

Monitoring CML Treatment: Addressing the Issues for the Community Hematologist/Oncologist
Upload
august-priceCategory
view
216download
0
2
ObjectivesAt the end of this chapter you will be able to:• Define anemia• Discuss the causes and clinical significance of different
categories of anemia• Describe the classification of anemia• Explain - Microcytic anemia
- Macrocytic anemia
- Normochromic normocytic anemia• Discuss the laboratory findings for each category of
anemia• Perform basic laboratory tests for the diagnosis of
anemia

3
Chapter Outline3.1. Definition of anemias
3.2. Classification of anemias 3.2.1. Hematologic Response to Anemia3.2.2. Signs of Accelerated Bone Marrow Erythropoiesis3.2.3. Physiologic Response to Anemia 3.2.4. Methods of classification 3.2.5. Anemia Diagnosis/Cause3.2.6. Lab Investigation of Anemia
3.3. Types of anemia. 3.3.1 microcytic hypochromic anemia
3.3.2. macrocytic normocytic anemias3.3.3. normocytic anemias3.3.4. normocytic anemias due to hemoglobinopathies

4
3.1. Introduction 3.1.1. Definition of Anemia• Anemia is a decrease in the RBC count, Hgb and/or
HCT values as compared to normal reference range for age and sex (Also determined by alteration in plasma volume)– ‘True’ anemia:
– decreased RBC mass and normal plasma volume
– Pseudo or dilutional anemia:
– normal RBC mass and increased plasma volume– An increase in plasma volume can occur in Pregnancy, volume
overload (IVs) congestive heart failure
– Low Hgb and HCT values

5
Definition of Anemia cont’d

6
3.1.1. Definition of Anemia cont’d…..
• Anemia must also relate to the level of hemoglobin the individual normally possesses. – If an adult male usually maintains a
hemoglobin level of 16g/dl, and over a period of days is noted to have decreased to 14g/dl, this must be considered significant even though both values are within the normal range for an adult male.

7
3.1.1. Definition of Anemia cont’d…..• Various diseases and disorders are associated
with decreased hemoglobin levels. These include:– Nutritional deficiencies– External or internal blood loss– Increased destruction of RBCs– Ineffective or decreased production of RBCs– Abnormal hemoglobin synthesis– Bone marrow suppression by toxins, chemicals, or
radiation & replacement by malignant cells– Infection

8
3.1.1. Definition of Anemia cont’d…..
Functionally anemia is defined as tissue hypoxia (inability of the body to supply tissue with adequate oxygen for proper metabolic function)– There is an abnormal hemoglobin with an
increased O2 affinity resulting in an anemia with normal or raised hemoglobin levels, hematocrit, or RBC count.
• Generally anemia is not a disease, but rather the expression of an underlying disorder or disease.

9
3.1.1. Definition of Anemia cont’d…..
• Anemia may develop:– When RBC loss or destruction exceeds
the maximal capacity of bone marrow RBC production or
– When bone marrow production is impaired

10
3.1.2.Hematologic Response to Anemia
• Tissue hypoxia causes increased renal release of erythropoietin (EPO) to accelerate bone marrow erythropoiesis
• The normal bone marrow can increase its activity 7-8 times normal – Marrow becomes hypercellular

11
Signs of Accelerated Bone Marrow Erythropoiesis
• The marrow becomes hypercellular due to a marked increase in RBC precursors (called erythroid hyperplasia) and the M:E ratio falls.
• Nucleated RBCs may be released into the blood circulation along with the outpouring of reticulocytes– NRBC number tends to correlate with the
severity of anemia– Increased polychromasia on the Wright's-
stained blood smear is seen due to increased number of circulating Retics.

12
• If demand exceeds maximal bone marrow activity, RBC production may occur in extramedullary sites, liver, spleen (hepatosplenomegaly).

13
3.1.3. Physiologic Response to Anemia
• Ability to adapt to anemia depends on:– Age and underlying disease– Cardio/pulmonary function– Rate at which anemia develops (BM can
compensate easier if the onset of anemia is slow),
– Underlying disease

14
3.1.4. Clinical features:
• Symptoms of hypoxia: decreased oxygen delivery to the tissues/organs
causes: – fatigue , faintness, weakness, dizziness,
headaches, dyspnea, poor exercise tolerance, leg cramps.

15
Signs of anemia• general signs include pallor of
mucous membrane, which occur if the Hgb concentration is less than 9g/dl,
• specific signs are associated with particular types of anemia, for example, jaundice in hemolytic anemia, leg ulcer in sickle cell anemia
3.1.4.Clinical features cont’d….:

16
3.1.5. Diagnosis of anemia
• Before making a diagnosis of anemia, one must consider:– Age– Sex– Geographic location– Presence or absence of lung disease

17
3.1.5. Diagnosis of anemia cont’d……
• How does one make a clinical diagnosis of anemia?A. Patient history
–Dietary habits–Medication–Possible exposure to chemicals and/or
toxins–Description and duration of symptoms

18
3.1.5. Diagnosis of anemia cont’d……
–Tiredness–Muscle fatigue and weakness–Headache and vertigo (dizziness)–Dyspnia (difficult or labored breathing)
from exertion–G I problems–Overt signs of blood loss such as
hematuria (blood in urine) or black stools
Patient history cont………..

19
3.1.5. Diagnosis of anemia cont’d….B. Physical exam
– General findings• Hepato or splenomegaly• Heart abnormalities• Skin pallor
– Specific findings • In vitamin B12 deficiency there may be signs of
malnutrition and neurological changes• In iron deficiency there may be severe pallor, a
smooth tongue, and esophageal webs• In hemolytic anemias there may be jaundice due to
the increased levels of bilirubin from increased RBC destruction

20
3.1.5. Diagnosis of anemia cont’d……
C. Lab investigations
1. A complete blood count, CBC– RBC count– Hematocrit (Hct) or packed cell volume– Hemoglobin determination– RBC indices calculation– Reticulocyte count
2. Blood smear examination to evaluate:– Poikilocytosis– Leukocytes or Platelets abnormalities

21
3.1.5. Diagnosis of anemia cont’d…..
Lab investigation cont’d……3. A bone marrow smear and biopsy to
observe:– Maturation of RBC and WBC series– Ratio of myeloid to erythroid series– Abundance of iron stores (ringed sideroblasts)– Presence or absence of granulomas or tumor
cells– Red to yellow ratio– Presence of megakaryocytes

22
4. Hemoglobin electrophoresis
3.1.5. Diagnosis of anemia cont’d…..

