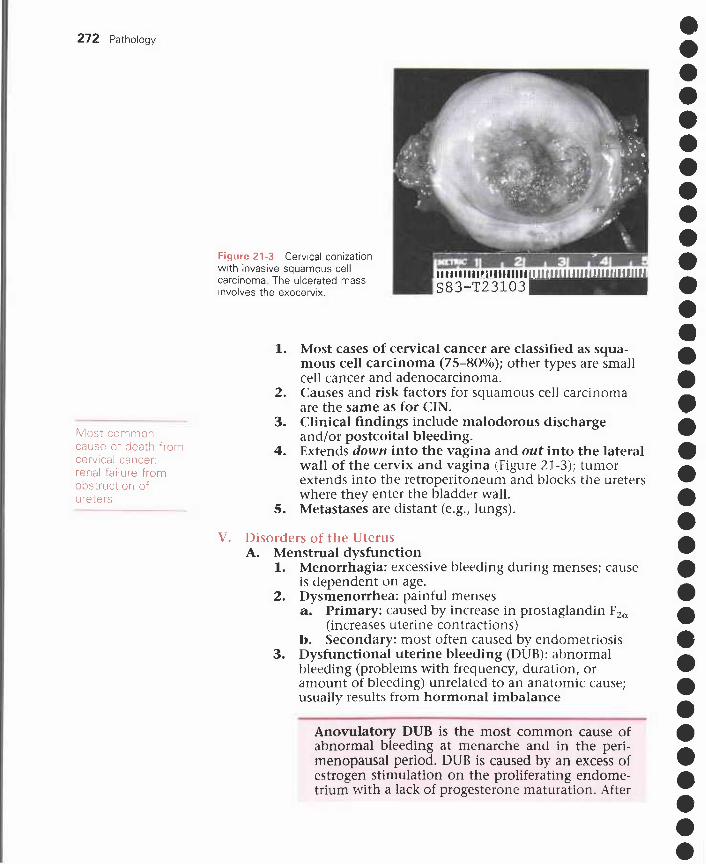
munnivonininS83–T23103
272 Pathology
Figure 21-3 Cervical conizationwith invasive squamous cellcarcinoma The ulcerated massinvolves the exocervix.
1. Most cases of cervical cancer are classified as squa-mous cell carcinoma (75-80%); other types are smallcell cancer and adenocarcinoma.
2. Causes and risk factors for squamous cell carcinomaare the same as for CIN.
3. Clinical findings include malodorous dischargeand/or postcoital bleeding.
4. Extends down into the vagina and out into the lateralwall of the cervix and vagina (Figure 21-3); tumorextends into the retroperitoneum and blocks the ureterswhere they enter the bladder wall.
5. Metastases are distant (e.g., lungs).
V. Disorders of the UterusA. Menstrual dysfunction
1. Menorrhagia: excessive bleeding during menses; causeis dependent on age.
2. Dysmenorrhea: painful mensesa. Primary: caused by increase in prostaglandin F2,,,
(increases uterine contractions)b. Secondary: most often caused by endometriosis
3. Dysfunctional uterine bleeding (DUB): abnormalbleeding (problems with frequency, duration, oramount of bleeding) unrelated to an anatomic cause;usually results from hormonal imbalance
Anovulatory DUB is the most common cause ofabnormal bleeding at menarche and in the peri-menopausal period. DUB is caused by an excess ofestrogen stimulation on the proliferating endome-trium with a lack of progesterone maturation. After
Most commoncause of death fromcervical cancer:renal failure fromobstruction ofureters

Chapter 21 Female Reproductive Disorders and Breast Disorders 273
a prolonged proliferative phase without a secretoryphase, the mucosa undergoes hyperplasia andsloughs off, often causing menorrhagia.
4. Amenorrheaa. Primary: absence of menses by 16 years of ageb. Secondary: absence of menses for 3 months after
previous establishment of normal mensesc. Mechanisms
(1) Hypothalamic and/or pituitary disorder: de-creased production of follicle-stimulatinghormone (FSH) and luteinizing hormone (LH),leading to decreased synthesis of estrogenand progesterone, such as in hypopituitarism
(2) Ovarian dysfunction: decreased productionof estrogen and progesterone, leading to anincrease in FSH and LH, such as in Turner'ssyndrome
(3) End-organ defects that prevent outflow ofblood: normal levels of FSH, LH, estrogen, andprogesterone, such as an imperforate hymen
B. Endometritis1. Acute: bacterial infection (e.g., group B streptococcus)
following delivery or miscarriage2. Chronic: retained placenta, tuberculosis, gonorrhea, in-
trauterine device (Actinomyces israelii)C. Adenomyosis
1. Invagination of the stratum basalis into the myo-metrium; endometrial glands and stroma cause in-creased thickening of myometrial tissue and an en-larged uterus.
2. Clinical findings: menorrhagia, dysmenorrhea, pelvicpain
D. Endometriosis1. Is present only during the reproductive years2. Caused by functional endometrial glands and stroma
outside the uterus that bleed cyclically3. Mechanisms: retrograde menses through the fallopian
tubes (most common), coelomic metaplasia, vascu-lar or lymphatic spread
4. Common sites: ovaries (blood-filled cysts, "chocolatecysts"), pouch of Douglas (posterior to uterus and ante-rior to rectum), round ligaments, intestine
5. Clinical findings: dysmenorrhea, intestinal obstruc-tion, painful defecation
E. Endometrial hyperplasia1. Caused by unopposed estrogen; lack of progesterone
to balance estrogen
•••••••••••••••1 •i ••••••••••••••••••••
Most commoncause of secondaryamenorrhea:pregnancy

274 Pathology
Figure 21 -4 Leiomyomas. Insagittal section. multiple well-circumscribed, gray-whitenodules (leiomyomas) are dis-persed throughout themyometrium
2. Risk factors: early menarche or late menopause, nulli-parity, obesity (increased aromatization of androgens toestrogen), polycystic ovarian syndrome, unopposedestrogen
3. Varies in severitya. Simple hyperplasia (increased number of dilated
glands)b. Complex hyperplasia (increased number of dilated
glands with branching)4. Clinical finding: excessive or irregular uterine bleeding5. May progress to endometrial carcinoma
F. Endometrial polyp1. No malignant potential2. Common cause of menorrhagia in women 20-45 years
of ageG. Leiomyomas ("fibroids"): benign tumors of myometrial
smooth muscle (Figure 21-4)1. More common in blacks than in whites2. Estrogen-sensitive tumors: increase in size during preg-
nancy; decrease in size during menopause3. Common cause of menorrhagia
H. Leiomyosarcoma: most common sarcoma of the uterus1. Does not arise from a leiomyoma2. Distinguished from a leiomyoma by atypical mitoses
and necrosisI. Endometrial carcinoma
1. Average age at diagnosis: 61 years2. Caused by unopposed estrogen stimulation3. Associated with increased risk of breast cancer4. Risk factors: same as for endometrial hyperplasia (see
section V E)S. Spreads down into the endocervix and out into the
uterine wall6. Characterized by postmenopausal bleeding
Most commonbenign tumor inwomen:leiomyomas
Most common gy-necologic cancer:endometrialcarcinoma

•
•Chapter 21 Female Reproductive Disorders and Breast Disorders 275
• VI. Fallopian Tube Disorders: PID
•A. Most often caused by C. trachomatis and N. gonorrhoeae
(see section I A 3)
• B. Inflammation often extends to ovaries (abscesses), fallo-• pian tubes (pyosalpinx, or pus-filled tubes), fallopian tubes
and ovaries (tubo-ovarian abscesses), and peritoneum
•(peritonitis).
C. Complications• 1. Ectopic pregnancy
•2. Sterility, hydrosalpinx (tube filled with watery fluid
after resorption of pus)• VII. Disorders of the Ovary• A. Ovarian cysts
• 3. Polycystic ovarian syndromea. Pathogenesis: excessive pituitary synthesis of LH
• and reduced synthesis of FSH• (1) Increased
cells, causingincreases
hirsutism.androgen synthesis by
stromal
•1. Follicular cyst: accumulation of fluid in follicle or pre-
viously ruptured follicle; rupture produces sterileperitonitis with pain. Most common
•2. Corpus luteum cyst: accumulation of fluid in corpus ass
luteum during pregnancy
follicular cystovarian m
•• Hirsutism or virilization caused by an
ovarian disorder such as polycystic ovar-• ian syndrome is due to an increase in
•testosterone synthesis by stromal cells.Hirsutism is excess hair in normal hair-
• bearing areas and is caused by overpro-• duction of androgens. Virilization is hir-
sutism in addition to other male
•secondary sex characteristics (e.g., in-creased muscle mass).•
•(2) Androgens are aromatized into estrogen in the
adipose, causing hyperestrinism and poten-
C(3) Increased estrogen has a positive feedback on
tial endometrial hyperplasia and cancer.
LH and negative feedback on FSH; lack of FSHcauses follicle degeneration and develop-
• ment of subcortical cysts that enlarge theovaries.
•b. Clinical findings: menstrual irregularities, hirsut-
ism, infertility, obesity• c. Laboratory findings: LH:FSH ratio > 2:1; increased
testosterone, androstenedione, and estrone• B. Ovarian m•
1. Most likely benign in women younger than 45 yearsof age••

TABLE 21-1 Classification of Ovarian Tumors
Tumor Characteristics
276 Pathology
Yolk sac tumor
Sex-Cord Stromal
Thecoma-fibroma
Granulosa-thecalcell tumor
Sertoli-Leydig cell
Krukenberg tumor
Endometrioid
Brenner tumor
Germ CellCystic teratoma
Dysgerminoma
Surface-Derived
Serous tumors
Mucinous tumors
Often bilateral; cysts lined by ciliated cellsSerous cystadenoma (benign)Serous cystadenocarcinoma has psammoma bodies (dystrophi-
cally calcified tumor cells)Lined by mucus-secreting cells; large, multiloculated tumors;
seeding produces pseudomyxoma peritoneiMucinous cystadenoma (benign)Mucinous cystadenocarcinomaUsually malignant; associated with endometrial carcinoma
(15-30%); often affects both ovaries
Usually benign; contain Walthard's rests (transitional-likeepithelium)
Usually benign; ectodermal differentiation (hair, sebaceousglands, teeth)
Immature malignant types contain mature and immature compo-nents (e.g., muscle, neuroepithelium)
Struma ovarii type has functioning thyroid tissue
Malignant germ cell tumor; associated with streak gonads ofTurner syndrome
Increased a-fetoprotein; most common ovarian cancer in girls< 4 years of age
Benign; associated with Meigs' syndrome (ascites, right-sidedpleural effusion)
Low-grade malignant; feminizing tumor (produces estrogen);contains Call-Exner bodies
Benign; masculinizing tumor (produces androgens); pure Leydigcell tumors contain cells with crystals of Reinke
May affect both ovaries; hematogenous spread of gastric cancer;contains signet-ring cells
••••••••••••••••••••••••••
Tumors Metastatic to Ovary
2. Risk factorsa. Nulliparity: increased number of ovulatory cycles
increases risk; use of oral contraceptives de-creases risk.
b. Genetic factors: mutations of BRCA1 and BRCA2suppressor genes, Lynch syndrome, Turner'ssyndrome (dysgerminoma)
3. Classification of ovarian tumors (Table 21-1)a. Surface epithelial: derived from coelomic
epitheliumb. Germ cell: derived from primitive cells that differ-
entiate along gonadal cell lines (e.g., dysgermi-
3••••••••
Most commonprimary benignovarian tumors:serouscystadenomas

Chapter 21 Female Reproductive Disorders and Breast Disorders 277
noma); somatic cell lines (e.g., teratoma); orextraembryonic lines (e.g., yolk sac tumor)
c. Sex cord-stromal: derived from stromal cells andmay be hormone-producing
d. Tumors metastatic to the ovary: derived frombreast cancer, stomach cancer (e.g., Krukenbergtumors)
4. Clinical and laboratory findingsa. Malignant surface-derived tumors often spread by
seeding, producing malignant ascites and increasedabdominal girth.
b. Cystic teratomas undergo torsion, leading to infarc-tion.
c. Signs of feminization (estrogen-secreting tumors)or masculinization (androgen-secreting tumors)may be present.
d. CA 125 is a marker for surface-derived tumors.
VIII. Gestational DisordersA. Infections
1. Most result from ascending bacterial infections (e.g.,group B streptococcus) associated with prematurerupture of membranes.
2. Also caused by congenital infections (e.g., cytomeg-alovirus)
B. Placental abnormalities1. Placenta previa: implantation over the cervical os; pro-
duces painless vaginal bleeding2. Abruptio placentae: retroplacental clot
a. Produces painful vaginal bleedingb. Associated with hypertension, smoking tobacco,
and cocaine use3. Placenta accreta: direct implantation into the myo-
metrium without intervening decidua; persistent bleed-ing requires hysterectomy.
4. Twin placentasa. Monochorionic: associated with identical twins;
derive from a single fertilized egg and are eithermonoamniotic (single amniotic sac) or diamniotic(separate amniotic sacs)
b. Dichorionic: either fraternal (separate fertilizedeggs) or identical twins, whether diamniotic orseparated
Most common ma-lignant ovariantumors: serouscystadenocarcinomas
C. Ectopic pregnancy1. Most commonly occurs in the fallopian tubes
Most common risk2. Mainly caused by scarring from previous PID3. Characterized by the extension of trophoblastic tissue factor for ectopic
(syncytiotrophoblast and cytotrophoblast) into thewall of the fallopian tubea. Produces a hematosalpinx (blood in the lumen)
pregnancy: PID

278 Pathology
b. May rupture into the peritoneal cavity, causingintraperitoneal hemorrhage with a potential forhypovolemic shock and death
4. Clinical findings: usually amenorrhea followed bysudden onset of lower abdominal pain
5. Most important initial screening test: serum 13-humanchorionic gonadotropin (hCG); transvaginal ultra-sound confirms the location of the ectopic pregnancy.
D. Preeclampsia-eclampsia (toxemia of pregnancy)1. Usually occurs in the third trimester (weeks 24-25);
more common in women older than 35 years of age2. Pathogenesis
a. Abnormal placentation causes mechanical orfunctional obstruction of the spiral arteries,leading to placental hypoperfusion.(1) Normal vasodilators are decreased (e.g., pros-
taglandin E2, nitric oxide)(2) Vasoconstrictors are increased (e.g., throm-
boxane A2, angiotensin II)b. Pathologic findings include premature aging, in-
farction of the placenta, and intimal atherosclerosisin the spiral arteries.
3. Clinical findingsa. Diastolic hypertension, proteinuria (often in the
nephrotic range), pitting edema. Preeclampsia isknown as eclampsia if seizures are present.
b. Renal disease: swollen endothelial cells inglomerulus
c. Liver disease: periportal necrosis with elevatedtransaminases
d. Hemolytic anemia and disseminated intravascu-lar coagulation; HELLP syndrome (hemolysis, el-evated liver enzymes, low platelet count)
E. Gestational trophoblastic neoplasms1. Normal chorionic villi are lined by trophoblastic
tissue.a. Syncytiotrophoblast: outer layer, which is exposed
to blood; synthesizes hCG and human placentallactogen
b. Cytotrophoblast: inner layer2. Hydatidiform moles: benign tumors of the chorionic
villusa. Complete mole
(1) Dilated, swollen villi without fetal bloodvessels (resembles a bunch of grapes; Figure21-5)
(2) Embryo absent: 46,XX (90%), with bothchromosomes of male origin. The egg has nochromosomes and is fertilized by twohaploid spermatozoa with X chromosomes.
(3) May transform into a choriocarcinoma
Eclampsia: treatwith magnesiumsulfate
Most common typeof hydatidiformmole: completemole
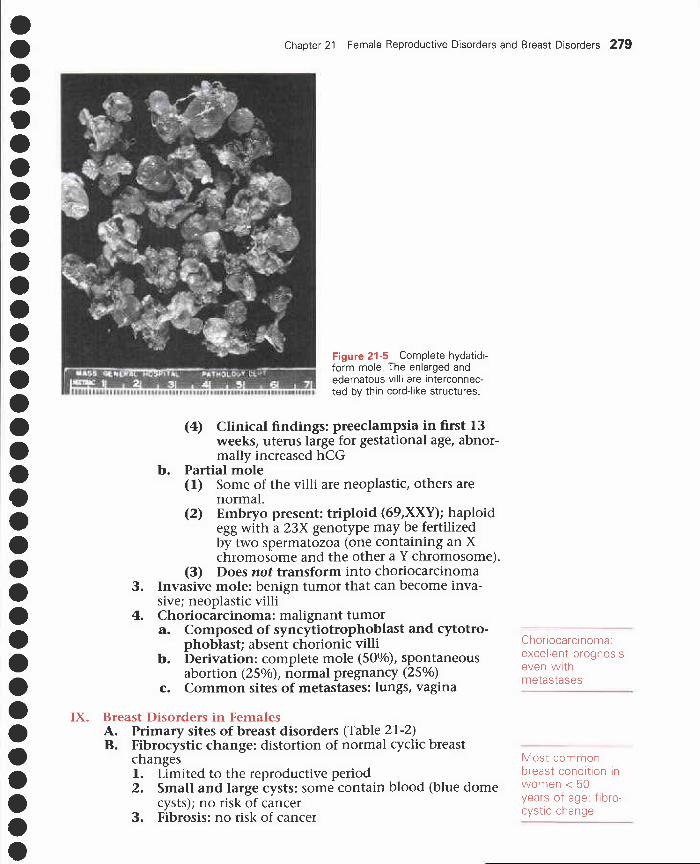
Chapter 21 Female Reproductive Disorders and Breast Disorders 279
Figure 21-5 Complete hydatidi-form mole The enlarged andedematous villi are interconnec-ted by thin cord-like structures.
(4) Clinical findings: preeclampsia in first 13weeks, uterus large for gestational age, abnor-mally increased hCG
b. Partial mole(1) Some of the villi are neoplastic, others are
normal.(2) Embryo present: triploid (69,XXY); haploid
egg with a 23X genotype may be fertilizedby two spermatozoa (one containing an Xchromosome and the other a Y chromosome).
(3) Does not transform into choriocarcinoma3. Invasive mole: benign tumor that can become inva-
sive; neoplastic villi4. Choriocarcinoma: malignant tumor
a. Composed of syncytiotrophoblast and cytotro-phoblast; absent chorionic villi
b. Derivation: complete mole (50%), spontaneousabortion (25%), normal pregnancy (25%)
c. Common sites of metastases: lungs, vagina
IX. Breast Disorders in FemalesA. Primary sites of breast disorders (Table 21-2)B. Fibrocystic change: distortion of normal cyclic breast
changes1. Limited to the reproductive period2. Small and large cysts: some contain blood (blue dome
cysts); no risk of cancer3. Fibrosis: no risk of cancer
Choriocarcinoma:excellent prognosiseven withmetastases
Most commonbreast condition inwomen < 50years of age: fibro-cystic change

280 Pathology
TABLE 21-2 Primary Sites of Breast Disorders
Site Breast Disorders
Nipple Paget's disease
Lactiferous duct, sinuses Intraductal papilloma
Major ducts Fibrocystic changeDuctal carcinoma in situInfiltrating ductal carcinoma
Terminal lobules Sclerosing adenosisLobular carcinomaTubular carcinoma
Stroma FibroadenomaPhyllodes tumor
4. Sclerosing adenosisa. Proliferation of small ductules and/or acini in
the terminal lobule; morphology is often confusedwith invasive ductal cancer.
b. Often contain microcalcifications; slight risk ofcancer
5. Ductal hyperplasiaa. Estrogen-sensitive ductsb. Pathologic findings: papillary proliferation, apo-
crine metaplasia, atypical hyperplasia (in-creased risk of cancer)
C. Inflammation1. Acute mastitis: caused by Staphylococcus aureus; most
often associated with breastfeeding2. Traumatic fat necrosis
a. Caused by trauma to breast tissueb. Lipid-laden macrophages with foreign body
giant cells; fibrosis and dystrophic calcificationc. Process may result in skin retraction, simulating
cancer.D. Benign breast tumors
1. Fibroadenoma: derived from stromal cellsa. Stromal cells proliferate and compress the ducts
(Figure 21-6).b. Discrete, movable mass; may increase in size
during menstrual cycle or pregnancy(estrogen-sensitive)
c. Not a precursor of breast cancer2. Phyllodes tumor: bulky tumor derived from stromal
cellsa. Lobulated tumor and cystic spaces with leaf-like ex-
tensions, often reaching massive sizeb. May be malignant (hypercellular stroma with
mitoses)3. Intraductal papilloma: develops in lactiferous ducts
or sinuses
Most commonbreast mass inwomen < 30 yearsof age:fibroadenoma
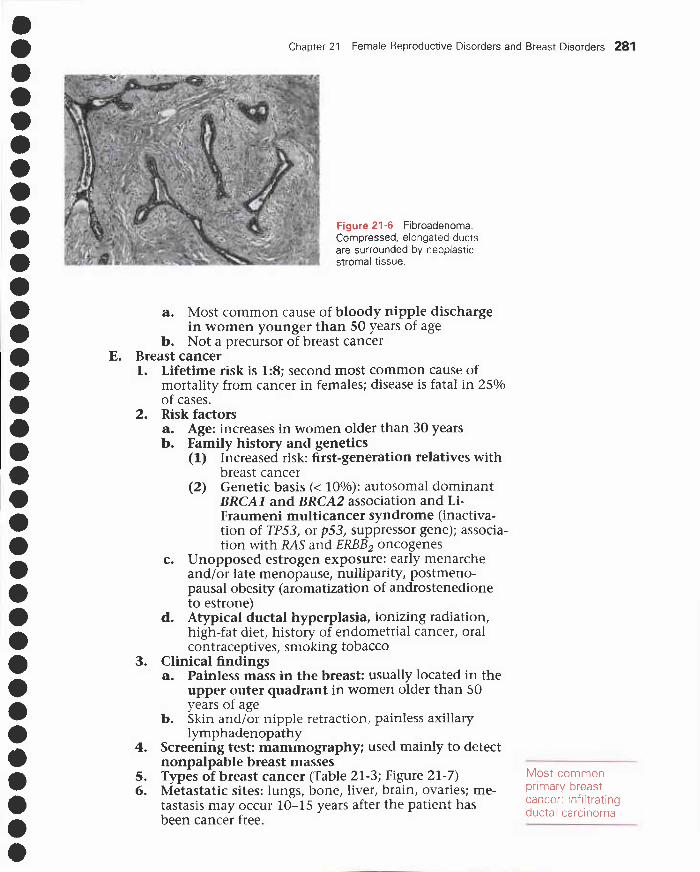
•••••••••
Chapter 21 Female Reproductive Disorders and Breast Disorders 281
Figure 21-6 Fibroadenoma.Compressed, elongated ductsare surrounded by neoplasticstromal tissue.
•••••a. Most common cause of bloody nipple discharge
in women younger than 50 years of ageb. Not a precursor of breast cancer
E. Breast cancer1. Lifetime risk is 1:8; second most common cause of
mortality from cancer in females; disease is fatal in 25%of cases.
2. Risk factorsa. Age: increases in women older than 30 yearsb. Family history and genetics
(1) Increased risk: first-generation relatives withbreast cancer
(2) Genetic basis (< 10%): autosomal dominantBRCA1 and BRCA2 association and Li-Fraumeni multicancer syndrome (inactiva-tion of TP53, or p53, suppressor gene); associa-tion with RAS and ERBB2 oncogenes
c. Unopposed estrogen exposure: early menarcheand/or late menopause, nulliparity, postmeno-pausal obesity (aromatization of androstenedioneto estrone)
d. Atypical ductal hyperplasia, ionizing radiation,high-fat diet, history of endometrial cancer, oralcontraceptives, smoking tobacco
3. Clinical findingsa. Painless mass in the breast: usually located in the
upper outer quadrant in women older than 50years of age
b. Skin and/or nipple retraction, painless axillarylymphadenopathy
4. Screening test: mammography; used mainly to detectnonpalpable breast masses
S. Types of breast cancer (Table 21-3; Figure 21-7)6. Metastatic sites: lungs, bone, liver, brain, ovaries; me-
tastasis may occur 10-15 years after the patient hasbeen cancer free.
•••••••••••••••••••••
Most commonprimary breastcancer: infiltratingductal carcinoma

282 Pathology
TABLE 21 -3 Types of Breast Cancer
Type
Comments
NoninvasiveDuctal carcinoma in situ
(DCIS)
Lobular carcinomain-situ
InvasiveInfiltrating ductal carci-
noma
Paget's disease of nipple
Medullary carcinoma
Inflammatory carcinoma
Invasive lobularcarcinoma
Tubular carcinoma
Colloid (mucinous)carcinoma
Patterns: cribriform (sieve-like), comedo (necrotic center);commonly contain microcalcifications
Terminal lobules distended with bland neoplastic cellsIncreased incidence of cancer in opposite breast
One-third overexpress ERBB2 oncogeneStellate shaped, indurated, gray-white tumor; gritty on cut
section; induration caused by reactive fibroplasia(desmoplasia)
Extension of DCIS into lactiferous ducts and skin of nipple(skin rash, nipple retraction); Paget's cells
Associated with BRCA1 mutationsBulky, soft tumor with large cells and lymphoid infiltrate
Erythematous breast with dimpling like an orange (peaud'orange); plugs of tumor blocking lumen of dermallymphatics cause localized lymphedema
Very poor prognosis
Neoplastic cells arranged in linear fashion or form concen-tric circles (bull's-eye appearance)
Develops in terminal ductules; increased incidence ofcancer in opposite breast
Usually occurs in elderly womenNeoplastic cells surrounded by extracellular mucin
Figure 21-7 Infiltrating ductalcarcinoma showing astellate-shaped scar (arrow) inthe fat tissue of the breast.

Chapter 21 Female Reproductive Disorders and Breast Disorders 283
7. Estrogen and progesterone receptor assays: morelikely to be positive in postmenopausal women; pres-ence of receptors correlates with a better prognosis andresponse to antiestrogen therapy.
X. Breast Disorders in MalesA. Gynecomastia
1. Benign glandular proliferation in the breast: subar-eolar mass; usually unilateral; caused by excess es-trogen stimulation
2. Physiologic gynecomastia: newborn, adolescent, andelderly males; surgery usually is not indicated.
3. Pathologic gynecomastia: caused by cirrhosis (inabil-ity to metabolize estrogen); Klinefelter's syndrome;drugs (e.g., spironolactone, digitalis)
B. Breast cancer1. Risk factors: BRCA2 suppressor gene, Klinefelter's
syndrome2. Usually poor prognosis

111.6611‘Endocrine Disorders
I. Overview of Endocrine DiseaseA. Nomenclature: primary disorders involve the target organ,
secondary disorders the anterior pituitary, and tertiarydisorders the hypothalamus.
B. Negative feedback loop increases or decreases hormoneproduction.• Increased serum thyroxine (T4) inhibits release of thyroid-
stimulating hormone (TSH) by negative feedback.C. Stimulation tests, such as the adrenocorticotropic hormone
(ACTH) stimulation test, are used to evaluate hypofunc-tion disorders.1. Autoimmune destruction is the most common cause of
hypofunction disorders.2. Other causes are infarction, decreased hormone stimula-
tion, enzyme deficiency, and infection.D. Suppression tests (e.g., dexamethasone suppression test;
DST) are used to evaluate hyperfunction disorders.1. Most hyperfunction disorders cannot be suppressed:
exceptions are prolactinoma and pituitary Cush-ing's disease.
2. Causes of hyperfunction: adenoma, hyperplasia, cancer
II. Pituitary GlandA. Anterior pituitary hypofunction
1. May be secondary to hypothalamic (e.g., autoimmunedestruction) or pituitary disease (most common); at least75% of the gland is destroyed.
2. Causes of anterior pituitary hypofunctiona. Nonfunctioning (null) pituitary adenoma: most
common cause in adults(1) Associated with multiple endocrine neoplasia
(MEN) I syndrome (pituitary adenoma, hyper-parathyroidism, pancreatic tumor such as (3-isletcell tumor, peptic ulcer)
Most commoncause of hyperfunc-tion disorders:adenoma
284

Chapter 22 Endocrine Disorders 285
(2) Expanding tumor causes loss of trophic hor-mones and hypofunction of target organs.
(3) Tumors cause an enlarged sella turcica, visualfield defects (e.g., bitemporal hemianopsia),and headache.
b. Sheehan's postpartum pituitary necrosis(1) Hypovolemic shock (e.g., blood loss) causes
anterior pituitary infarction.(2) Sudden cessation of lactation due to loss of
prolactin causes hypopituitarism.c. Craniopharyngioma: most common cause of hypo-
pituitarism in children(1) Benign pituitary tumor derived from remnants
of Rathke's pouch(2) Located superior to sella turcica; extends into
sella turcica and destroys the pituitary gland;cystic tumor with hemorrhage and calcification
3. Clinical findings resulting from trophic hormonedeficiencya. Decreased gonadotropins: delayed puberty in chil-
dren, amenorrhea in women, erectile dysfunctionin men
b. Growth hormone (GH) deficiency(1) Children have delayed growth and sexual
maturation.(2) Adults have hypoglycemia (GH is gluco-
neogenic).c. TSH deficiency: secondary hypothyroidism, de-
creased serum T4 and TSHd. ACTH deficiency: secondary hypocortisolism, hy-
poglycemia (cortisol is gluconeogenic)
The metyrapone test is a stimulation test ofpituitary ACTH reserve. Metyrapone blocks ad-renal 11-hydroxylase, which normally causesan increase in plasma ACTH (pituitary) and acorresponding increase in 11-deoxycortisol (ad-renal). In hypopituitarism, neither ACTH nor11-deoxycortisol is increased.
B. Posterior pituitary hypofunction1. Central diabetes insipidus: lack of antidiuretic hormone
(ADH)a. Etiology: hypothalamic disease (e.g., histiocytosis X),
transection of pituitary stalk (e.g., trauma), posteriorpituitary disease (e.g., metastasis)
b. Responds to ADH administration by increasing urineconcentration (urine osmolality)
2. Nephrogenic diabetes insipidus: collecting tubules re-fractory to ADH
Hypopituitarism: inadults, nonfunc-tioning adenoma;in children,craniopharyngioma

286 Pathology
a. Etiology: drug use (e.g., lithium), hypokalemia (vac-uolar nephropathy)
b. Does not respond to ADH administration; urineremains hypotonic.
3. Clinical findings: excessive thirst, polyuria4. Laboratory findings: hypernatremia and hypotonic
urine
The water deprivation test distinguishes central dia-betes insipidus from nephrogenic diabetes insipidus.Normally in water deprivation, plasma osmolality(Po.) is increased, which stimulates the release ofADH and increases urine osmolality (Uosm), indicat-ing normal urine concentration. In central diabetesinsipidus, Po. increases (hypernatremia) and Uosmdecreases. Uosm increases more than 50% after vaso-pressin (ADH) injection. In nephrogenic diabetesinsipidus, Uosm increases less than 50% after vaso-pressin injection.
C. Pituitary hyperfunction disorders1. Prolactinoma: benign adenoma; most common ante-
rior pituitary tumora. Clinical findings
(1) Women: secondary amenorrhea (prolactin in-hibits gonadotropin-releasing hormone) andgalactorrhea
(2) Men: erectile dysfunction without galactorrheab. Laboratory findings: prolactin usually > 200 ng/mL,
decreased gonadotropins
Other causes of increased prolactin include pri-mary hypothyroidism, drug use (e.g., estrogen),and stalk transection (loss of the inhibitory effectof dopamine on prolactin).
2. GH adenomaa. GH stimulates liver synthesis and release of insulin-
like growth factor (IGF)-I.(1) GH stimulates gluconeogenesis and amino acid
uptake in muscle.(2) IGF-I stimulates growth of bone (linear and
lateral), cartilage, and soft tissue.b. Clinical findings
(1) Causes gigantism in children: increased linearbone growth (epiphyses are not fused).
(2) Causes acromegaly in adults: increased lateralbone growth (jaw, hands, feet), frontal bossing,cardiomyopathy (cause of death)
Prolactinoma: sec-ondary amenor-rhea andgalactorrhea

TBG
TBG
0
000OGQFree T4 = 6 pgTotal Tr, = 12 pgTSH = normal
Chapter 22 Endocrine Disorders 287
c. Laboratory findings: increased GH and IGF-I(not suppressed by glucose administration),hyperglycemia
3. Syndrome of inappropriate ADH (SIADH): posteriorpituitary hyperfunctiona. Etiology: small cell carcinoma of lung, central
nervous system injury, drug use (e.g., chlorprop-amide), lung infection (e.g., tuberculosis)
b. Clinical findings: mental status changes due tocerebral edema
c. Laboratory finding: hyponatremia (serum M.+usually < 120 mEq/L)
d. Treatment: restrict water.
III. Thyroid GlandA. Thyroid function tests (Table 22-1)
1. Total serum T4 (Figure 22-1)a. Increase in total T4 is caused by increase in free T4
TABLE 22-1 Laboratory Findings in Thyroid Disease
Disorder Serum T, Serum TSH "11 Uptake
Graves' disease i 1 T
Patient taking excess hormone, or initialphase of thyroiditis
T 1 1
Primary hypothyroidism 1 T '1'
Secondary hypothyroidism 1 1 1
Increased TBG T Normal Normal
Decreased TBG 1 Normal Normal
T4, thyroxine; TBG, thyroid-binding globulin; TSH, thyroid-stimulating hormone.
Cause of SIADH:small cell carcinomaof lung
Figure 22-1 Schematic of totalserum thyroxine (T4) The bars repre-sent normal serum levels ofthyroid-binding globulin (TBG). The redcircles are T4 that is bound to TBG(6 pg/dL) and T4 that is unbound (freein serum; 6 pg/dL). The total serumT4 is the sum of the bound T4 and thefree T4 (12 pg/dL). The free T, ismetabolically active, and increased T4
inhibits release of thyroid-stimulating hormone (TSH) via nega-tive feedback An increase or decreasein TBG alters the total serum T4without affecting the free 14 or TSHlevels in serum (no signs of hyper-or hypothyroidism) An increase or de-crease in free T4 alters the total serumT4 and the serum TSH, causing hyper-or hypothyroidism

288 Pathology
(thyrotoxicosis) or increase in thyroid-bindingglobulin (TBG) (e.g., estrogen excess due to preg-nancy or oral contraceptive use).
b. Decrease in total T4 is caused by decrease in freeT4 (hypothyroidism) or decrease in TBG (e.g., due toanabolic steroid use).
2. Serum TSH: negative feedback relationship with free T4and free triiodothyronine (T3)a. Increased TSH: indicates primary hypothyroidismb. Decreased TSH: indicates thyrotoxicosis or second-
ary hypothyroidism3. Radioactive 131 1 uptake: evaluates thyroid gland syn-
thetic activity (iodide is used to synthesize thyroidhormone)a. Increased uptake when thyroid gland is synthesiz-
ing excess T4 (e.g., in Graves' disease)b. Decreased uptake when thyroid gland is inactive
(e.g., hypothyroidism or treatment with thyroidhormone)
c. Radioactive 131 1 uptake scan of thyroid detects"hot" (metabolically active; increased uptake of 1311)and "cold" nodules (metabolically inactive; de-creased uptake of 1311).
4. Thyroglobulin: marker of thyroid cancerB. Anatomic thyroid disorders
1. Lingual thyroid: failed descent of thyroid anlage frombase of tongue• Often represents total amount of thyroid tissue
2. Thyroglossal duct cyst: cystic midline massC. Thyroiditis
1. Acute thyroiditisa. Bacterial infection (e.g., Staphylococcus aureus)b. Initial thyrotoxicosis from gland destructionc. Decreased 131 1 uptake
2. Subacute granulomatous (de Quervain's) thyroiditisa. Viral infection (e.g., coxsackievirus)b. Initial thyrotoxicosis from gland destructionc. Decreased 131I uptake
3. Hashimoto's thyroiditisa. Autoimmune thyroiditis: HLA-DR3 and HLA-DR5
association(1) Cytotoxic T cells destroy parenchyma, causing
hypothyroidism.(2) Inhibitory anti-TSH receptor autoantibodies
decrease hormone synthesis.(3) Thyroid gland is enlarged and has a lympho-
cytic infiltrate and germinal follicles.b. Clinical findings: initial thyrotoxicosis from gland
destruction progresses to hypothyroidism.4. Reidel's thyroiditis: fibrous tissue replacement of the
thyroid gland and extension of fibrosis into surroundingtissue (tracheal obstruction)
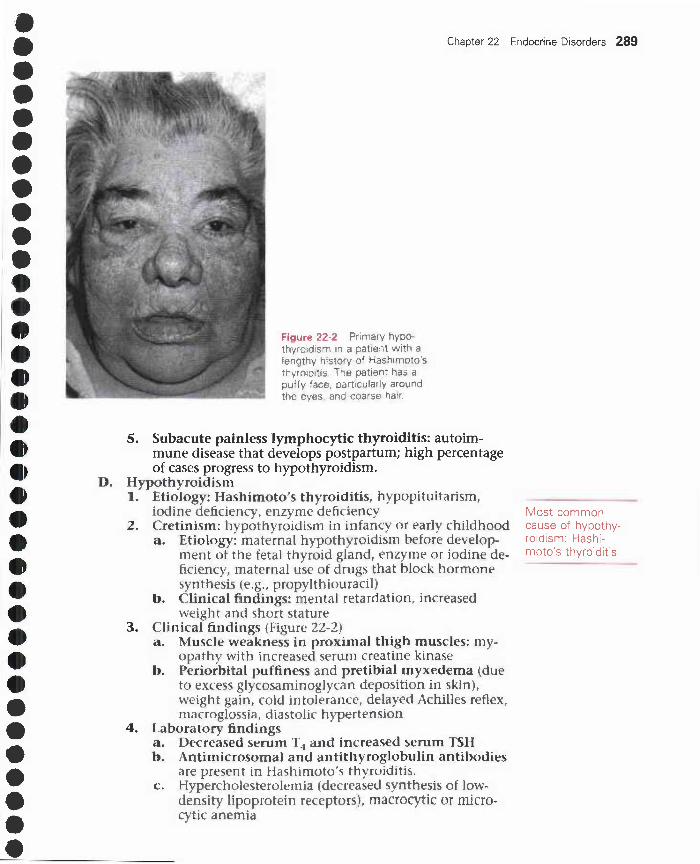
Chapter 22 Endocrine Disorders 289
Figure 22-2 Primary hypo-thyroidism in a patient with alengthy history of Hashimoto'sthyroiditis. The patient has apuffy face, particularly aroundthe eyes, and coarse hair.
5. Subacute painless lymphocytic thyroiditis: autoim-mune disease that develops postpartum; high percentageof cases progress to hypothyroidism.
D. Hypothyroidism1. Etiology: Hashimoto's thyroiditis, hypopituitarism,
iodine deficiency, enzyme deficiency2. Cretinism: hypothyroidism in infancy or early childhood
a. Etiology: maternal hypothyroidism before develop-ment of the fetal thyroid gland, enzyme or iodine de-ficiency, maternal use of drugs that block hormonesynthesis (e.g., propylthiouracil)
b. Clinical findings: mental retardation, increasedweight and short stature
3. Clinical findings (Figure 22-2)a. Muscle weakness in proximal thigh muscles: my-
opathy with increased serum creatine kinaseb. Periorbital puffiness and pretibial myxedema (due
to excess glycosaminoglycan deposition in skin),weight gain, cold intolerance, delayed Achilles reflex,macroglossia, diastolic hypertension
4. Laboratory findingsa. Decreased serum T4 and increased serum TSHb. Antimicrosomal and antithyroglobulin antibodies
are present in Hashimoto's thyroiditis.c. Hypercholesterolemia (decreased synthesis of low-
density lipoprotein receptors), macrocytic or micro-cytic anemia
Most commoncause of hypothy-roidism: Hashi-moto's thyroiditis

290 Pathology
Figure 22 -3 Graves' diseaseThe patient has exophthal-mos and a diffuse enlargementof the thyroid gland (goiter).
E. Increased thyroid hormone• Thyrotoxicosis connotes hormone excess regardless of
cause, whereas hyperthyroidism connotes increased syn-thesis of hormone (e.g., Graves' disease).
1. Graves' diseasea. Occurs most often in women (age of onset > 18
years); autoimmune disease with HLA-DR3association(1) Thyroid-stimulating (IgG) antibodies activate
the TSH receptor: type II hypersensitivity;hormone synthesis is increased.
(2) Symmetrical, nontender thyromegalyb. Clinical features (Figure 22-3)
(1) Infiltrative ophthalmopathy (exophthalmos):due to increased adipose and glycosaminogly-cans in orbital tissue
(2) Pretibial myxedema: from excess glycosami-noglycans in the dermis
2. Plummer's disease (toxic multinodular goiter)a. One or more nodules in a multinodular goiter
become TSH-independent.b. Lacks signs of exophthalmos and pretibial myxedema
3. Clinical findings in thyrotoxicosisa. Weight loss, heat intolerance, diarrhea, anxiety, lid
stare (due to increased sympathetic stimulation)b. Cardiac: sinus tachycardia, atrial fibrillation, systolic
hypertension, high-output failurec. Muscle weakness, osteoporosis (from increased bone
turnover), brisk reflexes4. Laboratory findings in thyrotoxicosis
a. Increased serum T4, decreased serum TSH
Graves' disease:autoantibodies stim-ulate TSH receptor

Chapter 22 Endocrine Disorders 291
b. Increased 131 1 uptake in Graves' disease and toxicmultinodular goiter
c. Decreased 131 1 uptake in thyroiditis and administra-tion of excess thyroid hormone
d. Hyperglycemia, hypocholesterolemia (due to in-creased LDL receptor synthesis), hypercalcemia (fromincreased bone turnover)
F. Nontoxic goiter: thyroid enlargement from excess colloid1. Endemic (due to iodide deficiency) or sporadic (due to
enzyme deficiency, puberty, pregnancy, goitrogenssuch as cabbage)
2. Absolute or relative deficiency of thyroid hormone3. Alternating hyperplasia and hypertrophy (attempt to
increase hormone synthesis) followed by gland invo-lution (failure of gland to sustain synthesis)
4. Thyroid initially diffusely enlarged, then becomesmultinodular
5. Complications: hemorrhage into cyst (sudden, painfulgland enlargement), hypothyroidism, toxic nodulargoiter
G. Solitary thyroid noduleI. Majority are "cold" nodules.2. In women: most are cysts in a goiter or a follicular
adenoma; I 5% of nodules are malignant.3. In men and children: same as for women, but nodule is
more likely to be malignant.4. Suspect malignancy when there is a history of irradia-
tion to the head and neck.H. Follicular adenoma
1. Most common benign thyroid tumor; "cold" nodule2. About 10% progress to follicular carcinoma.
I. Papillary carcinoma1. Most common primary thyroid cancer: most often
occurs in women; associated with radiation exposure;good prognosis
2. Multifocal tumor: papillary fronds intermixed with fol-licles; presence of psammoma bodies (dystrophically cal-cified cancer cells); lymphatic invasion
3. Metastasizes to cervical lymph nodes, then lungFollicular carcinoma1. Most often occurs in women; intermediate prognosis2. Encapsulated or invasive: neoplastic follicles invade
blood vessels (and spare lymph nodes); metastasizes tolung and bone
K. Medullary carcinoma1. Sporadic (80%) or familial (20%)2. Tumors derive from parafollicular C cells.
a. C cells synthesize calcitonin: tumor marker; mayproduce hypocalcemia (inhibits osteoclasts); calcito-nin is converted to amyloid.
b. C-cell hyperplasia is a precursor of cancer.
J.
Papillary carcinoma:associated withradiation exposureand psammomabodies
Medullary carci-noma: only thyroidcancer with amyloiddeposition

c. Genetic testing is available for familial cases: detec-tion of mutation of RET proto-oncogene.
3. Familial type is associated with autosomal dominantMEN IIA and IIB syndromes.a. MEN HA syndrome: medullary carcinoma of the
thyroid, hyperparathyroidism, pheochromocytomab. MEN IIB variant: medullary carcinoma, mucosal
neuromas involving lips and tongue, pheo-chromocytoma
L. Primary malignant lymphoma: most often arises fromHashimoto's thyroiditis
IV. Parathyroid GlandsA. Overview
1. Parathyroid hormone (PTH)a. Increases renal reabsorption of calcium in early
distal tubuleb. Decreases bicarbonate and phosphorus reabsorp-
tion in proximal tubulec. Maintains ionized calcium level in blood via bone
resorption and renal reabsorptiond. Stimulated by hypocalcemia; suppressed by
hypercalcemia2. Total serum calcium
a. Total calcium is bound calcium (40% to albumin,13% to phosphate and citrates) plus free, ionizedcalcium (47°/0; metabolically active fraction).
b. Hypoalbuminemia: decreases total serum calciumwithout altering the free, ionized level; therefore,there is no evidence of tetany.
c. Effect of respiratory or metabolic alkalosis(1) Increases negative charges on albumin: albumin
binds more calcium by removing some from thefree, ionized fraction.
(2) Total serum calcium is normal, but ionizedcalcium is decreased, causing tetany.
d. Tetany: ionized calcium decreased(1) Partial depolarization of nerves and muscle: less
stimulus is required to initiate an actionpotential.
(2) Clinical findings: carpopedal spasm (thumbflexes into palm); Chvostek's sign (tappingover the facial nerve produces a facial twitch);stridor
B. Hypoparathyroidism• One of the many causes of hypocalcemia (Table 22-2)1. Etiology
a. Previous thyroid surgery (most common cause)b. Autoimmune hypoparathyroidismc. DiGeorge syndrome: failure of descent of third and
fourth pharyngeal pouches; absence of thymusd. Hypomagnesemia: required for PTH activation
•292 Pathology ••••••••••0
••••••••••••••••••••••••

Some Causes of Hypocalcemia
Comments
•
•
• TABLE 22-2
• Disorder
•
•
•
•Acute pancreatitis
Hypovitaminosis D
• PTH, parathyroid hormone.••Disorder
•Malignancy-induced
•
••• Sarcoidosis
•Thiazides
•
• Hypervitaminosis D
• PTH, parathyroid hormone,
•
•
•
•
•
•
•
•
•
•
•
•
•
••
Chapter 22 Endocrine Disorders 293
Most commoncause of hypercalce-mia in general pop-ulation: primaryhyperparathyroidism
Pseudohypoparathyroidism X-linked dominant diseaseEnd-organ resistance to PTHMental retardation, basal ganglia calcificationHypocalcemia, normal to increased PTH
Calcium bound to fatty acids in enzymatic fat necrosis
Chronic renal failure most common cause
TABLE 22-3 Some Causes of Hypercalcemia
Comments
Common cause of hypercalcemia in hospitalized patientsMechanisms: bone metastasis with activation of osteoclasts,
ectopic secretion of a PTH-related protein (squamous cellcarcinoma of lung, renal cell carcinoma), multiple myeloma(increased secretion of osteoclast-activating factor byplasma cells)
Hypercalcemia with decreased serum PTH
Mechanism: macrophages in granulomas synthesizela-hydroxylase, causing hypervitaminosis D
Mechanism: increased calcium reabsorption from urine;volume depletion increases proximal tubule reabsorption ofcalcium
Increases calcium reabsorption in jejunum and kidneys
2. Clinical findings: tetany, calcification of basal ganglia(due to hyperphosphatemia), cataracts
3. Laboratory findings: hypocalcemia, hyperphosphate-mia, decreased PTH
C. Hyperparathyroidism• One of the many causes of hypercalcemia (Table 22-3)1. Primary hyperparathyroidism
a. Most often occurs in women older than 50 years ofage; associated with MEN I and MEN HA syndromes
b. Etiology: adenoma (85%), hyperplasia, cancerc. Clinical findings
(1) Renal: calcium stones (most common presen-tation), nephrocalcinosis (causes polyuria andrenal failure)
(2) Gastrointestinal: peptic ulcer disease (calciumstimulates gastrin), acute pancreatitis (calciumactivates phospholipase), constipation

Corticosterone Cortisol
18-Hydroxylase -4-- Angiotensin II stimulates
294 Pathology
•
Cholesterol •
Desmolase •17-Hydroxylase
•Pregnenolone ) 17-Hydroxypregnenolone ----)-- Dehydroepiandrosterone
17-Hydroxylase i
1 N17 Ketosteroids (17 KS)•
•
3-(3-Hydroxysteroiddehydrogenase/isomerase
Progesterone ) 17-Hydroxyprogesterone ----0- Androstenedione •I
1, Oxidoreductase(pregnanetriol)
21 - Hydroxylase 21-Hydroxylase Testosterone
5a - Reductase
11-Deoxycorticosterone 11-Deoxycortisol Dihydrotestosterone DHT)(compound S)
11 -Hydroxylase 11 -Hydroxylase17-Hydroxycorticoids (17-OH)
••••••
Aldosterone
Zona glomerulosa Zona fasciculata
Zona reticularis(mineralocorticoids)
(glucocorticoids)
(sex hormones)
Figure 22-4 Adrenocortical hormone synthesis. The zona glomerulosa produces mineralocorticoids (e.g., aldosterone),the zona fasciculata produces glucocorticoids (e.g., cortisol), and the zona reticularis produces sex hormones (e.g.,testosterone). The 17-hydroxycorticoids (17-0H) are 11-deoxycortisol and cortisol and the 17-ketosteroids (17-KS, weakandrogens) are dehydroepiandrosterone and androstenedione. Testosterone is converted to dihydrotestosterone (DHT) by5a-reductase
(3) Bone: osteitis fibrosa cystica (cystic lesion withhemorrhage usually involving the jaw), sub-periosteal bone resorption of phalanges
(4) Diastolic hypertensiond. Laboratory findings: increased serum PTH and
calcium, decreased phosphorus and bicarbonate(normal anion gap metabolic acidosis)
2. Secondary hyperparathyroidism: parathyroid gland hy-perplasia develops as compensation for hypocalcemia.
V. Adrenal GlandsA. Overview
1. Adrenocortical hormone synthesis (Figure 22-4)2. Adrenal medulla: synthesizes catecholamines (epineph-
rine and norepineph rine)B. Adrenocortical hypofunction (hypocortisolism)
1. Acute adrenocortical insufficiencya. Etiology: abrupt withdrawal of corticosteroids,
Waterhouse-Friderichsen syndrome
••••••••••••••••••••••

Chapter 22 Endocrine Disorders 295
b. Waterhouse-Friderichsen syndrome(1) Usually associated with septicemia from Neis-
seria meningitides(2) Patients develop endotoxic shock, which
causes endothelial damage and precipitatesdisseminated intravascular coagulation.
(3) Bilateral adrenal hemorrhage from fibrin clotsin vessels (hemorrhagic infarction)
2. Addison's disease (chronic adrenal insufficiency)a. Etiology: autoimmune destruction (most
common), miliary tuberculosis, adrenogenital syn-drome, metastasis
b. Clinical findings(1) Weakness and hypotension: due to sodium
loss from mineralocorticoid deficiency(2) Diffuse hyperpigmentation: due to increase in
plasma ACTH, which has melanocyte-stimulating properties
c. Laboratory findings(1) Prolonged ACTH stimulation test: no increase
in cortisol or 1 7-hydroxycorticoids (17-OH)(2) Metyrapone test: increase in ACTH; no increase
in 11-deoxycortisol(3) Increased plasma ACTH(4) Electrolytes: hyponatremia, hyperkalemia,
metabolic acidosis from mineralocorticoiddeficiency
Aldosterone increases the exchange of Ma+for K' in the kidneys. Hence, its deficiencyleads to a hypertonic loss of Ne in theurine (hyponatremia) and retention of K+(hyperkalemia). Aldosterone also increasesthe loss of 11 + in the urine. Deficiency leadsto retention of H+ and metabolic acidosis.
(5) Fasting hypoglycemia (cortisol is gluconeo-genic), eosinophilia, neutropenia, lympho-cytosis
3. Adrenogenital syndromea. Autosomal recessive disorders with enzyme defi-
ciencies causing hypocortisolism with a correspond-ing increase in ACTH(1) Increase in ACTH causes adrenocortical hyper-
plasia and diffuse skin pigmentation.(2) Increase in 17-ketosteroids (17-KS), testoster-
one, and di hydrotestosterone (DHT) producesambiguous genitalia in females and preco-cious puberty in males.
Waterhouse-Friderichsen syn-drome: meningococ-cemia with bilateraladrenal hemorrhage
Most commoncause of Addison'sdisease in children:adrenogenitalsyndrome

296 Pathology
(3) Increase in mineralocorticoids causes hyper-tension; decrease causes sodium loss andhypotension.
b. Substrates proximal to the enzyme block increase,while those distal to the block decrease (seeFigure 22-4).
c. 21-Hydroxylase deficiency: most common enzymedeficiency(1) Clinical findings: ambiguous genitalia in
females; precocious puberty in males; hypo-tension due to sodium loss
(2) Laboratory findings: increase in 17-KS, testos-terone, and DHT; decrease in 17-OH andmineralocorticoids
d. 11-Hydroxylase deficiency(1) Clinical findings: ambiguous genitalia in
females; precocious puberty in males; hyper-tension due to sodium retention
(2) Laboratory findings: increase in 17-KS, testos-terone, and DHT; increase in 17-OH (11-deoxycortisol) and mineralocorticoids (11-deoxycorticosterone)
e. 17-Hydroxylase deficiency(1) Clinical findings: hypogonadism in females;
female external genitalia in males (malepseudohermaphrodites); hypertension due tosodium retention
(2) Laboratory findings: decrease in 17-KS, testos-terone, and DHT; decrease in 17-OH; increase inmineralocorticoids
C. Adrenocortical hyperfunction (hypercortisolism)1. Cushing's syndrome
a. Etiology(1) Prolonged corticosteroid therapy(2) Pituitary Cushing's syndrome (Cushing's
disease): 60% of cases; due to pituitaryadenoma; increased plasma ACTH and cortisol
(3) Adrenal Cushing's syndrome: 25% of cases;most often due to an adenoma; decreasedplasma ACTH and increased cortisol
(4) Ectopic Cushing's syndrome: 15% of cases;usually small cell carcinoma of the lung withectopic ACTH production; increased ACTH andcortisol
b. Clinical findings (Figure 22-5)(1) Weight gain: due to fat deposition in face
("moon facies"), upper back ("buffalo hump"),and trunk; fat distribution is due to hyperin-sulinism from hyperglycemia.
Most commoncause of Cushing'ssyndrome: pro-longed corticoste-roid therapy

Chapter 22 Endocrine Disorders 297
Figure 22-5 Cushing's syn-drome, showing "moon facies,"truncal obesity, and purple ab-dominal striae.
(2) Diastolic hypertension: due to increase inmineralocorticoids
(3) Hirsutism (increased androgens), purple ab-dominal striae (ruptured blood vessels fromhypercortisolism)
c. Laboratory findings(1) Increased free cortisol in urine: very high sen-
sitivity; excellent initial screening test(2) Low-dose DST (cortisol analogue): unable to
suppress cortisol in all types of Cushing's syn-drome (pituitary, adrenal, ectopic)
(3) High-dose DST: can suppress cortisol in pitu-itary Cushing's syndrome by inhibiting ACTH
(4) Hyperglycemia: due to hypercortisolism (corti-sol is gluconeogenic)
(5) Hypokalemic metabolic alkalosis: due to in-creased mineralocorticoids
2. Hyperaldosteronisma. Conn's syndrome (primary aldosteronism)
(1) Most often due to a benign adenoma in thezona glomerulosa
(2) Clinical findings: diastolic hypertension,muscle weakness, tetany
(3) Laboratory findings: hypernatremia, hypokale-mia, metabolic alkalosis, decreased plasma reninactivity (increased plasma volume)
High-dose DSTsuppresses onlypituitary Cushing'ssyndrome.

