
Learning the heuristic distribution by an evolutionary hyper-heuristic
date post
20-Dec-2015Category
view
219download
0
.
Class 4: Sequence Alignment II
Gaps, Heuristic Search

Alignment with Gaps – Example
1
2
A A C — A A T T A A G — A C T A C —G T T C A T G A C
A — C G A — T T A — G C A C — A C T G — T — A — G A —
A A C A A T T A A G A C T A C G T T C A T G A C — — —
A A C A A T T — — — — — — — — G T T C A T G A C G C A

Gaps
Both alignments have the same number of matches and spaces but alignment 2 seems better
Definition: A gap is any maximal, consecutive run of spaces in a single string. The length of a gap = the number of spaces in it
Example 1 has 11 gaps, example 2 only 2 gaps Idea: develop alignment scores that take gaps (not
spaces) into account

Biological Motivation
Number of mutational events: A single gap – due to a single event that
removed a number of residues Each gap – due to distinct, independent event
Protein structure: Protein secondary structure consists of alpha
helices, beta sheets and loops Loops of varying size can lead to very
similar structure

Biological Motivation

cDNA Mataching
cDNA is the sequence after splicing (introns have been removed) and editing
We expect regions of high similarity, separated by long gaps

Gap Penalty Models (I)
Constant model Gives each gap a constant score, spaces are free Maximize: Time: O(mn) Works well with cDNA matching
Affine model Penalty for starting a gap + penalty for each additional space Each gap costs: Wg + qWs
Maximize: Time: O(mn) Widely used
( [ ], [ ]) gaps gs i t j N W
( [ ], [ ]) gaps g spaces ss i t j N W N W

Gap Penalty Models (II)
Convex model Each extra space contributes less penalty Gap function is convex in its length Example: Ws + log(q)
Time O(mnlogm) A better model of biology
General model The weight of a gap is some arbitrary w(q) Time O(mn2 + nm2)

Example Revised
1
2
A A C — A A T T A A G — A C T A C —G T T C A T G A C
A — C G A — T T A — G C A C — A C T G — T — A — G A —
A A C A A T T A A G A C T A C G T T C A T G A C — — —
A A C A A T T — — — — — — — — G T T C A T G A C G C A

Indel Model
Score: -6
Score: -6
Scoring Parameters
Match: +1
Indel: -2
1
2
A A C — A A T T A A G — A C T A C —G T T C A T G A C
A — C G A — T T A — G C A C — A C T G — T — A — G A —
A A C A A T T A A G A C T A C G T T C A T G A C — — —
A A C A A T T — — — — — — — — G T T C A T G A C G C A

Constant Model
Scoring Parameters
Match: +1
Open gap: -2
Score: -6
Score: 12
1
2
A A C — A A T T A A G — A C T A C —G T T C A T G A C
A — C G A — T T A — G C A C — A C T G — T — A — G A —
A A C A A T T A A G A C T A C G T T C A T G A C — — —
A A C A A T T — — — — — — — — G T T C A T G A C G C A
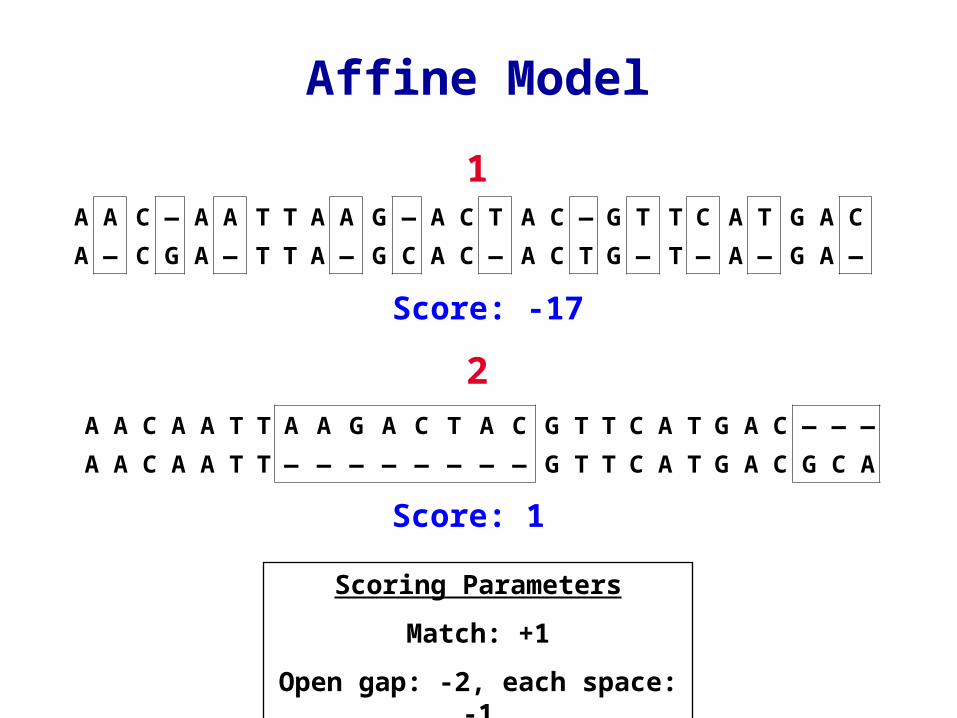
Affine Model
Scoring Parameters
Match: +1
Open gap: -2, each space: -1
Score: -17
Score: 1
1
2
A A C — A A T T A A G — A C T A C —G T T C A T G A C
A — C G A — T T A — G C A C — A C T G — T — A — G A —
A A C A A T T A A G A C T A C G T T C A T G A C — — —
A A C A A T T — — — — — — — — G T T C A T G A C G C A

