
Fast Heuristic Alignment
35
QB LECTURE #3: Fast Heuristic Alignment Nov. 19, 2015 Adam Siepel
Transcript of Fast Heuristic Alignment

QB LECTURE #3:Fast Heuristic Alignment
Nov. 19, 2015Adam Siepel

Plan for Today
Local alignment (Smith-Waterman)BLASTShort-read alignmentThe Burrows-Wheeler Transform
2

Local vs. Global Alignment
• The Global Alignment Problem tries to find the best end-to-end alignment between the two strings
– Only applicable for very closely related sequences
• The Local Alignment Problem tries to find pairs of substrings with highest similarity.
– Especially important if one string is substantially longer than the other
– Especially important if there is only a distant evolutionary relationship

Global vs Local Alignment Schematic
T
S
(0,0)
(n,m)
Max score for local
alignment
Nathan Edwards
Global alignment
always ends in the corner

Local vs. Global Alignment (cont’d)
• Global Alignment
• Local Alignment—better alignment to find conserved segment
--T—-CC-C-AGT—-TATGT-CAGGGGACACG—A-GCATGCAGA-GAC | || | || | | | ||| || | | | | |||| | AATTGCCGCC-GTCGT-T-TTCAG----CA-GTTATG—T-CAGAT--C
tccCAGTTATGTCAGgggacacgagcatgcagagac ||||||||||||
aattgccgccgtcgttttcagCAGTTATGTCAGatc
bioalgorithms.info

Smith-Waterman
If the expected score of a random match is negative, only a simple change is required
No source/sink: trace back from largest F(i,j)
F (i, j) = max
⎧
⎪
⎪
⎪
⎨
⎪
⎪
⎪
⎩
0
F (i − 1, j − 1) + sxi,yj
F (i − 1, j) − d
F (i, j − 1) − d
F (i, j) = 0, i = 0 or j = 0
6

Smith-Waterman Graph Interpretation
AA G T C C
G
C
C
T
GTCCG-CC
⇐⇒
7

Affine Gap PenaltiesHarder but still possible in O(nm)Key insight: penalty of new gap depends only on whether previous column of alignment is match (d), gap in x (e), or gap in y (e)The solution is to keep track of state by adding a 3rd dimension to the recurrence This can be represented by replacing F with three tables: M, Ix, and Iy
8

• Rapidly compare a sequence Q to a database to find all sequences in the database with an score above some cutoff S. – Which protein is most similar to a newly sequenced one? – Where does this sequence of DNA originate?
• Speed achieved by using a procedure that typically finds “most” matches with scores > S. – Tradeoff between sensitivity and specificity/speed • Sensitivity – ability to find all related sequences • Specificity – ability to reject unrelated sequences
Basic Local Alignment Search Tool
(Altschul et al. 1990)

Seed and Extend FAKDFLAGGVAAAISKTAVAPIERVKLLLQVQHASKQITADKQYKGIIDCVVRIPKEQGV F D +GG AAA+SKTAVAPIERVKLLLQVQ ASK I DK+YKGI+D ++R+PKEQGV
FLIDLASGGTAAAVSKTAVAPIERVKLLLQVQDASKAIAVDKRYKGIMDVLIRVPKEQGV
• Homologous sequence are likely to contain a short high scoring word pair, a seed. – However, BLAST *doesn't* make explicit guarantees
• BLAST then tries to extend high scoring word pairs to compute maximal high scoring segment pairs (HSPs). – Heuristic algorithm but evaluates the result statistically.