298 Pathology
In Conn's syndrome, hyperaldosteronismcauses increased exchange of Na + (hyper-natremia) for K± (hypokalemia). Na+ ex-changes with H± when K+ is depleted, caus-ing a loss of H + in the urine and an increasein bicarbonate reabsorption (metabolic al-kalosis).
b. Secondary aldosteronism(1) Aldosterone is released in response to activa-
tion of the renin-angiotensin-aldosteronesystem (e.g., decreased cardiac output).
(2) Plasma renin activity is increased.D. Adrenal medulla hyperfunction
1. Pheochromocytomaa. Approximately 90% of tumors are unilateral, benign
adenomas arising in the adrenal medulla: tumor isbrown and often necrotic.
b. Associated with neurofibromatosis, MEN IIA andMEN IIB, von Hippel-Lindau disease (often bilateraltumors)
c. Clinical findings: diastolic hypertension (sustainedwith occasional paroxysmal bursts), palpitations,anxiety, drenching sweats, headache
d. Laboratory findings(1) 24-hour urine collection: increased for vanil-
lylmandelic acid (VMA) and metanephrine(metabolic end products of epinephrine andnorepinephrine)
(2) Hyperglycemia (glycogenolysis), neutrophilicleukocytosis (inhibition of neutrophil adhesionmolecules)
2. Neuroblastomaa. Malignant tumor
(1) Primarily seen in children younger than 5years of age
(2) Occasionally located in posterior mediastinum(3) Small hyperchromatic cells
b. Clinical findings: diastolic hypertension, metastasisto skin and bones
c. Laboratory findings: increased urinary VMA, meta-nephrine, and homovanillic acid (metabolic endproduct of dopamine)
VI. PancreasA. Islet cell tumors
1. Insulinoma: benign tumor of 13-islet cellsa. Most common islet cell tumor; 80% of patients have
MEN I syndrome.b. Causes fasting hypoglycemia
Increased produc-tion of catechol-amines causeshypertension.
Excess catechol-amine + hyperten-sion -pheochromocytomain adults, neuro-blastoma in children

Chapter 22 Endocrine Disorders 299
c. Clinical findings: mental status abnormalities due tohypoglycemia C peptide: marker of
d. Laboratory findings: increase in serum insulin and endogenous syn-
C peptide (cleaved off proinsulin when insulin is thesis of insulin
produced) associated with hypoglycemia
In individuals who self-inject with insulin, se-rum insulin is increased. However, C peptide isdecreased, because exogenous insulin suppresses13-islet cell production of endogenous insulin.
2. Gastrinoma (Zollinger-Ellison syndrome)3. Glucagonoma: malignant tumor of ot-islet cells; associ-
ated with hyperglycemia and rash (necrolytic migratoryerythema)
4. Somatostatinomaa. Malignant tumor of 8-islet cells: somatostatin is an
inhibitory hormone.b. Clinical findings: achlorhydria (inhibition of
gastrin); cholelithiasis, steatorrhea (cholecysto-kinin); diabetes mellitus (gastric inhibitory peptide);steatorrhea (secretin)
5. VIPoma, or pancreatic choleraa. Malignant tumorb. Excessive secretion of vasoactive intestinal peptidec. Clinical and laboratory findings: secretory diar-
rhea; achlorhydria, hypokalemiaB. Diabetes mellitus
• Diabetes mellitus is a chronic disorder characterized by dis-turbances in carbohydrate, fat, and protein metabolism.
1. Classification: based on etiologya. Type 1: due to failure of insulin synthesis
(Table 22-4)b. Type 2: due to insulin resistance (see Table 22-4)c. Secondary: due to diseases (e.g., cystic fibrosis) or to
drug use (e.g., corticosteroids)d. Gestational diabetes: glucose intolerance due to
pregnancye. Impaired glucose intolerance: hyperglycemia that is
nondiagnostic2. Pathogenesis
a. Type 1 diabetes mellitus: insulin deficiency; pan-creas devoid of 13-islet cells(1) Autoimmune destruction of (3-islet cells: insuli-
tis; islet cell antibodies(2) Genetic susceptibility
b. Type 2 diabetes mellitus: insulin resistance(1) Defective 3-islet cell secretion of insulin; de-
creased insulin receptors; postreceptor defects(2) Obesity

300 Pathology
TABLE 22-4 Comparison Between Types 1 and 2 Diabetes Mellitus
Characteristic
Type 1
Type 2
Clinical findings
5-10%< 20 years
Rapid
Usually thinFamily history uncommonHLA-DR3 and HLA-DR4
Lack of insulinPancreas devoid of I3-islet cellsInsulitis; islet cell antibodies
Polyuria, polydipsia, polyphagia,weight loss
Ketoacidosis* (hyperglycemia,coma; production of ketonebodies)
90-95%
> 30 yearsInsidious
Usually obese (80%)
Family history commonNo HLA association
Insulin resistanceDecreased insulin receptors;
postreceptor defectsFibrotic I3-islet cells contain
amyloidPolyuria, polydipsia; patients
may develop hypertriglyceri-demia
Patients susceptible to hyperos-molar nonketotic coma
Prevalence
Age of onset
Speed of onset
Body habitus
Genetics
Pathogenesis
HLA, human leukocyte antigen*Ketoacidosis is not limited to diabetes; it also occurs in fasting and alcoholism.
c. Pathogenesis of complications(1) Nonenzymatic glycosylation: glucose com-
bines with amino acids in proteins.(2) Osmotic damage: aldose reductase in certain
tissues (e.g., lens, Schwann cells) convertsglucose into sorbitol, which draws glucose intothe tissue, causing damage.
3. Clinical findings (see Table 22-4)4. Complications
a. Insulin-induced hypoglycemia: most commoncomplication; may produce irreversible brain damage
b. Cardiovascular: atherosclerotic stroke, coronaryartery disease, peripheral vascular disease, hyalinearteriolosclerosis(1) Myocardial infarction is the most common
cause of death.(2) Gangrene of digits often requires limb
amputation.c. Renal: renal failure due to nodular glomerulo-
sclerosisd. Diabetic microangiopathy: diffuse thickening of
basement membranese. Ocular: cataracts; retinopathy (microaneurysms)
that may cause retinal detachment and blindness
Diabetes mellitus:organ damage corre-lates with lack ofglycemic control

Chapter 22 Endocrine Disorders 301
f. Neuropathy(1) Peripheral: osmotic damage to Schwann cells;
sensory and motor dysfunction; cause pressureulcers (inability to feel pain)
(2) Autonomic: gastroparesis, erectile dysfunction,cardiac arrhythmias
g. Infection: urinary tract, malignant external otitis(Pseudomonas aeruginosa), mucormycosis
5. Gestational diabetes mellitusa. Glucose intolerance develops during pregnancy
due to anti-insulin effect of human placentallactogen
b. Risks to newborn(1) Macrosomia: hyperglycemia in fetus causes
release of insulin; insulin increases fat stored inadipose and increases muscle mass by increas-ing amino acid uptake in muscle.Respiratory distress syndrome: insulin inhibitsfetal surfactant production.Neural tube defect, neonatal hypoglycemia (dueto hyperinsulinism)
c. Maternal risk: diabetes mellitus may develop.
•••••••••••••••••••••••
(2)
(3)
Glycosylated hemo-globin (HbA,c):evaluates glycemiccontrol during pre-vious 8-12 weeks
•••••••••••••

23
1•11111L MusculoskeletalDisorders
I. Bone DisordersA. Osteogenesis imperfecta ("brittle bone" disease)
1. Autosomal dominant disease with defective synthesisof type I collagen
2. Clinical findings: pathologic fractures at birth, bluesclera, deafness
B. Achondroplasia1. Autosomal dominant disease due to impaired enchon-
dral ossification and premature closure of the epiph-yseal plates of long bones
2. Clinical findings: normal-sized head and vertebralcolumn, shortened arms and legs
C. Osteopetrosis ("marble bone" disease)1. Autosomal recessive (severe) or autosomal dominant
(less severe): defect in osteoclasts causes overgrowth andsclerosis of cortical bone ("too much bone").
2. Clinical findings: pathologic fractures, anemia (replace-ment of marrow cavity), cranial nerve compression(visual and hearing loss)
D. Osteomyelitis1. Osteomyelitis in children
a. Metaphysis is most common site: hematogenousspread; most often due to Staphylococcus aureus
b. Draining sinus tracts to the skin surface: danger ofsquamous cell carcinoma developing at orifice ofsinus tract
2. Osteomyelitis in sickle cell disease: due to Salmonellaparatyphi
3. Tuberculous osteomyelitis: hematogenous spread from aprimary lung focus; targets vertebral column (Pott'sdisease)
Osteogenesis im-perfecta: blue scleradue to reflection ofunderlying choroi-dal veins
Pseudomonasaeruginosa osteo-myelitis: most oftendue to puncture offoot throughrubber footwear
302

Chapter 23 Musculoskeletal Disorders 303
E. Osteoporosis1. Most common metabolic abnormality of bone
a. Reduction in normal mineralized bone (decreasedbone mass and density): decreased thickness ofcortical and cancellous (trabecular) bone
b. Types(1) Primary: idiopathic, senile, postmenopausal(2) Secondary: due to an underlying disease (e.g.,
hypercortisolism) or drug use (e.g., heparin)2. Postmenopausal osteoporosis
a. Associated with estrogen deficiency: increased re-sorption of bone by osteoclasts and decreased forma-tion of bone by osteoblasts
b. Clinical findings: compression fractures of the ver-tebral bodies (most common); Colles' fracture ofdistal radius
c. Evaluation: dual-photon absorptiometry is a non-invasive test that evaluates bone density.
d. Prevention(1) Role of estrogen replacement is being
reevaluated.(2) Calcium, vitamin D, weight-bearing exercise
(excludes swimming, which decreases bonestress)
F. Avascular (aseptic) necrosis of bone1. Microcirculation is disrupted, leading to bone infarc-
tion: common sites include femoral head (fracture inelderly persons) and scaphoid bone.
2. Legg-Calve-Perthes diseasea. Type of aseptic necrosis involving the ossification
center in the femoral headb. Occurs most often in boys 3-10 years of agec. Pain in knee or limp: secondary osteoarthritis is
common.G. Osgood-Schlatter disease
1. Inflammation of proximal tibial apophysis at inser-tion of patellar tendon: affects physically active boys11-15 years of age
2. Produces permanent knobby-appearing kneesH. Paget's disease of bone (osteitis deformans)
1. Targets the pelvis, skull (enlarged), or femur2. Primarily occurs in elderly men; cause is unknown
(may be a virus).3. Early phase of osteoclastic resorption of bone: causes
shaggy-appearing lytic lesions in bone4. Late phase of increased osteoblastic bone formation
a. Increased alkaline phosphataseb. Production of thick, weak bone (mosaic bone)
without normal lamellar structure5. Clinical findings: pathologic fractures, risk of osteo-
genic sarcomas and high-output heart failure (due toarteriovenous connections in vascular bone)
Estrogen: inhibitsproduction of osteo-clasts; enhancesosteoblasts
Legg-Calve-Perthesdisease: asepticnecrosis of femoralhead in children

304 Pathology
I. Fibrous dysplasia1. Benign, nonneoplastic process of single (monostotic)
or multiple (polyostotic) bones: primarily targets ribs,femur, or cranial bones of children and young adults
2. Replacement of marrow by fibrous tissue; subject topathologic fracture
3. Polyostotic type is associated with Albright'ssyndrome.a. Café-au-lait spots on skinb. Precocious sexual development: due to a midline
hamartoma in the hypothalamusJ. Neoplastic disorders of bone
1. Metastasis is the most common malignancy of bone:the breast is the most common primary site.
2. Most common primary malignant tumors of bone, indescending order of frequency: osteogenic sarcoma,chondrosarcoma, Ewing's sarcoma
3. Summary of bone tumors (Table 23-1)
Osteochondroma:most commonbenign bone tumor
II. Joint DisordersA. Synovial fluid analysis
1. Joint lubricant: rich in hyaluronic acid2. Routine studies: white blood cell (WBC) count and dif-
ferential, crystal analysis, culture, Gram stainMonosodium urate 3. Crystal identificationcrystals: present a. Monosodium urate: needle shaped (monoclinic) byin gout special polarization (negative birefringence)
b. Calcium pyrophosphate: monoclinic or triclinic(rhomboid) by special polarization (positivebirefringence)
B. Osteoarthritis1. Noninflammatory joint disease
a. More common in women; universal after 65 yearsof age
Most common b. Secondary causes: obesity, traumadisabling jointdisease:osteoarthritis Ochronosis (alkaptonuria) is an autosomal re-
cessive disease caused by deficiency of homo-gentisic acid oxidase and accumulation of ho-mogentisic acid (urine turns black whenoxidized). Homogentisic acid deposits in the in-tervertebral disks, causing osteoarthritis andother systemic findings.
2. Progressive degeneration of articular cartilagea. Primarily targets weight-bearing jointsb. Hand involvement: distal interphalangeal (DIP) and
proximal interphalangeal (PIP) joints (Figure 23-1)c. Joint findings: erosion and clefts in articular carti-
lage, reactive bone formation at joint margins (osteo-phytes), subchondral cysts

BenignOsteochondroma
Osteoma
Osteoid osteoma
Osteoblastoma
Giant cell tumor
Malignant
Chondrosarcoma
Osteogenic sarcoma
Ewing's sarcoma
Males, 10-30 yearsof age
Solitary or multiple
Males, any age
Males, 10-20 yearsof age
Males, 10-20 yearsof age
Females, 20-40years of age
Males, 30-60 yearsof age
Males, 10-25 yearsof age
Risk factors: Paget'sdisease, familialretinoblastoma,irradiation
Males, 10-20 yearsof age
Metaphysis of distalfemur
Facial bones
Cortex of proximalfemur
Vertebra
Epiphysis of distal femuror proximal tibia
Pelvic bones, proximalfemur
Metaphysis of distalfemur, proximal tibia
Pelvic girdle, diaphysisand metaphysis ofproximal femur or rib
Chapter 23 Musculoskeletal Disorders 305
TABLE 23-1 Tumors of Bone
Tumor Type
Epidemiology Most Common Location
Characteristics
Outgrowth of bone (exostosis)capped by benign cartilage
Associated with Gardner's poly-posis syndrome
Radiographic finding: radiolucentfocus surrounded by scleroticbone
Nocturnal pain relieved by aspirin
Similar to osteoid osteoma
Reactive multinucleated giantcells resemble osteoclasts
Neoplastic mononuclear cells
Grade determines biologicbehavior
Metastasizes to lungs
Malignant osteoidRadiographic findings: "sunburst"
appearance (spiculated patternfrom calcified malignantosteoid), Codman's triangle(tumor lifting periosteum)
Metastasizes to lungs
Small, round cell tumorRadiographic finding: "onion
skin" appearance around bone(periosteal reaction)
Possible fever and anemia
Figure 23-1 Osteoarthritis.There are bony protuberances(Heberden's nodes) from thebase of the terminal phalanx(distal interphalangeal joints).These lesions represent osteo-phytes at the margin of the joint.
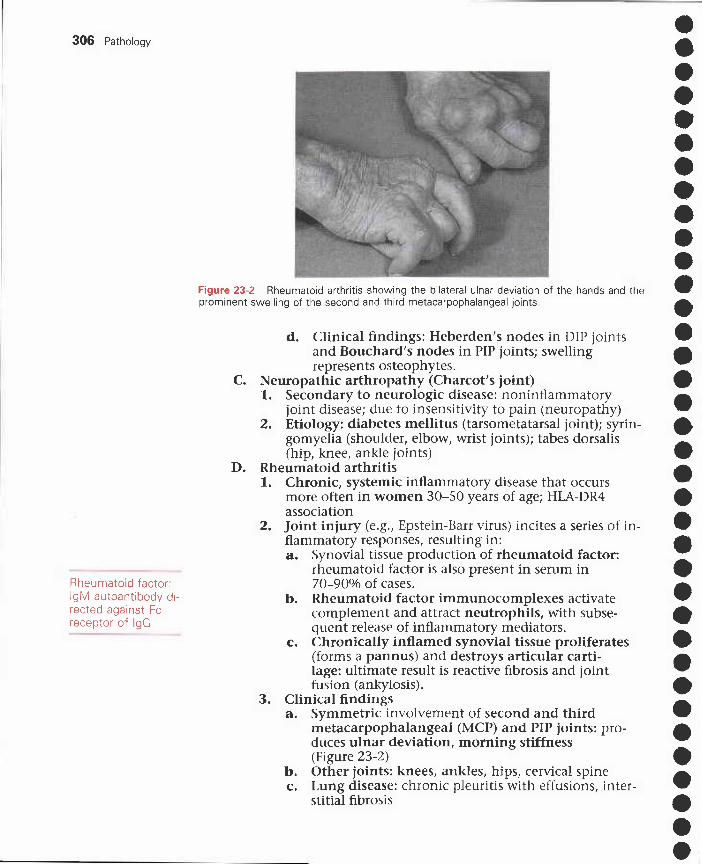
306 Pathology
Figure 23-2 Rheumatoid arthritis showing the bilateral ulnar deviation of the hands and theprominent swelling of the second and third metacarpophalangeal joints
d. Clinical findings: Heberden's nodes in DIP jointsand Bouchard's nodes in PIP joints; swellingrepresents osteophytes.
C. Neuropathic arthropathy (Charcot's joint)1. Secondary to neurologic disease: noninflammatory
joint disease; due to insensitivity to pain (neuropathy)2. Etiology: diabetes mellitus (tarsometatarsal joint); syrin-
gomyelia (shoulder, elbow, wrist joints); tabes dorsalis(hip, knee, ankle joints)
D. Rheumatoid arthritis1. Chronic, systemic inflammatory disease that occurs
more often in women 30-50 years of age; HLA-DR4association
2. Joint injury (e.g., Epstein-Barr virus) incites a series of in-flammatory responses, resulting in:a. Synovial tissue production of rheumatoid factor:
rheumatoid factor is also present in serum in70-90% of cases.
b. Rheumatoid factor immunocomplexes activatecomplement and attract neutrophils, with subse-quent release of inflammatory mediators.
c. Chronically inflamed synovial tissue proliferates(forms a pannus) and destroys articular carti-lage: ultimate result is reactive fibrosis and jointfusion (ankylosis).
3. Clinical findingsa. Symmetric involvement of second and third
metacarpophalangeal (MCP) and PIP joints: pro-duces ulnar deviation, morning stiffness(Figure 23-2)
b. Other joints: knees, ankles, hips, cervical spinec. Lung disease: chronic pleuritis with effusions, inter-
stitial fibrosis
Rheumatoid factor:IgM autoantibody di-rected against Fcreceptor of IgG

•• Chapter 23 Musculoskeletal Disorders 307
• d. Hematologic disease: anemia of chronic disease
•(most common), Felty's syndrome (autoimmuneneutropenia and splenomegaly)
e. Carpal tunnel syndrome: entrapment of median
•nerve under transverse carpal ligament; numbness inthumb, second, third, and radial side of fourth finger
•f. Sjogren's syndrome: autoimmune destruction of lac-
rimal and minor salivary glands that may be associ-ated with rheumatoid arthritis
•(1) Clinical findings: keratoconjunctivitis (dry
eyes), xerostomia (dry mouth)
• (2) Laboratory findings: positive serum antinu-• clear antibody, anti-SS-A (Ro) and anti-SS-B (La)
antibodies
•g. Rheumatoid nodules (forearm), vasculitis, pericardi-
tis and aortitis, popliteal cyst (outpouching of jointspace)
•• Caplan's syndrome: rheumatoid nodules in the
lung plus pneumoconiosis
• E. Juvenile rheumatoid arthritis1. Occurs in children younger than 16 years of age; more
• common in girls; rheumatoid factor is usually absent.
•2. Still's disease: commonly presents as an "infectious
disease" with fever, rash, polyarthritis, generalizedlymphadenopathy, and neutrophilic leukocytosis
•3. Polyarticular: disabling arthritis predominates.4. Pauciarticular: arthritis in a few joints, uveitis with the
potential for blindnessF. Gouty arthritis
• 1. Multifactorial inheritance pattern; occurs more often inmen older than 30 years of age
2. Secondary causes: increased nucleated cell turnover (e.g.,
•leukemia), decreased renal excretion (e.g., lead poison-ing, alcoholism)
• 3. Recurrent acute arthritis: initial attack usually occurs in
•the first metatarsophalangeal joint (great toe; podagra)with fever and neutrophilic leukocytosis.
• 4. Chronic gout
•a. Tophi are produced.
(1) Deposits of monosodium urate in soft tissuearound joints
(2) Granulomatous reaction with multinucleated
• giant cellsb. Tophi destroy subjacent bone: erosive arthritis
5. Clinical conditions associated with gout: urate ne-• phropathy, renal stones, hypertension, coronary artery
disease, lead poisoning• 6. Laboratory findings: hyperuricemia is usually, but not
• G.
always, present; joint aspiration is confirmatory.
Chondrocalcinosis (pseudogout)
• 1. Pseudogout is a degenerative joint disease (usually of the
Gout: most casesdue to underexcre-tion of uric acid
••

308 Pathology
knee) with crystals producing linear deposits in articu-lar cartilage.
2. Crystals phagocytosed by neutrophils show positivebirefringence.
H. Seronegative spondyloarthropathies1. Characteristics: rheumatoid factor negative; usually
HLA-B27 positive; more common in men younger than40 years of age; sacroiliitis with or without peripheralarthritis
2. Types: ankylosing spondylitis; Reiter's syndrome; arthri-tis associated with inflammatory bowel disease (ulcerativecolitis, shigellosis); psoriasis
3. Ankylosing spondylitisa. Initially targets sacroiliac joint: bilateral sacroiliitis
with morning stiffnessb. Eventually targets vertebral column: fusion of ver-
tebrae ("bamboo spine") causes kyphoscoliosis.c. Aortitis with aortic regurgitation; uveitis (blurry
vision)4. Reiter's syndrome: urethritis (due to Chlamydia tra-
chomatis), arthritis, conjunctivitis (noninfectious)I. Septic arthritis
1. Staphylococcus aureus is the most common nongono-coccal cause of septic arthritis.
2. Neisseria gonorrhoeae causes disseminated gono-coccemia.a. Most common cause of septic arthritis in urban
populations; more common in young womenb. Clinical findings: septic arthritis (knee), tenosyno-
vitis (wrists and ankles), dermatitis (pustules onwrists or ankles)
3. Lyme diseasea. Caused by Borrelia burgdorferi (spirochete) and
transmitted by Ixodes tick; white-tailed deer is themost common animal reservoir for the tick.
b. Early disease: rash (erythema chronicum migrans)develops at tick bite site; red, expanding lesion withconcentric circles ("bull's eye").
c. Late disease: disabling arthritis (usually of theknee), bilateral Bell's palsy, myocarditis andpericarditis
d. Babesiosis: secondary infection transmitted byIxodes; often presents concurrently with Lyme disease(1) Intraerythrocytic protozoal disease due to
Babesia microti(2) Fever, headache, hemolytic anemia
e. Diagnosis of Lyme disease: serologic tests, culturebiopsy specimens, silver stains of synovial biopsyto identify spirochetes
4. Septic arthritis and tendinitis due to cat bite: causal or-ganism is Pasteurella multocida.
Reiter's syndrome:Achilles tendon peri-ostitis is a confir-matory radiologicsign.
Disseminatedgonococcemia: com-plement deficien-cies in C5–C9predispose todissemination.
Cause of Lymedisease: Borreliaburgdorferi, a gram-negative spirochete

Chapter 23 Musculoskeletal Disorders 309
III. Muscle DisordersA. Muscle fibers
1. Type I: slow-twitch (red) fibers rich in mitochondria andoxidative enzymes
2. Type II: fast-twitch (white) fibers poor in mitochondriaB. Etiology of muscle weakness: abnormalities in motor
neuron pathways (e.g., poliomyelitis), neuromuscular synapse(e.g., myasthenia gravis), muscle (e.g., muscular dystrophy)1. Neurogenic atrophy: motor neuron or its axon degener-
ates, leading to atrophy of both type I and type II fibers.2. Duchenne's muscular dystrophy
a. X-linked recessive; deficiency of dystrophin(which normally anchors actin to membrane glyco-protein); Becker's type has defective dystrophin.
b. Progressive degeneration of type I and type II fibers:fibrosis and infiltration of muscle tissue by fattytissue (pseudohypertrophy of calf muscles)
c. Clinical findings(1) Symptoms occur between 2 and 5 years
of age.(2) Weakness and wasting of pelvic muscles;
child places hands on the knees for help instanding (Gower's sign).
(3) Death usually occurs by 20 years of age.d. Laboratory findings: serum creatine kinase (CK)
increased at birth; progressively declines as muscledegenerates; female carriers have increased levelsof serum CK.
3. Myotonic dystrophya. Autosomal dominant: trinucleotide repeat disor-
der; selective atrophy of type I fibersb. Clinical findings
(1) Presents in adolescence as facial weakness(2) Myotonia: inability to relax muscles (sustained
grip)(3) Frontal balding, cataracts, testicular atrophy,
cardiac involvementc. Laboratory finding: increased serum CK
4. Myasthenia gravisa. Autoimmune disease with autoantibodies against
acetylcholine (ACh) receptorsb. More common in women 20-40 years of agec. Thymic hyperplasia with germinal follicles (site of
antibody synthesis) in 85% of patients; some pa-tients develop thymomas (usually benign) that arelocated in the anterior mediastinum.
d. Clinical findings: ptosis, diplopia, muscle weaknessthat improves with rest
e. Laboratory findings: antibodies against ACh recep-tors and striated muscle
Duchenne's mus-cular dystrophy:waddling gait due toweakness of pelvicmuscles
Most commonmuscular dystrophyin adults: myo-tonic dystrophy
Myasthenia gravis:formation of autoan-tibodies againstACh receptors is atype II hypersensitiv-ity reaction.
Embryonal rhabdo- Genitourinary tractmyosarcoma
Leiomyoma
Uterus, stomach
Leiomyosarcoma
Lipoma
Liposarcoma
Malignant fibroushistiocytoma
Rhabdomyoma
GI tract, uterus
Trunk, neck, proximalextremities
Thigh, retroperito-neum
Retroperitoneum,thigh
Heart, also tongueand vagina
310 Pathology
TABLE 23-2 Selected Soft Tissue Tumors
Tumor Type
Location
Comment
Grape-like mass protrudes from penisor vagina
Most commonly located in uterusMost common benign tumor in GI
tract
Most common sarcoma of GI tract anduterus
Arises in subcutaneous tissueMost common benign soft tissue
tumor
Lipoblasts identified with fat stainsMost common malignant soft tissue
tumor
Associated with radiation therapy andscarring
Most common sarcoma in childrenBenign heart tumor associated with
tuberous sclerosis
GI, gastrointestinal.
f. Tensilon (edrophonium) test facilitates diagnosis:inhibits acetylcholinesterase, which increases AChand reverses muscle weakness
IV. Soft Tissue DisordersA. Fibromatosis
• Fibromatosis is a non-neoplastic, proliferative connectivetissue disorder in which fibrous tissue infiltrates tissue(usually muscle).
1. Dupuytren's contracture: fibromatosis involving palmarfascia; causes contraction of fingers; associated withalcoholism
2. Desmoid tumor: fibromatosis of the anterior abdominalwall in women; associated with previous trauma andGardner's polyposis syndrome
B. Selected soft tissue tumors (Table 23-2)

244
Skin Disorders
I. Terminology (Table 24-1)
TABLE 24-1 Terms Used to Describe Skin Disorders
Term
Definition Example
Macroscopic
Macule
Papule
Nodule
Plaque
Vesicle
Bulla
Pustule
Wheal (hive)
Scales
Microscopic
Hyperkeratosis
Parakeratosis
Papillomatosis
Acantholysis
Pigmented or erythematous flat lesionon epidermis
Peaked or dome-shaped surface eleva-tion < 5 mm diameter
Elevated, deep, dome-shaped lesion> 5 mm diameter
Flattened, elevated area on epidermis> 5 mm diameter
Fluid-filled blister < 5 mm diameter
Fluid-filled blister > 5 mm diameter
Fluid-filled blister with inflammatorycells
Edematous, transient plaque caused byinfiltration of dermis by fluid
Excessive number of dead keratinocytesproduced by abnormal keratinization
Increased thickness of stratum corneumproduces scaly appearance of skin
Persistence of nuclei in stratumcorneum layer
Spire-like projections from surface ofskin or downward into papillarydermis
Tinea versicolor
Acne vulgaris
Basal cell carcinoma
Psoriasis
Varicella (chickenpox)
Bullous pemphigoid
Impetigo
Urticaria
Seborrheic dermatitis
Psoriasis
Psoriasis
Verruca vulgaris
Loss of cohesion between keratinocytes Pemphigus vulgaris
311
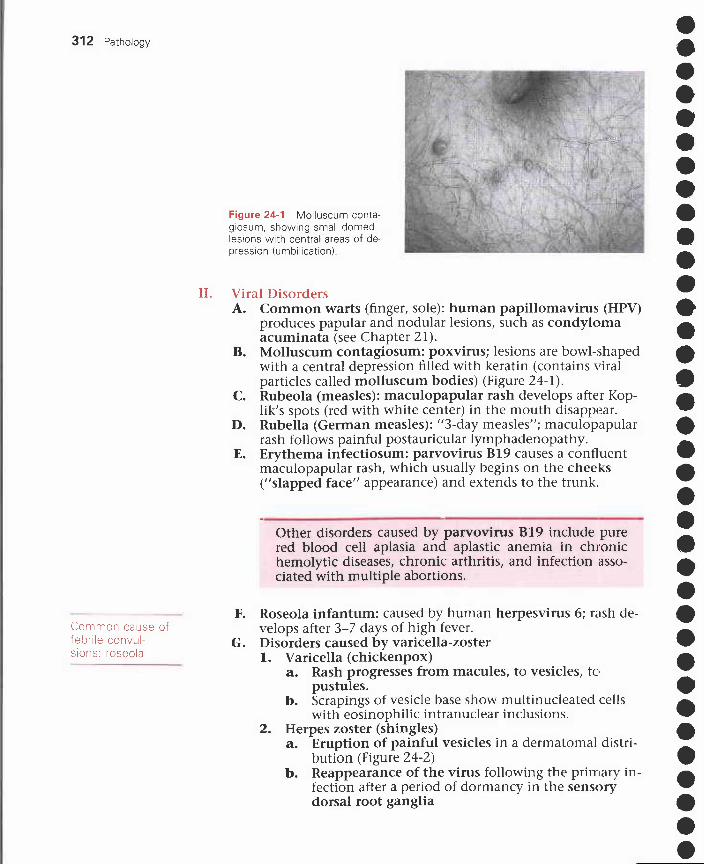
312 Pathology
Figure 24-1 Molluscum conta-giosum, showing small domedlesions with central areas of de-pression (umbilication).
II. Viral DisordersA. Common warts (finger, sole): human papillomavirus (HPV)
produces papular and nodular lesions, such as condylomaacuminata (see Chapter 21).
B. Molluscum contagiosum: poxvirus; lesions are bowl-shapedwith a central depression filled with keratin (contains viralparticles called molluscum bodies) (Figure 24-1).
C. Rubeola (measles): maculopapular rash develops after Kop-lik's spots (red with white center) in the mouth disappear.
D. Rubella (German measles): "3-day measles"; maculopapularrash follows painful postauricular lymphadenopathy.
E. Erythema infectiosum: parvovirus B19 causes a confluentmaculopapular rash, which usually begins on the cheeks("slapped face" appearance) and extends to the trunk.
Other disorders caused by parvovirus B19 include purered blood cell aplasia and aplastic anemia in chronichemolytic diseases, chronic arthritis, and infection asso-ciated with multiple abortions.
F. Roseola infantum: caused by human herpesvirus 6; rash de-velops after 3-7 days of high fever.
G. Disorders caused by varicella-zoster1. Varicella (chickenpox)
a. Rash progresses from macules, to vesicles, topustules.
b. Scrapings of vesicle base show multinucleated cellswith eosinophilic intranuclear inclusions.
2. Herpes zoster (shingles)a. Eruption of painful vesicles in a dermatomal distri-
bution (Figure 24-2)b. Reappearance of the virus following the primary in-
fection after a period of dormancy in the sensorydorsal root ganglia
Common cause offebrile convul-sions: roseola
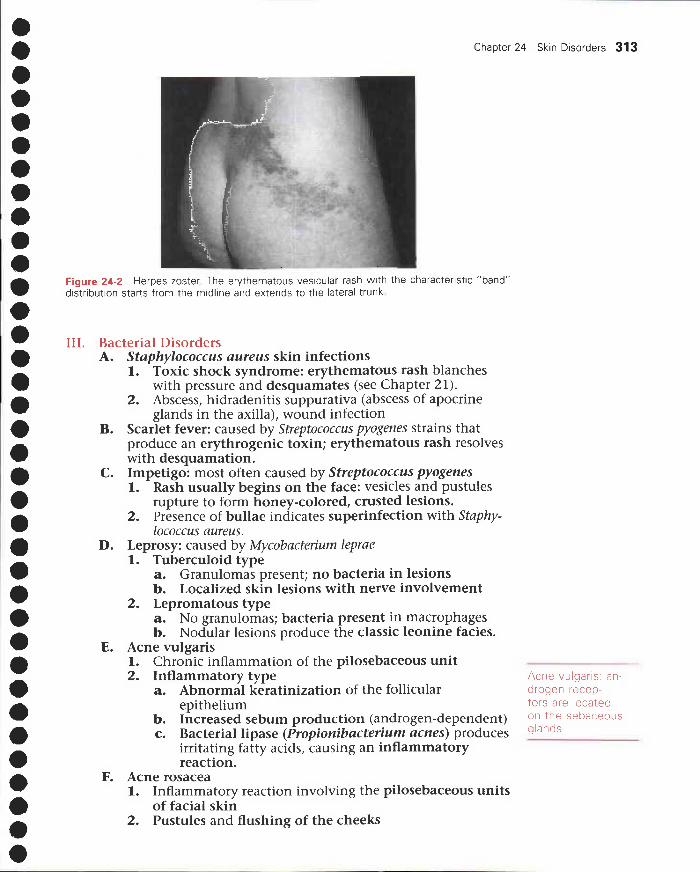
Chapter 24 Skin Disorders 313
Figure 24 -2 Herpes zoster The erythematous vesicular rash with the characteristic "band"distribution starts from the midline and extends to the lateral trunk
III. Bacterial DisordersA. Staphylococcus aureus skin infections
1. Toxic shock syndrome: erythematous rash blancheswith pressure and desquamates (see Chapter 21).
2. Abscess, hidradenitis suppurativa (abscess of apocrineglands in the axilla), wound infection
B. Scarlet fever: caused by Streptococcus pyogenes strains thatproduce an erythrogenic toxin; erythematous rash resolveswith desquamation.
C. Impetigo: most often caused by Streptococcus pyogenes1. Rash usually begins on the face: vesicles and pustules
rupture to form honey-colored, crusted lesions.2. Presence of bullae indicates superinfection with Staphy-
lococcus aureus.D. Leprosy: caused by Mycobacterium leprae
1. Tuberculoid typea. Granulomas present; no bacteria in lesionsb. Localized skin lesions with nerve involvement
2. Lepromatous typea. No granulomas; bacteria present in macrophagesb. Nodular lesions produce the classic leonine facies.
E. Acne vulgaris1. Chronic inflammation of the pilosebaceous unit2. Inflammatory type
a. Abnormal keratinization of the follicularepithelium
b. Increased sebum production (androgen -dependent)c. Bacterial lipase (Propionibacterium acnes) produces
irritating fatty acids, causing an inflammatoryreaction.
F. Acne rosacea1. Inflammatory reaction involving the pilosebaceous units
of facial skin2. Pustules and flushing of the cheeks
••••••••••••••••••••••••••••••••••
Acne vulgaris: an-drogen recep-tors are locatedon the sebaceousglands

314 Pathology
Figure 24 -3 Tnea corporis. Theannular lesions have erythema-tous margins and clear centers.
IV. Fungal DisordersA. Superficial mycoses (dermatophytoses): skin diseases that
are confined to the stratum corneum or its adnexalstructures
Wood's lamp and potassium hydroxide (KOH)–treatedskin scrapings from lesions are commonly used for diag-nosis of the dermatophytoses. Wood's lamp (ultravioletA light) detects fluorescent metabolites produced by or-ganisms (e.g., fungi, some bacteria). KOH preparationsidentify yeasts and hyphae in the stratum corneum.
1. Tinea capitis: circular or ring-shaped patches (ringworm)of alopecia (hair loss) with erythema and scaling aremost often caused by Trichophyton tonsurans.
2. Tinea corporis (body surface), tinea pedis (foot; "ath-lete's foot"), and tinea cruris (groin; "jock itch") are mostoften caused by T. rubrum (Figure 24-3).
3. Tinea versicolora. Caused by Malassezia furfur: KOH shows "spa-
ghetti" (hyphae) and "meatball" (yeasts) fungalmorphology.
b. Affected skin is hyperpigmented and hypo-pigmented.
4. Cutaneous infections caused by Candida albicans: in-tertrigo (erythematous rash in body folds, such as diaperrash), onychomycosis (nail infection)
5. Seborrheic dermatitis: scaly, often greasy dermatitis onthe scalp (dandruff), eyebrows, and nasal creases;caused by M. furfur
M. furfur. producesacid that inhibitsmelanin transfer tokeratinocytes

Chapter 24 Skin Disorders 315
B. Sporotrichosis: caused by Sporothrix schenckii1. Traumatic implantation of fungus (e.g., thorn punc-
ture): associated with rose gardening or sphagnum mosspacking material
2. Lymphocutaneous disease: chain of suppurating subcu-taneous nodules
V. Benign Noninfectious DisordersA. Ichthyosis vulgaris
1. Autosomal dominant disorder: defect in keratinizationcauses increased thickness of the stratum corneum.
2. Hyperkeratotic, dry skin; involves palms, soles, and ex-tensor areas
B. Eczema1. Group of inflammatory derinatoses characterized by
pruritus2. Acute eczema: weeping, erythematous rash with
vesicles3. Chronic eczema: dry, thickened skin (hyperkeratosis)
caused by continual scratching4. Atopic dermatitis
a. Children: dry skin; eczema on cheeks and extensorand flexural surfaces
b. Adults: dry skin; eczema on hands, eyelids, elbows,and knees
5. Contact dermatitis: inflammatory disorder associatedwith exposure to various antigens and irritatingsubstancesa. Allergic contact dermatitis: type IV hypersensitivity
(e.g., poison ivy, nickel in jewelry)b. Contact photodermatitis: ultraviolet (UV) light
reacts with drugs that have a photosensitizing effect(e.g., tetracycline).
C. Autoimmune skin disorders1. Systemic lupus erythematosus (see Chapter 3)
a. DNA—anti-DNA immunocomplex deposition in thebasement membrane(1) Degeneration of basal cells and hair shafts
(alopecia)(2) Positive band of immunofluorescence test
shows immunocomplexes along the basementmembrane.
b. Causes epidermal atrophyc. Clinical findings
(1) Erythematous maculopapular eruption, usuallyover the malar eminences and the bridge ofthe nose ("butterfly" rash)
(2) Skin lesions are exacerbated by UV light.2. Pemphigus vulgaris
a. IgG antibodies are directed against intercellular at-tachment sites between keratinocytes.
Most common in-herited skin dis-order: ichthyosisvulgaris
Atopic dermatitis:type I IgE-mediateddisease

316 Pathology
Figure 24-4 Actinic (solar)keratosis.
b. Vesicles and bullae develop on skin and oralmucosa.
c. Intraepithelial vesicles are located above the basallayer (suprabasal).(1) Basal cells resemble a row of tombstones.(2) Acantholysis of keratinocytes in the vesicle
fluid3. Bullous pemphigoid
a. IgG antibodies are directed against the basementmembrane.
b. Vesicles are subepidermal: no acantholytic cells;develop on skin and oral mucosa.
4. Dermatitis herpetiformisa. IgA—anti-IgA immunocomplex deposition at the
tips of the dermal papillae: subepidermal vesicleswith neutrophils
b. Strongly correlated with celiac disease: increase inantireticulin and endomysial antibodies
D. Premalignant skin disorders1. Actinic (solar) keratosis: associated with UV light
exposurea. Precursor (squamous dysplasia) of squamous cell
carcinoma: occurs in 2-5% of casesb. Hyperkeratotic, pearly gray-white appearance: seen
on the face, back of the neck, and dorsum of handsand forearms (Figure 24-4)
2. Lichen planus: may be associated with hepatitis Ca. Intensely pruritic, scaly, violaceous, flat-topped
papules: usually located on the wristsb. Oral mucosa often involved: produces a fine, white,
net-like lesion (Wickham's striae); minimal risk ofdeveloping squamous cell carcinoma
E. Psoriasis1. Squamous hyperplasia: unregulated proliferation of
keratinocytes; genetic factors and infection (e.g., strepto-coccal pharyngitis) are implicated.
In actinic keratosis,lesions recur afterbeing scraped off.
Pemphigus vulgarisand bullous pem-phigoid: type II hy-persensitivityreactions
•••••••••••••••••••••••••••••••••••

Chapter 24 Skin Disorders 317
Figure 24-5 Psoriasis.
2. Well-demarcated, flat, elevated salmon-colored plaques(Figure 24-5)a. Covered by adherent white to silver-colored scales;
pinpoint areas of bleeding occur when scales arescraped off (Auspitz sign).
b. Rash develops in areas of trauma (Koebnerphenomenon).
3. Sites: scalp; pressure areas, such as the elbows and lowerback
4. Pitting of the nails5. Microscopic findings: hyperkeratosis and parakeratosis,
elongation of rete pegs (downward extensions of basallayer), neutrophil collections in the stratum corneum
F. Pityriasis rosea1. Initially presents as a single, oval-shaped plaque on the
trunk ("herald patch")2. Days or weeks later, a papular eruption develops on the
trunk that follows the lines of cleavage ("Christmastree" distribution).
G. Erythema multiforme1. Hypersensitivity reaction against infectious organisms
such as Mycoplasma pneumoniae and drugs (e.g., sulfona-mides)
2. Vesicles and bullae have a "bulls-eye" appearance:located on the palms, soles, and extensor surfaces
3. Stevens-Johnson syndrome: involves the skin andmucous membranes
H. Erythema nodosum1. Raised, erythematous, painful nodules, usually located
on the anterior portion of the shins2. May accompany coccidioidomycosis, histoplasmosis,
streptococcal infection, sarcoidosis, or tuberculosis
Most common in-flammatory lesion ofsubcutaneous fat:erythema nodosum

318 Pathology
I. Porphyria cutanea tarda1. Genetic or acquired disease involving porphyrin metab-
olism; deficiency of uroporphyrinogen decarboxylasea. Urine is wine-red color on voiding; uroporphyrin I
increased in urineb. Associated with hepatitis C and excessive alcohol
intake2. Clinical findings
a. Photosensitive bullous skin lesions: caused by por-phyrin metabolites deposited in the skin
b. Hyperpigmentation, fragile skin, increased amountsof lanugo (fine, downy hair)
J. Urticaria1. Pruritic elevations of the skin caused by mast cell
release of histamine2. Etiology
a. IgE-mediated reactions: associated with certainfoods (e.g., peanuts), insect bites (e.g., fire ant), ordrugs (e.g., penicillin, morphine)
b. Immunocomplex-mediated reactions (e.g., serumsickness prodrome in hepatitis B)
3. Clinical findingsa. Dermatographism: urticaria develops in areas of me-
chanical pressure on skin.b. Angioedema: diffuse subcutaneous swelling (e.g., C1
esterase inhibitor deficiency)K. Acanthosis nigricans: verrucoid, pigmented skin lesion com-
monly present in the axilla; phenotypic marker for stomachadenocarcinoma
Albinism: defi-ciency of tyrosinase;absent melanin inmelanocytes
VI. Benign Melanocytic DisordersA. Solar lentigo
1. Brown, raised lesions on sun-exposed areas ("liverspots"); caused by UV light
2. Increased melanocytes; not precancerousB. Vitiligo
1. Autoimmune destruction of melanocytes causing skindepigmentation
2. Common in African Americans3. Often associated with other autoimmune conditions
(e.g., Hashimoto's thyroiditis)C. Chloasma
1. Flat, hyperpigmented lesions on forehead and cheeks2. Occurs in females taking oral contraceptives or who are
pregnant ("pregnancy mask")
VII. Neoplastic Skin DisordersA. Seborrheic keratosis
1. Benign pigmented epidermal tumor: coin-like, macularto raised lesion with "stuck-on" appearance(Figure 24-6)

Chapter 24 Skin Disorders 319
Figure 24-6 Seborrheic kerato-sis on the breast
2. Occurs in individuals older than 50 years of age3. Leser-Trelat sign: rapid increase in number of keratoses;
phenotypic marker for stomach adenocarcinomaB. Keratoacanthoma
1. Rapidly growing, crateriform benign tumor with acentral keratin plug
2. More common in men; develops in sun-exposed areas ofthe body
3. Mimics a well-differentiated squamous cell cancer: re-gresses spontaneously with scarring
C. Nevocellular nevus: neoplastic melanocytic disorder1. Benign tumor of neural crest—derived nevus cells:
nevus cells are modified melanocytes.2. Begins in early childhood as junctional nevus: flat, pig-
mented lesion with nests of nevus cells along the basalcell layer
3. Junctional nevi develop into compound nevus: usuallyoccurs in children and adolescents.a. Nevus cells extend into superficial dermis: both
junctional and intradermal componentsb. Raised, pigmented, verruca-like lesions
4. Intradermal nevus: compound nevus loses its junctionalcomponent. Most common
5. Dysplastic nevus: predisposes to malignant melanoma nevus in adults: in-
a. Arises sporadically or in association with dysplastic tradermal nevus
nevus syndrome (autosomal dominant; > 100 nevion skin)
b. Characteristics: > 5 mm diameter, irregular borders,irregular distribution of melanin
D. Basal cell carcinoma Most common ma-1. Caused by chronic exposure to UV light lignant skin tumor:
2. Raised papule or nodule with a central crater: sides of basal cell carcinoma
lesion surfaced by telangiectatic vessels3. Occurs in sun-exposed areas of the body: inner canthus
of the eye, upper lip4. Locally aggressive, infiltrating cancer that does not
metastasize

320 Pathology
a. Arises from basal cell layer of the epidermisb. Cords of basophilic-staining basal cells infiltrate the
underlying dermis.E. Squamous cell carcinoma
1. Risk factorsa. Excessive exposure to UV light (most common)b. Actinic keratosis, arsenic exposure, third-degree burn,
orifice of draining sinus tract, immunosuppressivetherapy (most common cancer complicatingimmunosuppression)
2. Scaly to nodular lesions: nodules often ulcerated; mostoften occur in sun-exposed areas of the body (e.g.,ears, lower lip, dorsum of the hands)
3. Usually well differentiated: minimal risk of metastasisF. Malignant melanoma: malignant tumor of melanocyl es
1. Most rapidly increasing cancer worldwide: morecommon in whites than in African Americans (except foracral lentiginous melanoma)
2. Risk factorsa. Exposure to excessive sunlight at an early age:
single most important risk factorb. Dysplastic nevus syndrome, melanoma in first- or
second-degree relative, xeroderma pigmentosum3. Radial growth phase: initial phase of invasion
a. Melanocytes proliferate laterally within the epider-mis, along the dermoepidermal junction, orwithin the papillary dermis.
b. No metastasis4. Vertical growth phase: final phase of invasion
a. Malignant cells penetrate the underlying reticulardermis.
b. Potential for metastasis5. Types
a. Superficial spreading melanoma: develops onlower extremities and back
b. Lentigo maligna melanoma: extension of lentigomaligna (intraepidermal lesion) into the derrnis;occurs in elderly individuals on the parts of the facemost exposed to the sun
c. Nodular melanoma: directly invades the dermis;poor prognosis
d. Acral lentiginous melanoma: located on the palm,sole, or beneath the nail; may occur in AfricanAmericans; poor prognosis
6. Depth of invasion best determines biologic behavior.a. Lesions < 0.76 mm invasion do not metastasize.b. Lesions > 1.7 mm invasion have the potential for
lymph node metastasis.7. Prevention: sunscreen > 15 SPF
Signs of mela-noma: Asymmetry;Borders irregular;Color changes; Di-ameter increased
Most common typeof malignant mela-noma: superficialspreading mela-noma

Nervous SystemDisorders
I. Cerebral Edema, Herniation, and HydrocephalusA. Cerebral edema (intracranial hypertension)
1. Intracellular: causes include tissue hypoxia andhyponatremia.
2. Extracellular: increased vessel permeability; causesinclude inflammation, metastasis, trauma, and respira-tory acidosis.
A patient with head trauma is purposely hyperven-tilated to produce respiratory alkalosis, whichcauses cerebral vessel constriction. This decreasesthe risk of increased vessel permeability and cere-bral edema. Respiratory acidosis causes increasedcerebral vessel permeability, hence enhancing cere-bral edema.
3. Clinical findingsa. Papilledema (swelling of the optic disk)b. Headache, projectile vomiting, sinus bradycardia,
hypertensionc. Potential for herniation
B. Cerebral herniation• Portions of the brain under increased pressure are dis-
placed through openings of dural partitions or into open-ings of the skull (e.g., foramen magnum).
1. Subfalcine herniation: the cingulate gyrus herniatesunder the falx cerebri, causing compression of the an-terior cerebral artery.
Papilledema: sign ofcerebral edema
321

322 Pathology
2. Uncal herniation: the medial portion of the temporallobe herniates through the tentorium cerebelli. Compli-cations include:a. Compression of midbrain, which produces
Duret's hemorrhagesb. Oculomotor nerve palsy
(1) Eye is deviated down and out.(2) Pupil is mydriatic (dilated).
c. Compression of posterior cerebral artery, whichcauses hemorrhagic infarction of the occipital lobe
3. Tonsillar herniation: cerebellar tonsils herniate intothe foramen magnum, causing cardiorespiratory arrest.
C. Hydrocephalus• Increase in the volume of cerebrospinal fluid (CSF) causes
enlargement of the ventricles.1. CSF is produced in the ventricles and then enters the
subarachnoid space.2. Communicating (nonobstructive) hydrocephalus is
an open communication of CSF between the ven-tricles and subarachnoid space caused by:a. Increased CSF production (e.g., choroid plexus
papilloma)b. Obstruction in the reabsorption of CSF by the
arachnoid granulations (e.g., by postmeningealscarring)
3. Noncommunicating (obstructive) hydrocephalusis an obstruction of CSF flow out of the ventriclescaused by:a. Stricture of the aqueduct of Sylvius: paralysis of
upward gaze (Parinaud's syndrome)b. Tumor in the fourth ventricle (e.g., ependymoma)c. Scarring at the base of the brain (e.g., tuberculous
meningitis), colloid cyst in the third ventricle4. Clinical findings
a. Newborns: ventricles dilate and enlarge the headcircumference.
b. Adults: progressive dementia, gait disturbance,urinary incontinence
5. Hydrocephalus ex vacuo: dilated appearance of theventricles when the brain mass is decreased (e.g., Alz-heimer's disease)
II. Developmental DisordersA. Neural tube defects
1. Caused by failure of fusion of the lateral folds of theneural plate or by rupture of a previously closed neuraltube
2. Associated with increase in maternal a-fetoprotein inserum or amniotic fluid
3. Anencephaly: complete absence of brain; frog-like ap-pearance; maternal polyhydramnios
Protection againstneural tube defectsfolate level mustbe adequate beforepregnancy
Most commoncause of hydroceph-alus in newborns:blockage of aque-duct of Sylvius
Uncal herniation:midbrain hemor-rhage, oculomotornerve palsy,mydriasis
•••••••••••••••S•••••••••••••••••••

Chapter 25 Nervous System Disorders 323
4. Spina bifida occulta: defect in closure of posterior ver-tebral arch; dimple or tuft of hair in the skin overly-ing LS-S1
5. Meningocele: cystic mass containing spinal meninges;most common in lumbosacral region
6. Meningomyelocele: cystic mass containing spinal me-ninges and spinal cord; most common in lumbosa-cral region
B. Arnold-Chiari malformation1. Caudal extension of the medulla and cerebellar
vermis through the foramen magnum2. Noncommunicating hydrocephalus3. Platybasia (flattening of base of skull), meningomyelo-
cele, syringomyeliaC. Dandy-Walker malformation
1. 1-lypoplasia of the cerebellar vermis2. Cystic dilation of the fourth ventricle3. Noncommunicating hydrocephalus
D. Syringomyelia1. Fluid-filled cavity (syrinx) within the cervical spinal
cord produces cervical cord enlargement; cavityexpands and causes degeneration of spinal tracts.
2. Associated with Arnold-Chiari malformation3. Clinical findings
a. Disruption of the crossed lateral spinothalamictracts: loss of pain and temperature sensation inhands (patient can burn hands without being awareof the burn)
b. Destruction of anterior horn cells: atrophy of in-trinsic muscles of the hands; may be confusedwith amyotrophic lateral sclerosis (ALS)
E. Phakomatoses• Neurocutaneous syndromes characterized by disordered
growth of ectodermal tissue associated with malforma-tions or tumors of the central nervous system (CNS)
1. 'Tuberous sclerosis: autosomal dominant disordera. Mental retardation and seizures beginning in
infancyb. Adenoma sebaceum (angiofibromas on the face),
hypopigmented lesions ("ash leaf" lesions)c. Astrocyte proliferations (hamartomas) in sub-
ependymal portions of the brain ("candlestick drip-pings" in the ventricles)
d. Rhabdomyoma in the heart, angiomyolipomas(hamartomas) in the kidneys
2. Neurofibromatosis: autosomal dominant disordera. Cafe"-au-lait macules, pigmented neurofibromasb. Lisch nodules: hamartomas in irisc. Optic nerve glioma, meningioma, acoustic
neuroma
Syringomyelia:pain and tempera-ture sensation inhands

11
Epidural hematoma:temporoparietalskull fracture andtear of middle men-ingeal artery
Subdural hema-toma: tear of bridg-ing veins produc-ing venous bloodclot
324 Pathology
Figure 25-1 Subdural hema-toma. The reflected dura showsthe outer membrane of an or-ganized venous clot covering theconvexity of the brain.
d. Pheochromocytoma, kyphoscoliosis, Wilms'tumor, neurofibrosarcoma (usually involving largenerve trunks)
3. Sturge-Weber syndrome: autosomal dominant disor-der; port-wine nevus on the face and ipsilateral arterio-venous malformation in the meninges
III. Head TraumaA. Cerebral contusion
1. Permanent damage to small blood vessels and thesurface of the brain
2. Most often is secondary to an acceleration-deceleration injury
3. Coup injuries occur at the site of impact.4. Contrecoup injuries occur on the opposite side of the
brain from the site of impact, usually at the tips of thefrontal and temporal lobes.
B. Acute epidural hematoma1. Arterial bleeding creates a blood-filled space between
the bone and dura.2. Caused by a fracture of the temporoparietal bone
with severance of the middle meningeal artery, whichlies between the dura and inner table of bone
3. Intracranial pressure increases, leading to herniationand death unless the clot is removed.
C. Subdural hematoma1. Venous bleeding between the dura and arachnoid
membranesa. Most often the result of blunt traumab. Bleeding is due to tearing of bridging veins
between the brain and dural sinuses (Figure 25-1).c. Slowly enlarging blood clot covers the convexity
of the brain.2. Usually occurs in the elderly or in chronic alcoholism
where there is brain atrophy3. Fluctuating levels of consciousness; herniation and
death may occur.