Convex Model
Scoring Parameters
Match: +1
Open gap: -2, gap length: -logn
Score: -6
Score: ~7
1
2
A A C — A A T T A A G — A C T A C —G T T C A T G A C
A — C G A — T T A — G C A C — A C T G — T — A — G A —
A A C A A T T A A G A C T A C G T T C A T G A C — — —
A A C A A T T — — — — — — — — G T T C A T G A C G C A

Affine Weight Model
We divide all possible alignments of the prefixes s[1..i] and t[1..j] into 3 types
s: i
t: j
s: i-----
t: j
s: i
t: j-----:),(
:),(
:),(
jiC
jiB
jiA

Affine Weight Model
s
sg
s
sg
WjiC
WWjiAjiC
WjiB
WWjiAjiB
jtisjiC
jtisjiB
jtisjiA
jiA
),1(
),1(),(
)1,(
)1,(),(
])[],[()1,1(
])[],[()1,1(
])[],[()1,1(
),(
Recurrence relations:

Affine Weight Model
sg
sg
jWWjCjA
iWWiBiA
),0(),0(
)0,()0,(Initial condition:
Optimal alignment:
Complexity:
Time: O(mn)
Space: O(mn)
)},(),,(),,(max{),( mnCmnBmnAnmV

Affine Weight Model
This model has a natural explanation as a finite state automata
A
B
C
S(i,j)
Wg+Ws
S(i,j)
Ws
Wg+Ws
S(i,j)
Ws

Alignment in Real Life
One of the major uses of alignments is to find sequences in a “database”
Such collections contain massive number of sequences (order of 106)
Finding homologies in these databases with the standard dynamic programming can take too long
Example: query protein : 232 AAs NR protein DB: 2.7 million sequences; 748 million AAs
m*n = ~ 1.7 *1011 cells !

Heuristic Search
Instead, most searches rely on heuristic procedures
These are not guaranteed to find the best match Sometimes, they will completely miss a high-
scoring match
We now describe the main ideas used by some of these procedures Actual implementations often contain additional tricks
and hacks

Basic Intuition
The main resource consuming factor in the standard DP is decision of where the gaps are. If there were no gaps, life was easy!
Almost all heuristic search procedures are based on the observation that real-life well-matching pairs of sequences often do contain long strings with gap-less matches.
These heuristics try to find significant local gap-less matches and then extend them.

Banded DP
Suppose that we have two strings s[1..n] and t[1..m] such that nm
If the optimal global alignment of s and t has few gaps, then path of the alignment will be close to the diagonal
s
t

Banded DP
To find such a path, it suffices to search in a diagonal region of the matrix
If the diagonal band has presumed width a, then the dynamic programming step takes O(an)
Much faster than O(n2) of standard DP in this cases
t a

Banded DP
Problem (for local alignment): If we know that t[i..j] matches the query s[p..q],
then we can use banded DP to evaluate quality of the match
However, we do not know i,j,p,q !
How do we select which sub-sequences to align using banded DP?

