







Languages
Pages
Legal


CRISPR-Cas9 Genome Editing
20160617 Lab Animal Pathology Rounds
Han Tan ([email protected]) TwiCer: @ekhtn

Why Genome Editing?
§ Accelerate basic research § Generate mulGple alleles § Generate mulGple gene mutaGons
§ Analysis of linked genes § Analysis of lethal genes
§ Disease modeling § Gene Therapy § Replace defecGve genes § Fix specific cell-‐types
§ Agriculture § Non-‐transgenic approaches to improve crops
§ Genome engineering of plants and animals
§ Biotechnology § Ecological control of vectors that transmit diseases
§ SyntheGc biology

Engineering POLLED Phenotype in Dairy Cows
480 VOLUME 34 NUMBER 5 MAY 2016 NATURE BIOTECHNOLOGY
confirmed the homozygous introgression of PC into RCI-001, RCI-002 (named Spotigy) and RCI-003 (named Buri), and heterozygous introgression into RCI-004 (RCI, Recombinetics, numbered in order of birth), whereas progenitor cells (2120) were negative for the PC allele (Fig. 1a,b). Two healthy, homozygous polled animals, Spotigy and Buri (Fig. 1c,d and Table 1), which are now more than 10 months old, were phenotypically polled.
To evaluate off-target effects in two distinct edited lines, we sequenced the genomes of RCI-001 and RCI-002, derived from clones HP14-B4 and HP7-P4-A1 respectively, along with those of their progenitor cell lines, 2122 and 2120 respectively (Table 1). We did not identify
costly but is also painful for the animals and has come under scrutiny owing to public concerns about farm animal welfare3,4. Numerous animal advocacy groups have campaigned for mandated anesthesia during dehorning or complete cessation of dehorning as a management practice. In response to this sentiment, retailers such as Wal-Mart, Starbucks, Nestle and Kroger prioritize dehorning in their animal welfare policies5. The increased use of polled Holstein sires has been impeded by their lower estimated breeding values (a genetic merit score) for milk production, likely a result of genomic drag due to POLLED introgression from non-dairy animals. Even if producers ignore the substantial value difference of $252 per lactation cycle between horned and polled animals, it would still take >20 years of classic breeding to reach a frequency of 50% polled animals6. Further attempts to increase the frequency of polled animals in dairy herds by cross-breeding with other polled breeds, such as Angus, is not feasible because the total genetic merit for milk production would suffer losses, and the genetic admixture would take many generations to unwind7.
Identification of the genetic cause of hornlessness in cattle has been the subject of intensive genetic and genomic research, culminating in the nomination of two different candidate neomutations on cattle chromosome 1 that are predicted to have arisen 500<1,000 years ago8: a complex allele of Friesian origin (PF), an 80,128 base pair (bp) duplication (1909352–1989480 bp)9, and a second, simple allele of Celtic origin (PC) corresponding to a duplication of 212 bp (chromosome 1 positions 1705834–1706045) in place of a 10-bp deletion (1706051–1706060).
We report the use of genome editing using transcription activator-like effector nucleases (TALENs) to introgress the putative PC POLLED allele into the genome of bovine embryo fibroblasts to try and produce a genotype identical to what is
achievable using natural mating, but without the attendant genetic drag and admixture7. In our previous studies, we used TALEN-stimulated homology-dependent repair to produce four cell lines either homozygous or heterozygous for the PC allele10 (Table 1). Each of the four lines were cloned by somatic cell nuclear transfer11, and full-term pregnancies were established for three of the four lines. In total, five live calves were produced, representing two different dairy genetic backgrounds, 2122 and 2120 (Table 1). When calves were born, a board-certified veterinarian inspected each of the live-born calves visually and by palpation for horn buds, and observed none, which suggested polledness. Analysis of the polled genotype using diagnostic PCR10
Table 1 Animal production statistics
Cell line Parental cells Genotype SCNT rep Blast rate (%)Embryos/ recipients
Pregnant at day 40
Pregnant at day 90 Liveborn Alive at 60 d
HP14-B6 2122 Homozygous POLLED 1 27/64 (42) 9/9 6 0 0 0
HP14-B4 2122 Homozygous POLLED 1 3/15 (20) 1/1 1 1 1a 0
HP7-P4-A1 2120 Homozygous POLLED 2 25/82 (30) 9/9 2 2 2 2b
HP-24.8 2120 Heterozygous POLLED 3 35/151 (23) 7/7 5 2 2a 0
Summary 70/295(24%)
26/26 14/26(54%)
5/26(19%)
5/26(19%)
2/26(7%)
aRCI-001 (HP14-B6), RCI-004 and RCI-005 (HP-24.8). Consistent with known cloning inefficiencies, these animals were not viable and were humanely euthanized within 24 h of birth11. bSpotigy (RCI-002) and Buri (RCI-003). SCNT rep, somatic cell nuclear transfer replicates.