23
3.1.5. Diagnosis of anemia cont’d…..
Lab investigation cont’d……
5. Antiglobulin testing
6. Osmotic fragility test

24
3.2. Methods of Anemia Classification• Several schemes of classifying anemias exist 1. Morphologic
– Based on RBC morphology– Anemia is divided into three groups mainly on
the basis of the MCV (RBC indices)2. Pathophysiologic
– Anemia is divided using three main causes/mechanisms I. Impaired erythrocyte formation (Aplastic
anemia, IDA, sideroblastic anemia, anemia of chronic diseases, megaloblastic anemia)– Retic count is low– The bone marrow fails to respond appropriately
due to disease or lack of essential supplies

25
Methods of Classification cont’dIII. Increased blood loss (Acute, Chronic)
– Retic count is typically high– Anemia results when red cell loss exceeds
the bone marrow’s capacity to increase its activity
IV. Increased destruction of RBCs (hemolytic anemias)
– Retic count is typically high– Anemia results when red cell destruction
exceeds the bone marrow’s capacity to increase its activity

26
Methods of Classification cont’d
Morphologic Categories of Anemia
1. Normocytic Normochromic anemia (normal red cell indices)• Blood loss anemia • Hemolytic anemia• Aplastic anemia• Chronic diseaes• Renal insufficciency

27
Morphologic Categories of Anemia
• Microcytic hypochromic
( low red cell indices)
• Iron deficiency anemia
• Sideroblastic anemia
• Lead poisoning
• Thallassemia
• Chronic diseases
Methods of Classification cont’d

28
Morphologic Categories of Anemia
• Macrocytic Normochromic
( high MCV and MCH, normal MCHC)
• Megaloblastic anemia
• Liver disease
• Post splenectomy
• Hypothyroidism
• Stress erythropoiesis
Methods of Classification cont’d

29
1 Microcytic/hypochromic
3
1 2
2 Macrocytic/Normochromic
3 Normocytic/Normochromic
Morphologic Categories of Anemia
N.B. The nucleus of a small lymphocyte (shown by the arrow) is used as a reference to a normal red cell size

30
3.2.2. Pathophysiological classification
1.
B.
2.
A.

31
1. Microcytic- Hypochromic Anemia

32
Microcytic- Hypochromic Anemia
• Many RBCs smaller than nucleus of normal lymphocytes
• Increased central pallor.• Includes
– Iron deficiency anemia– Thalassemia– Anemia of chronic disease – Sideroblastic anemia– Lead poisoning
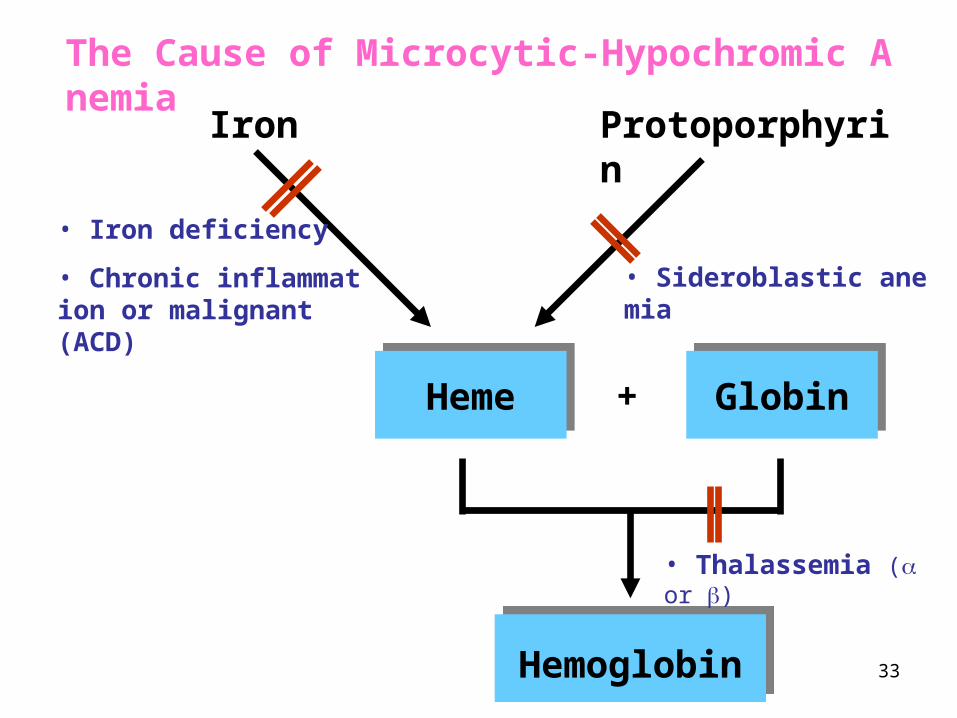
33
Iron Protoporphyrin
HemeHeme GlobinGlobin+
HemoglobinHemoglobin
• Iron deficiency
• Chronic inflammation or malignant (ACD)
• Thalassemia ( or )
• Sideroblastic anemia
The Cause of Microcytic-Hypochromic Anemia

34
RBC maturation in microcytic anemias
Normoblastic RBC maturation normocytic red cells
Abbott Manual
Microcytic/Hypochromic Anemias
Normal RBC maturation is shown for comparison

35
A. Iron Deficiency Anemia (IDA) • Is a condition in which the total body iron
content is decreased below a normal level
• This results in a reduced red blood cell and hemoglobin production
• More than half of all anemias are due to iron deficiency.

36
Iron Deficiency Anemia (IDA)
• Causes:– Nutritional deficiency– Malabsorption (insufficient or defective
absorption)– Inefficient transport, storage or utilization of
iron– Increased need– Chronic blood loss (GI bleeding, ulcer, heavy
menstruation, etc)

37
DIETARY SOURCES OF IRONDIETARY SOURCES OF IRON
Inorganic Iron eg lentils
Organic iron eg beef
DAILY IRON REQUIREMENT 10-15mg/day (5-10% absorbed)

38
Daily Iron cycle (Fig)

39
Adult men 0.5-1
Post menopausal female 0.5-1
Menstruating female 1-2
Pregnant female 1.5-3
Children 1.1
Female (age 12-15) 1.6-2.6
Estimated daily iron requirementsUnits are mg/day

40
Iron absorption, Transport and storage
• Iron absorbed from duodenum and jejunum in the GIT
• Moves via circulation to the bone marrow
• Incorporated with protoporpyirin in mitochondria of the erythroid precursor to make Heme

