







Languages
Pages
Legal


Abstract approved:
AN ABSTRACT OF THE THESIS OF
JOSE VELMONTE ZERRUDO for the DOCTOR OF PHILOSOPHY(Name of student) (Degree)
Wood Sciencein (Wood Chemistry) presented on -11,4 - 7, 7c-rn_
(Major) (Date)
Title: DOUGLAS-FIR BARK; WATER-SOLUBLE CARBOHYDRATES
AND ALKALINE DEGRADATION OF A XYLAN
Signature redacted for privacy.
Murray L. Laver
The inner bark of a Douglas-fir [Pseudotsuga menziesii (Mirb.)
Franco] was successively extracted with ethanol-water (4:1 qv),
benzene-ethanol (2:1 v/v), hot water, 0. 5% ammonium oxalate and
acidified sodium chlorite solution. Free glucose was detected in
the ethanol-water extract but the benzene-ethanol extract contained
no carbohydrates.
The hot-water-soluble fraction contained starch, protein,
tannins and hemicelluloses. The a-amylose portions of the starch,
the proteins, and the tannins were removed by a series of enzyme
hydrolyses followed by extensive dialyses. The polysaccharides
which remained were composed of the following ratio of sugar res-
idues: glucose (2. 9), arabinose (1. 3), galactose (1.0) and traces of
rhamnose, xylose and mannose. The glucose was considered to be from
amylopectin which did not hydrolyze with the a-amylase enzymes,

the arabinose and galactose were considered to be part of L-arabino-
D-galactan polysaccharides. Arabinogalactans have been found in
the hot-water-soluble extracts of wood and bark of several species
but have not been reported in the bark of Douglas-fir.
A xylan was isolated from the acidified sodium chlorite in-
soluble fraction (holocellulose). The xylan was composed of the
following ratio of sugar residues: xylose (4. 5), arabinose (1. 0),
glucuronic acid (1.0). The intrinsic viscosity of the polysaccharide
in molar diethylenediamine copper II reagent at 25° was 0. 42 dl/g
which corresponded to a degree of polymerization of 89. The
molecular weight of the xylan as analyzed by end-group analysis was
1.8 x 104 which, in combination with gas-liquid chromatographic
analysis, showed a degree of polymerization of 90. The molecular
size distribution of the xylan was not broad as shown by gel per-
meation chromatography.
The xylan was oxidized with periodate anion. It consumed
133. 5 moles of periodate anion and released 22. 5 moles of formic
acid per mole of xylan. The oxidized xylan was hydrolyzed and
analyzed by gas-liquid chromatography and showed the following
ratio of sugar residues: xylose (7. 0), arabinose (1. 0).
The above data are consistent with a polysaccharide xylan stru-c-
ture consisting of a backbone of 88 anhydro-D-xylopyranose units
hooked p-D-(1 -4) plus a reducing and non-reducing end-group on

the backbone. There are 20 anhydroglucuronic acid side chains and
20 anhydroarabinose side chains on these 90 units. At least ten of
the anhydroarabinose units are in the form of monoarabinose side
chains and up to 10 are in the form of arabinobiose or longer side
chains. A possible structure for the xylan has been proposed.
Alkaline degradation of the xylan in 0.1 N aqueous sodium
hydroxide at 100° showed a rate constant for end-group peeling of
k1 = k2 = 5.33 hour-1, and a termination rate constant of
k3 = 0.66 hour-1. The relatively high termination rate constant
compared to the peeling rate constant was demonstrated by the small
amount of degradation (19.81%) which actually occurred. The results
supported a possible reaction sequence involving mono- and di-anionic
end-group species as the intermediates leading to end-group elimi-
nation of reducing polysaccharides.

Douglas-fir Bark: Water-soluble Carbohydrates andAlkaline Degradation of a Xylan
by
Jose Velmonte Zerrudo
A THESIS
submitted to
Oregon State University
in partial fulfillment ofthe requirements for the
degree of
Doctor of Philosophy
June 1973

APPROVED:
Signature redacted for privacy.
Associate 'P rofessor of Forest Products Chemistryin charge of major
Signature redacted for privacy.
Head of Department of Forest Products
Signature redacted for privacy.
Dean of Graduate School.
Date thesis is presented '?/%`--t,^1---trat-et 7/, /5( 7 Z.-
Typed by Opal Grossnicklaus for Jose Velmonte Zerrudo

tn0
-Aoenpd Jo4 papepal ainT
euBis
ri,
Signature redacted for privacy.

ACKNOWLEDGEMENTS
I wish to express my sincere thanks to my major professor,
Dr. Murray L. Laver, for his ever present interest and guidance
in the research program and for his many constructive criticisms
in preparing the thesis manuscript.
Special thanks are extended to Mr. Chung-Hsien Chen, gradu-
ate student in the Department of Forest Products, Oregon State Uni-
versity, for his original isolation of the holocellulose, the starting
material for much of the research reported :rt this thesis.
Acknowledgement is also made to Dr: Robert L. Krahmer,
Associate Professor, Department of Forest Products, for the pho-
tograph used to produce Figure 1 (page 5 ) of the thesis and for his
counsel on the bark anatomy description.
Appreciation is also extended to the Fullbright-Hayes program
of the Department of State of the United States Government which
provided travel funds from and to the Philippines.
Special acknowledgement is made to Commissioner Manuel R.
Monsalud and the Forest Products Research and Industries Develop-
ment Commission of the Philippines for providing official time
to the author in order that he might complete the program.
This research was supported in part by the Environmen'al
Protection Agency of the United States Government under grant

number EP-00276-04, and in part by the Department of Forest
Products, Oregon State University.

TABLE OF CONTENTS
INTRODUCTION 1
HISTORICAL REVIEW 4
EXPERIMENTAL 35
A. Collection of Bark Samples 35
B. Sample Preparation and Solvent Extraction 35
Extraction of the Hot-Water-Soluble Solids 35Extraction of the )ylan 37
C. Characterization of the Hot-Water-Soluble Solids 39
Elemental Analyses for Nitrogen, Sulfur,Phosphorus, and the Halogens 39
Test for Tannins and Starch 41
Strong Acid Hydrolysis 41
Mild Acid Hydrolysis 42Qualitative Amino Acid Analysis by PaperChromatography 42Carbohydrate Analysis by Paper Chromatography 43
Purification of the Hot-Water-Soluble Solids 44Enzyme Hydrolysis to Remove Starch 46Enzyme Hydrolysis to Remove Protein 47Carbohydrate Analysis by Gas-Liquid-Chromatography 48
D. Characterization of the Xylan 50
Ash Determination of the Crude Xylan 50
Precipitation of Excess Barium and Dialysis of theCrude Xylan 51
Optical Rotation of the Xylan 52Qualitative Uronic Acid Analysis by Color Reaction 52
Paper Chromatography and Gas-Liquid Chromatog-raphy of the Xylan Hydrolyzate 53
Reducing End-Group Analysis (Somogyi Method) 54
Viscosity Measurements 56Gel Permeation Chromatography - Determinationof Molecular Weight Distribution 57
E. Periodate Oxidation of the Xylan 58
Acidity of the Xylan 58
Analysis of Periodate Consumed 59
Analysis of Formic Acid Released 59
Complete Hydrolysis of the Polyalcohol 60
F. Alkaline Degradation of the Xylan 61
1. Reaction in Sodium Hydroxide Solution 61

2. Phenol-Sulfuric Acid Method of Analysis of theXylan 62
IV. RESULTS AND DISCUSSION 64
A. Collection of Bark Samples 64B. Sample Preparation and Solvent Extraction 64
Extraction of the Hot-Water-Soluble Solids 64Extraction of the Xylan 67
C. Characterization of the Hot-Water-Soluble Solids 68Elemental Analysis for Nitrogen, Sulfur, Phosphorus,and the Halogens 68Test for Tannins and Starch 70
Strong Acid Hydrolysis 71
Mild Acid Hydrolysis 71Qualitative Amino Acid Analysis by PaperChromatography 72Carbohydrate Analysis by Paper Chromatography 74Purification of the Hot-Water-Soluble Solids 76
Enzyme Hydrolysis to Remove Starch 80Enzyme Hydrolysis to Remove Protein 81Carbohydrate Analysis by Gas-LiquidChromatography 84
D. Characterization of the Xylan Fraction 89Ash Determination of the Crude Xylan 89Precipitation of Excess Barium and Dialysis ofthe Crude Xylan 89
Optical Rotation of the Xylan 90Qualitative Uronic Acid Analysis by Color Reactions 90Paper Chromatography and Gas-LiquidChromatography of the Xylan Hydrolyzate 91Reducing End-Group Analysis (Somogyi Method) 96Viscosity Measurements 100
Gel Permeation Chromatography Determinationof Molecular Weight Distribution 108
E. Periodate Oxidation of the Xylan 111
Acidity of the Xylan 111
Analysis of Periodate Consumed 112Analysis of Formic Acid Released 117Complete Hydrolysis of the Polyalcohol 118
F. Alkaline Degradation of the Xylan 124Reaction in Sodium Hydroxide Solution 124Phenol-Sulfuric Acid Method of Analysis forthe Xylan 139

V. SUMMARY AND CONCLUSIONS 142
LITERATURE CITED 146

LIST OF FIGURES
Figure Page
Anatomy of Douglas-fir bark. 5
The classical base-catalyzed transformation of analdose (Lobry de Bruyn and Alberda van Ekenstein). 17
p Elimination in a carbohydrate under alkalineconditions. 18
Formation of two products after p elimination. 20
5, Alkaline degradation of a disaccharide (where Ma =mannopyranosyl). 21
The alkaline degradation of (1--4-4)-linkedand D-mannoglycans (where R = the remaining portionof the polysaccharide molecule). 22
The formation of stable, metasaccharinate end-groupsin (1-4-4) glycans (where R = the remaining portion ofthe polysaccharide molecule). 24
The formation of stable, ordinary saccharinate end-groups in (1-.4) glycans (where R = the remainingportion of the polysaccharide molecule). 25
Proposed mechanism for uronic acid decomposition. 29
Two dimensional paper chromatogram of the aminoacids in the hydrolyzate of the hot-water-solublesolids of Douglas-fir inner bark. Spray: ninhydrinin n-butanol. 73
Paper chromatogram of the acid hydrolyzate of thehot-water-soluble fraction of the inner bark ofDouglas-fir. Solvent: ethyl acetate-pyridine-water (8:2:1 v/v/v). 75
Two dimensional paper chromatogram of the aminoacids in the acid hydrolyzate of Fraction B. Spray:ninhydrin in n-butanol. 79

Figure Page
Paper chromatogram of the acid hydrolyzate ofFraction D. Solvent: ethyl acetate-pyridine-water(8:2:1, v/v/v). 83
Gas-liquid chromatographic separation of the alditolacetates from the acid hydrolyzate of Fraction B. 85
Gas-liquid chromatographic separation of alditolacetates from the acid hydrolyzates of Fraction D. 86
Spectrum of the reaction products from the treatmentof the xylan hydrolyzate with carbazole-sulfuric acid. 92
Paper chromatogram of the acid hydrolyzate of thexylan. Solvent: ethyl acetate-pyridine-water (8:2:1v/v/v). 93
Gas-liquid chromatogram of alditol acetates fromxylan. 95
Standard curve for Somogyi titration of xylose. 98
Relationship between the reduced viscosity and con-centration in three solvents of the xylan from Douglas-fir inner bark, at 25.00 ±0. 01° . 105
Gel permeation chromatography of xylan from theinner bark of Douglas-fir. 109
Oxidation of a branched xylan with periodate anion. 114
Rate of consumption of periodate on periodate oxida-tion of the xylan from Douglas-fir inner bark. 116
Paper chromatogram of the acid hydrolyzed polyalcoholfrom the oxidation products of the periodate oxidationof xylan from Douglas-fir inner bark. Solvent:ethyl acetate-pyridine-water (8:2:1 v/v/v). 119

_Figure
Gas-liquid chromatographic separation of the alditolacetates from the acid nydrolyzate of the polyalcohol(oxidation products of the periodate oxidation of ,;:ylo,n). 120
26. Proposed structure for a xylan from the inner barkDouglas -fir. 121
Reaction sequence leading to end-group eliminationof reducing polysaccharides in alkaline solutions. 126
Rate curves for the degradation of xylan in aqueoussodium hydroxide at pH values of 12.0 to 14.0. 131
Rate curves for the degradation of the xylan in aqueoussodium hydroxide at pH values of 14.6 and 14.8. 132
The effect of pH on the initial reaction rates of thealkaline degradation of the xylan. 133
Standard curve for xylose in phenol and concentratedsulfuric acid. Measured at 480 nrn. 141

LIST OF CHARTS
Chart Page
Isolation of carbohydrate fractions from Douglas-firinner bark. 66
Extraction and purification of xylan from holocelluloseA of Douglas-fir inner bark. 69
Purification of the polysaccharides extracted by hotwater. 77

LIST OF TABLES
Table Page
Compositional analysis of bast fibers and Douglas-fir wood. 8
Chemical composition of barks. 12
Calculated values of rate constants for glucansdegraded in sodium hydroxide solutions. 34
Viscosity measurements in distilled water. 102
Viscosity measurements in M aqueous sodium chloridesolution. 103
Viscosity measurements in M-diethylenediaminecopper II. 104
7, Millimoles of periodate anion consumed by the xylanfrom Douglas-fir inner bark. 115
8. Rate data for the degradation of the xylan in aqueoussodium hydroxide at various pH values. 130

DOUGLAS-FIR BARK: WATER-SOLUBLE CARBOHYDRATESAND ALKALINE DEGRADATION OF A XYLAN
I. INTRODUCTION
More than 3.2 million dry tons of bark (28, p. 15) are produced
in Oregon annually. Only about 50% of this material is used com-
mercially; the rest has to be disposed of, mostly by open burning in
"wigwam" type burners. Every year the amount of waste bark in-
creases and disposal by burning is no longer feasible due to the more
stringent requirements for control of air pollution. A real problem
of solid waste disposal thus confronts the forest industries. To
alleviate the problems of disposal and pollution, new and better
means of bark utilization must be found. Before this can be done,
however, it is necessary that the chemical constituents and physical
properties of this potentially valuable source of raw material are
studied and understood thoroughly.
The bark of Douglas-fir [Pseudotsuga menziesii (Mirb. ) Franco]
represents the greatest volume of bark generated in Oregon. Thus
the present work is concerned with Douglas-fir bark. The chemical
composition of Douglas-fir bark is extremely complex and not well
understood. It is known, however, that carbohydrates are the major
constituents (60-70%) of the inner bark just as they are the major
constituents (70-80%) in the wood (39, 61, 65). However, little

2
attention has been devoted to the carbohydrates present in the bark
of trees. In contrast, the carbohydrates in the wood of trees have
been extensively studied. One reason for this has undoubtedly been
the greater economic importance of wood as compared with bark.
Another is probably to be found in the occurrence in bark of non-
carbohydrate constituents such as suberin, tannins, phlobaphenes,
and various phenolic compounds, all of which make the isolation of
polysaccharides from bark difficult. Wood contains none or few of
these components.
Some of the interfering substituents may be removed by extrac-
tion with an azeotropic mixture of ethanol and benzene (2:1 v/v).
Benzene, a hydrophobic solvent, removes part of the lipid extrac-
tives, but not completely, since it is not able to penetrate all parts
of the plant hydrophobic structure. Ethanol, a hydrophilic solvent,
removes part of the lipid materials plus some other constituents
such as some lignin and low molecular weight carbohydrates. A
mixture of these two solvents combines the advantages of each, and
provides complete penetration of the bark tissue. Moreover, the
solvent action of the mixture is now limited to non-lignin, non-
carbohydrate materials, (113, p. 114).
A hot-water treatment after the above extraction removes the
readily soluble polysaccharides. However, little attention has been
devoted to understanding the chemical and physical properties of

3
these easily obtained natural polymers from Douglas-fir bark.
To isolate the water-insoluble polysaccharides of Douglas-fir
bark requires a delignification with acidified sodium chlorite. The
residue remaining (holocellulose) yields a xylan on treatment with
barium hydroxide followed by extraction with 10% potassium hydrox-
ide. This xylan has not been hitherto characterized.
The work herein reported is a detailed chemical investigation
of the hot-water-soluble polysaccharides and the xylan from the inner
bark of Douglas-fir. The investigation was undertaken to determine
the exact amounts present and the characteristics of the polysac-
charides present in these fractions. The study involves the use of
modern techniques of separation, purification, analyses and charac-
terization. The advent of these methods allows a more comprehen-
sive and complete understanding of bark materials than was previ-
ously possible.

II. HISTORICAL REVIEW
Authors have referred to Douglas-fir as Pseudotsuga taxifolia
(Poir. ) Britt. and other names. However, the presently preferred
botanical name is Pseudotsuga menziesii (Mirb. )Franco. The names
all refer to the same genus and species. Mention is made of this to
avoid confusion about the exact species investigated.
The present work is concerned with Douglas-fir inner bark.
This is a specific anatomical part of the bark and a brief description
of bark anatomy is included for purposes of definition.
For a detailed anatomical description of Douglas-fir bark,
reference is made to Grillos (41), Grillos and Smith (42), Chang
(23), and Ross and Krahmer (88). Briefly, however, bark can be
considered to consist of inner bark and outer bark (Figure 1, page 5).
The inner bark (phloem cells) is the portion from the vascular cam-
bium to the innermost cork layer. The outer bark ( hYtidome)
is everything to the outside of the innermost cork Cambium (Figure
1-, page 5).
The inner bark comes from the vascular cambium, that layer
of living cells between the wood and bark which divide to form wood
to the inside and bark to the outside. The inner bark is composed
mainly of sieve cells, axial and ray parenchyma, and sclereids
(Figure 1, page 5). Much of the inner bark is living in the living
4

-wok:
418/1,41014,9
418
loyet44-0, (cop,.--pk(
-stwopi".44)... 4%)ceps
,41)
leCrefis
44 V
An.atAr4yoo Dceigla..-,,,f01 bark.4 . ctur e supplied: by DT.. 1:21. K rahlal&kJ, As:sociAte.- Professor, School of For-estrY.
Oregkop.. State, Univer.slty.
a9
r944 taii4 4 *
elp.1t)ift 4 10,4,A
PASWrite,A;00
le,"';441%*r Te-,tiolbit4
-tor 0 ,L1,010
?-111",foofitt0:4.'6'410 VVP,
tenft'
11)%40:4? 14,
1,11;z1 014,ge,
of; ) 1,4114w
biti+.44401111611 u0004,11 104 4 r.1)
-440611
)111,1
g
t..V.Ator.
tor'
parenchymasieve cell
010%
ptikat-.
4Wrlierilott$
4.16vm PI/11
' 06,..;;;!:r,'4Aiii-1,11*VbP
..11.`1Z., "btb\%
41404te 471,i
try'l
/ corksclereid
Figure 1 a
Associate

6
tree because many of the parenchyma and sieve cells remain alive
as long as they are components of the inner bark.
Douglas-fir sclereids are short, sharply pointed, spindle-
shaped fibers of a red brown color (Figure 1, page 5). They are
often referred to as bast fibers. They are lignified cells and develop
from axial parenchyma cells some distance from the vascular cam-
bium. In becoming sclereids, axial parenchyma cells approximately
0.1 to 0. 5 mm in length elongate to 1 to 2 mm by apical intrusive
growth, and form thick walls. The sclereids are commonly straight
and somewhat cigar-shaped. Kiefer and Kurth (61) and Ross and
Krahmer (88) describe and illustrate the general appearance and
position of the sclereids in Douglas-fir bark.
The outer bark of Douglas-fir consists of layers of cork in
which growth increments are usually visible, as shown by Ross and
Krahmer (88). Interspersed among the corky layers are areas of
phloem tissue that contain the sclereids and other cell types found
in the inner bark (Figure 1, page 5). The cork layers form from
the cork cambia which are riving cells that were once living
parenchyma cells of the inner bark. New cork cambia form in
the inner bark and cut away part of the inner bark, which now be-
comes part of the outer bark. The cork cambium produces cork
cells to the outside and a few storage cells to the inside. Cork cells
have thin cellulose walls which are coated with suberin. Suberin is

7
essentially an ester condensation polymer of hydroxylated, saturated
and unsaturated straight-chain fatty acids (95). The cork layers
may also contain tannin, dihydroquercetin and starch. All cells
outside the innermost cork cambium are dead because no food supply
can pass through this layer of cork cells. This then results in an
outer bark composed of cork cells and dead phloem cells, which
were once inner bark.
The most easily obtained carbohydrates from bark are those
solubilized by treatment with hot water. Kiefer and Kurth ( 61) report
the extraction of a hot-water-soluble fraction from the bast fibers,
or sclereids, of Douglas-fir bark. Whole bark was collected, ground
and screened. All fractions larger than 40-mesh were reground and
rescreened. The fibers were obtained in an almost pure state by
stirring the crude fiber fraction in five times its volume of distilled
water at room temperature. By virtue of their high specific gravity,
the fibers readily sank to the bottom of the container, whereas cork
and other impurities remained on the surface and were skimmed off.
The percentage composition of the bast fibers was determined
and is presented in Table 1 (page 8). For the sake of comparison,
analyses of Douglas-fir wood, taken from the literature (39), are
included. The amount of hot-water-soluble materials in bast fibers
and in wood appears to be quite similar. However, no further frac-
tionation or identification of components was reported. The overall

Values based on oven-dry extractive-free material
aFrom: Kiefer, H. J. and E. F. Kurth. The chemical compositionof the bast fibers of Douglas-fir bark. Tappi 36:14-19. 1953.
bFrom: Graham, H. M. and E. F. Kurth. Constituents of extrac-tives from Douglas-fir. Industrial Engineering Chemistry 41:409-414, 1949.
cCorrected for lignin content.
8
Table 1. Compositional analysis of bast fibers and Douglas-fir wood.
Ash 0.60 0.17
Lignin 44.80 30.15
Holocellulose 54.58c 71.40
Pentosans 8.62 10.11
Methoxyl group 3.89 4.75
Acetyl group 2.39 0.59
Uronic acid anhydride 4.62 2.80
Methoxyl on lignin 7.16 15.20
Fibera Woodb
Ether soluble 2.92 1.32
Alcohol soluble 8.65 5.46
Hot water soluble 2.58 2.82
Sum of three extractives 14.15 9. 60

9
data of Table I indicate that the bast fibers may be a lignocellulosic
material with a composition similar to that of wood.
Although the hot-water-soluble carbohydrates of Douglas-fir
bark seem to have been largely ignored to date, similar fractions
from other bark species have been investigated. Some of the more
pertinent work is included for comparison purposes and because the
experimental techniques were of use in the present study.
It appears that prior to 1960 no systematic attempt had been
made to divide the total polysaccharide fraction of a bark into its
components, although it had long been recognized that barks were
rich in polysaccharides, some of them hot-water-soluble and easily-
hydrolyzable hemicelluloses.
In 1930, Schwalbe and Neumann (93) detected large amounts of
readily hydrolyzable hexosans and pentosans in the inner barks of
spruce, pine and red beech; and in 1938 Buston and Hopf (18) reported
the presence in ash bark of 20% of hemicellulosic material which,
upon hydrolysis, gave galactose, mannose, and arabinose.
In addition to hemicelluloses, many barks appeared to be very
rich in pectic substances (18, 93) which in the main were hot-water-
soluble. Ash bark (93) contained 7% of pectic material, and balsam
bark was found by Hay and Lewis (51) to contain 14% of a "water-
soluble mucilage" in addition to other carbohydrates. In 1938-39,
Sharkov and co-workers (96, 97, 98) published a series of papers

10
on pectic materials in the inner barks of pine, fir, and birch. Pine
bast was reported to contain up to 35% pectin.
Anderson and co-workers (4, 5) also studied the pectin compo-
nents of both inner bark and the adjacent cambial zone; 10% of pectic
material was isolated from the inner bark of black spruce (82). In
1956, Kotasek (63) found that hydrolysis of an aqueous extract of
spruce bark yielded D-galacturonic acid, in addition to arabinose,
glucose, galactose, xylose, and rhamnose.
The carbohydrate gums constitute another group of polysac-
charides which can be associated with barks (59) and which for the
most part are hot-water-soluble. The best known of these are the
"gum exudates" which in many cases arise from mechanical damage
to, or parasitic infection of, the exterior of the tree, although in
some instances exudation appears to take place spontaneously. It
can perhaps be argued that the gum exudates are not necessarily
"normal" components of bark, but similar polysaccharides are
known to be present in some barks, where they undoubtedly fill a
normal physiological role. A well known example is "slippery elm
mucilage," which occurs in the inner bark of Ulmus fulva Michs, now
known as Ulmus rubra Muhl., to the extent of 16% or more (3). It is
secreted in the bark "in sac-like membranes, considerably larger
than the surrounding cells, and scattered irregularly throughout the
tissue" (37). This mucilage has been shown by Anderson (3), and

11
by Hirst and co-workers (36, 37, 55) to contain residues of D-galac-
turonic acid, D-galactose, 3-0-methyl-D galactose, and L-rhamnose.
Another interesting gum was isolated from the inner bark of
red fir by Becker and Kurth (9). Upon hydrolysis, it yielded glucur-
onic acid, glucurone, an aldobiouronic acid, galactose, arabinose,
and two other sugars which were tentatively identified as 6-deoxy-
glucose and 6-deoxy-idose, respectively.
Many inner barks also contain starch, the most of which is
extracted with hot water. Larsen and Lynn (70) detected starch in
the bark of western larch, and Anderson and Pigman (4) have reported
its presence in the inner bark of black spruce. Histological studies
show very clearly that starch granules, similar to those of cereal
starches, are present in the parenchymatus cells of bark (22, 74)
where they undoubtedly act as a food-reserve.
In 1961, Time11 (106) reported analyses of the carbohydrates
occurring in the barks of several gymnosperms, each representing
a different genus. After exhaustive extraction with ethanol-benzene,
the products were analyzed by standard methods (104). The results
are presented in Table 2 (page 12). It should be noted that the sum
of the cold-water-soluble and the hot-water-soluble materials repre-
sents 40.6% of the inner bark of Ginkgo (Ginkgo biloba L.) and that the
inner bark of lodgepole pine (Pinus contorta Dougl. ) contains a
total of 36.7% of cold-water and hot-water-soluble materials. This

Table 2. Chemical composition of barks,a, b
aAll values in percent of extractive-free bark.Fir: Am abilis fir [Abies amabilis (Dougl. )Forbes]Spruce: Englemann spruce (Picea engelmanni Parry)Pine: Lodgepole pine (Pinus contort a Dougl. )Ginkgo: (Ginkgo bilob a L.)
From: Timell, T. E. Isolation of polysaccharides from the bark of gymnosperms. Svensk
papperstidning 64:651-661. 1961,
12
Component Fir SprucePine Ginkgo
Inner Outer Inner Outer
(Summative Data)
Lignin 38. 1 39. 4 24. 6 46. 9 7. 5 45. 6
Ash 2.1 4.0 3.3 2.2 24,4 10,1
Acetyl 0,8 0.5 0.2 0.8 0.2 0.3
Uronic anhydride 5. 6 8. 0 9. 9 7. 7 11. 5 9. 4
Residues of
Galactose 1. 6 2. 4 4. 3 4, 2 4. 5 1. 7
Glucose 37. 4 35, 7 40. 9 26. 8 38, 3 23. 8
Mannose 8. 0 2. 9 2. 5 2. 5 3.4 2. 1
Arabinose 3, 2 3. 3 10. 6 5, 5 6. 2 3. 5
Xylose 3. 2 3. 8 3. 7 3.4 4.0 3. 5
(Other Data)
Pentos an 9.2 11.3 19.3 13.0 11.6 8.9
Material solublein cold water 4.8 9.0 13.7 5.4 19.5 8.7
Material solublein hot water 8. 4 20. 3 22. 7 12.7 21, 1 14. 0
Material soluble in1% sodium hydroxide 35,4 51.6 47,0 56.5