Figure 1 Phenotypic and genotypic confirmation of POLLED introgression in Spotigy and Buri. (a,b) Diagnostic PCRs for the Pc allele using primer pairs btHP-F1 + btHP-R2 (a) and HP1748-F1 + HP1594_ 1748-R1 (b) (Supplementary Methods), respectively, confirmed homozygous introgression in RCI-001, Spotigy (RCI-002) and Buri (RCI-003), and heterozygous introgression in RCI-004 relative to the donor cell line that is negative. The identity of PCR products was confirmed by Sanger sequencing. The positive control (p1748) was a plasmid containing the Pc allele10. (c) Photograph of Spotigy at 2 months of age, so named after the black spots where horn buds would have developed. (d) Photograph of Buri (left) and Spotigy at 2 months of age.
900 3,0002,5002,000
1,500
1,000
700600500400
300
RCI-001
RCI-002
RCI-003
RCI-004
Holstein2120p1748
RCI-001
RCI-002
RCI-003
RCI-004
Holsteint2120p1748
a b
c d
Size (bp)
Size (bp)
CORRESPONDENCE
(Carlson et al., Nature Biotech. 2016)
*Instant introgression directly into elite breeds – bypasses tradiGonal breeding

Outline
§ Genome EdiGng
§ The CRISPR-‐Cas9 System
§ The many flavors of Cas9
§ Genome EdiGng in the lab and beyond

What is Genome Editing?
§ Targeted modificaGon to DNA
How?
§ Using customized sequence-‐specific nucleases
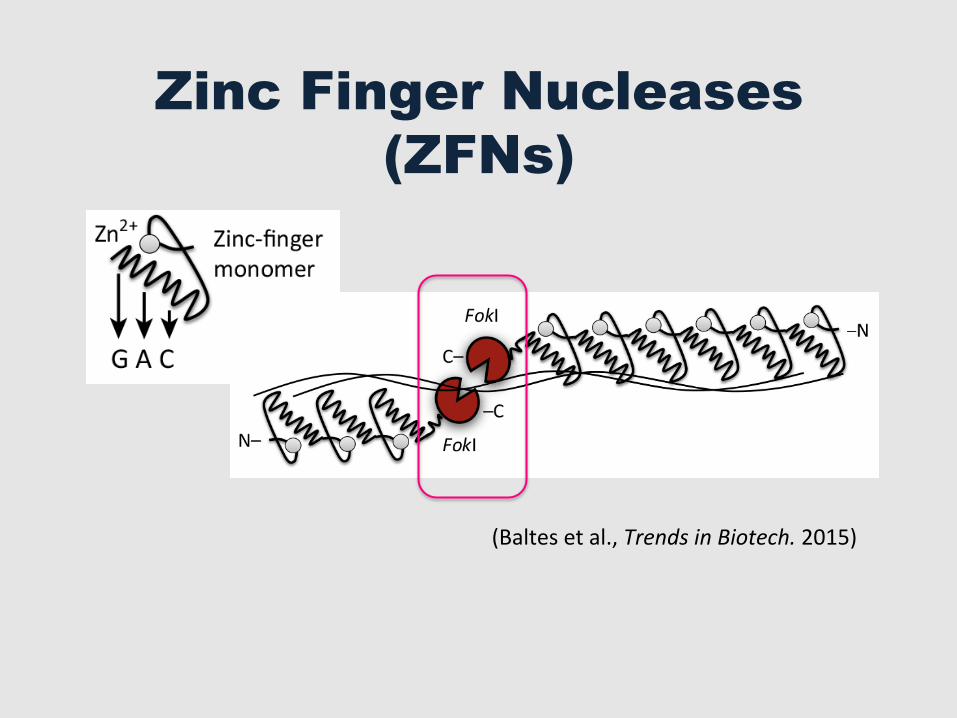
Zinc Finger Nucleases (ZFNs)
(Baltes et al., Trends in Biotech. 2015)
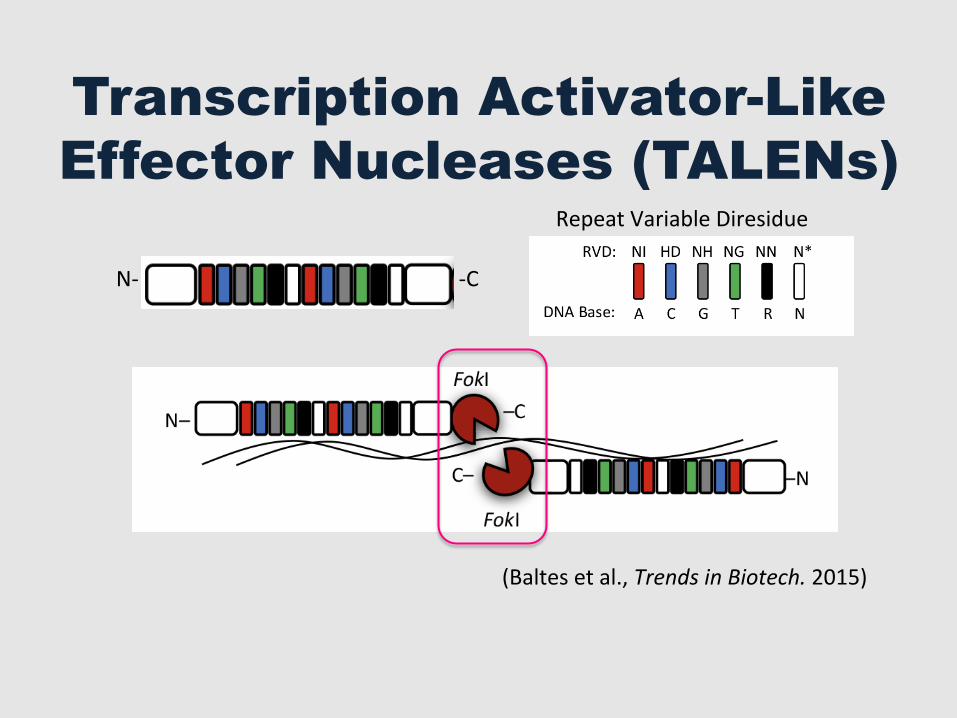
Transcription Activator-Like Effector Nucleases (TALENs)
(Baltes et al., Trends in Biotech. 2015)
N-‐ -‐C
Repeat Variable Diresidue
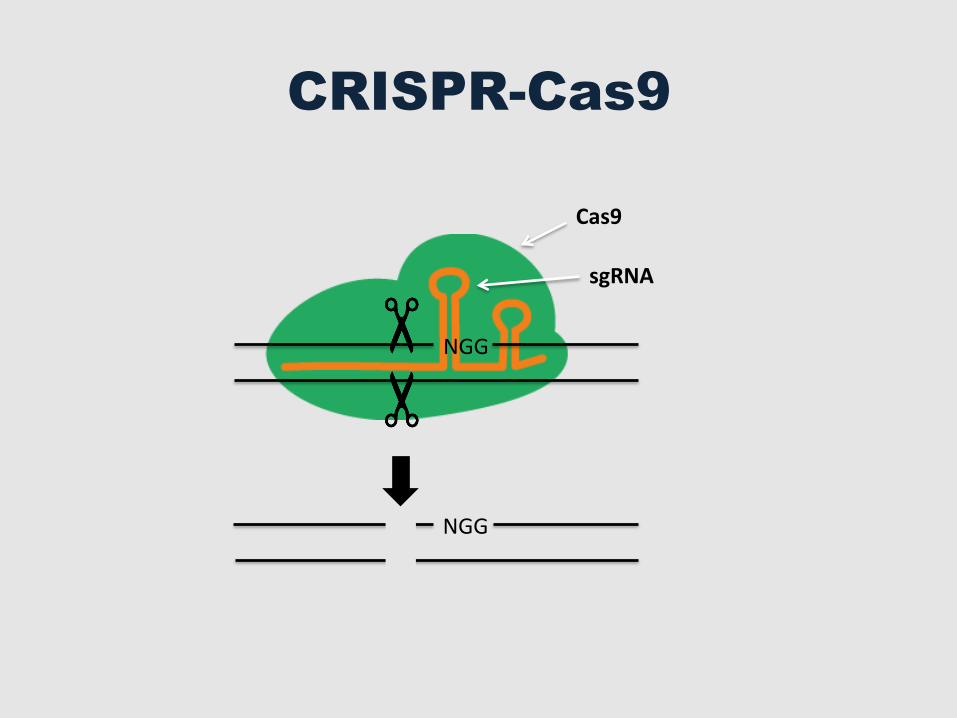
Cas9
CRISPR-Cas9
sgRNA
NGG
NGG
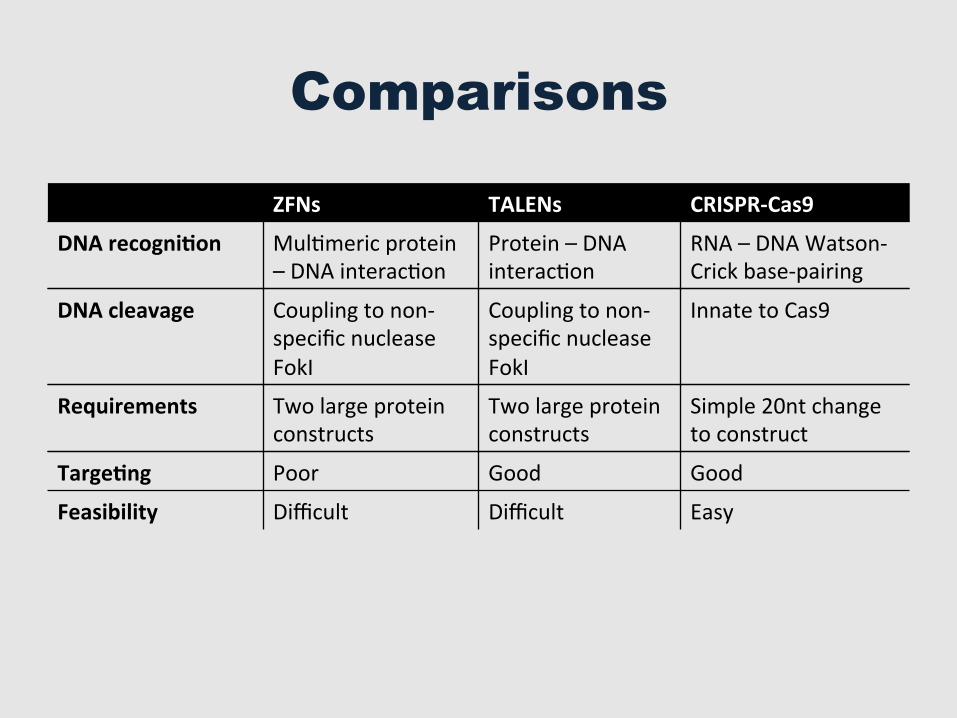
Comparisons
ZFNs TALENs CRISPR-‐Cas9
DNA recogni=on MulGmeric protein – DNA interacGon
Protein – DNA interacGon
RNA – DNA Watson-‐Crick base-‐pairing
DNA cleavage Coupling to non-‐specific nuclease FokI
Coupling to non-‐specific nuclease FokI
Innate to Cas9
Requirements Two large protein constructs
Two large protein constructs
Simple 20nt change to construct
Targe=ng Poor Good Good
Feasibility Difficult Difficult Easy
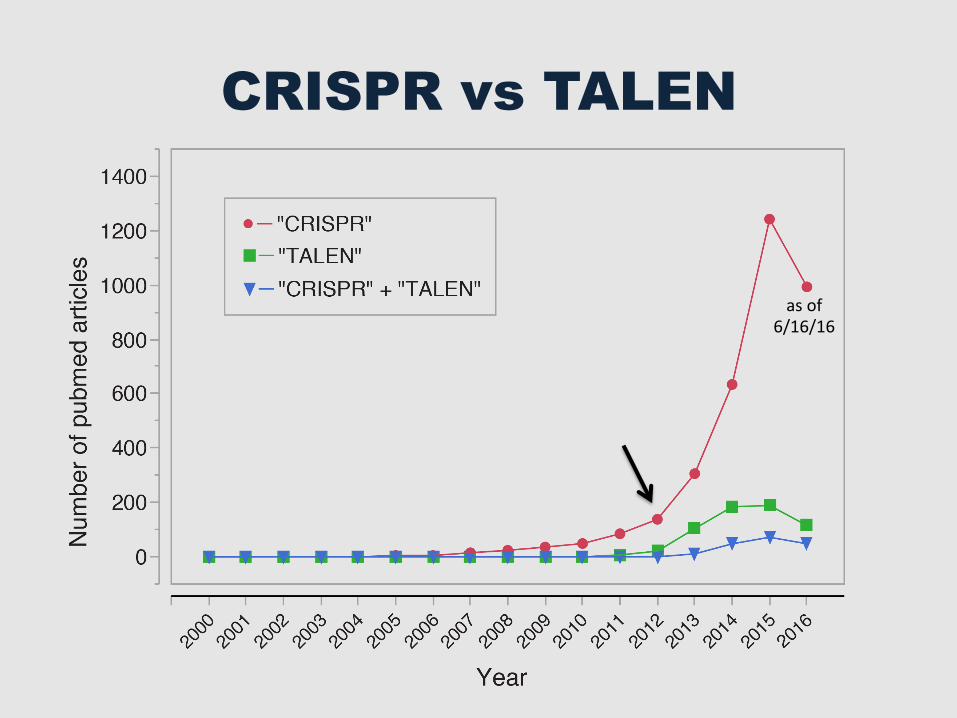
CRISPR vs TALEN
as of 6/16/16

Outline
§ Genome EdiGng
§ The CRISPR-‐Cas9 System
§ The many flavors of Cas9
§ CRISPR-‐Cas9 in the lab and beyond

Discovery of CRISPR-Cas