11
We have dynamic programming, what else do we need?
Running time: O(n2) on two sequences of length n
How much is that in practice?
(simple implementation, random sequences, modern desktop computer)
n time
100 0.0008s
1,000 0.08s
10,000 8s
100,000 13m (*)
1,000,000 22h (*)
10,000,000 3months (*)
100,000,000 25years (*)
We need more efficient algorithm, particularly for comparative genomics
Memory: basic implementation O(n2), but can be done in O(n)
4-a
Heuristic alignment
• Trade sensitivity for speed (some alignments not found)
• Reduce the search to “promising” parts of the matrix
Heuristic local alignment
BLASTN [Altschul et al 1990], FASTA [Pearson 1988]
• Find short exact matches of length w (hits)
• Extend hits along diagonal to ungapped alignments
• Connect alignments on nearby diagonals to gapped alignment
• Possibly optimize by dynamic programming
5
Heuristic local alignment
Example: Take w = 2 (hits are matching words of length 2)
C A G T C C T A G A
0 0 0 0 0 0 0 0 0 0 0
C 0 1 0 0 0 1 1 0 0 0 0
A 0 0 2 1 0 0 0 0 1 0 0
T 0 0 1 1 2 1 0 1 0 0 0
G 0 0 0 2 1 0 0 0 0 1 0
T 0 0 0 0 3 2 1 1 0 0 0
C 0 1 0 0 0 4 3 0 0 0 0
A 0 0 2 1 0 3 3 2 1 0 1
T 0 0 1 1 2 2 2 4 3 2 1
A 0 0 1 0 1 1 1 3 5 4 3
1. find hits
2. ungapped
3. gapped
6
How to find short exact matches?
• Build a “dictionary” of words of length w from the first sequence
• Look up each word from the second sequence in the dictionary
Example: CAGTCCTAGA vs CATGTCATA
Dictionary:
AG 2, 8
CA 1
CC 5
CT 6
GA 9
GT 3
TA 7
TC 4
Lookup:
CA → 1
AT → --
TG → --
GT → 3
TC → 4
CA → 1
AT → --
TA → 7
7

BLAST - Algorithm -
• Step 1: Preprocess Query Compile the short-high scoring word list from query. The length of query word, w, is 3 for protein scoring Threshold T is 13

BLAST - Algorithm -
• Step 2: Construct Query Word Hash Table
Query: LAALLNKCKTPQGQRLVNQWIKQPLMD
Word list
Hash Table
BLAST - Algorithm -
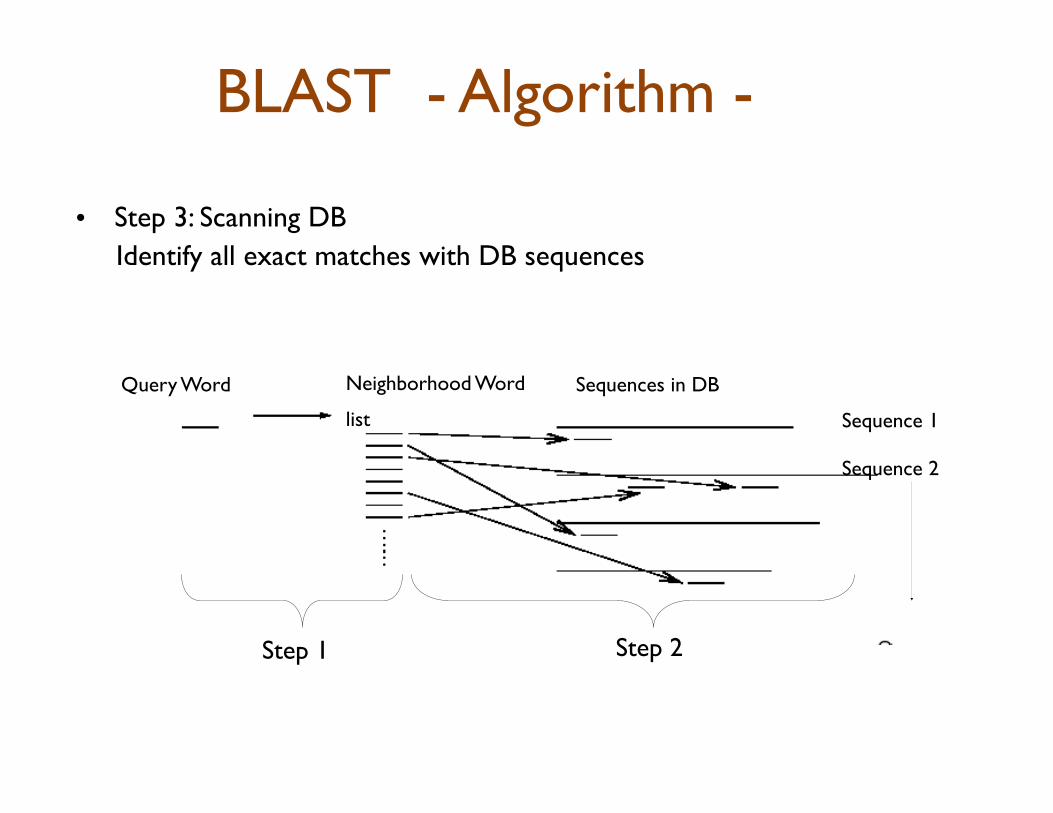
• Step 3: Scanning DB Identify all exact matches with DB sequences
Query Word Neighborhood Word
list
Sequences in DB
Step 1 Step 2
Sequence 1
Sequence 2

