







Languages
Pages
Legal


HGMHybrid networks with
Gene Evolution
Ali Tofigh, KTH
Jens Lagergren, KTH
Bengt Sennblad

Why?
• Lateral gene transfer– Important process in prokaryote evolution
– Less common in eukaryotes• Polyploidic hybridization, e.g., in plants• Endosymbionts -- mitochondria &chloroplasts
– Source of incongruence among gene trees

What?
• An integrated model for:
– Species evolution through speciation and polyploidic hybridization
• Yields species networks
– Gene evolution in species networks by gene duplication and loss
• Yields binary gene trees

A hybrid evolution model
Species evolution through speciation and polyploidic hybridization
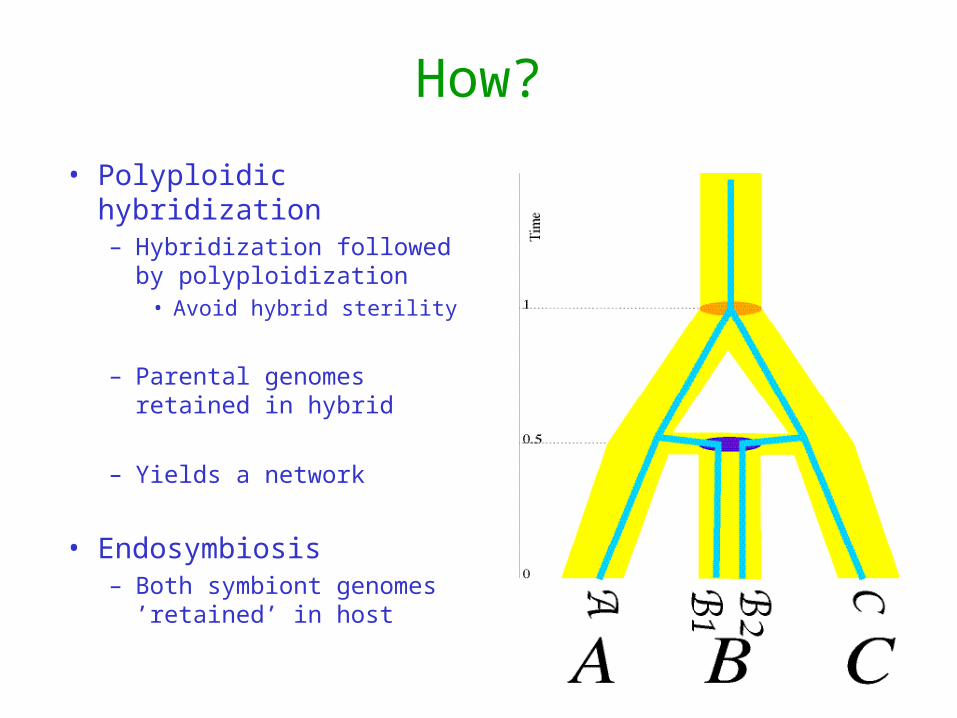
How?
• Polyploidic hybridization– Hybridization followed by
polyploidization• Avoid hybrid sterility
– Parental genomes retained in hybrid
– Yields a network
• Endosymbiosis– Both symbiont genomes
’retained’ in host