§ CRISPR = Clustered Regularly Interspersed Short Palindromic Repeats [DNA repeats]
§ Cas = CRISPR associated [Protein coding sequences]
§ Discovered in 1987 from the analysis of E. coli genomes (Ishino et al., J. Bacteriol. 1987)
§ Is important for adapGve immunity in bacteria and archaea
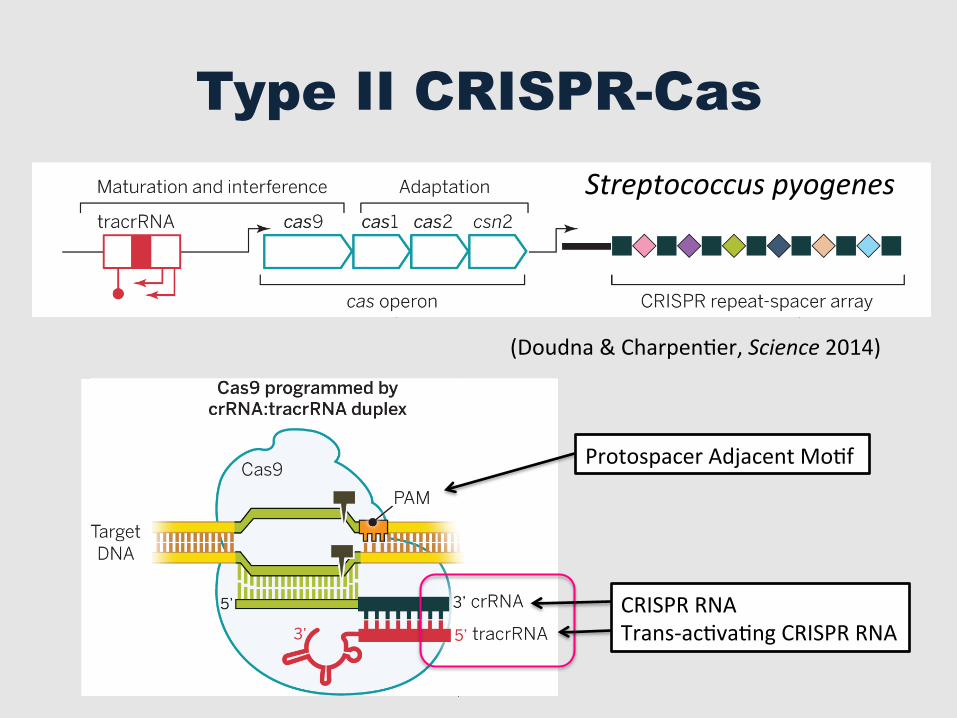
Type II CRISPR-Cas
activity, whereasmutating both domains (dCas9;Asp10 ! Ala, His840 ! Ala) results in an RNA-guidedDNAbinding protein (64, 65). DNA targetrecognition requires both base pairing to thecrRNA sequence and the presence of a short se-quence (PAM) adjacent to the targeted sequencein the DNA (64, 65) (Fig. 2).The dual tracrRNA:crRNA was then engineered
as a single guide RNA (sgRNA) that retains twocritical features: the 20-nucleotide sequence atthe 5! end of the sgRNA that determines theDNAtarget site by Watson-Crick base pairing, and thedouble-stranded structure at the 3! side of theguide sequence that binds to Cas9 (64) (Fig. 2).This created a simple two-component system inwhich changes to the guide sequence (20 nucleo-tides in the native RNA) of the sgRNA can beused to program CRISPR-Cas9 to target any DNAsequence of interest as long as it is adjacent toa PAM (64). In contrast to ZFNs and TALENs,which require substantial protein engineering foreach DNA target site to be modified, the CRISPR-Cas9 system requires only a change in the guideRNA sequence. For this reason, the CRISPR-Cas9
technology using the S. pyogenes system has beenrapidly and widely adopted by the scientific com-munity to target, edit, or modify the genomes of avast array of cells and organisms. Phylogeneticstudies (69–71) as well as in vitro and in vivoexperiments (64, 71, 72) show that naturallyoccurring Cas9 orthologs use distinct tracrRNA:crRNA transcripts as guides, defined by thespecificity to the dual-RNA structures (69–71) (Fig.3). The reported collection of Cas9 orthologs con-stitutes a large source of CRISPR-Cas9 systems formultiplex gene targeting, and several orthologousCRISPR-Cas9 systems have already been appliedsuccessfully for genome editing in human cells[Neisseria meningitidis (73, 74), S. thermophilus(73, 75), and Treponema denticola (73)].Although the CRISPR acronym has attracted