13
demonstrates that an appreciable amount of bark can be extracted
into water when exhaustive techniques are used.
In 1969, Beveridge, Stoddart, Szarek and Jones (11) reported
some outstanding work on the structural features of slippery elm
mucilage. This is the mucilage first isolated by Anderson (3) in
1933 and previously studied by Hirst, Gill, Jones, and Hough (36,
37, 55). The 1969 work continued the studies on the fine structure
of this complex mucilage. These workers concluded that the poly-
saccharide contains chains of 3-0-methyl-D-galactose residues
attached to the C4 positions of certain L-rhamnose residues, and
that 3-0-methyl-D-galactose residues occur in some cases as non-
reducing end-groups. D-galactose is attached as single residues or
as 4-0-substituted residues to the C3 positions of some L-rhamnose
residues. The evidence indicates that the polysaccharide is more
highly branched than was at one time supposed. This recent work
shows the continued interest in the chemistry of bark carbohydrates
by researchers in a number of laboratories.
The xylan studied in the present work was isolated from the
acidified sodium chlorite insoluble holocellulose fraction of Douglas-
fir inner bark. Numerous methods have been described for the
isolation of holocelluloses from plant materials with the aim of
minimum alteration of the carbohydrates present. These methods
can all be considered modifications of one of three principal proced-
ures. The first was that of Ritter and Kurth (66, 87) who subjected

14
wood to repeated chlorinations and subsequent extractions with a solu-
tion of pyridine in alcohol. The second method involved the in situ
generation of chlorine dioxide by use of an aqueous solution of acetic
acid and sodium chlorite (57). This in itself was a modification of
the work of Schmidt and Graumann (92) who delignified wood with
gaseous chlorine dioxide. However, chlorine dioxide is very haz-
ardous and the in situ generation was a great improvement. The
technique was further developed by Wise, Murphy and TY Addieco
(116) and again modified by Whistler, Bachrach and Bowman (11 4).
This is the procedure most often used today (112, p. 449).
The third procedure used for the preparation of holocellulose
involves peracetic acid followed by mild sodium borohyd ride reduc-
tion (72). However, this method of oxidative delignification has not
been generally applied.
The delignification reaction used in the present work was the
acidified sodium chlorite method (114). The method has several
obvious advantages. It requires a relatively cheap and stable ox-
idizing agent, it gives a product of unusual brightness, and it can
easily be used for delignifying both small and large quantities of
material.
The xylan was extracted from the holocellulose by the method
of Beelik, Conca, Hamilton and Partlow (10). The procedure
involved the impregnation of the holocellulose with 1-2% aqueous

15
barium hydroxide solution followed by extraction with 10% aqueous
potassium hydroxide solution. Xylans readily dissolve in this med-
ium, while the dissolution of manno se-containing polymers is largely
suppre s s ed.
Much of the pulp fiber in this country is produced by the kraft
process which involves treatment of wood with sodium hydroxide
and sodium sulfide. Although the effects of these alkaline conditions
on cellulose have been extensively studied, little work has been done
on the hemicelluloses. No work has been reported on the degrada-
tion of hemicelluloses from bark under alkaline conditions. There-
fore, this work was undertaken to determine some of these effects
in anticipation of future isolation and utilization of bark carbo-
hydrates.
A brief historical review of the effect of alkaline conditions
on carbohydrates is presented for purposes of clarification.
Alkalis (and acids) induce sugars to mutarotate under very
mild conditions. When more vigorous conditions are employed the
a-carbon (enolization) and 13-carbon (p elimination) are attacked.
Other reactions such as fragmentation of the chain may also occur.
Carbonyl compounds having an a-hydrogen atom undergo
enolization in acid-base catalyzed reactions. Sugars may therefore
be expected to enolize under suitable conditions. However, enolic
forms of the sugars have not been isolated and have probably not

16
been detected in solution (83, p. 165), although this has been claimed
(7). The evidence for enolization of sugars is indirect and comes
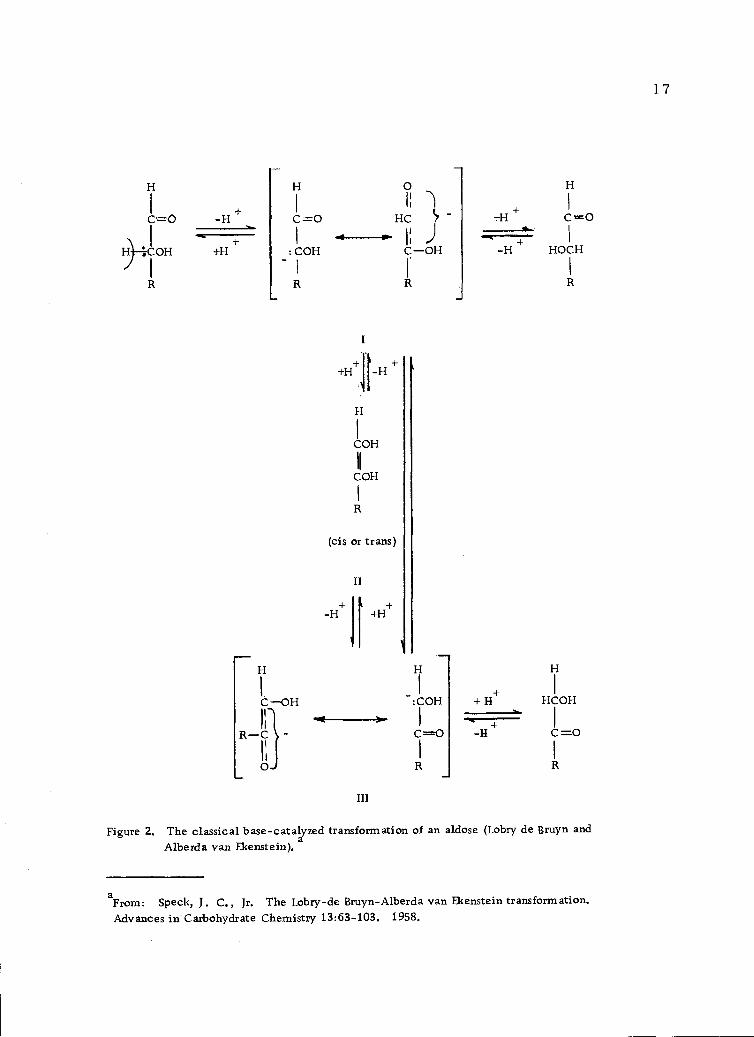
mainly from the Lobry de Bruyn-Alberda van Ekenstein transfor-
mation (Figure 2, page 17). This transformation includes the
epimerization of both aldoses and ketoses as well as aldose-ketose
isomerization. They proceed readily in alkaline solution (101). In
the absence of protecting groups, mixtures of aldoses and ketoses
are obtained. By further keto-enediol tautomerisms, the carbonyl
group of the sugar can move down the carbon chain. It may be noted
that enolization occurs after an attack by the base at the a-hydrogen
atom and that the actual formation of an enediol (II, Figure 2, page
17) is not necessary to produce the transformation (31).
The elimination of an hydroxyl or alkoxyl group in a p position
relative to a carbonyl group is a type of p elimination. In the case
of sugars, prior or simultaneous enolization is required before p
elimination can take place. Isbell (56) has proposed detailed mech-
anisms involving p eliminations for carbohydrates. In alkaline solu-
tions, enolization is rapid, but p elimination is slow (Figure 3,
page 18) and takes place from the enediol (VII) ionized at Cl (6, 56)
and ultimately, saccharinic acids are formed (89).
There may be more than one product formed, because in the
process of enolization, the carbonyl group can first migrate down
the carbon chain and also because elimination can be from two
COH
II
COH
(cis or trans)
II
-H+ 114 +H+
III
Figure 2. The classical base-catalyzed transformation of an aldose (Lobry de Bruyn andAlberda van Ekenstein) a
aFrom: Speck, J. C., Jr. The Lobry-de Bruyn-Alberda van Ekenstein transformation.Advances in Carbohydrate Chemistry 13:63-103. 1958.
17
H 0
+-H
I
C=70II
HC +H C=0
."1"---77--E. I44---
I
+H ..: COH -H HOCHCI OH
I'R
H
- I +
H
I
COH :COH +H HCOH
I+
RCi0
C =--0
I
-II C=0I
R R

H I H H
I II -
C=0 C=0 Cf-7-- 0
. I+
-H I I:FI),- -- COH -:COH -4-0- COH
I +H
HII
HO C H HOC H HO C HI I I
R R R
VII
HC0
HO- COH
CH
+H+
-H
Figure 3. p Elimination in a carbohydrate under alkaline conditions. a
COH11COH
I -HOC H
H C=0
C=0
CH12
aAnet, E. F. L. J. 3-Deoxyglucosuloses (3-Deoxyglycosones) and the degradation ofcarbohydrates. Advances in Carbohydrate Chemistry 19:181-218. 1964.
18
IV V (cis or trans)VI

19
positions if the enediol is not terminal (Figure 4, page 20).
Lindberg and co-workers (73) studied the alkaline degradation
of three disaccharides, representing the main linkages in the three
predominant wood polysaccharides, namely, cellulose, glucomannan,
and xylan. The reaction scheme proposed by these researchers is
shown in Figure 5 (page 21). They found that the elimination of the
hydroxyl group at C3, resulting in an alkali-stable glycosyl-
metasaccharinic acid (XVIII) and the elimination of the glycosyl
residue at C4 as an isosaccharinic acid (XVII) both proceeded through
a common step, the enediolate ion (XIV). Mannobiose reacted slower
than cellobiose, indicating a slower formation of the enediolate ion.
Xylobiose, on the other hand, was degraded at a considerably higher
rate than cellobiose and had a higher percentage of stopping reaction.
However the paper does not present any rate constants.
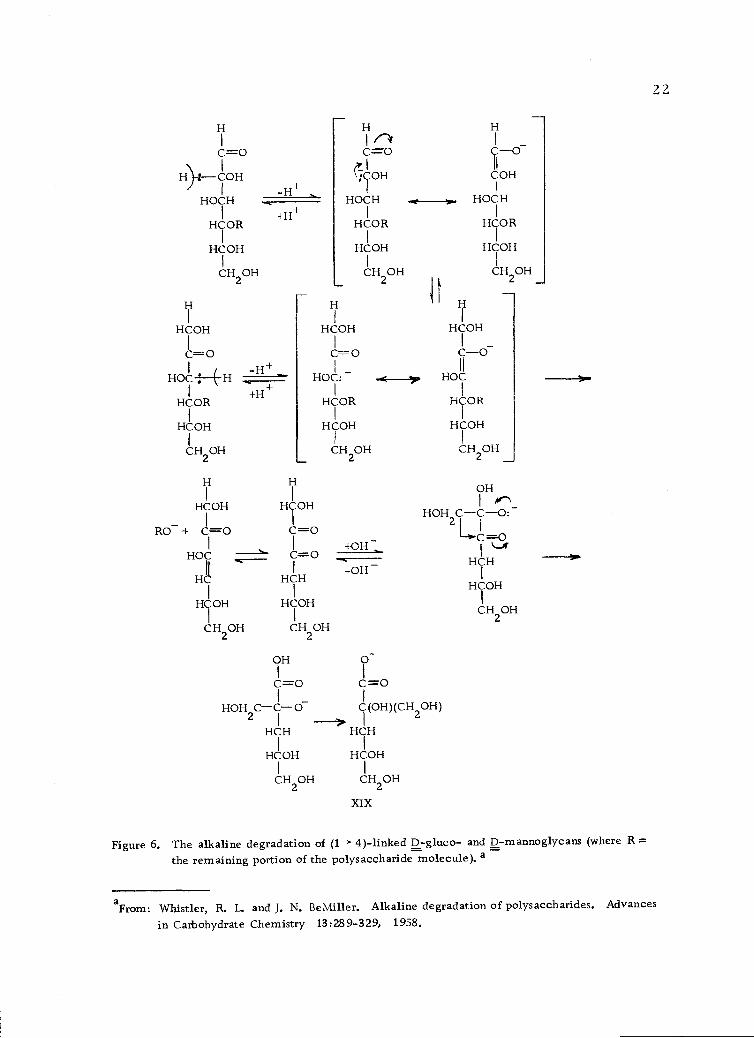
The alkaline degradation of polysaccharides (Figure 6, page 22)
proceeds by a peeling process in which the reducing end group is
liberated from a chain by elimination of the rest of the chain as a
glycoxy anion (110), similar to the degradation of the disaccharides.
Elimination takes place when the chain is in the position 13 to a
carbonyl group of the reducing end (56). Elimination of alkoxy
anions takes place under alkaline conditions when there is a carbonyl
13 to the alkoxide or when there is an easily removable proton on the
carbon atom in the position a to the alkoxide group. An alkoxy or

CH2Il
OHIC=0
HO- +iHOH
CHOHI
CH OH
+ f
2
-H +H
CH3I
C=0I
C=0I
CHOHI
CHOHI
CH2OH'II
HO *O0. C-''
tCH COH3 1
CHOHI
CHOHI
CM OH
CH2OH
I
C OH
T- OH
THOHCHOH
OH OH
X
XI XII
CH OH2
C=0
C OH16
CH + HO -I
CHOH1
CH OHI, 2
CH OHI 2
C=0
CH2
THOHCH OH
2HO
HOH CC OH2
CH2
CHOH
CHOH
Figure 4. Formation of two products after 1 elimination.
20

Hr-OHOIHHOCH
I-1?-0Ma
HCOH
CH2OH
-H+
+H
HCO
HO
H071-I
107via
HCOH
CH OH
HCOH- IIOC
1
HOCH
HCOMa
HTH
CH OH
+H
-H
CH2 OH
HOCH
110Ma
FITH
CH20H
aXIII XIV a XIV b XV
i' -H+1 [
+H +
COOHCH2
OH
I I
CHOH CO -I II
CH2HOC
I
I
HHCOMa 0Ma
HTHCHOH
H2 OH CH20HXVI
XVIII
00H
HOH2 C COH
MaOH 4- CH21
HCOH
CH OH
XVII
aFigure 5. Alkaline degradation of a disaccharide (where Ma = mannopyranosyl).
aFrom: Lindberg, B., 0. Theander and E. UddegIrd. Alkaline degradation of cellobiose,4-0-p-D-xylopyranosyl-D-xylose. Svensk papperstidning 69:360-363. 1966.
21
H H1 --4 7yio-C=O CO-,----
I
H): LH \:COH COHI -H+ I
HOCH --- HOCH ..r--v.. HOCH
I +HI- I
HCOR HCOR H ORI I
HCOH HCOH FITH
CH2OH
CH2OH CH20H
1 ,_,
11
II1
1
HCOH HCOH 10Hk-----0 Cr---0
I
..--0--
Hi : (+
HOi:-...4.--". Hoy-H+
+H
IORIOR HyoR
HTOH Hy0H HrH
2 2CH OH
2CH OH CH OH
11 7 OH
HrH HCOHI
I aHOH CC-0: -e
RO- + Cr=0 r0_
211I--4..0 =0
+OH , *.fHOC ------'- C=0HCH
Fi '-OHg ' HCH
-'
HI
COH HCOHI I
CHOH CH OH2 2
CrIf
=c) =0HOH CC-0- C (OH)(CH20H)21 ---> I
1HHr
FITHHCOH
CH20H CH2OH
XIX
Figure 6. The alkaline degradation of (1-)-4)-linked D-gluco- and D-mannoglycans (where R =the remaining portion of the polysaccharide molecule). a
HOH
CH2OH
aFrom: Whistler, R. L. and J. N. BeMiller. Alkaline degradation of polysaccharides. Advances
in Carbohydrate Chemistry 13:28 9-3 29, 1958.
22

23
glycoxy ion is more easily eliminated by the ionized enediol than is
a hydroxy ion, and the pyranose rings of the released glycoxy anions
will open readily because of the tendency of the negatively charged
oxygen to form a double bond with carbon. The released end groups
form 2, 3-diketone structures which rearrange by an intra-molecular
type of Cannizzaro reaction which is commonly referred to as a
benzilic acid rearrangement. These yield saccharinates (XIX)
(Figure 6, page 22).
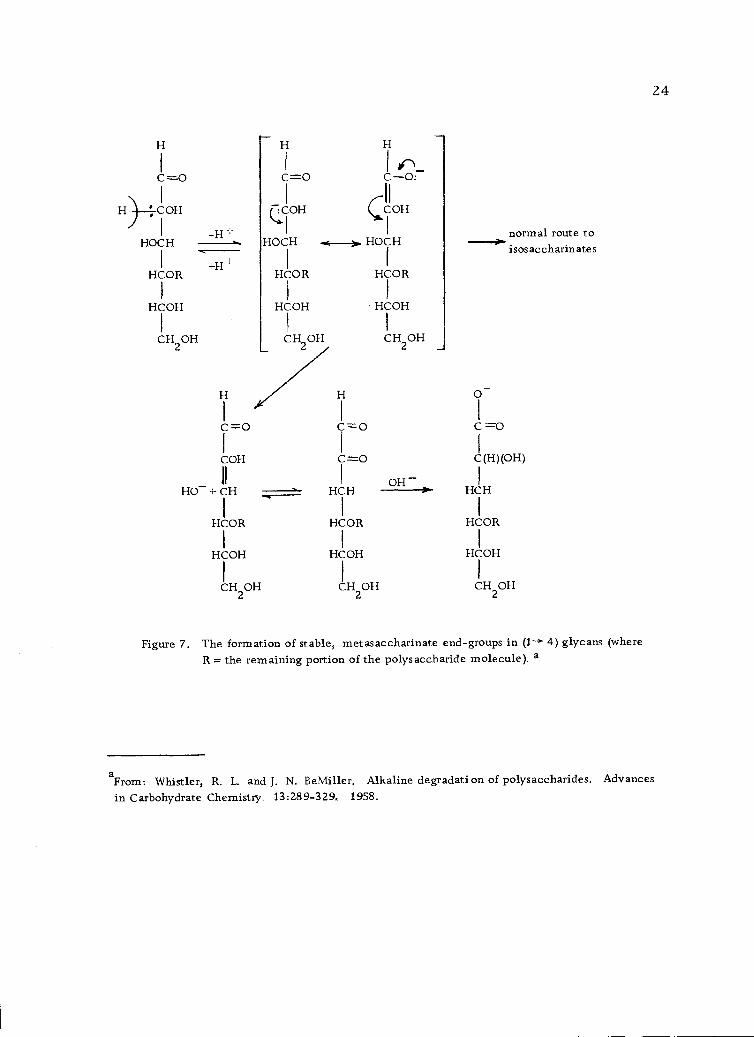
Termination of the peeling process may be caused by (a) an
alkali-resistant linkage in the polysaccharide, (b) the formation of
stable, metasaccharinate end-groups in (1- 4)-linked glycans (Fig-
ure 7, page 24) or, less likely, (c) formation of stable, ordinary
saccharinate end-groups in 4)-linked glycans (Figure 8, page
25) (110).
The alkaline degradation of cellulose has been extensively
studied. During alkaline digestion, cellulose is degraded in at least
two ways: (a) peeling off of the monomers at the reducing end and
(b) by the scission of glucosidic linkages (91, p.441). Non-volatile
acids (26, 75, 76) which are mostly D-glucoisosaccharinates and
low-molecular-weight acids are produced. The peeling reaction
does not proceed to completion. The cellulose becomes alkali stable
after about 50 glucose units have been peeled off consecutively, each
time exposing a new reducing end group in each chain before an
H H
I I 1 _.,CO.---- CO= CO:
I1
II
H) :COH (ICIOH CCOH
I
HOCH HOCH .------)._______...-H ' I normal route to
-. HOCHI1I isosaccharinates+H ±
HCOR HCOR HCOR
/ I I
HCOH HCOH - HCOH
I I I
CH2OH CH20H CH20H
HI /// H 0-
1 1 I
C=0 C=0 C=0
I I I
COH
.,..,
C---=-0 C(H)(OH)
II I OH- I
HO- + CH HCH --OP'. HCH
I I
HCORI
HCOR HCOR
I I I
HCOH HHCOH COH
I 1I
CH OH2
CH OH CH OH2 2
Figure 7. The formation of stable, metasaccharinate end-groups in (1--+ 4) glycans (whereR = the remaining portion of the polysaccharide molecule). a
aFrom Whistler, R. L. and J. N. BeMiller. Alkaline degradation of polysaccharides. Advances
in Carbohydrate Chemistry. 13:289-329, 1958.
24

H H
1 I_(COH
II C\ _
:COH
cl ,__C-0: C= 0
I I
HOCH -*----). HOCH
I I
HCOR , HCOR
1 I
HCOH HCOH
I I
CH20H CH20H
normal route to isosaccharinates
r 100 I r\
I(Hi:COH - COH
1
HOCH HOCH
HCOR HCOR
HCOH HCOH
CH20H CH20H
HC
COH
C=0
HCOR
HCOH
CH20H
CH2
OH
CH2
OH
I r 1
COH :COH
ll I
4HCI-0 o- -4-- c-----o
I
---4-
HCOR
I 1
HCOH HCOH
I I
CH20H CH20H
CH3
o-1 I
c=o c=oI o_ Ic=o ---- c
(CH3
)(OH)
I I
HCOR HCOR
I I
HCOH HCOH
I I
CH2OHCH2OH
Figure 8. The formation of stable, ordinary saccharinate end-groups in (1-' 4) glycans (whereR = the remaining portion of the polysaccharide molecule), a
aFrom: Whistler, R. L. and J. N. BeMiller. Alkaline degradation of polysaccharides. Advancesin Carbohydrate Chemistry 13:28 9-3 29. 1958.
25
H 4COH
-H+HOCH
+H+HCOR
HCOH
CH20H

26
alkali resistant metassacharinic acid is formed that remains attached
to the polymer chain (27, 85, 86, 90).
The reaction rate of non-cellulosic carbohydrates such as the
xylans, glucomannans, galactoglucomanans and others in alkaline
pulping, varies with the accessibility, branching, type of sugars,
and glycosidic bonds involved. However, the chemical behavior of
isolated hemicelluloses has been the subject of limited investigations
(19, 44). Studies on wood hemicelluloses have shown that this group
of compounds is composed primarily of two families; those contain-
ing mainly xylose and those composed primarily of mannose (35, 60).
The reaction in alkali of both glucomannans (46) and galactomannans
(19) have been examined.
Although studies on the alkaline degradation of xylan have been
carried out mostly on materials isolated from wood (20, 44, 47, 79),
some of the earliest work was done by Whistler and Corbett (111)
who treated a xylan preparation from corn cob with lime water.
They found that the degradation of the hemicellulose in lime water
at 25° was by way of the 13 elimination mechanism, and passed through
the following stages:
Xylotriose xylotriulose xylobiose xylobiu1ose-4-
xylose + saccharinic acid

27
However, no real evidence of this mechanism was presented. They
also reported that the xylan was stable to hot sodium hydroxide solu-
tion.
Meier (79) investigated the behavior of 4-0-methyl-glucurono-
arabinoxylan from spruce [Picea abies (L. )Karst.] under different
conditions used in technical pulping processes. The glucuronoarabino-
xylan showed a slow but significant loss of arabinose residues under
alkaline heating. The uronic acid content in the xylan was decreased
both when the xylan was heated in a homogeneous medium and in a
heterogeneous medium in the pulp. The presence of an arabino-
furanose unit at C3 on some of the xylose residues is assumed to be
the cause of a hindered peeling reaction during the alkaline digestion.
Hamilton (44) points out that studies on a 4-0-methylglucurono-
xylan indicate the selective removal of 4-0-methylglucuronic acid
under alkaline conditions. The actual mechanism of the uronic acid
removal in alkaline high temperature pulping is not clearly under-
stood.
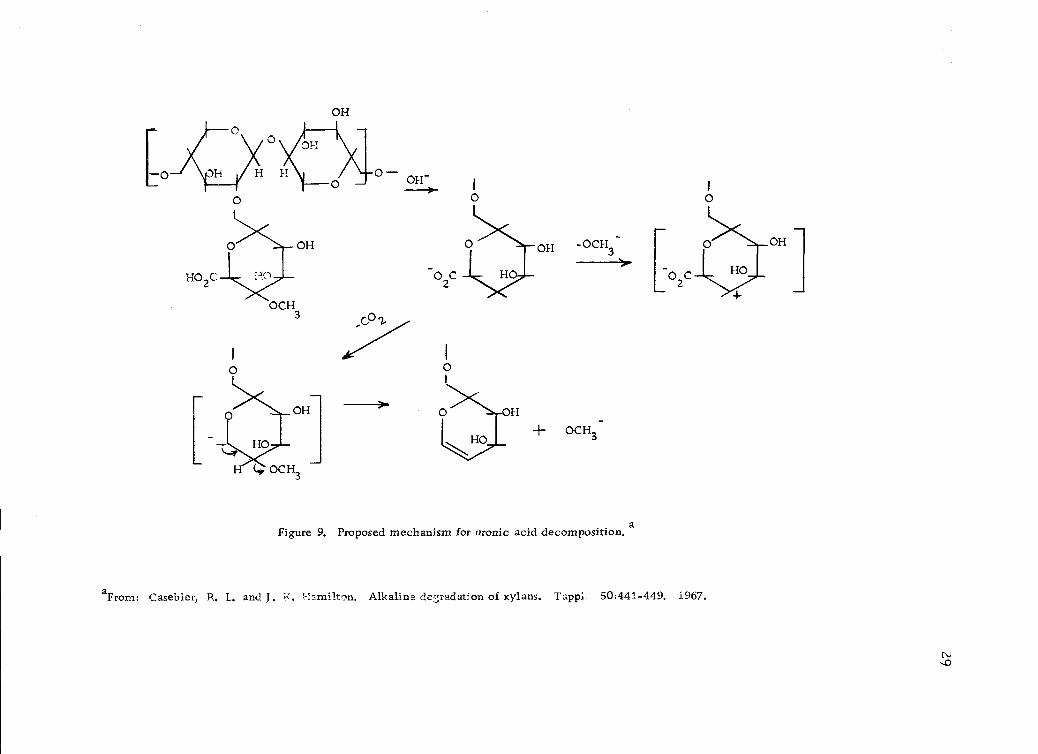
It has long been observed that xylan is adsorbed (precipitated,
crystallized) on cellulose fiber (21, 44,- 78, 91) during kraft pulping.
This crystallization of xylan polymers is thought to be related to
the removal or partial removal of (a) uronic acid, (b) removal,
partial removal, or translocation of acetyl groups and (c) removal
or partial removal of the arabinose residue. A mechanism of

4-0-methyl-D-glucuronic acid removal (Figure 9, page 29) is
proposed by Casebier and Hamilton (20).
Wood polysaccharides have been stabilized towards alkaline
peeling by changing their end groups from aldehyde (hemiacetal) to
alcohol or aldonic acid structures by use of sodium borohydride as
a reducing agent, or chlorite and polysulfide ions as oxidizing agents
(46, 49). Birch xylan is completely stabilized to alkaline de-
gradation at 100° by pre-treatment with sodium borohydride. De-
gradation occurs at 170° with the rate of splitting of the uronic acid
substituents highly dependent on the alkali concentration. The pres-
ence of air has a great influence on the degradation. The rate of
the peeling reaction doubled at 60° in air (47, 48) compared to the
reaction rate in an inert atmosphere.
The common view that a substituent at the C2 position stops
the peeling reaction (110) is refuted by Hartler and Svensson (50).
Studies on birch xylan [p ( 1 4)-linked D-xylose units, substituted
at approximately every tenth xylose unit in the C2 position with
4-0-methyl-a-D-glucuronic acid residues] showed that when alkaline=PC
solutions of birch xylan are kept at 1000 for several hours, the
amount of residue which can be isolated decreases to approximately
65 percent, and the degree of polymerization decreases. That the
degradation is due to primary peeling only, is shown by the fact
that birch xylan, when reduced with sodium borohydride and treated
28
I-102C
0
OH
3
OH-
0 C2
H
OH
4- OCH3-
Figure 9. Proposed mechanism for uronic acid decomposition. a
aFrom: Casebier, R. L. and J. K. Hamilton. Alkaline degradation of xylans. Tappi. 50:441-449. 1967.
0-c