Chapter 25 Nervous System Disorders 325
IV. CNS Vascular DisordersA. Global hypoxic injury to the brain
1. Causesa. Most commonly due to ischemia secondary to
atherosclerosis of the carotid arteryb. Carbon monoxide causes necrosis of globus palli-
dus; chronic hypoxemia (e.g., chronic lung disease).
Repeated episodes of hypoglycemia have thesame effects on the brain as does hypoxic in-jury. Hypoglycemia most commonly occurs intype 1 diabetes mellitus.
2. Complicationsa. Cerebral atrophy: due to apoptosis of neurons in
layers 3, 5, and 6 of the cerebral cortex (laminarnecrosis); neurons are most susceptible to hypoxia.
b. Watershed infarcts occur at the junctions of arte-rial territories (e.g., border between the anterior andmiddle cerebral arteries).
c. Cerebrovascular accident (e.g., stroke)B. Cerebrovascular accidents
1. Atherosclerotic (thrombotic) stroke: most commontypea. Usually pale infarcts (liquefactive necrosis)
(1) Platelet thrombus develops over an inflamma-tory plaque in the middle cerebral artery orin the internal carotid artery near thebifurcation.
(2) Reperfusion usually does not occur, and theinfarct remains pale.
b. Most occur in the distribution of the middle cere-bral artery.
c. Gross and microscopic findings(1) Wedge-shaped area of infarction develops at
the periphery of the cerebral cortex.(2) Swelling of the brain with loss of demarcation
between gray and white matter; myelin beginsto break down.
(3) Cystic area develops after 10 days to 3 weeks.(4) Gliosis: astrocytes proliferate at the margins
of the infarct; microglial cells (macrophages)remove lipid debris.
d. Clinical findings(1) Most atherosclerotic strokes are preceded by
transient ischemic attacks; transient neuro-logic deficits last less than 24 hours.
(2) Deficits that do not resolve within 24 hoursare called strokes.
Atheroscleroticstroke: pale infarc-tion extending to pe-riphery of cerebralcortex

326 Pathology
Figure 25-2 Embolic strokeThe arrow points to a hemor-rhagic infarction along the periph-ery of the cerebral cortex
2. Embolic strokea. Emboli most often originate from the heart or from
atherosclerotic plaques in proximally locatedarteries.
b. A hemorrhagic infarction usually occurs in thedistribution of the middle cerebral artery: hemor-rhage is due to reperfusion of the vessel after lysisof the embolic material (Figure 25-2).
3. Intracerebral hemorrhagea. Most often secondary to vascular changes related
to hypertension(1) Stress on penetrating branches of the lenticu-
lostriate vessels produces Charcot-Bouchardmacroaneurysms.
(2) Rupture of aneurysms results in intracerebralhemorrhage; the clot pushes the brain paren-chyma aside (Figure 25-3).
(3) Treatment of hypertension reduces the inci-dence of stroke by more than 40%.
b. Sites: basal ganglia (35-50% in the putamen), thal-amus (10%), pons and cerebellar hemispheres(10%)
4. Subarachnoid hemorrhagea. Most (800/o) are secondary to rupture of a congeni-
tal berry aneurysm; bleeding from an arteriove-nous malformation is a less common cause.
b. Congenital berry aneurysm(1) May develop from normal hemodynamic
stress or hypertension
Embolic stroke:hemorrhagic infarc-tion extending to pe-riphery of cerebralcortex
Intracerebral hem-orrhage: compli-cation of hyper-tension
Subarachnoid hem-orrhage: ruptureof congenital berryaneurysm

Chapter 25 Nervous System Disorders 327
Figure 25-3 Intracerebral hem.orrhage, showing a large bloodclot within the basal ganglia areaof the brain
(2) Rupture releases blood into the subarach-noid space, covering the surface of the brainwith blood.
c. Clinical findings(1) Sudden onset of severe occipital headache:
described as "worst headache ever"(2) About 50% of patients die soon after the hem-
orrhage or develop complications (e.g., subse-quent hemorrhage, hydrocephalus, neuro-logic deficit).
5. Lacunar infarctsa. Cystic areas of microinfarction < 1 cm in diameterb. Caused by hyaline arteriolosclerosis secondary to
either hypertension (most common) or diabe-tes mellitus
c. Produce pure motor strokes with or without dys-arthria if the posterior limb of the internal capsuleis involved
d. Produce pure sensory strokes if the thalamus isinvolved
V. CNS InfectionsA. Pathogenesis: hematogenous spread (most common),
traumatic implantation, local extension from nearby infec-tion, ascent of peripheral nerve
B. Meningitis1. Inflammation of the pia mater covering the brain2. Usually due to hematogenous spread3. Symptoms include nuchal rigidity and headache.4. Viral meningitis: increase in CSF protein and total CSF
leukocyte count (primarily lymphocytes), normal CSFglucose
5. Bacterial and fungal meningitis: increase in CSFprotein and total CSF leukocyte count (primarily neu-trophils and mononuclear cells), decreased CSF glucose
C. Encephalitis1. Inflammation of the brain
Meningitis: T CSFprotein (viral, bacte-rial, fungal); L CSFglucose (bacterial,fungal)
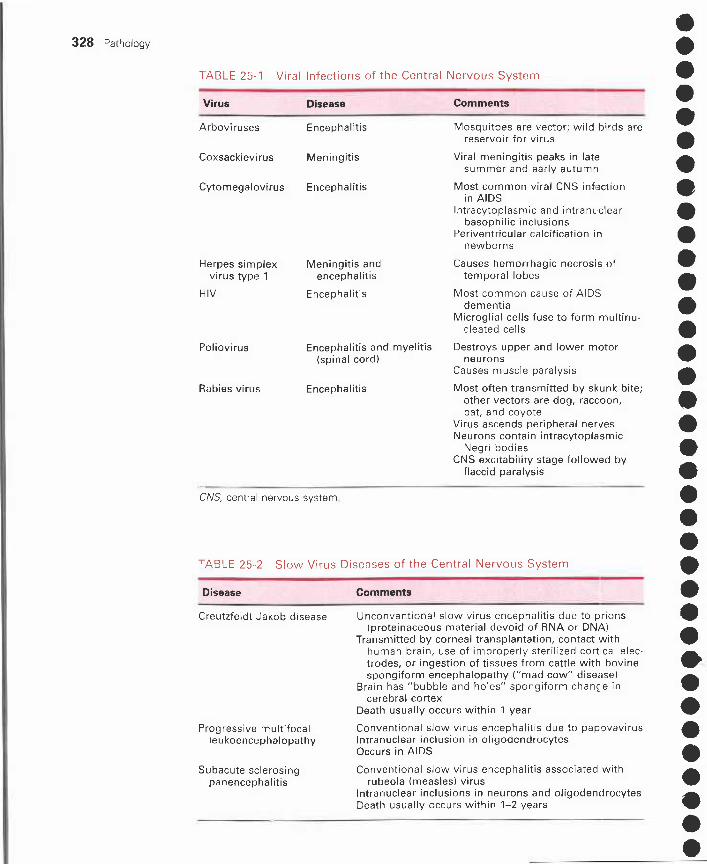
328 Pathology
TABLE 25-1 Viral Infections of the Central Nervous System
Virus Disease Comments
Arboviruses Encephalitis
Coxsackievirus Meningitis
Cytomegalovirus Encephalitis
Mosquitoes are vector; wild birds arereservoir for virus
Viral meningitis peaks in latesummer and early autumn
Most common viral CNS infectionin AIDS
Intracytoplasmic and intranuclearbasophilic inclusions
Periventricular calcification innewborns
Herpes simplex Meningitis and Causes hemorrhagic necrosis ofvirus type 1 encephalitis temporal lobes
HIV Encephalitis Most common cause of AIDSdementia
Microglial cells fuse to form multinu-cleated cells
Poliovirus Encephalitis and myelitis Destroys upper and lower motor(spinal cord) neurons
Causes muscle paralysis
Rabies virus Encephalitis Most often transmitted by skunk bite;other vectors are dog, raccoon,bat, and coyote
Virus ascends peripheral nervesNeurons contain intracytoplasmic
Negri bodiesCNS excitability stage followed by
flaccid paralysis
CNS, central nervous system
TABLE 25-2 Slow Virus Diseases of the Central Nervous System
Disease Comments
Creutzfeldt Jakob disease Unconventional slow virus encephalitis due to prions
(proteinaceous material devoid of RNA or DNA)Transmitted by corneal transplantation, contact with
human brain, use of improperly sterilized cortical elec-trodes, or ingestion of tissues from cattle with bovinespongiform encephalopathy ("mad cow" disease)
Brain has "bubble and holes" spongiform change incerebral cortex
Death usually occurs within 1 year
Progressive multifocal Conventional slow virus encephalitis due to papovavirusleukoencephalopathy Intranuclear inclusion in oligodendrocytes
Occurs in AIDS
Subacute sclerosing Conventional slow virus encephalitis associated withpanencephalitis rubeola (measles) virus
Intranuclear inclusions in neurons and oligodendrocytesDeath usually occurs within 1-2 years
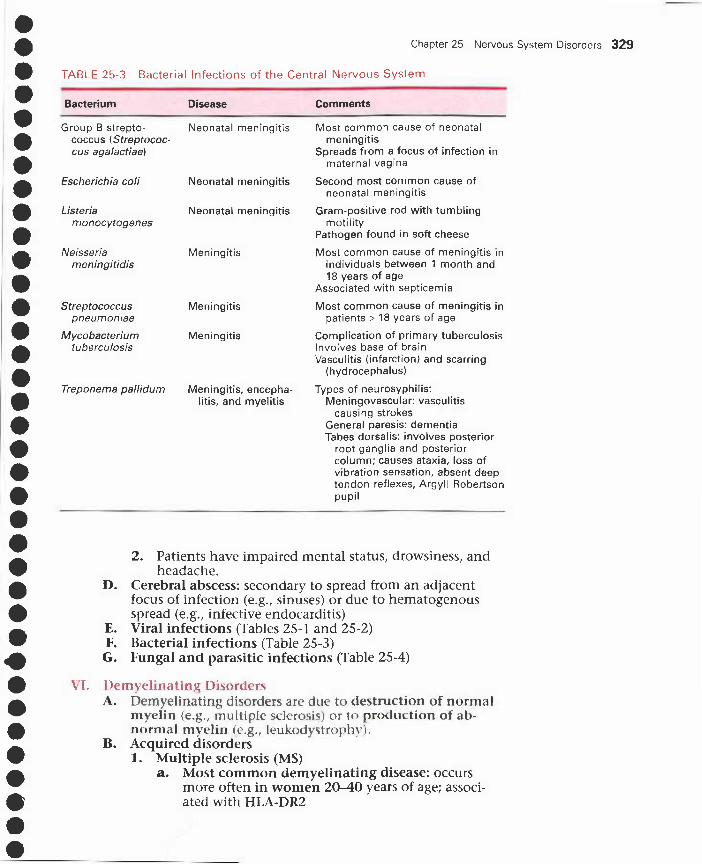
•
•
Chapter 25 Nervous System Disorders 329
• TABLE 25-3 Bacterial Infections of the Central Nervous System
•
Bacterium
Disease
Comments
Group B strepto- Neonatal meningitis Most common cause of neonatal
coccus (Streptococ- meningitiscus agalactiae)
Spreads from a focus of infection inmaternal vagina
Escherichia coli
Neonatal meningitis
Second most common cause ofneonatal meningitis
Listeria
Neonatal meningitis Gram-positive rod with tumbling
monocytogenes motilityPathogen found in soft cheese
Neisseria
Meningitis Most common cause of meningitis in
meningitidis
individuals between 1 month and18 years of age
Associated with septicemia
Streptococcus
Meningitis Most common cause of meningitis in
pneumoniae patients > 18 years of age
Mycobacterium
Meningitis
Complication of primary tuberculosistuberculosis
Involves base of brainVasculitis (infarction) and scarring
(hydrocephalus)
Treponema pallidum
Meningitis, encepha- Types of neurosyphilis:litis, and myelitis
Meningovascular: vasculitiscausing strokes
General paresis: dementiaTabes dorsalis: involves posterior
root ganglia and posteriorcolumn; causes ataxia, loss ofvibration sensation, absent deeptendon reflexes, Argyll Robertsonpupil
2. Patients have impaired mental status, drowsiness, andheadache.
D. Cerebral abscess: secondary to spread from an adjacentfocus of infection (e.g., sinuses) or due to hematogenousspread (e.g., infective endocarditis)
E. Viral infections (Tables 25-1 and 25-2)F. Bacterial infections (Table 25-3)G. Fungal and parasitic infections (Table 25-4)
Demyelinating DisordersA. Demyelinating disorders are due to destruction of normal
myelin (e.g., multiple sclerosis) or to production of ab-normal myelin (e.g., leukodystrophy).
B. Acquired disorders1. Multiple sclerosis (MS)
a. Most common demyelinating disease: occursmore often in women 20-40 years of age; associ-ated with HLA-DR2
•••••••••••••••••••••I.•••••S••

Cryptococcusneoformans
Mucor species
Naegleria fowleri
Taenia solium
Meningitis andencephalitis
Frontal lobe abscess
Meningoencephalitis
Cysticercosis
Toxoplasma gondii
Encephalitis
330 Pathology
TABLE 25-4 Fungal and Parasitic Infections of the Central Nervous System
Fungus/Parasite
Disease
Comments
Occurs in immunocompromised hostBudding yeasts visible with India ink
Occurs in diabetic ketoacidosis
Involves frontal lobesContracted by swimming in fresh-
water lakes
Patient (intermediate host) ingestsfood or water containing eggs;eggs develop into larval forms(cysticerci) that invade brain,producing calcified cysts causingseizures
Most common CNS space-occupyinglesion in AIDS
Congenital toxoplasmosis producesbasal ganglia calcification
CNS, central nervous system
b. Pathogenesis(1) Autoimmune disease with destruction of
myelin sheaths and/or oligodendrocytes,which synthesize myelin, by CD8 T-cell cyto-toxicity and release of cytokines from T cells
(2) Antibodies are directed against myelin basicprotein in oligodendrocytes.
c. Gross and microscopic findings(1) Demyelinating plaques develop primarily
in the white matter of the brain andspinal cord.
(2) Plaques have a perivenous distribution.d. Clinical findings
(1) Episodic course punctuated by acute relapsesand remissions
(2) Sensory and motor dysfunction: paresthe-sias, muscle weakness
(3) Optic neuritis: inflammation of optic nerve,blurry vision or sudden loss of vision
(4) Cerebellar ataxia(5) Scanning speech, intention tremor, nystag-
mus, bilateral internuclear ophthalmoplegia(demyelination of medial longitudinalfasciculus)
e. Laboratory findings(1) Increase in CSF leukocyte count: primarily T
lymphocytes(2) Increase in CSF protein: primarily an increase
in 7-globulins
MS: autoimmunedestruction ofmyelin sheath andoligodendrocytes
Bilateral internuclearophthalmoplegia:pathognomonicfor MS

Chapter 25 Nervous System Disorders 331
(3) Increase in CSF myelin basic protein: indi-cates active disease
(4) Normal CSF glucose(5) Electrophoresis shows oligoclonal bands,
discrete bands in the 7-globulin region that area sign of demyelination.
2. Central pontine myelinolysisa. Most often seen in alcoholism with hypo-
natremiab. Rapid intravenous correction of hyponatremia
causes demyelination within the basis pontis.3. Viral infection with direct infection of oligodendro-
cytes (e.g., subacute sclerosing panencephalitis, progres-sive multifocal leukoencephalopathy)
C. Hereditary disorders• I.eukodystrophies are due to inborn errors of
metabolism.1. Adrenoleukodystrophy
a. X-linked recessive disorderb. Enzyme deficiency in 13-oxidation of fatty acids
in peroxisomes; results in accumulation of long-chain fatty acids
c. Causes generalized loss of myelin in brain andadrenal insufficiency
2. Metachromatic leukodystrophya. Autosomal recessive disorderb. Deficiency of arylsulfatase A results in accumula-
tion of sulfatides.3. Krabbe's disease
a. Autosomal recessive disorderb. Galactocerebroside p-galactocerebrosidase defi-
ciency leads to accumulation of galactocerebrosidein large, multinucleated, histiocytic cells (globoidcells) in the CNS.
VII. Degenerative Disorders• Degenerative diseases of the CNS involve degeneration of
neurons in the brain and/or spinal cord.A. Alzheimer's disease
1. Most common cause of dementia: either presenile(< 65 years of age) or senile (> 65 years of age); usuallysporadic
2. Role of 13-amyloid (A(3) proteina. Amyloid precursor protein (APP) is normally
coded on chromosome 21.b. Defects in the degradation of APP by proteases
cause an increase in Af3.c. Trisomy 21 (Down syndrome) is associated with
a greater increase in APP, causing greater produc-tion of Ap; most patients with Down syndromehave Alzheimer's disease by 40 years of age.
Central pontine my-elinolysis: due torapid intravenouscorrection ofhyponatremia
Alzheimer's disease:13-amyloid de-
struction of neurons

••••
332 Pathology
d. A(3 deposits in neurons are neurotoxic: they alsodeposit in the walls of cerebral blood vessels.
e. Apolipoprotein gene E, allele €4, located onchromosome 19 codes for a product with a highbinding affinity for A[3, causing a familial late-onsetAlzheimer's disease.
3. Mutations on chromosome 14 produce a hyperphos-phorylated tau protein, causing formation of neurofi-brillary tangles.
4. Gross and microscopic findingsa. Cerebral atrophy with dilation of ventricles (hy-
drocephalus ex vacuo) is due to loss of neurons inthe temporal, frontal, and parietal lobes.
b. Presence of neurofibrillary tangles in the cyto-plasm of neurons(1) Pairs of protein-rich neurofilaments coiled
like a DNA helix(2) Best visualized with silver stains; also seen in
elderly patients without dementia and inHuntington's disease
c. Senile plaques: have a core of A13 surrounded bydistended neurites (distal neuronal cell processes)containing tau protein
d. Amyloid angiopathy: Al3 is present in cerebralvessels.
e. Increased density and widespread distribution ofneurofibrillary tangles and senile plaques in apatient with dementia are confirmatory for Alzhei-mer's disease (usually an autopsy diagnosis).
5. Clinical findingsa. General impairment of higher intellectual func-
tion without focal neurologic deficitsb. Patients usually die of an infection such as inter-
current bronchopneumonia.B. Parkinsonism
1. Parkinsonism refers to any disease that alters dopami-nergic pathways connecting the substantia nigra tothe basal ganglia.a. The striatal system is involved in voluntary
muscle movement; it consists of the substantianigra, caudate, putamen, globus pallidus, subthala-mus, and thalamus.
b. Dopamine is the principal neurotransmitter inthe nigrostriatal tract connecting the substantianigra with the caudate and putamen.
2. Idiopathic Parkinson's diseasea. Occurs between 45 and 65 years of age: distribu-
tion is equal in men and women; most cases aresporadic, but some cases are familial.
b. Degeneration and depigmentation of neurons inthe substantia nigra and locus ceruleus(1) Causes deficiency of dopamine
Alzheimer's disease:I density of neuro-fibrillary tanglesand amyloid plaques
Idiopathic Parkin-son's disease: de-pigmentation of sub-stantia nigraneurons
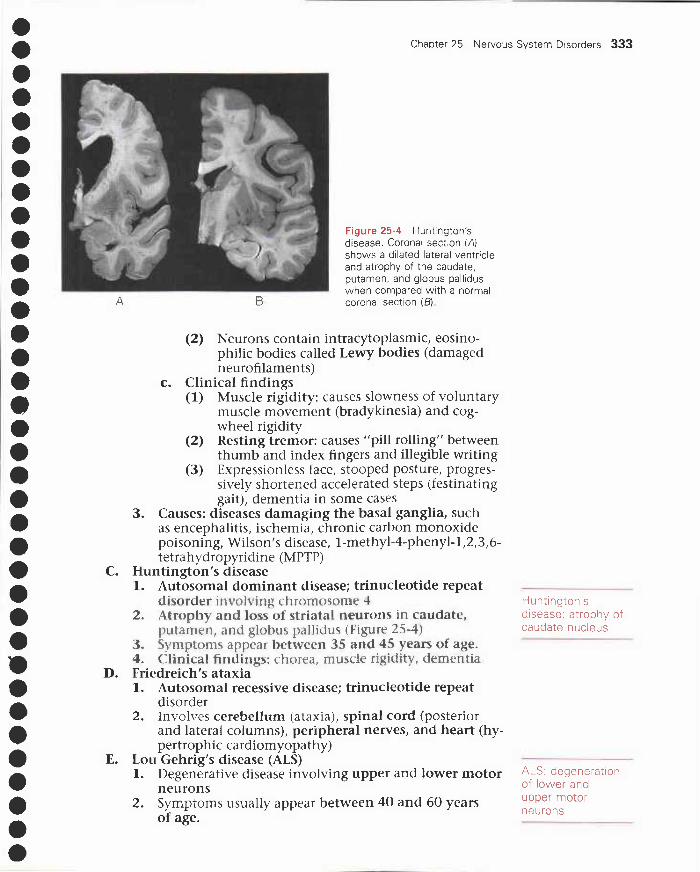
A
B
Chapter 25 Nervous System Disorders 333
Figure 25-4 Huntington'sdisease. Corona) section (A)shows a dilated lateral ventricleand atrophy of the caudate,putamen, and globus palliduswhen compared with a normalcoronal section (B)
(2) Neurons contain intracytoplasmic, eosino-philic bodies called Lewy bodies (damagedneurofilaments)
c. Clinical findings(1) Muscle rigidity: causes slowness of voluntary
muscle movement (bradykinesia) and cog-wheel rigidity
(2) Resting tremor: causes "pill rolling" betweenthumb and index fingers and illegible writing
(3) Expressionless face, stooped posture, progres-sively shortened accelerated steps (festinatinggait), dementia in some cases
3. Causes: diseases damaging the basal ganglia, suchas encephalitis, ischemia, chronic carbon monoxidepoisoning, Wilson's disease, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
C. Huntington's disease1. Autosomal dominant disease; trinucleotide repeat
disorder involving chromosome 4 Huntington's2. Atrophy and loss of striatal neurons in caudate,
putamen, and globus pallidus (Figure 25-4)disease: atrophy ofcaudate nucleus
3. Symptoms appear between 35 and 45 years of age.4. Clinical findings: chorea, muscle rigidity, dementia
D. Friedreich's ataxia1. Autosomal recessive disease; trinucleotide repeat
disorder2. Involves cerebellum (ataxia), spinal cord (posterior
and lateral columns), peripheral nerves, and heart (hy-pertrophic cardiomyopathy)
E. Lou Gehrig's disease (ALS)1. Degenerative disease involving upper and lower motor
neurons2. Symptoms usually appear between 40 and 60 years
of age.
ALS: degenerationof lower andupper motorneurons

334 Pathology
a. Most cases are sporadic.b. Familial cases involve mutations on chromosome
21 that lead to defective production of superoxidedismutase 1, which leads to superoxide freeradical injury of neurons.
3. Clinical findingsa. Upper motor neuron signs: spasticity, Babinski's
signb. Lower motor neuron signs: muscle weakness,
usually beginning with atrophy of intrinsic musclesof the hands and forearms
c. Average survival is 3-5 years.4. Werdnig-Hoffmann disease: upper and lower motor
neuron disease that occurs in children
VIII. Toxic and Metabolic DisordersA. Wilson's disease
1. Autosomal recessive disease: defect in copper excre-tion in bile, leading to liver cirrhosis and excess freecopper in blood (see Chapter 18)
2. CNS findings: signs of parkinsonism, chorea, and de-mentia; atrophy and cavitation of basal ganglia, par-ticularly the putamen
B. Acute intermittent porphyria1. Autosomal dominant disorder; defect in porphyrin
metabolism2. Deficiency of uroporphyrinogen synthase
a. Proximal increase in porphobilinogen and6-aminolevulinic acid (ALA)
b. When urine is exposed to light, porphobilinogen isoxidized to porphobilin, which produces a port-wine color.
c. Heme has a negative feedback relationship withALA synthase (rate-limiting reaction of porphyrinmetabolism).
d. Drugs enhancing liver cytochrome P-450 system(e.g., alcohol) precipitate porphyric attacks bydecreasing heme.
3. Clinical findingsa. Neurologic dysfunction, resulting in recurrent
bouts of severe abdominal pain simulating acuteabdomen; when mistaken for a surgical abdomen,a "bellyful of scars" results.
b. Psychosis, peripheral neuropathy, dementiaC. Vitamin B 12 deficiency: subacute combined degeneration
of the spinal cord with posterior column and lateral corti-cospinal tract demyelination; dementia
D. CNS findings associated with alcohol abuse1. Cortical and cerebellar atrophy2. Central pontine myelinolysis3. Wernicke-Korsakoff syndrome
a. Most often due to thiamine deficiency
Wilson's disease:cystic degenerationof putamen
Acute intermittentporphyria: "belly-ful of scars"

Chapter 25 Nervous System Disorders 335
b. Gross and microscopic findings(1) Ring hemorrhages with hemosiderin deposits
in mamillary bodies (orange-colored) andwall of the third and fourth ventricles
(2) Neuronal loss, gliosis, and vessel hemorrhagein mamillary bodies and ventricles
c. Wernicke's encephalopathy: reversible clinicalfindings include confusion, ataxia, nystagmus, andophthalmoplegia (eye muscle weakness).
d. Korsakoff's psychosis(1) Advanced irreversible stage of Wernicke's en-
cephalopathy that targets the limbic system(2) Anterograde amnesia (inability to form new
memories) or retrograde amnesia (inability torecall old memories)
IX. CNS TumorsA. Primary brain tumors in adults
1. About 70% occur above the tentorium cerebelli.2. In order of decreasing frequency: glioblastoma multi-
forme, meningioma, ependymomaB. Primary brain tumors in children
1. About 70% occur below the tentorium cerebelli.2. In order of decreasing frequency: cystic cerebellar as-
trocytoma, medulloblastoma, brainstem glioma,ependymoma
C. Risk factors: Turcot's syndrome, neurofibromatosisD. Astrocytorna
1. Accounts for about 70% of all neuroglial tumors;usually involves frontal lobe in adults and cerebellumin children
2. Glioblastoma multiforme is a high-grade astrocytoma.a. Arises de novo or by malignant transformation of a
low-grade astrocytomab. Hemorrhagic tumors with multifocal areas of ne-
crosis and cystic degeneration; commonly crossthe corpus callosum
c. May seed the neuraxis via the CSF; rarely metas-tasize outside the CNS
E. Meningioma1. Most common benign brain tumor in adults; more
common in women2. Derived from arachnoidal membrane; usually has a
parasagittal location3. Associated with neurofibromatosis4. Gross and microscopic findings
a. Firm tumors: may indent (not invade) the surface ofbrain and infiltrate overlying bone, causing in-creased bone density
b. Contain swirling masses of meningothelial cellsencompassing psammoma bodies (calcified bodies)
••••••••••••••••••••••••••••••••••
Wernicke-Korsakoffsyndrome: hemor-rhage in mamillarybodies
Most commonprimary CNS tumorin adults: glioblas-toma multiforme
Childhood tumors:cystic astrocy-toma and medullo-blastoma, both incerebellum

336 Pathology
5. Common cause of new-onset focal seizures due to im-pingement of the tumor on the brain surface
F. Ependymoma1. Benign tumor derived from ependymal cells2. Arises in cauda equina in adults and fourth ventricle
in childrenG. Medulloblastoma
1. Malignant small cell tumor that arises from the exter-nal granular cell layer of the cerebellum; primarily de-velops in children
2. Often seeds the neuraxis and invades the fourthventricle
H. Oligodendroglioma1. Benign tumor derived from oligodendrocytes in adults2. Frontal lobe tumor that frequently calcifies
I. CNS lymphoma1. Most lymphomas are metastatic high-grade non-
Hodgkin's lymphomas of B-lymphocyte origin.2. Primary CNS lymphomas
a. Most often associated with AIDSb. Epstein-Barr virus—mediated B-cell lymphomas
J. Metastasis1. Most common brain malignancy2. In order of decreasing frequency, metastasis originates
from the lungs, breast, skin (melanoma), kidney, andgastrointestinal tract.
X. Peripheral Nervous System and Pineal Gland DisordersA. Peripheral neuropathies
1. Neuropathies are associated with demyelination and/oraxonal degeneration.a. Demyelination is often segmental: produces
sensory changes (e.g., paresthesias), often in a"glove and stocking" distribution
b. Axonal degeneration is associated with musclefasciculations leading to muscle atrophy.
2. Charcot-Marie-Tooth disease: most common heredi-tary neuropathya. Autosomal dominant diseaseb. Peroneal nerve neuropathy: causes atrophy of
muscles of lower legs; legs have an "invertedbottle" appearance.
3. Guillain-Barre syndrome: most common acute periph-eral neuropathya. Autoimmune demyelination syndrome involving
peripheral and spinal nervesb. Often preceded by Mycoplasma pneumoniae pneu-
monia or Campylobacter jejuni enteritisc. Rapidly progressive ascending (may be descend-
ing) motor weakness; danger of respiratory muscleparalysis and death
d. Increased CSF protein and lymphocytes
GuiIlain-Barre syn-drome causesascending paralysis
Most common brainmalignancy:metastasis
••••••••••••••••••••••••••••••••••••

Chapter 25 Nervous System Disorders 337
e. Most patients recover after a few weeks to monthswith or without permanent disability.
4. Diabetes mellitus: most common cause of peripheralsensorimotor neuropathy; due to osmotic damageof Schwann cells
5. Toxin-associated neuropathies: alcohol, heavy metals,diphtheria
6. Idiopathic Bell's palsya. Lower motor neuron palsy causing unilateral
facial paralysisb. Inflammatory reaction involving the facial nerve
(CN VII) near the stylomastoid foramen or in thebony facial canal
c. May be associated with HIV, sarcoidosis, or Lymedisease (usually bilateral)
d. Clinical findings: drooping of the corner of themouth with drooling, difficulty speaking, inabilityto close the eye
B. Schwannoma (neurilemoma)1. Benign tumor derived from Schwann cells; may
involve the trigeminal nerve (CN V) and the acousticnerve (CN VIII; most common), spinal nerve roots, orperipheral nerves
2. Acoustic neuroma: schwannoma of CN VIIIa. Majority are located in the cerebellopontine
angle: encapsulated tumors; microscopic examina-tion shows alternating dark and light areas.
b. Clinical findings(1) Associated with neurofibromatosis: unilat-
eral or bilateral tumors(2) Tinnitus (ringing in the ears) and sensorineu-
ral deafness(3) Sensory changes in CN V from tumor im-
pingement on CN VC. Pineal gland tumors
1. Midline location above the quadrigeminal platebehind the posterior commissure; calcify with agea. Majority are germ cell tumors.b. May produce Parinaud's syndrome (paralysis of
upward gaze), obstructive hydrocephalus, preco-cious puberty
2. Interrupt site for melatonin production: important insleep, moods, and circadian rhythm
XI. Selected Eye and Ear DisordersA. Eye
1. Conjunctivitis may be caused by:a. Infection: ophthalmia neonatorum due to Chia-
mydia trachomatis or Neisseria gonorrhoeaeb. Chemical: erythromycin eyedrops in newborns
used to prevent bacterial infectionc. Allergy: seasonal itching of eyes
Idiopathic Bell'spalsy: facial muscleparalysis due toinflammation ofCN VII
Acoustic neuroma:tinnitus and sensori-neural hearing loss

338 Pathology
2. Optic neuritis: inflammation of the optic nerve (CN H),producing blurry vision or loss of vision; causes includeMS and methyl alcohol poisoning.
3. Amaurosis fugax: sudden unilateral loss of vision dueto a retinal embolus of atheromatous plaque mate-rial; described as a "curtain" going down and thencoming up
4. Glaucoma: increased intraocular pressure due to de-creased rate of aqueous outflow into the canal ofSchlemm or narrowing of the anterior chamber angle
5. Optic nerve atrophy: pale optic disk; most commonlydue to optic neuritis and glaucoma
6. Uveitis: inflammation of the uveal tract (iris, ciliarybody, choroid), producing blurry vision; causes includesarcoidosis, ulcerative colitis, and ankylosingspondylitis.
7. Macular degeneration: disruption of Bruch's mem-brane in the retina; cause of blindness in the elderly
8. Cataract: opacity in the lens; occurs most often in theelderly, in diabetes mellitus, infection (e.g., rubella),and with corticosteroid use
9. Malignant tumor: retinoblastoma in children, malig-nant melanoma in adults
B. Ear1. Meniere's disease: increased endolymph in inner ear
and loss of cochlear hairs; causes vertigo, tinnitus,and sensorineural hearing loss
2. Presbycusis: most common cause of sensorineuralhearing loss in the elderly; due to degeneration of co-chlear hairs
3. Otosclerosis: most common cause of conductiondeafness in the elderly; due to fusion of middle earossicles
4. Otitis media: most common cause of conductiondeafness in children; most commonly due to Strepto-coccus pneumoniae
Glaucoma: T intra-ocular pressure -optic atrophy
Most commoncause of permanentvisual loss in theelderly: maculardegeneration

••••••••••••••••••••••••••••••••••••
Tests

340 Pathology
COMMON LABORATORYVALUES
Test Conventional Units SI Units
Blood, Plasma, SerumAlanine aminotransferase
(ALT, GPT at 30°C)Amylase, serumAspartate aminotransferase
(AST, GOT at 30°C)Bilirubin, serum (adult)
Total // DirectCalcium, serum (Ca")Cholesterol, serumCortisol, serum
Creatine kinase, serum
Creatinine, serumElectrolytes, serum
Sodium (Na*)Chloride (CI-)Potassium (1(*)Bicarbonate (HCO31Magnesium (Mg")
Estriol, total, serum (inpregnancy)24-28 wk // 32-36 wk
28-32 wk // 36-40 wk
Ferritin, serum
Follicle-stimulatinghormone, serum/plasma (FSH)
Gases, arterial blood(room air)pHPco2Poe
Glucose, serum
Growth hormone–argininestimulation
Immunoglobulins, serumIgAIgEIgGIgM
8-20 U/L
25-125 U/L8-20 U/L
0.1-1.0 mg/dL //0.0-0.3 mg/dL
8.4-10.2 mg/dLRec: < 200 mg/dL8:00 AM: 6-23 gg/dL //4:00 PM: 3-15 gg/dL8:00 PM: 5_ 50% of 8:00 AM
Male: 25-90 U/LFemale: 10-70 U/L0.6-1.2 mg/dL
136-145 mEq/L95-105 mEq/L3.5-5.0 mEq/L22-28 mEq/L1.5-2.0 mEq/L
30-170 ng/mL //60-280 ng/mL
40-220 ng/mL //80-350 ng/mL
Male: 15-200 ng/mLFemale: 12-150 ng/mLMale: 4-25 rnIU/mLFemale: premenopause
4-30 mIU/mLmidcycle peak
10-90 mIU/mLpostmenopause
40-250 mIU/mL
7.35-7.4533-45 mm Hg75-105 mm HgFasting: 70-110 mg/dL2 hr postprandial:
< 120 mg/dLFasting: < 5 ng/mL
provocative stimuli:> 7 ng/mL
76-390 mg/dL0-380 IU/mL650-1500 mg/dL40-345 mg/dL
8-20 U/L
25-125 U/L8-20 U/L
2-17 tmol/L // 0-5 timol/L
2.1-2.8 mmol/L< 5.2 mmol/L170-630 nmol/L //
80-410 nmol/LFraction of 8:00 AM: 0.5025-90 U/L10-70 U/L53-106 tmol/L
135-145 mmol/L95-105 mmol/L3.5-5.0 mmol/L22-28 mmol/L1.5-2.0 mmol/L
104-590 // 208-970 nmol/L
140-760 // 280-1210 nmol/L
15-200 gg/L12-150 gg/L4-25 U/L
4-30 U/L
10-90 U/L
40-250 U/L
(Hi 36-44 nmol/L4.4-5.9 kPa10.0-14.0 kPa3.8-6.1 mmol/L
< 6.6 mmol/L< 5 gg/L
> 7 gg/L
0.76-3.90 g/L0-380 kIU/L6.5-15 g/L0.4-3.45 g/L

•••••
COMMON LABORATORYVALUES cont'd
Common Laboratory Values 341
••••••••
•
•••••••••••••••••••••
Test
IronLactate dehydrogenase,
serum (LDH)Luteinizing hormone,
serum/plasma (LH)
Osmolality, serumParathyroid hormone,
serum, N-terminalPhosphatase (alkaline),
serum (p-NPP at 30°C)Phosphorus (inorganic),
serumProlactin, serum (hPRL)Proteins, serum
Total (recumbent)AlbuminGlobulin
Thyroid-stimulatinghormone, serum orplasma (TSH)
Thyroidal iodine (1231)uptake
Thyroxine (T4 ), serumTriglycerides, serumTriiodothyronine (T3),
serum (RIA)Triiodothyronine (T 3) resin
uptakeUrea nitrogen, serum (BUN)Uric acid, serum
Cerebrospinal Fluid (CSF)
Cell countChlorideGamma globulinGlucosePressureProteins, total
Hematology
Bleeding time (template)Erythrocyte count
Erythrocyte sedimentationrate (Westergren)
Conventional Units
Male: 6-23 mIU/mLFemale: follicular phase
5-30 mIU/mLmidcycle 75-150 mIU/mLpostmenopause
30-200 mIU/mL275-295 mOsm/kg230-630 pg/mL
20-70 U/L
3.0-4.5 mg/dL
< 20 ng/mL
6.0-8.0 g/dL3.5-5.5 g/dL2.3-3.5 g/dL0.5-5.0 uU/mL
8%-30% of administereddose/24 hr
4.5-12 ug/d L35-160 mg/dL115-190 ng/dL
25%-38%
7-18 mg/dL3.0-8.2 mg/dL
0-5 cells/mm3118-132 mEq/L3%-12% total proteins50-75 mg/dL70-180 mm H20< 40 mg/dL
2-7 minMale: 4.3-5.9 million/mm3Female: 3.5-5.5 mil-
lion/mm3Male: 0-15 mm/hrFemale: 0-20 mm/hr
SI Units
9-30 umol/L45-90 U/L
6-23 U/L
5-30 U/L75-150 U/L
30-200 U/L275-295 mOsm/kg230-630 ng/L
20-70 U/L
1.0-1.5 mmol/L
< 20 ug/L
60-80 g/L35-55 g/L23-35 g/L0.5-5.0 mU/L
0.08-0.30/24 hr
58-154 nmol/L0.4-1.81 mmol/L1.8-2.9 nmol/L
0.25-0.38
1.2-3.0 mmol urea/L0.18-0.48 mmol/L
0-5 x 106/L118-132 mmol/L0.03-0.122.8-4.2 mmol/L70-180 mm H20< 0.40 g/L
2-7 min4.3-5.9 x 1012/L3.5-5.5 x 1012/L
0-15 mm/hr0-20 mm/hr
Continued
Blood, Plasma, Serum—cont'd50-170 ug/dL45-90 U/L

342 Pathology
COMMON LABORATORYVALUES cont'd
Test
Conventional Units
SI Units
Hematology—cont'dHematocrit (Hct)
Hemoglobin AlcHemoglobin, blood (Hb)
Hemoglobin, plasmaLeukocyte count and
differentialLeukocyte countSegmented neutrophilsBandsEosinophilsBasophilsLymphocytesMonocytes
Mean corpuscular hemo-globin (MCH)
Mean corpuscular hemo-globin concentration(MCHC)
Mean corpuscularvolume (MCV)
Partial thromboplastin time(activated) (aPTT)
Platelet countProthrombin time (PT)Reticulocyte countThrombin time
VolumePlasma
Red cell
SweatChloride
UrineCalciumCreatinine clearance
Estriol, total (in pregnancy)30 wk35 wk40 wk
17-Hydroxycorticosteroids
17-Ketosteroids, total
OsmolalityOxalateProteins, total
Male: 40%-54%Female: 37%-47%5. 6%Male: 13.5-17.5 g/dLFemale: 12.0-16.0 g/dL1-4 mg/dL
4500-11,000/mm354%-62%3%-5%1% -3%0%-0.75%25%-33%3%-7%25.4-34.6 pg/cell
31%-37% Hb/cell
80-100 pm3
25-40 sec
150,000-400,000/mm312-14 sec0.5%-1.5% of red cells<2 sec deviation from
control
Male: 25-43 mL/kgFemale: 28-45 mL/kgMale: 20-36 mL/kgFemale: 19-31 mL/kg
0-35 mmol/L
100-300 mg/24 hrMale: 97-137 mL/minFemale: 88-128 mL/min
6-18 mg/24 hr9-28 mg/24 hr13-42 mg/24 hrMale: 3.0-9.0 mg/24 hrFemale: 2.0-8.0 mg/24 hrMale: 8-22 mg/24 hrFemale: 6-15 mg/24 hr50-1400 mOsm/kg8-40 p.g/mL< 150 mg/24 hr
0.40-0.540.37-0.47
0.06%2.09-2.71 mmol/L1.86-2.48 mmol/L0.16-0.62 mmol/L
4.5-11.0 x 109/L0.54-0.620.03-0.050.01-0.030-0.00750.25-0.330.03-0.070.39-0.54 fmol/cell
4.81-5.74 mmol Hb/L
80-100 fl
25-40 sec
150-400 x 109/L12-14 sec0.005-0.015< 2 sec deviation from
control
0.025-0.043 L/kg0.028-0.045 L/kg0.020-0.036 L/kg0.019-0.031 L/kg
0-35 mmol/L
2.5-7.5 mmol/24 hr
21-62 p.mo1/24 hr31-97 pmo1/24 hr45-146 1.tmol/24 hr8.2-25.0 pmo1/24 hr5.5-22.0 pmo1/24 hr28-76 pmo1/24 hr21-52 pmo1/24 hr
90-445 pmol/L< 0.15 g/24 hr

TEST 1 S
DIRECTIONS: Each numbered item or incomplete statement is followed by options ar-ranged in alphabetical or logical order. Select the best answer to each question. Someoptions may be partially correct, but there is only ONE BEST answer.
1. A 58-year-old man with a history ofalcohol abuse has tenderness in the rightupper quadrant. Physical examinationshows tender hepatomegaly. Serum bili-rubin is normal, but serum transaminases,particularly aspartate aminotransferase,are slightly elevated. The figure shows asection of the liver obtained on biopsy.Which of the following best explains thepathogenesis of the liver disease?
O A. Decreased hydrolysis of adiposeO B. Decreased synthesis of fatty acidsO C. Increased concentration of glycerol
3-phosphateO D. Increased 13-oxidation of fatty acidsO E. Increased protein intake
2. Physical examination of a 2-year-oldchild with severe mental retardation showsa flat occiput, epicanthal folds, and a flatnasal bridge. There is a simian crease inboth palms. Chromosome analysis shows46 chromosomes. Which of the follow-ing types of genetic mutation is responsiblefor this condition?
O A. Balanced translocationO B. Frameshift mutationO C. MicrodeletionO D. NondisjunctionO E. Point mutation
3. A 26-year-old man is scuba diving off thecoast of Bermuda in water 30-60 feet deepwhen he develops problems with his airtank and must ascend quickly to thesurface. One hour later, he has pain inthe muscles and joints in the legs. Whichof the following is most likely responsi-ble for these symptoms?
O A. Deep venous thrombosisO B. Disseminated intravascular
coagulation•D C. Fat embolizationO D. Hemorrhage into muscles and jointsO E. Nitrogen gas embolism
343
0 0211W r)ti 4„%cat ciRr.i. w:q)
o • 0- •-Fteco°00
0c.41 • ;4 .0
0 bo 0 -0-,s". vigio 0 VIP * qz,
'0
0 -1G .3%-■
CF.Q.
344 Pathology
A
B
4. The figure shows two children, each witha different nutritional disorder. Which ofthe following findings would most likely bereported only in child A?
O A. Decrease in serum albuminO B. Decrease in somatic protein storesO C. Decrease in subcutaneous fatO D. Decrease in total calorie intake
5. A 52-year-old woman has a history ofchronic left-sided and right-sided heartfailure. The autopsy specimen shows athickening of the mitral valve leaflets andfusion of the commissures. Which of thefollowing is the most likely cause of mitralvalve disease in this patient?
O A. Immune reaction in systemic lupuserythematosus (SLE)
O B. Ischemic heart diseaseO C. Myxomatous degenerationO D. Recurrent bacterial endocarditisO E. Recurrent immune reaction against
group A streptococci
6. A 48-year-old man has fever, weight loss,sweating, and a dragging sensation in theabdomen. Physical examination showsgeneralized lymphadenopathy and massivehepatosplenomegaly. Laboratory studiesshow a normocytic anemia and throm-bocytopenia, and a WBC count of 110,000/mm3 . A bone marrow biopsy showshypercellularity and the presence of neutro-phils at all stages of development; less than2% of the WBCs are myeloblasts. Thefigure shows a peripheral blood smear.Which of the following laboratory findingswould most likely be positive?
O A. Leukocytes for alkaline phosphataseO B. Leukocytes for CD10O C. Leukocytes for Philadelphia
chromosomeO D. Leukocytes for tartrate-resistant acid
phosphataseO E. Leukocytes for terminal deoxy-
nucleotidyl transferase
Test 1 345
7. A 56-year-old woman who has smokedtwo packs of cigarettes daily for 35 years haschronic dyspnea. She has an emaciated ap-pearance and pink discoloration of the skin.It is difficult to hear heart and lung soundsbecause of the increase in the anteropos-tefior diameter of the chest cavity. A chestradiograph shows hyperinflation in bothlung fields, depression of both diaphragms,and a vertically oriented cardiac silhou-ette. Which of the following results of pul-monary function tests would be reported?
O A. Decreased functional residual capacityO B. Decreased total lung capacityO C. Increased FEV 1 :FVC ratioO D. Increased residual volumeO E. Increased tidal volume
8. A 46-year-old man develops pneumoniashortly after repainting a bridge. A silver-stained specimen from the lung shows yeastforms with narrow-based buds. Which ofthe following pathogens is the most likelycause of this patient's pneumonia?
O A. Aspergillus fumigatusO B. Coccidioides immitisO C. Cryptococcus neoformansO D. Histoplasma capsulatumO E. Pneumocystis carinii
9. The arrow in the figure points to anintraluminal mass in a left anterior descend-ing coronary artery. Which of the follow-ing drugs is most likely to be used toprevent the formation of such a mass?
O A. AspirinO B. Glycoprotein Hb/IIIa inhibitorO C. HeparinO D. Tissue plasminogen activator (tPA)O E. Warfarin

346 Pathology
10. Physical examination of a 72-year-oldman shows severe hypertension, an epi-gastric bruit, and diminished amplitudeof pedal pulses. Plasma renin activity isincreased. Angiogram of the renal arteryshows decreased uptake of dye and a smallkidney on the left side and normal uptakeof the dye and a normal-sized kidney onthe right side. Which of the following bestdescribes the pathogenesis of the hyper-tension?
) A. Adrenal tumor that produces excessaldosterone
0 B. Adrenal tumor that produces excesscatecholamines
0 C. Essential hypertension with bilateralnephrosclerosis
3 D. Unilateral renal artery stenosis causedby atherosclerosis
1) E. Unilateral renal artery stenosis causedby fibromuscular hyperplasia
11. Shortly after a trip to the desert inthe southwestern United States, a 58-year-old archaeologist develops a dry cough,fatigue, and painful nodules in the lowerextremities. A biopsy of the lung showsgranulomatous inflammation, with numer-ous spherules containing endospores.Which of the following is the most likelycause of the respiratory disorder?
) A. AspergillosisB. Coccidioidomycosis
0 C. Cryptococcosis) D. Histoplasmosis,) E. Tuberculosis
12. A 35-year-old woman complains ofcolicky abdominal pain and vomiting. Shehas had surgery for Crohn's disease. A plainabdominal radiograph is taken of thepatient in an erect position and shows mul-tiple air-fluid levels with a stepladder con-figuration. Which of the following is themost likely cause of the abdominal pain?
) A. Direct inguinal hernia) B. Intussusception) C. Large bowel infarction) D. Small bowel adhesions) E. Volvulus
I 1 `•• _ ••• - %• • - I, ' -.1" •. .
4 . . . , d .... i 1: c ••. . 41,;•••-v. r:
•
:1:
,. ..., , .-,Tils,1.'. .....■ iz jtt. ,.e.••••-`
• ' i' • k. .."1.q./..C•If .."1":"•,': '.....S. ;If: i■ :Ir"...• ;:fria:t • •. 6' 0 1... \ 11;! : . i:" • : Il #...: 5 . k i. • .1"( :, lilt! !4 ..c.,44; • 4 . r '6 o ! _ . ... .
- .4 .,.e-A . ' 47; 4"' •.•
. • j• .II..j.:1. .or ;:t??,... -2 :1:11Y. 1 :4:.1.1...'4\1" :14 .11!, * • .- 1... i.- i ... e , -e , ' ■• 'riot
... • 1 .,;%' • • • %.• • • •..... , :.- • . ^ m• '
13. A 28-year-old man has fever, fatigue,difficulty breathing, and substernal chestpain while walking or at rest. The patienthas a history of alcohol abuse. Physicalexamination shows bibasilar rales, disten-tion of the jugular neck vein, hepato-megaly, and dependent pitting edema.A chest radiograph shows generalizedcardiac enlargement. Laboratory studiesshow an increase in cardiac-specifictroponins. The figure shows a biopsy speci-men of myocardial tissue. Which of thefollowing is the most likely cause of thispatient's heart disease?
) A. Congestive cardiomyopathy) B. Coronary artery thrombosis) C. Ischemic heart disease
D. Rheumatic feverE. Viral myocarditis
Test 1 347
14. A 25-year-old man develops hemop-tysis. A few weeks later, he experiencessudden onset of acute renal failure and dies.Prior to his death, urinalysis shows mildproteinuria, hematuria, and RBC casts.The figure shows an immunofluorescencestudy of a representative glomerulus in asection of kidney removed at autopsy.Which of the following is the most likelydiagnosis?
O A. Diffuse membranous glomerulo-nephritis
O B. Focal segmental glomerulosclerosisO C. Goodpasture's syndromeO D. IgA glomerulonephritisO E. Minimal change disease
15. A 56-year-old man with a lengthyhistory of alcohol abuse has chronic epi-gastric pain and chronic diarrhea associatedwith bloating and fatty stools. Physicalexamination shows malnutrition, abdomi-nal distention, and tender hepatomegaly.A plain radiograph of the abdomenshows irregular densities in the left upperquadrant. Which of the following is themost likely diagnosis?
O A. Celiac diseaseO B. Chronic pancreatitis0 C. Crohn's diseaseO D. Pancreatic carcinomaO E. Peptic ulcer disease
16. A 23-year-old African-American womanwith a history of dysfunctional uterinebleeding complains of fatigue when exercis-ing. Laboratory studies show a mild micro-cytic anemia and increased RBC distributionwidth. The figure shows a peripheral bloodsmear. Which of the following investiga-tive studies is most useful in this diagnosis?
O A. Bone marrow aspiration biopsyO B. Hemoglobin electrophoresisD C. Osmotic fragility testO D. Serum ferritin testO E. Sickle cell screening
17. The retina of a 35-year-old man whohas a 15-year history of type 1 diabetes mel-litus shows microaneurysms and retinalhemorrhages. Which of the following is thepathogenesis of the lesions in the retina?
O A. Increased intraocular pressureO B. Inflammation of the optic nerveO C. MicroangiopathyD D. Nonenzymatic glycosylation3 E. Osmotic damage

•••••••••••••••••••••••••••••••••
O A. AcuteB. Chronic
) C. GranulomatousD. PseudomembranousE. Suppurative
•••
• vore.,.11IoP,.."•1,:ilik";*A-A! t'
348 Pathology
18. The figure shows the uterus, fallo-pian tubes, and cervix removed from a55-year-old woman who experiencedpostmenopausal bleeding. Which of thefollowing is the most likely pathogenesisof the lesion in the endometrial cavity?
O A. AdenomyosisO B. Herpes simplex virus type 2 (HSV-2)
infectionO C. Human papillomavirus (HPV)
infectionO D. MultiparityO E. Unopposed estrogen exposure
19. A febrile 42-year-old man has pain andswelling in the meta tarsophalangeal jointof the great toe. He has a 20-year history ofalcohol abuse. A complete blood cell countshows an absolute neutrophilic leukocy-tosis with > 10% band neutrophils. Whichof the following microscopic findings ismost likely to be present in the synovialfluid?
O A. Neutrophils with phagocytosedgram-negative diplococci
O B. Neutrophils with phagocytosedgram-positive cocci
O C. Neutrophils with phagocytosednegative birefringent crystals
O D. Neutrophils with phagocytosedpositive birefringent crystals
O E. Neutrophils with phagocytosedrheumatoid factor immunocomplexes
h... ....N--. •_:4111 .,- It:Wkol...--"vv,
i'd• N ' ••• *ft 4 •-6%. s• .":,"\•
, * % * *kw e• 9 to. W
s? 4; 410$ ,1P 14,14;+ . 4k41\ 49,1114f• • • .011.• 1■°,0** IN.:74 rA ,. .1
viez s. I l a ■•:. • oibr; ot....4....iiid-4411 Vic 4 % ,••■4.'• '411.0
. 40 ,•••
Ate. i• so • • .-
;VirieAMVP. • al aVa„ ,11-4
o
20. A 52-year-old woman has a 10-yearhistory of progressively worsening arthritisin the hands and knees, which has led toankylosis. The figure shows a section of sy-novial tissue removed from the knee joint.Which of the following types of inflam-mation is evident in this tissue?
21. A sexually active 19-year-old womancomplains of frequency and a burning sen-sation upon urination. Pelvic examina-tion shows inflammation of the exocervixand an exudate in the cervical os. Urinalysisshows numerous neutrophils but no bacte-ria. A cervical Pap smear shows metaplas-tic squamous cells with a cytoplasmicphagosome containing an inclusion. Whichof the following pathogens is the causalagent?
A. Candida albicansO B. Chlamydia trachomatisO C. Human papillomavirus (HPV)O D. Neisseria gonorrhoeaeD E. Trichomonas vaginalis

•• Test 1 349
22. A 48-year-old man complains ofextreme fatigue, weakness, and lightheaded-ness when he stands up quickly from aseated position. The patient is normoten-sive when lying down; when sitting, hisblood pressure drops and his pulse rate in-creases. There is diffuse brown pigmenta-tion of the buccal mucosa. The physiciansuspects a disorder involving the adrenalglands. Which of the following laboratoryfindings would most likely be reported?
O A. Decreased plasma ACTH and11-deoxycortisol after metyraponestimulation
O B. Decreased serum sodium and serumpotassium
O C. Increased plasma ACTH and de-creased serum cortisol
0 D. Increased plasma ACTH and11-deoxycortisol after metyraponestimulation
O E. Increased urine 17-hydroxycorticoidswith prolonged ACTH stimulation
23. A 45-year-old woman has fever,night sweats, and weight loss. The figureshows a biopsy specimen of lung tissue withevidence of necrosis. Which of the follow-ing best describes this necrosis?
O A. Gaseous necrosisa B. Coagulation necrosis0 C. Enzymatic fat necrosisO D. Fibrinoid necrosis0 E. Liquefactive necrosis
24. A centrally located lung mass isremoved from a 58-year-old man who hassmoked two packs of cigarettes a day for30 years. An H&E-stained section of themass shows round- to spindle-shaped baso-philic cells, increased mitotic activity, andnecrotic foci. Which of the following endo-crinopathies is frequently associated withthis type of tumor?
O A. Carcinoid syndromeO B. HypercalcemiaO C. HypocalcemiaO D. Inappropriate antidiuretic hormone
secretionO E. Polycythemia
25. A 23-year-old woman states that she isextremely tired and has drooping eyelidsand double vision toward the end of theworkday. She also has difficulty swallowingsolids and liquids and states that foodseems to "stick near my Adam's apple."Which of the following terms best describesthis patient's clinical disorder?
O A. DemyelinatingO B. Electrolytea C. Motor neuronO D. NeuromuscularO E. Primary muscular
26. A 10-year-old boy has muscle weaknessthat became symptomatic 5 years ago.When placed in a prone position, he must"walk" his hands to his feet and up thefront of his legs to stand. Which of the fol-lowing is the pathogenesis of this clinicaldisorder?
O A. Antibodies against acetylcholinereceptors
O B. Deficiency of dystrophinO C. Demyelinating disorderO D. Inflammatory myopathyO E. Motor neuron disorder
•••••••••••••••
••••••••••••••••

350 Pathology
27. A 65-year-old man has a ruptured ab-dominal aortic aneurysm and is in hypo-volemic shock. Which of the following is areversible cellular event that is most likelyto occur in the straight portion of the proxi-mal tubular cells of the kidneys?
O A. Ca2+ moving into the cytosolO B. Cytochrome c diffusing out of the
mitochondriaO C. Intracellular pH increasingO D. Na' and H 20 moving into the cytosolO E. Phospholipase damaging the cell
membrane
28. A 22-year-old woman with a historyof chronic diarrhea describes her stools asgreasy. She recently developed a pruriticvesicular lesion on her elbow. The figureshows an endoscopic biopsy specimen ofthe jejunum. Which of the following will bemost useful in identifying the cause of thediarrhea?
O A. Antigliadin antibodiesO B. Antinuclear antibodiesO C. Fecal smear for leukocytesO D. Stool for ova and parasitesO E. Stool osmotic gap
29. An afebrile 48-year-old man has a25-year history of lower back pain thatbegan in the sacroiliac joints. He has beentaking nonsteroidal anti-inflammatorydrugs intermittently. Physical examinationshows a stooped posture and tendernessin all the vertebral joints. A high-pitchedearly diastolic murmur is heard in thesecond intercostal space on the right. Aradiograph of the spine shows forwardcurvature of the spine with fusion of thelumbar vertebrae. Which of the followinglaboratory findings is most likely to bereported?
O A. Antibodies against Borrelia burgdorferiO B. HLA-B27 genotypeO C. HyperuricemiaO D. Positive blood cultureO E. Rheumatoid factor
30. A 12-year-old boy has an episodichistory of developing pink-staining urineshortly after an upper respiratory infection.The patient is normotensive and afebrile.The urine dipstick test is positive for bloodand shows mild to moderate amounts ofprotein. The anti–streptolysin 0 titer, theanti–DNAse B titer, and the serum anti-nuclear antibody (ANA) test are all negative.Urinalysis shows RBCs and RBC casts.Which of the following is the most likelydiagnosis?
O A. Diffuse membranous glomerulo-nephritis
O B. Glomerulonephritis in systemic lupuserythematosus (SLE)
O C. IgA glomerulonephritisO D. Minimal change diseaseO E. Poststreptococcal glomerulonephritis
•••••••••••••••••S••••••••••••••••S•