media attention and is widely used in the scien-tific and popular literature, nearly all genomeediting applications are based on the use of theprotein Cas9 together with suitable sgRNAs. Asdiscussed above, CRISPR refers to the repetitivenature of the repeats in the CRISPR arrays thatencode crRNAs, and the term does not relate
directly to genome engineering. Nonetheless weprefer to use “CRISPR-Cas9” in a way that is lessrestrictive than other nomenclatures that havebeen used in the field (76).
Mechanism of CRISPR-Cas9–mediatedgenome targeting
Structural analysis of S. pyogenes Cas9 has re-vealed additional insights into the mechanismof CRISPR-Cas9 (Fig. 3). Molecular structures ofCas9 determined by electron microscopy andx-ray crystallography show that the protein un-dergoes large conformational rearrangementupon binding to the guide RNA, with a furtherchange upon association with a target double-stranded DNA (dsDNA). This change creates achannel, running between the two structurallobes of the protein, that binds to the RNA-DNAhybrid as well as to the coaxially stacked dual-RNA structure of the guide corresponding tothe crRNA repeat–tracrRNA antirepeat interac-tion (77, 78). An arginine-rich a helix (77–79)bridges the two structural lobes of Cas9 and ap-pears to be the hinge between them, in addition
SCIENCE sciencemag.org 28 NOVEMBER 2014 • VOL 346 ISSUE 6213 1258096-3
Fig. 2. Biology of the type II-A CRISPR-Cas system. The type II-A system from S. pyogenes is shown as an example. (A) The cas gene operon withtracrRNA and the CRISPR array. (B) The natural pathway of antiviral defense involves association of Cas9 with the antirepeat-repeat RNA (tracrRNA:crRNA) duplexes, RNA co-processing by ribonuclease III, further trimming, R-loop formation, and target DNA cleavage. (C) Details of the natural DNAcleavage with the duplex tracrRNA:crRNA.
RESEARCH | REVIEW
on
May
20,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
(Doudna & CharpenGer, Science 2014)
to playing a central role in binding the guideRNA–target DNA hybrid as shown by mutagen-esis (77, 78). The conformational change in Cas9may be part of the mechanism of target dsDNAunwinding and guide RNA strand invasion,although this idea remains to be tested. Mech-anistic studies also show that the PAM is criticalfor initial DNA binding; in the absence of thePAM, even target sequences fully