30
similarly, can be isolated in 100 % yield with almost the same
degree of polymerization. If the uronic acid substituents impede
the peeling, a random or even distribution of the substituents would
result in between 2 and 5 percent degradation as the number average
degree of polymerization (DPn) of birch xylan is approximately 200.
Fractionation experiments on xylan have shown that polysaccharides
of moderately low glucuronic acid content can be isolated, but no
fractions free from glucuronic acid could be found. In view of this,
a decrease in yield of 35 percent seems extremely unlikely if it is
assumed that the glucuronic acid substituents impede the peeling.
If it is assumed that when the glucuronic acid side-chain is
on the C2 position of a reducing-end unit of the xylan, the bond
attaching this glucuronic acid is less in stability than those glucuronic
acid side-chains attached to other xylose units in the main chain,
these glucuronic acids are hydrolyzed from the reducing-end unit
and the peeling reaction can continue by the mechanism proposed
in Figure 6 (page 22). Thus, the glucuronic acid substituents would
not impede the peeling and the observed extent of the peeling can be
explained.
There is a dearth of information relating to the reaction
kinetics of the degradation of hemicelluloses in alkaline media.
Rate constants are extremely difficult to find. A kinetic analysis
for the alkaline degradation of cellulose at 170° has been provided

31
by Samuelson and co-workers (34, 90). A mathematical expression
was derived for the ratio between the rates of propagation and
termination reactions which occur during alkaline degradation. A
value of 65 was obtained for this ratio.
Besides the studies conducted by Samuelson and co-workers,
mentioned above, Sarkanen and his group at the University of Wash-
ington seem to be the only researchers currently engaged in a study
of the kinetics of alkaline degradation of carbohydrates. The first
of these studies dealt with the kinetics of alkaline degradation of
cotton hydrocellulose in 5 % sodium hydroxide solution at various
temperatures (43). An activation energy of 24 kcal/mole was found
for endwise degradation while termination to a stable metasaccharinic
acid end unit was 32 kcal/mole. Consequently the number average
degree of polymerization (DP) of the degradable chain length is
highly dependent on the reaction temperature, being 1000 at 65° and
140 at 132°.
The kinetics of degradation of cotton cellulose has also been
studied (68). It was found that the number of peeled glucose units
for each reducing end group was approximately 68 and was independent
of temperature in the range from 65° to 120°. This finding suggests
that the submicroscopic structure exerts a dominating influence on
the termination process of the endwise degradation reaction.
Studies on the degradation of amylose (68, 69) in various

32
alkaline concentrations and at various temperatures showed that the
extent of degradation was profoundly affected by the concentration
of the alkali. At 100° in the 0.01-0.1 N sodium hydroxide solutions,
amylose was degraded completely. This can only mean that the
peeling reaction proceeds through all units in the amylose chain and
the process represents an end-wise degradation without termination.
As the alkali concentration is increased to the 1 N level, the rate of
peeling increases but becomes constant thereafter. The ultimate
amount of degradation is reduced from 100% at dilute alkali concen-
trations down to a 45% level at 1 N alkali concentration. The rate
of termination, on the other hand, continues to increase beyond 1 N,
levelling off finally to a constant value at about 1.5 N alkali concen-
tration. As a consequence, the ratio of peeling to termination is
sufficiently high in the 0.01-0.1 N sodium hydroxide concentrations
to effect total degradation of amylose.
Young, Sarkanen, Johnson and Allan (119) give a general kinetic
expression for the rate of alkaline degradation of linear polysac-
charides in terms of mono- and di-anionic species formed from the
reducing end groups. The reaction is presumed to occur only via
the anionic species, which are the mono- and di-anionic forms of
the end group. Both mono- and di-anions are reactive towards
peeling, whereas end-group stabilization, which occurs in (1-* 4)
linked polymers only, is achieved through the di-anionic species

33
by conversion into a metasaccharinic end-group. Specific rate
constants for the end-wise depolymerization of (1 --->3)-f3-D-glucans
(laminaran, laricinan and pachyman) are given (Table 3, page 34).
Similar data for amylase degradation are given for comparison.
The rate constants of degradative chain-propagation via the mono-
and di-anion intermediate are shown to be essentially equal.
The recent advances in chromatographic separations coupled
with the physical techniques now available to characterize materials
allows for a thorough study of Douglas-fir bark carbohydrates. It
is possible to determine the amount of carbohydrates solubilized
by water extraction and to ascertain some of their structural char-
acteristics. It is also possible to learn something of the alterations
in the hemicellulosic molecules after alkaline extraction of these
natural polymers. The experimental work reported here represents
an effort to clarify many of these questions.

aL00 is the concentration at infinite time' kJ.' k k3 are rate constants, h is hours.
From: Young, R. A., K. V. Sarkanen, P. G. Johnson and G. G. Allan. Marine plant polymers. Part III. A kinetic analysis of the alkalinedegradation of polysaccharides with specific reference to (1 3) -(3 -D -glucans. Carbohydrate Research 21:111-122, 1972.
Jj
Glucan Temperature NaOHconcentration (M)
Loo D. p.-1
k1 = k2 h k2h-1
Amylose
Larninaran
Laricinan
Pachym an
56
100
78
56
40
56
56
1.25
0.1
1.0
0.1
0.1
0.1
0.21
0.45
0.42
0.30
0.45
820
25
165
700
5.95
323
298
74.2
10.9
2.10
11.5
29.6
0.53
1,33
Table 3. Calculated values of rate constants for glucans degraded in sodium hydroxide solutions.a, b

III. EXPERIMENTAL
A. Collection of Bark Samples
The inner bark used in this study was taken from a standing
Douglas-fir tree in the George T. Gerlinger State Experimental
Forest, located near Black Rock, Oregon, U.S.A. and operated
by the School of Forestry, Oregon State University, in cooperation
with the State Forestry Department of Oregon. Outer bark was
chipped off the tree at breast height in May 1969. The inner bark
was then carefully stripped off and immediately brought to the Labora-
tory where the adhering cambium layer was separated from the speci-
men. The cambium-free inner bark (4832.0 g, moisture content
44. 9%, based on green weight, hot-air oven at 1100) was immersed in
95% ethanol. Water was added later to provide a solution of ethanol-
water (4:1 v/v), with adjustments being made for the moisture content.
The tree was cut after the inner bark was stripped and, by count
of the annual rings, was found to be 130 years old.
B. Sample Preparation and Solvent Extraction
1. Extraction of the Hot-Water-Soluble Solids
After three days soaking in the ethanol-water (4:1 v/v) solution,
the inner bark was recovered by filtration and washed well with fresh
35

36
ethanol-water (4:1 v/v). The filtrate and washings were then com-
bined and concentrated to about 2.0 liters on a rotary evaporator.
The solution was tested for the presence of monosaccharides by
paper chromatography using the solvent system ethyl acetate-
pyridine-water (8:2:1 v/v/v). A trace of glucose was detected by
spraying the chromatograms with o-aminodiphenyl reagent (0.4 g
o-aminodiphenyl dissolved in a solution prepared from 100.0 ml of
glacial acetic acid and 20.0 ml of distilled water) and heating at
100±2° 2' in an oven for 5 minutes (108).
The residue of inner bark (air-dried)was ground in a Wiley
Mill and fractionated according to particle size by screening with a
series of "Tyler" screens. All material (1612.8 g) between -20 and
+100 mesh was used. In addition 238.3 g out of the 390.0 g of the
-100 mesh material was included. None of the +20 mesh material
(23.6 g) was used.
A part (1500.0 g dry weight) of the recombined bark was
divided into three portions and each portion extracted with benzene-
ethanol (2:1 v/v) for 37.5 hours (a minimum of 50 solvent exchanges)
in a Soxhlet extractor. The residues were combined and air-dried
for 4 days.
The air-dried bark residues were divided into four portions
and each portion was added to 3.0 liters of distilled water and the

37
mixture was kept at a constant temperature of 50-600 with intermit-
tent stirring. After 24 hours, the mixture was separated by filtra-
tion using a Bachner funnel and the residue was washed with distilled
water. The filtrate and washings were collected, combined, concen-
trated under vacuum, and freeze-dried to yield a light-tan-colored,
fluffy material; weight 202. 5 g.
Z. Extraction of the Xylan
The bark residue from the hot-water extraction, after being
dried in a hot-dry room for 4 days, was divided into four equal
portions. Each portion was extracted with 3.0 liters of 0.5% aque-
ous ammonium oxalate for 26 hours at a constant temperature of
70-800. The mixture was separated by filtration using a Bachner
funnel. The residue was washed with distilled water, dried in the
hot-dry room for 3 days, and further air-dried for another 3 days.
The dried residue was divided into six batches and each batch
was stirred into 3.0 liters of distilled water at 75-800 . Nitrogen
was bubbled through the mixture to prevent the accumulation of
gases during the following reaction. Glacial acetic acid (20.0 ml),
followed by sodium chlorite (80.0 g) were added at one hour intervals
until a total of 3 additions had been made (106, 114, 116). At the
end of 4 hours, the yellow solids were recovered by filtration using
a Bachner funnel with Whatman No. 1 filter paper, and they were

38
washed well with distilled water. The yellow solids were dialyzed
for one week, washed with distilled water, dried with ethanol and
finally dried in the air for 2 weeks; weight, 797.6 g (dry-weight
basis).
The yellow color of the solids indicated incomplete delignifica-
tion. Therefore, the acidified sodium chlorite treatment was repeat-
ed. The residue was recovered by filtration, dialyzed for 1 week
and freeze-dried. The white-colored, acidified-sodium-chlorite-
insoluble solids which were thus obtained were termed "Holocellulose
A"; weight 540. 0 g (dry-weight basis).
A portion of the freeze-dried Holocellulose A (50. 0 g dry
weight) was slurried in aqueous barium hydroxide solution (64.0 g
barium hydroxide octahydrate in 782.0 g solution) with intermittent
stirring at 25° (10, 4 5 ), At the end of 20 minutes, 18. 5% aqueous
potassium hydroxide solution (925.0 g) was added to the slurry and
stirring was continued intermittently for another 20 minutes. At
the end of this period, the mixture was separated by filtration using
a sintered glass funnel and the residual holocellulose was washed
with an aqueous solution of barium hydroxide and potassium hydrox-
ide (250. 0 ml) having the same concentration as the extracting
liquor.
The washings were added to the filtrate and the combined
liquor was acidified with acetic acid. Three volumes of methanol

39
were added and the precipitate which resulted was allowed to settle.
The precipitate was recovered by centrifugation and washed 4 times
with 70% aqueous methanol. The precipitate was dissolved in water,
traces of methanol were removed by evaporation under vacuum in
a rotary evaporator, and the solids were recovered by freeze drying.
The freeze-dried material had a white, fluffy appearance and was
labeled "crude xylan."
C. Characterization of the Hot-Water-Soluble Solids
1. Elemental Analyses for Nitrogen, Sulfur,Phosphorus, and the Halogens (109, p. 1039)
A small piece of freshly cut sodium was wiped thoroughly to
remove all traces of kerosene and placed in a small glass test tube.
The tube was gently heated in a flame until the sodium melted and
the vapors rose 1-2 cm up the walls of the tube. A small amount
of the hot-water-soluble solids was added to the molten sodium and
the tube was heated strongly over an open flame. Heating was con-
tinued for 1 to 2 minutes. After the entire end of the tube was red
hot, the tube was plunged into an evaporating dish which contained
about 10.0 ml of distilled water. The hot end of the tube shattered
and the resulting mixture was heated to boiling, the insolubles
removed by filtration, and the filtrate recovered for elemental
analyses.

40
An aliquot (2.0-3.0 ml) of the filtrate was added to powdered
ferrous sulfate (0.1 g) in a test tube. The solution was heated
gently with shaking until it boiled. Sufficient dilute sulfuric acid
was added to dissolve the iron hydroxides and to give an acid solu-
tion. A precipitate of Prussian blue formed, which indicated the
presence of nitrogen. For purposes of comparison, alanine was
fused with sodium, and tested for nitrogen. A Prussian blue color
formed which showed the presence of nitrogen.
A second aliquot (2.0 ml) of the filtrate was acidified with
dilute acetic acid, and a few drops of lead acetate were added. No
precipitate formed, indicating that sulfur was not present. As a
control for the analysis of sulfur, cystine was fused with sodium
and analyzed exactly as described above. A yellow precipitate
formed which confirmed the presence of sulfur.
A third aliquot (1.0 ml) of the filtrate was acidified with con-
centrated nitric acid (3.0 ml) and boiled for 1 minute. The solution
was cooled and an equal volume of ammonium molybdate reagent was
added. The solution was warmed to 40-50° and allowed to stand.
No precipitate formed, indicating that phosphorus was not present.
Glucose-l-phosphate was fused with sodium and tested for
phosphorus. A yellow precipitate (ammonium phosphomolybdate)
formed which indicated the presence of phosphorus.
A fourth aliquot (2.0 ml) of the filtrate was acidifecl with

41
dilute sulfuric acid, boiled gently to remove any hydrogen cyanide
which might be present, and the solution was treated with a few drops
of aqueous silver nitrate. No precipitate formed indicating that none
of the halogens were present.
A small sample of the hot-water-soluble solids was quanti-
tatively analyzed for nitrogen (Kjeldahl, 3.63%; Pascher and Pascher,
53 Bonn, Buschstrasse 54, West Germany).
Test for Tannins and Starch
A small amount of the hot-water-soluble solids was dissolved
in distilled water. An aliquot (1.0 ml) of this solution was treated
with a few drops of a ferric chloride-potassium ferricyanide solution
(15, p. 227) (1% solutions are mixed prior to use). A blue color
developed which indicated the presence of phenolics.
A second aliquot (5.0 ml) of the solution was treated with a
few drops of iodine indicator. A deep blue color developed which
indicated the presence of starch.
Strong Acid Hydrolysis
A portion of the hot-water-soluble solids (0.07 g) was treated
with 72% sulfuric acid (0. 9 g) and allowed to stand for 45 minutes at
room temperature. Water (20.1 ml) was added slowly with stirring
to provide a final concentration of 3.0% acid. The solution was

42
refluxed for 5 hours, cooled to room temperature, and neutralized
to pH 5.0 with saturated aqueous barium hydroxide solution. The
resulting barium sulfate precipitate was removed by centrifuge,
washed well with water and the washings were added to the decantate.
The combined decantate was concentrated under vacuum on a rotary
evaporator to about 50.0 ml.
Mild Acid Hydrolysis
A portion of the hot-water-soluble solids (0.32 g) was dissolved
in 3.0% sulfuric acid (96.0 ml) and the solution refluxed for 5 hours
After cooling to room temperature, the solution was neutralized to
pH 5.0 with saturated aqueous barium hydroxide solution. The
precipitate of barium sulfate was removed by centrifuge and washed
with water. The decantate plus the washings were concentrated to
about 100.0 ml under vacuum on a rotary evaporator (71).
Qualitative Amino Acid Analysis by Paper Chromatography
The hydrolyzates from the mild acid hydrolysis were subjected
to ascending two-dimensional paper chromatography (12, 25, p. 93)
on Whatman No. 1 paper, using water-saturated phenol as a developer
in one direction in an atmosphere of ammonia, and n-butanol-formic
acid-water (20:6:5 v/v/v) as developer in the second direction. A
solution of ninhydrin (1.0 g) dissolved in n-butanol (500.0 ml) was

used as the spray reagent.
Carboh drate Anal sis b Pa er Chromato ra h
43
A portion of the hot-water-soluble solids was dissolved in
distilled water, spotted on Whatman No. 1 filter paper and paper
chromatographed as described below to determine if free sugars were
present. No free sugars were detected.
The hydrolyzates from the strong acid hydrolysis and the mild
acid hydrolysis were applied at intervals of about 1 inch along one
edge of a Whatman No. 1 filter paper, 18 X 22.5 inches, in such
amounts as to produce a colored spot easily detectable with the
naked eye in a natural light or under ultraviolet light. A standard
solution containing about 1. 0% each of the known monosaccharides,
glucose, mannose, galactose, arabinose, xylose and rhamnose was
also spotted on the filter paper. The sugars were separated by
descending development with ethyl acetate-pyridine-water (8:2:1
v/v/v) (54) as developer. The solvent was allowed to migrate almost
to the bottom of the papers at which time they were removed from
the tank and air-dried for at least 6 hours. Some of the papers were
returned to the tank and developed as before (repeated up to 3 or 4
times) in order to obtain a better separation.
The paper chromatograms were sprayed with o-aminodiphenyl
reagent (0.4 g o-aminodiphenyl dissolved in 100.0 ml of glacial

acetic acid and 20.0 ml of distilled water) and heated at 100±2° in
an oven for 5 minutes (84, 108). The spots were outlined in pencil
under ultraviolet light. The rates of movement of the hydrolyzate
sugars were compared with those of authentic samples when run
simultaneously on the same chromatograms.
The o-aminodiphenyl was purified by recrystallizing the
technical grade material twice from aqueous ethanol. Activated
charcoal was used as a decolorant. The purified crystals were
dried at room temperature under vacuum.
7. Purification of the Hot-Water-Soluble Solids
A portion of the hot-water-soluble solids (3.0-4.0 g) was
stirred into a small amount of distilled water until it was thoroughly
wetted. Distilled water at room temperature was then added slowly
with stirring until the mixture had a concentration of 1.0%. Stirring
was continued for at least 2 hours at room temperature. The mix-
ture was allowed to stand and was then centrifuged. The supernatant
liquid was recovered. The undissolved solids were again stirred
into water and the whole process repeated 3 more times. The liquor
from the fourth washing was colorless and clear.
The undissolved solids which remained were freeze-dried;
yield 29.8% of the original hot-water-soluble solids. These were
labeled "Fraction A." A portion of "Fraction A" was tested for
44

45
starch (positive) and tannins (negative) as described in section
III-C-2 (page 41). A second portion of "Fraction A" was hydrolyzed
by strong acid as described in section III-C-3 (page 41) and the
hydrolyzate was examined on paper chromatograms (see section
III-C-6, page 43). The chromatograms showed glucose only. A
large sample of Fraction A was later prepared for additional research
purposes.
All of the liquors from the above treatments were combined
and concentrated to about 1.0 liter under vacuum at less than 300
temperature in a rotary evaporator. Ethanol (95%) was added to
provide a final solution of 70.0% ethanol. The flocculent precipitate
which resulted was recovered by centrifugation. The precipitate
was washed 4 times with 70.0% ethanol (1.0 liter each time). After
the fourth washing, the precipitate was dissolved in water and traces
of ethanol were removed under vacuum in the rotary evaporator.
The polysaccharide was then recovered by freeze-drying and labeled
"Fraction B"; yield 25.9% of the original hot-water-soluble solids.
A portion was tested for starch (positive) and tannins (faintly posi-
tive) as described in section III-C-2 (page 41). Another portion
was hydrolyzed under mild acid conditions (see section III-C-4,
page 42) and examined by paper chromatography (see sections
III-C-5 and III-C-6, pages 42 and 43). The chromatograms showed the
presence of amino acids and sugars.

46
The mother liquor and washings from "Fraction B" were
combined, concentrated, and freeze-dried. This sample was labelled
"Fraction C". The yield of Fraction C (by difference) was 44.3%
of the original hot-water-soluble solids. A portion of this sample
was tested for starch (negative) and tannins (positive) as described
in section III-C-2 (page 41).
8. Enzyme Hydrolysis to Remove Starch
The enzymes used were two commercial preparations
purchased from Marschall Division, Miles Laboratories, Elkhart,
Indiana. The first enzyme, HT-1000 (81),is a mixture of amylolytic
and proteolytic enzymes, capable of faster and more economical
liquifactions of starch than many other a-amylases. It has been
derived from Bacillus subtiles, and is in the form of a white, dry
powder. The second enzyme (commercial name, Diazyme L 30) is
an amyloglucosidase (80). It is sold in liquid form.
A part (15.0 g) of Fraction B was dissolved in distilled water
(85.0 ml) and the pH was adjusted to 5. 5-7.0 with sodium carbonate
solution (1.0 N). HT-1000 (7.5 mg, 0.05% based on sample weight)
dissolved in a small amount of distilled water was added to the
sample solution. The mixture was heated to 75° in a water bath
with continuous agitation and held at that temperature for 15 minutes.
Heating was then continued until the temperature reached 85-87° and

47
held at this level for 30-40 minutes. At the end of this time, the
sample and water bath were cooled to 600, and the pH of the sample
was adjusted to 3.8-4.2 with hydrochloric acid (O. 1 N). Diazyme
(0.09 ml, equivalent to 80 units/lb starch) was added directly to
the cooled carbohydrate solution. The mixture was incubated at 60°
with stirring for 72 to 96 hours. At the end of the incubation period,
the mixture was transferred to a dialysis bag and dialyzed for two
days in distilled water, one week in running tap water and another
day in distilled water. The non-dialyzable portion was concentrated
in a rotary evaporator at less than 40° and the concentrate was
freeze-dried; yield 9. 9 % of the original hot-water-soluble solids.
The materials passing out of the dialysis bag during the first
two days were collected, concentrated, and freeze-dried. A portion
of the freeze-dried sample was dissolved in water, spotted on
Whatman No. 1 filter paper and paper chromatographed (see
section III-C-6, page 43). The chromatograms showed glucose
only. Another portion of the sample was hydrolyzed under mild
acid conditions and the hydrolyzate was tested by paper chromatog-
raphy (see section III-C-6, page 43). The chromatograms showed
glucose only.
9. Enzyme Hydrolysis to Remove Protein
The non-dialyzable, freeze-dried material recovered from

48
the enzyme hydrolysis to remove starch was dissolved in 1. 5 liters
of distilled water. Tris (hydroxymethylamino)-methane was added
to the solution and the pH was adjusted to 8. 5 with 0.1 N hydrochloric
acid. Chymotrypsin (160 mg) and trypsin (150 mg), dissolved in a
small amount of water, were added to the buffered solution and the
volume adjusted to 2. 0 liters. The solution was placed in a hot-
water bath and heated at a constant temperature of 30-40° with
constant stirring for 12 days. At the end of the incubation period,
the solution was transferred to a dialysis bag and dialyzed for 12
days against running tap water. The precipitate which had formed
during the reaction was separated from the mother liquor by centrifu-
gation and washed three times with distilled water. The mother
liquor and the washings were combined, concentrated in a rotary
evaporator and freeze-dried (117). The freeze-dried material had
a tan, fluffy appearance. This sample was labelled "Fraction D";
yield 1.5% of the original hot-water-soluble solids. Fraction D
was hydrolyzed with 3. 0% sulfuric acid (see section III-C-4, page 42)
and investigated by paper chromatography (see section III-C-6,
page 43). The chromatograms showed the presence of galactose,
glucose, arabinose and traces of rhamnose, xylose and mannose.
10. Carbohydrate Analysis by Gas-Liquid-Chromatography
The gas-chromatograph used was a Hewlett-Packard 5751B

49
Research Chromatograph (Hewlett-Packard Company, Palo Alto,
California) equipped with dual flame ionization detectors. The
conditions were: column, 6.5% ECNSS-M on Gas Chrom Q 100/120
mesh, 6 ft X 1/8 in. 0.D, stainless steel; injection port 200°, detec-
tor 235°; column temperature 175° isothermal; helium flow 30 ml/
min; range setting 102; attenuation setting 16.
The various samples were hydrolyzed and the derivatives
prepared for injection into the gas chromatograph as follows. The
polysaccharide sample (0.32 g) was dissolved in 3. 0% sulfuric acid
(96.0 ml) and refluxed for 5 hours (mild acid hydrolysis). The
solution was cooled and authentic myo-inositol (0.1000 g) was added.
The hydrolyzate was then neutralized to pH 5.0 with a saturated
aqueous barium hydroxide solution. The resulting barium sulfate
precipitate was removed by centrifuge. An aliquot (25. 0 ml) of the
clear supernatant solution was transferred to a round-bottomed flask
(100.0 m1). Sodium borohydride (0.08 g) was added to the flask and
allowed to react for 2 hours at room temperature (1, 13).
The excess sodium borohydride was decomposed by adding
acetic acid until gas evolution ceased. The solution was concentrated
to a sirup in a rotary evaporator, and methanol (10.0 ml) was added
and re-evaporated. The addition and removal of methanol was
repeated five times (2). The resulting sirup was dried in an oven
at 105° for 15 minutes to ensure complete removal of water.