41
• There are three proteins important for transporting and storage of iron:
–Transferrin, –Transferrin receptors and –Ferritin
Transport: • Transferrin: transports iron from the
plasma to the erythroblasts in the marrow for erythropoiesis
• The transferrin will bind to transferrin receptor on the erythrocyte membrane

42
Storage • Hgb contains about two third of the body iron
• At the end of their life, RBCs are broken down in the macrophage of reticuloendothelial system and then iron is released from Hgb enters plasma and provided to transferrin.
• Some stored in reticuloendothelial cell as ferritin soluble protein – iron complex) and hemosiderin (37%) (degraded form of ferritin insoluble)
• iron is also found in muscles as myoglobin and in other cells as iron containing enzymes

43
Amount of iron Male Female %
in average adult (g) (g) of total
Hb 2.4 1.7 65
ferritin & hemosiderin 1.0 0.3 30
Myoglobin 0.15 0.12 3.5
Heme enzyme 0.02 0.15 0.5
Transferrin-bound 0.004 0.003 0.1
iron
The distribution of body iron

44
Iron Deficiency Anemia (IDA) • Sequence of iron depletion
When iron loss or use exceeds absorption, there is a sequence of iron depletion in the body:
1.Storage iron decreases/ low serum ferritin; serum iron & TIBC are normal, no anemia, normal red cells.
2.Serum iron decreases/TIBC increases (increased transferrin); no anemia, normal red cells.
3.Anemia with microcytic/hypochromic red cells = IDA.

45
CLINICAL FEATURES IRON DEFICIENCYCLINICAL FEATURES IRON DEFICIENCY
• Symptoms eg. fatigue, dizziness, headache
• Signs eg. pallor, Tongue atrophy/ glossitis -
raw and sore, angular cheilosis (Stomatitis)
Koilonychia Glossitis
Angular Cheilosis or Stomatitis

46
• Clinical signs and symptoms– Spoon‑shaped nails (koilonychia), brittle
nails and hair.

47
Lab Investigation of IDA
• Iron tests►Used to differentiate microcytic
hypochromic anemia's or detect iron overload (hemochromatosis)– Iron circulates bound to the transport protein
transferrin• Transferrin is normally ~33% saturated with iron
• Iron tests include serum iron, Total Iron Binding Capacity (TIBC), serum ferritin

48
Lab Investigation cont’d• Serum iron level
– measures the amount of iron bound to transferrin– Does not include the free form of iron
• Total Iron Binding Capacity (TIBC)– Is an indirect measure of the amount of transferrin
protein in the serum– Inversely proportional to the serum iron level
• If serum iron is decreased, total iron binding capacity of transferrin increased (transferrin has more empty space to carry iron)

49
Lab Investigation cont’d• Serum ferritin
– indirectly reflects storage iron in tissues – found in trace amount in plasma – It is in equilibrium with the body stores– Variation in the quantity of iron in the storing compartment is
reflected by plasma ferritin concentratione.g. Plasma ferritin is decreases in IDA
Plasma ferritin increases in ACD
Limitation: During infection or inflammation Serum Ferritin increases like other acute phase proteins, and then it is not an accurate indicator in such situations.

50
Bone marrow iron (Tissue iron)
• Tissue biopsy of bone marrow
• Prussian blue stain
• Type of iron is hemosiderin

51
ABSENT IRON STORES IN BONE ABSENT IRON STORES IN BONE MARROW IN IRON DEFICIENCYMARROW IN IRON DEFICIENCY
Iron deficiencyNormal control

52
Iron Deficiency Anemia
• Lab findings– Low RBC, Hgb, Hct– Low MCV, MCH, MCHC– Normal WBC and PLT
Blood smear

53
Iron Deficiency Anemia• RBC morphology
– Hypochromia– Microcytosis– Anisocytosis– Poikilocytosis
• Pencil cells (cigar cells)• Target cells
– no RBC inclusions
• Iron parameters– Low serum iron, – High TIBC, – Low serum ferritin
Blood smear

54
Wright’s stained blood smear
Ovalocytes - Pencil forms No RBC inclusions
Iron Deficiency

55
IDA cont’d
• Treatment
– Identify the underlying cause– Oral iron is given; see increased Retic
count post-therapy.– May see dimorphism following treatment
• a dual red cell population with older microcytic red cells along with the newly produced normocytic red cells.

56
B. Sideroblastic Anemia (SA)
• This group of anemias are characterized by defective protoporphyrin synthesis (blocks) resulting in iron loading and a hypochromic anemia due to deficient hemoglobin synthesis.
• Block(s) in protoporphyrin synthesis leads to iron overload and microcytic/hypochromic anemia

57
Sideroblastic Anemia (SA)

58
Terms:• Siderocytes are mature RBCs in the blood
containing iron granules called Pappenheimer bodies....abnormal.
• Sideroblasts are immature nucleated RBCs in the bone marrow containing small amounts of iron in the cytoplasm....normal.

59
SA• Sideroblastic anemia is characterized by the
– Accumulation of iron in the mitochondria of immature nucleated RBCs in the bone marrow;
– Iron forms a ring around the nucleus these are called ringed sideroblasts....abnormal.
• The iron accumulation in the mitochondria is the result of blocks in the protoporphyrin pathway.

60
SA cont’dLab findings:• Microcytic/hypochromic red cells, low MCV and MCHC;
variable anemia, low retic.• RBC inclusions: Basophilic stippling and Pappenheimer
bodies (siderocytes). (May see target cells).• High serum iron and high serum ferritin (stores); low
TIBC and high % saturation. • *Decreased transferrin synthesis occurs in iron
overload states.• Bone marrow: ringed syderoblasts (Hall mark of
Sideroblastic Anemia)

6161
RBC with iron Wright’s stain
NRBC with iron Prussian blue stain
NRBC with ring of iron Prussian blue stain
Pappenheimer bodies
Blood Bone marrowBone marrow
SideroblastRinged Sideroblast
Sideroblastic Anemia (SA)

62
Pappenheimer bodies Wright’s stain
Blood
Basophilic stippling/stippled RBCs
Blood
Pappenheimer bodies Prussian blue iron stain
Blood
Sideroblastic Anemia (SA)

63
100x
Ringed Sideroblasts Prussian blue iron stain
Bone marrow 10x
Increased stainable iron Prussian blue iron stain
Bone marrow
Sideroblastic Anemia (SA)Bone marrow findings (if done):
1. *Ringed sideroblasts demonstrated with Prussian blue stain.
2. Increased stainable iron in macrophages.