Test 1 351
31. The figure shows painless and non-pruritic lesions on the left side of the neckof an afebrile 25-year-old man with AIDS.Similar lesions are also present on the hardpalate. Which of the following is the mostlikely causal organism?
) A. BartowIla henselae) B. Cytomegalovirus
O C. Epstein-Barr virus) D. Herpesvirus 8
E. HIV
32. A 48-year-old man with alcoholic cir-rhosis has ascites and dependent pittingedema in the lower legs. Fluid accumulationin the peritoneal cavity and legs occurs bywhich of the following mechanisms?
O A. Decreased plasma oncotic pressureO B. Increased plasma hydrostatic pressure,_) C. Increased vessel permeability due to
histamineO D. Lymphatic obstruction with
lymphedemaE. Movement of water into the intra-
cellular compartment
33. A 53-year-old man with type 1 diabetesmellitus and cirrhosis also has chronicdiarrhea. The patient has a 58-year-old sisterwith similar problems. His skin is pale grayin color, and this is most obvious on hishands. Which of the following laboratoryfindings would most likely be reported?
D A. Decreased serum ceruloplasmin) B. Decreased serum ironD C. Decreased small bowel reabsorption
of o-xyloseO D. Increased serum ferritin• E. Increased total iron-binding capacity
34. The figure shows a valvular lesion onthe left side of the heart of a 25-year-oldwoman. Which of the following heartsounds is most likely to be associated withthis lesion?
D A. Diastolic blowing murmur after S2D B. Midsystolic click followed by a
murmur) C. Opening snap followed by a
mid-diastolic rumbling murmur• D. Pansystolic murmur at the apex7 E. Systolic ejection murmur

352 Pathology
35. A 9-month-old girl has an infectionon her face that began as erythematousmacules. She later develops pustules thatrupture and cause honey-colored crustedlesions. The girl's 5-year-old brother devel-ops similar lesions. Which of the follow-ing is the causal agent?
O A. Group A 0-hemolytic streptococcusO B. Herpes simplex virus type 1O C. Malassezia furfurO D. Propionibacterium acnesO E. Trichophyton rubrum
36. A 42-year-old man complains of head-aches and dyspnea. Physical examina-tion shows enlargement of the nose andsupraorbital ridge and jutting out of thelower jaw. The hands and feet are enlarged.A chest radiograph shows generalized en-largement of the heart. Which of thefollowing is the most sensitive screeningtest for this patient's disorder?
O A. Serum cortisolO B. Serum glucoseO C. Serum insulin-like growth factor—IO D. Serum prolactinO E. Serum thyroid-stimulating
hormone (TSH)
0 0 O n
6
■ 0 •Normal Affected Normal Affected
male male female female
37. Which of the following clinical dis-orders is most compatible with the distribu-tion of affected patients shown in thispedigree?
O A. Congenital spherocytosisO B. Glucose-6-phosphate dehydrogenase
(G6PD) deficiencyO C. HemochromatosisO D. NeurofibromatosisO E. Type 2 diabetes mellitus
38. For the past 15 years, a 72-year-oldwoman has experienced a loss in heightand has had a chronic backache, resultingin forward curvature of the spine. Radio-graphs of the vertebral column show de-creased bone density and wedge-shapedflattening of the vertebral bodies in themidthoracic region. Which of the followingis the pathogenesis of this clinicalcondition?
O A. Decreased estrogenO B. Genetic defect in osteoclastsO C. Hypovitaminosis DO D. Metastasis to bone

Test 1 353
39. A 1-year-old boy has painful vesicularlesions on the upper and lower lips. One ofthe vesicles was unroofed, and a scrapingof cells from the base of the lesion wassmeared onto a glass slide. The materialon the slide was stained with a Pap stain.Which of the following best describes thecytologic findings in this smear?
O A. Dysplastic squamous cells with in-creased nuclear chromatin
O B. Enlarged individual squamous cellswith intranuclear inclusions
O C. Multinucleated squamous cells withintranuclear inclusions
O D. Neoplastic squamous cells with atypi-cal mitotic figures
O E. Normal basal cells without intra-nuclear inclusions
40. A febrile 30-year-old woman sees herphysician because of a rash on the thigh(see figure) that developed at the site whereshe was bitten by an insect when on acamping trip about 4 weeks ago. Thewoman lives in the northeastern UnitedStates. Which of the following is the mostlikely causal pathogen?
O A. Babesia micronO B. Borrelia burgdorferiO C. Borrelia recurrentisO D. Ehrlichia chaffeensisO E. Rickettsia rickettsii
41. A 36-year-old man has a history ofchronic liver disease and a movement disor-der. Physical examination shows a rim ofbrown pigment around the perimeter of thecornea. Which of the following laboratoryfindings would most likely be reported?
O A. Decreased serum ceruloplasminO B. Decreased serum ironO C. Increased serum ferritinO D. Increased total serum copperO E. Normal serum prothrombin time
42. A 25-year-old man develops a pruriticlesion on the trunk. The lesion has an ovalshape with an erythematous margin, andthe central area has fine white scales. AKOH examination of skin scrapings takenfrom the leading edge of the lesion is nega-tive for fungal organisms. Two weeks later,the patient develops a more widespreadtruncal eruption that follows the lines ofcleavage of the skin. Which of the followingis the most likely diagnosis?
O A. EczemaO B. Pityriasis roseaO C. Secondary syphilisO D. Tinea corporisO E. Tinea versicolor

354 Pathology
6o • A
B
Normal Affected Normal Affectedmale male female female
43 Which of the following clinical dis-orders is most compatible with the distribu-tion of affected patients shown in thispedigree?
O A. Alport's syndromeO B. Familial hypercholesterolemiaO C. Familial polyposisO D. Leber's optic neuropathyO E. McCardle's disease
44 A 3-year-old boy develops a rash on hisface 24 hours after the onset of a mildfever and sore throat. The maculopapularrash causes erythema of the cheeks. Thecausal agent of this disorder may also causewhich of the following complications?
O A. Acute lymphocytic leukemiaO B. Acute myelogenous leukemiaO C. Aplastic anemiaO D. B-cell malignant lymphomaO E. Disseminated intravascular
coagulation
45. A 65-year-old man with an expres-sionless face and stooped posture shuffleswhen walking. He has constant tremorof the thumbs and index fingers of bothhands at rest. Physical examination showsincreased rigidity of the arm muscleswhen resisting extension of the arm. Alldeep tendon reflexes are normal. Figure Ashows a midbrain from a patient withsimilar signs and symptoms. Figure B showsa normal midbrain for comparison. Whichof the following best explains the pathogen-esis of this disorder?
O A. Degeneration of dopaminergic sub-stantia nigra neurons
0 B. Increased y-secretase activityO C. Loss of striatal neurons in the caudate
nucleusO D. Presence of apolipoprotein gene E,
allele €4O E. Presence of hyperphosphorylated tau
protein

Test 1 355
46. A 26-year-old woman has retro-orbitalpain and blurry vision in the right eye.Physical examination shows flame hemor-rhages around the disk vessels and a swollenoptic disk. After treatment with systemiccorticosteroids, the patient's vision is re-stored to normal. A few months later, thepatient has slurred speech, an ataxic gait,and weakness and paresthesias in the armsand legs that eventually remit withoutsequelae. Which of the following findingsis most likely present in the cerebrospi-nal fluid (CSF)?
O A. Decreased glucoseO B. Increased neutrophilsO C. Normal proteinO D. Oligoclonal bandsO E. Positive Gram stain
47. For the past 3 years, a 55-year-oldwoman has had multiple pigmented, pe-dunculated tumors and flat, oval, coffee-colored skin patches. She has complainedof episodic attacks of headache, palpita-tions, and profuse perspiration. She has re-cently experienced substernal chest painduring the attacks. Her pulse is 160/min;the average of three blood pressure readingsis 180/120 mm Hg. Which of the follow-ing laboratory studies is most likely to beuseful in the diagnosis of this patient?
D A. Complete urinalysisO B. Serum electrolytesD C. Urine for free cortisol, 24 hO D. Urine for 17-ketosteroids, 24 hO E. Urine for metanephrines, 24 h
48. For the past few months, a 26-year-oldman with AIDS has experienced progressiveloss of visual acuity in both eyes. Intraoc-ular pressure is normal. The CD4 helperT-cell count is 48 cells/mm3 . Examinationof the retinas of both eyes shows whiteareas with indistinct borders. Which of thefollowing pathogens is the most likelycausal agent?
O A. Candida albicansO B. CytomegalovirusO C. Herpes simplex virus type 1O D. HIVO E. Toxoplasma gondii
49. A 22-year-old woman who is anxiousabout her upcoming wedding has numb-ness and tingling at the tips of her fingers.Her right hand shows adduction of thethumb into the palm when provoked by in-flating the sphygmomanometer cuff. Whichof the following is most likely responsiblefor these findings?
O A. Diabetic ketoacidosisO B. Nephrotic syndromeO C. Primary hyperparathyroidismO D. Respiratory alkalosisO E. Sarcoidosis
50. An 11-year-old boy has enlarged,nontender testicles, a long face with aprominent jaw, a high arched palate, andprotruding ears. The child is in a specialeducational program at school. Which ofthe following studies is most useful in eval-uating this patient?
O A. Buccal smearO B. DNA analysis of the X chromosomeO C. Human chorionic gonadotropinO D. Serum gonadotropinsO E. Testicular biopsy


ANSWERS AND DISCUSSIONS
1. C (increased concentration of glycerol 3-phosphate) is correct.Glycerol 3-phosphate concentration increases during alcoholmetabolism because of increased production of NADH. Thefigure shows triacylglycerol as clear spaces in the cytosol of hepa-tocytes. The increase in glycerol 3-phosphate, the glycolytic in-termediate used to synthesize triacylglycerol in the liver, leads tothe accumulation of triacylglycerol. The fatty change shown inthe figure is typical of alcoholic liver disease. The hepatomeg-aly and slight elevation in serum transaminase found in thispatient are also characteristic.
A (decreased hydrolysis of adipose) is incorrect. Hydrolysis ofadipose is enhanced by alcohol and would increase (not de-crease) in a patient with a history of alcohol abuse.
B (decreased synthesis of fatty acids) is incorrect. Because theprimary substrate for fatty acid synthesis (acetyl CoA) is aby-product of alcohol metabolism, the synthesis of fatty acidswould increase (not decrease) in a patient with a history ofalcohol abuse.
I) (increased [3-oxidation of fatty acids) is incorrect. TheB-oxidation of fatty acids decreases during alcohol metabolismbecause of an increase in malonyl CoA, which inhibits therate-limiting enzyme (carnitine acyltransferase) and preventsfatty acid metabolism. Thus, the fl-oxidation of fatty acidswould decrease (not increase) in a patient with a history ofalcohol abuse.
E (increased protein intake) is incorrect. Increased protein intakedoes not produce the fatty change in the liver shown in thefigure.
2. A (balanced translocation) is correct. The child has Down syn-drome (epicanthal folds, flat nasal bridge, simian creases). Thepresence of 46 chromosomes in the child indicates that a trans-located chromosome, inherited from one of the parents, is re-sponsible. Translocation occurs when one part of a chromosomeis transferred to a nonhomologous chromosome. In balanced(robertsonian) translocation, the translocated fragment is func-tional. In this case, the long arm of chromosome 21 was translo-cated onto chromosome 14 in the mother, creating one longchromosome. The translocated chromosome was inherited bythe child as a second chromosome 14 that functions as a thirdchromosome 21.
B (frameshift mutation) is incorrect. The child's facial featuresare characteristic of Down syndrome, which is not caused bya frameshift mutation. In a frameshift mutation, nude-
357

358 Pathology
otides are inserted into or deleted from a DNA strand, resultingin synthesis of an abnormal protein product (e.g., Tay-Sachsdisease).
C (microdeletion) is incorrect. The child's facial features arecharacteristic of Down syndrome, which is not caused bymicrodeletion. Microdeletion involves the loss of a smallportion of one chromosome, which can be detected only byhigh-resolution techniques.
L) (nondisjunction) is incorrect. Nondisjunction refers tounequal separation of chromosomes in the first meiotic phase,resulting in an egg or a sperm with 22 or 24 chromosomes.Nondisjunction is responsible for most numeric chromosomedisorders (e.g., trisomy 21), but because this patient has46 chromosomes, a different type of mutation is responsible.(point mutation) is incorrect. A point mutation involves thesubstitution of a single nucleotide base.
3. E (nitrogen gas embolism) is correct. The patient has decom-pression sickness (gas embolism), which is a complication ofscuba diving. As a diver descends, the atmospheric pressure in-creases by 1 atmosphere (760 mm Hg) for every 33 feet of water.Under increased pressure, nitrogen gas moves from the alveolithrough the blood and dissolves in tissue and blood. Rapidascent forces nitrogen to move out of tissue and blood asbubbles. It forms gas emboli in the blood that obstruct bloodvessels, causing ischemic damage to bone (e.g., aseptic necrosisof the femoral head), spinal cord (hemiparesis), and othertissues. Gas bubbles within skeletal muscle and supportingtissues around joints cause a painful condition called the bends(decompression sickness). Treatment involves recompressionin a compression chamber to force the gas bubbles back intosolution followed by slow decompression to prevent them fromre-forming.
A (deep venous thrombosis) is incorrect. Increased atmosphericpressure under water may cause stasis and thrombus forma-tion in the deep veins of the leg, potentially causing pulmo-nary thromboembolism. Leg pain in deep venous thrombosisdevelops while under water.
B (disseminated intravascular coagulation) is incorrect. Dissemi-nated intravascular coagulation (DIC) is caused by in vivo acti-vation of the coagulation system, resulting in the formationof fibrin clots throughout the microvasculature. DIC is nota complication of scuba diving.(fat embolization) is incorrect. Fat embolization is most oftencaused by traumatic fractures of the long bones (e.g., femur)and pelvis. Microglobules of fat from the bone marrow andadipose lodge in the microvasculature throughout the body,producing ischemic damage to tissue.

Answers and discussions 359
D (hemorrhage into muscles and joints) is incorrect. Hemor-rhage into muscles and joints is a complication of a severecoagulation factor deficiency (e.g., hemophilia A). Such hem-orrhage is not associated with nitrogen gas embolism.
4. A (decrease in serum albumin) is correct. Child A has kwashior-kor, and child B has marasmus. Both conditions are examples ofprotein-energy malnutrition. Kwashiorkor is caused by de-creased protein intake, which decreases the serum albumin level.However, the total calorie intake is normal because of increasedintake of carbohydrates. A decrease in albumin decreases theplasma oncotic pressure, resulting in dependent edema (seefigure). In marasmus, serum albumin levels are usually normalin spite of diminished intake of calories.
(decrease in somatic protein stores) is incorrect. Somaticprotein stores represent the protein in muscle. In marasmus,somatic protein stores are decreased, resulting in musclewasting in the extremities (see figure). in kwashiorkor, somaticproteins are relatively intact; however, the visceral proteinstores (e.g., in the liver) are decreased.
C. (decrease in subcutaneous fat) is incorrect. In marasmus,subcutaneous fat is absent: in kwashiorkor, it is normal.
D (decrease in total calorie intake) is incorrect. In marasmus,the total calorie intake is decreased; in kwashiorkor, the totalcalorie intake is normal but deficient in protein.
5. E (recurrent immune reaction against group A streptococci) iscorrect. The patient had chronic rheumatic fever and developedmitral stenosis after recurrent infections by group A strepto-cocci. The most common cause of mitral stenosis is rheumaticfever. Antibodies against the M proteins of group A streptococcicross-react with antigens present in valvular tissue. Repeatedimmune reactions lead to stenosis of the valve, with eventualleft-sided and right-sided heart failure.
A (immune reaction in systemic lupus erythematosus) is incor-rect. SLE produces sterile vegetations on the mitral valve(Libman-Sacks endocarditis), but this does not cause mitralstenosis.
B (ischemic heart disease) is incorrect. Ischemic heart diseasedoes not cause valvular disease.
C (myxomatous degeneration) is incorrect. Myxomatous degen-eration is the characteristic histologic finding in rnitral valveprolapse, which is associated with mitral regurgitationrather than mitral stenosis.
D (recurrent bacterial endocarditis) is incorrect. Recurrent bacte-rial infection of the mitral valve leads to its destruction, result-ing in mitral regurgitation rather than mitral stenosis.

360 Pathology
6. C (leukocytes for Philadelphia chromosome) is correct.This patient has chronic myelogenous leukemia (CML). CMLoccurs in patients between 40 and 60 years of age. It is causedby translocation of the ABL proto-oncogene on chromosome9 to chromosome 22 (Philadelphia chromosome), where itforms a fusion gene. The Philadelphia chromosome is presentin > 95% of patients with CML. The presence of neutrophils inall stages of development and a myeloblast count of < 10% inthe bone marrow indicate a chronic (not an acute) leukemia.The peripheral blood smear shows a marked increase in thenumber of leukocytes and basophils (cells with dark granules);segmented and band neutrophils, myelocytes, and metamy-elocytes are prominent. Leukemias commonly metastasize tothe lymph nodes (lymphadenopathy), liver, and spleen(hepatosplenomegaly).
A (leukocytes for alkaline phosphatase) is incorrect. Benignneutrophils contain alkaline phosphatase (neoplastic neu-trophils do not). This patient has CML, in which leukocytesare negative for alkaline phosphatase.
B (leukocytes CD10) is incorrect. CD10 in leukocytes is themarker for the common acute lymphoblastic leukemia antigen(CALLA), which is present in early pre-B-cell types of acutelymphoblastic leukemia.
D (leukocytes for tartrate-resistant acid phosphatase) is incorrect.Leukemia cells positive for tartrate-resistant acid phosphataseare present in hairy cell leukemia, a type of B-cell leukemia.The leukemia cells have hair-like cytoplasmic projections inthe peripheral blood and do not contain Auer rods.
E (leukocytes for terminal deoxynucleotidyl transferase) is incor-rect. Terminal deoxynucleotidyl transferase is present in earlypre-B-cell types of acute lymphoblastic leukemia.
7. D (increased residual volume) is correct. The patient has aclassic history and findings of emphysema (chronic dyspnea,hyperinflated lungs, depressed diaphragms) related to a longhistory of smoking cigarettes. Emphysema is a chronic obstruc-tive pulmonary disease involving permanent enlargement ofall or part of the respiratory unit (respiratory bronchioles, alveo-lar ducts, and alveoli). Elastic tissue destruction in these airwayscauses trapping of air and distention of the distal air space. Thisincreases the residual volume, which is the volume of air leftin the lung after maximal expiration. An increase in residualvolume automatically increases total lung capacity, which causeshyperinflation of the lungs, an increase in the anteroposteriordiameter, depression of the diaphragms, and vertical orientationof the heart.
A (decreased functional residual capacity) is incorrect. The func-tional residual capacity is the total amount of air in the lungsat the end of a normal expiration. It is the sum of the expi-

Answers and Discussions 361
ratory reserve volume (amount of air forcibly expelled at theend of a normal expiration) and the residual volume. Anincrease in residual volume increases the functional residualcapacity.(decreased total lung capacity) is incorrect. An increase inresidual volume automatically increases total lung capacity.
C (increased FEV,:FVC ratio) is incorrect. In emphysema, lungcompliance (ability to fill the lung with air) is increased andelasticity (recoil of the lung) is decreased because of de-struction of elastic tissue. The FEV,, or the amount of air ex-pelled from the lungs in 1 second after a maximal inspiration,is decreased (e.g., to 1 L from the normal 4 L) because of thetrapping of air in the distended distal airways. The FVC, ortotal amount of air expelled after a maximal inspiration, isalso decreased (e.g., to 3 L from the normal 5 L). Therefore,the ratio of FEV, to FVC is decreased in emphysema.(increased tidal volume) is incorrect. The patient has emphy-sema, with increased residual volume. As the residual volumeincreases, the tidal volume (volume of air that enters orleaves the lungs during normal quiet respiration) is decreased.
8. C (Cryptococcus neoformans) is correct. This patient has crypto-coccosis caused by C. neoformans. Cryptococcosis is commonlycontracted by exposure to pigeon excreta, which is oftenfound under bridges where pigeons roost. C. neoformans pro-duces granulomatous inflammation with caseous necrosis inthe lungs.
A (Aspergillus llnigatus) is incorrect. A. finnigatus producesnarrow-angled septate hyphae and fruiting bodies withchains of parallel aligned conidia, unlike the yeast formsof C. neofornums.
B (Coccidioides immitis) is incorrect. En the lungs, C. immitis pro-duces spherules containing endospores, unlike the yeast formsof C. neoformans. C. immitis pneumonia is contracted primar-ily in the desert regions of the southwestern United States,particularly in Arizona, New Mexico, and southern California.
D (Histoplasma capsulation) is incorrect. The yeast forms ofH. capsulatum are phagocytosed by macrophages, unlikethe yeast forms of C. neoformans. Histoplasmosis is commonin immunocompromised patients (e.g., AIDS).
E (Pneumocystis carinii) is incorrect. On silver staining, P. cariniiappears as cysts with centrally located dots, unlike the yeastforms of C. neoformans. P. carinii usually produces pneumoniain patients with HIV when the CD4 T-cell count is about200/mm3.

362 Pathology
9. A (aspirin) is correct. The arrow in the figure points to a plate-let thrombus, which is composed of aggregated platelets boundtogether by fibrin. Aspirin prevents platelet aggregation by inhib-iting platelet cyclooxygenase activity. This prevents the produc-tion of prostaglandin H2 and its conversion to thromboxaneA2, a vasoconstrictor and potent platelet aggregator.
B (glycoprotein IIb/IIIa inhibitor) is incorrect. The arrow pointsto a platelet thrombus, which is composed of aggregatedplatelets held together by fibrin. Glycoprotein IIb/IIIa inhibi-tors prevent the attachment of fibrinogen to receptors onthe platelet membrane and thus prevent platelet aggregation.Glycoprotein IIb/IIIa inhibitors are most often used toprevent thrombosis after angioplasty.
C (heparin) is incorrect. The arrow points to a platelet thrombus,which is composed of aggregated platelets held together byfibrin. Heparin is an anticoagulant that inhibits the formationof fibrin clots, but it does not prevent platelet aggregation orthe formation of platelet thrombi.
D (tissue plasminogen activator) is incorrect. The arrow points toa platelet thrombus, which is composed of aggregated plate-lets held together by fibrin. A tPA is used primarily to break upan existing platelet thrombus by producing plasmin, whichbreaks up the fibrin strands holding a thrombus together, al-lowing reperfusion of the heart and preventing further ex-tension of an area of infarction. A tPA is not used to preventthrombus formation.
E (warfarin) is incorrect. The arrow points to a platelet throm-bus, which is composed of aggregated platelets held togetherby fibrin. Warfarin is an anticoagulant that inhibits the forma-tion of venous clots, and it does not prevent platelet aggrega-tion or the formation of a platelet thrombus.
10. D (unilateral renal artery stenosis caused by atherosclerosis)is correct. The patient has renovascular hypertension. Thiscondition is most often caused by an atherosclerotic plaquenarrowing the orifice of the renal artery in men older than50 years of age. A decrease in renal blood flow activates therenin-angiotensin-aldosterone system, resulting in hypertensioncaused by renal retention of sodium by aldosterone and vaso-constriction of the peripheral resistance arterioles by angiotensinII. Plasma renin activity is increased in the affected kidney anddecreased in the contralateral kidney. Narrowing of the orifice ofthe renal artery produces an epigastric bruit, causing atrophy ofthe affected kidney.
A (adrenal tumor that produces excess aldosterone) is incorrect.An adrenal adenoma that produces aldosterone (primary aldo-steronism) causes hypertension due to retention of sodium.Plasma renin activity is decreased, and an epigastric bruit isnot present.

Answers and Discussions 363
(adrenal tumor that produces excess catecholamines) is incor-rect. An adrenal tumor that produces catecholamines causinghypertension of adults is a pheochromocytoma, a benigntumor arising from the adrenal medulla. Classic findings asso-ciated with pheochromocytomas, such as sweating, anxiety,and paroxysms of hypertension, are not present in thispatient.
C (essential hypertension with bilateral nephrosclerosis) is incor-rect. Nephrosclerosis is the renal disease associated with essen-tial hypertension. It is caused by hyaline arteriolosclerosis ofarterioles in the kidneys that leads to loss of tubules andglomeruli, causing atrophy of both kidneys. It is not associ-ated with an epigastric bruit.
F (unilateral renal artery stenosis caused by fibromuscular hyper-plasia) is incorrect. Fibromuscular hyperplasia of the renal ar-teries is the primary cause of renovascular hypertension inwomen between the ages of 30 and 50 years.
11. B (coccidioidomycosis) is correct. This patient has coccidioido-mycosis caused by Coccidioides immitis. This infection occurs indesert regions of the southwestern United States, particularlyin Arizona, New Mexico, and southern California. It is con-tracted by inhalation of arthrospores in the dust. Erythemanodosum produces painful nodules in the lower extremities.The lesions are associated with inflammation of subcutaneousfat and are commonly associated with coccidioidomycosis.
A (aspergillosis) is incorrect. Aspergillus tiimisatus producesnarrow-angled septate hyphae with fruiting bodies, unlikethe spherules containing endospores of C. immitis.
C (cryptococcosis) is incorrect. Ciyptococcus neofirmans has yeastforms with narrow-based buds, unlike the spherules contain-ing endospores of C. immitis.
D (histoplasmosis) is incorrect. Histoplasma capsulatum has yeastforms phagocytosed by macrophages, unlike the spherulescontaining endospores of C. immitis. Histoplasmosis isendemic in the Ohio River and central Mississippi Rivervalleys.
E (tuberculosis) is incorrect. Mycobacterium tuberculosis is anacid-fast bacterium that is usually phagocytosed by alveolarmacrophages. Acid-fast stains are necessary to visualizeM. tuberculosis.

364 Pathology
12. D (small bowel adhesions) is correct. The patient's history ofcolicky pain (pain followed by a pain-free interval) is characteris-tic of a small bowel obstruction. The history of Crohn's diseaseand intestinal adhesions resulting from abdominal surgeryare the most likely cause of the obstruction. A radiograph insmall bowel obstruction shows multiple air-fluid levels with astepladder configuration.
A (direct inguinal hernia) is incorrect. Direct inguinal herniasproduce a bulge in the middle of the triangle of Hesselbach,which is located above the inguinal ligament. The bulgeappears when the patient is standing and disappears when thepatient is lying down. This type of hernia is not associatedwith entrapment of bowel leading to small bowel obstruction.
• (intussusception) is incorrect. An intussusception, or telescop-ing of a portion of bowel into another portion of bowel, is un-common in adults. The nidus for intussusception in adultsusually is a polyp or cancer. Obstruction and ischemic damagewith bloody diarrhea usually are present.(large bowel infarction) is incorrect. Large bowel infarctionscause bloody diarrhea and localized, noncolicky abdominalpain. Atherosclerosis of a mesenteric artery is the mostcommon cause of large bowel infarction.(volvulus) is incorrect. Volvulus occurs when bowel (sigmoidcolon or cecum) twists around the mesenteric root, resulting inobstruction and strangulation. The affected bowel is dis-tended and visible on a plain abdominal radiograph.
13. E (viral myocarditis) is correct. The specimen shows lympho-cytic infiltrate and dissolution of myocardial fibers, which arecharacteristic of a viral-induced acute myocarditis. Clinical find-ings include left-sided heart failure (dyspnea, bibasilar rales);right-sided heart failure (neck vein distention, hepatomegaly,dependent pitting edema); and myocardial damage (increasedcardiac troponin levels). Coxsackievirus is the most commonviral cause of myocarditis.
41)•••••••••••••••••••••••
A (congestive cardiomyopathy) is incorrect. Congestive cardio- •myopathy is characterized by an enlarged heart with dilation •and may be caused by alcohol abuse, but it is not associatedwith the presence of a lymphocytic infiltrate on biopsy or withan increase in cardiac troponin levels.
• (coronary artery thrombosis) is incorrect. Histologic features ofthrombosis leading to a myocardial infarction include coagula-tion necrosis (loss of nuclei and cross-striations) and a neu-trophilic infiltrate (not shown in the figure).
• (ischemic heart disease) is incorrect. Chronic ischemic heartdisease is associated with replacement of myocardial tissue bycollagen, which is not shown in the figure.
r) (rheumatic fever) is incorrect. A patient with rheumatic feverwould have a history of group A streptococcal infection,
•••••••••

•
•,Nr,swers D,6cL.ssions 365
• which this patient does not have. Other features of acute
•rheumatic fever, such as heart murmur, polyarthritis, and ery-thema marginatum, are also not present.•
• 14. C (Goodpasture's syndrome) is correct. Goodpasture's syn-drome is more common in men and is associated with IgG anti–basement membrane antibodies that are directed against pul-
• monary capillary and glomerular capillary basement membranes.The figure shows uninterrupted (linear) smooth imrnunofluo-
• rescence along the glomerular basement membrane. Pulmonary
•involvement with hemoptysis usually occurs before renalfailure. Renal failure is most often due to crescentic glomerulo-
• nephritis, which is associated with a nephritic presentation(hematuria, RBC casts, mild proteinuria), as in this case.•
•A (diffuse membranous glomerulonephritis) is incorrect. Diffuse
membranous glomerulonephritis is the most common cause of
•the nephrotic syndrome in adults (pitting edema, fatty casts).It is an immunocomplex disorder with deposition of immuno-
• complexes in a subepithelial location, which produce granu-• lar immunofluorescence.
li (focal segmental glomerulosclerosis) is incorrect. Focal seg-• mental glomerulosclerosis is most often associated with AIDS
and with heroin addiction. It causes the nephrotic syn-drome. Glomerular injury is due to cytokine damage of the
•visceral epithelial cells.
D (IgA glomerulonephritis) is incorrect. IgA glomerulonephritis
• is an immunocomplex type of glomerulonephritis associated
•with episodic bouts of microscopic or macroscopic hematuria.The immunocomplexes produce granular i mmunofluores-
• cence primarily located in the mesangium.(minimal change disease) is incorrect. Minimal change diseaseis the most common cause of the nephrotic syndrome in chil-
• dren. Cytokine damage to the basement membrane causes aloss of the negative charge, resulting in a selective loss of
• albumin in the urine. Patients usually are normotensive.
•• ls. It (chronic pancreatitis) is correct. The irregular densities in theleft upper quadrant of the radiograph represent foci of dystro-
• phic calcification (calcification of damaged tissue) in the paren-chyma of the pancreas. This is presumptive evidence of
• chronic pancreatitis, which in most cases is associated with
•chronic alcohol abuse. In chronic pancreatitis, recurrent attacksof acute pancreatitis lead to repair by fibrosis and loss of both
• exocrine and endocrine function. Loss of the pancreatic enzymesresults in malabsorption, which is the cause of the patient's
• chronic diarrhea and malnutrition.
• A (celiac disease) is incorrect. Celiac disease is an autoimmune
•disease that destroys intestinal villi, leading to malabsorptionof fat, proteins, and carbohydrates. The radiograph showsI
•

366 Pathology
dystrophic calcification in the parenchyma of the pancreas,which is not associated with celiac disease.
C (Crohn's disease) is incorrect. Crohn's disease is a chronic ul-cerative inflammatory disease that involves the terminal ileumand portions of the large intestine. The radiograph showsdystrophic calcification in the parenchyma of the pancreas,which is not associated with Crohn's disease.
D (pancreatic carcinoma) is incorrect. Most pancreatic carcino-mas involve the head of the pancreas, with subsequent ob-struction of the common bile duct. This leads to jaundice,which is not present in this patient.(peptic ulcer disease) is incorrect. Peptic ulcer disease due toduodenal ulcers that penetrate posteriorly may produce acutepancreatitis. However, this is an unlikely cause of chronicpancreatitis.
16. D (serum ferriti c) test) is correct. The patient's clinical andlaboratory findings are consistent with the diagnosis of an iron-deficiency anemia, which can be confirmed by the serum ferritintest. The peripheral blood smear shows RBCs with notablecentral areas of pallor, which indicate decreased hemoglobinconcentration. Strong evidence of iron deficiency is given by theincreased RBC distribution width (increased size variation inRBCs) and the patient's history of dysfunctional uterine bleeding(menorrhagia), which is the most common cause of iron defi-ciency in women younger than 50 years of age. The serum ferri-tin test is the best screening test for iron-related disorders,because serum ferritin levels correlate with the amount of ironstored in the bone marrow.
A (bone marrow aspiration biopsy) is incorrect. The patient has amicrocytic anemia. A bone marrow aspiration biopsy is rarelyindicated in workups for microcytic anemias, because serumtests (e.g., ferritin) are available to identify the most commonsuch anemias.
B (hemoglobin electrophoresis) is incorrect. Hemoglobin electro-phoresis identifies changes in the concentration of normal andabnormal forms of hemoglobin. The patient does not havethalassemia (normal RBC distribution width); therefore, ahemoglobin electrophoresis is not indicated.
C (osmotic fragility test) is incorrect. The osmotic fragility test isused to confirm a diagnosis of hereditary spherocytosis, inwhich the osmotic fragility of RBCs is increased. Hereditaryspherocytosis is a normocytic (not microcytic) anemia, andspherocytes are not present in the peripheral blood.
L (sickle cell screening) is incorrect. Although the patient is ofAfrican descent, sickle cell anemia is unlikely, because it isusually normocytic (not microcytic) and because sickle cellsare not present in the peripheral blood.

Answers and Discussions 367
17. E (osmotic damage) is correct. Microaneurysms are due toosmotic damage of the pericytes that surround the vessel. Peri-cytes contain aldose reductase, which converts glucose tosorhitol. Sorbitol is osmotically active and draws water into thepericyte, leading to its destruction. This weakens the vessel wall,causing formation of a microaneurysm. Rupture of aneurysmscan cause retinal detachment and neovascularization of theretina, resulting in blindness.
A (increased intraocular pressure) is incorrect. An increase in in-traocular pressure occurs in glaucoma. It may be due to anarrow anterior chamber angle or to abnormal drainage of theaqueous fluid through the trabecular meshwork. Pathologiccupping of the optic nerve usually is seen in long-standingglaucoma. This change is not present in the retina.
B (inflammation of the optic nerve) is incorrect. Optic neuritis(inflammation of the optic nerve) is most commonly due tomultiple sclerosis. The optic disk is swollen, and flame hemor-rhages are seen surrounding the disk. These changes are notpresent in the retina.
C (microangiopathy) is incorrect. Microangiopathy is a findingin patients with diabetes mellitus and signals the deposition ofincreased type IV collagen in the basement membrane ofsmall vessels. These changes usually occur in the kidneysrather than in the retina.
D (nonenzymatic glycosylation) is incorrect. Nonenzymatic gly-cosylation occurs in patients with diabetes mellitus and marksthe binding of glucose to amino acids in protein within thebasement membrane of vessels and in proteins such as hemo-globin. In blood vessels, it causes increased vessel permeabil-ity in arterioles, leading to hyaline arteriolosclerosis. Thesechanges are most prominent in the kidneys and are not re-sponsible for producing microaneurysms in the retina.
18. E (unopposed estrogen exposure) is correct. The figure shows ahemorrhagic and necrotic tumor filling the endometrial cavityand extending through the wall of the uterus. This patient hasendometrial carcinoma, which is most often due to unop-posed estrogen exposure. Excessive estrogen stimulation of theendometrial mucosa initially causes endometrial hyperplasia,which becomes atypical and progresses to cancer. Early menar-che and late menopause, obesity, taking estrogen withoutprogesterone, and niiil i parity are potential risk factors for endo-metrial adenocarcinoma.
A (adenomyosis) is incorrect. Adenomyosis is the presence ofnormal endometrial glands and stroma within the myo-metrium. It is due to invagination of the stratum basalis tothe myometrial tissue. It causes thickening of the myometrialtissue, leading to menstrual irregularities.
B (herpes simplex virus type 2 infection) and C (human papillo-

368 Pathology
mavirus infection) are incorrect. High-risk types of HPV (e.g.,types 16 and 18) predispose females to squamous cell carci-noma of the vulva, vagina, and cervix. HSV-2 is not an onco-genic virus.
D (multiparity) is incorrect. Multiparity reduces the risk ofcancer because progesterone opposes hyperestrinism. Referto the discussion for E.
19. ( (neutrophils with phagocytosed negative birefringent crys-tals) is correct. The patient has acute gouty arthritis (inflamedmetatarsophalangeal joint in the great toe). In gout, there is de-position of monosodium urate (MSU) crystals in the joint. MSUcrystals are phagocytosed by neutrophils, which causes themto release inflammatory mediators, leading to inflammation ofsynovial tissue. Negative birefringence is defined by colorchanges in the crystals that occur when examined under a mi-croscope with compensated polarized light. MSU crystals areyellow when they are aligned parallel to the slow ray of the com-pensator in the microscope. Excess alcohol intake frequentlyprecipitates acute gouty arthritis, because products of alcoholmetabolism lead to underexcretion of uric acid by the kidneys.
A (neutrophils with phagocytosed gram-negative diplococci) isincorrect. Septic arthritis due to Neisseria gonorrhoeae, a gram-negative diplococcus, usually involves the knee.
B (neutrophils with phagocytosed gram-positive cocci) is incor-rect. Staphylococcus aureus, a gram-positive coccus, is the mostcommon nongonococcal cause of septic arthritis. Both thehistory of alcohol abuse and the location of the inflammatoryarthritis in the great toe are contrary to a diagnosis of septicarthritis.
D (neutrophils with phagocytosed positive birefringent crystals)is incorrect. Calcium pyrophosphate crystals have positivebirefringence. The crystals are either needle- or rhomboid-shaped. Under compensated polarized light, the crystals areblue when aligned parallel to the slow ray of the compensator.Arthropathy associated with these crystals usually involvesthe knee.
E (neutrophils with phagocytosed rheumatoid factor immuno-complexes) is incorrect. Rheumatoid arthritis does not involvethe metatarsophalangeal joint of the great toe.

Answers and Discussions 369
20. B (chronic) is correct. A chronic inflammatory infiltrate isshown in the figure. It consists of reactive plasma cells with pe-ripherally located nuclei and perinuclear clearing, as well as lym-phocytes with round nuclei and scant cytoplasm. A few of theplasma cells are multinucleated. The patient's clinical history in-dicates rheumatoid arthritis, a chronic inflammatory diseaseassociated with destruction of articular cartilage caused by anovergrowth of hyperplastic synovial tissue (pannus).
A (acute) is incorrect. Acute inflammation is characterized by thepresence of neutrophils, which have multilobed nuclei andgranular cytoplasm. Neutrophils are not present in thespecimen.
C (granulomatous) is incorrect. Granulomatous inflammation isassociated with the formation of well-circumscribed granulo-mas that contain macrophages, lymphocytes, and multinucle-ated giant cells. Granulomas are not present in the specimen.
D (pseudomembranous) is incorrect. Pseudomembranous in-flammation is mucosal damage induced by bacterial toxins,which produce a shaggy membrane (pseudomembrane) com-posed of necrotic tissue. Pseudomembrane formation is notevident in the specimen.
E (suppurative) is incorrect. Suppurative inflammation is a typeof acute inflammation caused by bacterial pathogens and char-acterized by the excessive production of exudate (pus), whichis not shown in the specimen.
21. B (Chlamydia trachomatis) is correct. The patient has cervicitisdue to C. trachomatis. Two distinct forms of the organism thatdevelop in metaplastic squamous cells are the elementary body(metabolically inert but infective) and the reticulate body (meta-bolically active but not infective). Binary fission of the reticu-late bodies in the phagosome of the infected cell results in theproduction of numerous elementary bodies, which are visible asan inclusion in the phagosome. Urinalysis findings suggestacute urethral syndrome, which is also due to C. trachomatis.
A (Candida albicans) is incorrect. Candida causes vaginal mucosalinflammation with a white discharge containing yeasts andpseudohyphae. Irritation of the urethra can cause dysuria.
C (human papillomavirus) is incorrect. HPV produces koilocy-totic atypia in squamous cells, which is characterized by ahalo surrounding a pyknotic (dense) nucleus.
D (Neisseria gonorrhoeae) is incorrect. N. gonorrhoeae causes cervi-citis and urethritis. The gram-negative diplococci are notusually visible with Pap stains.
E (Trichomonas vaginalis) is incorrect. Trichomonas causes cervici-tis and urethritis. The pear-shaped organisms have flagella.

370 Pathology
22. C (increased plasma ACTH and decreased serum cortisol) iscorrect. Hyperpigmentation of the buccal mucosa, plus a historyof fatigue, weakness, and signs of hypovolemia when supine(decreased blood pressure and increased pulse rate), suggests adiagnosis of Addison's disease. Most cases of Addison's diseaseare due to autoimmune destruction of the adrenal cortex.This produces deficiencies of mineralocorticoids (e.g., aldoste-rone), glucocorticoids (e.g., cortisol), and sex hormones (e.g.,androstenedione). Hypocortisolism causes an increase in plasmaACTH due to a negative feedback relationship. ACTH hasmelanocyte-stimulating properties that increase the synthesisof melanin on the skin and mucosal surfaces. Hypovolemia isrelated to the loss of sodium in the urine due to aldosteronedeficiency.
A (decreased plasma ACTH and 11-deoxycortisol aftermetyrapone stimulation) and D (increased plasma ACTHand 11-deoxycortisol after metyrapone stimulation) are incor-rect. Metyrapone is a drug that blocks 11-hydroxylase in theadrenal cortex. This enzyme is normally responsible forconversion of the glucocorticoid 11-deoxycortisol to cortisol.Hence, a normal response to metyrapone is a decrease in corti-sol with a subsequent increase in ACTH and 11-deoxycortisolproximal to the enzyme block. If both plasma ACTH and11-deoxycortisol are decreased, then hypopituitarism causeshypocortisolism.
Li (decreased serum sodium and serum potassium) is incorrect.Aldosterone normally maintains a Na +-K+ pump that increasessodium reabsorption from urine in exchange for potassium,which is lost in the urine. In hypoaldosteronism, Na + is lost inthe urine (which causes hyponatremia) and there is reten-tion of K+ (which causes hyperkalemia).
E (increased urine 17-hydroxycorticoids with prolonged ACTHstimulation) is incorrect. The adrenal cortex is destroyed;therefore, urine 17-hydroxycorticoids (cortisol and 11-deoxycortisol) remain decreased after prolonged ACTH stimu-lation if the patient has Addison's disease. Patients with hy-popituitarism have increased 17-hydroxycorticoids in theirurine after prolonged ACTH stimulation.

Answers and Discussions 371
23. A (caseous necrosis) is correct. Caseous necrosis typicallyappears as a well-circumscribed granuloma with amorphousmaterial in the center, as shown in the specimen. The granulomais surrounded by an inflammatory infiltrate consisting of lym-phocytes, macrophages, and a few multinucleated giant cells,which can be seen at the top of the specimen. Caseous necrosisoccurs primarily in patients with mycobacterial infection(tuberculosis in this patient) or systemic fungal infection(e.g., histoplasmosis).
B (coagulation necrosis) is incorrect. Coagulation necrosis isusually associated with cessation of arterial blood flow totissue, often evidenced by the presence of an infarct and thepersistence of cellular outlines in the dead tissue. Thesefeatures are not present in the specimen.
C (enzymatic fat necrosis) is incorrect. Enzymatic fat necrosisoccurs in the pancreas, in adipose tissue in and around anarea of inflammation (as in acute pancreatitis). It does notoccur in the lung.
D (fibrinoid necrosis) is incorrect. Fibrinoid necrosis occurs insmall muscular arteries, arterioles, venules, and glomerularcapillaries. It is often associated with small-vessel vascul it is inhypertension or with deposits of antigen-antibody com-plexes in vessel walls in immunocomplex disease.
E (liquefactive necrosis) is incorrect. Liquefactive necrosisappears as an area of soft tissue with a liquid center, unlike thelesion shown in the specimen. The softening of tissue occursin liquefactive necrosis because of the release of hydrolyticenzymes from neutrophils, usually in acute bacterial infection.
24. D (inappropriate antidiuretic hormone secretion) is correct.The patient has small cell carcinoma of the lung (round- tospindle-shaped basophilic cells), which presents as a centrallylocated lung mass. Small cell carcinomas are neuroendocrinetumors that derive from Kulchitsky cells. These tumors maysecrete antidiuretic hormone or adrenocorticotrophic hormoneectopically, causing hyponatremia or hypercortisolism, re-spectively. Small cell carcinomas have a strong association withsmoking and have usually metastasized widely by the timethey are discovered.
A (carcinoid syndrome) is incorrect. Carcinoid syndrome iscaused by secretion of serotonin from a carcinoid tumorthat has metastasized to the liver. In most cases, the tumoris located in the terminal ileum. Bronchial carcinoids maysecrete serotonin without metastasis, but this is very uncom-mon. They are not associated with smoking.
B (hypercalcemia) is incorrect. Primary squamous carcinomas ofthe lung secrete parathyroid hormone–related peptide ectopi-cally, causing hypercalcemia. These tumors are centrallylocated and strongly associated with smoking, but H&E-

372 Pathology
stained sections show keratin pearls and eosinophilic-stainingcells, which are not present in this patient.
C (hypocalcemia) is incorrect. No primary cancer of the lungsecretes calcitonin to produce hypocalcemia.
E (polycythemia) is incorrect. No primary cancer of the lungsecretes erythropoietin to produce polycythemia.
25. D (neuromuscular) is correct. This patient has myastheniagravis, which is characterized by drooping eyelids, history oftiredness, diplopia (double vision), and dysphagia for solids andliquids in the upper esophagus. This autoimmune disorder ischaracterized by the production of IgG antibodies that reactagainst acetylcholine receptors in the neuromuscular junction ofstriated muscle. The most common initial presentation is muscleweakness involving the ocular muscles, resulting in ptosis anddiplopia toward the end of the day. Other muscle groups eventu-ally become involved. The IgG antibodies are produced in thethymus, where there are prominent germinal follicles (B-cell hy-perplasia). Dysphagia for solids and liquids occurs in the upperesophagus, because the primary muscle for motility is striatedmuscle.
A (demyelinating) is incorrect. Multiple sclerosis is the mostcommon demyelinating disorder. It is not associated withdrooping eyelids or motor problems in the esophagus.
It (electrolyte) is incorrect. Hypokalemic periodic paralysis is agenetic disease that causes muscle weakness and paralysis.Attacks produce a sudden onset of generalized muscle weak-ness and often are provoked by strenuous exercise or ahigh-carbohydrate meal.
C (motor neuron) is incorrect. Amyotrophic lateral sclerosis isan example of an upper and lower motor neuron disorder.Muscle weakness begins in the hands and progresses through-out the body.
IL (primary muscular) is incorrect. A primary muscular disorder(e.g., muscular dystrophy with a deficiency of dystrophin)mainly involves defects in skeletal muscle, rather than defectsin the transmission of the nerve impulse to muscle.
26. B (deficiency of dystrophin) is correct. The patient hasDuchenne's muscular dystrophy, an X-linked recessive disorderwith a deficiency of dystrophin. Dystrophin normally anchorsactin to membrane glycoprotein. To move into a standing posi-tion from a prone position, he must "walk" his hands to hisfeet and up the front of his legs and then move to a standingposition. There is generalized muscle atrophy and pseudohyper-trophy of the calf muscles. Weakening and wasting of proxi-mal pelvic muscles produce a waddling gait.
A (antibodies against acetylcholine receptors) is incorrect. Anti-bodies against acetylcholine receptors occurs in myasthenia

Answers and Discussions 373
gravis, an autoimmune disease that causes progressive muscleweakness and is commonly seen in women. IgG antibodiesare directed against acetylcholine receptors in the neuromus-cular junction of striated muscle.
C (demyelinating disorder) is incorrect. Demyelinating disordersare uncommon in children. They cause muscle weakness andsensory changes (paresthesias).
D (inflammatory myopathy) is incorrect. Polymyositis and der-matomyositis are examples of inflammatory myopathies thatcause pain and muscle atrophy. These diseases are uncommonmuscle diseases in children.
I (motor neuron disorder) is incorrect. Degeneration of upper orlower motor neurons is uncommon in children.
27. D (Nat and H20 moving into the cytosol) is correct. Blood lossdue to a ruptured abdominal aortic aneurysm causes tissuehypoxia (inadequate oxygenation of tissue). The straight portionof the proximal tubule is the part of the nephron that is mostsensitive to hypoxia, and it responds to loss of 0 2 first by de-creasing the synthesis of ATP in the mitochondria. This decreasein ATP affects the Na±, IC-ATPase pump by allowing I•la' andH20 to enter the cytosol, which results in cellular swelling. Intissue hypoxia, this swelling is reversible.
A (Ca2÷ moving into the cytosol) is incorrect. Lack of ATP causesCa2+ to move out of the mitochondria and the interstitialtissue into the cytosol of the cell. Ca2± activates enzymes inthe cell membrane (phospholipase), cytosol (proteases), andnucleus (endonucleases), irreversibly damaging the cell.
B (cytochrome c diffusing out of the mitochondria) is incorrect.Movement of cytochrome c out of the mitochondria stimu-lates apoptosis (death) of the cell.
C (intracellular pH increasing) is incorrect. In tissue hypoxia,anaerobic glycolysis is the key biochemical process usedby cells to produce ATP. Lactic acid is the final product ofanaerobic glycolysis and reduces the intracellular pH. Intra-cellular acidosis denatures enzymatic proteins in the cell,leading to coagulation necrosis.
E (phospholipase damaging the cell membrane) is incorrect.Activation of phospholipase by Ca 2+ irreversibly damages thecell membrane.

374 Pathology
28. A (antigliadin antibodies) is correct. This patient has celiacdisease. The figure shows villous atrophy (flat mucosa) and hy-perplastic glands with an increased number of chronic in-flammatory cells in the lamina propria. These findings, plus thehistory of chronic diarrhea with greasy stools, are features ofceliac disease, an autoimmune disease that has antibodiesagainst the gliadin fraction in gluten, which is present in wheatproducts. The vesicular lesion on the patient's elbow is derma-titis herpetiformis, an autoimmune skin disease that has analmost 100% correlation with underlying celiac disease. Otherantibodies present in celiac disease include antiendomysial andantireticulin antibodies.
B (antinuclear antibodies) is incorrect. The antibodies in celiacdisease are not directed against nuclear proteins.
C (fecal smear for leukocytes) is incorrect. A fecal smear forleukocytes is used for evaluating diarrhea that may becaused by invasive microbial pathogens (e.g., Campylobacterjejuni, Shigella sonnei). The presence of leukocytes presumes aninvasive enterocolitis.
L) (stool for ova and parasites) is incorrect. Testing the stoolfor ova and parasites usually is recommended in the workupof a patient with chronic diarrhea. Giardiasis is the mostcommon cause of chronic diarrhea associated with malabsorp-tion; however, the characteristic pear-shaped organisms arenot present in the figure.
E (stool osmotic gap) is incorrect. A stool sample to calculate theosmotic gap is used for high-volume diarrheal states when asecretory or osmotic type of diarrhea is suspected. Secretory di-arrheas are characterized by isotonic diarrheal fluid (e.g., dueto certain types of laxatives, enterotoxigenic bacteria), whereasosmotic diarrheas are characterized by hypotonic stool becauseof the presence of osmotically active solutes (e.g., lactase defi-ciency with an excess of lactose).
29. B (HLA-B27 genotype) is correct. The radiograph of the spineshowing forward curvature of the spine (kyphosis) with ankylo-sis (fusion) of the lumbar vertebrae, plus a history of lowerlumbar pain beginning in the sacroiliac joints, suggest ankylos-ing spondylitis. This disorder, which commonly occurs in males,is part of a related group of disorders called seronegativespondyloarthropathies. These disorders are characterized by astrong association with the HLA-B27 genotype, involvement ofthe sacroiliac joints, and arthritis that may involve peripheraljoints. The heart murmur in this patient is due to aortitis involv-ing the ascending aorta, which is often associated with aorticregurgitation (high-pitched diastolic murmur).
A (antibodies against Borrelia burgdorferi) is incorrect. Arthritisdue to B. burgdorferi (causative agent of Lyme disease) doesnot involve the vertebral joints.

Answers and Discussions 375
(hyperuricemia) is incorrect. Gouty arthritis usually involvesthe peripheral joints (e.g., first metatarsophalangeal joint),where it causes an erosive type of arthritis.
D (positive blood culture) is incorrect. The aortitis in ankylosingspondylitis does not have an infectious etiology.
E (rheumatoid factor) is incorrect. Rheumatoid factor usually isnot present in ankylosing spondylitis.
30. C (IgA glomerulonephritis) is correct. RBC casts, hematuria,and proteinuria are characteristic of a nephritic type of glomeru-lonephritis. The episodic history of hematuria following upperrespiratory infections and the absence of hypertension are char-acteristic of IgA glomerulonephritis, which is the most commontype of glomerulonephritis.
A (diffuse membranous glomerulonephritis) and D (minimalchange disease) are incorrect. These are associated with a ne-phrotic type of glomerular disease, which is characterized bymassive proteinuria (> 3.5 g/24 h), pitting edema, and fattycasts.
B (glomerulonephritis in systemic lupus erythematosus) is incor-rect. A negative serum ANA test excludes glomerulonephritisin SLE.
E (poststreptococcal glomerulonephritis) is incorrect. Negativeanti–streptolysin 0 and DNAse B titers and the absence of hy-pertension exclude poststreptococcal glomerulonephritis.
31. D (herpesvirus 8) is correct. The lesions shown are typical ofKaposi sarcoma, a vascular malignancy closely associated withherpesvirus 8. Kaposi sarcoma is the most common malignancyin patients with AIDS. Lesions appear most often on the skin butmay also occur in the intestinal tract, particularly on the hardpalate.
A (Bartonella henselae) is incorrect. B. henselae is a gram-negativebacterium that causes bacillary angiomatosis, a disease thatoccurs almost exclusively in patients with AIDS. It produceshighly vascular skin lesions that can mimic the lesions ofKaposi's sarcoma. Systemic signs of the infection include fever,lymphadenopathy, and hepatomegaly; these findings are notpresent in this patient.
li (cytomegalovirus) is incorrect. Cytomegalovirus (CMV) is notan oncogenic virus and does not produce vascular lesions onthe skin or in the oral cavity. In patients with AIDS, CMV isthe most common cause of blindness, binary tract disease, andpancreatitis.

376 Pathology
C (Epstein-Barr virus) is incorrect. In patients with AIDS,Epstein-Barr virus does not cause vascular skin lesions, al-though it may cause hairy leukoplakia (glossitis), primarycentral nervous system lymphoma, and Burkitt's lymphoma.
F. (HIV) is incorrect. HIV is not oncogenic and does not producevascular skin lesions. In patients with AIDS, it is associatedwith generalized lymphadenopathy, destruction of CD4 helperT cells, and many central nervous system findings (e.g., AIDSdementia).
32. A (decreased plasma oncotic pressure) is correct. Edema is theaccumulation of fluid in body cavities (e.g., ascites) and in theinterstitial space (e.g., peripheral edema). Edema caused bycirrhosis of the liver involves alterations in vascular hydrostaticpressure and in oncotic pressure. An increase in hydrostatic pres-sure and/or a decrease in plasma oncotic pressure (hypoalbu-minemia) cause outflow of a protein-poor fluid (< 3 g/dL) intobody cavities and interstitial spaces. This transudate (protein-poor fluid) is devoid of inflammatory cells. In cirrhosis, theportal vein encounters increased resistance to emptying bloodinto the liver sinusoids (intrasinusoidal hypertension) due tocompression of the sinusoids by regenerative nodules and fibro-sis. This causes increased hydrostatic pressure (portal hyper-tension) that contributes to ascites formation. The syntheticfunction of the liver is compromised in cirrhosis, and thus hypo-albuminemia occurs, which decreases the plasma oncotic pres-sure, further contributing to ascites and peripheral edema(dependent pitting edema).
B (increased plasma hydrostatic pressure) is incorrect. Increasedhydrostatic pressure is involved only in ascites formation.
C (increased vessel permeability due to histamine) is incorrect.Increased vessel permeability due to histamine, a marker ofacute inflammation, causes a nonpitting type of peripheraledema. The edema fluid is a protein-rich exudate (> 3 g/dL)that contains polymorphonuclear leukocytes. Exudatesalso accumulate in body cavities (e.g., pleural effusion inpneumonia).
D (lymphatic obstruction with lymphedema) is incorrect. Ob-struction of lymphatic channels causes leakage of lymphaticfluid into the interstitial space (e.g., filariasis), producing anonpitting lymphedema. Lymphatic fluid accumulates inbody cavities (e.g., chylous effusions in the pleural cavitiescaused by a tear in the thoracic duct).
E (movement of water into the intracellular compartment) isincorrect. Movement of water between the extracellular fluidcompartment (ECF) and the intracellular fluid compart-ment (ICF) is called osmosis. Alterations in the serum Na±concentration in the ECF compartment is the primary causeof water movement between the compartments. In hypo-natremia, water moves from the ECF into the ICF compart-

Answers and Discussions 377
ment, whereas in hypernatremia, water moves from the ICFinto the ECF compartment.
33. D (increased serum ferritin) is correct. The patient has hemo-chromatosis. Features of hemochromatosis include personaland family history of type 1 diabetes mellitus, cirrhosis, chronicdiarrhea, and a pale gray skin color. Hemochromatosis is anautosomal recessive disease in which increased reabsorptionof iron from the small intestine leads to iron overload in theliver (where it causes cirrhosis), pancreas (where it causes diabe-tes and malabsorption leading to diarrhea), and skin (where it in-creases the production of melanin). The term "bronze diabetes"is often applied to this condition.
A (decreased serum ceruloplasmin) is incorrect. Patientswith Wilson's disease have decreased serum ceruloplasmin.Wilson's disease, an autosomal recessive disorder, is associatedwith chronic liver disease and a movement disorder causedby defective secretion of copper into bile and reduced cerulo-plasmin synthesis in the liver. Excess copper deposition intissue does not cause skin discoloration or pancreatic insuffi-ciency; however, it does cause cirrhosis.
B (decreased serum iron) is incorrect. Patients with iron overloaddiseases have an increase in serum iron, ferritin, and percentsaturation of transferrin.
C (decreased small bowel reabsorption of D-xylose) is incorrect.D-Xylose absorption is an excellent screening test for smallbowel disease as a cause of malabsorption. Small bowel disease(e.g., celiac disease) is characterized by inability to reabsorborally administered D-xylose into the blood. The mechanismof malabsorption in hemochromatosis is deficiency of pancre-atic enzymes (e.g., lipase).
E (increased total iron-binding capacity) is incorrect. Patientswith iron overload diseases have an increase in serum iron,ferritin, and percent saturation of transferrin. However, thetotal iron-binding capacity is decreased because increased ironstores are associated with decreased synthesis of transferrin(binding protein of iron) in the liver.
34. B (midsystolic click followed by a murmur) is correct. Thefigure shows redundancy of the mitral valve, particularly inthe posterior leaflet on the right. A systolic click occurs whenthe valve prolapses into the left atrium during systole and issuddenly restrained by the chordae tendineae; the murmur iscaused by mitral regurgitation. Most patients with mitral valveprolapse are asymptomatic.
A (diastolic blowing murmur after S2) is incorrect. A diastolicblowing murmur after S2 characterizes aortic regurgitation,which causes volume overload of the left ventricle. The aorticvalve is not shown in the specimen.