50
Acetic anhydride (7.5 ml) and concentrated sulfuric acid (0. 5
ml) were added to the sirup and the solution was heated for 1 hour
at 50-60° in a water bath. After cooling for 5 minutes, the acetyla-
tion mixture was poured slowly with stirring into about 70.0 ml of
ice-water. The mixture was transferred to a separatory funnel
and the alditol acetates were extracted with three successive amounts
of freshly distilled methylene chloride (25.0 ml, 15.0 ml and 10.0
ml). The methylene chloride extract was concentrated to dryness on
a rotary evaporator at 750. Distilled water (1. 0 ml) was added to
the residue and re-evaporated. The alditol acetates were dissolved
in 2.0 ml of freshly distilled methylene chloride and about 4.0 41
of the solution were injected into the gas chromatograph (13, 99).
The peaks in the resulting spectra were identified by comparison
of retention times with authentic known alditol acetates and by peak
enhancement techniques. The areas under the peaks in the resulting
spectra were measured by means of a planimeter.
D. Characterization of the Xylan
1. Ash Determination of the Crude Xylan
The ash content of the crude xylan was determined according
to a modified TAPPI standard method (102).
Duplicate samples (O. 20000 g dry weight) were weighed into

51
previously ignited and weighed porcelain crucibles. The crucibles
were placed in a muffle furnace and heated gently (200° ) until char-
ring occurred. The temperature was increased to 6000 and heating
continued until ashing was complete. The ashed samples were placed
in a dessicator to cool and were weighed after 1.0 hour. The samples
were then reignited and reweighed until constant weight was obtained.
The ash content was 27.9 %
2. Precipitation of Excess Barium andDialysis of the Crude Xylan
For purposes of calculation, it was assumed that the ash con-
tent of the crude xylan was due entirely to the presence of barium as
barium oxide. From this value, the percent barium (25.0%) present
in the sample was calculated. The large barium residue was due
to the use of barium hydroxide in the isolation of the crude xylan.
A part (5.06 g) of the crude xylan was dissolved in distilled
water (100.0 m1). A slight excess of 2 M sulfuric acid (5.0 ml) was
added to precipitate all of the barium in the sample. The precipitate
was allowed to settle, recovered by filtration and washed several
times with hot distilled water (33, p. 134). The washings and the
filtrate were recombined, placed in a dialysis bag and dialyzed
against tap water for 5 days. The material left in the dialysis bag
was concentrated in a rotary evaporator and freeze-dried (yield,

52
69.8%). A portion of the freeze-dried material was ignited to deter-
mine its ash content (4.4%). This purified, freeze-dried material
was labelled "xylan."
3. Optical Rotation of the Xylan
A part (0.18235 g) of the xylan was dissolved in water (10.0 m1).
Ten different readings were taken on the polarimeter (Rudolf and
Sons) at 25°. The specific rotation was calculated according to
the relationship (115, p. 415):
rai25 100a, 100abc
where [a]25 = specific rotation
a = optical rotation in degrees
b = cell length in dm = 1
c = concentration in g/100 ml.
The specific rotation was -30.5, (c 1,74 g/100 ml, water)
(average of 10 readings).
4. Qualitative Uronic Acid Analysis by Color Reaction
Concentrated sulfuric acid (6.0 ml) was added slowly to an
aqueous solution (1.0 ml) of the 3.0% sulfuric acid hydrolyzate ofxylan
( see section III-C-4, page 42) and cooled under tap water. The
reaction mixture was heated for 20 minutes in a boiling water bath

and cooled. An aliquot (0. 2 ml) of a 0.1% ethanolic solution of
carbazole was added and the test sample allowed to stand for 2
hours at room temperature. The development of a purple color
indicated that the xylan contained a uronic acid. The solution was
scanned in the visible region and showed maximum absorption at
535 nm indicating the presence of a uronic acid (29, p. 497). Color
tests were also run on authentic galacturonic acid (positive test),
oxalic acid (negative test), and benzoic acid (negative test).
5. Paper Chromatography and Gas-LiquidChromatography of the Xylan Hydrolyzate
A part (0.30 g) of the dialyzed xylan was hydrolyzed in 3.0%
sulfuric acid (mild acid hydrolysis) for 5 hours, cooled to room
temperature, neutralized to pH 5.0 with saturated aqueous barium
hydroxide and centrifuged to remove the barium sulfate precipitate.
An aliquot of the supernatant liquor was concentrated under vacuum
in a rotary evaporator. The concentrated liquor was spotted on
Whatman No. 1 filter paper. A solution of known sugars was also
spotted on the same paper. The paper chromatogram was developed
and sprayed as described in section 111-C-6 (page 43). The sugars
detected were xylose (strong), arabinose (weak), galactose (trace),
glucose (trace), and rhamnose (trace).
Another aliquot (25.0 ml) of the hydrolyzate was reduced with
53

54
sodium borohydride and alditol acetates were prepared as described
in section III-C-10 (page 48). The alditol acetates were injected
into a Hewlett Packard Gas Chromatograph using the same column
and conditions as described in section III-C-10 (page 48). The
sugars detected were tit same as those reported above for paper
chromatography. The areas under the peaks were determined by
planime try.
6. Reducing End-Group Analysis (Somogyi Method)
A Somogyi determination of the reducing end group was carried
out on a portion of the dialyzed xylan. The alkaline copper reagent
(1945) was prepared as follows: Rochelle salt (40.0 g), disodium
hydrogen phosphate dodecahydrate (71.0 g)(or 53.0 g of the hepta-
hydrate), and 1 N sodium hydroxide (100.0 ml). A solution containing
cupric sulfate pentahydrate (8.0 g) was added with stirring followed
by a solution of potassium iodate (0.372 g in 100 ml water). This
amount of potassium iodate is good for determining up to 1.25 mg
glucose. Finally, anhydrous sodium sulfate (180.0 g) was dissolved;
the solution was diluted to 1.0 liter and allowed to stand 3 days so
that the impurities would settle out. The clear supernatant solution
was further clarified by filtration through a fritted glass filter. The
pH of the solution was about 9. 5 as reported in the directions for
preparation. The solution is supposed to be stable for at least one

55
year (52, p. 383).
A sample 8.40 mg of the unhydrolyzed xylan in solution (5.0
ml) was placed in a 25 X 200 mm test tube. Alkaline copper reagent
(5.0 ml) was added by pipette and mixed thoroughly. The tube was
closed with a glass bulb and placed in a rack. Blanks were prepared
with water (5.0 ml). Standard xylose solutions (1. 25 mg, 0. 75 mg
and 0. 25 mg) were also added to aliquots (5.0 ml) of the alkaline
copper reagent. The rack of tubes was immersed in a vigorously
boiling water bath to a depth of about 5.0 cm above the solution inside
the tubes. The solutions were heated for 30 minutes, removed from
the bath and allowed to cool. Care was taken that the tubes were
not agitated during the heating or cooling periods. Triplicate deter-
minations were carried out in all cases.
Potassium iodide (2.5%, 2.0 ml) was added to each tube without
mixing. From a fast flowing buret, 2 N sulfuric acid (1. 5 ml) was
run into each tube with shaking so that the liberated iodine would
oxidize all reduced copper. After 5 minutes, the tubes were re-
shaken. The excess of liberated iodine not reduced by cuprous ions
was then titrated with sodium thiosulfate (0.005 N). When the
solution was light yellow, two drops of starch indicator (1.0%) and
two drops of phenol red indicator were added, and the titration
continued until the starch-iodine blue disappeared. The difference
in the amount of titer consumed by the blank and the xylan was

attributed to the reducing end group of the polysaccharide.
7. Viscosity Measurements
A part (0.19156) of the dialyzed xylan was weighed into a
stoppered volumetric flask (25.0 ml) which had been previously
swept free of air by a stream of nitrogen. Diethylenediamine copper
II ion (cupriethylenediamine) (General Chemical Division, Allied
Chemical, Columbia Road and Park Ave., Morristown, NJ or Ecusta
Paper Division, Olin, P. 0. Box 200, Pisgah Forest, NC) at 25°
was added and the solution was shaken until the xylan was completely
dissolved. The flask was filled to the mark with the solvent and
mixed thoroughly. The filled flask was carefully weighed to deter-
mine the density of the solution in g/25 ml. An aliquot (10.0 ml)
of the solution was transferred to a Cannon-Ubbelhode dilution
viscometer previously placed in a water bath at 25°±0. 010 and
flushed with nitrogen. After 5 minutes the solution was drawn into
the bulb of the viscometer by applying pressure with nitrogen. The
time for the miniscus to pass between the two calibration marks was
measured to 0. 1 seconds. Duplicate measurements were made until
they agreed to within ±0. 3% (16, p. 537, 100, 103).
The solution was diluted directly in the viscosimeter with a
suitable amount of solvent and mixed by stirring with a stream of
nitrogen. Measurements were taken with each dilution. A total
56

57
of six concentrations were measured. The viscosity of the pure
solvent was also measured. The intrinsic viscosity was found to
be 0.42 dl/g in diethylenediamine copper II ion.
The intrinsic viscosity was similarly determined in M aqueous
sodium chloride (0.48 dl/g) and in distilled water (084 di/g)
8. Gel Permeation Chromatography - Determinationof Molecular Weight Distribution
A chromaflex column (Kontes Glass Co., Vineland, NJ) 400 X
1000 mm with a total bed volume (vi) of 1250.0 ml was used for the
determination of the molecular weight distribution of the xylan.
Dry sephadex G-75 (40-120 4) was stirred into a 4-liter beaker
with distilled water and swollen for 30 hours. During this time the
suspension was decanted four times to remove the fine particles. The
suspension was well stirred and was transferred through a funnel into
the vertically mounted column which had previously been filled with
water. After a layer of a few centimeters had formed, the capillary
outlet at the bottom of the tube was opened to release a slow stream
of water. When all of the gel had settled, the eluant was allowed
to flow through the bed overnight to complete the stabilization of the
column ( 40). The void volume (Vo)of 369 ml was determined by use
of Blue Dextran 2000 (Mw 2, 000, 000).
A part (500 mg) of the dialyzed xylan was dissolved in water in

58
a 50.0 ml volumetric flask. After the xylan had completely dissolved
the solution was diluted to the 50 ml mark and mixed thoroughly.
The xylan solution (20.0 ml) was added dropwise to the top of the
column with great care and when it had disappeared below the
surface a small quantity of water was applied to wash the surface.
Then more water was added to a height of 2-3 cm to start the elution.
The elution was carried out under a low hydrostatic pressure at a
flow rate of 0.5-0.7 ml/minute. The concentration of the eluted
fractions (10.0 ml each) was estimated by the phenol-sulfuric acid
method to be described later (see section Ill-F- 2, page 62 ).
E. Periodate Oxidation of the Xylan
1. Acidity of the Xylan
Three samples (49.90 mg, 81.76 mg, 133.43 mg) of the xylan
were each dissolved in separate aliquots (10.0 ml each) of distilled
water. An aliquot (10.0 ml) of aqueous sodium hydroxide solution
(0.0526 N) was added to each of the three samples. Each solution
was back titrated to a methyl red end point with hydrochloric acid
solution (0.0406 N). Three blank solutions were similarly titrated.
The differences between the titers of the blanks and the titers of the
xylan samples in the order of the weights given above were: 0.15 ml,
0.25 ml, 0.35 ml.

Analysis of Periodate Consumed
A portion (0. 26959 g) of the dialyzed xylan was placed in a
volumetric flask (50.0 ml), dissolved in water (10.0 ml), after
which, 0. 5 M sodium periodate solution (25. 0 ml) was added and
finally water was added to bring the volume up to the 50.0 ml mark.
The solution was mixed thoroughly. A blank having the same period-
ate concentration was also prepared. Both reaction mixtures were
incubated in a constant temperature bath at 5° in the dark (94).
The time at which the periodate was added was taken as time
zero. Aliquots (1.0 ml) of each reaction mixture were withdrawn
at timed intervals and added to 250. 0 ml iodine flasks containing 1 N
sulfuric acid (5.0 ml) and 20. 0% potassium iodide solution (5.0 ml).
The liberated iodine was titrated with 0.1 N sodium thiosulfate solu-
tion until only a pale yellow color remained. Starch indicator (1.0
ml, 1% solution) was added and the titration continued until the blue
color disappeared.
Analysis of Formic Acid Released
When the consumption of periodate had stopped, aliquots (5. 0
ml) of each reaction mixture were added to Florence flasks (150.0
ml) containing ethylene glycol (1. 0 m1). The mixture was allowed
to stand at room temperature for 5 minutes with occasional
59

60
swirling until the unreacted periodate was consumed. The solution
was titrated with 0.05 N sodium hydroxide solution using methyl red
as the indicator.
4. Complete Hydrolysis of the Polyalcohol
The oxidized xylan from the periodate reaction was placed in
a dialysis bag and dialyzed against running tap water for two days.
At the end of this period, it was taken out and concentrated under
vacuum in a rotary evaporator to about 100.0 ml. The concentrate
was neutral to litmus paper.
An excess of sodium borohydride (1.5 g) was added to the con-
centrate and the reduction was allowed to proceed at room tempera-
ture for 24 hours with occasional shaking. At the end of the reduc-
tion period the reaction mixture was returned to the dialysis bag and
dialyzed against running tap water for 3 additional days (100).
The polyalcohol was hydrolyzed under mild acid hydrolysis
conditions as described previously (see section 111-C-4, page 42).
The hydrolyzate was spotted on Whatman No. 1 filter paper and paper
chromatographed (see section III-C-6, page 43). The sugars in an-
other aliquot (25.0 ml) of the hydrolyzate were converted to their
alditol acetates and analyzed by gas-liquid chromatography as de-
scribed previously (see section III-C-10, page 48). The spectrum
showed xylitol pentaacetate and arabinitol pentaacetate and three

61
small unidentified peaks which came out early. The areas under the
peaks were measured with a planimeter.
F. Alkaline Degradation of the Xylan
1. Reaction in Sodium Hydroxide Solution
A part (4.34 g) of the dialyzed xylan was placed in a volumetric
flask (25.0 ml) which had previously been evacuated and flushed with
nitrogen and dissolved in sodium hydroxide solution (1.0 N). After
the xylan was completely dissolved the solution was diluted to the
mark with sodium hydroxide solution and shaken vigorously to attain
complete mixing. The time when the sodium hydroxide was added
was recorded.
Glass ampules (5.0 ml) were evacuated and filled with nitrogen
in a glove box. Aliquots (1.0 ml each) of the xylan solution were
placed in the ampules. The ampules were taken out of the glove box,
sealed, and placed in boiling water (68, 69). The ampules were
withdrawn from the constant temperature bath at definite time inter-
vals (30, 60, 90, 120, 180 minutes, and so on). Each withdrawn
ampule was allowed to cool for 1 to 2 minutes and placed in ice water
to stop the reaction. Normal hydrochloric acid (1.0 ml) was added
to the contents of the ampule to neutralize the sodium hydroxide
reaction mixture.

The degradations were also carried out at 78.5° (bp 95%
ethanol) and 64.5° (bp methanol).
2, Phenol-Sulfuric Acid Method of Analysis of the Xylan
The analysis for the undegraded xylan was carried out by the
phenol-sulfuric acid method of Smith and coworkers (8, 30, 62). A
Beckman Model DB spectrophotometer was used for this investiga-
tion.
Reagent grade sulfuric acid (95.5%, specific gravity 1.84) con-
forming to ACS specifications was used. Phenol (80% by weight) was
prepared by adding glass distilled water (20. 0 g) to redistilled reagent
grade phenol (80.0 g). This mixture formed a water-white liquid
which was readily pipetted and has been known to stay the same color
after one year of storage (30). A pale yellow color developed after
several weeks but this does not interfere in the determination since
a blank is included.
A fast delivery pipette (5.0 ml) was used to deliver the concen-
trated sulfuric acid. The pipette was prepared by cutting a portion
off the tip of a standard 5.0 ml pipette. A high maximum tempera-
ture was desired because it increased the sensitivity of the reagent.
Water (1.7 ml) and 80% phenol (0.05 ml) were added to an
aliquot (0.3 ml) of the neutral xylan solution in a test tube. Con-
centrated sulfuric acid (5.0 ml) was added rapidly. The stream of
62

63
acid was directed against the liquid surface in order to obtain good
mixing. Intense heat was generated at this time. The tubes were
allowed to stand for 10 minutes, then shaken and placed in a water
bath at 25 to 300 for another 10 to 15 minutes. The test tubes were
shaken again and samples were transferred to spectrophotometer
cells (1.0 cm light path) and readings taken at 480 nm. The color
developed was stable for several hours. The weight of hydrolyzed
pentoses and uronic acids was expressed as xylose equivalents by
using percent transmission in conjunction with a standard curve
prepared from known xylose.

IV. RESULTS AND DISCUSSION
A. Collection of Bark Samples
A sample of inner bark free from outer bark and cambium was
desired because it provided a relatively homogeneous starting mater-
ial. Outer bark contains considerable cork material which would
interfere with experimental studies of the carbohydrates. The
cambium layer contains proteins as well as carbohydrates and the
two are best separated at the outset.
In the spring of the year from April through June, the outer
bark of Douglas-fir is easily separated from the inner bark by
simply chipping it away. The inner bark for the present work was
taken from a standing tree to reduce contamination from other
sources. The cambium layer was then carefully separated from
the inner bark and a relatively homogeneous sample resulted.
B. Sample Preparation and Solvent Extraction
1. Extraction of the Hot-Water-Soluble Solids
The inner bark after the ethanol-water extraction was carefully
ground, screened and reground to ensure that most of the fibers were
separated and that the surface areas were exposed. Some of the
fine material was included in the fraction to provide a large starting
64

65
weight to ensure that sufficient polysaccharides would be isolated
for later studies. All of the fines were not included because pre-
liminary experiments showed that they plugged the extraction appa-
ratus and filter systems and made laboratory procedures time-
consuming and difficult.
It is desirable, before proceeding with polysaccharide separa-
tion, to remove as much as possible of the low molecular weight
materials (simple sugars, organic acids, lipids, waxes, and so on)
present in the bark. Some of these materials are readily oxidized
and would interfere with the delignification reaction.
Chart 1 (page 66) outlines the scheme followed in the treatment
of the inner bark and the isolation of the different fractions. The
yields for each extraction are shown (24). None of the extraction
procedures shown in this chart accomplished a clear-cut separation
of one type of material from another, but in general each step in
the sequence performed a definite function.
The ethanol-water (4:1 v/v) extraction served two purposes.
It prevented possible enzyme action which might change the natural
materials from their native state, and it also solubilized the simple
sugars which could be identified to provide a more detailed investi-
gation of the total carbohydrate fraction in the bark. The ethanol-
water (4:1 v/v) soluble solids were shown to contain the free sugar,
glucose, by paper chromatography.

Douglas- fir Inner Bark(100.0 g dry weight)
Extraction withethanol-water (4:1 v/v)
Extract Residue(15. 4 g) (84.6g)
Extraction withbenzene-ethanol (2:1 v/v)
Extract Residue(4.4 g) (80. 2 g)
Extraction withwater (50-60°)
Extract Residue(11. 1 g) (69. 1 g)
IAdditional fractionation Extraction with
shown in Chart 3, page 0. 5% aqueous ammonium oxalate
Extract Residue
(2.8 g) (66.3 g)
Sodium chlorite
CH3COOH
Extract Residue
(22.0 g) (44. 3 g)
Extract(14.3 g)
Additional fractionationshown in Chart 2, page
Chart 1. Isolation of carbohydrate fractions from Douglas-fir inner bark, a
Sodium chlorite
CH1COOHHolocelitlose A
(30.6 g)
aFrom: Chen, C. H. Douglas-fir bark; isolation and characterization of a holocellulose fraction.
Doctoral thesis in preparation. Corvallis, Oregon State University, 1972.
66

67
The benzene-ethanol (2:1 v/v) azeotrope removed lipids and
waxes (64) which would interfere with later reactions. The extract
contained no free sugars when investigated by paper chromatography.
Having de-fatted the bark it was possible to carry out a hot water
extraction and remove a water-soluble fraction which amounted to
11.1% of the original bark sample.
2. Extraction of the Xylan
Not all of the pectic material of plants is extractable with
water. The insoluble part, protopectin (calcium pectate) was re-
moved from the water-insoluble residue of Douglas-fir inner bark
by extraction with a 0.5% ammonium oxalate solution (Chart 1, page 66.
Presumably a cation exchange occurred in which insoluble calcium
oxalate was formed along with soluble ammonium pectate. The latter
was extracted with the filtrate.
Through all of the extractions the bark retained its character-
istic brown color. However, the strong oxidizing conditions of the
acidified sodium chlorite reaction bleached the reaction mixture to
a pale yellow color. Fumes of yellow gases, undoubtedly chlorine
and chlorine dioxide, were visible in the reaction vessel above the
mixture. These gases emphasize the necessity of performing the
delignification reaction in a fume hood and of bubbling nitrogen through
the reaction to sweep away these toxic materials. Fresh sodium

68
chlorite was added to the reaction at one hour intervals rather than
all of it at the beginning to prevent an excess of gas formation and
to give the reaction time to proceed. The residue from the second
delignification, termed "Holocellulose Al (Chart 1, page 66) was
freeze-dried to a white, fluffy material.
The isolation of a xylan material from Holocellulose A is shown
in Chart 2 (page 69). The Holocellulose A material was impregnated
with aqueous barium hydroxide to suppress dissolution of the mannan
containing polysaccharides prior to the extraction of the xylan (Chart
2, page 69). The mannose units react with the barium ions to form
a precipitate, and thus prevent the mannan containing polysaccharides
from being extracted with the xylan. In addition, aqueous potassium
hydroxide has been found to be excellent for the extraction of xylan
(10, 45). Mannans and other polysaccharides are less soluble in
this solvent.
C. Characterization of the Hot-Water-Soluble Solids
1. Elemental Analysis for Nitrogen, Sulfur,Phosphorus, and the Halogens
Compounds isolated from natural sources often contain one or
more of the above elements. Therefore, it is best to qualitatively
determine if they are present.
The sodium fusion test failed to detect the presence of sulfur,
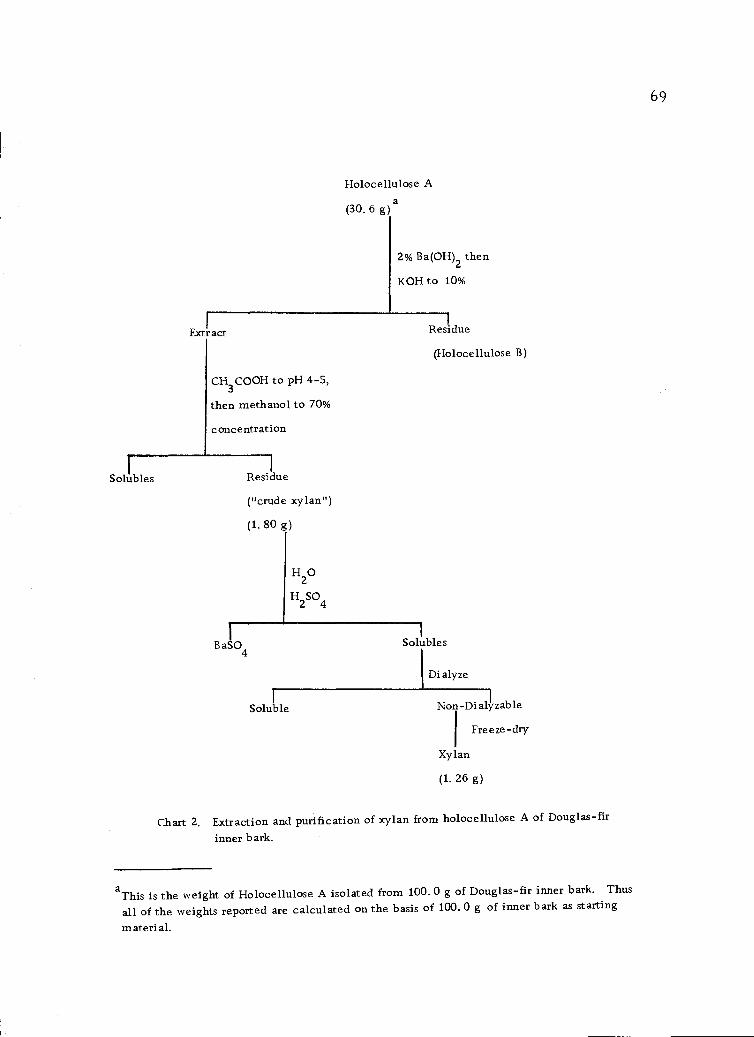
Holocellulose A
(30, 6 g) a
2% Ba(OH)2 then
KOH to 10%
Extr act Residue
(Holocellulose B)
CH3COOH to o pH 4-5,
then methanol to 70%
concentration
Solubles Residue
("crude xylan")
(1. 80 g)
H20
H2SO4
BaS04Solubles
1 Dialyze
Non-Dialyzable
Xylan
(1, 26 g)
Chart 2. Extraction and purification of xylan from holocellulose A of Douglas-fir
inner bark.
Freeze -dry
aThis is the weight of Holocellulose A isolated from 100. 0 g of Douglas-fir inner bark. Thus
all of the weights reported are calculated on the basis of 100. 0 g of inner bark as starting
material.
69
1
Soluble

70
phosphorus, and the halogens. Nitrogen was shown to be present as
evidenced by the formation of a precipitate of Prussian blue with
ferrous sulfate.
A portion of the sample was sent to an analytical laboratory
(Pascher and Pascher, Mikroanalytisches Laboratorium 53 Bonn,
Buschstrasse 54, West Germany) for analysis of nitrogen by the
Kjeldahl method, and was found to contain 3. 63% nitrogen. A part
of this nitrogen is in the form of proteins as discussed later (section
IV-C-5, page 74.
2. Test for Tannins and Starch
The ferric chloride-potassium ferricyanide test produced a
dark-blue color. This color (Turnbull's blue) was due to the forma-
tion of a 1:1 complex of ferric ion and phenolic hydroxyl (14) which
gives the ferrous reaction. The iodine test likewise resulted in a
blue color characteristic of a starch-iodine complex.
These strongly positive color tests showed the presence of
considerable amounts of tannins and starch. Kiefer and Kurth (61)
indicated the possibility of phenolic materials in Douglas-fir inner
bark and starch has been reported in the hot-water-soluble fractions
from several inner barks (4, 22, 69, 73). Therefore, starch was
expected to be present in Douglas-fir bark.

Strong Acid Hydrolysis
A fundamental aspect of polysaccharides is the component mono-
saccharides which are linked together to form the polymer chain.
Often polysaccharides contain linkages which are resistant to acid
cleavage. Thus, to ensure complete hydrolysis, the hot-water-
soluble solids were treated with strong acid according to the pro-
cedure of Laver, Root, Shafizadeh and Lowe (71). A portion of the
hot-water-soluble solids would not dissolve in 72.0% sulfuric acid.
After hydrolysis, the insoluble portion had a dark-red color and
presumably were phenolic acids. This precipitate was not investi-
gated. The hydrolyzate was analyzed for monosaccharides as dis-
cussed below (section IV-C-6, page 74).
Mild Acid Hydrolysis
If the polysaccharides under investigation are water soluble
and if hydrolysis can be accomplished under mild acid conditions,
treatment with strong acids as discussed above (section IV-C-3,
this page)is not desirable because of clegradative side reactions.
Since the hot-water-extracted solids were water soluble, a mild
acid hydrolysis (3.0% sulfuric acid) was compared to the 72.0%
sulfuric acid hydrolysis. The hydrolyzate was analyzed for amino
acids and monosaccharicles as discussed below (section IV-C-6,
71

72
page 74). As was the case in the strong acid hydrolysis, the insol-
uble portion of the extract had a reddish appearance. This precipi-
tate was found to persist later even after purification of the water-
soluble solids.
5. Qualitative Amino Acid Analysis by Paper Chromatography
Quantitative analysis showed that the hot-water-soluble solids
contained 3. 63% nitrogen. Bark has been known to contain both
protein and non-protein nitrogen (58, ID. 625), as well as free amino
acids. At least 5 water-soluble proteins have been detected. The
cambium layer of Douglas-fir bark has also been shown to contain
protein (53), and possibly some cambium layer may have remained
with the inner bark during the sample preparation.
The hydrolyzate from the mild acid hydrolysis (3.0% sulfuric
acid) of the hot-water-soluble solids was examined for amino acids
by two-dimensional paper chromatography.
The chromatograms showed several purple spots but the spots
were not well resolved (Figure 10, page 73). Lai (68) found that
3. 0% sulfuric acid hydrolysis did not appear to be strong enough to
hydrolyze the proteins into their component amino acids. The strong
acid treatment (72.0% sulfuric acid) better hydrolyzed the proteins
into their component amino acids. The evidence in the present work
shows that proteins were extracted by the hot-water treatment.

0Origin
Solvent Front
2. n-Butanol-Formic acid-water (20:6:5, v/v/v)
73
Figure 10. Two dimensional paper chromatogram of the amino acids in the hydrolyzateof the hot-water-soluble solids of Douglas-fir inner bark. Spray: ninhydrinin n-butanol.