BLAST - Algorithm -
• Step 4 (Search optimal alignment) For each hit-word, extend ungapped alignments in both directions. Let S be a score of hit-word
• Step 5 (Evaluate the alignment statistically) Stop extension when E-value (depending on score S) become less than
threshold. The extended match is called High Scoring Segment Pair.
E-value = the number of HSPs having score S (or higher) expected to occur by chance. ! Smaller E-value, more significant in statistics Bigger E-value , by chance
E[# occurrences of a string of length m in reference of length L] ~ L/4m

Expected Occurrences The expected number of occurrences (e-value) of a given sequence in a
genome depends on the length of the genome and inversely on the length of the sequence – 1 in 4 bases are G, 1 in 16 positions are GA, 1 in 64 positions are GAT, … – 1 in 16,384 should be GATTACA – E=n/(4m) [183,105 expected occurrences]

Very Similar Sequences
Query: HBA_HUMAN Hemoglobin alpha subunit Sbjct: HBB_HUMAN Hemoglobin beta subunit
Score = 114 bits (285), Expect = 1e-26 Identities = 61/145 (42%), Positives = 86/145 (59%), Gaps = 8/145 (5%)
Query 2 LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF------DLSHGSAQV 55 L+P +K+ V A WGKV + E G EAL R+ + +P T+ +F F D G+ +V Sbjct 3 LTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKV 60
Query 56 KGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPA 115 K HGKKV A ++ +AH+D++ + LS+LH KL VDP NF+LL + L+ LA H Sbjct 61 KAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGK 120
Query 116 EFTPAVHASLDKFLASVSTVLTSKY 140 EFTP V A+ K +A V+ L KY Sbjct 121 EFTPPVQAAYQKVVAGVANALAHKY 145

Quite Similar Sequences
Query: HBA_HUMAN Hemoglobin alpha subunit Sbjct: MYG_HUMAN Myoglobin
Score = 51.2 bits (121), Expect = 1e-07, Identities = 38/146 (26%), Positives = 58/146 (39%), Gaps = 6/146 (4%)
Query 2 LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF------DLSHGSAQV 55 LS + V WGKV A +G E L R+F P T F F D S + Sbjct 3 LSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDL 62
Query 56 KGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPA 115 K HG V AL + + L+ HA K ++ + +S C++ L + P Sbjct 63 KKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHPG 122
Query 116 EFTPAVHASLDKFLASVSTVLTSKYR 141 +F +++K L + S Y+ Sbjct 123 DFGADAQGAMNKALELFRKDMASNYK 148

Not similar sequences
Query: HBA_HUMAN Hemoglobin alpha subunit Sbjct: SPAC869.02c [Schizosaccharomyces pombe]
Score = 33.1 bits (74), Expect = 0.24 Identities = 27/95 (28%), Positives = 50/95 (52%), Gaps = 10/95 (10%)
Query 30 ERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAH 89 ++M ++P P+F+ +H + + +A AL N ++DD+ +LSA D Sbjct 59 QKMLGNYPEV---LPYFNKAHQISL--SQPRILAFALLNYAKNIDDL-TSLSAFMDQIVV 112
Query 90 K---LRVDPVNFKLLSHCLLVTLAAHLPAEF-TPA 120 K L++ ++ ++ HCLL T+ LP++ TPA Sbjct 113 KHVGLQIKAEHYPIVGHCLLSTMQELLPSDVATPA 147

Blast Versions
Program Database Query
BLASTN Nucleotide NucleotideBLASTP Protein Protein
BLASTX Protein Nucleotide translated into protein
TBLASTN Nucleotide translated into protein Protein
TBLASTX Nucleotide translated into protein
Nucleotide translated into protein