complemen-tary to the guide RNA sequence are not rec-ognized by Cas9 (80). A crystal structure of Cas9in complex with a guide RNA and a partiallydsDNA target demonstrates that the PAM lieswithin a base-paired DNA structure (81). Argininemotifs in the C-terminal domain of Cas9 interactwith the PAM on the noncomplementary strandwithin the major groove. The phosphodiestergroup at position +1 in the target DNA strandinteracts with the minor groove of the duplexedPAM, possibly resulting in local strand separa-tion, the so-called R-loop, immediately upstreamof the PAM (81). Single-molecule experiments alsosuggest that R-loop association rates are affectedprimarily by the PAM, whereas R-loop stabilityis influenced mainly by protospacer elementsdistal to the PAM (82). Together with single-molecule and bulk biochemical experimentsusing mutated target DNAs, a mechanism canbe proposed whereby target DNA melting startsat the level of PAM recognition, resulting in di-rectional R-loop formation expanding towardthe distal protospacer end and concomitant RNAstrand invasion and RNA-DNA hybrid forma-tion (80–82).To assess the target-binding behavior of Cas9
in cells, researchers used chromatin immuno-precipitation and high-throughput sequencing(ChIP-seq) to determine the numbers and typesof Cas9 binding sites on the chromosome. Re-sults showed that in both human embryonic kid-ney (HEK293) cells (83) and mouse embryonicstem cells (mESCs) (84), a catalytically inactiveversion of Cas9 bound to many more sites thanthose matching the sequence of the sgRNA usedin each case. Such off-target interactions withDNA, typically at sites bearing a PAM and par-tially complementary to the guide RNA se-quence, are consistent with established modes
of DNA interrogation by Cas9 (80). Active Cas9rarely cleaves the DNA at off-target binding sites,implying decoupled binding and cleavage eventsin which nearly perfect complementarity betweenthe guide RNA and the target site are necessaryfor efficient DNA cleavage. These observations areconsistent with results obtained for Cas9–guideRNA complexes in single-molecule experiments(80). Furthermore,Cas9binding events occurmoredensely in areas of open chromatin as comparedto regions of compact, transcriptionally inactivechromatin. However, because the method involvescross-linking cells for ~10 min before quenchingthe reaction, transient and long-lived binding in-teractions cannot be distinguished. It is possiblethat many of the apparent off-target DNA in-teractions in fact reflect brief encounters thatwould not normally trigger strand invasion bythe guide RNA.