6. Carbohydrate Analysis by Paper Chromatography
Paper chromatography of the original extract failed to detect
free monosaccharides. Therefore, the carbohydrates present in
the hot-water-soluble fraction are considered to be in the form of
polysaccharides.
The mixture of monosaccharide sugars resulting from acid
hydrolysis of the hot-water-soluble solids was well resolved by
paper chromatography. The chromatograms showed a very large
amount of glucose, a moderate amount of arabinose, a slight amount
of galactose, trace amounts of xylose and rhamnose and almost no
mannose (Figure 11, page 75).
The hydrolyzates from both the strong acid hydrolysis and
mild acid hydrolysis contained what appeared to be identical mono-
saccharides by paper chromatography. Therefore, strong acid
hydrolysis was not necessary, and to avoid degradation, the 3.0%
sulfuric acid method was used in all subsequent hydrolyses.
C. H. Chen (24) has isolated the monosaccharides glucose,
galactose, mannose, arabinose and xylose from acid hydrolyzates
of Holocellulose A (Chart 1, page 66) of Douglas-fir inner bark.
He has prepared crystalline derivatives of these compounds and has
thus definitely proven their presence.
The five major sugars, glucose, mannose, galactose,
74

Galactose
Glucose
Mannose
Arabinose
Xylose
Rhamnose
o O0 ° 00
000 0
0
Figure 11. Paper chromatogram of the acid hydrolyzate of the hot-water-solublefraction of the inner bark of Douglas-fir. Solvent: ethyl acetate-pyridine-water (8:2:1 v/v/v).
75
a aa4-Jcd a>
60 0)
cd
x
6 Ti0 ct
_0,00
ct al
:4a..V)
0' 'r<C
' -',.
2"0
X
6tivsr.d0
ct,'",0040
06czd
....... --I --- -- -.... 0 -. 0 -.-- --- -- ar

76
arabinose and xylose are those ordinarily found in wood and wood
pulp (106). Rhamnose in Douglas-fir bark was first reported by Lai
(68) in the acidified acid hydrolyzate of the sodiu.m-chlorite-soluble
fraction. Its identification in the acid hydrolyzate of the hot-water-
soluble solids supports its finding in the polysaccharides of Douglas-
fir inner bark.
7. Purification of the Hot-Water-Soluble Solids
Chart 3 (page 77) outlines the scheme followed for the purifica-
tion of the polysaccharides extracted by hot water. When the freeze-
dried sample was redissolved in distilled water at room temperature,
it was found that 29.8% of the sample would not dissolve. The in-
soluble portion (Fraction A) gave a strong positive reaction to the
iodine test showing the presence of starch and a negative test to the
ferric chloride-potassium ferricyanide solution showing the absence
of phenolics. Strong acid hydrolysis and paper chromatography
indicated the presence of glucose only. No traces of other mono-
saccharides could be detected. Microscopic examination of this
sample showed some fiber debris and irregularly shaped materials
which were presumably starch granules. Since only glucose was
detected and the most obvious sources of this monosaccharide were
starch and cellulose, this fraction was not investigated further.
Ethanol was added to the water-soluble portion to provide a

Hot-Water-Soluble Solids from Douglas-fir Inner Bark(11.1 g from 100.0 g of original inner bark, see Chart 1, page
Extraction with waterat room temperature
Water solubles 7.8 g Residue 3.3 gFraction A
Precipitation with70% ethanol thencentrifuge
70% Ethanol Solubles Residue 2.9 g
4.9 g, "Fraction C" "Fraction B"
HT-1000 thenDiazyme L 30then dialysis
Dialyzables Non-dialyzables 1.1 g
Chymotrypsin thentrypsin then dialysis
Dialyzables
77
Chart 3. Purification of the polysaccharides extracted by hot water.
Precipitate Non-dialyza les 0.16 g"Fraction D"

78
solution 70.0% in ethanol. A precipitate formed which amounted to
25.9% of the original water-soluble solids. These precipitated solids
were labelled E raction B (Chart 3, page 77). Those solids which
remained soluble in the 70. 0% ethanol solution (44. 3% of the original
water-soluble solids) were labelled Fraction C (Chart 3, page 77).
Fraction C was found to react strongly with ferric chloride-potassium
ferricyanide indicating the presence of phenolics but gave no reaction
with iodine indicating the absence of starch. This fraction was not
studied further. Polysaccharides which may be in this fraction would
be expected to possess a very low molecular weight.
The fraction insoluble in the 70.0% ethanol-water mixture
(Fraction B) gave a positive reaction with iodine indicating the
presence of starch. It also gave a slightly positive reaction with
ferric chloride-potassium ferricyanide, thus indicating that separa-
tion of the polysaccharides from the phenolics was not complete.
Hydrolysis of Fraction B and two-dimensional paper chro-
matography of the hydrolyzate showed the presence of amino acids
(Figure 12, page 79). Six major spots, seven light spots, five very
light spots and two large smears showed on the paper chromato-
gram. The identification of these amino acids was not investigated
further because the main objective of this study was the carbo-
hydrates. However, the paper chromatogram (Figure 12, page 79)
definitely showed the presence of protein in the inner bark of

2. n-Butanol-formic acid-water (20:6:5, v/v/v)
Figure 12. Two dimensional paper chromatogram of the amino acids in the acidhydrolyz ate of Fraction B. Spray: ninhydrin in n-butanol.
79

80
Douglas-fir.
Paper chromatography of the monosaccharides showed the same
relative amounts of sugars as found in the whole hot-water-soluble
fraction (Section IV-C-6, page 74).
8. Enzyme Hydrolysis to Remove Starch
At the end of the hydrolysis the reaction mixture was dialyzed
(Chart 3, page 77). The dialyzate showed the presence of glucose
only, both before and after hydrolysis with acid. No other mono-
saccharides were detected.
The enzymes used in this investigation were a-amylases and
therefore specific for the hydrolysis of the 4-0-a-D-glucopyranosyl-
type bond. The fact that glucose was released proves the presence
of an a-D-(l--4)-glucan, undoubtedly the amylose portion of starch.
This represents the first time that starch has been shown by chemical
analysis to be present in the inner bark of Douglas-fir. The starch
undoubtedly acts as a food reserve for the living cells in the inner bark.
The non-dialyzables from the enzyme hydrolysis showed a
blue color with iodine indicator. Thus the a-amylases did not
hydrolyze all of the starch. This was undoubtedly due to the pres-
ence of some amylopectin in the starch molecule. Amylopectins
possesses some a-D-(1 -.6)-glucan branch chains which are not
cleaved by a-amylases. The residue was, therefore, expected to

contain some residual starch molecules.
9. Enzyme Hydrolysis to Remove Protein
The original hot-water-soluble solids were found to have a
nitrogen content of 3.63%. The protein content of natural products
is generally calculated as the product of the nitrogen percentage and
the factor 6. 25, which is the usual factor used for conversion of
nitrogen content to protein content. In accordance with this calcu-
lation the hot-water-soluble solids of the inner bark of Douglas-fir
contained 21.8% protein. This appeared to be a very high protein
content, but when it was corrected to the original bark weight (Chart
1, page 66) the value was 2.42% protein in the inner bark of Douglas-
fir. Values for bark proteins which have been reported are 5.0%
for the inner bark of birch and an average of 21.6% for the inner
bark of black locust (58, p. 625). Therefore, in comparison,
Douglas-fir bark appears quite low in protein content.
The quantitative nitrogen content (Kjeldahl) of Fraction D
(Chart 3, page 77) was 1. 57%. This proved to be a reversal in
nitrogen content. The original water-soluble solids contained 3.63%
nitrogen as previously mentioned. Extraction with water at room
temperature and precipitation with 70% ethanol provided Fraction
B (Chart 3, page 77) which contained 0.28% nitrogen. Apparently
in the re-precipitation of the water-soluble solids, a large amount
81

82
of the protein remained in solution with the phenolic compounds.
In an effort to obtain purer polysaccharide samples, and in particu-
lar to remove the starch, Fraction B was treated with starch
hydrolyzing enzymes (HT-1000 and Diazyme L30) and with protein
hydrolyzing enzymes (chymotrypsin and trypsin) (Chart 3, page 77).
The nitrogen content of the final product (Fraction D) increased to
1.57%. It would appear from this increase in nitrogen content that
not all of the enzymes were removed by dialysis. Therefore, the
net result of the treatments was to introduce protein impurities into
the sample.
Fraction D was hydrolyzed with 3.0% sulfuric acid and paper
chromatographed (Figure 13, page 83). The paper chromatogram
showed a large reduction in the amount of glucose present, relative
to the amount of the other monosacchatides. This is evident when
Figure 11 (page 75) is compared to Figure 13 (page 83). Glucose
is still the major monosaccharide present, although the spots for
arabinose and galactose are now more prominent relative to the
glucose spot. A part of the glucose residues may come from
residual starch that was not destroyed by the enzyme (Section IV-C-8,
page 80). It was thought, however, that another treatment with the
starch enzyme would not completely remove the starch and that it
might even prove detrimental to the other polysaccharides that were
present. Starch enzyme treatment was carried out at a pH of 3.8-4.2

Galactose
Glucose
Mannose
Arabinose
Xylose
Rhamnose
'0 (1), 0cd 0 0
'17...
cd cd
V) ei:.
0
Figure 13. Paper chromatogram of the acid hydrolyzate of Fraction D.Solvent: ethyl acetate-pyridine-water (8: 2:1, v/v/v).
83

and at this slightly acidic condition, there was a distinct possibility
that acid hydrolysis might destroy the polysaccharides that were
the object of the purification process.
10. Carbohydrate Analysis by Gas-Liquid Chromatography
Carbohydrates are neither heat resistant nor volatile and so
derivatives must be prepared which will volatilize without degrada-
tion in order to perform gas-liquid chromatographic analysis. There
are numerous derivatives which have been tried but the ones most
commonly used today are the "alditol acetates." In the preparation
of the alditol acetates the monosaccharides are first treated with
sodium borohydride to reduce the aldehyde function to the alcohol
function. This has the result of preventing ring isomerization of
the pyranose and furanose forms and so prevents the formation of
alpha and beta forms of the sugars. The end result is only one form,
the alditol, for each of the monosaccharide sugars, rather than three,
four or even five isomers. The acetates are synthesized from the
alditols to make a volatile, heat resistant derivative.
The gas chromatographic resolutions of the alditol acetates
prepared from Fractions B and D (Chart 3, page 77) were excellent
(Figures 14 and 15,pages 85, 86). The areas under the peaks of the
different sugars may be compared to each other to give relative
weight values. The areas under the peaks of the other sugars are
84

30
Retention Time, MinutesFigure 14. Gas-liquid chromatographic separation of the alditol acetates from the acid hydrolyzate of Fraction E. Peak "a" is from rhamnose, "b"
from arabinose, "c" from xylose, "d" from galactose, "e" from glucose. Conditions: Column, 6.5% ECNSS-M on 100/120 mesh GasChrom Q, 6 ft x 1/8 in 0.D. stainless steel; injection port 200°; detector 230°; column temp. 175° isothermal; helium flow 30 ml/min;range setting 102, attenuation setting 16.
40 500 10 20
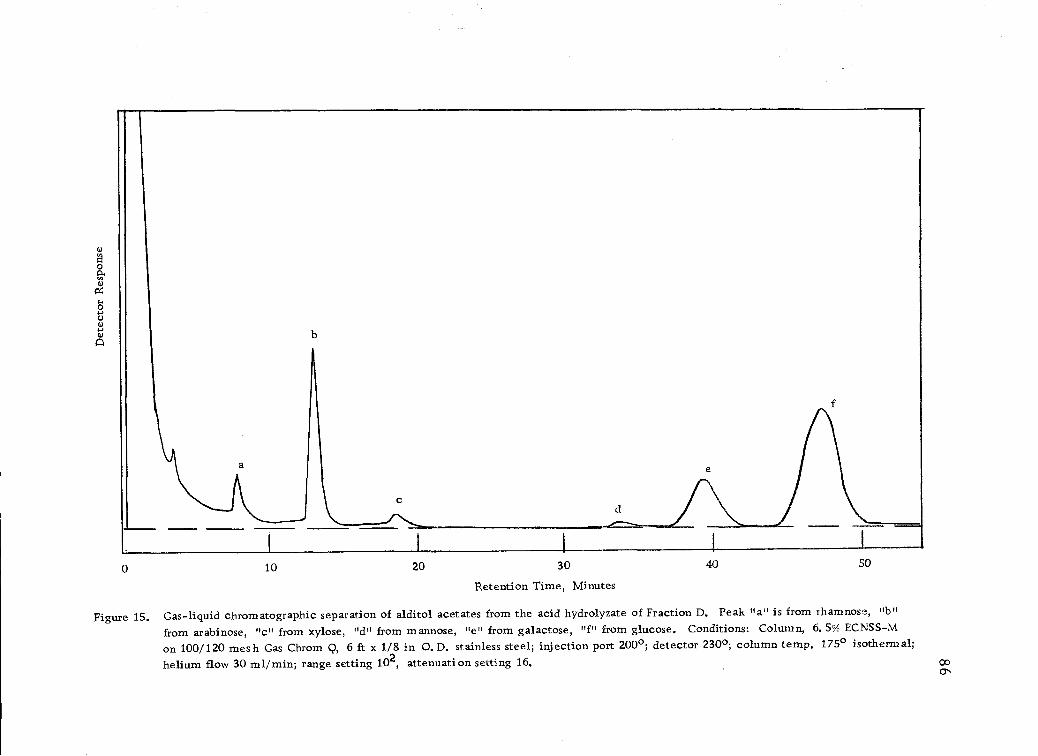
0 10 20 30 40 50
Retention Time, Minutes
Figure 15. Gas-liquid chromatographic separation of alditol acetates from the acid hydrolyzate of Fraction D. Peak "a" is from rhamnose,
from arabinose, "c" from xylose, "d" from mannose, "e" from galactose, "f" from glucose. Conditions: Column, 6.5% ECNSS-M
on 100/120 mesh Gas Chrom Q, 6 ft x 1/8 in 0.D. stainless steel; injection port 200°; detector 230°; column temp. 175° isothermal;helium flow 30 ml/min; range setting 102, attenuation setting 16.

87
compared to the area of the derivative for galactose which was taken
as 1.0. For Fraction B (Chart 3, page 77) the areas under the curves
were: galactitol hexaacetate, 1.0; arabinitol pentaacetate, 1.4; and
glucitol hexaacetate, 32. J. Only traces of rhamnitol pentaacetate
(0.1), xylitol pentaacetate and mannitol hexaacetate were found.
For Fraction D (Chart 3, page 77) the areas were galactitol hexa-
acetate, 1.0; arabinitol pentaacetate, 1.3; glucitol hexaacetate, 2.9;
and traces of rhamnitol pentaacetate, xylitol pentaacetate and mannitol
hexaacetate. These relative areas are in agreement with the rela-
tive area and intensity of color developed by the spots on the paper
chromatograms of both samples (Figures 11 and 13, pages 75,83).
The starch enzyme treatment resulted in a decrease of glucitol
hexaacetate from 32.4 units to 2.9 units when compared with galac-
titol hexaacetate. The ratio (1. 4:1.0) of arabinose to galactose in
Fraction B is the same as in Fraction D (1.3:1.0), allowing for
errors in measurements. The results show that the starch enzyme
was effective in reducing the amount of starch in the sample. In
section III-C-8 (page 46) it was mentioned that the paper chromato-
gram of the dialyzate (hydrolyzed and unhydrolyzed) detected the
presence of glucose only. The almost constant ratio of arabinose
to galactose in Fraction B and Fraction D confirms what the paper
chromatograms have already shown, that the starch enzymes acted
to hydrolyze the starch only. All other polysaccharides were left

88
intact by the starch enzyme treatment. Therefore, it is concluded
that the major carbohydrate component of the hot-water-extract is
starch.
The arabinose and galactose monosaccharides released by
acid hydrolysis undoubtedly exist in the inner bark as part of L-
arabino-D-galactan polysaccharides. Water-soluble, highly branched
L-arabino-D-galactans are known to be present in the wood of con-=ifers (113, p. 203). These polysaccharides are the only major
wood glycans that can be isolated in good yield by extraction of
wood with water before delignification. Their ease of extraction
and their useful qualities as gums have brought them into com-
mercial production marketed as the commercial gum, Stractan.
Since the L-arabino-D-galactans are common to the wood in==
conifers, it is considered possible that the arabinose and galactose
containing polysaccharides isolated in the present work are similar.
This represents the first time that possible L-arabino-D-galactan
polysaccharides have been reported in Douglas-fir bark. Because
of the large volumes of Douglas-fir bark which are waste products,
it is possible that the bark could become of interest as a raw-material
source for these polymers since they have already been marketed
as commercial gums.

89
D. Characterization of the Xylan Fraction
Ash Determination of the Crude Xylan
The crude xylan had an ash content of 29. 7% on a dry weight
basis. This value is similar to the ash content (30. 7%) of a xylan
that Beelik and co-workers (10) isolated from a softwood. The ash
was probably composed mostly of barium and potassium oxides.
Barium hydroxide had been used to prevent polysaccharides which
contained mannose units from solubilizing in solutions of aqueous
alkali, while potassium hydroxide solution was used as the solvent
for the extraction of the xylan (Chart 2, page 69). No further effort
was made to analyze the ash.
Precipitation of Excess Barium andDialysis of the Crude Xylan
The ash content (29. 7%) of the crude xylan was high and it was
desirable that the amount of inorganic material in the xylan be reduced
to a lower level before further investigations were performed. The
barium ions were precipitated as their sulfate salts by the addition
of excess sulfuric acid. The potassium ions were removed by
dialysis as was the excess sulfuric acid. The resulting sample was
labelled xylan. The yield based on the moisture-free crude xylan
was 69.8%. The ash content was now 4. 4%.

Optical Rotation of the Xylan
The xylan sample was completely soluble in water and there-
fore optical rotation readings were taken in an aqueous medium.
The xylan was found to have a specific rotation of -30.5°. The
negative rotation of the polymer indicated that the glycosidic linkage
was p-D (118). Beelik, Conca, Hamilton and Partlow (10), whose
extraction procedure was modified for this study, obtained a xylan
from western hemlock [Tsuga heterophylla (Raf. ) Sarg.] with a spe-
cific rotation of -34°. Therefore, the xylans may be very similar
in structure. However, the authors proposed no structure for their
xyl an.
Qualitative Uronic Acid Analysis by Color Reactions
Hexuronic acids react with carbazole to form 5-carboxy-2-
formylfuran which, when treated with concentrated sulfuric acid,
yields a colored product with an absorption maximum at 535 nm.
The carbazole-sulfuric acid color reaction is claimed to be reason-
ably specific for hexuronic acids (29). This was verified by testing
monosaccharides, oxalic acid, and benzoic acid. These compounds
showed no reaction, indicating that the simple sugars, and carboxylic
acid functions other than those in uronic acids, did not interfere with
the color reaction.
90

9 1
The 3.0% sulfuric acid hydrolyzate of the xylan showed a pos-
itive purple color, indicating the presence of hexuronic acids. The
reaction showed a maximum absorption in the visible region at
535±1 nm (Figure 16, page 9 2 ) as described for the test.
The presence of hexuronic acids in the xylan is in accordance
with the findings of other researchers. Few xylans contain D-xylose
residues only. Most bear side chains of other sugars. The most
common side chain is 4-0-methyl-a-D-glucopyranosyluronic acid (20,
100, 105, 107). The major softwood hemicellulose that contains
D-xylose residues is an L-arabino-(4-0-methyl-D-g1ucurono)-D-
xylan. Sometimes unmethylated D-glucuronic acid occurs. Each
of these residues is usually joined to the D-xylopyranose chain
residues by a-D-(1 -.2) linkages. Xylans isolated from bark also
contain D-glucuronic acid side chains as evidenced by the xylans
reported by Timell (106) in his assay of the polysaccharides from
the bark of gymnosperms.
5. Paper Chromatography and Gas-LiquidChromatography of the Xylan Hydrolyzate
The paper chromatogram of the hydrolyzed xylan (Figure 17,
page 93) showed the presence of a large amount of xylose, a mod-
erate amount of arabinose, and trace amounts of glucose, galactose,
and rhamnose. No trace of niannose could be detected, thus showing

oo
100
80
60En
0
ELI 40. '
20
--a
-
350 400 450 500 550 600 650 700 750
Wavelength (nm)
Figure 16. Spectrum of the reaction products from the treatment of the xylan hydrolyzate with carbazole-sulfuric acid.
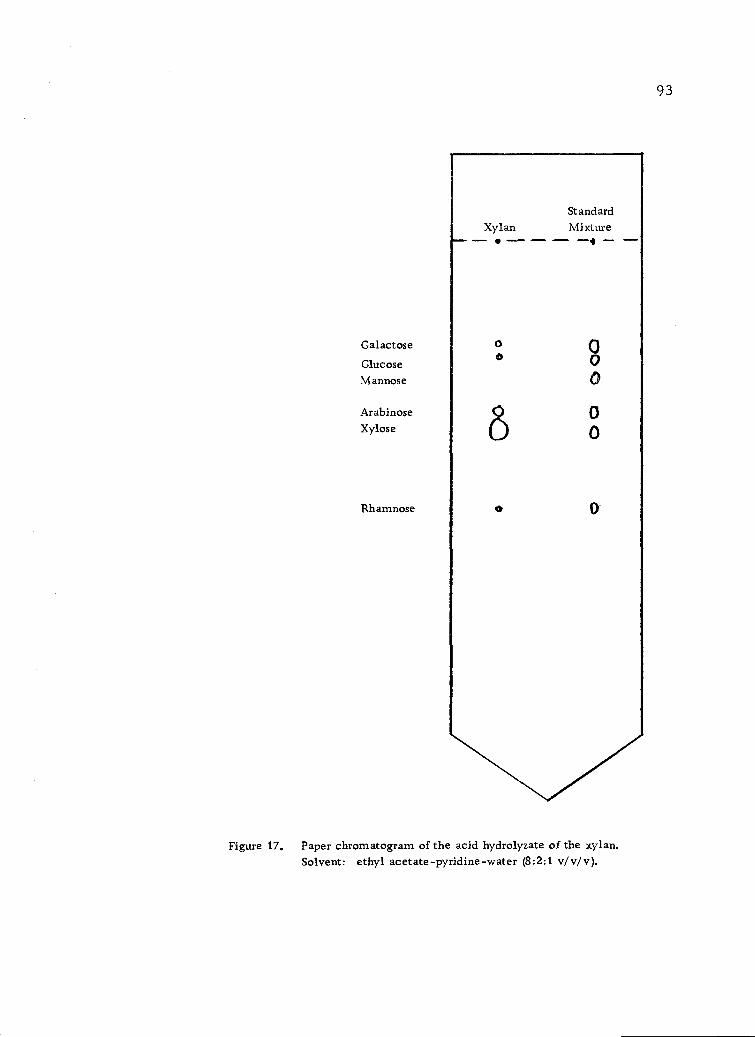
Galactose
GlucoseMannose
ArabinoseXylose
Rhamnose
StandardXylan Mixture
o
0
Figure 17. Paper chromatogram of the acid hydrolyzate of the xylan.Solvent: ethyl acetate -pyridine -water (8:2:1 v/ v/ v ).
93

94
that the impregnation of the holocellulose with barium hydroxide
solution was successful in preventing the manno se-containing poly-
saccharides from being extracted with the xylan. The use of potas-
sium hydroxide further reduced the possibility that mannose-contain-
ing polysaccharides would be co-extracted with the xylan. The xylan
sample, therefore, appeared to be relatively free of other polysac-
charides.
The gas-liquid-chromatographic spectrum (Figure 18, page
95) showed that the major peak was that of xylitol pentaacetate
(relative area, 4. 5) followed by glucitol hexaacetate (relative area,
1. 2) and arabinitol pentaacetate (relative area, 1.0). There was
a trace of galactitol pentaacetate and very slight traces of rhamnitol
pentaacetate and mannitol hexaacetate. Since the peak area is pro-
portional to the weight (77), the relative area of glucitol hexaacetate
was corrected to a molecular basis. This was done by multiplying
the area (1. 2) by the factor 362/434 (xylitol pentaacetate, MW =
362; glucitol hexaacetate, MW = 434). This calculation resulted in
a molecular ratio of xylose to glucuronic acid of 4.5:1 from the
parent xylan polymer. The molecular ratio of xylose to arabinose
from the parent xylan polymer is the same as the peak area ratio
(4. 5:1) of their alditol acetates.
The presence of hexuronic acids in the xylan was demonstrated
in section IV-D-4 (page 90). The preparation of the alditol acetates

Retention time in minutes
Figure 18. Gas-liquid chromatogram of alditol acetates from xylanReak "a" is from rhamnose; "b" from arabinose; "c" fromxylose; "d" from galactose; "e" from glucose. Conditions: Column, 6.5% ECNSS-M on 100/120 mesh Gas ChromQ, 6 ft x 1/8 in. 0.D. stainless steel; injection port 200°; detector 230°; column temp. 1800 isothermal; heliumflow 30 ml/min; range setting 102, attenuation setting 16.
1 1 j I 1 1
0 4 8 1.2 16 20 24 28 32 36 40 44 48 52 56

RCHO +++ 2 Cu + 5 OH-xylose-4- RCO-2 +Cu20 + 3 H20
2
103+ 5 I + 6H+ -4- 3 12 + 3 H20
96
for gas-liquid chromatographic analysis involved a reduction with
sodium borohydride (section III-D-5, page 53). The hexuronic acids
were reduced to the primary alcohols and appeared, after acetyla-
tion, as hexitol hexaacetate peaks on the spectrum from the gas-
liquid chromatograph. Since the only hexitol hexaacetate peak to
appear on the spectrum in any quantity (Figure 18, page 95), was
glucitol hexaacetate, it was concluded that the hexuronic acid in the
xylan was glucuronic acid.
Therefore, the results of the gas-liquid chromatographic analy-
sis showed that the xylan polysaccharide contained xylose, arabinose,
and glucuronic acid residues in a ratio of 4. 5:1. 0:1. 0.
6. Reducing End-Group Analysis (Somogyi Method)
The Somogyi method is based on the ability of certain sugars
(reducing sugars) to act as reducing agents. The sugar reacts with
Cu++ in the aqueous alkaline medium to produce cuprous oxide. The
cuprous oxide is oxidized by iodine back to Cu++ and the excess iodine
is titrated with thiosulfate. The reactions are:

Cu20 + 2H+ +12
2Cu++ + 21 + H20
12 + 2S203 -* 21-
This method has been widely and successfully applied on both
milligram and microgram quantities, both titrimetrically and colori-
metrically. Its accuracy over a wide range of sugar concentrations,
the ease and rapidity of operation, and its proven reliability place
it above other micro-oxidation methods (52).
The curve for the reducing power of xylose is shown in Figure
19 (page 98). The weight of xylose is plotted on the abscissa. The
titer of thiosulfate (0.005 N) for the xylose solution subtracted from
the titer for the blank solution is located on the ordinate. The differ-
ence in titer for the blank and the xylan is projected to the xylose
curve and the equivalent of reducing xylose is obtained on the
abscissa.
The results of the present Somogyi analysis (triplicate samples)
showed an average reducing power of 0.07 mg of xylose equivalents
for a sample weight of 8.40 mg of starting xylan.
The calculation of the molecular weight of the xylan from the
reducing-end-group analysis is as follows:
Let A = reducing end group of xylan in terms of mg xylose = 0.07 mg
W = weight of xylan in sample = 8.40 mg
97

10
8
mg Xylose
Figure 19. Standard curve for Somogyi titration of xylose.
R = volume thiosulfate (.005 N) used to titrate blank.
R = volume thiosulfate (.005 N) to titrate reaction mixture,
98
2
0
0 o. s 1.0 1.5 2.0

(MW)XR = molecular weight of the reducing-end group of the xylan
= 149
(MW)X = molecular weight of xylan
MA= moles reducing-end group
A - 0.07MA (MW)XR 149
(MW)X = MA
W(MW)x -A
8. 40 8.40 X 1490.07 - 0.07
(MW)x = 1.8 x 104
According to this end-group analysis, the xylan isolated from
Douglas-fir bark has an average molecular weight of 1.8 X 104.
From this molecular weight and the analysis by gas-liquid chro-
matography a degree of polymerization (DP) for the xylan can be
calculated.
The ratio of sugar residues by gas-liquid chromatography is
anhydroxylose-anhydroarabinose-anhydroglucuronic acid (4.5:1:1
or 9:2:2).
The molecular weight of anhydroxylose is 132.
The molecular weight of anhydroarabinose is 132.
The molecular weight of anhydroglucuronic acid is 176.
Therefore, the molecular weight of an average repeating unit is
(9X132) + (2X 132) + (2X 176) = 1804.
149
99