64
Types of Sideroblastic anemia cont’d
• Primary ‑ cause unknown (can't identify blocks) and are not reversible....called Idiopathic or primary Sideroblastic anemia.
1.Elderly, responds to no treatment. Requires transfusion support if severe anemia.
2.Characterized by a dimorphic red cell population - micro/hypo red cells with
3.normocytic and/or macrocytic red cells....MCV is variable and RDW is high.
4.Primary type of sideroblastic anemia is one of myelodysplastic syndromes called Refractory Anemia with Ringed Sideroblasts; may terminate in leukemia

65Wright’s stained blood smear
Stippled RBCs – Lead poisoning
Secondary Types of SA• Alcohol inhibits vitamin B6/pyridoxine
• Anti-tuberculosis drugs inhibit vitamin B6
• Lead causes multiple blocks
– Inhaled or ingested
– Abnormal lead level
– Neurologic problems
– Lead line (gums)
– Chelation therapy (EDTA).

66

67
C. Anemia of chronic disease• Anemia of chronic disease (ACD) – inability to use
iron and decreased response to EPO – Very common anemia, #2
• Associated with systemic disease, including chronic inflammatory conditions:
–Rheumatoid arthritis–Chronic renal disease –Thyroid disease –Malignancies –Tuberculosis –Chronic fungal infections etc

68
ACD pathogenesis
• Lactoferrin is an iron biding protein in the granules of neutrophils
• Its avidity for iron is grater than transferrin• During infection or Inflammation, neutrophil-
lactoferrin released into plasma and Scavenges available iron
• Bind to macrophage and liver cells (because they have receptor for lactoferrin
• Cytokines: Produced by macrophages during inflammation and contribute to ACD by inhibiting erythropoiesis

69
Lab Diagnosis
• Blood findings– Early stage: normocytic normochromic– Late stage: hypochromic microcytic,
– Low serum iron, low TIBC, normal or high serum ferritin
• Leukocytosis
• Abundant storage of iron in macrophage (Prusian blue)

7070Target cells/Codocytes
Beta
Alpha
D. Thalassemias • Inherited decrease in alpha or beta globin chain synthesis
needed for Hgb A; quantitative defect– All have microcytic/hypochromic RBCs and target cells
• Genetic mutations classified by:– ↓ beta chains = beta thalassemia…Greek/Italian– ↓ alpha chains = alpha thalassemia…Asian

71
Haemoglobin Molecule
Hgb A = 2α & 2β Hgb A2 = 2α & 2δ Hgb F = 2α & 2γ
• Consists of 4 globin chains + 4 heme groups• Normally, each individual inherits 2α, 1β, 1γ,
and 1δ gene from each parent.....so 4α, 2β, 2γ, and 2δ genes are inherited.
97% 2% 1%

72
Thalassemia • Impaired alpha or beta
globin synthesis results in an unbalanced number of chains produced that leads to:– RBC destruction in beta
Thalassemia major– Production of
compensatory Hgb types in beta thals
– Formation of unstable or non-functional Hgb types in alpha thals

73
Thalassemia• Severity ranges from lethal, to severe
transfusion-dependency, to no clinical abnormalities; severity depends on the number and type of abnormal globin genes inherited.
1.Major severe anemia; no α (or β) chains are produced, so cannot make normal hemoglobin (s).
2.Minor/trait mild anemia; slight decrease in normal hemoglobin types made.

74
Heinz bodies Excess alpha chains Supravital stain
Beta Thal Major (Homozygous)• Both beta genes abnormal
– Marked decrease/absence of beta chains leads to alpha chain excess…no Hgb A is produced
– Rigid RBCs with Heinz bodies destroyed in bone marrow and blood (ineffective erythropoiesis)

75
Stippled NRBC
NRBC
Target cell
Wright’s stained blood smear
HJB
Beta Thal Major (Homozygous)• Clinical findings• Lab findings
– Severe anemia, target cells, nucleated red cells– RBC inclusions– No hemoglobin A; compensatory Hgb F

76
Pap bodiesNRBC
Transfused RBC
Target cell
Hypercellular Bone Marrow (10x)
Blood smear
Howell-Jolly body
Target cells
Blood smear Transfused
RBC
Beta Thal Major (Homozygous)
• Treatment– Transfusion– Splenectomy– Iron chelation

77
Wright’s stained blood smear
Stippled RBC
Target cell
Beta Thal Minor (Heterozygous)
• One abnormal beta gene– Slight decreased rate of
beta chain production– Blood picture can look
similar to iron deficiency• Lab findings
– Mild anemia, target cells, no nRBCs, stippled RBCs
– No Heinz bodies– Normal iron tests– Compensates with
Hgb A2
Ovalocytes

78
Alpha Thal Major/Homozygous
• Deletion of all 4 alpha genes results in complete absence of alpha chain production– No normal hemoglobin types made
• Known as Barts Hydrops Fetalis– Die of hypoxia….Bart’s Hgb

79
Target cells
Wright’s stain blood smear
Heinz bodies Excess beta chains Supravital
stain
Alpha Thal Intermedia = Hgb H Disease
• Three alpha genes deleted– Moderate decrease in alpha chains leads to beta
chain excess…unstable Hgb H– Moderate anemia

80
Alpha Thal Minor (Heterozygous)• One or two alpha genes deleted (group)
– Slight decrease in alpha chain production – Mild or no anemia, few target cells– Essentially normal electrophoresis; many undiagnosed

81
Beta Thalassemias

82
Alpha Thalassemias

83
+
HGB Synthesis Defects
Differential Diagnosis of Microcytic Anemia

84
2. Macrocytic Normocytic Anemias

85
Macrocytic Normocytic Anemias
Characteristics ??????
Wright’s stained blood smear

86
A. MEGALOBLASTIC ANEMIA
• Vitamin B12 deficiency• Folate deficiency• Abnormal metabolism of folate and vit B12
B. Non megaloblastic anemia
• Liver disease• Alcoholism• Post splenoctomy• Neonatal macrocytosis• Stress erythropoiesis

87
A. Megaloblastic Anemia • Macrocytosis due to a deficiency of vitamin B12 or folic acid
that causes impaired nuclear maturation – Vitamin B12 & folate are DNA coenzymes necessary for
DNA synthesis and normal nuclear maturation– Results in megaloblastic maturation…nucleus lags
behind the cytoplasm and leads unbalanced growth called maturation asynchrony
• Both deficiencies cause enlarged fragile cells– Many cells die in the marrow (ineffective)– Show a similar blood picture and clinical findings
• Only vitamin B12 deficiency causes neurological symptoms…required for myelin synthesis