378 Pathology
(opening snap followed by a mid-diastolic rumbling murmur)is incorrect. An opening snap followed by mid-diastolic rum-bling characterizes mitral stenosis, in which the leaflets ofthe mitral valve appear fibrotic or calcified, unlike those in thespecimen. Mitral stenosis is most often caused by chronicrheumatic fever.
D (pansystolic murmur at the apex) is incorrect. A pansystolicmurmur at the apex characterizes mitral regurgitation, whichcauses volume overload of the left ventricle. The mitral re-gurgitation in mitral valve prolapse follows a systolic clickand does not occur throughout systole.(systolic ejection murmur) is incorrect. A systolic ejectionmurmur characterizes aortic stenosis, in which the area ofthe valvular orifice is reduced. The aortic valve is not shownin the specimen.
35. A (group A (3-hemolytic streptococcus) is correct. The child hasimpetigo, which causes honey-colored crusted lesions that covershallow ulcerations of the skin. A group A streptococcus (Strep-tococcus pyogenes) is the most common cause of this superfi-cial skin lesion. If the lesion has a bullous component, Staphylo-coccus aureus is the most common causative agent. Impetigo ishighly contagious, which explains why the child's brother devel-ops similar lesions.
B (herpes simplex virus type 1) is incorrect. Herpes simplex virustype 1 produces vesicles and pustules on the vermilion borderof the lip.
C (Malassezia furfur) and E (Trichophyton rubrum) are incorrect.M. furfur causes tinea versicolor and seborrheic dermatitis(dandruff). T. rubrum causes tinea corporis (body), tinea cruris(groin), and tinea pedis (foot). Both pathogens are superfi-cial dermatophytes.
D (Propionibacterium acnes) is incorrect. P. acnes is an anaerobeinvolved in producing the inflammatory reaction associatedwith acne vulgaris.

Answers and Discussions 379
36. C (serum insulin-like growth factor—I) is correct. The patienthas acromegaly. Clinical findings of acromegaly include enlarge-ment of the nose and supraorbital ridge, jutting out of the lowerjaw (prognathism), and enlarged hands and feet. Pituitary ade-nomas secreting excess growth hormone are often quite largeand extend out of the sella turcica, causing headache, visual fielddefects, and hydrocephalus. Gigantism occurs if the tumor ispresent before the epiphyses have fused, whereas acromegalydevelops if the epiphyses have closed. Excess growth hormonecauses hyperglycemia (growth hormone is gluconeogenic)and increased amino acid uptake in muscle and other tissues.Excess growth hormone also stimulates the liver to synthesizeand release excess amounts of insulin-like growth factor—I. Thishormone increases linear and lateral bone growth and alsocauses visceromegaly. Although growth hormone is used as ascreening test in cases of suspected acromegaly, insulin-likegrowth factor—I is a more sensitive test.
A (serum cortisol) is incorrect. Serum cortisol is a screening testfor adrenocortical hypofunction or hyperfunction disorders,neither of which produces the changes noted in this patient.
B (serum glucose) is incorrect. Serum glucose is a screening testfor diabetes mellitus, types 1 and 2.
D (serum prolactin) is incorrect. Serum prolactin is used toscreen patients who have galactorrhea to rule out a pro-lactinoma.
E (serum thyroid-stimulating hormone) is incorrect. TSH is usedto screen for thyroid hypofunction and hyperfunction. TSH isdecreased in hyperthyroidism and hypopituitarism and in-creased in primary hypothyroidism.
37. C (hemochromatosis) is correct. Hemochromatosis, a disorderinvolving excessive reabsorption of iron from the gastrointesti-nal tract, has an autosomal recessive inheritance pattern, asshown in the pedigree. In autosomal recessive disorders, diseaseis present only in patients who are homozygous for the abnor-mal allele (e.g., aa), and both parents must have the abnor-mal allele to transmit the disease to their children. In most cases,the parents are asymptomatic heterozygous carriers (e.g., Aa).They have a 25% chance of having a normal child (AA), a 50%chance of having children who are asymptomatic carriers(Aa), and a 25% chance of having a child with the disease (aa).
A (congenital spherocytosis) is incorrect. Congenital sphero-cytosis is an autosomal dominant disorder associated withsplenomegaly and hemolytic anemia. Autosomal dominantdisorders are characterized by a dominant allele that expressesitself in either the homozygous or the heterozygous state.Only one parent must have the abnormal allele for the diseaseto be transmitted to the children; a heterozygous parent with

380 Pathology
disease will transmit it to 50% of the children. The pedigreedoes not show this pattern of inheritance.
B (glucose-6-phosphate dehydrogenase deficiency) is incorrect.Deficiency of G6PD is an X-linked recessive disorder associatedwith hemolytic anemia induced by infection or oxidant drugs(e.g., primaquine). X-linked recessive disorders are expressed inmales, whereas females with the abnormal allele are usuallyasymptomatic carriers. The pedigree does not show thispattern of inheritance.(neurofibromatosis) is incorrect. Neurofibromatosis is an auto-somal dominant disorder characterized by café-au-lait spotsand pedunculated neurofibromas on the skin. Refer to thediscussion for A.
E (type 2 diabetes mellitus) is incorrect. Type 2 diabetes is anexample of multifactorial (polygenic) inheritance, which doesnot have the pattern shown in the pedigree. Multifactorialinheritance involves the additive effect of two or more genemutations of small effect conditioned by environmental andother nongenetic factors.
38. A (decreased estrogen) is correct. The patient has severe kypho-sis (anterior curvature of the thoracic vertebrae) and a history ofchronic backache, suggesting postmenopausal osteoporosis as-sociated with estrogen deficiency. Estrogen normally inhibits theformation and function of osteoclasts, and estrogen deficiencyresults in increased osteoclastic activity with greater break-down of bone than formation of bone by osteoblasts. There is adecrease in bone mass and bone density. Histologically, thereis loss of cortical thickness and a reduction in the number andsize of trabeculae in bone. Thoracic vertebral bodies becomebiconcave and develop anterior wedging caused by compressionor collapse of bone, resulting in kyphosis (dowager's hump) andnerve root compression (radicular pain).
B (genetic defect in osteoclasts) is incorrect. Osteopetrosis is agenetic defect in osteoclasts. Osteopetrosis is an autosomaldominant or recessive disorder characterized by a defect inosteoclast resorption of bone, resulting in overgrowth andsclerosis of bone.
C (hypovitaminosis D) is incorrect. This condition causes osteo-malacia, in which defective mineralization of bone is accom-panied by an increase in nonmineralized osteoid. Total bonemass eventually decreases. In osteoporosis, bone mass is alsodecreased; however, the mineral content of the remainingbone is normal.
D (metastasis to bone) is incorrect. The vertebral column isthe most common site for bone metastasis. Most metasta-ses occur in the lumbar vertebrae and result in osteolyticlesions (radiolucent) and/or osteoblastic lesions (radiodense).In osteoporosis, bone density is decreased (osteopenia), andcompression fractures of the vertebrae are the key finding.

Answers and Discussions 381
39. C mu I t i nucleated squamous cells with intranuclear inclusions)is correct. The patient has herpes simplex labialis ("fever blis-ter"). The process of unroofing of vesicular lesions and stainingof cells scraped from the base of the lesion is called a Tzanckpreparation. The main purpose of this procedure is to evaluatecells for viral inclusions. Herpes simplex virus type 1 and herpesgenitalis type 2 both stimulate the formation of multinucle-a ted squamous cells with "ground glass" nuclei or eosinophilicintranuclear inclusions.
A (dysplastic squamous cells with increased nuclear chromatin)and D (neoplastic squamous cells with atypical mitotic figures)are incorrect. Squamous dysplasia and cancer usually involvethe lower lip. Neither lesion produces pain and vesicleformation.
B (enlarged individual squamous cells with intranuclear inclu-sions) is incorrect. Cytomegalovirus is identified as enlargedcells with intranuclear inclusions; it does not produce vesicularskin lesions.(normal basal cells without intranuclear inclusions) is incor-rect. Herpes simplex infections invariably produce intercellu-lar edema and nuclear changes in basal cells and othersquamous cells in the epithelium.
40. B (Borrelia burgdorferi) is correct. The figure shows an erythema-tous expanding rash with concentric circles separated by clearspaces. This is erythema chronicum migrans, which is the patho-gnomonic skin lesion of early Lyme disease. The disease istransmitted to humans by the bite of an Ixodes tick, whichcarries the gram-negative spirochete B. burgdorferi.
A (Babesia microti) b incorrect. B. microti is a protozoan carriedby the Ixodes tick. It parasitizes RBCs and causes a mild hemo-lytic anemia that may occur concurrently with Lyme disease.
C (Borrelia recurrentis) is incorrect. B. recurrentis is a spirochetethat is transmitted to humans by the bite of a tick calledDermacentar anikrsoni. This tick also carries the rickettsialorganisms Elirlichia chaffeensis and Rickettsia rickettsii. B. recur-rentis is the causative agent of relapsing fever, which is asso-ciated with a high fever and a petechial rash that covers thetrunk and extremities.
• (Ehrlichia chaffeensis) is incorrect. E. chaffeensis is a tick-transmitted rickettsial pathogen that is the causal agent ofehrlichiosis. The organism parasitizes leukocytes and causesfever and multisystem disease. No rash is associated with theinfection.
E (Rickettsia rickettsii) is incorrect. R. rickettsii is a tick-transmittedpathogen that is the causative agent of Rocky Mountainspotted fever, which is characterized by fever, multisystemdisease, and a petechial rash that begins on the palms andspreads to the trunk.

382 Pathology
41. A (decreased serum ceruloplasmin) is correct. The patient hasWilson's disease. A Kayser-Fleischer ring (brown pigment aroundthe perimeter of the cornea), chronic liver disease, and a move-ment disorder are features of Wilson's disease, an autosomalrecessive disease. Wilson's disease is due to a defect in the hepa-tocyte transport system for copper secretion into bile. In addi-tion, copper cannot be incorporated into an a 2-globulin toproduce ceruloplasmin, which is the binding protein of copper.The total serum copper equals copper that is bound to cerulo-plasmin (95% of the total) plus copper that is unbound (free).In Wilson's disease, the total serum copper level is decreasedbecause ceruloplasmin is decreased. However, the free copperlevel in the serum and urine is increased due to defective excre-tion in the bile and subsequent accumulation of copper in theserum. Excess copper deposits in Descemet's membrane of theeye and in the basal ganglia, particularly the putamen, result inparkinsonism, or choreiform movements, in some cases.
B (decreased serum iron) and C (increased serum ferritin) areincorrect. Movement disorders and deposition of iron in thecornea are not associated with disorders of iron metabo-lism (e.g., iron deficiency, hemochromatosis).
D (increased total serum copper) is incorrect. In Wilson's disease,the total serum copper level is decreased because ceruloplas-min is decreased. Refer to the discussion for A.
E (normal serum prothrombin time) is incorrect. The patient haschronic liver disease (chronic hepatitis or cirrhosis); therefore,the prothrombin time is most likely increased due to de-creased synthesis of coagulation factors in the liver.
42. 13 (pityriasis rosea) is correct. The patient has pityriasis rosea, adermatitis of unknown etiology. This eruptive dermatitis beginswith an oval-shaped lesion ("herald patch") with an erythem-atous margin. The central area has fine white scales. Within 1-2weeks, a more widespread eruption follows the lines of cleav-age of the skin ("Christmas tree" distribution).
A (eczema) is incorrect. Eczema does not have a herald patch ora rash with a "Christmas tree" distribution.
C (secondary syphilis) is incorrect. The palms, soles, and mucousmembranes (condyloma latum) are affected in secondarysyphilis.
D (tinea corporis) and E (tinea versicolor) are incorrect. Tineacorporis is caused by Trichophyton rubrum, and tinea versi-color is caused by Malassezia furfur. These organisms are super-ficial dermatophytes. The KOH examination is negative; thus,these diagnoses are excluded.

Answers and Discussions 383
43. I) (Leber's optic neuropathy) is correct. Leber's optic neuropa-thy is a neurodegenerative disease in which progressive loss ofcentral vision eventually leads to blindness. It has a mito-chondrial DNA inheritance pattern like that shown in the pedi-gree. Mitochondrial DNA disorders generally involve enzymedeficiencies in oxidative phosphorylation in the mitochondria.Unlike sperm, ova do not lose their mitochondria on fertiliza-tion, so affected females transmit the abnormal allele to all theirchildren, but affected males do not transmit it to any of theirchildren.
A (Alport's syndrome) is incorrect. Alport's syndrome is anX-linked dominant disorder associated with hereditary nephri-tis and sensorineural hearing loss. X-linked dominant disor-ders are characterized by a dominant allele that causes bothmale and female carriers to express the disease. Affected malestransmit the abnormal allele to all their daughters, and carrierfemales transmit it to 50% of their sons and 50% of theirdaughters. This pattern is not shown in the pedigree.
B (familial hypercholesterolemia) is incorrect. Familial hypercho-lesterolemia is an autosomal dominant disorder involving adeficiency of low-density lipoprotein receptors that leads tosevere hypercholesterolemia. Autosomal dominant disordersare characterized by a dominant allele that expresses itselfin either the homozygous or the heterozygous (Aa) state. Onlyone parent must carry the abnormal allele to transmit thedisease to the children; a heterozygous parent with diseasetransmits it to SO% of the children. This pattern is not shownin the pedigree.
C (familial polyposis) is incorrect. Familial polyposis is an auto-somal dominant disorder characterized by the development ofpremalignant polyps in t he colon. The disorder eventuallyprogresses to cancer. Refer to the discussion for B.
L (McCardle's disease) is incorrect. McArdle's disease is an auto-somal recessive glycogenosis that involves a deficiency ofmuscle phosphorylase, which renders muscle incapable ofmetabolizing glycogen to glucose. In autosomal recessive dis-orders, disease is present only in patients who are homozygous(aa) for the abnormal allele. In most cases, both parents areasymptomatic heterozygous (Aa) carriers; approximately 25%of their children will express the disease. This pattern of distri-bution is not evident in the pedigree.

384 Pathology
44. C (aplastic anemia) is correct. The child has erythema infectio-sum, or fifth disease, which is caused by parvovirus B19. Therash causes erythema of the cheeks, giving the child a "slappedface" appearance. If a patient has chronic hemolytic anemia(e.g., congenital spherocytosis), the virus can infect either a tri-lineage stem cell, resulting in a self-limited aplastic anemia, oran erythroid stem cell, resulting in a pure red blood cell aplasia.
A (acute lymphocytic leukemia), B (acute myelogenous leuke-mia), and D (B-cell malignant lymphoma) are incorrect. Par-vovirus B19 has not been associated with the development ofany leukemia or malignant lymphoma.
L (disseminated intravascular coagulation) is incorrect. Activa-tion of the intrinsic or extrinsic coagulation system must occurto produce intravascular clotting. Parvovirus B19 does notcause tissue damage (release of tissue thromboplastin) or endo-thelial cell damage (activation of factor XII).
45. A (degeneration of dopaminergic substantia nigra neurons) iscorrect. The patient has Parkinson's disease. Figure A showsatrophy and depigmentation of the substantia nigra when com-pared with the normal midbrain (figure B). Degeneration ofneurons of the substantia nigra causes a loss of dopamine, theprincipal neurotransmitter of afferents in the nigrostriatal tractthat connects the substantia nigra with the caudate andputamen. This interrupts voluntary muscle movement, resultingin muscle rigidity (cogwheel rigidity) and other extrapyramidalsigns (resting tremor with "pill rolling" of the thumbs and indexfingers).
B (increased y-secretase activity), D (presence of apolipoprotei ngene F, allele €4), and (presence of hyperphosphorvlatedtau protein) are incorrect. All of these options describeAlzheimer's disease. An increase in y-secretase causes increasedcleavage of amyloid precursor protein into fragments that areconverted to (3-amyloid protein, which is toxic to neurons inthe brain. Apolipoprotein gene E, allele €4, produces a sub-stance that has a high affinity for (3-amyloid protein. This con-centrates the protein and causes late-onset Alzheimer's disease.Hyperphosphorylated tau protein increases the formation ofneurofibrillary tangles (protein-rich neurofilaments), which areprominent findings in the brain of patients with Alzheimer'sdisease.
C (loss of striatal neurons in the caudate nucleus) is incorrect.Loss of striatal neurons in the caudate nucleus occurs inHuntington's disease. This autosomal dominant trinucleotiderepeat disorder is characterized by chorea, extrapyramidalsigns, and dementia.

Answers and Discussions 385
46. D (oligoclonal bands) is correct. The patient has multiple scle-rosis, an autoimmune disease characterized by destruction of themyelin sheaths and antibodies directed against myelin basicprotein. The episodic course of acute relapses with optic neuritis(blurry vision), scanning speech, cerebellar ataxia, and sensoryand motor dysfunction, followed by remissions, are characteris-tic of this disease. The demyelinating plaques in multiple scle-rosis occur in the white matter of the cerebral cortex. Theplaques usually have a perivenular distribution and are accompa-nied by a perivascular lymphoid and plasma cell infiltrate withmicroglial cells containing phagocytosed myelin. CSF showsan increase in CSF protein. High-resolution electrophoresis ofCSF shows discrete bands of immunoglobulins in the y-globulinregion called oligoclonal bands. They are indicative of a de-myelinating process.
A (decreased glucose) is incorrect. The CSF glucose is normal inmultiple sclerosis.
B (increased neutrophils) is incorrect. T lymphocytes are in-creased in the CSF in multiple sclerosis.
C (normal protein) is incorrect. Refer to the discussion for D.F (positive Gram stain) is incorrect. Gram stain is negative for
microbial pathogens.
47. E (urine for metanephrines, 24 h) is correct. This patient hasneurofibromatosis complicated by hypertension due to a pheo-chromocytoma. The skin lesions in neurofibrom atosis includepigmented, pedunculated tumors (neurofibromas) and flat,oval-shaped, café-au-lait patches. Neurofibromatosis is an auto-somal dominant neurocutaneous disorder with increased in-cidence of pheochromocytoma (unilateral or bilateral) andcentral and peripheral nervous system tumors (e.g., meningi-oma, acoustic neuroma). The classic triad of headache, palpita-tions, and excessive perspiration is highly predictive. Cate-cholamine excess may cause subendocardial ischemia resultingin angina (as occurred in this patient). Hypertension is character-ized as sustained, sustained with paroxysms (most common), orparoxysmal only. A 24-hour urine test for metanephrines (mostsensitive test) and vanillylmandelic acid usually are used asscreening tests.
A (complete urinalysis) is incorrect. Although a urinalysis isuseful in the workup of a patient with hypertension relatedto renal disease, it does not provide adequate information inscreening for a pheochromocytoma.
B (serum electrolytes) is incorrect. Serum electrolytes are mostuseful in diagnosing primary aldosteronism. The benign adre-nocortical adenomas arise in the zona glomerulosa of theadrenal cortex. Excess aldosterone causes hypernatremia,hypokalemia, and metabolic alkalosis.
C (urine for free cortisol, 24 h) and D (urine for 17-ketosteroids,

386 Pathology
24 h) are incorrect. Increases in urine free cortisol and 17-ketosteroids (dehydroepiandrosterone and androstenedione)are characteristic of patients with Cushing's syndrome, whichis associated with hypertension. The patient does not haveabdominal striae and other findings to suggest this diagnosis.
48. B (cytomegalovirus) is correct. The patient has retinitis due tocytomegalovirus (CMV). Examination of the retinas shows whiteareas with indistinct borders (cotton wool exudates) that repre-sent retinal infarctions due to a CMV vasculitis. CMV retinitisis the most common cause of blindness in patients with AIDSand occurs when the CD4 helper T-cell count is < 50 cells/mm3.
1 (Candida albicans), C (herpes simplex virus type 1), and E(Toxoplasma gondii) are incorrect. C. albicans, herpes simplexvirus type 1, and T. gondii all can cause retinitis in AIDS.However, CMV is overall the most common cause of retinitis.(HIV) is incorrect. HIV has not been implicated as a cause ofretinitis leading to blindness.
49. D (respiratory alkalosis) is correct. The patient has developedtetany due to anxiety-induced respiratory alkalosis. Adductionof the thumb into the palm, plus numbness and tingling atthe tips of the fingers, is a classic sign of tetany. Tetany is due toa decreased concentration of ionized calcium in the blood. Thisincreases neuromuscular excitability by bringing the thresh-old potential of neuromuscular tissue closer to the resting mem-brane potential. Therefore, less of a stimulus is required to ini-tiate the action potential, which results in sustained musclecontractions. In an alkalotic condition, albumin has more nega-tive charges, causing some of the ionized calcium to bind toalbumin. In states of acidosis (e.g., diabetic ketoacidosis), lesscalcium is bound to albumin, causing an increase in ionizedcalcium and a corresponding decrease in neuromuscular excit-ability. Conditions associated with hypercalcemia (e.g., primaryhyperparathyroidism and sarcoidosis) also increase the ionizedcalcium level, causing a corresponding decrease in neuromuscu-lar excitability.
A (diabetic ketoacidosis) is incorrect. Refer to the discussionfor D.
B (nephrotic syndrome) is incorrect. In the nephrotic syndrome,serum albumin is markedly decreased due to loss of albuminin the urine. Although this decreases the total calcium level(calcium bound to albumin plus ionized calcium), the ionizedcalcium level remains unchanged and tetany does not occur.
C (primary hyperparathyroidism) and I (sarcoidosis) are incor-rect. Refer to the discussion for D.

Answers and Discussions 387
50. B (DNA analysis of the X chromosome) is correct. The boy hasfragile X syndrome. In most cases, inheritance of fragile X syn-drome is X-linked recessive, with a fragile site or gap at theend of the long arm of the X chromosome, where there are tri-nucleotide repeats (CGG). DNA analysis of the X chromosome inlymphocytes identifies the trinucleotide repeats and is consid-ered more sensitive than the fragile X chromosome study.
(buccal smear) is incorrect. A buccal smear is performed to ruleout deficient or extra X chromosomes. Normal females haverandom inactivation of one of the two X chromosomes.Hence, normal females have one Barr body, and normal maleshave no Barr bodies.(human chorionic gonadotropin) is incorrect. Human chori-onic gonadotropin is not present in males unless they havechoriocarcinoma of the testicle.
D (serum gonadotropins) is incorrect. The patient shows noclinical evidence of hypogonadism, such as in Klinefelter'ssyndrome, which is characterized by testicular atrophy andincreased serum gonadotropins.(testicular biopsy) is incorrect. Testicular biopsy is notwarranted, because the patient does not have a testicularneoplasm or signs of Klinefelter's syndrome, which is charac-terized by testicular atrophy.


TEST 2
DIRECTIONS: Each numbered item or incomplete statement is followed by options ar-ranged in alphabetical or logical order. Select the best answer to each question. Someoptions may be partially correct, but there is only ONE BEST answer.
1. A 59-year-old man has an acute ante-rior myocardial infarction (MI). Six weekslater, he see ,. his physician because of feverand precordial chest pain that is lesssevere when he leans forward. On physicalexamination, a friction rub is heard over theprecordium. The figure shows lesions onthe heart. Which of the following mecha-nisms is most likely involved in thepathogenesis of these lesions?
O A. Alteration in Starling's forcesO B. Immunologic reactionO C. Metastatic diseaseO D. Rupture of the anterior wallO E. Viral infection
2. A 4-year-old boy has a history of patho-logic fractures since birth. Physical exam-ination shows blue discoloration of thesclera in both eyes. Which of the followingis the most likely diagnosis?
O A. Marfan syndromeD B. Osteogenesis imperfecta) C. Osteopetrosis
D. Vitamin D—dependent rickets
3. A cholecystectomy is performed on a55-year-old woman, and the incision is nothealing properly. When asked about herdiet, the woman says that she consumesa diet high in protein but does not eat fruitsor vegetables. Which of the followingevents most likely accounts for the poorwound healing?
O A. Decreased synthesis of granulationtissue
O B. Decreased synthesis of type Illcollagen
) C. Decreased tensile strength of collagenO D. Defect in fibrillin in elastic tissue• E. Leukocyte adhesion molecule defect
389

10,1%
44:k 14-4 '400P.S.
390 Pathology
4. A 22-year-old man with cystic fibrosisand type 1 diabetes mellitus has chronicdiarrhea characterized by excessive bloatingand greasy stools. The figure shows a sec-tion of the pancreas obtained on biopsy.Which of the following alterations in cellgrowth is responsible for the morphologicchanges shown in this specimen?
) A. AtrophyB. Dysplasia
C3 C. Hyperplasia) D. Hypertrophy3 E. Metaplasia
5. The umbilical cord of a 6-week-old maleinfant has not sloughed on, and the cordis removed surgically. Histologic sectionsof the cord show an absence of neutrophilmargination along the umbilical vessels andabsence of neutrophil transmigration intointerstitial tissue. Which of the follow-ing defects of neutrophil function preventsseparation of the umbilical cord?
O A. Absent respiratory burstO B. Leukocyte adhesion molecule defectO C. Myeloperoxidase deficiencyO D. Opsonization defectO E. Phagocytosis defect
6. The figure shows a lung removed atautopsy from a 72-year-old woman whodied suddenly 6 days after undergoingsurgery for a total hip replacement. Whichof the following is the most likely causeof death?
O A. Acute pulmonary infarctionO B. Acute right-sided ventricular strain0 C. Aspiration of gastric contentsO D. Disseminated metastasis0 E. Nosocomial pneumonia
7. A routine physical examination of anasymptomatic 21-year-old African-Americanwoman is normal. Urinalysis shows RBCswith no casts. The patient says that she oc-casionally has had blood in her urine. Aurine culture is negative, a peripheral smearis normal, and renal ultrasonography isnormal. Laboratory studies show serumblood urea nitrogen 10 mg/dL, serum creati-nine 1.0 mg/dL, hemoglobin 14.0 g/dL,and mean corpuscular volume 82 i.tm3.Which of the following is the next best stepin the workup?
O A. Bone marrow examinationO B. CystoscopyO C. Renal biopsyO D. Serum ferritinO E. Sickle cell screen
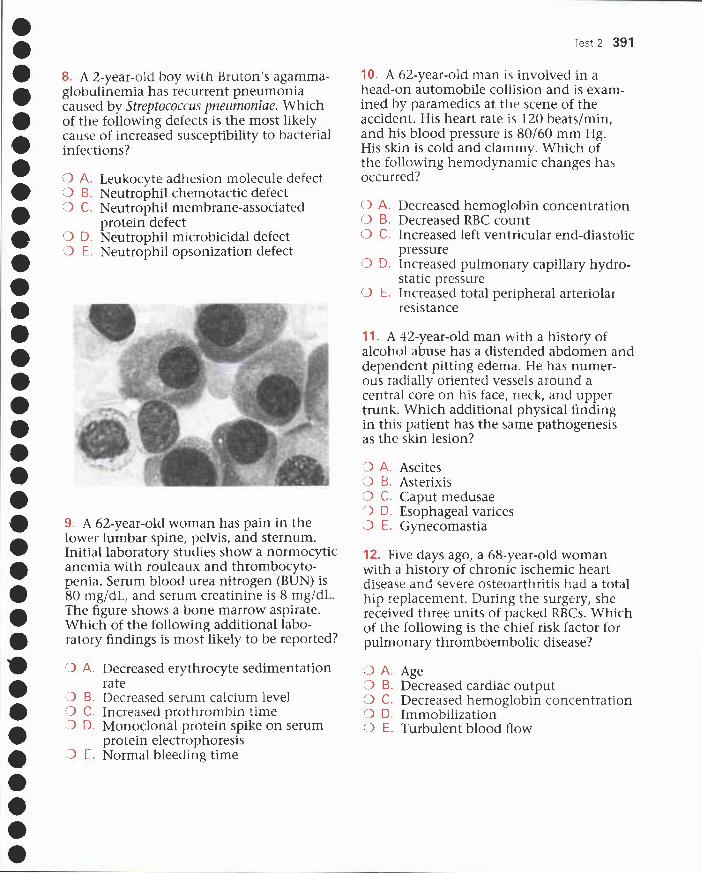
Test 2 391
8. A 2-year-old boy with Bruton's agamma-globulinemia has recurrent pneumoniacaused by Streptococcus pneumoniae. Whichof the following defects is the most likelycause of increased susceptibility to bacterialinfections?
O A. Leukocyte adhesion molecule defectO B. Neutrophil chemotactic defectO C. Neutrophil membrane-associated
protein defectO D. Neutrophil microbicidal defectO E. Neutrophil opsonization defect
9. A 62-year-old woman has pain in thelower lumbar spine, pelvis, and sternum.Initial laboratory studies show a normocyticanemia with rouleaux and thrombocyto-penia. Serum blood urea nitrogen (BUN) is80 mg/dL, and serum creatinine is 8 mg/dL.The figure shows a bone marrow aspirate.Which of the following additional labo-ratory findings is most likely to be reported?
O A. Decreased erythrocyte sedimentationrate
O B. Decreased serum calcium levelO C. Increased prothrombin timeO D. Monoclonal protein spike on serum
protein electrophoresisO E. Normal bleeding time
10. A 62-year-old man is involved in ahead-on automobile collision and is exam-ined by paramedics at the scene of theaccident. His heart rate is 120 beats/min,and his blood pressure is 80/60 mm Hg.His skin is cold and clammy. Which ofthe following hemodynamic changes hasoccurred?
O A. Decreased hemoglobin concentrationO B. Decreased RBC countO C. Increased left ventricular end-diastolic
pressureD D. Increased pulmonary capillary hydro-
static pressureO E. Increased total peripheral arteriolar
resistance
11. A 42-year-old man with a history ofalcohol abuse has a distended abdomen anddependent pitting edema. He has numer-ous radially oriented vessels around acentral core on his face, neck, and uppertrunk. Which additional physical findingin this patient has the same pathogenesisas the skin lesion?
O A. AscitesO B. AsterixisO C. Caput medusaeO D. Esophageal varicesO E. Gynecomastia
12. Five days ago, a 68-year-old womanwith a history of chronic ischemic heartdisease and severe osteoarthritis had a totalhip replacement. During the surgery, shereceived three units of packed RBCs. Whichof the following is the chief risk factor forpulmonary thromboembolic disease?
O A. AgeO B. Decreased cardiac outputO C. Decreased hemoglobin concentrationO D. ImmobilizationO E. Turbulent blood flow

392 Pathology
13. A 6-year-old boy has shortness of breathand itchy eyes whenever he plays with hiscat. His mother states that she can hear"wheezes" when he breathes. Which of thefollowing chemical mediators is most likelyresponsible for these respiratory findings?
) A. Bradykinin) B. Cony element C3a and C5a) C. Leukotrienes C4, D4, and E4
D. Prostaglandin 12) E. Thromboxane A2
14. A 58-year-old man with small cell carci-noma of the lung complains of a head-ache and blurry vision. Examination showsswelling of the optic nerve. MRI of thehead shows cerebral edema, but no there isevidence of metastatic disease. The serumM.+ level is 115 mEq/L. Which of thefollowing is the most appropriate nonphar-macologic treatment for this patient?
O A. Decrease intake of Na÷ and H20O B. Decrease intake of Na +, maintain
intake of H20O C. Decrease intake of H20, maintain
intake of NW-O D Increase intake of Na+ and H20O E. Maintain intake of Na+ and H20
15. A 62-year-old woman complainsthat her tongue is sore. She reports instabil-ity when walking. Physical examinationshows atrophy of the tongue papillae, de-creased vibratory sensation in both lowerextremities, and instability when she standsand closes her eyes. An endoscopic exami-nation shows chronic atrophic gastritisinvolving the body and fundus of thestomach. Laboratory studies identify asevere macrocytic anemia with pancytope-nia. The figure shows a peripheral bloodsmear. Which of the following findingswould most likely be reported?
O A. Decreased serum folateO B. Decreased serum gastrinO C. Decreased urine methylmalonic acidO D. Increased antigliadin antibodiesO E. Increased vitamin B 12 absorption after
addition of intrinsic factor

Test 2 393
16. A 58-year-old man complains of fre-quent headaches and generalized itchingafter bathing. Physical examination showscongestion of the retinal vessels, a ruddycomplexion, and splenomegaly. Initial labo-ratory studies show an RBC count of 8.0million/mm3 , a WBC count of 15,000/mm3,and a platelet count of 500,000/mm 3. TheWBC differential count shows an in-crease in normal-appearing segmentedneutrophils. Which of the following setsof laboratory results is most likely to bereported?
RBCmass
Plasmavolume
02saturation
EPOconcentration
A. Increased Increased Normal Decreased
B. Increased Normal Decreased Increased
C. Increased Normal Normal Increased
D. Normal Decreased Normal Normal
EPO, erythropoietin_
17. A 32-year-old African-American medicalmissionary who recently returned to theUnited States from a trip overseas is diag-nosed with malaria due to Plasmodiumvi vax. After 4 days of therapy with pri-maquine, he develops fever, chills, low backpain, and dark urine. A complete blood cellcount shows a hemoglobin of 6 g/dL, aWBC count of 15,000/mm 3, and a plateletcount of 450,000/mm • . A corrected reticulo-cyte count is 10%. A peripheral smearshows polychromasia and numerous RBCsthat are missing parts of their membrane.A urine dipstick test is positive for blood.Which of the following laboratory findingsis most likely to be reported?
O A. Abnormal hemoglobin electro-phoresis
O B. Decreased mean corpuscular hemo-globin concentration
O C. Decreased serum ferritin concen-tration
O D. Positive direct Coombs' testO E. Positive Heinz body preparation
18. A 25-year-old African-American manhas fever and a pain in the lower rightthigh. A radiograph of the thigh shows anirregular lucency in the metaphysis andthickening of the periosteum of the distalright femur. The figure shows a periph-eral blood smear. Which of the followingpathogens is most likely responsible for thisbone lesion?
O A. Pseudomonas aeruginosaO B. Salmonella paratyphiO C. Staphylococcus aureusO D. Streptococcus pneumoniaeO E. Streptococcus pyogenes

h,
AP,
394 Pathology
19. The arrow in the figure points to anormal cell in the alveolus. The vacuolecontains lamellar material. In which of thefollowing respiratory disorders is this ma-terial most likely to be absent?
O A. Bronchopneumonia1) B. Emphysema
C. Primary small cell carcinoma) D. Respiratory distress syndrome) E. Sarcoidosis
20. A 28-year-old man has a family historyof sudden cardiac death at a young age.Physical examination shows a systolicmurmur that decreases in intensity whenthe patient lies down and increases inintensity when he stands up. An echo-cardiogram shows abnormal movement ofthe anterior mitral valve leaflet against anasymmetrically thickened interventricu-lar septum (IVS). Which of the followingis the cause of the systolic murmur?
O A. Aortic regurgitationO B. Aortic stenosisO C. Hypertrophic cardiomyopathyO D. Mitral stenosisO E. Mitral valve prolapse
21. A 42-year old woman develops feverand dyspnea approximately 24 hours aftera cholecystectomy for acute gangrenouscholecystitis. Physical examination showsdullness to percussion, absent vocaltactile fremitus, and absent breath soundsin the right lower lobe of the lung. The dia-phragm is elevated, and an inspiratory lagis present on the right side. The tracheais shifted to the right. Which of the follow-ing is the most likely diagnosis?
0 A. AtelectasisO B. Lobar pneumoniaO C. Lung abscessO D. Pulmonary infarctionO E. Spontaneous pneumothorax
22. An afebrile 50-year-old man complainsof watery diarrhea and weight loss overthe past 6 months. He states that his faceoften becomes flushed. Physical examina-tion shows an enlarged, nodular liver. Apansystolic murmur along the parasternalborder that increases in intensity with inspi-ration is heard. Which of the followinglaboratory studies is most useful for con-firming the diagnosis?
O A. Blood culturesO B. Liver function testsO C. Serum electrolytesO D. Urine test for 5-HIAA
23. A 30-year-old woman states that shefeels no pain in her hands and frequentlyburns them. Physical examination showsdecreased pain and temperature sensationin the upper extremities, no deep tendon re-flexes in the upper extremities, and atrophyof the intrinsic muscles of the hands.Which of the following is the most likelydiagnosis?
O A. Amyotrophic lateral sclerosisO B. Multiple sclerosisO C. SyringomyeliaO D. Vitamin B 12 deficiency

•••••••••••••••••••••••••••••••••
24. A 39-year-old man who has recentlyreturned from a business trip to Mexico Citysees his physician because of fatigue andyellow discoloration of the eyes. The figureshows a section of the liver obtained onbiopsy. Which of the following is the mostlikely diagnosis?
O A. Alcoholic hepatitisO B. Metastatic liver diseaseO C. Obstructive liver diseaseO D. Primary liver cancerO E. Viral hepatitis
25. A 2-day-old newborn male infant withrespiratory distress syndrome (RDS) has acontinuous harsh murmur that is heardover the entire precordium. Which of thefollowing sets of oxygen saturation(Sao2) values in the cardiac chambers andvessels is most likely present in this patient?
RA RV PA PV LV Ao
NormalSao, 75 75 75 95 95 95
') A. 75 80 80 95 95 95
D B. 80 80 80 95 95 95
D C. 75 75 80 95 95 95
D D. 75 75 75 95 80 80
RA, right atrium; RV, right ventricle; PA, pulmonary artery;PV pulmonary vein; LV, left ventricle; Ao, aorta
Test 2 395
26. A 62-year-old man complains of fatigue.Physical examination shows a harsh systolicejection. murmur, grade 4/6, that radiatesinto the carotid arteries. Laboratory studiesshow a mild microcytic anemia. A urinedipstick test is positive for blood. The figureshows a peripheral blood smear. Which ofthe following laboratory studies would mostlikely be abnormal?
O A. Direct Coombs' testO B. Enzyme assay for pyruvate kinaseO C. Hemoglobin electrophoresisO D. Osmotic fragility testO E. Serum ferritin test
27. A 42-year-old woman with rheumatoidarthritis takes aspirin to relieve the painin her inflamed joints. Aspirin alleviates thepain by decreasing which of the following?
O A. Chemotaxis of leukocytesO B. Synthesis of bradykininO C. Synthesis of leukotrienesO D. Synthesis of prostaglandinsO E. Transmigration of leukocytes
•••

396 Pathology
28. A 25-year-old intravenous drug abuserdevelops fever, scleral icterus, and rightupper quadrant pain. Laboratory studiesshow an increase in serum aminotrans-ferases and an increase in the serum biliru-bin with approximately equal unconjugatedand conjugated bilirubin fractions. Thesuspected diagnosis is acute hepatitis B.Which of the following hepatitis B profilesis expected?
IgM- IgG-HBsAg HBeAg anti-HBc anti-HBc Anti-HBs
O A. Negative Negative Negative Negative Positive
O B. Negative Negative Negative Positive Positive
O C. Negative Negative Positive Negative Negative
O D. Positive Negative Negative Negative Negative
O E. Positive Positive Positive Negative Negative
Anti-HBs, anti-HBV surface antibody; HBeAg, hepatitis B eantigen; HBsAg, hepatitis B surface antigen; IgG-anti-HBc, anti-HBV core antibody IgG; IgM-anti-HBc, anti-HBV core antibody IgM
29. Physical examination of a 21-year-oldman shows a xanthoma of the Achillestendon in the right foot and bilateralyellow, raised patches on the eyelids. He hasa family history of premature death due tostroke and myocardial infarction by 30 to40 years of age. Which of the following bestexplains the pathogenesis of the tendonand skin lesions?
O A. Decreased activation of capillarylipoprotein lipase
O B. Deficiency of apolipoprotein C-IIO C. Deficiency of apolipoprotein EO D. Deficiency of low-density lipoprotein
(LDL) receptors0 E. Increased synthesis of very low
density (VLDL) lipoprotein
30. A 50-year-old man had an acute ante-rior myocardial infarction (MI) 4 daysago, and he now complains of substernalchest pain with radiation into the left armand jaw. Physical examination shows nocardiac or pulmonary abnormalities. Labo-ratory studies show increases in serumcreatine kinase MB (CK-MB), serumtroponin-I (cTn-I), and serum troponin-T(cTn-T). Which of the following is the mostlikely cause of the chest pain?
O A. Angina pectorisO B. PericarditisO C. ReinfarctionO D. Right ventricular infarctionO E. Rupture of the anterior wall
31. A 38-year-old man has a familyhistory of colectomies between 35 and 40years of age. The figure shows a portion of atotal colectomy specimen from this patient.Which of the following best characterizesthis disorder?
O A. Complication of Crohn's diseaseO B. Complication of ulcerative colitisO C. Inactivation of a suppressor geneO D. Oral mucosal pigmentationO E. X-linked recessive inheritance pattern

•••••••••••••••••••••S•••••••••••••
32. A 24-year-old woman in her first trimes-ter of pregnancy complains of heat intoler-ance. Physical examination shows aslightly enlarged, nontender thyroid gland.Thyroid function studies show a serumthyroxine (T4) level of 14 gg/dL and a serumthyroid-stimulating hormone (TSH) levelof 3.0 gl.J/mL. Which of the following bestexplains the results of these studies?
O A. Decreased peripheral conversion of T4
to triiodothyronine (T3)O B. Increased release of T4 from acute
thyroiditisO C. Increased synthesis of T3
O D. Increased synthesis of T4
O E. Increased synthesis of thyroid-binding globulin
33. A 50-year-old man with ischemic heartdisease has signs of both left- and right-sided heart failure. Which of the followingis characteristic of both types of heartfailure?
O A. Bibasilar inspiratory cracklesO B. Decreased cardiac outputO C. Dependent pitting edemaO D. Paroxysmal nocturnal dyspneaO E. Passive congestion in the liver
Test 2 397
34. A 62-year-old man dies of complica-tions from valvular disease. The figureshows the aortic side of an unopened aorticvalve. Which of the following complica-tions has most likely occurred?
O A. Acute myocardial infarctionO B. Aortic dissectionO C. Hemolytic anemiaO D. HypertensionO E. Stroke syndrome
35. A febrile 5-year-old child complains ofchest pain. Physical examination showspainful cervical adenopathy; dry, crackedlips; and erythema and swelling of thehands and feet. A desquamating rash devel-ops on the fingers and toes. Which of thefollowing is a potential complication of thepatient's disease?
O A. Acute myocardial infarctionO B. Aortic arch aneurysmO C. Aortic dissectionO D. Infective endocarditisO E. Mitral stenosis

•
398 Pathology
36. A 25-year-old woman with poorly con-trolled gestational diabetes mellitus givesbirth to a male infant who develops jitteri-ness and seizures 3 hours after birth. Whichof the following hormones is the mostlikely cause of these symptoms?
O A. CortisolO B. EpinephrineO C. GlucagonO D. Growth hormoneO E. Insulin
37. A 75-year-old man with a 7-year historyof progressive loss of intellectual functionrequires 24-hour custodial care. He developsa spiking fever and productive cough. Achest radiograph shows patchy areas of con-solidation in the right lower lobe, which isdiagnosed as bronchopneumonia. Thepatient dies 3 days later. The H&E stainedsection of brain taken from the frontal lobeshows a lesion (see arrow in figure). Whichof the following best describes this lesion?
O A. Amyloid angiopathyO B. GliosisO C. Neurofibrillary tangleO D. Senile plaqueO E. Spongiform encephalopathy
38. A 35-year-old man complains of recur-rent episodes of forgetfulness and tiredness.Laboratory studies show a serum glucoselevel of 20 mg/dL and increases in seruminsulin and serum C-peptide. Which of thefollowing is the most likely cause of thishypoglycemia?
O A. Benign tumor of 13-islet cellsO B. Ectopic secretion of an insulin-like
factorO C. Malignant tumor of a-islet cellsO D. Patient injection of human insulin
39. A 48-year-old man complains of feverand several fainting spells over the past fewmonths. He states that he does not havefainting spells when he is lying down, butthey occur when he stands up. Physicalexamination shows normal blood pressurewhen he is lying down or when he is sittingup. A late diastolic murmur is heard. Painin the left upper quadrant is aggravatedby inspiration. The spleen is enlarged andtender, and a splenic friction rub is present.There is right flank pain on percussion.A urine dipstick test is positive for blood,and RBCs are present in the urine sediment.Which of the following is the most likelydiagnosis?
O A. Calcific aortic stenosisO B. Hypertrophic cardiomyopathyO C. Left atrial myxomaO D. Mitral stenosisO E. Pericardial effusion
40. A newborn child coughs and becomescyanotic while breastfeeding. Thestomach is distended and tympanitic.The mother had polyhydramnios duringpregnancy. Which of the following is themost likely diagnosis?
O A. Choanal atresiaO B. Congenital pyloric stenosisO C. Duodenal atresiaO D. Esophageal webO E. Tracheoesophageal fistula
II

••••••
I •
•••••SoSoSo•••SoSoSo•••••■
41. A 22-year-old man has blood oozingfrom his nose and mouth. Physical exami-nation shows petechiae and ecchymosesover most of his body. There is generalizedlymphadenopathy and hepatospleno-megaly. Laboratory studies show a nor-mocytic anemia and thrombocytopeniaand a WBC count of 32,000/mm 3 . Thereare increases in n-dimers and prothrombinand partial thromboplastin times. Thefigure shows a peripheral blood smear.This patient is most likely to have whichchromosome translocation?
O A. t(8;14)O B. t(9;22)O C. t(14;18)O D. t(15;17)
Test 2 399
42. Colonoscopic studies show a 3-cmannular mass in the sigmoid colon of a62-year-old man. Multiple biopsies showa poorly differentiated adenocarcinoma.A colectomy is performed, and a fewnodular lesions on the surface of the liverare apparent. A frozen section shows apoorly differentiated adenocarcinoma.Gross and microscopic findings of the colec-tomy specimen show a mucosally derivedcancer that has invaded through the musclewall and out into the serosal fat. There ismetastasis in 3 of 15 mesenteric lymphnodes directly beneath the tumor. Which ofthe following most influences the patient'sprognosis?
O A. Differentiation of the tumorO B. Extent of invasionO C. Liver involvementO D. Lymph node involvement
43. A 22-year-old asymptomatic African-American woman has had a mild microcyticanemia since early childhood. Her men-strual history is normal. Physical examina-tion is normal. Laboratory studies show aslightly decreased hemoglobin concen-tration and an increased RBC count. Hemo-globin electrophoresis is normal. Serumferritin is normal. Which of the following isthe most likely diagnosis?
O A. Iron deficiencyO B. Sickle cell traitO C. Sideroblastic anemiaO D. a-ThalassemiaO E. 13-Thalassemia
0••e
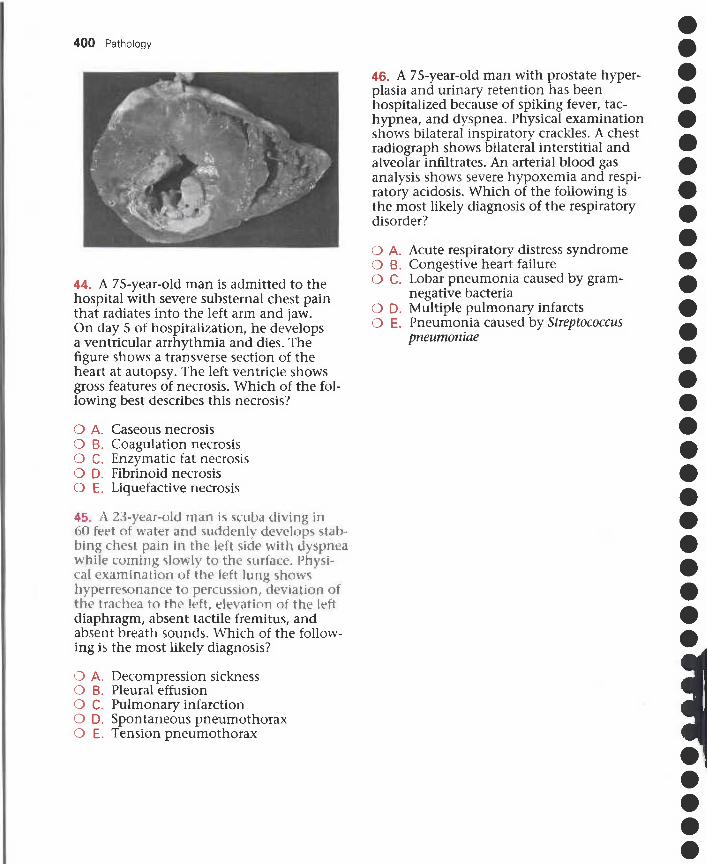
400 Pathology
44. A 75-year-old man is admitted to thehospital with severe substernal chest painthat radiates into the left arm and jaw.On day 5 of hospitalization, he developsa ventricular arrhythmia and dies. Thefigure shows a transverse section of theheart at autopsy. The left ventricle showsgross features of necrosis. Which of the fol-lowing best describes this necrosis?
O A. Caseous necrosisO B. Coagulation necrosisO C. Enzymatic fat necrosisO D. Fibrinoid necrosisO E. Liquefactive necrosis
45. A 23-year-old man is scuba diving in60 feet of water and suddenly develops stab-bing chest pain in the left side with dyspneawhile coming slowly to the surface. Physi-cal examination of the left lung showshyperresonance to percussion, deviation ofthe trachea to the left, elevation of the leftdiaphragm, absent tactile fremitus, andabsent breath sounds. Which of the follow-ing is the most likely diagnosis?
O A. Decompression sicknessO B. Pleural effusionO C. Pulmonary infarctionO D. Spontaneous pneumothoraxO E. Tension pneumothorax
46. A 75-year-old man with prostate hyper-plasia and urinary retention has beenhospitalized because of spiking fever, tac-hypnea, and dyspnea. Physical examinationshows bilateral inspiratory crackles. A chestradiograph shows bilateral interstitial andalveolar infiltrates. An arterial blood gasanalysis shows severe hypoxemia and respi-ratory acidosis. Which of the following isthe most likely diagnosis of the respiratorydisorder?
O A. Acute respiratory distress syndromeO B. Congestive heart failureO C. Lobar pneumonia caused by gram-
negative bacteriaO D. Multiple pulmonary infarctsO E. Pneumonia caused by Streptococcus
pneumoniae

47. A 45-year-old woman undergoes cho-lecystectomy for a gangrenous gallbladder.On day 4 postoperatively, the patientcomplains of chest pain on the right side,which increases on inspiration. Analysis ofthe first set of arterial blood gases drawnafter this complaint shows an increase in ar-terial pH and a decrease in arterial Po 2 . Onday 7 postoperatively, the patient dies.The figure shows a cut section from theright lower lobe of the lung obtained atautopsy. Which of the following best ex-plains the decrease in arterial Po 2 on day 4?
O A. Diffusion defectO B. Perfusion defectO C. Respiratory acidosisO D. Respiratory alkalosisO E. Ventilation defect
48. A 4-year-old boy has a history of fre-quent respiratory infections and greasystools. The child is below the normal per-centile for weight and height for age. Physi-cal examination shows nasal polyps andcoarse inspiratory crackles in both lungfields that clear with coughing. Which ofthe following laboratory studies is the nextstep in determining a diagnosis?
O A. Chromosome studyO B. Serum IgE levelO C. Stool cultureO D. Sweat chloride test
Test 2 401
49. A 2-year-old boy accidentally ingests ratpoison. Physical examination showsbleeding from the mouth and gastrointesti-nal tract. Which of the following resultsof coagulation studies is most likely to bereported?
Plateletcount
Bleedingtime PT aPTT
0 A. Decreased Prolonged Normal Normal
0 B. Normal Normal Normal Prolonged
0 C. Normal Normal Prolonged Prolonged
0 D. Normal Prolonged Normal Normal
0 E. Normal Prolonged Normal Prolonged
PT, prothrombin time; aPTT, activated partial thromboplas-tin time
50. A febrile 65-year-old man with prostatehyperplasia and urinary retention devel-ops endotoxic shock. Within 24 hours,he has oozing of blood from all needlepuncture sites, extensive ecchymoses andpetechiae, and gastrointestinal bleed-ing. Laboratory studies show hemoglobin9 g/dL, platelet count 75,000/mm 3, pro-thrombin time (PT) 20 sec, partial thrombo-plastin time, activated (aPTT) 50 sec,D-dimer positive. Which of the followingis the most likely diagnosis?
O A. Autoimmune thrombocytopeniaO B. Circulating anticoagulantO C. Disseminated intravascular
coagulationO D. Primary fibrinolysisO E. Thrombotic thrombocytopenic
purpura
••••••••••••••••••
•••••••••••••••

••••••••••••••••••••••••••••••••••••

•••• ANSWERS AND DISCUSSIONS fil4•••
1. B (immunologic reaction) is correct. The figure shows fibrinouspericarditis, in which a layer of fibrin covers the visceral surface
•of the heart. Because of the MI 6 weeks earlier, an immuno-logic reaction is the likely cause. In Dressler's syndrome (post–MIsyndrome), the patient develops antibodies against the pericar-
• dial tissue. The immunologic reaction causes increased vesselpermeability, loss of proteins, and production of a fibrinous
•exudate. Clinical manifestations of Dressler's syndrome includefever, precordial friction rub, and pain that increases on inspi-
• ration but lessens when the patient leans forward.
• A (alteration in Starling's forces) is incorrect. An alteration in
•Starling's forces signifies increased hydrostatic pressure ordecreased oncotic pressure within the vascular compartment.The transudate produced by this change is poor in proteins
•and cells, unlike the fibrinous exudate shown in the figure.C (metastatic disease) is incorrect. Metastatic disease involving
•the pericardium produces multiple nodular masses and a fibri-nous and hemorrhagic exudate, unlike the lesions shown in
• the figure.
•D (rupture of the anterior wall) is incorrect. A rupture of the an-
terior wall occurs 3 to 7 days after an acute MI, causing cardiac
• tamponade and death. This patient is seen 6 weeks post–MI.E (viral infection) is incorrect. Coxsackievirus is the most corn-
• mon cause of pericarditis. This patient's history of MI indicates
•that an immunologic cause is more likely.
• 2. B (osteogenesis imperfecta) is correct. This child has osteogene-• sis imperfecta (brittle bone disease), which is characterized by a
•history of pathologic fractures since birth. This autosomal domi-nant disease is associated with a defect in the synthesis of type
• I collagen. Blue discoloration of the sclera is due to visualizationof the choroidal veins beneath the collagen-deficient sclera.
• Bone is a type I collagen–based matrix that is mineralized with
•calcium, phosphorus, and magnesium. Decreased synthesis oftype I collagen results in structurally weak bone with a decreasein bone mass and density (secondary osteoporosis), resulting
•in pathologic fractures.
• A (Marfan syndrome) is incorrect. Marfan syndrome is an auto-somal dominant disorder due to a defect in fibrillin, a compo-
• nent of elastic tissue. The key ocular manifestation is dislo-cation of the lens. Pathologic fractures are not a feature.
• C (osteopetrosis) is incorrect. Osteopetrosis is an autosomal
•dominant or recessive disorder that is characterized by a defectin osteoclast resorption of bone, resulting in the overgrowth
•• 403

404 Pathology
and sclerosis of bone (marble bone disease). Primary clinicalfindings include pathologic fractures, anemia, and visual andauditory defects.
D (vitamin D—dependent rickets) is incorrect. Vitamin D—dependent rickets is an autosomal recessive disorder. It isassociated with a deficiency of la-hydroxylase, resulting ina deficiency of 1,25-(OH) 2 D3 , which is the active form ofvitamin D. Hypovitaminosis D, or rickets, is characterizedby defective mineralization of bone accompanied by an in-crease in nonmineralized osteoid. Pathologic fractures mayoccur; however, a blue sclera is not a feature of the disease.
3. C (decreased tensile strength of collagen) is correct. Thispatient has scurvy (vitamin C deficiency) caused by a dietlacking in fruits and vegetables. Lack of cross-linking in scurvycauses decreased tensile strength in collagen and poor woundhealing. Vitamin C (ascorbic acid) is important in the hydroxyl-ation of proline and lysine in the initial phases of collagensynthesis by fibroblasts. Normally, collagen is a triple helix ofcross-linked a-chains and has sufficient tensile strength.
A (decreased synthesis of granulation tissue) and B (decreasedsynthesis of type III collagen) are incorrect. Granulation tissueis produced in patients with scurvy. However, the type IIIcollagen produced lacks tensile strength.
D (defect in fibrillin in elastic tissue) is incorrect. A defect infibrillin is present in Marfan syndrome, causing weakness ofelastic tissue, thus weakening the aorta (e.g., aortic dissection)and ligaments.(leukocyte adhesion molecule defect) is incorrect. A defect inneutrophil adhesion molecules (integrins) prevents neutro-phils from adhering to endothelial cells and transmigratinginto tissue. Although this interferes with proper woundhealing, it usually presents as a congenital defect and is notassociated with poor nutrition.
4. A (atrophy) is correct. Atrophy is a decrease in tissue masscaused by cellular shrinkage or loss of cells. Glandular atrophyoccurs in cystic fibrosis because of a defect in the transmembraneregulator, which increases the viscosity of ductal secretions andthe ductal pressure. The figure shows a single layer of ductalepithelial cells, with obstruction and dilation caused by the in-creased pressure. Duct obstruction, in turn, causes back pressureon the proximally located exocrine glands and loss of ductalcells by apoptosis, which results in glandular atrophy. This pa-tient's chronic diarrhea with malabsorption of fat and other nu-trients (greasy stools) is caused by lack of exocrine secretions.
B (dysplasia) is incorrect. Dysplasia is atypical hyperplasia (in-crease in cell number) or atypical metaplasia (replacement ofone adult cell type by another). Dysplastic epithelial

Answers and Discussions 405
changes include increased mitoses, lack of cell orientation,and nuclear enlargement with atypical changes in chromatin.These changes are not shown in the specimen.
C (hyperplasia) is incorrect. Hyperplasia is an increase incell number. There is no evidence of hyperplasia in thespecimen.
D (hypertrophy) is incorrect. Hypertrophy is an increase in cellsize. There is no evidence of hypertrophy in the specimen.
E (metaplasia) is incorrect. Metaplasia is the replacement of oneadult cell type by another in response to injury. There is noevidence of metaplasia in the specimen.
5. B (leukocyte adhesion molecule defect) is correct. A congenitalleukocyte adhesion molecule defect prevents separation of theumbilical cord. Histologic sections of the cord show an absenceof neutrophil adhesion to vessel endothelium and an absenceof neutrophils in the interstitial tissue. During acute inflamma-tion, neutrophils must adhere to endothelial cells in venulesbefore they can transmigrate into interstitial tissue. Neutrophilsmigrate to spaces between the endothelial cells that haveexposed basement membrane, release collagenase, and trans-migrate into the interstitial tissue.
A (absent respiratory burst) is incorrect. The respiratory(oxidative) burst in neutrophils and monocytes is part ofthe 02-dependent myeloperoxidase (MPO) system for killingbacteria. Activated NADPH oxidase in the cell membraneoxidizes reduced NADPH, converting molecular 02 to super-oxide free radicals (02i). The respiratory burst is the energyreleased in this reaction. In chronic granulomatous disease,an X-linked recessive disease caused by absence of NADPHoxidase, the respiratory burst is absent, resulting in a defectin microbicidal activity.
C (myeloperoxidase deficiency) is incorrect. In the 02-dependentMPO system, O. is converted to peroxide (H 202) by superox-ide dismutase in the phagolysosomes of neutrophils andmonocytes. MPO catalyzes a reaction that combines H202with Cl to form hypochlorous free radicals that kill thephagocytosed bacteria. Deficiency of MPO results in a defectin microbicidal activity.
D (opsonization defect) is incorrect. IgG and C3b are opsonizingagents that bind to the surface of bacteria. Receptors for IgGand C3b are located on the plasma membranes of phagocyticleukocytes. Binding of the opsonized bacteria to leukocytesfacilitates phagocytosis of the bacteria, and a deficiency of IgGor C3 produces a detect in phagocytosis.
E (phagocytosis defect) is incorrect. Leukocyte phagocyticdefects include defects in opsonization and defects in the for-mation of phagolysosomes, which are produced by fusion oflysosomes containing hydrolytic enzymes with phago-somes. In Chediak-Higashi syndrome, a defect in lysosomal

406 Pathology
degranulation into phagosomes is present. Leukocytes containlarge azurophilic granules (lysosomes) in the cytosol, becausethe lysosomes have never been emptied.
6. B (acute right-sided ventricular strain) is correct. The figureshows a pulmonary artery thromboembolism (saddle embolus)with extension into all the main pulmonary artery trunks. Thesite of origin for an embolus of this size is usually one of thefemoral veins in the lower extremity. Thromboembolism iscommon in the postoperative setting, particularly after a totalhip replacement. Sudden occlusion of the mainstem pulmonaryarteries produces an acute increase in pulmonary artery pres-sure and acute right-sided ventricular strain, leading to suddendeath (acute cor pulmonale).
A (acute pulmonary infarction) is incorrect. The figure does notshow acute changes in the lung parenchyma, such as a hemor-rhagic pulmonary infarction.
C (aspiration of gastric contents) is incorrect. Aspiration ofgastric contents is common postoperatively. It often pre-cipitates acute respiratory distress syndrome, but it does notcause sudden death.
D (disseminated metastasis) is incorrect. There are no gray-whitelesions in the lung parenchyma to suggest either a primarylung cancer or metastatic lung disease.
E (nosocomial pneumonia) is incorrect. There are no patchyyellow-gray areas in the lung parenchyma to suggest ahospital-acquired pneumonia.
7. E (sickle cell screen) is correct. The patient most likely hassickle cell trait, which causes recurrent microscopic hematuria.In sickle cell trait, the percentage of sickle hemoglobin is40-45%, and the remainder of hemoglobin is hemoglobin A.There are no sickle cells in the peripheral smear in sickle celltrait; therefore, a sickle cell screen is required to induce sicklingof RBCs containing sickle hemoglobin. The oxygen tension inthe renal medulla is low enough to induce sickling of RBCs inthe peritubular capillaries. This causes microinfarctions in therenal medulla and the potential for renal papillary necrosis.
A (bone marrow examination) is incorrect. A bone marrow ex-amination is not warranted, because the patient does not havebone marrow–related anemia or evidence of intrinsic bonemarrow disease.
B (cystoscopy) is incorrect. If the sickle cell screen is negative,a cystoscopy may be necessary to determine the cause of thehematuria.
C (renal biopsy) is incorrect. If the sickle cell screen is negative,a renal biopsy may be necessary to rule out primary renaldisease, particularly IgA glomerulonephritis, which is com-monly associated with episodic hematuria.