C-4" 0
1
100
Therefore, the number of repeating units in any given molecular
weight of 1.8 X 104 is 1.8 X 104/1804 = 9.98. These data are inter-
preted to mean that there are 10 repeating units in any given xylan
molecule.
If the xylan from the inner bark of Douglas-fir is similar in
structure to the xylans so far isolated from the wood and bark of
gymnosperms, then its gross framework consists of a backbone of
repeating anhydroxylose units with the arabinose and glucuronic
acid residues attached as side chains (20, 100, 105, 107), Since
the number of repeating units in the xylan is 10 and the ratio of sugar
residues is anhydroxylose-anhydroarabinose-anhydroglucuronic acid
(9:2:2) then the DP of the anhydroxylose backbone is 90, and there
are 2 anhydroarabinose and 2 anhydroglucuronic acid side chains
for every 9 of these repeating anhydroxylose units.
7. Viscosity Measurements
The calculations of the viscosity data were made as follows:
= Kpt
= Tv%
= (11-r1 )/ri =sp o o r
Tisp/C = reduced viscosity
[rd lirn (1-1 /C)sp

101
where
= viscosity in centipoise (cp)
rio= viscosity in pure solvent
= relative viscosity
sp= specific viscosity
ird = intrinsic viscosity in deciliters/gram (dug)
K = viscometer constant
p = density at 25. 0° ±0. 01
t = time in seconds for solution to flow through the viscometer
c = concentration in g/dl
The data for the viscosity measurements are shown in Tables
4 (page 102), 5 (page 103), and 6 (page 104).
The plots of the reduced viscosities against the concentrations
are shown in Figure 20 (page 105). The values for the intrinsic vis-
cosities were obtained by extrapolating the plots to zero concentra-
tion.
The intrinsic viscosity of the xylan in water was 0.84 dl/g.
This value was much higher than the value of 0. 42 dl/g obtained in
M-diethylenediamine copper II solution. The reduced viscosity
(Figure 20, page 105) in water showed a marked increase with dilu-
tion. This behavior is typical of a polyelectrolyte (38). At high
concentration, the molecules overlapped one another, but upon
dilution, the polymer expanded as net charges developed. At very

Table 4. Viscosity measurements in distilled water.a
Conc. Density Time
(0(11) (g/m1) (secs) ( cp)
0.453 0.99722 138.4 1.1345
0.302 0.99506 128.4 1.0502
0.226 0.99398 125.6 1.0262
0.181 0.99333 122.7 1.0019
0.151 0.99290 121.0 0.9876
Water 0.99074c 109.8 0.8937c
aAverage of triplicate determinations.b calculated from the expression 11 = Kpt where K = 0.00822, a viscometer constant determined from
the known viscosity and density of water at 25±0.01°.
cPerry, J. H. Chemical Engineers' Handbook. 3rd edition. New York, N. Y. McGraw-Hill BookCo., Inc. 1950. p. 175.
71r71sp
/Csp(dug)
0.5948
0.5798
0.6561
0.6689
0.6955
1.2694 0.2694
1.1751 0.1751
1.1483 0.1483
1.1211 0.1211
1.1050 0.1050

Table 5. Viscosity measurements in M aqueous sodium chloride solution.b
Conc. Density Timer1 li ri 1 /Cr sp sp(g/dl) (g/ml) (secs) (cp) (dl/g)
aAverage of triplicate determinations.bi calculated from the expression ri = Kpt where K = 0.00822, a viscometer constant determined fromthe known viscosity and density of water at 25±0.01°.
0.477 1.03584 142.2 1.2108 1.2252 0.2252 0.4722
0.318 1.03573 132.1 1.1247 1.1381 0.1381 0.4342
0.238 1.03568 127.6 1.0863 1.0993 0.0993 0.4171
0.191 1.03564 125.0 1.0641 1.0768 0.0768 0.4022
0.119 1.03560 121.6 1.0351 1.0475 0.0475 0.3992
0.095 1.03558 120.8 1.0283 1.0406 0.0406 0.4272
o 1.03552 116.1 0.9882
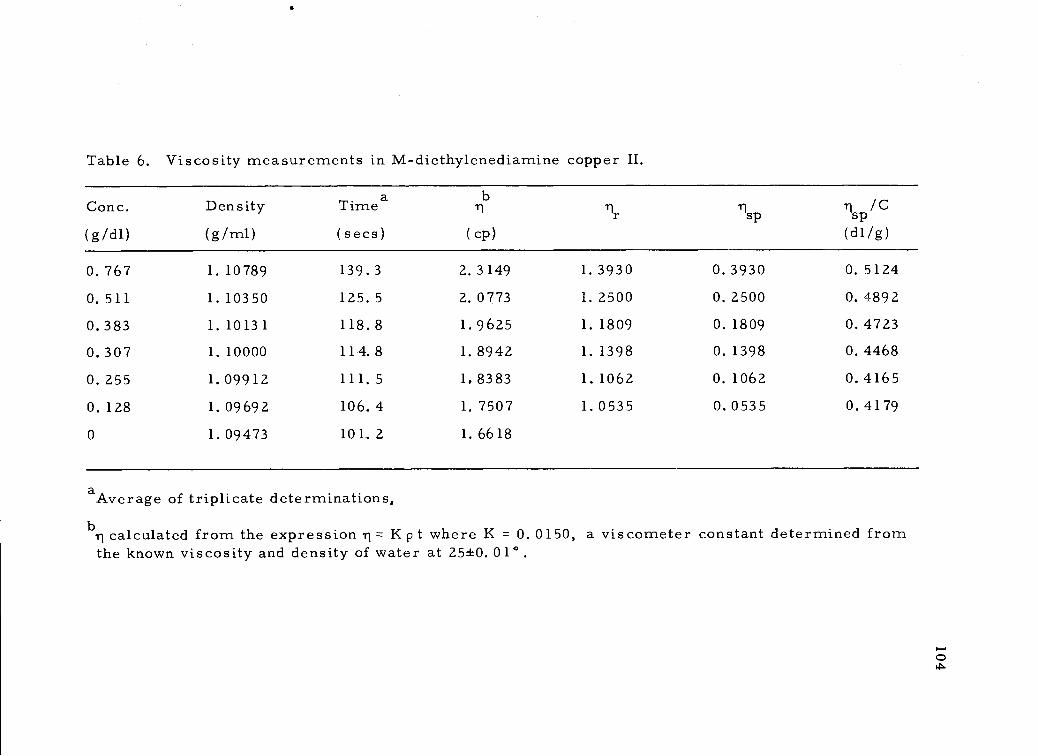
Table 6. Viscosity measurements in M-diethylenediamine copper II.
Conc. Density
(g/d1) (g/ml)
. meaTi
(secs)
aAverage of triplicate determinations,
calculated from the expression ii=Kpt where K = 0. 0150, a viscometer constant determined fromthe known viscosity and density of water at 25±0. 01° .
/Csp sp(dug)
0. 767 1. 10789 139.3 2.3149 1.3930 O. 3930 O. 5124
O. 511 1. 10350 125.5 2. 0773 1. 2500 0. 2500 0. 4892
0.383 1. 10131 118.8 1.9625 1. 1809 O. 1809 0. 4723
0.307 1.10000 114. 8 1. 8942 1. 1398 0. 1398 0. 4468
0.255 1.09912 111.5 1. 8383 1. 1062 O. 1062 0.4165
0.128 1.09692 106. 4 1. 7507 1.0535 0.0535 0.4179
0 1.09473 101.2 1.6618
1 rir( cp)
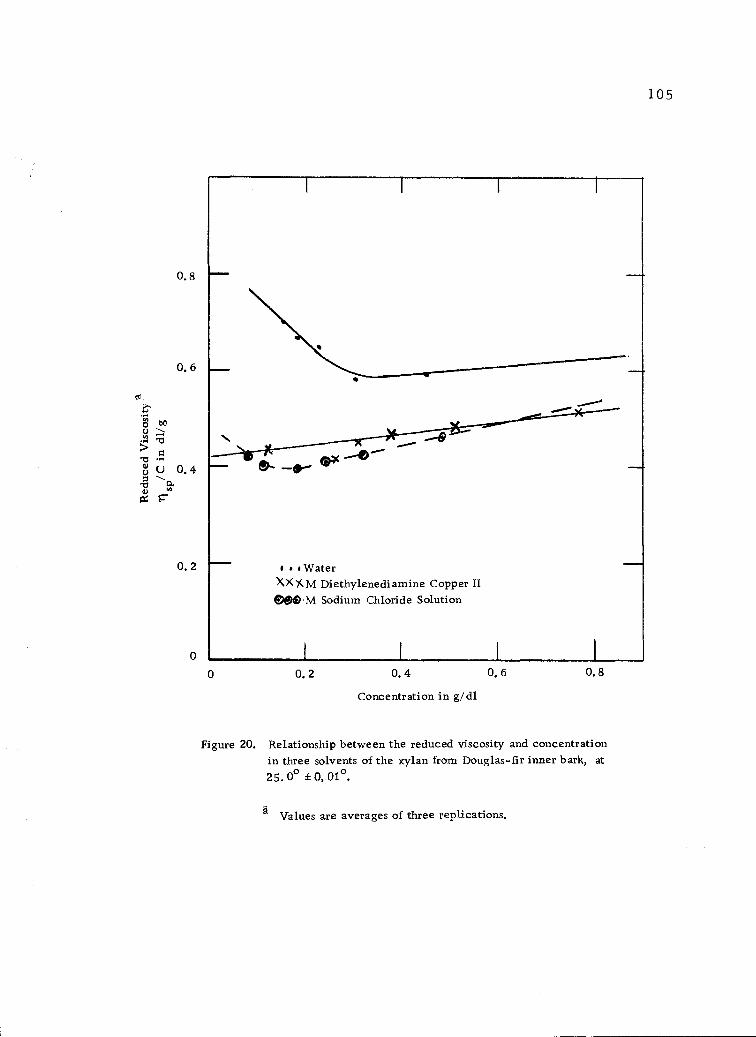
0. 8
0. 6
0.2
0
0
'WaterXX )( M Diethylenediamine Copper II&M Sodium Chloride Solution
0.2 0.4
Concentration in g/dl
Figure 20. Relationship between the reduced viscosity and concentrationin three solvents of the xylan from Douglas-fir inner bark, at
25. 00 # 0, 01°.
Values are averages of three replications.
0.6 08
105

106
high dilutions the polymer may have extended to its maximum length.
This expansion and extension of the polymer molecules were reflected
in the increasing reduced viscosity at high dilutions.
The addition of sodium chloride to the medium caused less of
an increase in reduced viscosity (H ---- 0.48 dl/g, Figure 20, page 105).
The reduced viscosity did increase at high dilution but the increase
was considerably less than in pure water, This is also typical of
a polyelectrolyte.
The behavior of the xylan as a polyelectrolyte could only be
due to the presence of uronic acids in the side chains. The presence
of hexuronic acids had previously been detected by color reactions
(section IV-D-4, page 90) and this was confirmed by the presence
of a much larger amount of glucitol hexaacetate in the gas-liquid
chromatographic results than had been evident in the paper chromato-
grams(section IV-D-5, page 9 1 ). The uronic acid side chains in the
xylan would start to give up their protons as the xylan solution
became more dilute in water. A net charge would start to develop
as the protons detached from the uronic acids and this would cause
the reduced viscosity to increase, Further dilution would increase
the net charge developed on the xylan, thus causing the molecules
to expand even more. The net result was reflected as a marked
increase in the reduced viscosity at low concentrations in both water
and aqueous sodium chloride solutions.

The reduced viscosity of the xylan in M-thethyleneciiamine
copper II solution decreased with increasing dilution which is normal
for this type of polymer (38), Extrapolation to zero concentration
provided an intrinsic viscosity [q: of 0. 42 dl/g.
Glaudemans and Time11 (38) determined the relationship be-
tween the intrinsic viscosities of glucuronoxylans in diethylene-
diamine copper II ions and their degrees of polymerization (DP) by
comparing osmotic and viscosity measurements. The relationship
is:
DPn -= K[rd
where DPn = number average degree of polymerization
[r] = intrinsic viscosity in dl/g
K = a constant
The value of the constant K has been found to be 212 for numer-
ous xylan polysaccharides (38, 105) and has been generally accepted
by researchers in the field (16, p. 708, 47, 120). Using the above
relationship with a value of 212 for the constant and the measured
value of 0.42 dl/g for the intrinsic viscosity, a number average
DP of 89 is obtained for the xylan isolated from the inner bark ofn
Douglas-fir.
The value of 212 for the constant K has been determined from
authentic xylans which possess a backbone of anhydroxylose units
107

108
with sidechains of both anhydroglucu onic acid and anhydroarabinose
residues. The DPn of 89 determined for the xylan from Douglas-fir
inner bark therefore means that the xylan possesses 89 anhydroxylose
units linked to form the backbone and the anhydroglucuronic acid and
anhydroarabinose units exist as side chains.
It has also been shown (107) that the acid side chains and the
anhydroarabinose side chains in xylans have no influence on the
intrinsic viscosity of these polysaccharides. These side chains are
short' and the intrinsic viscosity data are considered to result from
the linear part of the polysaccharides only. Therefore, the value
of 89 is considered to apply only to the linear part of the xylan
molecule.
The DPn of 89 by viscosity measurements is very close to the
DP of 90 obtained by the completely independent and quite different
reducing-end-group method discussed in section IV-D-6 (page 96).
A degree of polymerization of 90 for the anhydroxylose chain in the
xylan isolated from Douglas-fir inner bark is used in subsequent
calculations and structural determinations.
8. Gel Permeation Chromatography Determinationof Molecular Weight Distribution
The gel permeation chromatography results are shown on
Figure 21 (page 109). The xylan showed a small peak eluting at low

.3
.2
109
0 100 200 300
Elution Volume (ml)
Figure 21. Gel permeation chromatography of xylan from the inner barkof Douglas-fir.

volumes. This early peak indicated either polysaccharides of yet
higher molecular weights or acidic polysaccharides associated
through H-bonding at their carboxyl functions (94). The associa-
tion through H-bonding is a very distinct possibility since uronic
acids had previously been detected in the hydrolyzate of the xylan.
The void volume of the column was found to be 370 ml with
Blue Dextran 2000 (M = 2, 000, 000, 50.0 mg). The Blue Dextran
was found to elute out of the column completely at about 200 ml.
The xylan sample started to elute out of the column at 20 ml and by
360 ml (vo = 370 ml) was completely out of the column. The elution
curve (Figure 21, page 109) was not broad. This suggests that the
xylan may have a fairly narrow molecular-weight distribution. This
result is in agreement with results obtained on gel permeation of other
xylans (17, 32, 100). The molecular weight of 1.8 X 104 (section
IV-D-6, page 76) and the DP of 90 (section IV-D-7, page 100) there-
fore applies to most of the molecules in the xylan. The elution band
(less than 1 void volume) showed that the xylan was not too heteroge-
neous and not likely to be a mixture of polysaccharides because other
polysaccharides with similar molecular sizes would be coincidental
after the isolation and purification procedure used.
110

E. Periodate Oxidation of the Xylan
1. Acidity of the Xylan
The xylan (0.13343 g in 10 ml water) had a pH of 6.4 (Beckman,
Model 72 pH meter) (average of triplicate determinations on two
meters). The reaction with excess dilute aqueous sodium hydroxide
(0.0526 N) and the titration of the excess sodium hydroxide with
dilute hydrochloric acid (0.0406 N) showed that 2 moles of sodium
hydroxide were needed to neutralize one mole of the xylan.
The calculations (section IV-D-6, page 96) carried out on the
degree of polymerization based on end-group analysis and gas-liquid
chromatography, gave a xylan with 20 glucuronic acid side chains
per mole. The theoretical amount of sodium hydroxide that the xylan
should require, therefore, should be 20 moles. Since only 2 moles
of sodium hydroxide were consumed, 18 moles of glucuronic acid
must be bound in some way such that they are not available for the
neutralization reaction. This could only be correct if the glucuronic
acid side chains were in the form of an ester or a salt.
The ash content of the xylan (section IV-D-2, page 89) had been
previously found to be 4.4%. At the time the ash analysis was made,
this value could not be explained. The extraction procedure had
involved barium and potassium hydroxides (section III-B-2, page 37).
111

112
The barium had been precipitated as the sulfate salt and thus removed
and the xylan solution was dialyzed for 5 days (section 1II-B -2, page
37) to remove potassium and other ions plus low molecular weight
carbohydrates. The results of the acid-base titration now suggest
that the xylan was in the salt form wherein 18 of the glucuronic acid
side chains were bound to potassium. If this were so, the 4.4% ash
content of the xylan must come from the potassium bound to the
glucuronic acid.
To test the above hypothesis, a theoretical calculation was
made;A
18K+ + 9/2 0 -÷ 9 K 02 2
9K2= 9(94. 2) = 847. 8
847.8 X 100% ash as K 0 -2 41.8 X 10
- 4. 7% (theoretical value).
The theoretical value of the ash is very close to the experi-
mental value of 4.4%. In fact, if potassium were incorporated into
the molecular formula weight, a value of 4.5% ash is obtained. The
results of these calculations therefore substantiate the results of
the acid-base titration that the xylan is in salt form. It also explains
the source of the ash.
2. Analysis of Periodate Consumed
When a (1 4)-linked xylan with single glucuronic acid and

113
single arabinose side chains is oxidized with the periodate anion
the bonds in the xylan backbone and the sugar side chains are cleaved
where there are two adjacent hydroxyl groups (Figure 22, page 114).
One mole of periodate anion is consumed per bond cleaved. The
acetal linkage is stable to periodate oxidation. The glucuronic acid
side chain is prevented from over-oxidizing to formic acid and oxalic
acid by conducting the oxidation at a controlled temperature of 50 ,
in the dark, and at neutral pH (16, p. 68).
The periodate oxidation of the xylan from Douglas-fir inner
bark was essentially complete after 28 hours (Table 7, page 115;
Figure 23, page 116). The consumption of periodate anion was ob-
tained by extrapolating the rate curve back to zero time in order to
compensate for overoxidation by the periodate anion (100, 105, 120).
A value of 2.0 mM of periodate anion was consumed for a sample
weight of 0.26959 g (section III-E- 2 , page 59), the consumption
was 133. 5 moles of periodate anion per mole of xylan.
Since the xylan possessed a repeating unit of 9 anhydroxylose
units in the backbone with 2 anhydroglucuronic acid side chains and
2 anhydroarabinose side chains (section IV-D- 6, page 96), the
calculated consumption would be 11 moles of periodate consumed
per repeating unit. There were 10 repeating units so the periodate
consumed would be 110 moles plus one mole extra for the reducing-
end group plus one extra for the non-reducing end group, or a total

Figure 22. Oxidation of a branched xylan with periodate anion.
OH
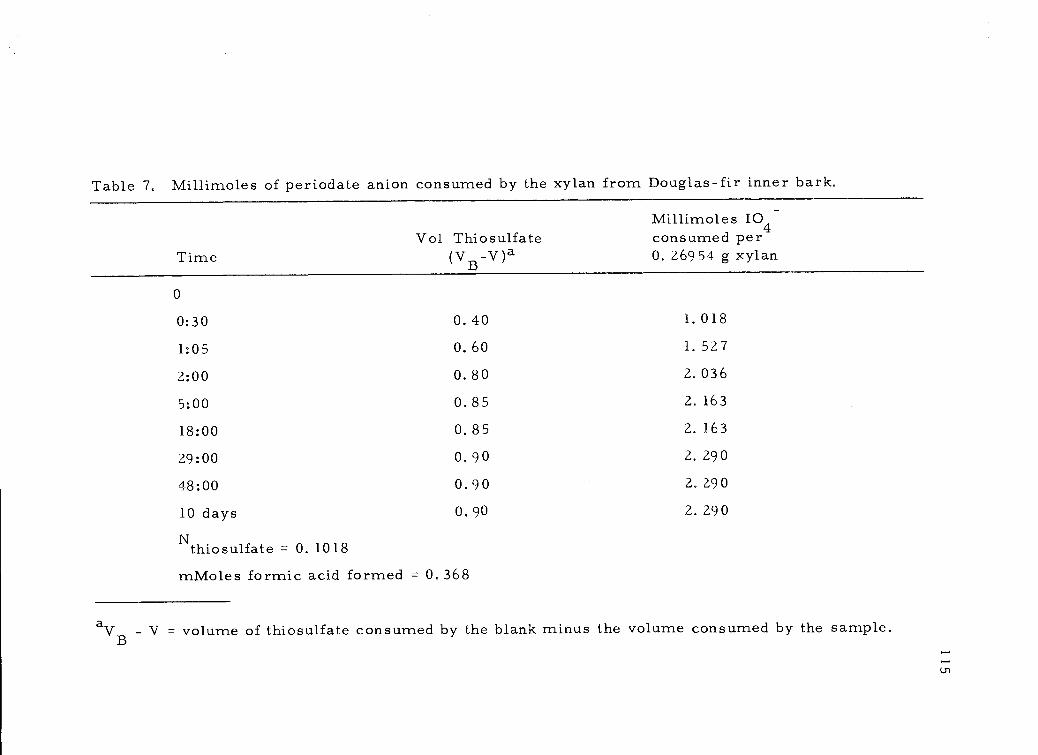
Table 7. Minim°les of periodate anion consumed by the xylan from Douglas-fir inner bark.
mMoles formic acid formed = 0.368
aVB - V = volume of thiosulfate consumed by the blank minus the volume consumed by the sample.
TimeVol Thiosulfate
(VB-V)a
-Millimoles 104consumed per0. 26954 g xylan
0
0:30
1:05
2:00
5:00
18:00
29:00
48:00
10 days
Nthiosulfate = 0. 1018
0.40
0.60
0.80
0.85
0.85
0.90
0.90
0.90
1.018
1. 527
2.036
2. 163
2. 163
2.290
2.290
2.290
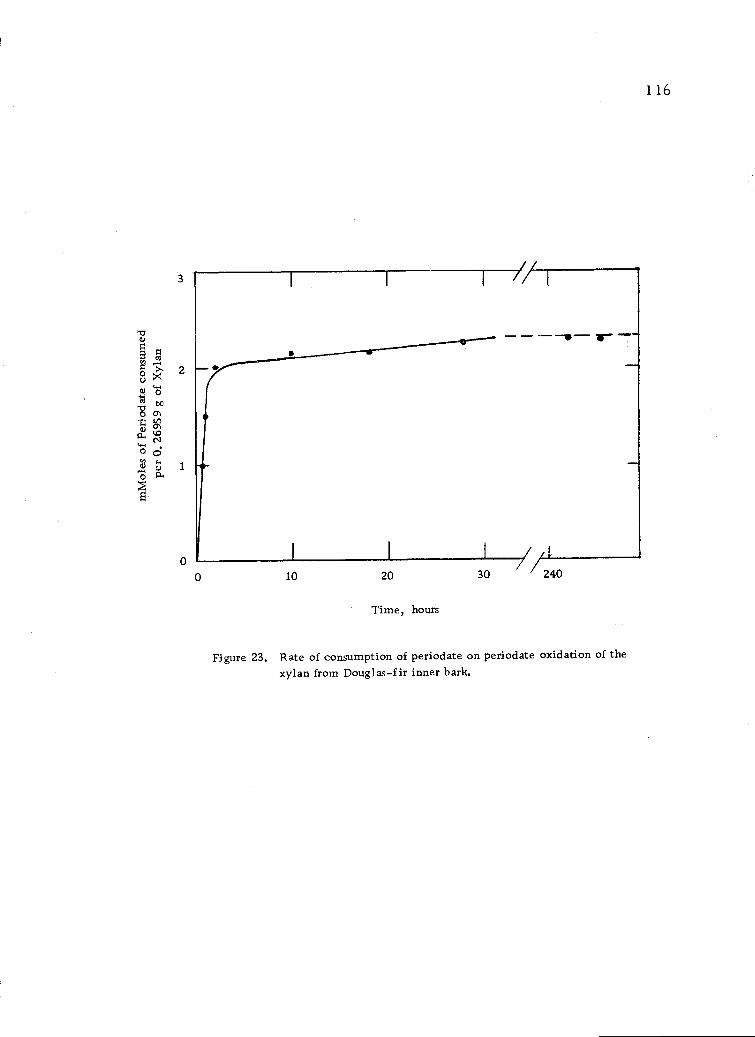
010 20 30 240
Time, hours
Figure 23. Rate of consumption of periodate on periodate oxidation of thexylan from Douglas-fir inner bark.
116

of 112 moles of periodate anion per mole of xylan.
The difference between the calculated value and the experimental
value is considered to be due to overoxidation by the periodate anion.
This tendency of overoxidation is well known for polysaccharides of
this type (38, 104, 120).
3. Analysis of Formic Acid Released
One of the products formed in the periodate oxidation of xylan
is formic acid (Figure 22, page 114). Two moles are produced from
the reducing-end group (1 mole by cleavage, 1 mole by hydrolysis)
and 1 mole each from the glucuronic acid side chains and the non-
reducing-end group. The amount of formic acid produced is thus a
measure of the number of glucuronic acid branches. This technique
has been used routinely for estimating the number of branches (100,
107, 120).
The amount of formic acid formed at the end of the oxidation
was 0.368 mM. This value corresponded to 24. 5 moles of formic
acid formed per mole of xylan (MW 1.8 X 104). The theoretical
formation of formic acid was calculated to be 23 moles per mole
of xylan. The unreacted xylan had previously been found to con-
sume 2 moles of sodium hydroxide per mole of xylan (section IV-
E-1, page 111). These 2 moles of sodium hydroxide would appear
as having titrated 2 moles of formic acid after the perioclate oxidation.
117

118
Therefore, the true amount of formic acid titrated was 22. 5 moles.
Thus the experimental value and the calculated value are well within
experimental error.
4. Complete Hydrolysis of the Polyalcohol
The paper chromatogram of the hydrolyzed polyalcohol showed
spots for xylose (strong) and arabinose (weak), and very slight traces
of galactose, glucose, and rhamnose, plus two other very light spots
which were migrating very fast (Figure 24, page 119). The gas-liquid--
chromatogram showed the presence of 3 unidentified peaks (Figure
25,page 120) which came out early. The major peaks were the xylitol
pentaacetate and arabinitol pentaacetate peaks. The areas under
the xylitol pentaacetate and the arabinitol pentaacetate peaks were
measured with a planimeter and were found to have a xylitol penta-
acetate-arabinitol pentaacetate ratio of 7:1.
A structure for the xylan from the inner bark of Douglas-fir
can now be proposed based on the experimental data. The structure for an
average repeating unit is shown on Figure 26 (page 121). The repeating
unit consists of a backbone of 18 anhydro-D-xylopyranose units with
p-D-( 1-3-4) linkages. There are seven branch points; one attached to a
possible arabinobiose unit, two to single arabinose units, and four to
single glucuronic acid units. The actual attachment of the branch
points are not known. However, it is reasonable to assume that the
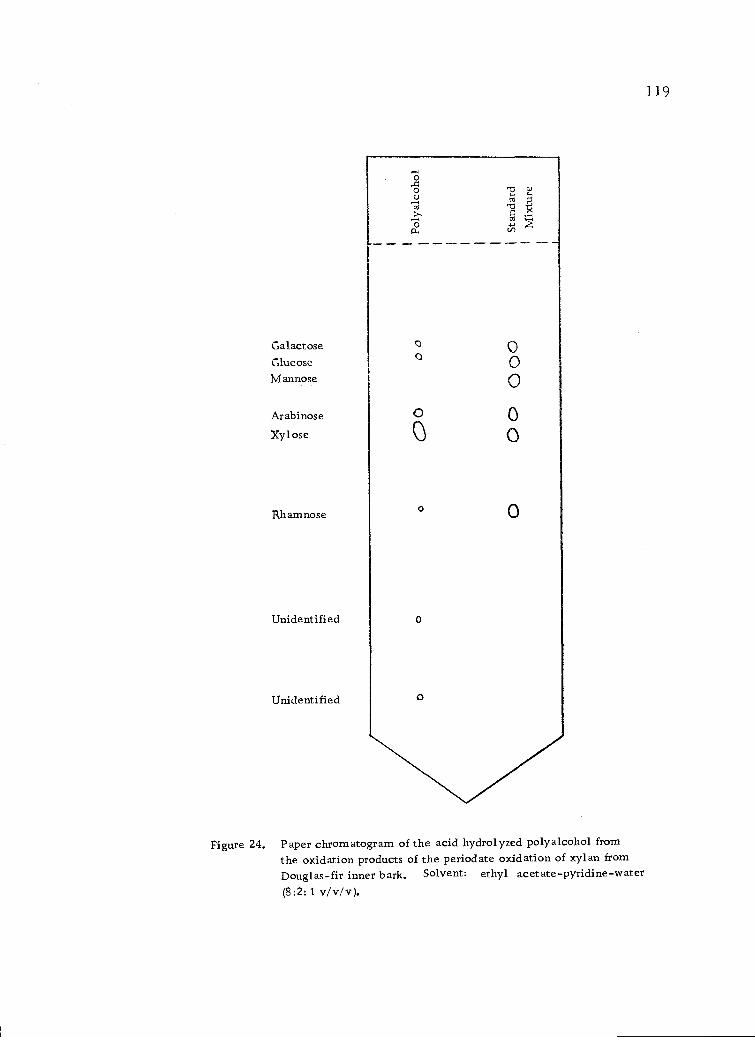
GalactoseGlucoseMannose
Arabinose
Xyl ose
Rhamnose
Unidentified
Unidentified
O.4o 7,1o--, coca -cs
4ca
o
aa
00
0
0
0
Figure 24. Paper chromatogram of the acid hydrolyzed polyalcohol fromthe oxidation products of the periodate oxidation of xylan fromDouglas-fir inner bark. Solvent: ethyl acetate-pyridine-water(8:2:1 v/v/v).
119

10 15
Retention time, minutes
Figure 25. Gas-liquid chromatographic separation of the alditol acetates from the acid hydrolyzate of the polyalcohol (oxidationproducts of the periodate oxidation of xylan). Peaks "a" are from unidentified products, "b" from arabinose, "c" isfrom xylose, "d" is from glucose. Conditions: Column 6.5% ECNSS-M on 100/120 mesh Gas Chrom Q, 6 ft x 1/8 in0.D. stainless steel; injection port 200°; detector 230°; column temp. 175° isothermal; helium flow 30 ml/min;range setting 102, attenuation setting 16.
4\140 45 50

OH OH OH OH OH
0COO
HOHOH2
Figure 26. Proposed structure for a xylan from the inner bark of Douglas-hr.