88
RBC maturation in microcytic anemias…IDA
Normoblastic RBC maturation normocytic red cells
Megaloblastic RBC maturation macrocytic red cells
Megaloblastic Anemia

89
Lab Findings of Megaloblastic Anemia
Mild to severe anemia, – Increased MCV & MCH, normal MCHC– Low RBC, HGB, WBC and PLT counts (fragile
cells) due to ineffective hematopoiesis.– Low reticulocyte count – Macrocytic ovalocytes and teardrops; – Marked anisocytosis and poikilocytosis – Schistocytes/microcytes - due to RBC breakage
upon leaving the BM– Erythroid hyperplasia - low M:E ratio (1:1) – Iron stores increased.
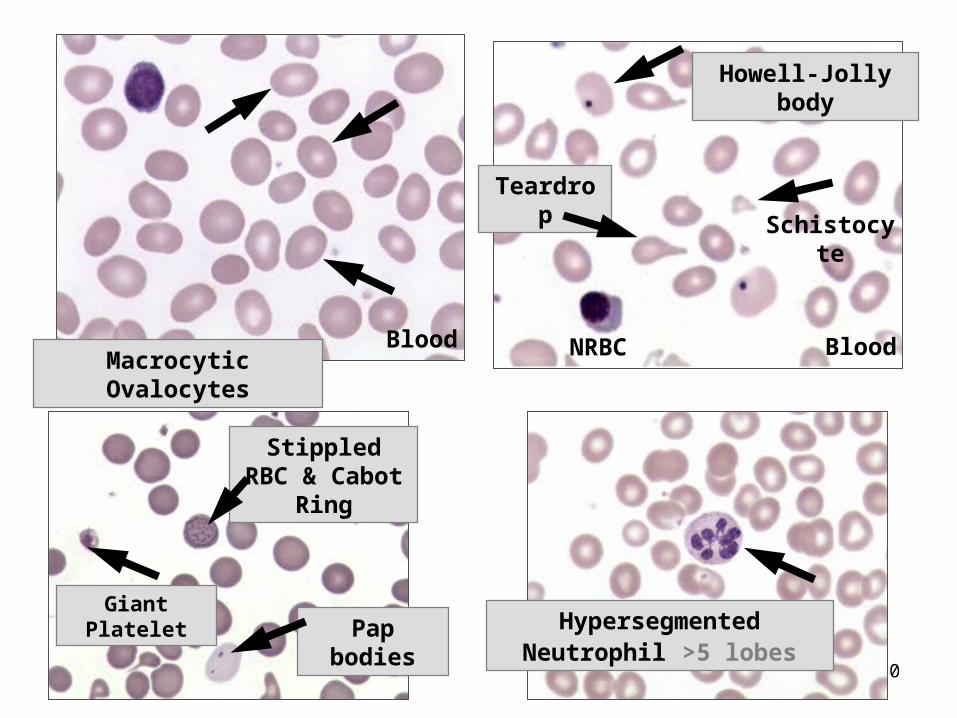
90
Macrocytic OvalocytesBlood NRBC Blood
Howell-Jolly body
Teardrop
Schistocyte
Stippled RBC & Cabot Ring
Giant PlateletPap bodies Hypersegmented Neutrophil >5
lobes

91
Vitamin B12 (Cobalamin) Deficiency

92
Vitamin B12 deficiencyOccur as a result of one of the following
conditions1. Nutritional Coballamin deficiency
• Strict vegetarianism
2. Abnormal intragasteric events ( i.e. inadequate proteolysis of food Coballamin)• atrophic gastritis.
3. Loss or atrophy of gastric mucosa ( deficient IF)• total or partial gasterectomy
– May develop B12 and iron deficiency with macro and micro red cells…a dual (dimorphic) RBC population
• pernicious anemia

93
Cont…..4. Abnormal events in the small bowel lumen
– Inadequate pancreatic protease– Competing agents like fish tape worms (D.latum)– Disorders of ileac mucosa– Diminished or absent of IF – Coballamin receptor– Drug effects– Metabolic disorders ( coballamin is not used by
cells)

94
Folate (Folic acid) Deficiency: • Deficient intake. • Increased needs: pregnancy,
infant, rapid cellular proliferation, and cirrhosis
• Malabsorption (congenital and drug-induced)
• Inherited DNA Synthesis Disorders: Deficient thiamine and factors (e.g. enzymes) responsible for folate metabolism.
• Toxins and Drugs:
Two RBC populations Dimorphism
Macrocytic RBCs
Microcytic RBCs

95
1. Pernicious Anemia
• it is defined as anemia resulting from defective secretion of IF associated with autoimmune attack on the gastric mucosa leading to atrophy of the stomach or Abs that block IF action.
• Abs block the site of IF where vit B12 binds.
• The diagnosis is confirmed by low serum B12 level and typically abnormal results of schilling test

96
Schilling test
• Used to diagnose pernicious anemia and determine if IF is available.
• If absorbed a portion of oral dose of vit B12 (not
used by the body) ---- excreted in urine
• If not absorbed (malabsorption)…….. Not detected in urine but pass out in feces

97
Stage I. To diagnose malabsorption• give the patient a radio active cobalt labeled vit B12 orally
and • Non labeled vit B12 intramuscularly to saturate liver
• If IF is present in the stomach (no other disease) vit B12 is absorbed and labeled vit B12 detected in urine
• If absorption is impaired labeled vit B12 not detected in urine instead in stool
• Diagnose malabsorption if it is not appear in urine by proceeding stage II.

98
Stage II. Differentiate cause of malabsorption
• Oral dose labeled vit B12 + IF
• Appear in urine…… pernicious anemia
(give IF)
• If not appear in urine it is other cause of malabsorption

99
• Performed to determine if the patient suffer from mal absorption of the IF – B12 complex secondary to small intestinal bacterial overgrowth.
• If this is the condition then tetracycline should normalize vit B12 absorption.
Stage III

100
Polychromatophilic RBCs Wright’s stain
NRBC
B. Non-Megaloblastic Anemia
• Macrocytosis that is NOT due to vitamin B12 or folate deficiency
• Accelerated erythropoiesis– Regenerating marrow or marked reticulocyte
response following recent blood loss

101
Stomatocytes, Alcoholic Stomatocytes, Alcoholic EchinocytesAcanthocytes
Target cells
Non-Megaloblastic Anemia• Liver disease and alcoholism
– Complex & multiple problems– Degree of anemia varies, round macrocytes– Target cells/acanthocytes - due to abnormal lipid metabolism.– Echinocytes are also commonly found on the smear in liver disease.