A hybrid evolution model• Extended BD model
– -- extinction rate– = +– -- speciation rate – -- hybridization parent 1
parent 2 ~U([n]), n=#lineages at ti
• Generation simple• Reconctruction Pr[S] non-trivial
– Dependencies– Ghosts
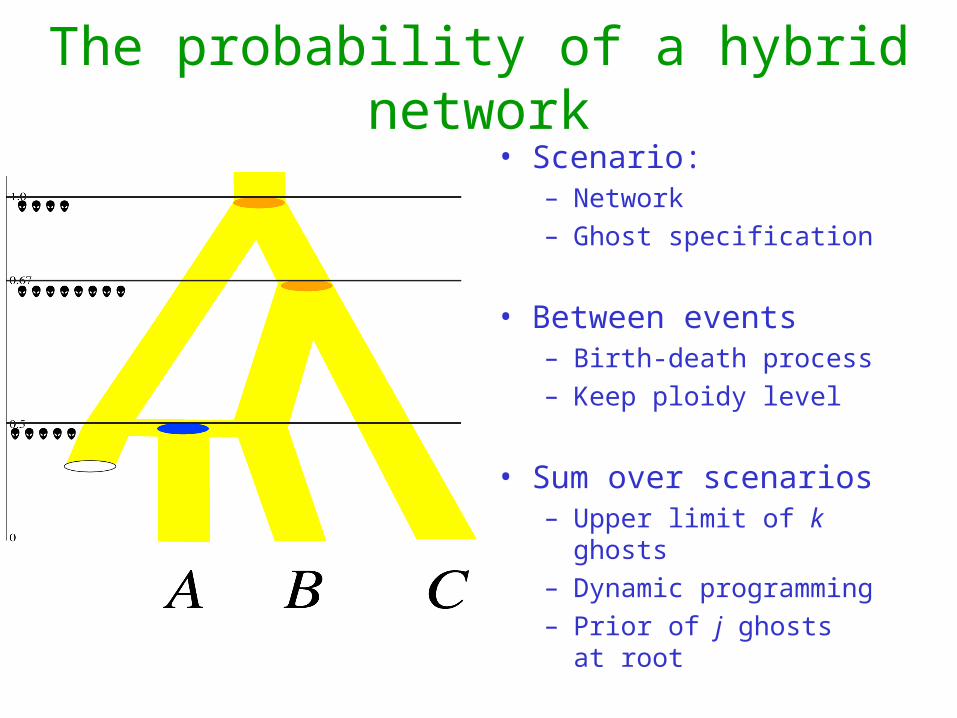
The probability of a hybrid network• Scenario:
– Network– Ghost specification
• Between events – Birth-death process– Keep ploidy level
• Sum over scenarios– Upper limit of k ghosts– Dynamic programming– Prior of j ghosts at root

Summary
• Algorithm for Pr[S] given maximmum k ghosts– Event-based model– Efficient o(nk3)
• Approximation– k 100 good approximation

Hybrid species network Gene evolution Model
Gene evolution in hybrid networks
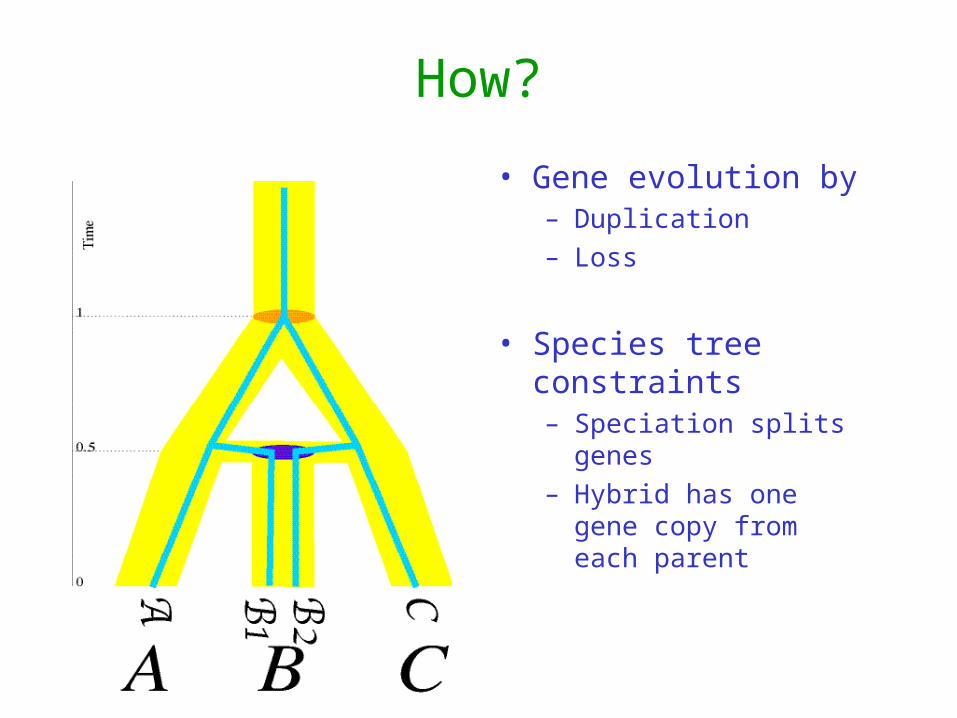
How?
• Gene evolution by– Duplication– Loss
• Species tree constraints– Speciation splits genes– Hybrid has one gene
copy from each parent