122
arabinose units have (1-.3) linkages while the glucuronic acid units
have (1-.2) linkages. This assumption is based on the structures of
xylans presented in the literature (44, 47, 107, 113, 120). The side
chains are probably in random rather than in ordered distribution.
The molecule has an average degree of polymerization in the back-
bone of 88 anhydroxylopyranose units plus a reducing-end group and
a non-reducing-end group. The xylan possesses an average molecu-
lar weight of 1.8 X 104.
A summary of the evidence for the structure of the xylan iso-
lated from the inner bark of Douglas-fir is as follows:
The presence of xylose was demonstrated by paper and gas-
liquid chromatography of the acid hydrolyzate.
The presence of arabinose was demonstrated by paper and
gas-liquid chromatography of the acid hydrolyzate.
The presence of a hexuronic acid was detected by a color
reaction. This hexuronic acid was shown to be glucuronic
acid because the acid hydrolyzate showed only traces of
glucose by paper chromatography but after reduction and
acetylation a large amount of glucitol hexaacetate was dem-
onstrated by gas-liquid chromatography. The presence of
a uronic acid was also suggested by the ash and acidity
analyses, by the viscosity measurements in water and aque-
ous sodium chloride solution, and by gel permeation

123
chromatography.
The p -D-(1-.4) glycosidic linkages were deduced from the
negative optical rotation.
The basic repeating unit of the xylan was demonstrated by
gas-liquid chromatographic analysis (xylose-arabinose-
glucuronic acid ratio of 9:2:2).
The molecular weight and degree of polymerization were shown
by end-group analysis and viscosity measurements.
The number of branches of glucuronic acid was shown by the
amount of formic acid released on oxidation with periodate
anion.
The presence of arabinobiose units was shown by the presence
of undegracled arabinose in the products after periodate oxida-
tion.
The total number of branch points was determined by gas-
liquid chromatography of the periodate oxidation products and
by the formic acid formation. The repeating unit was revised
to 18 anhydroxylose units in the backbone with 7 branch points.
The position of the linkages of the glucuronic acid and arabinose
side chains were based on the structures of xylans previously
isolated from the wood and bark of gymnosperms.
There was no evidence for methoxy groups on the C4 position
of the glucuronic acid residues. There were no mixed

124
methoxy-acetate derivatives of glucitol detected by gas-liquid
chromatography of alditol acetates. The data also clearly
showed that each glucuronic acid residue produced one mole
of formic acid during periodate oxidation. This would not be
possible if the C4 hydroxyl group was methylated.
The structure differs from xylans previously reported because
it has a monoarabinose side chain alternating with arabinobiose or longer
side chains. The structure of most of the xylans thus far isolated
from the wood and bark of gymnosperms have only the monoarabinose
side chains (44, 47, 107, 1.13, 120). The amount of glucuronic acid
is also two times higher than most of the xylans previously reported.
The conclusion is that the inner bark of Douglas-fir contains a xylan
that is quite different from the xylans previously found in the wood
and bark of gymnosperms.
F. Alkaline Degradation of the Xylan
1. Reaction in Sodium Flydroxide Solution
Young, Sarkanen, Johnson and Allan (119) have recently inves-
tigated the alkaline degradation of polysaccharides with specific
reference to (1 --.3)-p-D-glucans. They propose that the rate of
alkaline degradation of linear polysaccharides is best explained in
terms of mono- and di-anionic species formed from the reducing

125
end-groups. Their conclusions result in part from the investigations
of Haas, Hrutfiord, and Sarkanen (43) on the alkaline degradation of
cotton hydrocellulose, and the investigations of Lai and Sarkanen
(69) on the alkaline degradation of amylose. Their work on (1 -.3)-
(3-D-glucans supports the mono- and di-anionic concept.
Polysaccharides linked (1 -.4) have long been known to undergo
alkaline degradation by p-alkoxy elimination via an enolate ion inter-
mediate as reviewed in Figure 6 (page 22). However, it has also
long been known that the (1 -.4)-linked polysaccharides do not degrade
to completion. There occurs a termination or "stopping" reaction
that probably involves end-group conversion into metasaccharinic
acid moieties as reviewed in Figure 7 (page 24). The other possi-
bilities of chain termination, namely an alkali resistant linkage in
the polysaccharide or the formation of stable, ordinary saccharinate
end-groups (Figure 8, page 25) are unlikely under the relatively
strong alkali conditions and the elevated temperatures used in the
present work.
The description for the rate of alkaline degradation of poly-
saccharides proposed by Young, Sarkanen, Johnson, and Allan (119)
takes into account the termination reaction in (1 -.4)-linked poly-
saccharides. They consider that the probable sequence leading to
end-group elimination of reducing polysaccharides can be repre-
sented by the scheme illustreated in Figure 27 (page 126).

(Polys. )-Gr K1 ,.r
- + 2 , , 2- , +o Lys. )-G .r+ H olys )- +1-1
k\End-group
stabilizationEndwise degradation
(chain termination)
Where:
(Polys. )-Gr represents a polysaccharide with a reducing end-
group; K1 and K2 are equilibrium constants k1, k2 andk3
are rate constants for the reactions shown.
Figure 27. Reaction sequence leading to end-group elimina-tion of reducing polysaccharides in alkaline solu-tions.a
126
aFrom: Young, R. A. , K. V. Sarkanen, P. G. Johnson andG. G. Allan. Marine Plant Polymers Part III, A kinetic analysisof the alkaline degradation of polysaccharides with specific refer-ence to (1 -.3)-p -g-glucans. Carbohydrate Research 21:111-122. 1972.

127
It is assumed that the reaction occurs only via the anionic species,
in this case the mono- and di-anionic forms of the end-group.
According to this scheme an equilibrium is approached between
neutral, mono- and di-anionic end-group species, characterized by
the equilibrium constants K1 and K2. Both mono- and di-anions are
reactive towards peeling, whereas end-group stabilization, which
occurs in (1 -4)-linked polymers only, is achieved via the di-anionic
species by conversion into a metasaccharinic acid end-group.
Young, Sarkanen, Johnson, and Allan (119) derive their kinetic
expression from the reaction scheme outlined in Figure 27 (page 126).
The degradative chain-propagation and chain-termination are ex-
pressed by equations la and lb.
d[Ge]/dt = k1[Gr-] +
k2[Gr2-] = dL/dt la
-d[Gd/dt k3[Gr2-1 lb
where t represents the reaction time; L, the weight fraction of
polysaccharide degraded after time t; [Ge], the mole fraction of
peeled-off end-groups at time t; [Gr], the total mole fraction of
remaining end-groups at time t; [Gr ] and [Gr21, the mole frac-
tions of relevant mono- and di-ionized end-groups; while kr k2,
and k3 are the rate constants for the reactions depicted in the scheme
in Figure 27 (page 126).

128
From the reaction sequence (Figure 27, page 126) it follows
that the first and second ionization constants, K1 and K?, can be
represented by:
K1 = [Gri[H+]/{[Gr] ([Gr-] + [Gr2-])} 2
andK2
[Gr2-][H+1i[Gr ], respectively 3
Using equation 3 to eliminate the term [ 2-] from equation
2 shows that
[Gr]- = K1[Gr][H+1/ ([1-1+12 +
K1[H+ +
K1K2).
Similar elimination of the term [Gr-] from equation 3 by
using equation 4 indicates that
[Gr2-] = K1K2[Gr]/([H+]2 + K1[F1+] + K1K2).
Combination of equations 4, 5 and 1 yields the rate expression
dL = [Gr] { (k1K 1[H+I + k2K1K)/([H+]2 + K1 [H+I + K1K2)}dt
Then if s =K1
+2 + K1[H+I + K1K2), equation 6 becomes
dL- s[Gr](k1 [H+] + k2K2).dt
Thus, from equations 5 and 1, the rate of chain termination can
be written as-d[Grj/dt = sk3K2[Gr] which, on integration, becomes
[Cr] = [Gr]o exp(-sk3K2t) 8
4
5
7

where [Grio represents the mole fraction of end groups at zero
time, and hence from e a ons 7 and 8
dL - s(kdt 1
+ k2K2)[Gr]o exp (-sk3K2t)
which is a general expression for the rate of alkaline degradation
of linear polysaccharides. Evaluation of equation 9 by integration
demonstrates that
L {s(ki[H+] + k2K2)[Gr]o} {1-exp(-sk3K2t)} /sk3K2 10
and so at infinite time
L.= {Gr}o (k1[H+] + k2K2)/k3K2 11
Table 8 (page 130) shows the rate data obtained for the degrada-
tion of the xylan at various sodium hydroxide concentrations. The
phenol-sulfuric acid reaction was used to measure the total residual
undegraded xylan remaining after the times indicated. These data
are plotted in Figure 28 (page131) and Figure 29 (page 132). The
initial rates of reaction (dL/dt) were obtained from the experimental
curves and are plotted in Figure 30 (page 133)versus the pH of the
sodium hydroxide solution. The shape of this curve is almost iden-
tical to that published by Young, Sarkanen, Johnson, and Allan
(119) for the alkaline degradation of laminaran. The similarity of
these curves for two widely different polysaccharide structures
129
9

Table 8. Rate data for the degradation of the xylan in aqueous sodium hydroxide at various pH values.
aConsiderable time was required to perform the test so the samples were removed at various time intervals.
Time(hours )a
Xylan remaining in mg/nilpH 11.0 pH 12.0 pH 13,0 pH 14.0 pH 14.6 pH 14.8
298 0, 303 0. 303 O. 142 0,306 0.251
0. 5 0. 303 0, 304 0, 140 0. 290 0. 244
1.0 0.308 0, 293 0. 287 0. 134 0. 288 0. 220
1.5 O. 132
2. 0 0.300 0.292 0. 279 O. 131
2. 5 0. 279 0. 235
3.0 0. 297
4. 0 0. 265 0. 128 0. 272 0. 209
5.0 0.297 0. 287 0. 268 O. 122
5.5 0. 270 0. 216
7.0 0. 262 0. 228
8.0 0. 275 0. 268
10.0 0. 245
12.0 0. 275
14.0 0,262
14. 5 0.184
21.0 0.117

21
18
12
6
21
18
0
I I I I 1 I i 1 1
2 4 6 8 10 12 14 16 18
Time in hours
Time in hours
pH = 13.0
pH = 14.0
0 2 4 6 8 10 12 14 16 18
Time in hours
Figure 28. Rate curves for the degradation of the xylan in aqueous sodium hydroxideat pH values of 12.0 - 14.0.
13 1
2 4 6 8 10 12 14 16 18
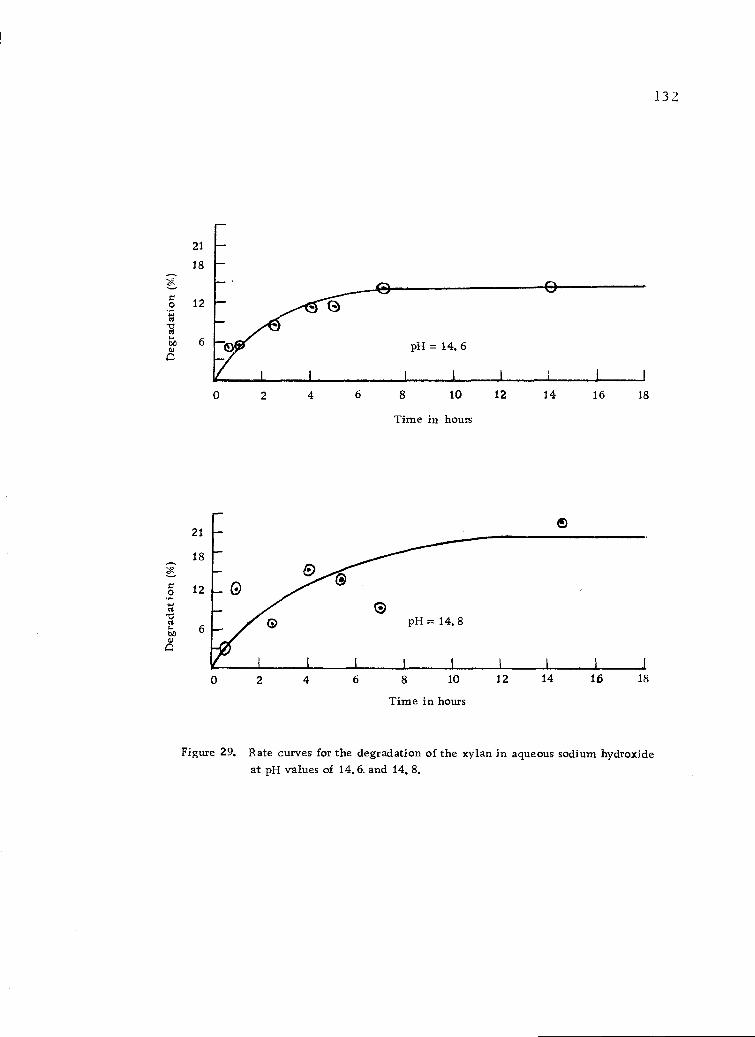
132
0 2 4 6 8 10 12 14 16 18
Time in hours
Figure 29. Rate curves for the degradation of the xylan in aqueous sodium hydroxideat pH values of 14.6. and 14. 8.
0 2 4 6 8 10 12 14 16 18
Time in hours
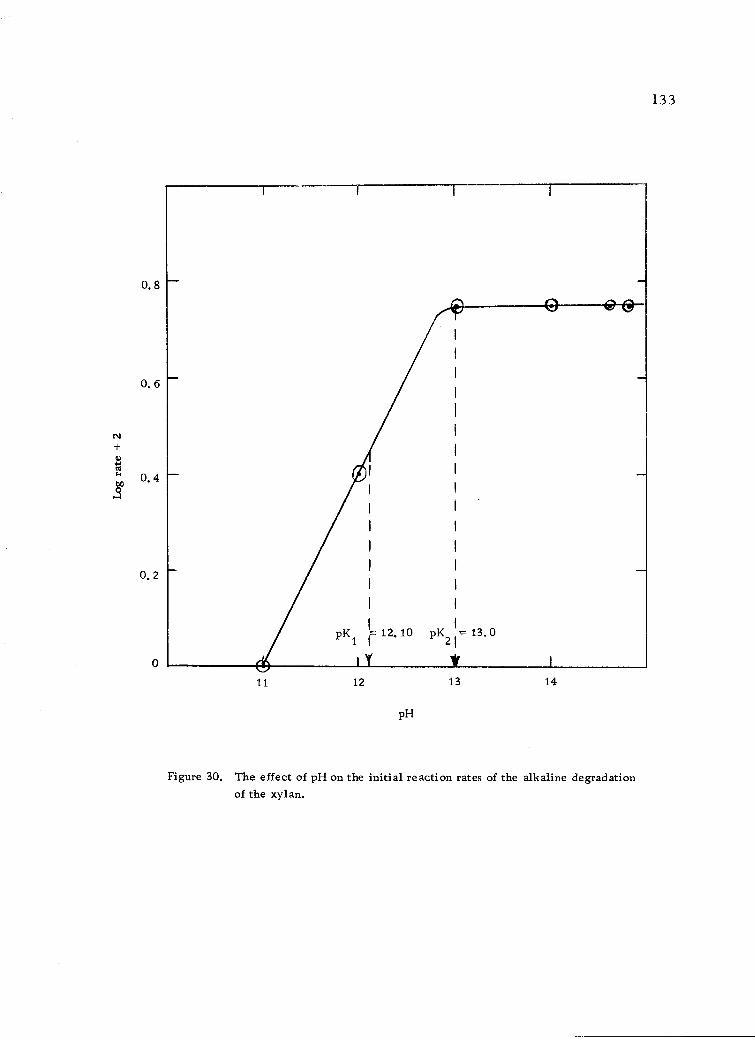
0.8
0. 6
c\I
4.44)
0. 4
0. 2
11
pKI
12.10pK21=
13,0
Y
12
pH
Figure 30. The effect of pH on the initial reaction rates of the alkaline degradationof the xylan.
13 14
00
133

134
supports the general reaction scheme outlined in Figure 27 (page 126)
and the general rate expression for the alkaline degradation of linear
polysaccharides (Equation 9, page 129).
The magnitudes of the dissociation constants K1 and K2 were
obtained from the pKa values of this plot. At one-half the maximum
rate for degradation of the xylan, the pK1 value for the first ionization
to monoanion was determined as 12. 10 (K1 = 9. 54x 10-13). This
number is in good agreement with the pK1 value of 12. 2 reported by
Young, Sarkanen, Johnson, and Allan (119) for the alkaline degrada-
tion of laminaran. It is also in good agreement with the literature
data reported by Young, Sarkanen, Johnson, and Allan (119) for the
dissociation constants for D-glucose (5. 75x 10-13), a-D-glucose
(3. 45x 10-13), and p-D-glucose (6. 75x 10-13). The maximum rate
of degradation is reached at pH 13.0 (Figure 30, page 133). This
represents the second ionization to the dianion and so the dissociation
constant pK2is 13.0 (K2 = lx 10-13).
The initial rates of degradation do not increase after pH 13
(Figure 30, page 133) and so subsequent calculations will be consid-
ered to apply to the alkaline degradation of the xylan at pH 13 (0. 1N
in sodium hydroxide) and 1000. From the data now available the
constant s can be calculated from the following relationship (page
128):
s = K1 /([H+]2 +K1
[H+] + K1K2) = 4. 75 x 1012

page 131).
135
Young, Sarkanen, Johnson, and Allan (119) showed that since
there was no increase in the rate of degradation of larninaran in the
upper pH range where the dianionic species was the major reaction
intermediate, then the rate of 13-elimination from both mono- and
di-anions was essentially the same. Thus the rate constants k1 and
k2 were considered equal and were calculated for a single value.
Similarly, there was no increase in the rate of degradation of the
xylan in the present work (Figure 30,page 133) at higher pH ranges
and so k1 and k2 were calculated for a single value.
With k1 = k2the general expression developed for the rate of
alkaline degradation (Equation 9, page 129) becomes:
dL =s (k [H+] + k1K2)[Gr]o exp ( - sk3 K2t )dt 1
At time t = 0 the expression exp( -sk3K2t) becomes 1.0 and
the expression remaining is:
dL = s (k [H+] + kK2)[Gdodt 1
where [Gdo is the fraction of reducing end groups in comparison to
the total number of repeating monosaccharides in the chain. For
the xylan under present investigation [Gr] = = 0.0111 = 1.11x 10-2o 90
The initial rate, dLidt, for the alkaline degradation at pH 13.0
and a temperature of 100° was 5.625 x 10-2 hour-1 (Figure 28,

Substituting in the values gives:
5.675x 10-2 hour' = (4. 75x1012)(kx10-13-Fk1x10-13)(1.11x10-2)-
k = k2 = 5.33 hours-11
This is considerably smaller than the rate constant of 323
hour-1 reported for amylose (119) under the same conditions. The
slower rate is undoubtedly due to the branch points on the present
xylan (Figure 26,page 121). In order for peeling to occur on a
(1 --'.4)-linked polysaccharide it is necessary that a ketone group
be formed on C2 (Figure 6, page 22). Therefore, in the present
xylan it is necessary that the glucuronic acid groups be cleaved
from C2 (Figure 26,page 121). Hamilton (44) has shown that glucu-
ronic acid derivatives are removed from the C2 position of wood
xylans under alkaline conditions and Casebier and Hamilton (20)
have discussed the reaction in considerable detail. Hartler and
Svensson (50) have also shown that substituents on the C2 position
of xylans do not prevent alkaline peeling. Although no kinetic data
have been presented for the removal of the glucuronic acid moieties,
the reaction undoubtedly slows the rate of degradation of the xylan
backbone.
The constantk3
is the rate constant for the end-group stabil-
ization (chain termination) reaction depicted in Figure 27 (page 126).
136

It is this reaction which causes degradation of the polysaccharide
chain to cease. The constant can be obtained from Equation 9 (page
129) once k1 andk2
have been calculated.
The rate of reaction, dL/dt, at 1000, pH 13. 0 and a time
lapse of 2.0 hours was determined as 0.03 hour-1 from Figure 28
(page131). Substituting this into Equation 9 (page 129) the expres-
sion becomes:
3.0 x 10-2 = [4.751 x 1012] [(5.333)(10-13)
-+ (5.333)(10-13)] [1. 11 x 10 2] exp-sk3K2t
Solving the equation fork3
results in a termination rate constant-
of k3 = 0.66 hour'.
This rate constant is smaller than the rate constant (k3 =
1.33 hour-1) reported (119) for amylose. However, the relative
rate of termination compared to the rate of the peeling reaction
itself is much greater for the xylan than for amylose. That is,O. 66 - 1the fraction
k3 /k1 5.33- - L2 x 10 for the xylan is much greater1. 33than the same fraction k /k - - 4. 1 x 10-3 for amylose. The
3 1 323
branch points on the xylan undoubtedly are responsible for the faster
relative termination rate. This faster relative termination rate is
reflected in the lower final percentage of degradation (19.81%) of
the xylan compared to the final percentage degradation (45. 0%) of
amylose. The value of 19.81% degradation is supported by the value
137

138
of 15% degradation reported by Hans son and Hartler (47) for a
xylan isolated in a similar manner (sodium chlorite holocellulose)
from white birch.
The polymer chain is terminated only from the dianion (Figure
27, page 126). In order for the dianion to form an alkali stable
metasaccharinic acid and thus terminate the reaction it is neces-
sary for the substituent on C3 to leave, whether it be an hydroxyl
group or another substituent (Figure 7, page 24). The structure of
the present xylan (Figure 26, page121) shows several anhydroarabi-
nose side chains attached to C3 of the xylan backbone. It is known
(119) that glycosyloxy anions are better leaving groups than hydroxide
anions. Therefore, the presence of anhydroarabinose side chains
on C3 would be expected to increase the relative rate of termination
beyond that of an unbranched polymer. The value of the relative
rate of termination for the xylan (k3/k1 = 3. Ox10-1) compared to
-3the relative rate of termination for amylose (k3/k1 = 1. 6x10 ) is
consistent with this view. These results are supported by the work
of Hansson and Hartler (47). They attributed the greater stability
of a pine xylan over the stability of a birch xylan in alkaline media
as being the result of a more extensive arrest of degradation due to
the presence of arabinose substituents on the pine xylan. However,
no rate data were presented.
The data from the alkaline degradation studies support the

structure proposed for the xylan in Figure 26 (page 121). The low
amount of degradation (19. 81%) is consistent with the (1-4) glycosidic
linkage because it is known that (1-4)-linked polysaccharides must
rearrange to a ketose form before 13-elimination can take place (119).
This allows a greater possibility for termination to occur than in
polysaccharide linked (1-.3) for example. The high relative rate of
termination compared to the rate of peeling supports the presence of
branch points. The glucuronic acid moieties must be cleaved before
rearrangement to the ketone form at C2 is possible and this slows
the rate of peeling. On the other hand the presence of arabinose
moieties on C3 enhances the rate of termination because they are
good leaving groups and thus speed up the formation of terminating
metasaccharinic acid end-groups. The combination of these two
positions of the branch points on the xylan backbone describes the
slow rate of end-group elimination (peeling), the relatively high rate
of end-group stabilization (termination), and the small amount of
xylan degradation (19.81%).
2. Phenol-Sulfuric Acid Method of Analysis for the Xylan
Phenol in the presence of sulfuric acid can be used for the
quantitative microdetermination of sugars and their methyl deriva-
tives, oligosaccharides, and polysaccharides (104). The reagent is
inexpensive and stable and a given solution requires only one standard
139