102
Blood smear
Differential Diagnosis of Macrocytic Anemia
• Megaloblastic and non-Megaloblastic
– Perform B12 and folate levels
– Specific morphology

103
3. Normocytic Normochromic Anemia

104
3. Normocytic Normochromic Anemia
• It includes• Aplastic anemia
due to BM failure
• Blood loss anemia
• Hemolytic anemia
Is a condition in which the size & Hgb content of RBCs is normal but the number of RBCs is decreased.

105
A. Aplastic Anemia
– Condition of blood pancytopenia caused by bone marrow failure…decreased production of all cell lines and replacement of marrow with fat.
– Due to damaged stem cells, damaged bone marrow environment or suppression
– No extramedullary hematopoiesis

106
Types of aplastic anemia
–Primary/idiopathic = 50%
–Secondary/acquired….chemicals, drugs, infections, radiation = 50%
–Congenital….Fanconi’s • Aplasia plus dwarfism, skeletal
abnormalities, mental retardation, abnormal skin pigmentation.

107Bone marrow, decreased # Bone marrow, decreased #
precursor cellsprecursor cells
10X10X
Normal RBCs No Platelets
Blood
Lab diagnosis of Aplastic Anemia
• Normochromic –Normocytic RBC (normal MCV & MCH)• Low reticulocyte count & Hgb • Pancytopenia • No abnormal cells • Hypoplasia Bone marrow•Normal Serum iron, vitamin B12 and folate levels

108
B. Hemolytic anemia
• Result from an increase in the rate of pre mature red cell destruction.
• Compensated hemolytic disease
• Uncompensated hemolytic disease
• It leads to– Erythropoietic hyperplasia – BM produces red cells 6 to 8X the normal rate– Marked reticulocytosis

109
Hemolytic anemia
• Two main mechanisms for RBC destruction in HA–Intravascular hemolysis: in the
circulation
–Extravascular hemolysis: in RE system (reticuloendothelial system)

110
Extravascular hemolysis
Aged RBC 120 day
Abnormal RBC
During destruction RBC releases Hgb
Hgb
Exstravascularly removed by Macrophage (RES) in BM, liver and spleen
Iron reabsorbed
Globin
Amino acid
Protein synthesis
Protoporphyrin
Unconjugated bilirubin liver (glucuronic acid)
conjugated bilirubin gut reabsorbed &
Excreted as urobilin & urobilinogen

111
Extravascular hemolysis
• Lab features –Increased RBC break down
• Serum bilirubin increase
• Stool urobilinogen increase
• Blood urobilinogen increase
• Urine urobilinogen increase

112
Intravascular hemolysis • Red cells are destroyed in blood vessels and Hgb is released into
the circulation:
Free Hgb
Saturates plasma haptoglobin
Excess free Hgb is filtered by the glomerules (kidney)(if rate of hemolysis saturates renal reabsorption capacity)
Free Hgb enters urine
Fe is released in bladder tubule
Renal tubule loaded with hemosiderin

113
Intravascular hemolysis
• Lab features– Hemoglobinemia and hemoglobinuria– Hemosiderin uria– Reduced/absent serum haptoglobin

114
1. Hereditary hemolytic anemia• This is a congenital hemolytic anemia. some of
which present at birth and other later in life, while still others may remain silent unless a physiological stress is super imposed
• Result of intrinsic red cell defects – Membrane defect (Hereditary Shperocytosis,
Elliptocytosis and sickle cell anemia)
– Metabolic defect : G6PDH and PK defic
– Hgb chain defect (hemoglobinopatheis) : sickle cell anemia

115
Spherocytes
A. Hemolytic Anemias due to Membrane Defects
• Most common is Hereditary Spherocytosis (HS)– Membrane defect is
decreased spectrin and increased permeability of membrane to sodium ions
• Lab findings– Anemia varies– Few to many
spherocytes on smear, high MCHC
– Increased OF test

116
H Ovalocytosis
Normocytic ovalocytes
H Ovalocytosis/Elliptocytosis• Membrane defect is
polarization of cholesterol or hemoglobin at ends and increased sodium permeability
• Over 25% ovalocytes– Most asymptomatic– Mild anemia in 10-
15%

117
H Stomatocytosis
Hereditary Stomatocytosis• Membrane defect is
abnormal permeability to sodium and potassium
• Caused by edema • 20-30% stomatocytes
on blood smear– Mild to severe
hemolytic anemia

118
H Acanthocytosis = Abetalipoproteinemia
H Acanthocytosis
• Defect is increased membrane cholesterol due to abnormal plasma lipids
• Numerous acanthocytes on smear– Mild anemia – Also known as abetalipoproteinemia

119
Hypotonic
Osmotic Fragility Test (OF)• Most commonly used to diagnose Hereditary
Spherocytosis– Red cells are placed in hypotonic solutions

120
Osmotic Fragility Test (OF)
Decreased Surface: Volume Ratio “Easy
to Lyse”
Increased Surface: Volume Ratio “Hard to Lyse”

121
B. Defect red cell metabolism (Enzyme defect)
• G6PD deficiency – G6PD is the only source of NADPH in red cell – NADPH is reduced for the production of reduced
Glutathione. – Hgb and RBC membrane are usually protected from
oxidant stress by reduced glutathione (GSH)– In G6PD deficiency NADPH and GSH synthesis is
impaired, rendering the red cells vulnerable to oxidant stress.
– Most individuals with G6PD are asymptomatic except during oxidant stress resulting from drugs or other causes.