Engineering cells and model organisms
Following the 2012 publication of Jinek et al.(64), three studies in January 2013 demonstratedthat CRISPR-Cas9 represents an efficient tool toedit the genomes of human cells (75, 85, 86).The “humanized” versions of S. pyogenes Cas9(75, 85, 86) and S. thermophilus Cas9 (75) werecoexpressed with custom-designed sgRNAs(75, 85, 86) or with tracrRNA coexpressed withcustom-designed crRNAs (75) in human embry-onic kidney, chronic myelogenous leukemia, orinduced pluripotent stem cells (75, 85, 86) as wellas in mouse cells (75). The expected alterationsin the target DNA were observed, indicatingthat site-specific DSBs by RNA-guided Cas9 hadstimulated gene editing by nonhomologous endjoining repair or gene replacement by homology-directed repair (Fig. 4). Targeting with multiplesgRNAs—referred to as multiplexing—was alsosuccessfully achieved (75, 86). RNA-programmableS. pyogenes Cas9-mediated editing has now beenapplied to various human cells and embryonicstem cells [(87–90); for reviews, see (91–93)]. Al-though direct comparisons can be difficult toassess because of differences in target sites andprotein expression levels, some analyses showthat CRISPR-Cas9–mediated editing efficienciescan reach 80% or more depending on the target,
which is as high as or higher than levels observedusing ZFNs or TALENs (89, 94).These initial studies were only the beginning
of what has become an incredibly fast-paced fieldin which laboratories around the world have usedCRISPR-Cas9 to edit genomes of a wide range ofcell types and organisms (summarized in Fig. 5). Asof this writing, more than 1000 papers have beenpublished that include the CRISPR acronym inthe title or abstract, with the majority of thesepublished since the beginning of 2013. Many ofthese applications have been discussed in re-cent reviews (91–93). Here we highlight a fewexamples that illustrate the power of the tech-nology (Fig. 6). The first example is the precisereproduction of tumor-associated