140
curve for each sugar. The color produced is permanent and it is
not necessary to pay special attention to the control of the conditions.
Several investigators (103, 105) have investigated this method and
have found it satisfactory. Lindberg, Theander and UddegA.rd (49)
have demonstrated that saccharinic acid products do not interfere
with the color reactions.
The curve for xylose is shown in Figure 31 ( page 141) Measure-
ments were taken at 480 nm. Pentoses, methyl pentoses, and uronic
acids have an absorption maximum at this wavelength. Hexoses and
their methylated derivatives absorb at 485-490 nm maximum. Cer-
tain of the methylated pentose sugars and their methyl glycosides
show selective absorption at 415 to 420 nm.
The xylan is converted to furfural by the sulfuric acid and the
furfural reacts with the phenol to form a complex that absorbs at
480 nm. Dubois, Gilles, Hamilton, Rebers, and Smith (104) found
that xylose is converted to furfural under conditions of the test to
93% that predicted by theory. The intensity of the color depends on
the amount of phenol added. As the amount of phenol is increased,
the absorbance increases to a maximum and then falls off. Under
proper conditions, the method is reported to be accurate to ±2%.
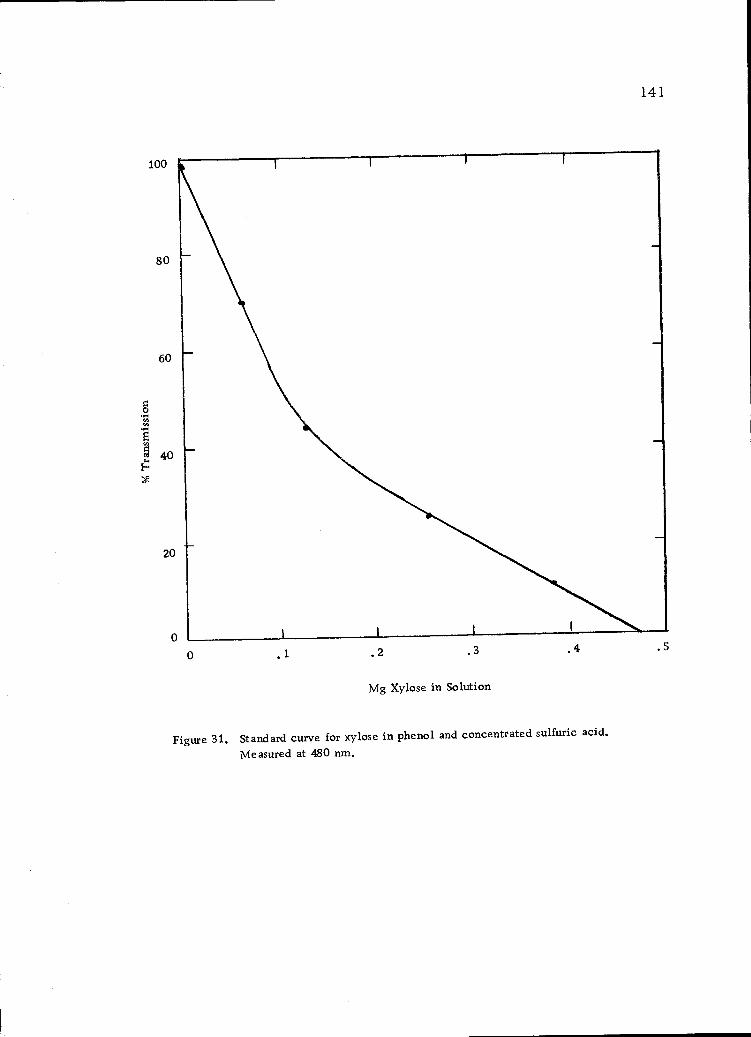
100
80
60
40
20
0
141
0 .2 .3
Mg Xylose in Solution
Figure 31. Standard curve for xylose in phenol and concentrated sulfuric acid.
Measured at 480 nm.
.4 .5

V. SUMMARY AND CONCLUSIONS
A sample of Douglas-fir inner bark was successively extracted
with ethanol-water (4:1 v/v), benzene-ethanol (2:1 v/v), hot
water, 0.5% ammonium oxalate, and acidified sodium chlorite
solution.
The ethanol-water extract contained free glucose but the
benzene-ethanol extract contained no carbohydrates.
The hot-water-soluble solids contained nitrogen (0.4% of whole
inner bark), but no sulfur, phosphorus or halogens.
The hot-water-soluble solids showed positive tests for proteins,
tannins (4.9% of whole inner bark), starch (no more than 5.1%)
and hemicelluloses.
The proteins, tannins and the a-amylose portion of the starch
were removed by a series of enzyme hydrolyses followed by
extensive dialyses.
The polysaccharides which remained were composed of the
following ratio of sugar residues; glucose (2. 9), arabinose (1. 3),
galactose (1.0) and traces of rhamnose, xylose and mannose.
The glucose was considered to be amylopectin which did not
hydrolyze with the a-amylase enzymes used to reduce the
starch content.
The arabinose and galactose residues were considered to exist
in the inner bark as part of L-arabino-D-galactan
142

polysaccharides. Water-soluble, highly branched, L-arabino-
D-galactans have been reported in the wood of conifers but have
never before been reported in the bark of Douglas-fir.
A xylan polysaccharide was isolated from the insoluble fraction
(holocellulose) remaining after treatment of the bark residue
with acidified sodium chlorite olation.
The xylan possessed a specific rotation of -30. 50 in aqueous
solution indicative of p-D-glycosidic linkages.
The xylan showed a positive carbazole-sulfuric acid color test
for the presence of hexuronic acids.
Gas-liquid chromatographic analysis showed that the xylan was
composed of the following ratio of sugar residues: xylose
(4.5), arabinose (1.0), glucuronic acid (1.0).
End-group analysis by the Somogyi copper reduction method
showed a molecular weight of 1.8 x 104. This in combination
with the gas-liquid chromatographic analysis showed a degree
of polymerization of 90.
The intrinsic viscosity of the xylan in molar diethylenediamine
copper II solution was 0.42 dl/g which corresponded to a degree
of polymerization of 89.
Gel permeation chromatography of the xylan indicated that the
molecular size distribution of the xylan was not too broad.

144
The xylan was oxidized with periodate anion. It consumed
133. 5 moles of periodate anion and released 22. 5 moles of
formic acid per mole of xylan.
The oxidized xylan was hydrolyzed and analyzed by gas-liquid
chromatography. The results showed the following ratio of
sugar residues: xylose (7. 0), arabinose (1. 0).
The data are consistent with a polysaccharide xylan structure
composed of a backbone of 88 anhydro-D-xylopyranose unifr,
attached ç3-D-(1 -.4) plus a reducing and non-reducing end-
group. There are 20 anhydroarabinose side chains and 20
anhydroglucuronic acid side chains on these 90 units. At least ten
of the anhydroarabinose units are in the form of monoarabinose
side chains and as many as 10 are in the form of arabinobiose or
longer side chains. This structure is quite unique and represents
a new xylan isolated from the wood and bark of gymnosperms.
The xylan was reacted at 1000 with aqueous sodium hydroxide
solutions ranging in concentration from 0. 001N to 3.676N.
The rate of degradation did not change at alkali concentrations
above 0. 1 N in sodium hydroxide.
Alkaline degradation of the xylan in 0. 1N aqueous sodium
hydroxide at 1000 showed a rate constant for end-group peeling
of k1 = k2 = 5.33 hour 1, and a termination rate constant of
k3 = 0.66 hour-1.

145
O. The peeling reaction stopped when 19.81% of the xylan had
been degraded. This small amount of degradation reflected
the relatively high termination rate constant compared to the
peeling rate constant.
21. The results of the alkaline degradation supported a possible
reaction sequence involving mono- and di-anionic end-group
species as the intermediates leading to end-group elimination
of reducing polysaccharides.

LITERATURE CITED
Abdel-Ahker, M., J. K. Hamilton and T. Smith. The reduc-tion of sugars with sodium borohydride. Journal of the Amer-ican Chemical Society 73:4691-4692. 1961.
Albersheim, P., D. J. Nevins, P. D. English, and A. Karr.A method for the analysis of sugars in plant cell-wall poly-saccharides by gas-liquid chromatography. CarbohydrateResearch 5:300-345. 1967.
Anderson, E. The mucilage from slippery elm bark. Journalof Biological Chemistry 104:163-170. 1933.
Anderson, E. and W. W. Pigman. A study of the inner barkand cambial zone of black spruce (Picea mariana B. S. P. ).Science 105:601-602. 1947.
Anderson, E., L. E. Wise and E. K. Ratliff. Note on thechemistry of the cambial zone of black spruce. Tappi 37:422-424. 1954.
Anet, E. F. L. J. 3-Deoxyglucosulose5 (3-Deoxyglycosones)and the degradation of carbohydrates. Advances in Carbo-hydrate Chemistry 19:181-218. 1964.
Angyal, S. J. and D. 3. McHugh. Interaction energies ofaxial hydroxyl groups. Chemistry and Industry (London)1147-1148. 1956.
Barnett, A. J. G. and G. A. Tawab. A rapid method for thedetermination of lactose in milk and cheese. Journal of Sci-entific Food Agriculture 8:437-441. 1957.
Becker, E. S. and E. F. Kurth. The chemical nature of theextractives from the bark of red fir. Tappi 41:380-384. 1958.
Begik, A., R. J. Conca, J. K. Hamilton and E. V. Partlow.Selective extraction of hemicelluloses from softwoods. Tappi
50:78-81. 1967.
Beveridge, K. J., J. F. Stoddart, W. A. Szarek and J. K. N.Jones. Some structural features of the mucilage from thebark of Ulmus fulva (slippery elm mucilage). CarbohydrateResearch 9:429-439, 1969.
146

147
Black, R. J., R. LeShange and G. Zweig. Paper chromatog-raphy, a laboratory manual. New York, N. Y. AcademicPress, Inc. 1952. 195 p.
Borchardt, L. G. and C. V. Piper. A gas chromatographicmethod for carbohydrates as alditol-acetates. Tappi 53:257-260. 1970.
Brourriand, H. and J. H. Smith. Composition of the complexesof iron (III) with phenols and enols. Journal of the AmericanChemical Society 74:1013-1016. 1952.
Browning, B. L. Methods of wood chemistry,Vol. I. NewYork, N. Y. Interscience Publishers. 1967. 384 p.
Browning, B. L. Methods of wood chemistry, Vol. II. NewYork, N. Y. Interscience Publishers, 1967. p. 384-882.
Bryant, R. W., T. E. Timell, M. Zinbo, D. A. M. Goring,and W. Q. Yean. Polymolecularity of an aspen xylan. Cellu-lose Chem. Techn. 2:269-273. 1968.
Buston, H. E. and H. S. Hopf. Carbohydrate constituents ofbark of ash (Fraxinus excelsior). Biochemical Journal 32:44-46. 1938.
Casebier, R. L. and J. K. Hamilton. Alkaline degradationof glucomannans and galactomannans. Tappi 48:664-669.1965,
Casebier, R. L. and J. K. Hamilton. Alkaline degradation ofxylans. Tappi 50:441-449. 1967.
Caulfield, D. F. On questioning x-ray evidence of crystalliz-ability of xylan in situ. Tappi 51:371-372. 1968.
Chang, Y. P. Anatomy of common North American pulpwoodbarks. Tappi Monograph No, 14. 1954,
Chang, Y. Bark structure of North American conifers. (U.S.Dept. of Agriculture, Technology Bulletin 1095), 1954. 86 p.
Chen, C. H. Doctoral thesis in preparation. Corvallis,Oregon State University. 1972.

148
Clark, J. M., Jr. Experimental biochemistry. San Francisco,Cal. W. H. Freeman and Co. 1964. 228 p.
Corbett, W. M. and G. N. Richards. The degradation of cellu-lose by aqueous sodium hydroxide at 1700, Svensk Papperstid-ning 60:79l-79. 1957.
Corbett, W. M., J. Kenner and G. N. Richards. The degrada-tion of carbohydrates by alkali. X. Acetal derivatives. Jour-nal of the Chemical Society 11709-11711. 1955,
Currier, R. A. Physical consideration. In: Converting barkinto opportunities. Proceedings. Compiled by A. C. VanVliet, Corvallis, Oregon State University. Nov. 1971. 110 p.
Dische, S. Color reactions of hexuronic acids. In: Methods
of carbohydrate chemistry. Vol. 1. ed. R, L. Whistler andM. L. Wolfrom, New York, N. Y. Academic Press, 1962.589 p.
Dubois, M., K. Gilles, J. K. Hamilton, P. A. Rebers, andF. Smith. Colorirnetric method for determination of sugarsand related substances. Analytical Chemistry 28:350-356.
1956.
Edward, J. T. Stability of glycosides to acid hydrolysis.Chemistry and Industry (London) 1102-1104. 1955.
Eriksson, K. , R. A. Pettersson, and B. Steenberg. Gel
filtration chromatography of hemicelluloses and carboxy-methylcellulose in cadoxen solution. Svensk Papperstidning71:695-698. 1968.
Fischer, R. B. Quantitative chemical analysis. 2nd ed.
Philadelphia, Penn. W. B. Saunders Co. 1961. 501 13.
Franzon, 0. and 0. Samuelson. Degradation of cellulose byalkali cooking. Svensk Papperstidning 60:872-877. 1957.
Garegg, P. J. Studies on some partially substituted glycosidesand polysaccharides. Svensk Kemisk Tidskrift 77:28-50. 1965.
(Abstracted in Abstract Bulletin of the Institute of Paper Chem-
istry 36:440. 1942. )

Gill, R. E., E. L. Hirst and J. K. N. Jones. Constitutionof the mucilage from the bark of Ulmus fulva (slippery elm).I. The aldobionic acid obtained by hydrolysis of the mucilage.Journal of the Chemical Society (London) 1469-1471. 1939.
Gill, R. E., E. L. Hirst and J. K. N. Jones. Constitutionof the mucilage from the bark of Ulmus fulva (slippery elmmucilage). II. The sugars formed in the hydrolysis of themethylate of mucilage. Journal of the Chemical Society(London) 1025-1029. 1946.
Glaudemans, C. P. J. and T. E. Time11. The polysaccharidesof white birch (Betula papyrifera). Svensk Papperstidning 61:1-9. 1958.
Graham, H. M. and E. F. Kurth. Constituents of extractivesfrom Douglas-fir. Industrial Engineering Chemistry 41:409-414. 1949.
Granath, K. A. Gel Filtration. In: Methods in carbohydratechemistry. Vol. V. ed. R. L. Whistler. New York, N. Y.
Academic Press. 1965. 463 p.
Grillos, S. 3. Structure and development of the bark ofDouglas-fir, Pseudotsuga menziesii (Mirb. ) Franco. Doc-toral thesis. Corvallis, Oregon State University, 1956. 67
numb. leaves.
Grillos, S. J. and F. H. Smith. The secondary phloem ofDouglas-fir. Forest Science 5:377-388. 1959.
Haas, D. W., B. F. Hrutfiorcl and K. V. Sarkanen. Kineticstudy on the alkaline degradation of cotton hydrocellulose.Journal of Applied Polymer Science 11:587-600. 1967.
Hamilton, J. K. The behaviour of wood carbohydrates intechnical pulping processes. Pure and Applied Chemistry 5:197-217. 1962.
Hamilton, J. K. and G. R. Quimby. The extractive power oflithium, sodium and potassium hydroxide solutions for thehemicelluloses associated with wood cellulose and holocellu-lose from western hemlock. Tappi 40:781-786. 1957.
149

Hans son, J. A. and N. Hartler. Alkaline degradation of pineglucomannan. Holzforschung 24:54-59. 1970.
Hansson, J. A. and N. Hartler. Alkaline degradation of xylansfrom birch and pine. Svensk Papperstidning 71:358-365, 1968.
Hansson, J. A. and N. Hartler. Effects of reducing and ox-idizing agents on the resistance of xylans from birch and pinetowards alkaline degradation. Svensk Papperstidning 71:669-673. 1968.
Hansson, J. A., N. Hartler, I. Szabo, and A. Teder. Effectof borohydride concentration on the reduction of carbohydrates.Svensk Papperstidning 72:78-81. 1969.
Hartler, N. and I. L. Svensson. Alkali stability of someuronic acids and its implications in borohydride and poly-sulfide cooking. Industrial and Engineering Chemistry,Product research development 4:80-82. 1965.
Hay, K. D., and H. F. Lewis. TI-e chemical compositionof balsam bark. Paper Trade Journal 111:39-43. 1940.
Hodge, J. E. and B. T. Hofreiter. Determination of reducingsugars and carbohydrates. In: Methods in carbohydrate chem-istry. Vol. I. ed. R. L. Whistler and M. L. Wolfrom. NewYork, N. Y. Academic Press, 1962. 589 p.
Holmes, G. W. and E. F. Kurth. The chemical compositionof the newly formed inner bark of Douglas-fir. Tappi 44:893-898. 1961.
Hough, L. and J. K. N. Jones. Chromatography on paper.In: Methods in carbohydrate chemistry.Vol. I. ed. R. L.Whistler, M. L. Wolfrom. New York, N. Y. AcademicPress. 1962. 589 p.
Hough, L., J. K. N. Jones and G. L. Hirst. Chemical con-stitution of slippery elm mucilage: isolation of 3-methyl-D-galactose from the hydrolysis products. Nature 165:34-35.1950.
Isbell, H. S. Interpretation of some reactions in the carbo-hydrate field in terms of consecutive electron displacement.Journal of Research of the National Bureau of Standards. 32:
45-49. 1944.
150

Jayme, G. and G. Hanke. The sodium chlorite oxidationproducts and constitution of native spruce lignin. Cellulose-Chemie 21:127-140. 1943.
Jensen, W., K. E. Fremer, P. Sierila and V. Wartiovaara.The chemistry of bark. In: The chemistry of wood. ed.B. L. Browning. New York, N. Y. Interscience Publishers1963. 689 p.
Jones, J. K. N. and F. Smith. Plant gums and mucilages.Advances in Carbohydrate Chemistry 4:243-291. 1949.
Katz, G. The location and significance of the 0-acetyl groupsin a glucomannan from Parana pine (Araucaria angustifolia).Tappi 48:34-41. 1965.
Kiefer, H. J. and E. F. Kurth. The chemical composition ofthe bast fibers of Douglas-fir bark. Tappi 36:14-19. 1953.
Kock, R. B., W. F. Geddes, and F. Smith. The carbohydratesof gramineae. I. The sugars of the flour of wheat (Triticumvulgare ) Cereal Chemistry 28:424-430. 1951.
Kotasek Z. Polyuronides in water extracts of spruce bark.Veda a vyzku.rn v prumyslu kozedelnem 1:47-61 1956: Ab-stracted in Chemical Abstracts 51:12525. 19 57. )
Kurth, E. F. Separation of wood extractives into simplercomponents. Industrial and Engineering Chemistry. Analyti-cal Edition 11:203-205. 1939.
Kurth, E. F. The chemical composition of bark. ChemicalReviews 40:33-49. 1947.
Kurth, E. F. and G. J. Ritter. Spruce holocellulose and thecomposition of its easily hydrolyzable fraction. Journal ofthe American Chemical Society 56:2720-2723. 1934.
Lai, L. Y. C. Douglas fir bark; carbohydrates solubilizedby the acidified sodium chlorite delignification reaction.Masters thesis. Corvallis, Oregon State University, 1972.62 numb. leaves.
Lai, Y. A. Kinetics of degradation of carbohydrates. Doctoralthesis. Seattle, University of Washington, 1968. 97 numb. leaves.
151

152
Lai, Y. Z. and K. V. Sarkanen. Kinetic study on the alkaline
degradation of amylose. Journal of Polymer Science, Part C,
No. 28:15-26. 1969.
Larsen, E. E. and E. V. Lynn. The bark of American larch.
Journal of the American Pharmaceutical Association 26:288-
290. 1937. (Abstracted in Chemical Abstracts 31:4451, 1937. )
Laver, M. L., D. F. Root, F. Shafizadeh and J. C. Lowe.
An improved method for the analysis of the carbohydrates of
wood pulps through refined conditions of hydrolysis, neutral-ization, and monosaccharide separation. Tappi 50:618-622.
1967.
Leopold, B. Chemical composition and physical properties of
wood fibers I. Preparation of holocellulose fibers from lob-
lolly pine wood. Tappi 44:230-240. 1961.
73, Lindberg, B., 0. Theander and E. Uddegard. Alkaline de-
gradation of cellobiose, 4-0-13-11-xyloPyranosyl-a-xylose.Svensk papperstidning 69:360-363. 1966.
Lyness, W. I. and D. L. Opdyke. Paper presented at 133rdMeeting of the American Chemical Society, San Francisco.
Apr il, 1958.
Machell, G. and G. N. Richards. Mechanism of saccharinic
acid formation. I. Competing reactions in the alkaline de-gradation of 4O-methyl-P- glucose, maltose, amylose, and
cellulose. Journal of the Chemical Society 1924-1931. 1960.
Machell, G., G. N. Richards and H. H. Sephton. Alkaline
degradation of cellulose. Chemistry and Industry (London)
467-469. 1957.
McNair, H. M. and E. J. Bonelli. Basic gas chromatog-raphy. Berkeley, California. 1969. 306 p.
Marchessault, R. H. , W. Settineri and W. Winter. Crystal-
lization of xylan in the presence of cellulose. Tappi 50:55-59.
1967.
Meier, H. On the behavior of wood hemicelluloses under dif-
ferent pulping conditions. Svensk Papperstidning 65:589-597.
1962.

Miles Laboratories, Inc. Diazyrne, an amyloglucosidaseenzyme for conversion of starch and/or dextrins to glucose.Technical Bulletin No. 9 - 24 5. Elkhart, Indiana. 1963.
Miles Laboratories, Inc. HT-1000. Product Information.Elkhart, Indiana. No date.
Pigman, W., E. Anderson and R. L. Leaf. Occurrenceof pectic materials in wood. Journal of the American Chem-ical Society 70:432-433. 1948.
Pigman, W. and E. F. L. J. Anet. Mutarotations and actionsof acids and bases. In: The carbohydrates. Vol. IA. 2nd edition.ed. W. Pigman and D. Horton. New York, N. Y. AcademicPress, 1972. 642 p.
Piper, C. V. and L. J. Bernardin. A spectrophotometricmethod for the chromatographic analysis of sugars. Tappi41:16-18. 1958.
Richtzenhain, H. and B. Abrahamsson. The alkaline degrada-tion of polysaccharides. II. Degradation of hemicellulose.Svensk Papperstidning 57:538-541. 1954.
Richtzenhain, H., B. 0. Lindgren, B. Abrahamsson, andK. Holmberg. The alkaline degradation of polysaccharides.I. Degradation of cotton cellulose. Svensk Papperstidning57:363-366. 1954.
Ritter, G. J. and E. F. Kurth. Holocellulose, total carbo-hydrate fraction of extractive-free maple wood. Industrialand Engineering Chemistry 25:1250-1253. 1933.
Ross, W. D. and R. L. Krahrner. Some sources of variationin structural characteristics of Douglas-fir bark. Wood andFiber 3:35-46. 1971.
Rowell, R. M. and J. W. Green. Oxidative alkaline degrada-tion of 3-deoxyaldosuloses. Carbohydrate Research 15:197-203. 1970.
Samuelson, 0. and A. Wennerblom. Degradation of celluloseby alkali cooking. I. Formation of carboxyl groups. SvenskPapperstidning 57:827-830. 1954.
153

154
Sanyer, N. and G. H. Chidester. Manufacture of wood pulp.
In: The chemistry of wood. ed. B. L. Browning. New York,
N. Y. Interscience Publishers, Inc. 1963. 689 p.
Schmidt, E. and E. Grau.mann. Incrusting substances of plants.
I. Methods for the preparation of pure plant skeletal substances.Deusche Chemische Gesellschaft Berichte 54:1860-1873. 1921.
Schwalbe, C. G. and K. E. Neumann. New bark (and sap) of
wood of spruce, pine and red beech Cellulosechemie 11:113-128. 1930.
Sears, K. D., A. Beelik, R. L. Casebier, R. J. Engen, J. K.Hamilton, and H. L. Hergert. Southern pine prehydrolyzates:Characterization of polysaccharides and lignin fragments.Journal of Polymer Science. Part C. 425-443. 1971.
Segall, G. H. and C. B. Purves. Chemical composition ofwood barks. Pulp and Paper Magazine of Canada 47:149-161.1946.
Sharkov, V. I. and A. Girchits. The chemical composition of
wood bark. VIII. The pectin substances in pine bark. Lesok-him Prom. No. 2, 9-12. 1939; Khim Referat Zhur. 9:113.1939. (Abstracted in Chemical Abstracts 34:6324, 1940.)
Sharkov, V. I., A. Girchits, and F. A. Sartaniya. The chem-ical composition of bark. IX. The pectin substances of pine
and fir bast. Lesokhim Prom. No. 3:40-46. 1939; Khim
Referat Zhur. 8:113. 1939. (Abstracted in Chemical Abstracts34: 6324, 1940. )
Sharkov, V. I., Z. A. Tyzgunova and V. N. Kolpina. The
chemical composition of wood bark. Lesokhim. Prom. 6,No. 2. 9-10. 1938: Khim Referat Zhur. 2:123. 1939.
(Abstracted in Chemical Abstracts 34:1050. 1940. )
Sloneker, J. H. Gas-liquid chromatography of alclitol acetates.In: Methods in carbohydrate chemistry. VI. General carbo-hydrate methods, ed. R. L. Whistler and J. N. BeMiller.New York, N. Y. Academic Press. 1972. 603 p.
Song, M. J. The structure of a xylan from Tilia americana L.
Master's thesis. Syracuse, N. Y. State University College of
Forestry at Syracuse University, 1970. 97 numb. leaves.

155
101. Speck, J. C., Jr. The Lobry de Bruyn-Alberda van Ekensteintransformation. Advances in Carbohydrate Chemistry 13:63-103. 1958.
102, Technical Association of the Pulp and Paper Industry. Ash inpulp. TAPPI Standards and Suggested Methods. T 211 M-58.New York, N. Y. 1958.
Technical Association of the Pulp and Paper Industry. Cu.pri-ethylenediamine disperse viscosity of pulp. Tappi Standardsand Suggested Methods. T 230 SU-66. New York, N. Y. 1966.
Timell, T. E. Carbohydrate composition of ten North Amer-ican species of wood. Tappi 40:568-572, 1957,
Timell, T. E. Isolation and properties of an 0-acety1-4-0-methyl-glucuronoxyloglycan from the wood of white birch(Betula papyrifera). Journal of the American Chemical Soci-ety 82:5211-5215. 1960.
Timell, T. E. Isolation of polysaccharides from the bark ofgymnosperms. Svensk papperstidning 64:651-661. 1961.
Timell, T. E. and M. Zinbo. Polysaccharides in the wood ofeastern hemlock: structure and degree of branching of anarabino-4-0-methylglucuron0xylan. Tappi 50:196-198. 1967.
Timell, T. E., C. P. Glaudemans and A. C. Currie. Spec-trophotometric method for determination of sugars. AnalyticalChemistry 28:1916-1920. 1956.
Vogel, I. A. Textbook of practical organic chemistry includ-ing qualitative organic analysis. 3rd edition, New York, N. Y.Longmans, Green and Co. 1956. 1188 p.
Whistler, R. L. and J. N. BeMiller. Alkaline degradation ofpolysaccharides. Advances in Carbohydrate Chemistry 13:
289-329. 1958.
Whistler, R. L. and C. M. Corbett. Behavior of xylan inalkaline solution: The isolation of a new C-5 saccharinic acid.Journal of the American Chemical Society 78:1003-1005. 1956.
Whistler, R. L. and E. L. Richards. Hemicelluloses. In: Thecarbohydrates chemistry and biochemistry, Vol. IIA. 2nd edition.

ed. W. Pigman and D. Horton. New York, N. Y. Aca-demic Press, Inc. 1970. 469 p.
Whistler, R. L. and C. L. Smart. Polysaccharide chemistry.New York, N. Y. Academic Press, Inc. 1953. 493 p.
Whistler, R. L., J. Bachrach, and D. R. Bowman. Prepara-tion and properties of corn cob holocellulose. Archives ofBiochemistry 19:25-33. 1928.
Willard, H. H., L. L. Merritt, Jr. and J. A. Dean. Instru-mental methods of analysis. 4th edition. Princeton, N.J. D.VanNostrand Co., Inc. 1965. 784 p.
Wise, L. E., M. Murphy and A. A. DIAddieco. Chlorite holo-cellulose, its fractionation and bearing on summative woodanalysis and on studies of the hemicelluloses. Paper TradeJournal 122:35-43. 1946.
Wolfrom, M. L. and D. L. Patin. Carbohydrates of the coffeebean. IV. An arabinogalactan. Journal of Organic Chemistry30:4060-4063. 1965.
Wolfrom, M. L., M. L. Laver, and D. L. Patin. Carbo-hydrates of the coffee bean. II. Isolation and characteriza-tion of a mannan. Journal of Organic Chemistry. 26:4533-4535. 1961.
Young, R. A., K. V. Sarkanen, P. G. Johnson and G. G.Allan. Marine plant polymers. Part III. A kinetic analysisof the alkaline degradation of polysaccharides with specificreference to (1 ->3)-D-glucans. Carbohydrate Research 21:111-122. 1972.
Zinbo, M. and T. E. Timell. The degree of branching ofhardwood xylans. Svensk Papperstidning 68:647-662. 1965.
156
Top Related