Idea: treat genomes individually and use gene evolution model
1) Extract binary homeolog tree from the hybrid species network
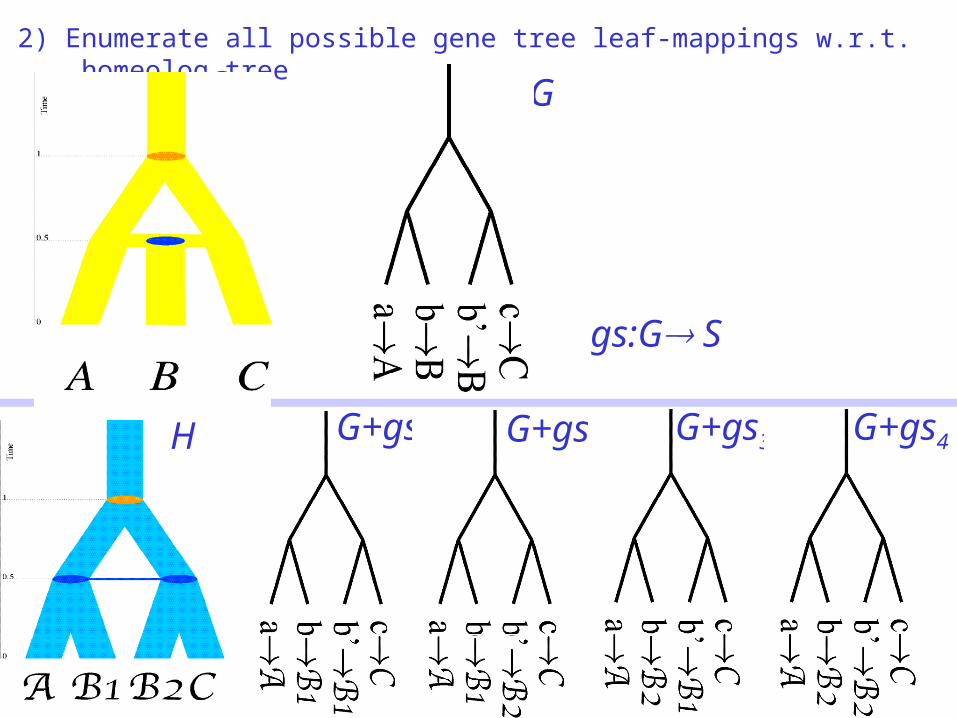
2) Enumerate all possible gene tree leaf-mappings w.r.t. homeolog tree
S G
H G+gs1 G+gs2 G+gs3 G+gs4
gs:G S

Probability of gene tree in hybrid network
• For each enumerated pair (G,gsi)– Compute probability Pr[G, gsi|H]
using the gene evolution model
• Probability of original gene tree is
i.e., the expectation over enumerated trees

Summary
• Naive brute force algorithm for Pr[G|S]– Enumeration of gs-maps exponential
• Reasonable for small problems, bad for larger
– Can be done efficiently with DP
• Model extensions– Gene loss probabilities after hybridization– Use prior information about ploidy level