FASTA Overview
Main idea:
Find (fast!) “good” diagonals and extend them to complete matches
Suppose that we have a relatively long gap-less local match (diagonal):
…AGCGCCATGGATTGAGCGA…
…TGCGACATTGATCGACCTA… Can we find “clues” that will let us find it quickly?

Signature of a Match
Assumption: good matches contain several “patches” of perfect matches
AGCGCCATGGATTGAGCGA
TGCGACATTGATCGACCTA
s
t

FASTA
Given s and t, and a parameter k Find all pairs (i,j) such that s[i..i+k] and t[j..j+k]
match perfectly Locate sets of pairs that are on the same diagonal
by sorting according to i-j thus… Locating diagonals that contain
many close pairs. This is faster than O(nm) !
s
t
i i+k
jj+k

FASTA
Extend the “best” diagonal matches to imperfect (yet ungapped) matches, compute alignment scores per diagonal. Pick the best-scoring matches.
Try to combine close diagonals to potential gapped matches, picking the best-scoring matches.
Finally, run banded DP on the regions containing these matches, resulting in several good candidate alignments.
Most applications of FASTA use very small k (2 for proteins, and 4-6 for DNA)

BLAST Overview
FASTA drawback is its reliance on perfect matches
BLAST (Basic Local Alignment Search Tool) uses similar intuition, but relies on high scoring matches rather than exact matches
Given parameters: length k, and threshold T Two strings s and t of length k are a high scoring
pair (HSP) if d(s,t) > T

High-Scoring Pair
Given a query string s, BLAST construct all words w (“neighborhood words”), such that w is an HSP with a k-substring of s.
Note: not all k-mers have an HSP in s

BLAST: phase 1
Phase 1: compile a list of word pairs (k=3) above threshold T
Example: for the following query:
…FSGTWYA… (query word is in green)
A list of words (k=3) is: FSG SGT GTW TWY WYA YSG TGT ATW SWY WFA FTG SVT GSW TWF WYS

GTW 6,5,11 22neighborhood ASW 6,1,11 18word hits ATW 0,5,11 16> threshold NTW 0,5,11 16
GTY 6,5,2 13 GNW 10
neighborhood GAW 9word hitsbelow threshold
(T=11)
scores
BLAST: phase 1

BLAST: phase 2
Search the database for perfect matches with neighborhood words. Those are “hits” for further alignment.
We can locate seed words in a large database in a single pass, given the database is properly preprocessed (using hashing techniques).

Extending Potential Matches
Once a hit is found, BLAST attempts to find a local alignment that extends it.
Seeds on the same diagonal tend to be combined (as in FASTA) s
t

An improvement: look for 2 HSPs on close diagonals
Extend the alignment between them Fewer extensions considered There is a version of BLAST,
involving gapped
extensions. Generally faster then FASTA,
arguably better.
Two HSP diagonal
s
t

Blast Variants
blastn (nucleotide BLAST)
blastp (protein BLAST)
tblastn (protein query, translated DB BLAST)
blastx (translated query, protein DB BLAST)
tblastx (translated query, translated DB BLAST)
bl2seq (pairwise alignment)

Biological Databases
Today, most of the biological information can be freely accessed on the web.
One can: Search for information on a known gene Check if a sequence exists in a database Find a homologous protein, helping us guess:
Structure Function

Databases and Tool
Important gateways: National Center for Biotechnology (GenBank)
http://www.ncbi.nlm.nih.gov/
European Bioinformatics Institue (EMBL-Bank) http://www.ebi.ac.uk/
Expert Protein Analysis System (SwissProt) http://www.expasy.org/
→ Different tools and DBs to allow biologists a rich suite of queries

Database Types
Nucleotide DBs (GenBank, EMBL-Bank): Contain any and every type of DNA fragment:
Full cDNA, ESTs, repeats, fragments
“Dirty” and redundant
Protein DBs (SwissProt): Contain amino-acid sequences for full proteins High quality, strict screening process Lots of annotated information on each protein







![Informed [Heuristic] Search - University of Delawaredecker/courses/681s07/pdfs/04-Heuristic...Informed [Heuristic] Search Heuristic: “A rule of thumb, simplification, or educated](https://static.fdocuments.us/doc/165x107/5aa1e13c7f8b9a84398c48b6/informed-heuristic-search-university-of-delaware-deckercourses681s07pdfs04-heuristicinformed.jpg)