122
B. Hemolytic Anemias due to Enzyme Defects
• Inherited enzyme deficiencies that lead to premature RBC death

123
PK Deficiency
Echinocytes
Hemolytic Anemias due to Enzyme Defects
• PK deficiency– ↓ATP impairs cation pump– Severe hemolytic anemia– Echinocytes
• G-6-PD Deficiency– Unable to protect Hgb due to
decreased NADPH – No clinical problems unless
exposed to oxidants– Exposure to oxidants induce
Heinz body formation and RBC destruction Normal RBCs if no
exposure to oxidant
G-6-PD Deficiency

124
G-6-PD deficiency after exposure to oxidant
Heinz bodies - denatured Hgb Supravital stain
G-6-PD deficiency Hemolytic episode
Damaged RBCs Wright’s stain
G-6-PD Deficiency• Blood findings after oxidant exposure:
– Mod to severe anemia– Schistocytes, spherocytes due to pitting out of Heinz
bodies by spleen• Enzyme assay

125
Target cells/Codocytes
C. Normocytic anemias due to hemoglobinopathies
• Inherited hemoglobin defect with production of structurally abnormal globin chains;– All have target cells
• Beta chain amino acid substitution = variant Hgb – Hgb S = valine substituted for glutamic acid @ 6th of ß – Hgb C = lysine substituted for glutamic acid @ 6th of ß
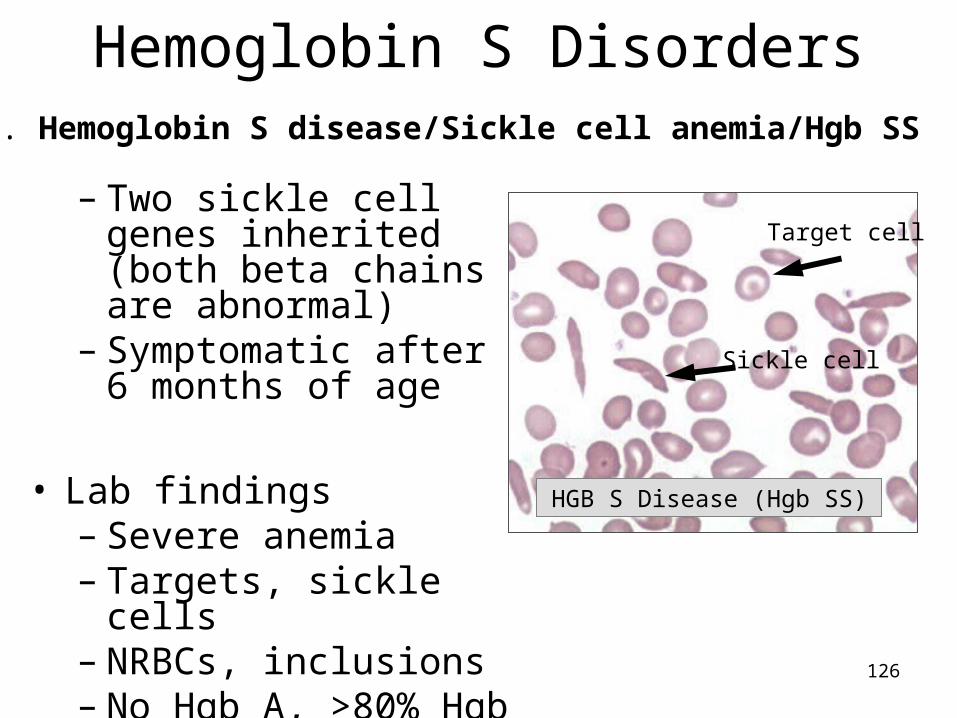
126
HGB S Disease (Hgb SS)
Sickle cell
Target cell
Hemoglobin S Disorders
– Two sickle cell genes inherited (both beta chains are abnormal)
– Symptomatic after 6 months of age
• Lab findings– Severe anemia – Targets, sickle cells– NRBCs, inclusions– No Hgb A, >80% Hgb S,
↑ F
A. Hemoglobin S disease/Sickle cell anemia/Hgb SS

127
Target cells only NO Sickle cells
HGB S Trait (Hgb SA)
Hemoglobin S Disorders
– One sickle cell gene inherited
• Lab findings– Asymptomatic, targets only– No anemia or sickle cells– ~60% Hgb A, ~40% Hgb S
• Potential problems if hypoxic
B. Hemoglobin S trait/Sickle cell trait/Hgb SA

128
C crystals
HGB C Disease (Hgb CC)
Target cell
Hemoglobin C Disorders
• Lab findings– Mild anemia – Many target cells – Intracellular C crystals– No Hgb A, >90% Hgb C– Decreased OF
A. Hemoglobin C disease/Hgb CCTwo C genes inherited (both β chains are abnormal)C crystals polymerize differently and look like blocky Hgb packed rods in the red cells....intracellular.

129
HGB C Trait (Hgb CA)
Target cells only NO C crystals
Hemoglobin C Disorders
B. Hemoglobin C trait/Hgb CA– One C gene inherited
• Lab findings– Asymptomatic, no anemia– Targets, no C crystals– ~60% Hgb A, ~40% Hgb C– Normal Hgb A2 and F

130
SC Crystals
Target cells
HGB SC Disease (Hgb S & Hgb C)
Hemoglobin SC Disease
• Lab findings– Intermediate in severity
between Hgb SS & SA – Several target cells – Many SC crystals– No Hgb A, ~50% Hgb
S, ~50% Hgb C, ↑ F
Hemoglobin SC disease/Hgb SC• One sickle gene and one C gene inherited• Double heterozygote‑ inherit sickle gene (S) from one parent and C gene from other parent; • Both β chains are abnormal

131
2. Acquired hemolytic anemia
• A variety of acquired conditions result in shortened survival of previously normal red cells. These include immune mediated destruction, red cell fragmentation disorders, acquired membrane defects, spleen effects
• Result of extrinsic causes– Immune HA; warm HIHA, cold AIHA– Drug associated– Infection associated

132
Spherocytes & polychromasia
Blood
Warm Autoimmune HA (WAIHA)• Altered immune response causes production of an
IgG warm autoantibody against ‘self’ RBC antigens– Antibody/complement attaches to RBC antigen…
partially phagocytosed (loss of membrane) spherocytes
• Cause: Primary (idiopathic) or secondary to disease

133
Ingestion of coated RBC
RBC
Electron Microscopy
Blood
Monocyte with ingested RBC
RBC
Warm Autoimmune HA (WAIHA)
• Lab findings– Mod to severe anemia, spherocytes, high MCHC– Erythrophagocytosis – Looks similar to H spherocytosis but positive DAT– Increased OF, bilirubin– Erythroid hyperplasia

134
50x
RBC Agglutination
100x
Cold Autoimmune HA (CAIHA)• Altered immune response causes production of an IgM
cold autoantibody against ‘self’ RBC antigens– Antibody/C3 attaches to RBC antigen agglutination
(lysis by complement or macrophage)• Primary (idiopathic) or secondary to disease

135
Cold Autoimmune HA (CAIHA)
• Lab findings– Agglutination of red cells in
extremities....ears, toes, nose tissue damage gangre
– Severity varies with seasons….avoid the cold
– IgM antibodies cause RBC agglutination
– Reticulocytosis
– Positive Direct Antiglobulin Test (detects complement)