Answers and Discussions 407
1) (serum ferritin) is incorrect. Although serum ferritin is de-creased in the early stages of iron deficiency when anemia isnot present, hematuria usually is not associated with any ofthe microcytic anemias.
8. F. ( neutrophi 1 opsonization defect) is correct. Bruton's agamma-globulinemia is an X-linked disorder characterized by failure ofpre-B cells to develop into B cells. Deficiency of IgG has anopsonizing defect; the antigen recognition site of IgG attachesto the bacteria, and the Fc portion of the immunoglobulinattaches to receptors in the plasma membrane of phagocyticleukocytes. This facilitates the phagocytosis of bacteria by trig-gering engulfment of bacteria by pseudopods and eventualformation of a phagocytic vacuole.
A (leukocyte adhesion molecule defect) is incorrect. A defect inneutrophil adhesion molecules (integrins) prevents neutro-phils from adhering to endothelial cells and transmigratinginto tissue.
B (neutrophil chemotactic defect) is incorrect. Movement ofneutrophils toward the site of acute inflammation is chemo-taxis. Chemical mediators (e.g., C5a, leukotriene B4) bind toneutrophil receptors causing release of calcium, whichincreases neutrophil motility.
C (neutrophil membrane-associated protein defect) is incorrect.Such a defect (e.g., Chediak-Higashi syndrome) interferes withthe fusion of lysosomes that contain hydrolytic enzymes withphagosomes in the cytosol of phagocytic leukocytes, pro-ducing phagolysosomes.
D (neutrophil microbicidal defect) is incorrect. A leukocytedefect in killing bacteria usually involves a defect in theoxygen-dependent myeloperoxidase (MPO) system (e.g., defi-ciency of NADPH oxidase or MPO deficiency). Activation ofthis system eventually results in the formation of hypo-chlorous free radicals in phagolysosomes that kill bacteria.
9. D (monoclonal protein spike on serum protein electrophoresis)is correct. The patient has multiple myeloma complicated byrenal failure. The bone marrow aspirate shows immature plasmacells with eccentric nuclei and perinuclear clearing, and the cy-toplasm stains blue with a Wright-Giemsa stain, indicating

408 Pathology
increased synthesis of protein. A monoclonal spike in the-y-globulin region will be seen on serum protein electrophoresis.The spike is caused by a single immunoglobulin (usually IgG)and its corresponding light chain (usually lc), which are secretedby clones of a single neoplastic plasma cell. Renal failure, acommon complication of multiple myeloma, is present whenserum BUN and serum creatinine levels are raised and the ratioof these values is less than 15:1. The other plasma cell clonesare suppressed. Excess light chains, called Bence Jones proteins,are usually present in the urine. In this case, renal failure isdue to the formation of casts composed of Bence Jones proteinsblocking the tubular lumens and inciting a foreign body giantcell reaction.
A (decreased erythrocyte sedimentation rate) is incorrect. Theincreased weight of cells in rouleaux causes them to settle inplasma more rapidly and increases the erythrocyte sedimenta-tion rate.
B (decreased serum calcium level) is incorrect. The patient hasmultiple myeloma, which causes osteolytic lesions. Theselesions tend to increase serum calcium levels.
C (increased prothrombin time) is incorrect. In multiplemyeloma, coagulation studies, including prothrombin time,are usually normal.
D (normal bleeding time) is incorrect. In multiple myeloma, thebleeding time is usually prolonged because of thrombocyto-penia (too few platelets to aggregate) and interference of7-globulins with platelet aggregation.
10. E (increased total peripheral arteriolar resistance) is correct. Thepatient has hypovolemic shock caused by blood loss (tachycardiawith weak pulse; cold, clammy skin; decreased blood pressure).In the initial phase of acute blood loss, the hemoglobin and RBCcount are normal, because whole blood that contains both RBCsand plasma is lost. Within a few hours when plasma begins tobe replaced, the RBC count and the hemoglobin level drop. A de-crease in cardiac output causes underfilling of the aortic arch,which activates the sympathetic nervous system, subsequentlyreleasing catecholamines. Catecholamines cause venocon-striction, increased myocardial contraction, increased heart rate,
r

Answers and Discussions 409
and vasoconstriction of the smooth muscle cells of the peripheralresistance arterioles. Decreased renal blood flow activates therenin-angiotensin-aldosterone system, causing the release of an-giotensin II and peripheral arteriolar vasoconstriction. Vaso-constriction of arterioles in the skin shunts blood to more impor-tant areas of the body, causing cold, clammy skin.
A (decreased hemoglobin concentration) and B (decreased RBCcount) are incorrect. Hemoglobin concentration and RBCcount are normal in the early phase of hypovolemic shockresulting from blood loss.(increased left ventricular end-diastolic pressure) and D (in-creased pulmonary capillary hydrostatic pressure) are incor-rect. Left ventricular end-diastolic pressure and pulmonarycapillary hydrostatic pressure are decreased in hypovolemicshock.
11. E (gynecomastia) is correct. This patient has cirrhosis of theliver (history of alcohol abuse, protuberant abdomen, and de-pendent pitting edema). The skin lesions on the face, neck, andupper trunk are spider angiomas, which contain a central spiralarteriole with a group of small vessels radiating from the arte-riole. Spider angiomas are associated with hyperestrinism, whichis associated with cirrhosis. In cirrhosis, the dysfunctional liveris unable to metabolize estrogen or 17-ketosteroids (e.g., an-drostenedione), which are aromatized in the adipose to weakestrogen compounds. Hyperestrinism in men causes gyneco-mastia (development of breast tissue in males) and female sec-ondary sex characteristics (palmar erythema, soft skin, femalehair distribution).
A (ascites) is incorrect. Development of ascites in cirrhosis ismultifactorial. Factors that contribute to the development ofascites include portal hypertension (increase in hydrostaticpressure), hypoalbuminemia (decrease in oncotic pressure),secondary aldosteronism (salt retention), and increased lym-phatic drainage into the peritoneal cavity.
B (asterixis) is incorrect. Asterixis, or flapping tremor, refers tothe inability to sustain posture. It is a sign of hepatic enceph-

410 Pathology
alopathy, which is caused by an increase in ammonia and falseneurotransmitters (e.g., y-aminobenzoic acid).
C (caput medusae) is incorrect. Caput medusae, or dilated peri-umbilical veins, are associated with increased venous pressurecaused by portal hypertension.
D (esophageal varices) is incorrect. Esophageal varices are dilatedleft gastric coronary veins. These veins normally drain thedistal esophagus and proximal stomach and empty into theportal vein. An increase in portal vein pressure leads to di-lation of the gastric veins, which may rupture.
12. D (immobilization) is correct. The three main causes of intra-vascular thrombus formation are endothelial cell injury (e.g.,cigarette smoking), stasis of blood flow (e.g., postsurgery), andhypercoagulability (e.g., use of oral contraceptives, heredi-tary factor deficiencies). Stasis of blood flow is the most commoncause of venous thrombus formation, which most often occursin the deep veins below the knee. Stasis of blood causes endothe-lial cell injury and activation of the coagulation system, result-ing in the formation of an adherent, occlusive, firm, dark redfibrin clot that entraps RBCs, WBCs, and platelets. The clotpropagates toward the heart and may break off once it reachesthe femoral vein, which is the most common site of pulmonarythromboembolic disease.
A (age) is incorrect. Incidence of venous clots does not increasewith age.
B (decreased cardiac output) is incorrect. Decreased cardiacoutput may occur in ischemic heart disease complicated bycongestive heart failure (not present in this patient), whichcauses stasis of venous blood, predisposing the patient to deepvenous thrombosis.
C (decreased hemoglobin concentration) is incorrect. Decreasedhemoglobin concentration decreases the viscosity of bloodand reduces the risk of thrombosis.
E (turbulent blood flow) is incorrect. Turbulent blood flow con-tributes to endothelial damage causing arterial thrombosis.Arterial thrombi usually develop on top of atheroscleroticplaques that rupture. Thrombi are composed of platelets fusedby fibrin.

Answers and Discussions 411
13. C (leukotrienes C4 , D4 , and E4) is correct. Hydroxylation of ara-ch idonic acid by lipoxygenase yields the leukotrienes C4, D4 , andE4, which are known as the slow-reacting substances of anaphy-laxis. These leukotrienes cause vasoconstriction and broncho-spasm, and they increase vessel permeability. This child's asthmais a type I hypersensitivity reaction induced by exposure to anextrinsic allergen (e.g., cat dander). The leukotrienes contributeto luminal narrowing in the bronchi and terminal bronchi-oles by contracting smooth muscle cells and increasing vesselpermeability, causing inspiratory and expiratory wheezing.
1 (bradykinin) is incorrect. Bradykinin causes vasodilation ofarterioles, increased vessel permeability, and pain in acute in-flammation. Currently, it is not believed to play a pivotalrole in producing airway narrowing in asthma.
13 (complement C3a and C5a) is incorrect. Complement C3a andC5a are anaphylatoxins that directly stimulate the mast cellsto release histamine. Activation of the complement systemdoes not occur in type I hypersensitivity reactions.
D (prostaglandin 1 2) is incorrect. Prostaglandin 1 2 causes vaso-dilation and inhibition of platelet aggregation. It does notplay a pivotal role in asthma.
E (thromboxane A2) is incorrect. Thromboxane A 2 causes vaso-constriction, bronchospasm, and platelet aggregation. Cur-rently, it is not believed to play a pivotal role in producingairway narrowing in asthma.
14. C (decrease intake of H20, maintain intake of Na+) is correct.The patient has the syndrome of inappropriate antidiuretichormone (SIADH) caused by ectopic secretion of antidiuretichormone (ADH) by a primary small cell carcinoma of the lung.ADH normally concentrates urine by reabsorbing H 20 that isfree of electrolytes in the collecting tubules of the kidneys.Therefore, an excess of ADH causes increased reabsorption ofH20, which enters the extracellular fluid (ECF) and produces adilutional hyponatremia. In hyponatremia, an osmotic gradientbetween the ECF and intracellular fluid (ICF) causes 11,0 tomove into the ICF, producing cerebral edema. There is no excessof Na÷ in the ECF; the most effective nonpharmacologic treat-ment is to restrict H20 intake.
A (decrease intake of Na ± and H 20) is incorrect. In SIADH, hypo-natremia is caused by the addition of pure H20 to the ECF,not by a loss of Na+.
B (decrease intake of Na +, maintain intake of H 2O) is incorrect.Decreasing intake of Na+ and maintaining intake of H20 isused to treat hypernatremia due to a hypertonic gain of Nay.
D (increase intake of Ne and H 2O) and E (maintain intake ofNa+ and H20) are incorrect. Refer to the discussion for C.

412 Pathology
15. E (increased vitamin B 12 absorption after addition of intrinsicfactor) is correct. The patient has pernicious anemia, an auto-immune disease in which impaired intestinal absorption ofvitamin B 12 is caused by a lack of intrinsic factor. The figureshows a number of large, egg-shaped macro-ovalocytes; thearrow points to a hypersegmented neutrophil (with more thanfour nuclear lobes), a valuable and early marker of vitamin B12deficiency. Reabsorption of orally administered vitamin B 12 afterthe addition of intrinsic factor confirms the diagnosis of perni-cious anemia.
A (decreased serum folate) is incorrect. Because of the patient'sneurologic deficits, a folic acid deficiency (with decreasedserum folate) is unlikely.
B (decreased serum gastrin) is incorrect. The patient has achlor-hydria (absence of hydrochloric acid), which causes an in-crease (not a decrease) in serum gastrin levels.
C (decreased urine methylmalonic acid) is incorrect. VitaminB 12, unlike folic acid, is involved in propionate fatty acidmetabolism (odd-chain fatty acids). Propionyl CoA is con-verted to methylmalonyl CoA, and methylmalonyl CoA isconverted to succinyl CoA using an enzyme reaction that re-quires vitamin B 12 as a cofactor. Deficiency of vitamin B12causes an increase (not a decrease) in methylmalonic acidlevels in the urine.
D (increased antigliadin antibodies) is incorrect. Antigliadinantibodies are diagnostic of celiac disease, which may causemalabsorption of vitamin B 12 . However, in this patient, perni-cious anemia is the cause of the vitamin B 12 deficiency.
16. A is correct. The patient has polycythemia vera, a myeloprolif-erative disease involving a neoplastic proliferation of trilineagemyeloid stem cells in the bone marrow. In polycythemia vera,which is an absolute polycythemia, the total number of RBCs inthe body in mL/kg (RBC mass) is increased. The RBC count(RBCs/mm3 blood) is also increased. An increase in plasmavolume accompanies the increase in RBC mass, unlike in othertypes of polycythemia. There is no hypoxic stimulus for theincrease in RBC mass; the 02 saturation is normal. Therefore, theincrease in RBC mass is inappropriate. EPO is decreased becausethe 02 content of blood is increased. Absolute leukocytosis andthrombocytosis commonly accompany the increase in RBCs.This patient's pruritus is caused by the increase in histaminereleased from excess numbers of mast cells in the skin.
B is incorrect. These laboratory findings are signs of an appropri-ate type of absolute polycythemia related to a hypoxic stimu-lus for EPO release (decreased 02 saturation). Examples includeobstructive and restrictive lung disease, cyanotic congenitalheart disease, and living at high altitudes.

Answers and Discussions 413
• is incorrect. These laboratory findings are signs of an inappro-priate type of absolute polycythemia (normal oxygen satura-tion) related to ectopic or inappropriate secretion of EPO.Examples include renal disease (e.g., cancer, cystic disease,hydronephrosis) and ectopic secretion of EPO from a hepato-cellular carcinoma.
D is incorrect. These laboratory findings are signs of a relative, asopposed to absolute, type of polycythemia in which a decreasein plasma volume hemoconcentrates RBCs in the peripheralblood. This increases the RBC count without affecting RBCmass. Any cause of volume depletion (e.g., excessive sweating,severe diarrhea) leads to a relative polycythemia.
17. E (positive Heinz body preparation) is correct. The patient hasan acute hemolytic anemia caused by glucose-6-phosphate dehy-drogenase (G6PD) deficiency, an X-linked recessive disorder.G6PD deficiency is most common in persons of African-American and Mediterranean descent. G6PD deficiency leads todecreased synthesis of glutathione, which is necessary to neutral-ize H202, an oxidant product in RBC metabolism. Oxidantstresses that induce hemolysis include infection and drugs (e.g.,primaquine, dapsone). H 20, accumulation in the RBC damagesthe RBC membrane (intravascular hemolysis) and denatureshemoglobin, forming discrete inclusions called Heinz bodies.Splenic macrophages often remove damaged RBC membranes,leaving cells with membrane defects, called "bite cells," circulat-ing in the peripheral blood. The screening test of choice in acutehemolysis is a Heinz body preparation, which requires a specialsupravital stain to identify the Heinz bodies. Enzyme analysis forG6PD is the confirmatory test and must be performed whenactive hemolysis has subsided.
A (abnormal hemoglobin electrophoresis) is incorrect. Thepatient does not have a hemoglobinopathy, in which a de-crease in the synthesis of globin chains (e.g., thalassemia) orthe synthesis of an abnormal hemoglobin (e.g., sickle cellanemia) is present. The development of a hemolytic anemiashortly after beginning primaquine therapy rules out ahemoglobinopathy.(decreased mean corpuscular hemoglobin concentration) isincorrect. The mean corpuscular hemoglobin concentration,the average hemoglobin concentration in RBCs, is normalin G6PD deficiency, increased in congenital spherocytosis,and decreased in microcytic anemias.(decreased serum ferritin concentration) is incorrect. Serumterritin, which indicates the status of the iron stores in the
macrophages in the bone marrow, is normal in G6PD defi-ciency, decreased in iron deficiency, and increased in ironoverload diseases.
D (positive direct Coombs' test) is incorrect. The direct Coombs'test, which is used to detect IgG and/or complement on RBC

414 Pathology
membranes, is the screening test of choice for diagnosis of anautoimmune hemolytic anemia.
18. B (Salmonella paratyphi) is correct. This patient has Salmonellaosteomyelitis. The peripheral blood smear shows sickle cells(dense, boat-shaped RBCs) and numerous target cells (RBCswith a bull's-eye appearance). Sickle cell disease is the mostcommon hemolytic disease in African Americans. The radio-graphic findings in the lower right femur (a periosteal reactionand lytic lesion in the metaphysis) are indicative of osteomy-elitis, which, in sickle cell disease, is most often caused byS. paratyphi. Staphylococcus aureus is the usual pathogen in pa-tients without sickle cell disease.
A (Pseudomonas aeruginosa) is incorrect. P. aeruginosa is acommon cause of localized osteomyelitis associated withpuncture of the foot in patients wearing rubber footwear, butit is not a common cause of osteomyelitis in sickle cell disease.
C (Staphylococcus aureus) is incorrect. Refer to the discussionfor B.
D (Streptococcus pneumoniae) is incorrect. In sickle cell disease,S. pneumoniae is a common cause of septicemia when thespleen becomes dysfunctional from repeated infarctions,but it is not a common cause of osteomyelitis.(Streptococcus pyogenes) is incorrect. S. pyogenes is not the mostcommon cause of osteomyelitis in sickle cell disease.
19. D (respiratory distress syndrome) is correct. The arrow pointsto a type II alveolar cell. The lamellar material in the vacuole issurfactant (lecithin or phosphatidylcholine). It is secreted byalveolar cells to reduce the surface tension in small airways. Re-duction of surface tension decreases collapsing pressure, whichkeeps the small airways open during expiration. In respira-tory distress syndrome, surfactant is deficient or absent, causingmassive atelectasis (collapse) of the alveoli and widespread in-trapulmonary shunting of blood.
A (bronchopneumonia), B (emphysema), C (primary small cellcarcinoma), and E (sarcoidosis) are incorrect. Surfactant syn-thesis remains normal in bronchopneumonia, emphy-sema, primary small cell carcinoma, and sarcoidosis.

•
•Answers and Discussions 415
•• 20. C (hypertrophic cardiomyopathy) is correct. Hypertrophic car-
diomyopathy is the most common cause of sudden cardiac death
.in young people. In some cases, inheritance is autosomal domi-nant. In asymmetric hypertrophy of the interventricular
• septum (IVS), the anterior leaflet of the mitral valve is closer tothe septum than normal, thus narrowing the outlet channel
• for blood flow through the aorta. When systole occurs, the ante-
•rior leaflet of the mitral valve is drawn against the IVS and ob-structs blood flow, producing a systolic ejection murmur thatmay easily be confused with aortic stenosis. Aberrant myofibers
•in the hypertrophied septum and conduction system abnormali-ties also occur; the latter are responsible for a fatal ventricular
•arrhythmia and sudden death. Whenever left ventricular volume(preload) is increased, the intensity of the associated heart
• murmur decreases, indicating decreased obstruction. Lying down
•(increasing venous return to the right side of the heart) or usingdrugs that decrease cardiac contractility and heart rate (e.g.,
• (i-blockers, calcium channel blockers) also decreases murmur in-tensity. Standing reduces venous return to the heart, decreas-
• ing preload, and intensifies the murmur, indicating increased
•obstruction.
•\ (aortic regurgitation) is incorrect. Aortic regurgitation is char-
acterized by an early diastolic murmur directly after the• second heart sound.• B (aortic stenosis) is incorrect. Aortic stenosis is characterized by
a murmur intensity that decreases when the patient is lying
— down and increases when the patient is standing.
•
1) (mitral stenosis) is incorrect. Mitral stenosis is characterized bythe presence of an opening snap followed by a diastolic
•E rumble. It is a complication of recurrent rheumatic fever.
(mitral valve prolapse) is incorrect. Mitral valve prolapse pro-
' duces a midsystolic ejection click followed by a murmur. It
•does not cause sudden cardiac death except when associatedwith Marfan syndrome.•
•21. A (atelectasis) is correct. The most common cause of fever
within the first 24-36 hours after surgery is atelectasis, which• refers to either collapse of a previously inflated lung or to incom-• plete expansion of the lungs on inspiration. Postoperatively,
mucous plugs develop in the terminal bronchioles, allowing re-• sorption of air out of the distal respiratory unit through the
•pores of Kohn. The loss of lung mass causes ipsilateral elevationof the diaphragm and inspiratory lag, because the lung is not
•expanding properly on inspiration. In addition, tactile fremitusand breath sounds are absent, because no air is entering the
• lungs. The trachea also deviates to the ipsilateral side.
• 13 (lobar pneumonia) is incorrect. Postoperative pneumonia
•usually occurs 3-10 days after surgery. Signs of lung consolida-tion are present (e.g., increased tactile fremitus).•
•

416 Pathology
C (lung abscess) is incorrect. Aspiration of oropharyngeal con-tents, the most common cause of a lung abscess, occurs morethan 24 hours after surgery. There is usually a cough pro-ductive of foul-smelling sputum due to aerobes and anaerobesin the abscess.
D (pulmonary infarction) is incorrect. Pulmonary thrombo-embolism usually occurs 5-7 days after surgery.(spontaneous pneumothorax) is incorrect. Spontaneous pneu-mothorax involves the collapse of a portion of the lung, whichproduces hyperresonance to percussion.
22. D (urine test for 5-HIAA) is correct. The patient has the signsand symptoms of carcinoid syndrome (facial flushing, diarrhea,tricuspid regurgitation). In carcinoid syndrome, a carcinoidtumor that secretes serotonin metastasizes to the liver. Tumornodules release serotonin directly into tributaries of the hepaticvein, and the serotonin enters the systemic circulation. The mostcommon site of carcinoid tumors is the terminal ileum. Seroto-nin causes fibrosis of the tricuspid valve, leading to tricuspidregurgitation. 5-HIAA, a metabolic end-product of serotoninmetabolism in the liver, is increased.
A (blood cultures) is incorrect. Blood cultures would show thatthe patient's tricuspid regurgitation is not caused by infectiousendocarditis, which usually occurs in intravenous drug abuse.Blood cultures are not used to confirm the diagnosis of car-cinoid syndrome.
B (liver function tests) is incorrect. Liver function tests areusually normal in the presence of liver metastasis caused byfocal, rather than diffuse, involvement of the liver paren-chyma. Liver function tests are not used to confirm the diag-nosis of carcinoid syndrome.
C (serum electrolytes) is incorrect. Serum electrolytes usuallyshow hypokalemia and metabolic acidosis in secretory typesof diarrhea. Serum electrolytes are not used to confirm thediagnosis of carcinoid syndrome.
23. C (syringomyelia) is correct. The patient has syringomyelia,a degenerative disease that produces a fluid-filled cavity in thecervical spinal cord, causing cervical cord enlargement. As thecavity expands, it destroys the crossed lateral spinothalamictracts (loss of pain and temperature sensation) and anterior horncells (atrophy of intrinsic muscles of the hand).
A (amyotrophic lateral sclerosis) is incorrect. Amyotrophiclateral sclerosis is a degenerative disease that causes thedestruction of upper and lower motor neurons. Sensoryabnormalities do not occur.
B (multiple sclerosis) is incorrect. Multiple sclerosis is anautoimmune disease that causes destruction of myelin andmyelin-producing cells (e.g., oligodendrocytes, Schwann cells).

Answers and Discussions 417
Features include blurry vision (optic neuritis), paresthesias,spastic paraparesis, intention tremors, nystagmus, ataxia, andscanning speech. Loss of pain and temperature sensation andsigns of lower motor neuron disease do not occur.
D (vitamin B 12 deficiency) is incorrect. Vitamin B 12 deficiencyproduces a macrocytic anemia and subacute combined degen-eration of the spinal cord. Myelopathy involving the poste-rior columns causes a loss of proprioception (joint sensation)and vibratory sensation. Myelopathy involving the lateral cor-ticospinal tract causes upper motor neuron disease with signsof spasticity. Loss of pain and temperature sensation andatrophy of the intrinsic muscles of the hand do not occur.
24. E (viral hepatitis) is correct. Clinical manifestations of acuteviral hepatitis include fatigue and jaundice, which is indicatedby the yellow discoloration of the patient's eyes. Hepatitis A iscommonly associated with travel outside the United States.Characteristic histologic changes include evidence of apoptosis,swelling of hepatocytes, and the presence of lymphocytic in-filtrate, which are shown in the specimen. An apoptotic(Councilman's) body is apparent along the lower border of thespecimen; it has a dense black nuclear remnant surrounded bydense-staining eosinophilic cytoplasm and has moved awayfrom nearby cells. Swollen cells and a prominent lymphocyticinfiltrate, also indicative of hepatitis, are apparent in the subja-cent hepatic parenchyma.
A (alcoholic hepatitis) is incorrect. Histologic features of alco-holic hepatitis include a neutrophilic infiltrate, Mallory'sbodies (pink-staining keratin intermediate filaments in hepa-tocytes), and fatty change in hepatocytes. None of thesefeatures is present in the specimen.
U (metastatic liver disease) is incorrect. If metastatic liver diseasewere present, the biopsy specimen would show hyperchro-matic cells with atypical mitotic spindles.
C (obstructive liver disease) is incorrect. Obstructive liver diseaseis characterized by obstruction of intrahepatic or extrahepaticbile ducts. If the patient had obstructive liver disease asopposed to acute viral hepatitis, the specimen would showevidence of dilated ducts filled with bile.
D (primary liver cancer) is incorrect. Hepatocellular carcinoma(primary liver cancer) usually occurs in a setting of underlyingcirrhosis. There is no evidence of fibrosis to indicate cirrhosisor hyperchromatic cells to indicate hepatocellular carcinoma.

418 Pathology
25. C is correct. The patient has the classic machinery murmur(continuous murmur) of a patent ductus arteriosus (PDA). Thisneonate has hypoxemia (decreased arterial Poe) secondary toRDS; therefore, closure of the ductus is not stimulated. Whenoxygenated blood (Sao 2 95%) is shunted into a chamber or vesselwith venous blood (Sao2 75%), there is a step-up of Sao 2 (— 80%)in the venous blood; this is known as a left-to-right shunt.Similarly, when venous blood is shunted into a chamber orvessel with oxygenated blood, there is a step-down of the Sao,(— 80%) leading to clinical cyanosis; this is known as a right-to-left shunt. In PDA, there is a left-to-right shunt causing bloodto flow from the aorta (where pressure is high) through the PDAto the pulmonary artery (where pressure is low), which causesa step-up of Sao2 in the pulmonary artery.
A is incorrect. A step-up of Sao 2 in the right ventricle and pul-monary artery characterizes a ventricular septal defect (VSD).or left-to-right shunt, which is the most common type of con-genital heart disease. If a VSD is not corrected, cyanosis orEisenmenger's syndrome may occur.
B is incorrect. A step-up of Sao2 in the right atrium, right ventri-cle, and pulmonary artery characterizes an atrial septal defect(ASD, or left-to-right shunt), which is most often the resultof a patent foramen ovale. If an ASD is not corrected, cyanosisor Eisenmenger's syndrome may eventually occur.
I) is incorrect. A step-down of Sao 2 in the left ventricle and aortacharacterizes tetralogy of Fallot (right-to-left shunt), which isthe most common type of cyanotic congenital heart disease. Itconsists of an overriding aorta (least common defect), VSD,pulmonary stenosis, and right ventricular hypertrophy. Thedegree of pulmonic stenosis determines the severity of theright-to-left shunt. If the stenosis is not severe, then most ofthe venous blood enters the pulmonary artery and is oxygen-ated; hence the patient is often acyanotic. However, whenthe stenosis is severe, most of the venous blood is shuntedthrough the VSD into the left ventricle (right-to-left shunt),leading to cyanosis.
26. E (serum ferritin test) is correct. The serum ferritin test wouldidentify an iron deficiency, the probable cause of the patient'smicrocytic anemia. The systolic ejection murmur indicates aorticstenosis, and the fragmented RBCs (schistocytes) in the periph-eral blood indicate a macroangiopathic hemolytic anemia.Iron deficiency is suspected in aortic stenosis, because RBCs aredamaged when they pass through a stenotic valve. The damagedcells release hemoglobin directly into the blood, leading to he-moglobinuria (indicated by the findings on urinalysis) and even-tual depletion of iron stores in the bone marrow.
A (direct Coombs' test) is incorrect. The direct Coombs' testdetects the presence of IgG and C3b on the surface of RBCs.

Answers and Discussions 419
It is useful when autoimmune hemolytic anemia is suspected.Autoimmune hemolytic anemias are normocytic, and schis-tocytes are not present.(enzyme assay for pyruvate kinase) is incorrect. When anemiais caused by pyruvate kinase deficiency, the peripheral bloodsmear shows dehydrated RBCs with thorny projections, unlikethe cells shown in the figure.
C (hemoglobin electrophoresis) is incorrect. Hemoglobin electro-phoresis detects changes in the concentration of normal andabnormal forms of hemoglobin, such as hemoglobin S insickle cell disease, which is not present in this patient.
1) (osmotic fragility test) is incorrect. The osmotic fragility testis used to confirm a diagnosis of hereditary spherocytosis,in which the osmotic fragility of RBCs is increased. Hereditaryspherocytosis is a normocytic anemia, and schistocytes arenot present.
27. D (synthesis of prostaglandins) is correct. Cyclooxygenase con-verts arachidonic acid to prostaglandins. Aspirin and nonsteroi-dal anti-inflammatory drugs inhibit cyclooxygenase andprevent the synthesis of prostaglandins that may produce pain(e.g., prostaglandin E2).
A (chemotaxis of leukocytes) is incorrect. Chemotaxis refers tothe movement of neutrophils toward the site of acute inflam-mation. Chemical mediators (e.g., C5a, leukotriene B 4) bind toneutrophil receptors, causing release of calcium, which in-creases neutrophil motility.
B (synthesis of bradykinin) is incorrect. Activated coagulationfactor XII converts high-molecular-weight kininogen to bra-dykinin, which causes vasodilation of arterioles, increasedvessel permeability, and pain in acute inflammation.
C (synthesis of leukotrienes) is incorrect. Lipoxygenase convertsarachidonic acid to leukotrienes. Aspirin does not inhibitlipoxygenase.
E (transmigration of leukocytes) is incorrect. In acute inflamma-tion, neutrophils adhere to endothelial cells in venules andthen transmigrate through the basement membrane to theinterstitial tissue. Aspirin does not interfere with neutrophiladhesion or transmigration.

420 Pathology
28. E is correct. The presence of a mixed hyperbilirubinemia(increase in unconjugated and conjugated bilirubin) and anincrease in the serum transaminases are markers of the ictericphase of acute hepatitis B. HBsAg, HBeAg, and IgM-anti-HBcare positive. HBsAg first appears in the serum 2-8 weeks beforesymptoms develop and is the last antigen marker to disappearif recovery occurs. HBeAg is an infective particle that appearsshortly after HBsAg and disappears before it. IgM-anti-HBc is anonprotective antibody that appears shortly after HBsAg andpersists until no viral particles are present in the hepatocytes.
A is incorrect. The presence of anti-HBs and the absence ofHBsAg, HBeAg, IgM-anti-HBc and IgG-anti-HBc indicateimmunization with hepatitis B vaccine. Anti-HBs is a protec-tive antibody.
B is incorrect. The presence of anti-HBs and IgG-anti-HBc andthe absence of HBsAg, HBeAg, and IgM-anti-HBc indicate re-covery from hepatitis B. Anti-HBs is a protective antibody, andanti-HBc-IgG develops when all the viral particles in hepato-cytes have disappeared.
C is incorrect. The presence of IgM-anti-HBc and the absence ofHBsAg, HBeAg, IgG-anti-HBc, and anti-HBs indicate that thepatient is in the "window phase" of recovery from hepatitis Bwhen all the antigens have disappeared and their correspond-ing antibodies have not yet had time to develop.
D is incorrect. The presence of HBsAg and the absence of HBeAg,104-ant IgG-anti-HBc, and anti-HBs are characteristicof the earliest phase of acute hepatitis B.
29. D (deficiency of low-density lipoprotein receptors) is correct.The patient has familial hypercholesterolemia, an autosomaldominant disorder associated with a deficiency of LDL receptors,which is characterized by increased levels of serum cholesterol.Cholesterol deposits in the Achilles tendon (tendon xanthoma)and the eyelids (yellow patches called xanthelasma). Increasedcholesterol causes premature atherosclerosis, resulting in strokesand myocardial infarctions between 30 and 40 years of age.
A (decreased activation of capillary lipoprotein lipase) and B(deficiency of apolipoprotein C-II) are incorrect. Deficiencyof capillary lipoprotein lipase or apolipoprotein C-II, the acti-vator of capillary lipoprotein lipase, causes an increase inchylomicrons in children, not adults. Hyperchylomicronemialeads to hypertriglyceridemia, causing pancreatitis, hepato-splenomegaly, and papular skin lesions (eruptive xanthomas).
C (deficiency of apolipoprotein E) is incorrect. Chylomicronremnants and intermediate-density lipoproteins normallyhave apolipoprotein E on their surface. Receptors for apolipo-protein E in hepatocytes remove these remnants from theblood. Deficiency of apolipoprotein E leads to an increase inchylomicron remnants (remnant disease or dysbetalipo-

Answers and Discussions 421
proteinemia) and increased levels of both triacylglycerol andcholesterol. There may be yellow deposits of triacylglyceroland cholesterol in skin creases on the palms.
E (increased synthesis of very low density lipoprotein) is incor-rect. Increased synthesis of endogenous triacylglycerol in theliver causes increased levels of both LDI, fraction and tria-cylglycerol. There may he deposits of VLDL fraction and trig-lycerides in yellow papular skin lesions (eruptive xanthomas).
30. C (reinfarction ) is correct. After an uncomplicated MI, serumCK-MB peaks in 24 hours and returns to normal within 72hours. Therefore, the presence of CK-MB on day 4, after an MI,indicates reinfarction. Both cTn-I and cTn-T peak within 24hours and return to normal within 7 to 10 days following anuncomplicated MI. The presence of cTn-I and cTn-T on day 4is expected.
A (angina pectoris) is incorrect. Angina pectoris causes myocar-dial ischemia without causing myocardial cell injury. Anginapectoris does not lead to an increase in CM-MB, cTn-1, andcmn-T.
B (pericarditis) is incorrect. Pericarditis is inflammation of thesurface lining of the heart, causing precordial chest pain, andit is associated with a pericardial friction rub. Pericarditisdoes riot damage the myocardial tissue and lead to the releaseof cardiac enzymes.
D (right ventricular infarction) is incorrect. Isolated right ven-tricular infarction is extremely rare, because the blood vesselsperfusing the right ventricle are usually too small to developoccluding atheromas. Right ventricular infarction producessigns of right-sided heart failure (neck vein distention, depen-dent pitting edema), which are not present.
E (rupture of the anterior wall) is incorrect. Rupture caused bynecrosis of the myocardial wall usually occurs on days 3-7 fol-lowing an MI. Rupture of the anterior wall produces cardiactamponade with muffled heart sounds and neck vein disten-tion followed by rapid death.
31. C (inactivation of a suppressor gene) is correct. The figureshows the mucosal surface of the colon covered by numerous,sessile adenomas. These findings, plus the family history ofcolectomies between 35 and 40 years of age, suggest familialpolyposis. Familial polyposis is an autosomal dominant disordercharacterized by inactivation of the adenomatous polyposis colisuppressor gene on chromosome 5.
A (complication of Crohn's disease) is incorrect. Crohn's diseaseis a chronic granulornatous ulceroconstrictive disease that in-volves only the colon in 20% of cases. There is an increasedincidence of the disease in first-degree relatives. The inflamma-tion is transmural and produces skip lesions throughout the

422 Pathology
gastrointestinal tract. Therefore, a total colectomy at an earlyage is not a treatment option in Crohn's disease, althoughthere is minimal risk of developing colorectal cancer. Further-more, the nodules on the mucosal surface in the figure are truepolyps and do not represent cobblestoning (mucosa sur-rounded by areas of ulceration).
B (complication of ulcerative colitis) is incorrect. Ulcerativecolitis is a chronic ulceroinflammatory disease that begins inthe rectum and may spread continuously to involve the entirecolon up to the ileocecal valve. There is an increased inci-dence of the disease in first-degree relatives. Ulcerative colitisis characterized by extensive areas of mucosal and submucosalulceration. Islands of inflamed and hemorrhagic mucosa rep-resenting pseudopolyps are interspersed between the areasof ulceration. These findings are not present in the figure.
D (oral mucosal pigmentation) is incorrect. Peutz-Jegherssyndrome is an autosomal dominant polyposis with hamar-tomatous polyps located primarily in the small bowel andstomach. It is characterized by increased melanin pigmenta-tion of the lips and buccal mucosa. There is no increasedrisk of colorectal cancer.
E (X-linked recessive inheritance pattern) is incorrect. Familialpolyposis is an autosomal dominant disorder.
32. E (increased synthesis of thyroid-binding globulin) is correct.The total serum 14 level reflects free, unbound, metabolicallyactive T4 and metabolically inactive T4 bound to thyroid-bindingglobulin (TBG). Slight enlargement of the thyroid gland isnormal in pregnancy, and the increase in estrogen that normallyoccurs during this period stimulates liver synthesis of TBG. T4normally occupies one third of the binding sites on TBG, andthe additional TBG with its bound fraction increases the totallevel of T4 without affecting the free Hence, the patient isclinically euthyroid, and the serum TSH remains normal.
A (decreased peripheral conversion of T4 to triiodothyronine) isincorrect. The outer ring deiodinase that normally converts T4to 13 in the periphery is normal in pregnancy.
B (increased release of T4 from acute thyroiditis) is incorrect.The patient's thyroid gland is nontender, and there is no ev-idence of thyrotoxicosis. Furthermore, the serum TSH levelis normal, indicating that the patient is euthyroid.
C (increased synthesis of T3 ) and I) (increased synthesis of T4)are incorrect. Increased synthesis of 13 and 14 produce signsof thyrotoxicosis and suppression of serum TSH. There isno evidence of thyrotoxicosis.

Answers and Discussions 423
33. ti (decreased cardiac output) is correct. The heart fails whenit cannot pump blood delivered to it by the venous system.Cardiac output is decreased whether the heart failure is left-sidedor right-sided.
A (bibasilar inspiratory crackles) is incorrect. Bibasilar inspiratorycrackles are a sign of left-sided heart failure, a "forward" heartfailure causing a decreased cardiac output and backup ofblood in the left ventricle, left atrium, and pulmonary capillar-ies. Increased pulmonary capillary hydrostatic pressure causesfluid to enter the interstitium of the lung and eventually thealveoli (pulmonary edema). Air entering alveoli containingfluid produces inspiratory crackles that are best heard at thebase of both lungs.
C (dependent pitting edema) and F. (passive congestion in theliver) are incorrect. Dependent pitting edema and passivecongestion in the liver are both signs of right-sided heartfailure, a "backward" heart failure causing systemic venouscongestion. The increase in hydrostatic pressure in the venoussystem causes fluid (transudate) to leak into the interstitialspace through the venules, leading to dependent pittingedema. The hepatic vein empties into the inferior vena cava,and blood backs up into the central veins, leading to passivecongestion in the liver.
D (paroxysmal nocturnal dyspnea) is incorrect. Paroxysmalnocturnal dyspnea is a sign of left-sided heart failure, whichoccurs primarily at night when the patient is supine in bed.At this time, gravity does not impede blood flow to theright side of the heart, and fluid from the interstitial spaceenters the venous system. Excess blood enters the failed leftventricle and backs up into the lungs, causing pulmonaryedema and dyspnea, which awakens the patient. Symptomsresolve when the patient stands up and gravity decreasesvenous return to the right side of the heart.
34. C (hemolytic anemia) is correct. The figure shows a tricuspidaortic valve with severe aortic stenosis. The commissures havefused, and multiple fibrotic calcium deposits protrude into thesinuses of Valsalva. A common complication of aortic stenosisis an intravascular hemolytic anemia with schistocytes (frag-mented RBCs), which may lead to chronic iron-deficiencyanemia from loss of hemoglobin in the urine. Serum haptoglo-bin levels are often zero, because haptoglobin combines withfree hemoglobin in the plasma and is removed by splenicmacrophages.
A (acute myocardial infarction) is incorrect. The figure showssevere aortic stenosis. Acute myocardial infarction is not acommon complication of aortic stenosis.
B (aortic dissection) is incorrect. 'The figure shows severe aorticstenosis. Aortic dissection is associated with hypertension and

424 Pathology
connective tissue disorders leading to cystic medial degenera-tion. It is not a complication of aortic stenosis, in whichneither hypertension nor connective tissue disorders arepresent.
D (hypertension) is incorrect. The figure shows severe aorticstenosis, in which hypertension is unlikely. In aortic steno-sis, the diminished area of the stenotic valve orifice leads to adecrease in stroke volume and cardiac output, which reducessystolic pressure but has no effect on diastolic pressure.
E (stroke syndrome) is incorrect. The figure shows severe aorticstenosis. There is no increased incidence of stroke in patientswith aortic stenosis.
35. A (acute myocardial infarction) is correct. The patient hasKawasaki disease (mucocutaneous lymph node syndrome).Characteristic findings include painful cervical lymph nodes,swelling of the hands and feet, a desquamating rash involvingthe fingers, and vasculitis of the coronary arteries. Vasculitisoften leads to coronary artery thrombosis and acute myocardialinfarction.
B (aortic arch aneurysm) and k (aortic dissection) are incorrect.Thoracic aneurysms are most often caused by atherosclerosis.Kawasaki disease is not associated with inflammation of theaortic arch or the aortic arch vessels; aneurysm and aortic dis-section do not occur in Kawasaki disease.
D (infective endocarditis) and E (mitral stenosis) are incorrect.Infective endocarditis is most often caused by an infectiousorganism such as Staphylococcus aureus or Streptococcus viridans.Mitral stenosis is most often caused by chronic rheumaticmitral valvulitis. Infection of the heart valves does not occurin Kawasaki disease.
36. E (insulin) is correct. Neonatal hypoglycemia (jitteriness andseizures) is caused by hyperinsulinism related to the poor glyce-mic control in the mother who has gestational diabetes mellitus.After delivery, the source of the fetal hyperglycemia is removed,and the increased insulin levels cause a drastic reduction in theblood glucose levels. This emphasizes the need to infuse thesepatients with glucose after birth.
A (cortisol), B (epinephrine), C (glucagon), and D (growthhormone) are incorrect. These hormones are all increased innewborns because of the stress of the delivery. They are all glu-coneogenic and cause hyperglycemia, which does not producejitteriness or seizures.

Answers and Discussions 425
37. D (senile plaque) is correct. Alzheimer's disease is the mostcommon cause of senile dementia in patients older than 65 yearsof age. The figure shows a senile plaque containing a core ofpink-staining 13-amyloid protein surrounded by distended neu-rites (distal neuronal cell processes). Numerous plaques arepresent in the frontal, temporal, and parietal lobes in patientswith Alzheimer's disease. 13-Amyloid protein derives fromamyloid precursor protein, which is normally coded on chromo-some 21. 13-Amyloid protein is toxic to neurons and accountsfor loss of intellectual function.
1 (amyloid angiopathy) is incorrect. Amyloid angiopathy is acharacteristic finding in patients with Alzheimer's disease. Itdescribes the deposition of 13-amyloid protein in the walls ofleptomeningeal and parenchymal vessels, which are notevident in the figure.
B (gliosis) is incorrect. Gliosis is the repair process that occurswhen the brain is injured. It consists of a proliferation of as-trocytes that produce a dense network of cytoplasmic pro-cesses. Activated microglial cells commonly accompany thisreactive process.(neurofibrillary tangle) is incorrect. Neurofibrillary tangles arepresent in patients with Alzheimer's disease and in elderly in-dividuals who do not have dementia. The tangles are bestvisualized with silver stains and represent protein-rich fila-ments arranged in helical fashion within the cytoplasm ofneurons.(spongiform encephalopathy) is incorrect. "Spongiformchange" in the brain is a characteristic finding in Creutzfeldt-Jakob disease, which is caused by an infectious agent (prion)that is devoid of nucleic acids.
38. A (benign tumor of [3-islet cells) is correct. Benign tumors ofthe 13-islet cells (insulinomas) synthesize excess insulin, resultingin severe fasting hypoglycemia. When preproinsulin in thej3-islet cells is delivered to the Golgi apparatus, proteolytic reac-tions generate insulin and a cleavage peptide called C-peptide.Hence, C-peptide is a marker for endogenous synthesis ofinsulin. Both serum insulin and serum C-peptide levels are in-creased, thus confirming the presence of an insulinoma.
B (ectopic secretion of an insulin-like factor) is incorrect. Aninsulin-like factor that causes hypoglycemia is most oftenproduced by a hepatocellular carcinoma. Hypoglycemia sup-presses 13-islet cells, resulting in a decrease in serum insulinand C-peptide.
C (malignant tumor of a-islet cells) is incorrect. Tumors ofa-islet cells secrete glucagon, which produces hyperglyce-mia by stimulating gluconeogenesis.
D (patient injection of human insulin) is incorrect. Injection ofhuman insulin increases serum insulin and produces hypogly-

426 Pathology
cemia. Hypoglycemia suppresses fl-islet cells, causing a de-crease in endogenous synthesis of insulin and a correspondingdecrease in serum C-peptide.
39. C (left atrial myxoma) is correct. Cardiac rnyxomas are themost common primary cardiac tumors in adults. Symptomsinclude nonspecific complaints such as fever and malaise. Thetumor has a ball-valve effect that causes sudden blockage ofblood flow through the mitral valve, resulting in episodic faint-ing spells. A diastolic murmur similar to that of mitral stenosis isalso present. Peripheral embolization of tumor also occurs. Inf-arctions of the spleen cause pain in the left upper quadrantand friction rubs. Infarctions of the kidneys cause flank painand hematuria.
A (calcific aortic stenosis) is incorrect. Calcific aortic stenosis isassociated with a systolic ejection murmur. Angina with exer-cise occurs because of ischemia of the subendocardium in theconcentrically hypertrophied left ventricle. The decreasedcardiac output through the stenotic valve causes syncope withexercise. Peripheral embolization does not occur.
B (hypertrophic cardiomyopathy) is incorrect. Hypertrophic car-diomyopathy is characterized by asymmetric hypertrophy ofthe interventricular septum causing findings similar to thosedescribed for calcific aortic stenosis. Peripheral emboliza-tion does not occur.
D (mitral stenosis) is incorrect. Refer to the discussion for C.E (pericardial effusion) is incorrect. A pericardial effusion pro-
duces muffled heart sounds and is not associated with syncopeor with peripheral embolization.
40. F (tracheoesophageal fistula) is correct. In a tracheoesophagealfistula, the proximal esophagus ends blindly. The distal esopha-gus arises from the trachea, causing the stomach to distendwith air. When the infant breast-feeds, milk refluxes into thetrachea, causing coughing and cyanosis due to aspiration of milkinto the lungs. Polyhydramnios occurs during pregnancy,because the fetus cannot swallow the amniotic fluid and reab-sorb it in the duodenum.
A (choanal atresia) is incorrect. Choanal atresia is caused by abony septum between the nose and the pharynx, which forcesthe infant to breathe only through the mouth. The cyanosisthat develops when the infant is hreastfeeding ceases whenthe infant breaks from the breast and begins crying. Gastricdistention or polyhydramnios is not associated with choanalatresia.
B (congenital pyloric stenosis) is incorrect. Hypertrophy of thepylorus does not present at birth but at approximately 2-4weeks of life. Projectile vomiting of non–bile-stained fluid

Answers and Discussions 427
occurs. Polyhydramnios or cyanosis on breastfeeding are notassociated with congenital pyloric stenosis.
C (duodenal atresia) is incorrect. Duodenal atresia (lack of alumen) occurs just distal to the entry of the common bile ductinto the duodenum. Projectile vomiting of bile-stained fluidoccurs at birth. Air is present in the stomach and in theduodenum proximal to the opening of the common bile duct.Polyhydramnios occurs during pregnancy, because there isnot enough duodenum to reabsorb the amniotic fluid.
D (esophageal web) is incorrect. Esophageal webs are uncommonin newborns. They produce dysphagia for solids but notliquids. Polyhydramnios is not associated with esophagealwebs.
41. D (t(15;17)) is correct. This patient has acute promyelocyticleukemia (APL) complicated by disseminated intravascular coag-ulation (DIC). The peripheral blood smear shows a hyper-granular promyelocyte filled with multiple intertwining Auerrods. Auer rods are present only in variants of acute myeloge-nous leukemia. Patients with APL have a characteristic t(15;17)translocation, which causes an abnormality in retinoic acidmetabolism. DIC is invariably present in APL, because the releaseof procoagulant from the granules of the promyelocytes acti-vates the coagulation system cascade. DIC causes multiple coagu-lation factor deficiencies (prolonged prothrombin time andpartial thromboplastin time), thrombocytopenia (which causespetechiae and ecchymoses), and activation of the fibrinolyticsystem (increased D-dimers). Leukemias commonly metastasizeto the lymph nodes (generalized lymphadenopathy), liver, andspleen (hepatosplenomegaly). Treatment with retinoic acidcauses maturation of the promyelocytes; however, relapses arecommon.
A (t(8;14)) is incorrect. A t(8;14) translocation is associated withBurkitt's lymphoma, which is a malignancy involving B cells.
B (t(9;22)) is incorrect. A t(9;22) translocation is associated withchronic myelogenous leukemia, in which myeloblasts withAuer rods are not present.
C (t(14;18)) is incorrect. A t(14;18) translocation is associatedwith a follicular B-cell lymphoma.