chromosomaltranslocations, which come about during carcino-genesis through illegitimate nonhomologousjoiningof two chromosomes. The ability of CRISPR-Cas9 to introduce DSBs at defined positions hasmade it possible to generate human cell lines andprimary cells bearing chromosomal translationsresembling those described in cancers such aslung cancer (95), acute myeloid leukemia, andEwing’s sarcoma (96, 97). An improved methodto generate liver cancer or myeloid malignancymodels in mice facilitated by CRISPR-Cas9 wasrecently reported (98, 99). CRISPR-Cas9 thus pro-vides a robust technology for studying genomicrearrangements and the development and pro-gression of cancers or other diseases.A second example is the systematic analysis of
gene functions in mammalian cells. A genome-scale lentiviral sgRNA library was developedto generate a pooled loss-of-function geneticscreening approach suitable for both positiveand negative selection (100, 101). This approachwas also used to identify genes essential forcell viability in cancer and pluripotent stem cells(102). Although such studies have been attemptedusing RNA interference (RNAi) to reduce theexpression of genes, this strategy does not allowthe generation of gene knockouts and can suf-fer from substantial off-target effects. The useof CRISPR-Cas9 for genome-wide studies willenable large-scale screening for drug targets andother phenotypes and thus will expand the natureand utility of genetic screens in human and other
nonmodel cell types and organisms.Other pertinent examples of
CRISPR-Cas9 applications with rel-evance to human health includethe ability to correct genetic muta-tions responsible for inherited dis-orders. A dominant mutation inthe Crygc gene responsible forcataracts was successfully correctedin mice (103). Using cultured pri-mary adult intestinal stem cellsderived from cystic fibrosis patients,the CFTR locus responsible forcystic fibrosis was corrected byhomologous recombination, result-ing in the clonal expansion ofminiature organlike cell cultures(organoids) harboring the desired,exact genetic change (104). These
1258096-4 28 NOVEMBER 2014 • VOL 346 ISSUE 6213 sciencemag.org SCIENCE
Fig. 3. Evolution and structure of Cas9.The structure of S. pyogenes Cas9 in the unliganded and RNA-DNA–boundforms [from (77, 81)].