Combining the models

Integrated analysis -- primeHGM
• Aim: identify hybrid species network given a set of gene trees {G1, G2,…,Gn}
• Bayesian framework
– Pr[G|S] - Extended gene evolution model– Pr[S]- Model for hybrid networks

Search for best hybrid network• Ideally -- MCMC over S
– Branch-swapping on networks problematic
– Maximum a posteriori (MAP) comparison
– Probabilistic pseudo-enumeration
• Synthetic data

Probabilistic pseudo-enumeration• Generate a set S of networks from hybrid model
• Select ’true’ S’ from S and generate set G of gene trees
• For each S S– Compute MAP of Pr[S |G] over div. time space of S
• Evaluate rank of S’ w.r.t. MAP
• Repeat with different true S’ and compute frequencies of different ranks of S’
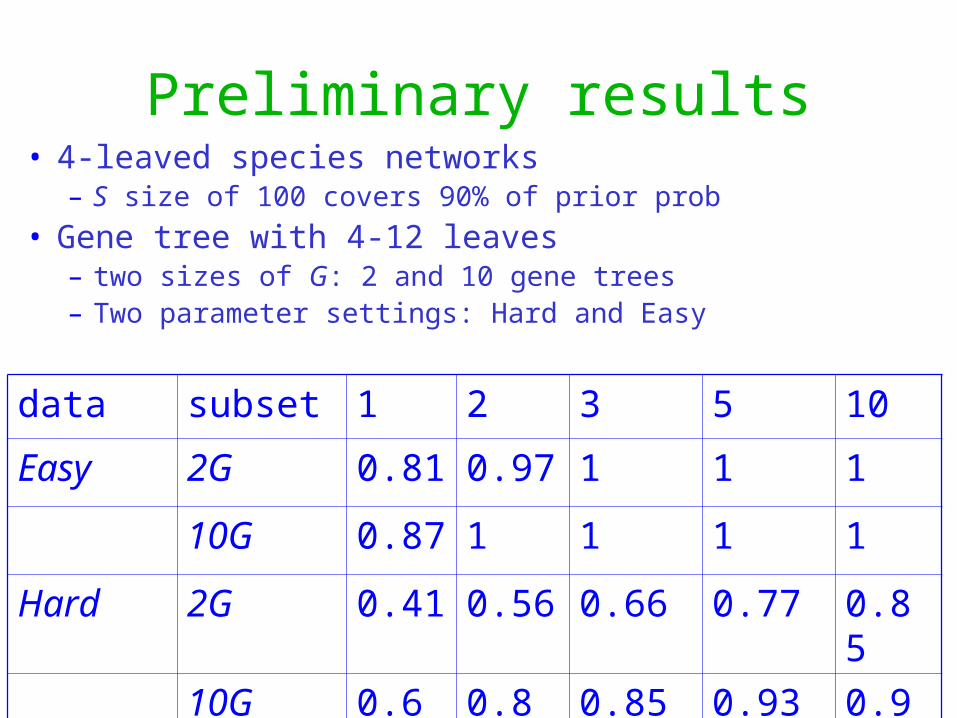
Preliminary results
data subset 1 2 3 5 10
Easy 2G 0.81 0.97 1 1 1
10G 0.87 1 1 1 1
Hard 2G 0.41 0.56 0.66 0.77 0.85
10G 0.6 0.8 0.85 0.93 0.99
• 4-leaved species networks– S size of 100 covers 90% of prior prob
• Gene tree with 4-12 leaves– two sizes of G: 2 and 10 gene trees– Two parameter settings: Hard and Easy

Summary
• primeHGM– Integrated model
• Hybrid species network• GEM in hybrid network
– MAP estimation of net work divergence times
• Future– include sequence data– Branchswapping– Inclusion of prior information


Acknowledgements
• Gene evolution model– Lars Arvestad, Ann-Charlotte Berglund-
Sonnhammer, Jens Lagergren, Örjan Åkerborg
• Hybrid species network model– Ali Tofigh, Jens Lagergren

’Pseudo-visible’ extinction vertices
Top Related