428 Pathology
42. C (liver involvement) is correct. The tumor-node-metastasis(TNM) system is used to stage cancers arising from epithelialtissues. T refers to the size and nuclear features of the tumor;N refers to the presence or absence of lymph node metastasis;and M refers to the presence or absence of metastasis to sitesother than lymph nodes, such as the liver. M is the mostimportant prognostic factor. The presence of distant metastasesimplies that the cancer has already infiltrated through re-gional lymph nodes draining the cancer and has entered thebloodstream.
A (differentiation of the tumor) and B (extent of invasion) areincorrect. Differentiation or grade of the tumor describes thehistologic appearance of the tumor. If the tumor has recogniz-able features, such as keratin in squamous cells or glands, thenthe tumor is well differentiated or low grade. If the tumor hasno histologic features with characteristics identifying thetissue of origin, then the tumor is poorly differentiated, ana-plastic, or high grade. The extent of differentiation and extentof invasion of the tumor are less important prognostic factorsthan the M of the TNM system.
D (lymph node involvement) is incorrect. Refer to the discussionfor C.
43. D (a-thalassemia) is correct. a-Thalassemia is an autosomal re-cessive disorder that involves decreased production of a-globinchains, causing a microcytic anemia with decreased synthesisof hemoglobin A (2a213), hemoglobin A2 (2a26), and hemoglo-bin F (2a27). For unexplained reasons, the RBC count is in-creased in all the thalassemias, and it is decreased in all othermicrocytic anemias (e.g., iron deficiency anemia). Hemoglobinelectrophoresis is normal, because the proportion of each ofthe hemoglobins remains the same. a-Thalassemia involves thesynthesis of globin chains; serum ferritin, which correlateswith bone marrow iron stores, is normal.
A (iron deficiency) is incorrect. Iron deficiency is the mostcommon cause of a microcytic anemia. RBC count is de-creased, and serum ferritin is decreased.
B (sickle cell trait) is incorrect. Sickle cell trait does not causeanemia.
C (sideroblastic anemia) is incorrect. Sideroblastic anemias are agroup of microcytic anemias that are caused by a defect in thesynthesis of heme (iron plus protoporphyrin) in the mito-chondria of developing RBC normoblasts. Iron accumulatesin the mitochondria of nucleated RBCs and produces ringedsideroblasts, which are easily identified in a bone marrow aspi-rate. RBC count is decreased, and serum ferritin is increased.
E ((-thalassemia) is incorrect. In 13-thalassemia, hemoglobin A(2a2(3) is decreased, because 3-globin chain synthesis is de-creased. There is a corresponding increase in hemoglobin A2

Answers and Discussions 429
(2a26) and hemoglobin F (2a2y), because a-, 6-, and y-chainsynthesis is normal. The hemoglobin electrophoresis is normalin this patient, thus excluding P-thalassemia.
4-4. B (coagulation necrosis) is correct. Coagulation necrosis occurswhen arterial blood flow suddenly ceases, which in this patientfollows thrombosis of the right coronary artery. An infarct is thegross manifestation of coagulation necrosis in underlying tissue.The specimen shows an extensive pale yellow infarct in the leftventricle, which involves the posterior wall and papillary muscle(the round structure projecting into the ventricular lumen). Theinfarct is pale because of the density of dead myocardial tissue,which prevents the infiltration of blood from necrotic bloodvessels.
A (caseous necrosis) is incorrect. Caseous necrosis has the grossappearance of soft, cheese-like material within a granuloma. Itoccurs primarily in mycobacterial infections (e.g., tuberculo-sis) and systemic fungal infections (e.g., histoplasmosis).
C (enzymatic fat necrosis) is incorrect. Fat necrosis appears aschalky white areas of saponified fat in an area of pancreaticinflammation (e.g., acute pancreatitis), not in cardiac muscle.
D (fibrinoid necrosis) is incorrect. Fibrinoid necrosis occurs insmall muscular arteries, arterioles, venules, and glomerularcapillaries. It is commonly associated with small-vessel vascu-litis in hypertension and with deposits of antigen-antibodycomplexes in vessel walls in immunocomplex disease.
I (liquefactive necrosis) is incorrect. Liquefactive necrosis typi-cally appears as an area of soft tissue with a liquid center.Unlike the coagulation necrosis shown in the specimen,liquefactive necrosis is associated with softening of tissuecaused by the release of hydrolytic enzymes from neutrophils(as in acute inflammation) or from the cells in tissue (as incerebral infarction).
45. 11 (spontaneous pneumothorax) is correct. A pulmonary com-plication that results from scuba diving is rupture of a preexist-ing intrapleural bleb or a subpleural bleb, causing a hole in thepleura and a spontaneous pneumothorax. A hole in the pleuraresults in a loss of negative pressure in the pleural cavity, whichcauses a collapse of the lung. Physical findings include hyper-resonance to percussion, tracheal deviation to the side of thecollapse, elevation of the diaphragm, and absent breath sounds.
A (decompression sickness) is incorrect. Decompression sickness(gas embolism) is a complication of scuba diving. As a diverdescends, nitrogen gas under increased pressure moves fromthe alveoli and dissolves in tissue and blood. Rapid ascentforces nitrogen to move out of the tissue and blood in theform of bubbles, causing ischemic damage.

430 Pathology
li (pleural effusion) and C (pulmonary infarction) are incorrect.Although a pulmonary thromboembolism leading to a pulmo-nary infarction and pleural effusion is a complication of scubadiving, it usually occurs when the diver is stationary and indeep water. Physical findings of a pleural effusion include dull-ness to percussion, deviation of the trachea to the contralat-eral side, and absent breath sounds.(tension pneumothorax) is incorrect. A tension pneumothoraxis not a common cause of dyspnea in scuba diving. Unlike aspontaneous pneumothorax, a tension pneumothorax is asso-ciated with a flap-like pleural tear. Inspiration causes the flapto open and allows air to enter the pleural cavity. However,the flap closes on expiration and prevents air from leavingthe cavity. Increased intrapleural pressure (greater than theatmosphere) causes compression of the lung (atelectasis) anddeviation of the trachea to the contralateral side. The dia-phragm is depressed.
46. A (acute respiratory distress syndrome) is correct. In thispatient, urinary retention due to prostate hyperplasia has ledto sepsis (most often due to Escherichia coli) and endotoxicshock, which is the most common cause of acute respiratorydistress syndrome. Alveolar macrophages release cytokines thatcause neutrophil adhesion to the pulmonary capillaries. Neu-trophils transmigrate into the alveoli and destroy type I and typeII pneumocytes. Destruction of type II pneumocytes causes lossof surfactant and collapse of the alveoli. An exudate leaks intothe interstitium and alveoli, where hyaline membranes areformed. Clinical findings include dyspnea, tachypnea, and inspi-ratory crackles. Chest radiography shows interstitial and alveo-lar infiltrates. Arterial blood gases show hypoxemia andrespiratory acidosis.
B (congestive heart failure) is incorrect. Congestive heart failureis most often caused by ischemic heart disease. It is not associ-ated with fever and septicemia.
C (lobar pneumonia caused by gram-negative bacteria) is incor-rect. A chest radiograph shows consolidation of a lobe in thelung rather than interstitial and alveolar infiltrates in bothlungs.
D (multiple pulmonary infarcts) is incorrect. Pulmonary infarc-tions produce hypovascular areas at the periphery of the lung,hypoxemia, and respiratory alkalosis, not acidosis.
E (pneumonia caused by Streptococcus pneurnoniae) is incorrect.S. pneumoniae is the most common cause of typical community-acquired, not nosocomial, pneumonia. Pneumonia caused byS. pneumoniae is not an outcome of urinary retention.

Answers and Discussions 431
• 47. B (perfusion defect) is correct. A perfusion defect occurs whenblood flow to the alveoli is obstructed. This interferes with the
•normal exchange of 02 between the alveoli and pulmonary cap-illaries and results in decreased arterial Po 2 . In this patient, the
•perfusion defect is caused by a pulmonary infarction, which pro-duces the wedge-shaped hemorrhagic infarct shown in the
• specimen. The base of the infarct extends to the pleural surface.
•Pulmonary infarctions are common in nonambulatory hospi-talized patients because of blood stasis in the lower extremities,
•leading to deep venous thrombosis.
A (diffusion defect) is incorrect. A diffusion defect occurs when
•blood flows to the alveoli, but diffusion of 02 into the pulmo-nary capillaries is inhibited by factors such as fibrous tissue
• (as in sarcoidosis) or fluid (as in pulmonary edema). In thispatient, a diffusion defect could not have caused the reduced
• Po2, because the specimen shows a pulmonary in farct, i ndicat-ing that blood flow to the alveoli had ceased.
C (respiratory acidosis) is incorrect. Respiratory acidosis (de-• creased arterial pH) is caused by retention of CO 2 with a subse-
quent increase in alveolar Pco,, decrease in alveolar Po2, and• decrease in arterial Po,. Respiratory acidosis does not occur
in a pulmonary infarction.D (respiratory alkalosis) is incorrect. Respiratory alkalosis
(increased arterial pH with a decrease in Prot) is the most
•common arterial blood gas abnormality in a pulmonary in-farction. Respiratory alkalosis does not cause a decrease in
•arterial Po2.(ventilation defect) is incorrect. A ventilation defect occurs
• when blood flows to the alveoli, but 02 is prevented from
•entering the alveoli (e.g., atelectasis with collapse of theairways). In such cases, perfusion to the lung without ventila-
•tion leads to a decrease in Po2 . In this patient, a ventilationdefect could not have caused the reduced P0 2, because atelecta-
• sis of the lung is not hemorrhagic or wedge-shaped.
• • 48. D (sweat chloride test) is correct. The patient has cystic fibrosis,
an autosomal recessive disease characterized by a deficiency of
• cystic fibrosis transport regulator that normally regulates sodium
•and chloride ions in secretions. The sweat chloride test detectsthe presence of defective chloride transport by measuring in-
•creased amounts of chloride in the sweat. Clinical findings ofcystic fibrosis include recurrent respiratory infection caused by
• thickened secretions and malabsorption caused by thickened se-• cretions blocking exocrine ducts in the pancreas. Nasal polyps
are frequently present in children.
• A (chromosome study) is incorrect. Chromosome studies are
• generally reserved for prenatal diagnosis of cystic fibrosis. The
•defect occurs on chromosome 7 (three-nucleotide deletion),
•

432 Pathology
and specialized techniques are required to identify the abnor-mal gene locus.
13 (serum IgE level) is incorrect. Allergic polyps occur in patientswith allergic rhinitis associated with IgE antibody-mediateddisease (type I hypersensitivity). Allergic polyps mainlydevelop in adults and are not seen in children. The patienthas no signs of allergic rhinitis (e.g., nasal stuffiness, seasonalvariation).
C (stool culture) is incorrect. The history of greasy stools in thepatient is a sign of malabsorption and not an invasive entero-colitis due to a microbial pathogen.
49. C is correct. Rat poison contains warfarin, an anticoagulantthat inhibits epoxide reductase, which normally converts inac-tive vitamin K to active vitamin K. Lack of active vitamin Kcauses vitamin K-dependent coagulation factors (e.g., prothrom-bin, factor VII, factor IX, and factor X) to be nonfunctional.Bleeding from the mouth and gastrointestinal tract is a sign ofexcess anticoagulation. Warfarin does not affect platelet produc-tion or function; thus, the platelet count and bleeding time (testof platelet function) are normal. The PT evaluates the activityof coagulation factors in the extrinsic system (factor VII) to theformation of a fibrin clot in the final common pathway (factorX, factor V, prothrombin, fibrinogen). The aPTT evaluates theactivity of the coagulation factors in the intrinsic system (factorXII, factor XI, factor IX, factor VIII) to the formation of a fibrinclot. Both the PT and aPTT are prolonged, because factor Xand prothrombin are present in the final common pathway.
A is incorrect. A decreased platelet count (thrombocytopenia)prolongs the bleeding time, because the end of the bleedingtime is marked by the formation of a temporary platelet plug.Signs of thrombocytopenia include petechiae and ecchymoses.Both PT and aPTT are normal in thrombocytopenia.
B is incorrect. A normal platelet count, normal bleeding time,normal PT, and a prolonged aPTT indicate a factor deficiencyin the intrinsic coagulation system (e.g., factor VIII deficiency).A normal PT indicates that there are no coagulation factor de-ficiencies in the final common pathway.
1-) is incorrect. A prolonged bleeding time associated with anormal platelet count, normal PT, and normal aPTT indicatesa defect in platelet function. The most common cause ofprolonged bleeding time is aspirin or other nonsteroidal anti-inflammatory drugs. Warfarin does not affect platelet pro-duction or function.
E is incorrect. The most common cause of a prolonged bleedingtime and prolonged aPTT is von Willebrand's disease. Warfarindoes not affect bleeding time.

Answers and Discussions 433
50. C (disseminated intravascular coagulation) is correct. The en-dotoxins in endotoxic shock (most often due to Escherichia colisepsis) damage tissue, causing the release of tissue thromboplas-tin. This activates the extrinsic coagulation system, causingDIC. Fibrin clots are produced in the microcirculation that ob-struct blood flow and consume coagulation factors, causingbleeding from needle puncture sites and the gastrointestinaltract. Prolongation of the PT and aPTT also occur. Platelets aretrapped in the fibrin clots leading to thrombocytopenia and pro-ducing petechiae and ecchymoses. The fibrinolytic system isactivated, and plasmin cleaves the fibrin strands holding thefibrin clots together. Fibrin strands are held together by cross-links, and cleaved fragments with cross-links are detected in theD-dimer assay.
A (autoimmune thrombocytopenia) and E (thrombotic throm-bocytopenic purpura) are incorrect. DIC is associated withfibrin clots, multiple coagulation factor deficiencies, activationof the fibrinolytic system, and thrombocytopenia. Therefore,conditions that produce thrombocytopenia (e.g., autoimmunethrombocytopenia and thrombotic thrombocytopenicpurpura) do not explain all of the clinical and laboratoryfindings that are present in this patient.
B (circulating anticoagulant) is incorrect. Circulating anti-coagulants are antibodies that destroy coagulation factors(e.g., factor VIII), causing prolongation of the aPTT and/orPT. However, these antibodies do not destroy platelets orproduce fibrin clots that obstruct the microcirculation, causingbleeding from the needle puncture sites and the gastrointesti-nal tract.
D (primary fibrinolysis) is incorrect. Primary fibrinolysis is veryrare. Only the fibrinolytic system is activated, and thus clini-cal findings are primarily those related to coagulation factordeficiencies (e.g., fibrinogen, factor V, factor VIII). The plateletcount is normal; n-dimers are not present, because there areno fibrin clots.

434
Goljan Rapid Review Pathology:Figure Acknowledgments-Chapters and QuestionsBouloux P-M: Self-Assessment Picture Tests: Medicine, Vol. 1. London,
1997, Mosby-Wolfe. Figures 28, p. 14; 90, p. 45; 163, p. 82. Vol. 3.Figures 33, p. 17; 75, p. 38.
Burger PC, Scheithauer BW, Vogel FS: Surgical Pathology of the NervousSystem and Its Coverings. New York, 2002, Churchill Livingstone.Figures 3-17, p. 121; 8-9, p. 428.
Clement PB, Young RH: Atlas of Gynecologic Surgical Pathology.Philadelphia, 2000, Saunders. Figures 2-15, p. 33; 3-11, p. 53; 5-19,p. 103; 10-1, p. 214.
Corrin B: Pathology of the Lungs. London, 2000, Churchill Livingstone.Figures 1.26, p. 15; 3.5, p. 85; 13.1.1A, p. 460; 13.1.16, p. 475.
Damjanov I: Pathology for the Health-related Professions, 2nd ed.Philadelphia, 2000, Saunders. Figures 4-11, p. 79; 5-23B, p. 117; 5-26,p. 120; 10-9A, p. 258; 10-10A, p. 259; 11-18, p. 303; 13-7, p. 330;13-8C, p. 331; 16-5B, p. 395; Figure on p. 415, right column; 21-7,p. 490.
Damjanov I, Linder J: Anderson's Pathology, 10th ed. St. Louis, 1996,Mosby. Figures 17-13, p. 374; 17-15, p. 375; 17-16, p. 375; 17-17,p. 375; 17-19, p. 376; 17-25, p. 384; 18-3, p. 388; 18-7B, p. 390; 41-61,p. 1105; 42-47A, p. 1145; 45-6B, p. 1268; 45-6C, p. 1268; 45-60,p. 1320; 77-120, p. 2750.
Damjanov I, Linder J: Pathology: A Color Atlas. St. Louis, 2000, Mosby.Figures 1-16, p. 11; 1-22, p. 13; 1-24, p. 13; 1-28A, p. 15; 1-47, p. 22;1-59, p. 25; 2-1A, p. 32; 3-16, p. 47; 4-8A, p. 52; 4-22B, p. 56; 4-24,p. 56; 4-26, p. 57; 4-27, p. 57; 4-51B, p. 65; 5-7, p. 75; 5-21, p. 79;5-26, p. 80; 5-35A and B, p. 83; 6-12A, p. 105; 6-23A, p. 110; 6-26,p. 111; 7-8, p. 124; 7-25A, p. 128; 7-30A, p. 131; 7-32, p. 131; 7-55,p. 138; 7-58, p. 139; 7-59, p. 139; 7-61, p. 139; 8-8 (left side), p. 146;8-38, p. 154; 8-42, p. 154; 8-70, p. 161; 8-78, p. 164; 9-6, p. 169;10-27, p. 191; 10-53, p. 198; 11-7, p. 212; 11-46, p. 224; 11-54, p. 226;11-64, p. 229; 11-79, p. 234; 12-31, p. 249; 12-32, p. 249; 12-38,p. 250; 13-8, p. 260; 13-10B, p. 261; 13-17A, p. 262; 13-32, p. 267;13-37, p. 268; 13-49, p. 271; 13-111A, p. 290; 14-14A, p. 304; 15-57A,p. 327; 17-35B, p. 369; 19-20, p. 405; 19-25, p. 408; 19-38, p. 411;19-41, p. 412; 19-43, p. 413; 19-48, p. 414; 19-67, p. 419; 19-82,p. 424; 19-107, p. 432; 20-39, p. 445.
Forbes CD, Jackson WF: Color Atlas and Text of Clinical Medicine, 2nd ed.London, 1997, Mosby-Wolfe. Figures 1.48, p. 16; 1.85, p. 29; 2.15,p. 87; 2.57, p. 97; 2.107, p. 110; 3.3, p. 121; 3.66, p. 137; 3.77, p. 140;3.81 and 3.86, p. 141; 4.184, p. 211; 5.8, p. 217; 6.54, p. 287; 6.56,p. 288; 6.68, p. 291; 7.61, p. 323; 7.72, p. 325; 7.104, p. 335; 7.138,p. 343; 8.22, p. 361; 8.28, p. 362; 8.110, p. 382; 10.10, p. 425; 10.27,p. 431; 10.112, p. 459.
Goldman L, Bennett JC (Eds): Cecil Textbook of Medicine. 2 Vols.Philadelphia, 2000, Saunders. Figure 102-8A, p. 557.
••••••••••••••••••••••••••••••••••••

Goljan Rapid Review Pathology: Figure Acknowledgments-Chapters and Questions 435
Goldstein BG, Goldstein AO: Practical Dermatology, 2nd ed. St. Louis,1997, Mosby Year Book. Figures 6-11, p. 69; 6-12, p. 69; 13-1, p. 140.
Goljan EF. Pathology: Saunders Text and Review Series. Philadelphia, 1998,Saunders. Figures 3-4, p. 41; 10-2A, p. 210; 10-2E, p. 211; 10-3A,p. 222, 10-5, p. 224; 16-2, p. 346.
Hart T, Shears P: Color Atlas of Medical Microbiology. London, 2000,Mosby -Wolfe. Figure 446, p. 271.
Henry JB: Clinical Diagnosis and Management by Laboratory Methods, 20thed. Philadelphia, 2001, Saunders. Plates 18-14, between pp. 376-377;18-31, between pp. 376-377; 19-7, between pp. 408-409; 26-2A,between pp. 552-553; 50-1, between pp. 1096-1097; 55-7C, betweenpp. 1208-1209; 55-8C, between pp. 1208-1209.
Hoffbrand AV, Pettit JE: Color Atlas of Clinical Hematology, 3rd ed.St. Louis, 2000, Mosby. Figures 1.43b, p. 18; 1.62, p. 22; 5.85a, p. 103;7.11a, p. 119; 7.54b, p. 128; 10.11, p. 179; 12.3b, p. 233; 13.22a,p. 251; 18.4c, p. 315.
Lookingbill DP, Marks JG Jr: Principles of Dermatology, 3rd ed.Philadelphia, 2000, Saunders. Figures 10-16A, p. 165; 14-2, p. 227;14-3, p. 227; 16-3, p. 259; 17-5A, p. 269.
MacSween RNM, Burt AD, Portmann BC, et al (Eds): Pathology of theLiver, 4th ed. London, 2002, Churchill Livingstone. Figures 6.2a,p. 276; 7.4, p. 317.
Naeim F: Atlas of Bone Marrow and Blood Pathology. Philadelphia, 2001,Saunders. Figures 2-22A, p. 27; 2-22D, p. 27; 2-22M, p. 27; 11-14B,p. 157; 11-17B, p. 159; 14-10B, p. 180.
Perkin GD: Mosby's Color Atlas and Text of Neurology. London, 1998,Mosby-Wolfe. Figures 4.3, p. 64; 8.19, p. 156; 9.4, p. 160; 11.1, p. 196.
Savin JA, Hunter JAA, Hepburn NC: Diagnosis in Color: Skin Signs inClinical Medicine. London, 1997, Mosby-Wolfe. Figures 2.47, p. 79;4.27, p. 104.
Silver MD, Gotlieb AI, Schoen FJ: Cardiovascular Pathology, 3rd ed.Philadelphia, 2001, Churchill Livingstone/Harcourt. Figure 4-29,p. 90.


AAbdominal aortic aneurysm, 92-93, 93fAbetalipoproteinemia, 91
malabsorption and, 214ABO blood groups, transfusions and, 174Abortion, spontaneous, 59Abrasion, 63Abruptio placentae, 277Abscess(es)
frontal lobe, 330tliver, causes and characteristics of, 228tlung, 188-189periappendiceal, 221peritonsillar, 202tpilonidal, 221
Acanathosis nigricans, 318Acantholysis, definition of, 311tAcanthosis nigricans, cancer associated with, 89tAcetaminophen
adverse effects of, 61free radicals from, 3-4
Acetylsalicylic acid overdose, adverse effects of, 61-62Achalasia, 205Achondroplasia, 302Acidosis
metabolic, complicating shock, 43respiratory
cerebral edema and, 321hypoxemia from, 1
Acne rosacea, 313Acne vulgaris, 313Acquired immunodeficiency syndrome (AIDS), 34-36Actinic keratosis, 316, 316fActinomycosis, cervicofacial, 202tAddison's disease, 295Adenocarcinoma, 75-76, 77f
clear cell, of vagina, 270esophageal, 206
Page numbers followed by f indicate figures; t, tables;b, boxes.
Adenocarcinoma—cont'dgallbladder, 236gastric, 209-210lung, 198tpancreatic, 238
Adenoma(s)bowel, 218, 219ffollicular, 291growth hormone, 286-287liver cell, 234pituitary, 284-285
Adenomyosis, 273Adenosine triphosphate (ATP), decreased synthesis of,
from hypoxic cell injury, 3Adhesions
in inflammation, 14-15small bowel obstruction from, 218
Adrenal gland disorders, 294-298adrenal medulla hyperfunction as, 298adrenocortical hyperfunction as, 296-298adrenocortical hypofunction as, 294-296
Adrenocortical hormone synthesis, 294fAdrenocortical hyperfunction, 296-298Adrenocortical hypofunction, 294-296Adrenogenital syndrome, 295-296Adrenoleukodystrophy, 331Adson's test, positive, in thoracic outlet syndrome, 96Adult respiratory distress syndrome, 192Aflatoxin, as carcinogen, 85tAgammaglobulinemia, Bruton's, 33tAgenesis, congenital, 58AIDS (acquired immunodeficiency syndrome),
34-36Airway, upper, disorders of, 180-181Alanine aminotransferase, in cell death, 13tAlanine transaminase, serum, significance of, 223tAlbumin, serum, significance of, 223tAlcohol
abuse of, CNS findings associated with, 334-335as carcinogen, 85tfolate deficiency from, 130
437

438 Index
Alcohol—cont'dliver disorders induced by, 230, 230tuse of, pathology from, 61
Alcoholic cirrhosis, 231, 232fAlcoholism
sideroblastic anemias and, 128thiamine deficiency from, 72
Aldosteronism, 297-298Alkaline phosphatase, serum, significance of,
223tAlkalosis, respiratory, production of, for head
trauma, 321Al kaptonuria, 47t, 304Alkylating agents, as carcinogens, 85tAllergic nasal polyps, 180Allergic reaction, transfusion, 176Allograft, 27a 1 -Antitrypsin deficiency, in cirrhosis, 234a-Fetoprotein
in genetic/developmental disorder diagnosis, 59maternal, increased, in neural tube defects, 322
a-Thalassemia, 126-127laboratory findings in, 129t
Alport's syndrome, 247ALS, 333-334Alzheimer's disease, 331-332Amaurosis fugax, 338Amenorrhea, 273Ammonia, serum, significance of, 223tAmniocentesis, in genetic/developmental disorder
diagnosis, 59Amniotic fluid embolism, 41Amylase, in cell death, 13tAmyloidosis, 36-37, 37t
primary, in multiple myeloma, 159Anaerobic glycolysis, in ATP synthesis, 3Anal carcinoma, 221Analgesic nephropathy, 253Anaphylactic shock, 26Anemia(s), 1
aplastic, 133-134pathogenesis of, 135t
cancer and, 88-89of chronic disease, 126
laboratory findings in, 129tpathogenesis of, 135t
classification of, 123fColley's, 127hemolytic, 134-141. See also Hemolytic anemias.iron-deficiency, 125-126, 126fmacrocytic, 130-133in malabsorption, 215megaloblastic, 72tmicrocytic, 125-129
laboratory findings in, 129tnormocytic, 133-141. See also Normocytic
anemias.
Anemia(s)—cont'dpernicious
laboratory findings in, 132tvitamin B 12 deficiency in, 132
sickle cell, 136-138, 138fpathogenesis of, 135t
sideroblastic, 127-128, 129f, 129tin small bowel disease, 210
Anencephaly, 322Aneurysm(s)
berry, congenital, 326-327ventricular, 108vessel, 92-95
Angelman's syndrome, 55-56Angina pectoris, 105-106Angiodysplasia, 212Angioedema
hereditary, 36, 36tin urticaria, 318
Angiomatosis, bacillary, 97tAngiomyolipoma, 256Angiomyolipomas, in kidneys in tuberous sclerosis,
323Angiosarcoma, 97tAngiotensin-converting enzyme inhibitors, in diabetic
nephropathy, 250Aniline dyes, as carcinogens, 85tAnorectal disorders, 221Anorexia nervosa, 67-68Anovulatory dysfunctional uterine bleeding, 272-273Anthracotic pigment, intracellular accumulations of, 6tAntibiotics, vitamin K deficiency and, 71Anticoagulants, in small blood vessels, 162Antigens, cancers associated with, 89tAorta, coarctation of, 111-112Aortic aneurysm, abdominal, 92-93, 93fAortic dissection, 94-95Aortic regurgitation, 115Aortic stenosis, 115Aphthous ulcers, 201Aplasia, congenital, 58Aplastic anemia, 133-134
pathogenesis of, 135tApnea, sleep, obstructive, 180-181Apolipoprotein B deficiency, 91Apoptosis, 12-13
activation of, by hypoxic cell injury, 3Apoptosis genes, mutations involving, 84Appendicitis, acute, 221Arachidonic acid, as inflammation mediator,
17, 18t, 19fArboviruses, CNS infections from, 328tArnold-Chiari malformation, 323Arrhythinias, from myocardial infarction, 107Arsenic
as carcinogen, 85ttoxic effects of, 64t

••
Arterial embolism, 40-41Arterial oxygen saturation (Sa02)
decreasing, methemoglobinemia from, 1definition of, factors contributing to, and
significance of, 2tArterial thrombi, 40Arterioles, in inflammation, 14Arteriolosclerosis, 92Arteriosclerosis, 91-92Arteritis
giant cell, 98tTakayasu's, 98ttemporal, 98t
Artery, hepatic, thrombosis of, 228Arthritis
gouty, 307rheumatoid, 306-307, 306fseptic, 308
Arthropathy, neuropathic, in osteoarthritis, 306Asbestos
as carcinogen, 85ttoxic effects of, 64t
Asbestosis, 193, 193fAscaris lumbricoides, diarrhea from, 213tAscites, in cirrhosis, 232-233Ascorbic acid, 74ASD (atrial septal defect), 110Aspartate aminotransferase, in cell death, 13tAspartate transaminase, serum, significance of, 223tAspergillus fumigatus, respiratory infections from, 187tAspirin
overdose of, adverse effects of, 61-62prolonged bleeding time from, 167t
Asthma, 195Astrocytoma, 335Ataxia, Friedreich's, 333Ataxia telangiectasia, 33t, 84Atelectasis, 181-183Atherosclerosis, 6t, 91-92Atherosclerotic stroke, 325Atopic dermatitis, 315ATP. See Adenosine triphosphate (ATP).Atresia, congenital, 58Atrial septal defect, 110Atrophic gastritis, 207-208Atrophy, in response to cell injury, 7Auspitz sign, in psoriasis, 317Autoantibodies, in autoimmune diseases, 29tAutograft, 27Autoimmune destruction, of melanocytes, 318Autoimmune diseases, 29-33
autoantibodies in, 29tdermatomyositis as, 32-33mechanisms of, 29polymyositis as, 32-33systemic lupus erythematosus as, 29-31systemic sclerosis as, 31-32
Index 439
Autoimmune hemolytic anemia, 139-140pathogenesis of, 135t
Autoimmune hepatitis, 227Autoimmune skin disorders, 315Autoimmune thrombocytopenic purura, chronic, 170tAutoimmune thyroiditis, 288Autosomal dominant disorders, 48-49, 49fAutosomal recessive disorders, 45-48, 46fAvascular necrosis of bone, 303Axonal degeneration, in peripheral neuropathies, 336Azotemia, 239-240
BB-cell lymphomas, 155, 156t
large, diffuse, 156tB cells, derivation, location, and function of, 24tBacillary angiomatosis, 97tBacillus cereus, diarrhea from, 213tBacteria
causing diarrhea, 213tCNS infections from, 329tmeningitis from, 327, 329tskin disorders from, 313vaginosis from, 269
Balantidium coli, diarrhea from, 213tBarrett's esophagus, 8, 206, 206fBasal cell carcinoma
oral, 202skin, 319-320from UV light, 86, 87f
Basophilia, 144Bell's palsy, 337Bence Jones proteinuria, in multiple myeloma,
253-254Benign prostatic hyperplasia, 264, 264fBenzene
as carcinogen, 85ttoxic effects of, 64t
Berger's disease, 244-245Beriberi, 72tBernard-Soulier syndrome, prolonged bleeding time
in, 167tBerry aneurysm, congenital, 326-327Berylliosis, 193Beryllium, as carcinogen, 85tb-Naphthylamine, as carcinogen, 85tb-Thalassemia, 127
laboratory findings in, 129tb-thalassemia major, 44Bile salt deficiency, malabsorption and, 214Biliary atresia, extrahepatic, 231Biliary cirrhosis, 233Biliary tract disorders, 235-236Bilirubin
conjugated, significance of, 223tintracellular accumulations of, 6t
•••••1a•••••••••••••••••••••••••

440 Index
Bilirubin—cont'dmetabolism of, disrupted, in liver cell injury,
222, 224furine, significance of, 223t
Bilirubin excretion tests, 223tBiopsy, renal, in glomerular disease evaluation, 243Biotin, 72t, 73Bladder
disorders of, 259, 260tumors of, 260
Blastomyces dermatitidis, respiratory infections from,187t
Bleeding time, 166prolonged, causes of, 166, 167t
Blindness, night, 70Blood
donor, tests performed on, 175loss of, acute, normocytic anemias from, 133
pathogenesis of, 135ttransfusion of
ABO blood groups and, 174disorders of, 174-179
hemolytic disease of newborn as, 177-179non-Rh antigen systems and, 174-175patient cross-match for, 175-176reactions to, 176-177Rh antigen systems and, 174therapeutic, 175-177
Blood cellsred, disorders of, 121-141. See also Red blood cell
disorders.white, disorders of, 142-152. See also White blood
cell disorders.Blood pressure, elevated, 100-102Blood urea nitrogen (BUN)
in hepatocyte function assessment, 223tserum, in renal function assessment, 239-240
Blood vesselsdisorders of, 90-102. See also Vascular disorders.in inflammation, 14tumors of, 97t
Bloom syndrome, 84Boerhaave's syndrome, 205Bone(s)
disorders of, 302-304neoplastic, 304, 305t
metastasis to, 80
Paget's disease of, 303tumors of, 305t
Bowel disorders, 210-220acute appendicitis as, 221anorectal, 221colorectal carcinoma as, 219-220congenital, 210-211diarrhea as, 212-214, 212t, 213tinfarction as, 211, 211finflammatory, 215, 216t, 217f
Bowel disorders—cont'dmalabsorption as, 214-215, 215tpolyps as, 218-219signs and symptoms of, 210small bowel cancer as, 219small bowel obstruction as, 218vascular, 211-212, 211f
Bowen's disease, 261Bradykinin, as inflammation mediator, 17, 18tBrain
global hypoxic injury to, 325tumors of, 335
Breast(s)disorders of
in females, 279-283fibrocystic, 279-280primary sites of, 280ttumors as, 280-283
in males, 283seborrheic keratosis of, 319f
Brenner tumor, 276t"Brittle bone" disease, 302Bronchial hamartoma, 198tBronchiectasis, 197-198Bronchitis, chronic, 197Bruise, 63Bruton's agammaglobulinemia, 33tBudd-Chiari syndrome, 229Buerger's disease, 98tBulimia nervosa, 68Bulla, definition of, 311tBullous pemphigoid, 316Burkitt's lymphoma, 155
translocations in, 84Burns, 64-65
CC6-C9 deficiency, 36tC-reactive protein
in atherosclerosis, 91-92in inflammation, 23
Cachexia, cancer, 88Cafe-au-lait macules, in neurofibromatosis, 323Caisson disease, 41-42Calcification, pathologic, 6Calcium, cytosolic, increased, from hypoxic cell
injury, 3Calcium-ATPase pump, impaired, ATP synthesis and, 3Calculi, renal, 256Calor, in inflammation, 14Campylobacter jejuni, diarrhea from, 213tCancer(s)
antigens associated with, 89tbreast
in female, 281-283, 282f, 282tin male, 283

Index 441
Cancer(s)—cont'dcervical, 271-272, 272fepidemiology of, 81grading of, 87lung, 198-200, 198tnomenclature of, 75-76obesity and, 69tpenile, 260-261prostate, 265-266renal pelvis, 257small bowel, 219staging of, 87testicular, 262, 263tfrom ultraviolet light B, 66
Candida albicans, respiratory infections from, 187tCandidal vaginosis, 269Canker sores, 201Capillary hemangioma, 97tCaplan's syndrome, 307Carbon monoxide (CO)
cytochrome oxidase inhibition by, 2poisoning by, 2toxic effects of, 64t
Carbon tetrachloride, free radical injury from, 4Carcinogenesis, 81-84Carcinogens
chemical, 84-85, 85tmicrobial, 85
Carcinoid heart disease, 116Carcinoid syndrome, 219Carcinoid tumor, small bowel, 219Carcinoma(s), 75-76
anal, 221basal cell
of mouth, 202of skin, 319-320from UV light, 86, 87f
breast, 281-283, 282f, 282tcolorectal, 219-220, 220fendometrial, 274hepatocellular, 236, 236flaryngeal, 181lung, 198-200, 198tmucoepidermoid, 203nasopharyngeal, 181pancreatic, 238renal cell, 256-257, 257fsquamous cell. See Squamous cell carcinoma.thyroid, 291-292transitional cell, of bladder, 260
Cardiac output, in septic shock, 43Cardiogenic shock, 42-43, 42tCardiomyopathy, 118-119, 119fCarditis, in rheumatic fever, 112Carpal tunnel syndrome, in rheumatoid arthritis, 307Caseous necrosis, 11
in tuberculosis, 17
Cat bite, septic arthritis from, 308Cat-scratch disease, 155Cataract, 338Cavernous hemangioma, hepatic, 234CD4, derivation, location, and function of, 24tCD8, derivation, location, and function of, 24tCeliac disease, malabsorption in, 21StCell(s)
death of, 10-13enzyme markers of, 13, 13t
of immune system, 24tinjury to, 1-13
adaptation to, 7-9free radical, 3-4hypoxic, consequences of, 3intracellular accumulations in, 5-6, 6t
organalles of, injury to, 4-5Cell-mediated cytotoxicity, antibody-dependent, 26Cellulitis, 19-20Central nervous system (CNS)
in AIDS, 35tdisorders of
degenerative, 331-334demyelinating, 329-331vascular, 325-327
infections of, 327-329, 328t, 329t, 330ttumors of, 335-336
Central pontine myelinolysis, 331Centriacinar emphysema, 196, 196fCentrilobular emphysema, 196, 196fCerebral atrophy, from atherosclerosis, 92Cerebral contusion, 324Cerebral edema, 321Cerebral herniation, 321-322Cerebrovascular accidents, 325-327Cervical intraepithelial neoplasia, 271Cervicitis, 271Cervicofacial actinomycosis, 202tCervix, disorders of, 271-272Chancroid, 268Charcot-Bouchard macroaneurysms, in intracerebral
hemorrhage, 326Charcot-Marie-Tooth disease, 336Charcot's joint, in osteoarthritis, 306Chediak-Higashi syndrome, 15Chemical carcinogens, 84-85, 85tChemicals
free radicals from, 3-4injury from, 60-63, 64tliver disorders induced by, 230, 230t
Chemotaxis, in inflammation, 15CHF. See Congestive heart failure (CHF).Chickenpox, 312
Reye's syndrome after, 227Children, osteomyelitis in, 302Chlamydia pneumoniae, respiratory infections from,
185t

442 Index
Chlamydia psittaci, respiratory infections from, 185tChlamydia trachomatis
respiratory infections from, 185tsexually transmitted disease from, 267-268urethritis from, 259-260
Chloasma, 318Choanal atresia, 180Cholangiocarcinoma, 236Cholangitis, sclerosing, primary, 231Cholecystitis, 236Cholelithiasis, 235-236
obesity and, 69tCholera, pancreatic, 299Cholestasis, tests for, 223tCholestatic liver disease, 230-231Cholesterol, intracellular accumulations of, 6tChondrocalcinosis, 307-308Chondrosarcoma, 305tChorea, Sydenham's, in rheumatic fever, 113Choriocarcinoma, 279
testicular, 263tChoristoma, 76Christmas disease, 171Chromium, as carcinogen, 85tChromosome instability syndromes, 84Chronic granulomatous disease of childhood, 16Churg-Strauss syndrome, 99tChylomicron, 90Cigarette smoke
emphysema and, 195-196lung cancer and, 198
Circulatory disorders, of liver, 228-229Cirrhosis, 231-234
postnecrotic, 233Clear cell adenocarcinoma, vaginal, 270Clear cell carcinoma, renal, 256-257Cleft lip, 201Cleft palate, 201Clostridium botulinum, diarrhea from, 213tClostridium difficile, diarrhea from, 213tClotting disorders, anticoagulants for, 165CN. See Cyanide (CN).CNS. See Central nervous system (CNS).CO. See Carbon monoxide (CO).Coagulation
defects of, in cirrhosis, 232disorders of, 162-173, 169-172
in cancer, 88classic von Willebrand's disease as, 171clinical findings in, 169disseminated intravascular coagulation as,
171-172hemophilia A as, 170-171laboratory findings associated with, 166-168normal hemostasis and, 162-166pathogenesis of, 169
Coagulation—cont'ddisorders of—cont'd
platelet disorders as, 168-169thrombotic disorders as, 172-173
disseminated intravascular, 171-172in acute myelocytic leukemia, 150laboratory findings in, 166t
Coagulation cascade, 163-165, 163fCoagulation necrosis, 10-11
complicating shock, 43Coal worker's pneumoconiosis, 6t, 192Coarctation of aorta, 111-112Cobalamin, 72t, 73Cocaine, toxic effects of, 62tCoccidioides immitis, respiratory infections from, 187tCold autoimmune hemolytic anemia, pathogenesis of,
135tColic, in lead poisoning, 128Colicky pain, in small bowel disease, 210Colitis
ischemic, 211-212ulcerative, 215, 217f
Crohn's disease compared with, 216tCollagen, production of, in tissue repair, 21Collagen vascular diseases, producing restrictive lung
disease, 194Colloid carcinoma, of breast, 282tColon
disorders of, 210-220. See also Bowel disorders.diverticulosis of, 215, 218
Colorectal carcinoma, 219-220, 220fCommon laboratory values, 340-342Complement
cytotoxic reactions dependent on, 26as inflammation mediator, 17, 18t
Complement system disorders, 36, 36tComplete blood cell count (CBC), 122-125Compression atelectasis, 182Condyloma acuminata, 267, 312Congenital anomalies, 57-58
of bowel, 210-21of heart, 109-112of kidneys, 240of lower urinary tract, 259of penis, 260
Congenital berry aneurysm, 326-327Congenital heart disease, 109-112Congenital immunodeficiency disorders, 33-34, 33tCongenital spherocytosis, 134, 136, 136f
pathogenesis of, 135tCongenital syphilis, oral features of, 202tCongestive cardiomyopathy, 118Congestive heart failure (CHF), 103-105
from myocardial infarction, 107Conjunctivitis, 337Connective tissue, repair by, 21-22

•••••••••••
II
p
•••••••••••••••••
Conn's syndrome, 297-298Contact dermatitis, 315Contraceptives, oral
adverse effects of, 62-63as carcinogens, 85t
Contrecoup injuries, 324Contusion, 63
cerebral, 324Cooley's anemia, 127Cor pulmonale, 191Corpus luteum cyst, 275Corrosive esophagitis, 206Cortical necrosis, diffuse, 255-256Coup injuries, 324Coxiella burnetii, respiratory infections from,
185tCoxsackievirus
CNS infections from, 328tmyocarditis and pericarditis from, 117
Craniopharyngioma, 285Creatine kinase MB, in cell death, 13tCreatinine
clearance of, in renal function assessment, 240serum, in renal function assessment, 239-240
CREST syndrome, in systemic sclerosis, 32Creutzfeldt-Jakob disease, 328tCri du chat syndrome, 53Crohn's disease, 215, 217f
ulcerative colitis compared with, 216tCryoglobulinemia, 99tCryptococcus neoforrnans
CNS infections from, 330trespiratory infections from, 187t
Cryptorchidism, 261Cryptosporidium parvum, diarrhea from, 213tCushing's syndrome, 296-297, 297f
cancer and ectopic hormone associated with, 89tCyanide (CN)
cytochrome oxidase inhibition by, 2poisoning by, oxidative phosphorylation and, 2toxic effects of, 64t
Cyanotic congenital heart disease, 110-111Cyclophosphamide, as carcinogen, 85tCyst(s)
Gartner's duct, 270ovarian, 275pilonidal, 221thyroglossal duct, 288
Cystic diseases, of kidney, 240, 242Cystic fibrosis, 46, 236-237
nasal polyps associated with, 180Cystic hygroma, 97tCystic teratoma, 276tCysticercosis, 330tCystitis, 252
acute, 259Cystocele, 270
Index 443
Cytochrome c, release of, in hypoxic cell injury, 3Cytokines, as inflammation mediators, 17, 18tCytomegalovirus
CNS infections from, 328tcongenital defects associated with, 58tdiarrhea from, 213trespiratory infections from, 185t
Cytoskeleton, injury to, 5Cytosolic calcium, increased, from hypoxic cell
injury, 3Cytotoxic hypersensitivity, 26-27
Dandy-Walker malformation, 323De Quervain's thyroiditis, 288Death
cell, 10-13enzyme markers of, 13, 13t
sudden cardiac, 106Decompression sickness, 41-42Deformations, congenital, 58Degenerative CNS disorders, 331-334Deletion, 52Dementia, in Alzheimer's disease, 331Demyelinating disorders, 329-331Demyelination, in peripheral neuropathies, 336Dendritic cells, derivation, location, and function
of, 24tDermatitis
atopic, 315contact, 315seborrheic, 314stasis, from deep vein thrombosis, 95
Dermatitis herpetiformis, 316Dermatographism, in urticaria, 318Dermatomyositis (DM), 32-33Dermatopathic lymphadenitis, 154Dermatophytoses, 314Desensitization therapy, 25Desmoid tumor, 310Developmental disorders, 44-59. See also
Genetic/developmental disorders.Diabetes insipidus, 285-286Diabetes mellitus, 299-201
"bronze," 233classification of, 299clinical findings in, 300tcomplications of, 300-301congenital anomalies and, 57gestational, 299, 301glycogen accumulations in, 6tpathogenesis of, 299-300peripheral neuropathies in, 337type 2, obesity and, 69ttypes 1 and 2 compared, 300t
Diabetic nephropathy, 249-250Diaper rash, 314

444 Index
Diarrhea, 212-214in bowel disease, 210microbial pathogens causing, 213ttypes of, 212t
Diastolic dysfunction, 104DIC. See Disseminated intravascular coagulation (DIC).Diethylstilbestrol, as carcinogen, 85tDiffuse cortical necrosis, 255-256Diffuse large B-cell lymphoma, 156tDiffusion defect, hypoxemia from, 1DiGeorge syndrome, 33t
hypothyroidism in, 292Dihydrofolate reductase, folate deficiency and,
130, 131Dihydrotestosterone, prostate development and, 262Diphtheria, oral features of, 202tDiphyllobothrium latum
diarrhea from, 213tvitamin B 12 deficiency and, 132
Disseminated intravascular coagulation (DIC),171-172
in acute myelocytic leukemia, 150laboratory findings in, 166t
Diverticulosis, 215, 217f, 218Diverticulum(a)
bladder, 259esophageal, 204Meckel's, 211
DNA polymerase chain reaction, in genetic/developmental disorder diagnosis, 59
DNA repair genes, 82DNA viruses, oncogenic, 86tDolor, in inflammation, 14Down syndrome, 52-53
duodenal atresia in, 210triple marker for, 59
Drug(s)autoimmune hemolytic anemia from, 139, 140free radicals from, 3-4liver disorders induced by, 230, 230tlupus erythematosus induced by, 30-31producing restrictive lung disease, 194therapeutic, adverse effects of, 61-63, 62t, 63ttubulointerstitial nephritis induced by, 252-253
Duchenne's muscular dystrophy, 309Duodenal atresia, 210Duodenal ulcers, gastric ulcers compared with,
208tDupuytren's contracture, 310Dysbetalipoproteinemia, familial, 90Dyscrasias, plasma cell, 158-159Dysfunctional uterine bleeding, 272-273Dysgerminoma, 276tDysmenorrhea, 272Dysphagia, in esophageal disease, 203Dysplasia, in response to cell injury, 8-9Dystrophic calcification, 6
EEar disorders, 338Eating disorders, 67-68Eaton-Lambert syndrome, cancer associated
with, 89tECG changes, in myocardial infarction, 108-109Echinococcosis, causes and characteristics of,
228tEctopic pregnancy, 277-278Eczema, 315Edema, 38-39
cerebral, 321pitting, in nephrotic syndrome, 247
Edrophonium test, for myasthenia gravis, 310Edward's syndrome, 53Ehlers-Danlos syndrome, 21
mitral valve prolapse in, 114Elastic artery vasculitis, 100Electrical injury, 65Embolic stroke, 326, 326fEmbolism, 40-42
pulmonary, in decompression sickness, 41-42Embolus, pulmonary, 190Embryonal carcinoma, testicular, 263tEmbryonal rhabdomyosarcoma, 260, 270, 310tEmphysema, 195-197, 196fEncephalitis, 327, 328t, 329, 329t
cryptococcal, 330ttoxoplasmal, 330t
Encephalopathyhepatic, in cirrhosis, 232in lead poisoning, 128Wernicke's, 335
Endocarditisinfective, 116-117Libman-Sacks, 117nonbacterial thrombotic, 117in rheumatic fever, 112thrombotic, nonbacterial, cancer associated
with, 89tEndocrine disorders, 284-301. See also specific gland,
e.g. Adrenal glands, Pancreas, Parathyroidglands, Pituitary gland, Thyroid gland.
Endocrinopathies, 89tEndodermal sinus tumor, testicular, 263tEndometrial carcinoma, 274Endometrial hyperplasia, 273-274Endometrial polyp, 274Endometrioid, 276tEndometriosis, 273Endometritis, 273Endonucleases, activation of, in hypoxic cell
injury, 3Endoplasmic reticulum, smooth, injury to, 4Endotoxic shock, 42t, 43Entamoeba histolytica, diarrhea from, 213tEnterobius vermicularis, diarrhea from, 213t

Index 445
Environmental pathology, 60-66chemical injury in, 60-63, 64tphysical injury in, 63-65radiation injury in, 65-66
Enzymatic fat necrosis, 11-12Enzyme deficiencies, in inborn errors of metabolism,
46-48, 47tEnzyme markers, of cell death, 13, 13tEosinopenia, 144Eosinophilia, 143Eosinophilic granulomas, 160Ependymoma, 336Epididymis, disorders of, 261-262Epididymitis, 261-262Epidural hematoma, acute, 324Epispadias, 260Epstein-Barr virus, infectious mononucleosis
from, 144Erectile dysfunction, 266Erythema infectiosum, 312Erythema multiforme, 317Erythema nodosum, 317Erythrocyte sedimentation rate (ESR),
in inflammation, 23Erythrocytes, disorders of, 121-141. See also
Red blood cell disorders.Erythroplakia, 201Erythroplasia of Queyrat, 261Erythropoiesis, 121-122Erythropoietin, erythropoiesis and, 121Escherichia coli
CNS infections from, 329tdiarrhea from, 213t
Esohpagitis, 205-206Esophageal diverticula, 204Esophageal varices, 205, 205fEsophageal web, 204Esophagus
Barrett's, 8, 206, 206fdisorders of, 203-207tumors of, 206-207
Estrogen, exogenous, adverse effects of, 62Ethylene glycol, toxic effects of, 64tEwing's sarcoma, 305tExtracellular fluid, 38Extramedullary hematopoiesis, 122Extranodal marginal zone lymphoma,
156tExudate, 39Exudative tonsillitis, 202tEye disorders, 337-338
FFactor III, in normal hemostasis, 163Factor IX deficiency, 171Fallopian tube disorders, 275
Familial dysbetalipoproteinemia, 90Familial hypercholesterolemia, 90Familial hype rtricglyceridemia, 90-91Familial polyposis syndromes, 219Fanconi's syndrome, 84Fat embolism, 41Fat necrosis, enzymatic, 11-12Febrile transfusion reaction, 176Feminization, testicular, 56-57Ferritin
intracellular accumulations of, 5-6, 6tserum, 125
Fibrinoid necrosis, 12Fibrinolysis, studies evaluating, 168Fibrinolytic system, 165Fibrinous inflammation, 20Fibrinous pericarditis, in myocardial infarction, 107Fibroadenoma, of breast, 280Fibrocystic change in breast, 279-280Fibroids, uterine, 274Fibromatosis, 310Fibrosis, in tissue repair, 21Fibrous dysplasia, 304Fibrous histiocytoma, malignant, 310tFish tapeworm, vitamin 13 12 deficiency and, 132Fistula, 20
tracheoesophageal, 204Fluid disorders, 38-43
edema as, 38-39embolism as, 40-42shock as, 42-43thrombosis as, 39-40
Folate deficiency, 130-131from alcohol abuse, 61laboratory findings in, 132t
Folic acid, 72t, 73Follicular adenoma, 291Follicular carcinoma, of thyroid, 291Follicular cyst, 275Follicular hyperplasia, 154Follicular lymphoma, 156t
translocations in, 84Fragile X syndrome, 50Frameshift mutation, 45Fredrickson's classification, of lipoprotein disorders,
90-91Free radical cell injury, 3-4Friedreich's ataxia, 333Frontal lobe abscess, 330tFrostbite, 65Fructose intolerance, hereditary, 47tFungal infections
CNS, 330tmeningitis as, 327respiratory, systemic, pathogens causing, 187tskin, 314-315

446 Index
GGalactosemia, 47tGallbladder
adenocarcinoma of, 236disorders of, 235-236
Gallstones, 235-236g-Glutamyltransferase, serum, significance of,
223tGangrene, 10, 10f
dry, 10, 10fwet, 10f, 11
Gardner's polyposis syndrome, 219, 310Gartner's duct cyst, 270Gastrinoma, 299Gastritis, 207-208Gastroesophageal reflux disease, 206Gastrointestinal system
in AIDS, 35tdisorders of, 201-221
acute appendicitis as, 221anorectal, 221bowel disorders as, 210-220. See also
Bowel disorders.esophageal, 203-207oral, 201-202salivary gland, 202-203stomach, 207-210
Gaucher's disease, 48tGenes, in carcinogenesis, 81-82, 83t, 84Genetic analysis, in genetic/developmental disorder
diagnosis, 59Genetic/developmental disorders, 44-59
chromosomal, 51-55congenital anomalies as, 57-58diagnosis of, 59genomic imprinting and, 55-56infant, 59Mendelian, 45-48mitochondrial DNA, 55, 55fmutations as, 44-45mutifactorial inheritance of, 55nervous system, 322-324perinatal, 59polygenic, 55of sex differentiation, 56-57
Genomic imprinting, 55-56German measles, 312Gestational diabetes, 299, 301Gestational disorders, 277-279Gestational trophoblastic neoplasms, 278-279Giant cell arteritis, 98tGiant cell tumor, of bone, 305tGiant cells, multinucleated, formation of, 20bGiardia laroblia, diarrhea from, 213tGland(s)
adrenal, disorders of, 294-298parathyroid, disorders of, 292-294
Gland(s)—cont'dparotid, tumors of, 202pineal, disorders of, 336-337pituitary, disorders of, 284-287prostate, disorders of, 262, 264-266.
See also Prostate.salivary, tumors of, 202-203thyroid, disorders of, 287-292. See also Thyroid
gland, disorders of.Glandular metaplasia, 8Glanzmann's disease, prolonged bleeding time in,
167tGlaucoma, 338Global hypoxic injury, to brain, 325Glomerular disorders, 242-251
evaluation of, 243mechanisms of, 242-243morphologic characteristics of, 245tnephritic syndrome as, 243-247nomenclature and description of, 243tterminology of, 243t
Glomerulonephritis, 242-243diffuse membranous, 249diffuse proliferative, in SLE, 246-247IgA, 244-245membranoproliferative, 249nomenclature and description of, 243tpostinfectious, 244fpoststreptococcal, 245-246, 246frapidly progressive, 247
Glomerulosclerosisfocal segmental, 243t, 248-249nodular, 249-250
Glomus tumor, 97tGlossitis, 201Glucagonoma, 299Glucocorticoids
healing and, 22in respiratory distress syndrome prevention, 182
Glucose intolerance, impaired, 299Glycogen, intracellular accumulations of, 6tGlycogen storage diseases, 47-48Glycogenosis, Von Gierke's, 6tGlycolysis, anaerobic, in ATP synthesis, 3Goiter
nontoxic, 291toxic multinodular, 290
Gonococcemia, disseminated, septic arthritisfrom, 308
Gonorrhea, 268Goodpasture's syndrome, 244f, 247Gouty arthritis, 307G6PD deficiency, 138-139
pathogenesis of, 135tGraft-versus-host (GVH) reaction, 28Granulation tissue, in tissue repair, 21