136
Hemolytic Transfusion Reaction
• Incompatible blood transfusion
– Recipient has antibodies to antigens on the donor red cells received
– Donor cells are destroyed
• ABO worst
– Intravascular hemolysis that is complement-induced lysis…immediate
– Can be life-threatening

137
Hemolytic Disease of the Newborn • Caused by maternal IgG antibodies directed against
baby RBC antigens– Antibodies cross placenta and destroy fetal red
cells• HDN due to Rh incompatibility
– Rh negative mother forms Rh antibody after exposure– HDN due to Rh
• Sever anemia• Many nucleated red cells
• HDN due to ABO incompatibility – Mother’s ABO blood type is O; baby is type A or B– HDN due to ABO
• Mild, no anemia• Spherocytosis

138
Schistocytes
Fibrin Strands
RBC
RBC fragmentation on fibrin strands
Hemolytic Anemias due to Trauma• Fragmentation syndromes…most common finding on smear
are schistocytes; anemia varies• Types of trauma
– Mechanical…prosthetic heart valves/cardiac abnormalities – Microangiopathic (MAHA)…small vessels
(DIC.........bleeding) – March hemoglobinuria…forceful contact…. Schistocytes

139
Normocytic/normochromic Hemolytic Anemias due to Trauma –
Fragmentation Syndromes
A.When RBCs are exposed to excessive trauma within the cardiovascular system, they may undergo fragmentation and lysis.Schistocytes are the most common finding on the smear; RBC destruction tests are abnormal. Severe trauma causes intravascular hemolysis.
B.Three types of trauma: 1. Mechanical trauma
A. Prosthetic heart valves or cardiac abnormalities fragment red cells. B. Mod to severe anemia with schistocytes and polychromasia.

140
Normocytic/normochromic Hemolytic Anemias due to Trauma –
2. Microangiopathic hemolytic anemia (MAHA) – trauma occurs in small vessels
A. Disseminated Intravascular Coagulation (DIC) is a widespread clotting disorder initiated by conditions such as OB obstetrical) complications or sepsis. In DIC, clotting factors and platelets form fibrin fibrin deposited in the microvessels fragment red cells. See anemia with schistocytes on smear, decreased platelets and depletion coagulation factors leads to severe bleeding; can be fatal..

141
2. Microangiopathic hemolytic anemia (MAHA) cont’d……..
B. Hemolytic uremic syndrome (HUS) - most often occurs in children following GI infection (E. coli); noted for renal failure fibrin damages kidney; hemolytic anemia with echinocytes and schistocytes, decreased platelets; often requires dialysis; can be fatal

142
Normocytic/normochromic Hemolytic Anemias due to Trauma –
3. March Hemoglobinuria A. Transient, occurs after forceful contact of
body with hard surfaces....joggers, soldiers after long march, bongo drum players.
B. Hemoglobinuria; schistocytes may be present on smear.
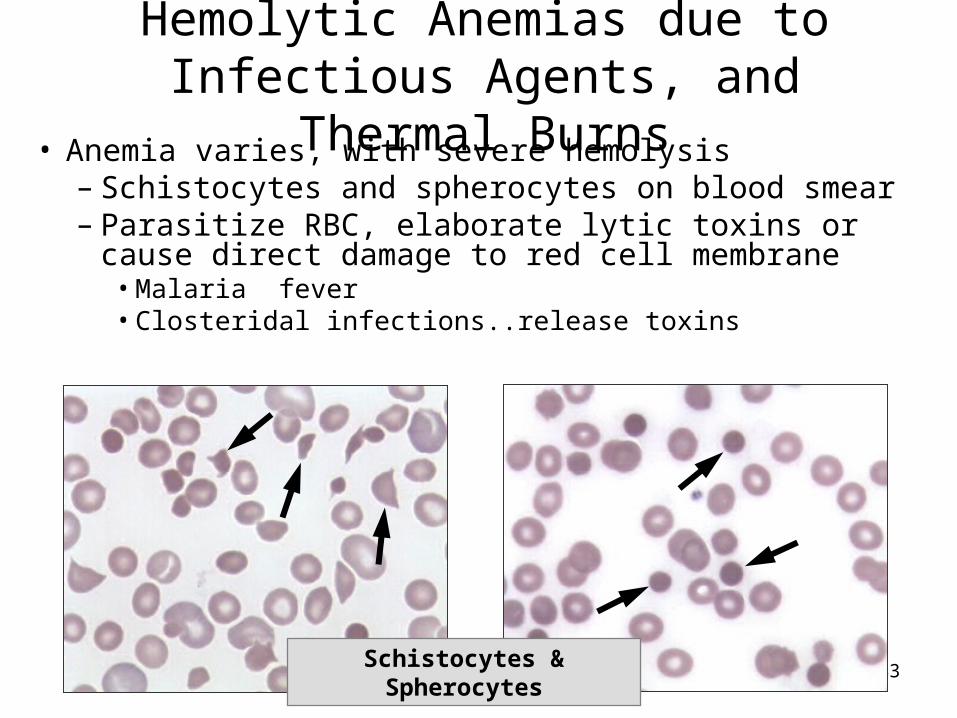
143Schistocytes & Spherocytes
Hemolytic Anemias due to Infectious Agents, and Thermal Burns
• Anemia varies, with severe hemolysis – Schistocytes and spherocytes on blood smear– Parasitize RBC, elaborate lytic toxins or cause direct
damage to red cell membrane• Malaria fever• Closteridal infections..release toxins

144
Normocytic/normochromic Hemolytic Anemia due to Infectious Agents
A. Malarial infection
1. Damage to membrane occurs when parasite is pitted out of red cell by splenic macrophages or the entire RBC is removed; chills and fever as red cells rupture.
2. P. falciparum causes severe hemolysis called Blackwater fever.

145
Normocytic/normochromic Hemolytic Anemia due to Infectious Agents
B. Clostridial infections - Clostridia elaborate toxins which damage RBC membrane causing severe intravascular hemolysis.
Both malaria and clostridial infections are characterized by schistocytes and spherocytes on the blood smear; other infectious organisms can cause hemolysis, e.g. Toxoplasma, Bartonella, Babesia, Ehrlichia.

146
END OF ANEMIA
















![The Hematologist - Final PDF of September October 2010[1]](https://static.fdocuments.us/doc/165x107/577d35911a28ab3a6b90cc96/the-hematologist-final-pdf-of-september-october-20101.jpg)