NCBI Blast
• Nucleotide Databases – nr: All Genbank – refseq: Reference
organisms – wgs: All reads
• Protein Databases – nr: All non-redundant
sequences – Refseq: Reference proteins

Parameters
• Larger values of w increases the number of neighborhood words, but decreases the number of chance matches in the database. – Increasing w decreases sensitivity.
• Larger values of T decrease the overall execution time, but increase the chance of missing a MSP having score ≥ S. – Increases T decreases the sensitivity

23
Performance of heuristic local alignment
Tradeoff: sensitivity vs. running time
Smallw
many spurious hits, slow
Largew
many alignments not found
12
Spaced seeds – different hashing strategy
PatternHunter [Ma, Tromp, Li 2002]
Spaced seed: required configuration of matches in a hit
Example:
“match—match—don’t care—match” denoted as 1101
GTGGTGCTCTCTGACAAAGCC| || || ||| ||||
ATTGTTCTTAATGAGAAAGAA1101 1101
1101
BLASTN hit definition (11 consecutive matches)
equivalent to seed 11111111111
13
Not all spaced seeds are equal
Weight of a seed: number of required matches
Every dot: performance of one seed in a simple probabilistic model
0 0.2 0.4 0.6 0.8 1
Predicted sensitivity
What portion of alignments found?
0
2E-06
4E-06
Pre
dic
ted
fals
e p
osi
tive
rate
How
man
y s
pu
riou
s h
its?
Weight 11
Weight 10
Weight 9
14
Why are spaced seeds more sensitive?
Assume: alignment length 64, probability of match 0.7 at every position
Generate 40 random alignments, find hits
11111111111
Sn.: 14/40, hits: 46
111010010100110111
Sn.: 18/40, hits: 35
15

Short Read Alignment
Can we do better when mapping short reads to a reference genome sequence?Yes! This is a well-defined special case of the alignment problem, which can be solved very efficientlyBecause reads are short, numbers of mismatches, indels can be bounded (typically at most 2-3 mismatches, 1 indel in 28–35 base read)
24

Widely Used ProgramsSOAP (SOAP2, SOAP3). Very fast, somewhat approximate.MAQ. Has full error model, provides quality scores. Slower.Bowtie. Very memory efficient (Burrows Wheeler Transform)BWA (MAQ with Burrows-Wheeler Transform)GEMStampy…
25

Typical ApproachIndex k-mers, either for query sequences or reference genomeMake use of bounds on mismatches/indels for efficient indexingFor example: 32mers with ≤2 mismatches. At least two of four 8-mers must map exactly. Consider all possibilities: (1001,1010,1100,0011,0101,0110). Then identify pairs, extend to full 32mer alignmentsIssues: index query or ref, mate pairs, multiple mappings, memory efficiency
26

Burrows-Wheeler Transform
Clever data structure allowing fast lookup of substrings with a small memory footprintFinds permutation of original string with excellent compression properties. Key is transformation is easily invertibleApplied to an FM-index, a suffix-array-like data structure allowing fast substring lookupsAllows indexing of entire genomes with ~1GB memory footprints (workstation size)Essentially combines the speed of a suffix array (O(log n)) with the size of a brute-force analysis (O(n))
27

BWT: Compressible permutation of input text
28
Wikipedia

How Does it Work?29
Wikipedia

How to Invert?30
Wikipedia

Optimization Tricks
Don’t explicitly represent table; use pointers to original stringCustomized sorting algorithms for encodingNo need to explicitly represent EOFMost importantly, in decoding, don’t need to explicitly sort!
31

Burrows-Wheeler Transform
• Recreating T from BWT(T) – Make use of an auxiliary last-to-first (LF) column
mapping function (details omitted but const time) – Start in the first row and apply LF repeatedly,
accumulating predecessors along the wayOriginal T

BWT & Suffix ArraysThere is a close relationship between the BWT and suffix arraysNotice that the full BWT table contains the sorted suffixes of the input textUsing various data-structure tricks, it is possible to access this suffix array implicitly and find the indices of all instances of a query string from the BWTThis is essentially what is known as the “FM-index” and used for fast BWT-based read mapping
33

Bowtie Overview
3. Evaluate end-to-end match
2. Lookup each segment and prioritize
1. Split read into segments

That’s All35


