Index 447
Granuloma(s)eosinophilic, 160tuberculous, formation of, 19, 20b
Granuloma inguinale, 268Granulomatosis, Wegener's, 98tGranulomatous disease of childhood, chronic, 16Granulomatous hepatitis, causes and characteristics
of, 228tGranulomatous inflammation, 17-19Granulomatous thyroiditis, subacute, 288Granulosa-thecal cell tumor, 276tGraves' disease, 290, 290f
laboratory findings in, 287tGrawitz' tumor, 256-257Growth alterations, in response to cell injury, 7-9Growth hormone deficiency, in anterior pituitary
hypofunction, 285Growth retardation, in lead poisoning, 128Guillain-Barre syndrome, 336-337Gunshot wounds, 64Gynecomastia, 283
cancer and ectopic hormone associated with, 89t
HHaemophilus influenzae, respiratory infections from,
184tHairy cell leukemia, 152Hairy leukoplakia, 202tHamartoma(s), 76
bronchial, 198tHamartomatous polyps, of bowel, 218Hand-Schuller-Christian disease, 160Hashimoto's thyroiditis, 288, 289fHbH disease, 127Head trauma, 321, 324Heart
disorders of, 103-120acquired valvular, 112-117cardiomyopathy as, 118-119, 119fcongenital, 109-112congestive heart failure as, 103-105ischemic, 105-109myocarditis as, 117pericarditis as, 117-118ventricular hypertrophy as, 103
reperfusion injury to, after MI, 4rhabdomyoma in, in tuberous sclerosis, 323tumors of, 119-120
Heart murmurin aortic regurgitation, 115in mitral stenosis, 113in mitral valve prolapse, 114-115in tricuspid regurgitation, 116
Heartburnin esophageal disease, 203in gastroesophageal reflux disease, 206
Heat injuries, 65, 65t
Heberden's nodes, in osteoarthritis, 305f, 306Helicobacter pylori, gastric adenocarcinoma and, 209Helminths, causing diarrhea, 213tHemangioma(s)
capillary, 97tcavernous, of liver, 234
Hematemesis, in small bowel disease, 210Iiematochezia, in large bowel disease, 210Hematogenous spread, of malignant tumors, 79fematomaepidural, acute, 324subdural, 324
Hematopoiesis, extramedullary, 122Hemochromatosis, 4
hereditary, in cirrhosis, 233Hemodynamic disorders, 38-43
edema as, 38-39embolism as, 40-42shock as, 42-43thrombosis as, 39-40
Hemoglobin, abnormalities related to, 1-2Hemoglobin Bart's disease, 127Hemoglobinuria, nocturnal, paroxysmal, 136
pathogenesis of, 135tHemolytic anemia(s), 134-141
autoimmune, 139-140pathogenesis of, 135t
congenital spherocytosis as, 134, 136, 136fG6PD deficiency and, 138-139macroangiopathic, pathogenesis of, 135tmicroangiopathic, pathogenesis of, 135tpyruvate kinase deficiency and, 139
Hemolytic disease of newborn, 177-179Hemolytic transfusion reaction, acute, 176-177Hemolytic uremic syndrome, 170tHemophilia A, 170-171
laboratory findings in, 166tHemophilia B, 171Hemorrhage
intracerebral, 326subarachnoid, 326-327
Hemorrhagic gastritis, 207Hemorrhoids, 212Hemosiderin, intracellular accumulations of, 6, 6tHemosiderosis, 233Hemostasis
disorders of, laboratory findings in, 166tlaboratory findings associated with, 166-168normal, 162-166
Henock-Schonlein purpura, 99tHeparin
for clotting disorders, 165in normal hemostasis, 162thrombocytopenia induced by, 170t
Hepatic artery thrombosis, 228Hepatic encephalopathy, in cirrhosis, 232Hepatic vein thrombosis, 229

448 Index
Hepatitisalcoholic, 230autoimmune, 227granulomatous, causes and characteristics of, 228tneonatal, 227viral, 223-227
Hepatitis A, 224transmission and clinical findings in, 226t
Hepatitis B, 224-227, 226ffrom intravenous drug use, 61serologic studies in, 227ttransmission and clinical findings in, 226t
Hepatitis C, 227lichen planus and, 316transmission and clinical findings in, 226t
Hepatitis D, 227transmission and clinical findings in, 226t
Hepatitis E, 227transmission and clinical findings in, 226t
Hepatitis G, transmission and clinical findings in,226t
Hepatobiliary systemin AIDS, 35tdisorders of, 222-237. See also Liver, disorders of.
Hepatocellular carcinoma, 236, 236fHepatocyte function tests, 223tHepatorenal syndrome, in cirrhosis, 233Hereditary angioedema, 36tHereditary fructose intolerance, 47tHereditary nonpolyposis syndrome, 84, 220Hereditary polycystic kidney disease, 242Hereditary telangiectasia, 97tHeredity, cancer and, 81Hermaphrodite, true, 56Hernia
hiatal, 204small bowel obstruction from, 218
Herniation, cerebral, 321-322Heroin, toxic effects of, 62tHerpes labialis, 202tHerpes simplex virus
CNS infections from, 328ttype 2
congenital defects associated with, 58tinfections from, 267
Herpes zoster, 312, 313fHerpesvirus 6, 312Hiatal hernia, 204Hirschsprung's disease, 210Hirsutism, 275Histamine, as inflammation mediator, 17, 18tHistiocytoma, fibrous, malignant, 310tHistiocytosis
Langerhans' cell, 159-160sinus, 155
Histoplasina capsulatutn, respiratory infections from,187t
HIV (human immunodeficiency virus), laboratorytests for, 35t
HIV thrombocytopenia, 170tHive, definition of, 311tHodgkin's lymphoma, 156-158Homocystinuria, 47tHomogentisic acid, accumulation of, 304Homogentisic acid oxidase, deficiency of, 304Hormonal imbalance, dysfunctional uterine bleeding
from, 272Hormone(s)
adrenocortical, synthesis of, 294fparathyroid, 292
Homer's syndrome, in lung cancer, 199Horseshoe kidney, 240Host, cancer effects on, 88Host defense, against tumors, 86Human chorionic gonadotropin, in genetic/
developmental disorder diagnosis, 59Human immunodeficiency virus (HIV)
CNS infections from, 328tlaboratory tests for, 35t
Human papillomavirus infections, 267, 312Huntington's disease, 333, 333fHurler's syndrome, 48tHyaline arteriolosclerosis, 92Hydatidiform moles, 278-279, 279fHydrocarbons, polycyclic, as carcinogens, 85tHydrocele, 261Hydrocephalus, 322Hydronephrosis, 256Hydroxyl ions, 311-Hydroxylase deficiency, 29617-Hydroxylase deficiency, 29621-Hydroxylase deficiency, 296Hygroma, cystic, 97tHyperaldosteronism, 297-298Hyperbilirubinemia, in liver cell injury, 222Hypercalcemia, 6
cancer and ectopic hormone associated with, 89tcauses of, 293t
Hypercholesterolemiafamilial, 90in nephrotic syndrome, 248
Hypercortisolism, 296-298Hyperestrinism, in cirrhosis, 233Hyperkeratosis
definition of, 311tin eczema, 315
Hypernephroma, 256-257Hyperparathyroidism, 293-294Hyperphosphatemia, 6Hyperplasia
dysplasia from, 8in response to cell injury, 8
Hyperplastic arteriolosclerosis, 92Hypersensitivity pneumonitis, 194

Index 449
Hypersensitivity reactions, 25-27, 26tHypersensitivity vasculitis, 100Hypersplenism, 160-161Hypertension, 100-102
from atherosclerosis, 92intracerebral hemorrhage and, 326intracranial, 321malignant, 255in nephrotic syndrome, 247obesity and, 69tportal, in cirrhosis, 232pulmonary, 190-191in renal failure, 254
Hyperthyroidism, 290Hypertriglyceridemia
familial, 90-91obesity and, 69t
Hypertrophic cardiomyopathy, 118, 119fHypertrophic osteoarthropathy, cancer associated
with, 89tHypertrophy, in response to cell injury, 7Hypervitaminosis D, hypercalcemia from, 293tHypoalbuminemia
in cirrhosis, 232in nephrotic syndrome, 248total serum calcium and, 292
Hypoaldosteronism, secondary, in cirrhosis, 232Hypocalcemia
cancer and ectopic hormone associated with, 89tcauses of, 293tin renal failure, 254
Hypocortisolism, 294-296Hypogammaglobulinemia, in nephrotic
syndrome, 248Hypoglycemia, cancer and ectopic hormone
associated with, 89tHypogonadism, male, 266Hypomagnesemia, hypothyroidism in, 292Hyponatremia, cancer and ectopic hormone
associated with, 89tHypoparathyroidism, 292-923Hypopituitarism, metyrapone test for, 285Hypoplasia, congenital, 58Hypospadias, 260Hypothyroidism, 289
laboratory findings in, 287tHypovitaminosis D, hypocalcemia in, 293tHypovolemic shock, 42, 42tHypoxemia, 1Hypoxia
definition of, 1tissue, 1-3
Ichthyosis vulgaris, 315IgA deficiency, 33tIgA glomerulonephritis, 244-245
IgE antibody production, in immediatehypersensitivity, 25
Ileus, meconium, 218Immediate hypersensitivity, 25-26Immune system, cells of, 24tImmunocomplex hypersensitivity, 27Immunocomplexes, in glomeruli, 243Immunodeficiency disorders, 33-36
congenital, 33tImmunology, transplantation, 27-28, 29tImmunopathology, 24-37
amyloidosis as, 36-37, 37tautoimmune diseases as, 29-33, 29t, 33t.
See also Autoimmune diseases.hypersensitivity reactions as, 25-27, 26timmunodeficiency disorders ass, 33-36transplantation immunology and,
27-28, 29tImpetigo, 313Inborn errors of metabolism, 46-48Incision, 63Inclusion (I)-cell disease, 5Infarction
bowel, 211, 211frenal, 255
Infarcts, lacunar, 327Infection(s)
CNS, 327-329, 328t, 329tcomplicating diabetes, 301dysplasia from, 9gestational, 277human papillomavirus, 267, 312interfering with healing, 22oral, 202trespiratory
bacterial, 184tfungal, systemic, 188
pathogens causing, 187tlung abscess as, 188-189pathogens causing, 185tpneumonia as, 183-186tuberculosis as, 187-188
Infectious mononucleosis, 144Infectious vasculitis, 99tInfective endocarditis, 116-117Inflammation, 14-23
acute, 14-17cellular events in, 14-17chemical mediators of, 17, 18t, 19foutcome of, 17vascular events in, 14
of breast, 280cardinal signs of, 14chronic, 17-19fibrinous, 20granulomatous, 17-19laboratory findings associated with, 23

450 Index
Inflammation—cont'dpatterns of, 19-20pseudomembranous, 20suppurative, 19tissue repair in, 20-22
Inflammatory bowel disease, 215, 216t, 217fInflammatory carcinoma, of breast, 282tInfluenza, Reye's syndrome after, 227Influenza virus, respiratory infections from, 185Injury, cell, hypoxic, consequences of, 3Insulinoma, 298-299Interleukin-1, as inflammation mediators, 18Interstitial disorders, 251-254Intestines, disorders of, 210-220. See also
Bowel disorders.Intracellular fluid, 38Intracerebral hemorrhage, 326Intracranial hypertension, 321Intraductal papilloma, 280-281Intraepithelial neoplasia
cervical, 271vulvar, 269-270
Intussusception, small bowel obstruction from, 218Invasive diarrhea, 212tInvasive lobular carcinoma, of breast, 282tIonizing radiation injury, 65-66Iron
deficiency of, 6tin large bowel disease, 210stages of, 126
intracellular accumulations of, 5-6serum, 124serum iron124
Iron deficiency anemia, 125-126, 126fcancer and, 88laboratory findings in, 129tpathogenesis of, 135t
Iron overload, 4Iron overload disorders, 6tIron studies, 124-125Irradiation, cancers induced by, 85-86Ischemia, 1Ischemic colitis, 211-212Ischemic heart disease, 105-109Islet cell tumors, 298-299Isogra ft, 27Isopropyl alcohol, toxic effects of, 64t
JJaundice
causes of, 225tin hemolytic disease of newborn, 177, 179from liver cell injury, 222
Jaw, disorders of, 201-202Job's syndrome, 142Joint disorders, 304-308Juvenile rheumatoid arthritis, 307
K
Kaposi's sarcoma, 97tin HIV, 35, 36f
Karyotyping, chromosome, in genetic/developmentaldisorder diagnosis, 59
Kawasaki disease, 98tKeloids, 22Keratoacanthoma, 319Keratosis
actinic, 316, 316fseborrheic, 318-319, 319f
cancer associated with, 89tKernicterus, 6tKidney(s)
in AIDS, 35tangiomyolipomas in, in tuberous sclerosis, 323biopsy of, in glomerular disease evaluation, 243disease of, anemia in, pathogenesis of, 135tdisorders of, 239-258
congenital, 240cystic, 240, 242glomerular, 242-251interstitial, 251-254obstructive, 256tubular, 251-254vascular, 255-256
failure ofchronic, 254prolonged bleeding time in, 167tfrom vitamin D deficiency, 71
free radical injury to, 4function of, assessment of, 239-240horseshoe, 240medullary sponge, 242tumors of, 256-258
Killer cells, natural, derivation, location, and functionof, 24t
Kimmelstiel-Wilson disease, 249-250Klebsiella pneumoniae, respiratory infections from,
184tKlinefelter's syndrome, 54-55, 54fKoebner phenomenon, in psoriasis, 317Korsakoff's psychosis, 61, 335Korsakoff's syndrome, 72tKrabbe's disease, 331Krekenberg tumor, 276tKwashiorkor, 67, 68f
LLaboratory values, common, 340-342Laceration(s), 63
esophageal, 204-205Lactase deficiency, 212, 214Lactiferous ducts/sinuses, intraductal papilloma in,
280-281Lacunar infarcts, 327Langerhans' cell histiocytoses, 159-160

•••••••••••••••••••••••••••••••••
Large bowel disorders, 210-220. See alsoBowel disorders.
Large cell carcinoma, of lung, 198tLaryngeal carcinoma, 181Lead
intracellular accumulations of, 6ttoxic effects of, 64t
Lead poisoningchronic, 253laboratory findings in, 129tsideroblastic anemias from, 128
Legg-Calve-Perthes disease, 303Legionella pneumophila, respiratory infections from,
184tLeiomyoma(s), 75, 209, 274, 310t
esophageal, 206Leiomyosarcoma, 274, 310tLentigo, solar, 318Leprosy, 313Lesch-Nyhan syndrome, 51Letterer-Siwe disease, 159-160Leukemia(s), 149-152
adult T-cell, 151-152hairy cell, 152from ionizing radiation, 85lymphoblastic, T-cell, precursor, 155lymphoid, 151-152myelogenous, chronic, 147-148promyelocytic, acute, 150from radiation injury, 66
Leukemoid reaction, 142-143Leukocyte adhesion molecule defect, 15Leukocytes
disorders of, 142-152. See also White blood celldisorders.
in inflammation, 23Leukocytosis, neutrophilic, 143Leukodystrophy, metachromatic, 331Leukoencephalopathy, progressive multifocal, 328tLeukoerythroblastic reaction, 143Leukoplakia, 201
hairy, 202tLeukotrienes, as inflammation mediators, 18Libman-Sacks endocarditis, 117Lichen planus, 316Lichen sclerosis, 269Lichen simplex chronicus, 269Lip, cleft, 201Lipase, in cell death, 13tLipid peroxidation, from free radical injury, 4Lipids, 90
disorders of, 90-91Lipoid nephrosis, 248Lipoma, 75
soft tissue, 310tLipoproteins, 90
Index 451
I,iposarcoma, soft tissue, 310tLiquefactive necrosis, 11I,isch nodules, in neurofibromatosis, 323Listeria monocytogenes, CNS infections from, 329tLiver
abscess of, causes and characteristics of, 228tcells of
injury to, laboratory evaluation in, 222necrosis of, tests for, 223t
disorders of, 222-235alcohol-related, 230cholestatic, 230-231circulatory, 228-229cirrhosis as, 231-234drug- and chemical-induced, 230, 230t
fatty changes in, 5free radical injury to, 4infectious diseases affecting, 228tpulsating, in tricuspid regurgitation, 116tumors of, 234-235
Liver function tests, 222, 223tLou Gehrig's disease, 333-334Low-density lipoproteins, increased levels of, obesity
and, 69tLung(s)
abscess of, 188-189carcinoma metastatic to, 198tdiseases of
obstructive, 194-198asthma as, 195bronchiectasis as, 197-198chronic bronchitis as, 197emphysema as, 195-197
restrictive, 191-194adult respiratory distress syndrome as, 192collagen vascular diseases producing, 194drugs producing, 194hypersensitivity pneumonitis as, 194pathogenesis of, 191-192pneumoconioses as, 192-193sarcoidosis as, 194
tumors of, 198-200, 198tvascular lesions of, 189-191
Lyme disease, 308Lymph nodes, spread of carcinomas through, 79Lymphadenitis
dermatopathic, 154reactive, 154-155
Lymphadenopathy, 153-154Lymphangitis, 96Lymphatic disorders, 96Lymphatic spread, of malignant tumors, 79Lymphedema, 39, 96Lymphoblastic leukemia, T-cell, precursor, 155Lymphoblastic lymphoma, t-cell, precursor, 155Lymphocyte predominant Hodgkin's lymphoma, 157t

452 Index
Lymphocytic lymphoma, small, 156tLymphocytosis, 144Lymphoid leukemia, 151-152Lymphoid tissue disorders, 153-161
Langerhans' cell histiocytoses as, 159-160lymphadenopathy as, 153-154lymphomas as, 155-158. See also Lymphoma(s).mast cell disorders as, 160plasma cell dyscrasias as, 158-159reactive lymphadenitis as, 154-155spleen disorders as, 160-161
Lymphoma(s)B-cell, 155, 156t
follicular, 84large, diffuse, 156t
Burkitt's, 155translocations in, 84
CNS, 336follicular, 156t
translocations in, 84gastric malignant, 210in HIV, 35Hodgkin's, 156-158lymphoblastic, T-cell, precursor, 155lymphocytic, small, 156tmalignant
testicular, 263tthyroid, 292
marginal zone, extranodal, 156tnon-Hodgkin's, 155-156T-cell, 155-156
Lymphopenia, 144-145Lynch syndrome, 84Lyon hypothesis, 51-52Lysosomal storage diseases, 48, 48tLysosomes, injury to, 4-5
MMacroaneurysms, Charcot-Bouchard, in intracerebral
hemorrhage, 326Macroangiopathic hemolytic anemia, 140
pathogenesis of, 135tMacrocytic anemia(s), 130-133
cancer and, 88Macroglobulinemia, WaldenstrOm's, 159tMacrophages
in chronic inflammation, 17derivation, location, and function of, 24tin host defense against tumors, 86
Macular degeneration, 338Macule(s)
cafe-au-lait, in neurofibromatosis, 323definition of, 311t
Maculopapular rash, 312Major histocompatibility complex (MHC), 25Malabsorption, 214-215, 21StMalar butterfly rash, 30, 30f
Malaria, 141, 141fpathogenesis of, 135t
Male hypogonadism, 266Malformations, congenital, 57-58Malignancy-induced hypercalcemia, 293tMalignant fibrous histiocytoma, 310tMalignant hypertension, 255Malignant lymphoma, testicular, 263tMalignant melanoma, 320
of vulva, 270Mallor-Weiss syndrome, 205Mallory bodies, 5Malnutrition, protein-energy, 67MALToma, 210Maple syrup urine disease, 47tMarasmus, 67, 68f"Marble bone" disease, 302Marfan syndrome
cardiovascular abnormalities in, 94mitral valve prolapse in, 114
Marginal zone lymphoma, extranodal, 1561Margination, in inflammation, 14Marijuana, toxic effects of, 62tMast cells
activation of, in immediate hypersensitivity, 25disorders of, 160
Mastitis, 280Mastocytosis, 160McArdle's disease, 47tMean corpuscular HB concentration (MCHC), 124Mean corpuscular volume (MCV), 123Measles, 312Meckel's diverticulum, 211Meconium ileus, 218Medullary carcinoma
of breast, 282tof thyroid, 291-292
Medullary sponge kidney, 242Medullohlastoma, 336Megacolon, congenital, 210Megaloblastic anemia, 72tMelanin, intracellular accumulations of, 6tMelanocytic disorders, benign, 318Melanoma, malignant, 320
of vulva, 270Melena, in small bowel disease, 210Membranes, rupture of, premature, infections
and, 277Membranoproliferative glomerulonephritis, 249Mendelian disorders, 45-48Menetrier's disease, 208Meniere's disease, 338Meningiomas, 335-336Meningitis, 327, 328t, 329t
cryptococcal, 330tMeningocele, 323
Meningoencephalitis, 330t

Meningomyelocele, 323Menorrhagia, 272Menstrual dysfunction, 272-273Mercury, toxic effects of, 64tMetabolic acidosis, complicating shock, 43Metabolic disorders, healing and, 22Metabolic nervous system disorders, 334-335Metabolism, inborn errors of, 46-48Metachromatic leukodystrophy, 331Metalloproteinases, in scar tissue remodeling, 21Metaplasia
dysplasia from, 9in response to cell injury, 8
Metastasis, 79-80, 80fto CNS, 336to lung, 198tto ovary, 276t
Metastatic calcification, 6Methanol, toxic effects of, 64tMethemoglobinemia, 1-2Methotrexate, folate deficiency and, 130Methyldopa, autoimmune hemolytic anemia
from, 139Methylene blue, for methemoglobinemia, 2Metyrapone test, for ACTH reserve, 285MHC. See Major histocompatibility complex (MHC).Microabscesses, Pautrier's, 156Microangiopathic hemolytic anemia, 140
pathogenesis of, 135tMicroangiopa thy, diabetic, 300Microbes, as carcinogens, 85Microcytic anemias, 125-129
laboratory findings in, 129tMicroscopic polyangiitis, 99tMigratory thrombophlebitis, superficial, cancer
associated with, 89tMissense mutations, 44Mitochondria, injury to, 4Mitochondrial DNA disorders, 55, 55fMitotic spindle defects, 5Mitral regurgitation, 113-114Mitral stenosis, 113, 114fMitral valve prolapse, 114-115
mitral regurgitation from, 113Mixed-cellularity Hodgkin's lymphoma, 157tMole, hydatidiform, 278-279, 279fMolluscum bodies, 312Molluscum contagiosum, 312, 312fMOnckeberg medial sclerosis, 91Monoclonal origin of neoplasms, 78Monocytes, in chronic inflammation, 17Monocytosis, 145Mosaicism, 52Motor vehicle collisions, 64Mouth, disorders of, 201-202MPTP, toxic effects of, 62tMucinous carcinoma, of breast, 282t
Index 453
Mucinous tumors, 276tMucoepidermoid carcinoma, 203Mucor species
CNS infections from, 330trespiratory infections from, 187t
Multifactorial inheritance, 55Multifocal leukoencephalopathy, progressive, 328tMultiorgan dysfunction, complicating shock, 43Multiple endocrine neoplasia (MEN) I syndrome,
anterior pituitary hypofunction in, 284Multiple myeloma, 158-159, 253-254Multiple sclerosis, 329-331Mumps, oral features of, 202tMural thrombus, in myocardial infarction, 107Muscle disorders, 309-310Muscular artery vasculitis, 100Muscular dystrophy, Duchenne's, 309Musculoskeletal disorders, 302-310Mutations, 44-45
involving apoptosis genes, 84involving proto-oncogenes, 82, 83tinvolving suppressor genes, 82, 83t, 84
Myasthenia gravis, 309Mycobacterium tuberculosis
CNS infections from, 329tdiarrhea from, 213trespiratory infections from, 184t
Mycoplasma pneumoniae, respiratory infections from,185t
Mycoses, superficial, 314Mycosis fungoides, 155-156Mycotic aneurysm, 93Myelinolysis, central pontine, 331Myelitis
bacterial, 329tviral, 328t
Myelodysplastic syndrome, 148-149Myelofibrosis, 148Myelogenous leukemia, chronic, 147-148Myeloid disorders, neoplastic, 145-149Myeloid metaplasia, 148Myeloperoxidase system, oxygen-dependent,
15-17Myelophthisic anemia, cancer and, 88-89Myeloproliferative disorders, chronic, 145-148Myocardial infarction (MI)
reperfusion injury to heart after, 4Myocarditis, 117
in rheumatic fever, 112Myotonic dystrophy, 309Myxoma, 119-120
NNaegleria fowleri, CNS infections from, 330tNasal polyps, 180Nasopharyngeal carcinoma, 181
••••••••••••••••••
•••
••
••••••••••••

454 Index
Natural killer cellsderivation, location, and function of, 24tin host defense against tumors, 86
Necator americanus, diarrhea from, 213tNecrosis, 10-12
tubular, acute, ischemic, complicatingshock, 43
Neisseria meningitidis, CNS infections from, 329tNeoplasia, 75-89. See also Cancer; Tumor(s).Neoplasms, respiratory, 198-200, 198tNeoplastic disorders of bone, 304Neoplastic myeloid disorders, 145-149Nephritic syndrome, 243-247. See also
Glomerulonephritis.morphologic characteristics of, 245t
Nephritis, tubulointerstitial, 251-254Nephrocalcinosis, in multiple myeloma, 159Nephrogenic diabetes insipidus, 285-286Nephropathy
analgesic, 253diabetic, 249-250, 301sickle cell, 255urate, 253
Nephrosclerosis, benign, 255Nephrosis, lipoid, 248Nephrotic syndrome, 247-251
morphologic characteristics of, 245tNerve, optic, atrophy of, 338Nervous system
central. See Central nervous system (CNS).disorders of, 321-338
degenerative, 331-334demyelinating, 329-331developmental, 322-324in head trauma, 324hereditary, 331infectious, 327-329, 328t, 329t, 330tmetabolic, 334-335toxic, 334-335vascular, 325-327
peripheral, disorders of, 336-337Neural tube defects, 322-323Neurilemoma, 337Neuritis, optic, 338Neuroblastoma, 298Neurocutaneous syndromes, 323Neuroendocrine tumor, 219Neurofibromatosis, 323-324Neuropathic arthropathy, in osteoarthritis, 306Neuropathy(ies)
peripheral, 336-337in lead poisoning, 128
toxin-associated, 337Neutropenia, 143Neutrophilic leukocytosis, 143Neutrophils, in inflammation, 14-15Nevocellular nevus, 319
Nevusmelanin accumulation in, 6tnevocellular, 319
Newbornhemolytic disease of, 177-179hepatitis in, 227respiratory distress syndrome in, 182-183, 183f
NHL. See Non-Hodgkin's lymphoma (NHL).Niacin, 72t, 73Nickel, as carcinogen, 85tNicotinic acid, 72t, 73Niemann-Pick disease, 48tNight blindness, 70Nitric oxide, as inflammation mediator, 17, 18tNitrosamines, as carcinogens, 851Nocturnal hemoglobinuria, paroxysmal, 136
pathogenesis of, 135tNodular glomeruloscierosis, 249-250Nodular sclerosing Hodgkin's lymphoma, 157tNodule(s)
definition of, 311tLisch, in neurofibromatosis, 323rheumatoid, 307thyroid, solitary, 291
Non-Hodgkin's lymphoma (NHL), 155-156Non-Rh antigen system, transfusions and, 174-175Nonbacterial thrombotic endocarditis, cancer
associated with, 89tNondisjunction, 52Nonionizing radiation, 66Nonsense mutations, 44Nonsteroidal anti-inflammatory drugs (NSAIDs)
hemorrhagic gastritis from, 207nasal polyps associated with, 180prolonged bleeding time from, 167t
Normocytic anemias, 133-141Norwalk virus, diarrhea from, 213tNosocomial pneumonia, 186NSAIDs. See Nonsteroidal anti-inflammatory drugs
(NSAIDs).Nutritional deficiencies, healing arid, 22Nutritional disorders, 67-74
eating disorders as, 67-68fat-soluble vitamin-related, 69-72obesity as, 68-69protein-energy malnutrition as, 67
0Oat cell carcinoma, of lung, 198tObesity, 68-69Obstipation, in small bowel disease, 210Obstructive liver disease, 230-231Obstructive lung disease, 194-198Obstructive lymphedema, 96Obstructive sleep apnea, 180-181Ochronosis, 304Oligodendroglioma, 336

••••••••••••••••••••••••••••••••••
Oligohydramnios, deformations from, 58Oncogenic viruses, 86tOncology, clinical, 86-89Opsonization, in inflammation, 15Optic nerve atrophy, 338Optic neuritis, 338Oral cavity
disorders of, 201-202infections of, 202t
Oral contraceptivesadverse effects of, 62-63as carcinogens, 85t
Oral thrush, 202tOrchitis, 261Organophosphates, toxic effects of, 64tOsgood- Schlatter disease, 303Osmotic diarrhea, 212tOsteitis deformans, 303Osteitis fibrosa cystica, 254Osteoarthritis, 304, 305f, 306
obesity and, 69tOsteoarthropathy, hypertrophic, cancer associated
with, 89tOsteoblastic metastases, 80Osteoblastoma, 305tOsteochondroma, 305tOsteogenesis imperfecta, 302Osteogenic sarcoma, 305tOsteoid osteoma, 305tOsteolytic metastases, 80Osteoma, 305t
osteoid, 305tOsteomalacia, in renal failure, 254Osteomyelitis, 302Osteopetrosis, 302Osteoporosis, 303
in renal failure, 254Otitis media, 338Otosclerosis, 338Ovarian disorders, 275-277, 276tOxidative phosphorylation, abnormalities in, 2-3Oxidizing agents, methemoglobinemia from, 1Oxygen-dependent myeloperoxidase system, 15-17Oxygen (02) content, definition of, factors contribut-
ing to, and significance of, 2tOxygen toxicity, 4
Paget's diseaseof bone, 303extramammary, 270of nipple, 282t
Palate, cleft, 201Palpable purpura, 168Panacinar emphysema, 196-197, 196fPancoast's tumor, in lung cancer, 199Pancreas, disorders of, 237-238, 298-301
Index 455
Pancreatic cholera, 299Pancreatic insufficiency
in cystic fibrosis, 237malabsorption and, 214
Pancreatitisacute, 237-238
hypocalcemia in, 293tchronic, 238
vitamin B 12 deficiency in, 132enzymatic fat necrosis and, 11
Panencephalitis, sclerosing, subacute, 328tPa02 . See Partial pressure of arterial oxygen (Pa02).Pap smear, cervical, 271Papillary carcinoma, of thyroid, 291Papillary cystadenoma lymphomatosum, 203Papilledema, in cerebral edema, 321Papilloma, intraductal, 280-281Papillomatosis, definition of, 311tPapillomavirus infection, human, 267, 312Papule, definition of, 311tParacortical hyperplasia, 154Parainfluenza virus, respiratory infections from, 185tParakeratosis, definition of, 311tParaneoplastic syndromes, 88, 89t
in lung cancer, 199-200Paraseptal emphysema, 196f, 197Parasitic infections, CNS, 330tParathyroid glands, disorders of, 292-294Parathyroid hormone, 292Parenchymal cell regeneration, 20-21Parkinsonism, 332-333Parotid gland tumors, 202Paroxysmal nocturnal hemoglobinuria, 136
pathogenesis of, 135tPartial pressure of arterial oxygen (Pa0 2), definition
of, factors contributing to, and significanceof, 2t
Partial thromboplastin time, 167Parvovirus B19, 312l'atau's syndrome, 53Patent ductus arteriosus, 110Pautrier's microabscesses, 156Pellagra, 72t, 73Pelvis, renal, cancers of, 257l'emphigoid, bullous, 316l'emphigus vulgaris, 315-316Penicillin
autoimmune hemolytic anemia from, 139tubulointerstitial nephritis induced by, 252
Penisdisorders of, 260-261erectile dysfunction of, 266
Peptic ulcer disease, 208-209Perfusion defect, hypoxemia from, 1Periappendiceal abscess, 221Pericarditis, 117-118
fibrinous, in myocardial infarction, 107

456 I ndex
Peripheral nervous system disorders, 336-337Peripheral neuropathy(ies), 336-337
in lead poisoning, 128Peripheral vascular disease, from atherosclerosis, 92Peritonitis, spontaneous, causes and characteristics of,
228tPeritonsillar abscess, 202tPernicious anemia
laboratory findings in, 132tvitamin B i z deficiency in, 132
Peroxidation, lipid, from free radical injury, 4Peroxides, 3Peutz-Jeghers polyps, 218Peyronie's disease, 260pH, intracellular, decreased, in ATP synthesis, 3Phagocytosis, in inflammation, 15Phagolysosome formation, in inflammation, 15Phakomatoses, 323-324Pharyngitis, 202tPhenylketonuria (PKU), 47tPhenytoin, folate deficiency from, 130Phcochromocytoma, 298
in neurofibromatosis, 324Phimosis, 260Phlebothrombosis, 95Phosphofructokinase, activation of, in ATP
synthesis, 3Phospholipase activation, in hypoxic cell injury, 3Phosphorylation, oxidative, abnormalities in, 2-3Photodermatitis, contact, 315Phyllodes tumor, of breast, 280Pilonidal cyst and abscess, 221Pineal gland disorders, 336-337Pitting edema, in nephrotic syndrome, 247Pituitary gland
disorders of, 284-287hyperfunction of, 286-287hypofunction of, 284-286
Pityriasis rosea, 317PKU (phenylketonuria), 47tPlacenta accreta, 277Placenta previa, 277Placentas, twin, 277Plaque, definition of, 311tPlasma cell dyscrasias, 158-159Platelet count, 124Platelets
disorders of, 168-169in normal hemostasis, 163
Pleura, disorders of, 200Pleural effusion, 200Plummer-Vinson syndrome, 126, 204Plummer's disease, 290Pneumoconiosis(es), 192-193
coal worker's, 6tPneumocystis carinii, respiratory infections from,
187t
Pneumonia, 183-186community-acquired
atypical, 185-186typical, 183, 185
in immunocompromised hosts, 186nosocomial, 186
Pneumonitis, hypersensitivity, 194Pneumothorax, 200
in decompression sickness, 41Point mutations, 44Poisoning
carbon monoxide, 2cyanide, oxidative phosphorylation and, 2lead, 6t
chronic, 253laboratory findings in, 129tsideroblastic anemias from, 128-129
Poliovirus, CNS infections from, 328tPolyangiitis, microscopic, 99tPolycyclic hydrocarbons, as carcinogens, 85tPolycystic kidney disease, 240, 242Polycystic ovarian syndrome, 275Polycythemia(s), 145-146
laboratory findings in, 146tsecondary, cancer and ectopic hormone associated
with, 89tPolycythemia vera, 146-147Polygenic inheritance, 55Polymyositis (PM), 32-33Polyp(s)
bowel, 218-219cervical, 271endometrial, 274gastric, 209-210nasal, 180
Polysomnography, in sleep apnea documentation, 180Polyvinyl chloride, toxic effects of, 64tPompe's disease, 47tPorphyria, acute intermittent, 334Porphyria cutanea tarda, 318Portal hypertension, in cirrhosis, 232Portal vein thrombosis, 228Postmenopausal osteoporosis, 303Postmortem clot, 40Potter's facies, 58Poxvirus, 312Prader-Willi syndrome, 55-56Preeclampsia-eclampsia, 278Pregnancy
cholestasis induced by, 231ectopic, 277-278toxemia of, 278
Prehn's sign, 262Preneoplastic disorders, 82tPresbycusis, 338Priapism, 260Prinzmetal's angina, 105

Index 457
Procoagulants, in normal hemostasis, 162-163Progressive multifocal leukoencephalopathy, 328tProlactin, increased, in pituitary hyperfunction, 286Prolactinoma, 286Promyelocytic leukemia, acute, 150Propionate metabolism, vitamin B 12 and, 133Prostaglandin(s)
as inflammation mediators, 18in normal hemostasis, 162
Prostatebenign hyperplasia of, 264, 264fcancer of, 265-266disorders of, 262, 264-266
Prostatitis, 262Proteases, activation of, in hypoxic cell injury, 3Protein(s)
C, in normal hemostasis, 162C-reactive
in atherosclerosis, 91-92in inflammation, 23
S, in normal hemostasis, 162synthesis of, decreased, ATP synthesis and, 3
Protein-energy malnutrition, 67Proteinuria
Bence Jones, in multiple myeloma, 253-254in nephrotic syndrome, 247
Prothrombin time, 166-167significance of, 223t
Proto-oncogenes, 81mutations involving, 82, 83t
Protozoa, causing diarrhea, 213tPseudogout, 307-308Pseudohermaphrodite, 56Pseudohypoparathyroidism, hypocalcemia in,
293tPseudomembranous inflammation, 20Pseudomonas aeruginosa, respiratory infections from,
184tPseudomonas aeruginosa osteomyelitis, 302Psoriasis, 316-317, 317fPsychosis, Korsakoff's, 61, 335Pulmonary embolus, 190
in decompression sickness, 41-42Pulmonary hypertension, 190-191Pulmonary thromboembolism, 40, 189-190Pulseless disease, 98tPurpura
Henock-Schonlein, 99tpalpable, 168senile, 168thrombocytopenic, types of, 170t
Purulent inflammation, 19Pustule, definition of, 311tPyelonephritis
acute, 251-252chronic, 252
Pyloric stenosis, 207Pyridoxine, 72t, 73
deficiency of, sideroblastic anemias and, 128Pyruvate kinase deficiency, pathogenesis of, 135tPyuria, "sterile," 260
Q fever, 185t
RRabies virus, CNS infections from, 328tRadiation injury, 65-66Radioactive 1311 uptake, in thyroid function
testing, 288Raynaud's disease, 98tRaynaud's phenomenon, 98t
inn systemic sclerosis, 31, 32fRectal prolapse, 221Red blood cell disorders, 121-141
anemia as, 125-141. See also Anemia(s).complete blood cell count and, 122-125erythropoiesis as, 121-122extramedullary hematopoiesis as, 122
Red blood cell indices, 123-124Reed-Sternberg cell, in Hodgkin's lymphoma,
156-157, 157fReflux disease, 206Regurgitation
aortic, 115mitral, 113-114tricuspid, 116
Reidel's thyroiditis, 288Rejection, transplantation, 27-28Renal calculi, 256Renal dysplasia, 240Renal infarction, 255Renal osteodystrophy, 254Renal pelvis, cancers of, 257Reproductive disorders
female, 267-283cervical, 271-272fallopian tube, 275gestational, 277-279ovarian, 275-277sexually transmitted diseases as, 267-269uterine, 272-274vaginal, 270vulvar, 269-270
male, 259-266Resorption atelectasis, 181-182Respiratory acidosis
cerebral edema and, 321hypoxemia from, 1
Respiratory alkalosis, production of, for headtrauma, 321
Respiratory distress syndrome, 182-183, 183fadult, 192

458 Index
Respiratory infections, systemic, 188Respiratory syncytial virus, respiratory infections
from, 185tRespiratory system
in AIDS, 35tdisorders of, 180-200
atelectasis as, 181-183infectious, 183-189
bacterial, 184tfungal, systemic, 187t, 188lung abscess as, 188-189pathogens causing, 185tpneumonia as, 183-186tuberculosis as, 187-188
neoplastic, 198-200, 198tpleural, 200restrictive lung disease as, 191-194upper airway, 180-181vascular lung lesions as, 189-191
Restriction fragment length polymorphism, ingenetic/developmental disorder diagnosis, 59
Restrictive cardiomyopathy, 119Restrictive lung disease, 191-194Retardation, growth, in lead poisoning, 128Reticulocyte count, in erythropoiesis, 121-122Retinoblastoma suppressor genes, 82Retinoic acid, teratogenicity of, 57Retinopathy, diabetic, 300Reye's syndrome, 227-228Rh antigen system, transfusions and, 174Rhabdomyoma, 120, 310t
in heart in tuberous sclerosis, 323Rhabdomyosarcoma, embryonal, 260, 310tRheumatic fever, mitral disorders in, 112Rheumatoid arthritis, 306-307, 306fRheumatoid nodules, 307Rhinovirus, respiratory infections from, 185tRiboflavin, 72t, 73Rigor mortis, mechanism of, 5RNA viruses, oncogenic, 86tRobertsonian translocation, 52Roseola infantum, 312Rotavirus, diarrhea from, 213tRubella, 312
congenital defects associated with, 58tRubeola, 312
respiratory infections from, 185tRubor, in inflammation, 14
SSalivary gland tumors, 202-203Salmonella species, diarrhea from, 213tSarcoidosis, 194
hypercalcemia in, 293tSarcoma(s), 76, 77f
Ewing's, 305tKaposi, 97t
Sarcoma(s)—cont'dKaposi's, in HIV, 35, 36fosteogenic, 305t
Sarcoma botryoides, 270Scales, definition of, 311tScar tissue, remodeling of, 21Scarlet fever, 313Schistosomiasis, causes and characteristics of, 228tSchwannoma, 337Scleroderma, 31-32Sclerosing cholangitis, primary, 231Sclerosing panencephalitis, subacute, 328tScrotal disorders, 261-262Scurvy, 74, 74f
prolonged bleeding time in, 167tSeborrheic dermatitis, 314Seborrheic keratosis, 318-319, 319f
cancer associated with, 89tSecretory diarrhea, 212tSeeding, of malignant tumors, 80Seminoma, 263tSenile purpura, 168Septic arthritis, 308Septic shock, 42t, 43Seronegative spondyloarthropathies, 308Serotonin, as inflammation mediator, 17, 18tSerous tumors, 276tSertoli-Leydig cell tumor, 276tSerum alanine transaminase, significance of, 223tSerum albumin, significance of, 223tSerum alkaline phosphatase, significance of, 223tSerum ammonia, significance of, 223tSerum aspartate transaminase, significance of, 223tSerum g-glutamyltransferase, significance of, 223tSerum sickness, 99tSerum total iron-binding capacity (TIBC), 124-125Sex differentiation, disorders of, 56-57Sexually transmitted diseases (STDs), 267-269Sexually transmitted urethritis, 259-260Sezary syndrome, 156Sheehan's postpartum pituitary necrosis, 285Shigella species, diarrhea from, 213tShingles, 312, 313fShock, 42-43
anaphylactic, 26complications associated with, 43
Sialadenitis, 202tSickle cell anemia, 136-138, 138f
pathogenesis of, 135tSickle cell disease, osteomyelitis in, 302Sickle cell nephropathy, 255Sickle cell trait/disease, 44Sideroblastic anemias, 127-128, 129f, 129tSIDS (sudden infant death syndrome), 59Sildenafil, for erectile dysfunction, 266Silent mutations, 44Silica, as carcinogen, 85t

index 459
Silicosis, 193Sinus, urachal, persistent, 259Sinus histiocytosis, 155Sinusitis, 181Sjogren's syndrome(s), 155
in rheumatoid arthritis, 307Skin
in AIDS, 35tdisorders of, 311-320
autoimmune, 315bacterial, 313benign melanocytic, 318benign noninfectious, 315-318descriptive terms for, 311tfungal, 314-315neoplastic, 318-320premalignant, 316viral, 312, 312f, 313f
SLE. See Systemic lupus erythematosus (SLE).Sleep apnea, obstructive, 180-181Small bowel
disorders of, 210-220. See also Bowel disorders.obstruction of, 218
Small cell carcinoma, of lung, 198tSmall lymphocytic lymphoma, 156tSmoke, cigarette
emphysema and, 195-196lung cancer and, 198
Smooth endoplasmic reticulum, injury to, 4Sodium, potassium-ATPase pump, impaired,
ATP synthesis and, 3Soft tissue disorders, 310Solar keratosis, 316, 316fSolar lentigo, 318Somatostatinoma, 299Spherocytosis, congenital, 134, 136, 136f
pathogenesis of, 135tSpider telangiectasia, 97tSpina bifida occulta, 323Spleen disorders, 160-161Splenomegaly, 160Spondyloarthropathies, seronegative, 308Spontaneous abortion, 59Spontaneous pneumothorax, 200Sporotrichosis, 315Squamous cell carcinoma, 75, 76f
of bladder, 260of esophagus, 206-207of lung, 198tof mouth, 201-202of penis, 261of renal pelvis, 257of skin, 320of vagina, 270of vulva, 270
Squamous metaplasia, 8
Stable angina, 105Staphylococcus aureus
diarrhea from, 213trespiratory infections from, 184tseptic arthritis from, 308skin infections from, 313
Stasis dermatitis, from deep vein thrombosis, 95Steatorrhea, in malabsorption, 214Stillbirth, 59Stomach
disorders of, 207-210polyps of, 209tumors of, 209-210ulcers of, duodenal ulcers compared with, 208t
Streptococcus, group B, CNS infections from, 329tStreptococcus pneumoniae
CNS infections from, 329trespiratory infections from, 184t
Stress ulcers, 201Stroke
atherosclerotic, 325embolic, 326, 326f
Strongyloides stercoralis, diarrhea from, 213tSturge-Weber syndrome, 97t, 324Subacute sclerosing panencephalitis, 328tSubarachnoid hemorrhage, 326-327Subdural hematoma, 324Subfalcine herniation, 321Sudden cardiac death, 106Sudden infant death syndrome (SIDS), 59Superficial migratory thrombophlebitis, cancer
associated with, 89tSuperficial mycoses, 314Superior vena cava syndrome, 96
in lung cancer, 199Superoxides, 3Suppressor genes, 82
mutations involving, 82, 83t, 84Suppurative inflammation, 19Surfactant, loss of, atelectasis from, 182-183Sydenham's chorea, in rheumatic fever, 113Syndrome of inappropriate antidiuretic hormone
(SIADH), 287Syngeneic graft, 27Synovial fluid analysis, 304Syphilis, 268-269
congenital, oral features of, 202tcongenital defects associated with, 58t
Syphilitic aneurysm, 93-94Syringomyelia, 323Systemic lupus erythematosus (SLE), 29-31
autoimmune hemolytic anemia in, 139congenital anomalies and, 57diffuse proliferative glomerulonephritis in,
246-247skin lesions in, 315

460 I ndex
Systemic sclerosis, 31-32Systolic dysfunction, 104
T-cell lymphoblastic leukemia, precursor, 155T-cell lymphoblastic lymphoma, precursor, 155T-cell lymphomas, 155-156T cells, derivation, location, and function of, 24tTaenia solium, CNS infections from, 330tTakayasu's arteritis, 98tTapeworm, fish, vitamin B 12 deficiency and, 132Tay-Sachs disease, 45, 48tTelangiectasia
ataxia, 33t, 84hereditary, 97tspider, 97t
Temporal arteritis, 98tTendinitis, from cat bite, 308Tensilon test, for myasthenia gravis, 310Tension pneumothorax, 200Teratogens, 57, 57tTeratoma(s), 75, 76f
cystic, 276ttesticular, 263t
Testiclecancer of, 262, 263ttorsion of, 262
Testicular feminization, 56-57Testis, disorders of, 261-262Tetany, total serum calcium and, 292Tetralogy of Fallot, 111, 111fThalassemias, 126-127
laboratory findings in, 129tThecoma-fibroma, 276tThermal injury, 64-65Thiamine, 72, 72t
deficiency of, from alcohol abuse, 61Thiazides, hypercalcemia from, 293tThoracic outlet syndrome, 96Thromboangiitis obliterans, 98tThrombocythemia, essential, 148Thrombocytopenia, 168-169
laboratory findings in, 166tprolonged bleeding time in, 167ttypes of, 170
Thrombocytosis, 169Thromboembolism, pulmonary, 40, 189-190Thrombophlebitis, 96
migratory, superficial, cancer associated with, 89tThrombosis, 39-40
from atherosclerosis, 92hepatic artery, 228hepatic vein, 229portal vein, 228
Thrombotic disorders, 172-173Thrombotic endocarditis, nonbacterial, 117
cancer associated with, 89t
Thrombotic thrombocytopenic purpura, 170tThromboxane A2, in normal hemostasis, 162Thromoboxane A2 , as inflammation mediators, 18Thrush, oral, 202tThyroglobulin, in thyroid function testing, 288Thyroglossal duct cyst, 288Thyroid-binding globulin, altered levels of, 287tThyroid gland
disorders of, 287-292anatomic, 288carcinoma as, 291-292follicular adenoma as, 291hypothyroidism as, 289laboratory findings in, 287tnontoxic goiter as, 291primary malignant lymphoma as, 292solitary thyroid nodule as, 291thyroiditis as, 288-289thyrotoxicosis as, 290-291
function tests for, 287-288Thyroid-stimulating hormone (TSH), serum,
in thyroid function testing, 288Thyroiditis, 288-289, 289tThyrotoxicosis, 290-291Thyroxine (T4), total serum, in thyroid function
testing, 287-288Tinea capitis, 314Tinea corporis, 314, 314fTinea versicolor, 314Tissue hypoxia, 1-3Tissue plasminogen activator, in normal
hemostasis, 162Tissue repair, 20-22Tissue thromboplastin, in normal hemostasis, 163TNM staging, of cancer, 87Tobacco
as carcinogen, 84use of, pathology from, 60
Tongue, inflammation of, 201Tonsillar herniation, 322Tonsillitis, exudative, 202tTORCH syndrome, 58Torsion, testicular, 262Total body water, 38Total iron-binding capacity, serum, 124-125Toxemia, of pregnancy, 278Toxic multinodular goiter, 290Toxic nervous system disorders, 334-335Toxic shock syndrome, 269
skin rash in, 313Toxicity, oxygen, 4Toxin, neuropathies associated with, 337Toxoplasma gondii, CNS infections from, 330tToxoplasmosis, congenital defects associated with, 58tTracheoesophageal fistula, 204Transfusion, blood, disorders of, 174-179. See also
Blood, transfusion of.
••••••••••••••••••••••••••••••••••••

Index 461
Transitional cell carcinomaof bladder, 260of renal pelvis, 257
Translocation, 52Transplantation immunology, 27-28, 29tTransplants, types of, 29tTransudate, 39Trauma, head, 321, 324Treponema pallidum, CNS infections from, 329tTrichomoniasis, 269Trichuris trichiura, diarrhea from, 213tTrimethoprim, folate deficiency and, 130Trinucleotide repeat disorders, 45Trisomy 13, 53Trisomy 18, 53Trisomy 21, 52-53Tuberculosis, 187-188
caseous necrosis in, 11granulomatous inflammation in, 17
Tuberculous granuloma, formation of, 19, 20bTuberculous osteomyelitis, 302Tuberous sclerosis, 323Tubular adenomas, 218, 219fTubular carcinoma, of breast, 282tTubular disorders, 251-254Tubular necrosis, acute, ischemic, complicating
shock, 43Tubulointerstitial nephritis, 251-254Tubulovillous adenomas, 218Tumor(s)
benign, 75properties of, 77-80
bladder, 260bone, 305tbreast, benign, 280-281CNS, 335-336components of, 77Desmoid, 310differentiation of, 77-78esophageal, 206-207of eye, malignant, 338gastric, 209-210glomus, 97tgrowth rate of, 78of heart, 119-120host defense against, 86liver, 234-235local invasion of, 78-79malignant, 75-76
properties of, 77-80metastasis of, 79-80, 80fmonoclonality of, 78of mouth, malignant, 201-202nuclear features of, 78ovarian, 275-277, 276tpineal gland, 337renal, 256-258
Tumor(s)—cont'dsalivary gland, 202-203telomerase activity of, 78vaginal, 270vascular, 97tof vulva, 269-270Warthin's, 203Wilms', 258
Tumor, in inflammation, 14Tumor-like conditions, 76Tumor markers, 88, 89tTumor necrosis factor, as inflammation mediator, 18Turcot's syndrome, 219Turner's syndrome, 53-54, 53f
coarctation of aorta in, 111lymphedema in, 96
Twin placentas, 277
UUlcer(s)
peptic, 208-209stress, 201
Ulceration, 20Ulcerative colitis, 215, 217f
Crohn's disease compared with, 216tUltrasound, in genetic/developmental disorder
diagnosis, 59Ultraviolet light
cancers induced by, 86dysplasia from, 9
Ultraviolet light B, injury from, 66Uncal herniation, 322Unstable angina, 105-106Upper airway disorders, 180-181Urachal sinus, persistent, 259Urate nephropathy, 253Urethra, disorders of, 259-260Urethral syndrome, acute, 259-260Urethritis, 252
sexually transmitted, 259-260Urinalysis, laboratory tests in, 241tUrinary tract, lower, disorders of, 259-261Urine bilirubin, significance of, 223tUrine urobilinogen, significance of, 223tUrobilinogen, urine, significance of, 223tUrolithiasis, 256Urticaria, 318Urticaria pigmentosa, 160Uterus, disorders of, 272-275Uveitis, 338
VVagina, disorders of, 270Vaginosis, 269Valvular heart disease, acquired, 112-117Varicella, 312
congenital defects associated with, 58t
•••••••

••••••••••••••••••••••••
••••
••462 Index
Varicocele, 261Varicose veins, 95Varicosities, from phlebothrombosis, 95Vascular disorders, 90-102
arteriosclerosis as, 91-92bowel, 211-212, 211fCNS, 325-327collagen, producing restrictive lung
disease, 194hypertension as, 100-102lipids and, 90-91lymphatic, 96renal, 255-256vasculitic, 97, 98-99t, 100venous system, 95-96
Vascular lung lesions, 189-191Vascular tumors, 97tVasculitic disorders, 97, 98-99t, 100Vasculitis
hypersensitivity, 100infectious, 99t
Vasoconstriction, in inflammation, 14Vasodilation, in inflammation, 14Vegan diet, vitamin B 12 deficiency from, 132Vein(s)
hepatic, thrombosis of, 229portal, thrombosis of, 228varicose, 95
Venereal warts, 267Venous system disorders, 95-96Venous thrombi, 39-40Ventilation defect, hypoxemia from, 1Ventricular aneurysm, 108Ventricular hypertrophy, 103Ventricular septal defect (VSD), 109, 110fVesicle, definition of, 311tVesicoureteral reflux, acute pyelonephritis
and, 251Vessel aneurysms, 92-95Vibrio cholerae, diarrhea from, 213tVillous adenomas, 218VIPoma, 299Virilization, 275Virus(es)
causing diarrhea, 213tCNS infections from, 327, 328tDNA, oncogenic, 86thepatitis, 223-227, 224-227meningitis from, 327, 328trespiratory infections from, 185tRNA, oncogenic, 86tskin disorders from, 312, 312f, 313f
Vitamin(s)A, 69-70, 70tB1, 72, 72tB2, 72t, 73B3, 72t, 73
Vitamin(s)-cont'dB 6 , 72t, 73
deficiency of, sideroblastic anemias and, 128B 12, 72t, 73
deficiency of, 131-133, 334laboratory findings in, 132t
C, 74D, 70-71, 70tdeficiencies of
from alcohol abuse, 61in malabsorption, 214
E, 70t, 71fat-soluble, 69-72, 70tK, 70t, 71-72water-soluble, 72-74, 72t
Vitiligo, 318Volvulus, small bowel obstruction from, 218Von Gierke's disease, 47tVon Gierke's glycogenosis, 6tVon Hippel-Lindau syndrome, 97tVon Willebrand's disease, 49, 171
laboratory findings in, 166tprolonged bleeding time in, 167t
Von Willebrand's factor, in normal hemostasis, 162VSD (ventricular septal defect), 109, 110fVulva, disorders of, 269-270Vulvar intraepithelial neoplasia, 269-270
Waldenstrom's macroglobulinemia, 159tWarfarin, for clotting disorders, 165Warm autoimmune hemolytic anemia, pathogenesis
of, 135tWarthin's tumor, 203Warts, 312
venereal, 267Water, total body, 38Water deprivation test, for diabetes insipidus, 286Waterhouse-Friderichsen syndrome, 295Weakness, muscle, etiology of, 309Wegener's granulomatosis, 98tWeight loss, in malabsorption, 214Wernicke's encephalopathy, 335Wernicke's syndrome, 61, 72tWheal, definition of, 311tWhipple's disease, malabsorption in, 215tWhite blood cell count and differential, 124White blood cell disorders, 142-152
benign qualitative, 142-143benign quantitative, 143-145neoplastic myeloid disorders as, 145-149
Wilms' tumor, 258Wilson's disease, 234, 334Wiskott-Aldrich syndrome, 33tWood's lamp, in dermatophytoses diagnosis, 314Wound healing, 22, 22b

Index 463
XX-linked dominant disorders, 51, sitX-linked recessive disorders, 50-51, 50tXanthelasma, 6tXenograft, 27Xeroderma pigmentosum, 84XYY syndrome, 55
YYersinia enterocolitica, diarrhea from,
213t
Yocardial infarction (MI), 106-109acute, C-reactive peptide predicting, 92
Yohimbe, for erectile dysfunction, 266Yolk sac tumor, 276t
testicular, 263t
Zenker's diverticulum, 204Zinc deficiency, healing and, 22Zollinger-Ellison syndrome, 209

www.elsevierhealth.comELSEVIERMOSBY
•••••••••
THE RAPID REVIEW SERIES
MOSBY'S RAPID REVIEW SERIES....NEW USMLE REVIEW PRODUCTS THAT
RESPOND DIRECTLY TO THE NEEDS OF MEDICAL STUDENTS LIKE YOU!
Mosby's Rapid Review Series is designed for today's busymedical student preparing for Boards. Each book in theseries is packed full of useful features PLUS a CD-ROM •that simulates the USMLE Step 1.
•LOOK FOR THE FOLLOWING RAPID REVIEW TITLES: •• Behavioral Science (ISBN: 0-323-02007-0) •• Biochemistry (ISBN: 0-323-00835-6) •
• Neuroscience (ISBN: 0-323-02261-8) •
• Gross and Developmental Anatomy (ISBN: 0-323-01201-9) ••
• Pharmacology (ISBN: 0-323-00838-0) •• Physiology (ISBN: 0-323-01991-9) •• Hi stology and Cell Biology (ISBN: 0-323-00834-8) •• Microbiology and Immunology (ISBN: 0-323-00840-2) •
•• USMLE Step 1 (ISBN: 0-323-00841-0) •
http://www.mosby.com/MERLIN/rapidreview/••••Recommended
Shelving Classification •Review— Patholo,
ISBN-13: 978-0-323-02393.ISBN-10: 0-323-02393-2
9"780323"023931" 4,


