RESEARCH | REVIEW
on
May
20,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
Streptococcus pyogenes
Protospacer Adjacent MoGf
CRISPR RNA Trans-‐acGvaGng CRISPR RNA
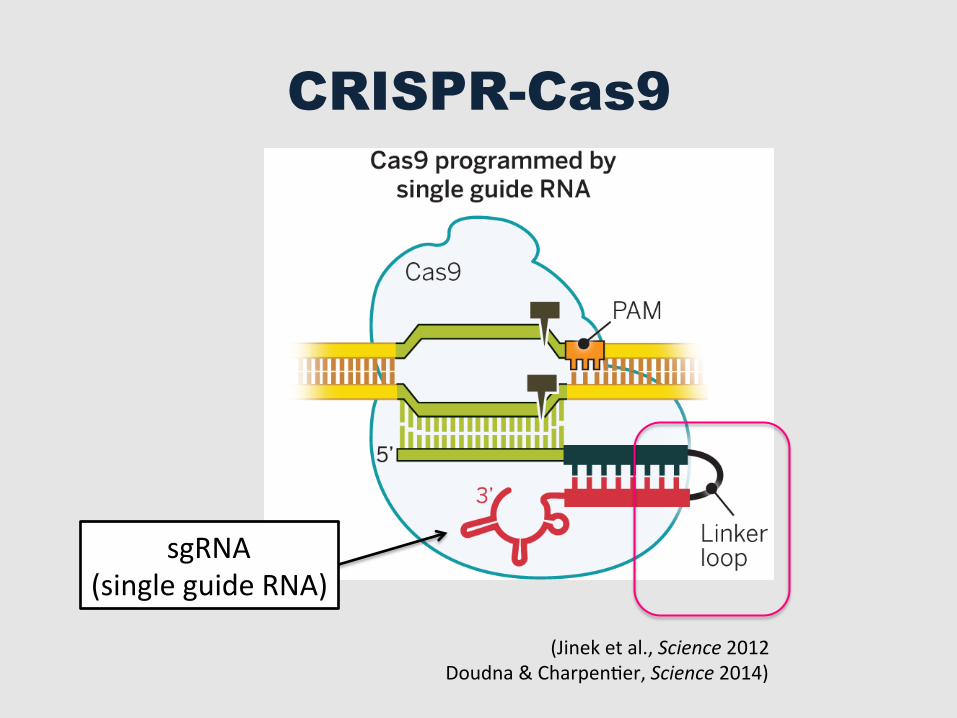
CRISPR-Cas9
to playing a central role in binding the guideRNA–target DNA hybrid as shown by mutagen-esis (77, 78). The conformational change in Cas9may be part of the mechanism of target dsDNAunwinding and guide RNA strand invasion,although this idea remains to be tested. Mech-anistic studies also show that the PAM is criticalfor initial DNA binding; in the absence of thePAM, even target sequences fully complemen-tary to the guide RNA sequence are not rec-ognized by Cas9 (80). A crystal structure of Cas9in complex with a guide RNA and a partiallydsDNA target demonstrates that the PAM lieswithin a base-paired DNA structure (81). Argininemotifs in the C-terminal domain of Cas9 interactwith the PAM on the noncomplementary strandwithin the major groove. The phosphodiestergroup at position +1 in the target DNA strandinteracts with the minor groove of the duplexedPAM, possibly resulting in local strand separa-tion, the so-called R-loop, immediately upstreamof the PAM (81). Single-molecule experiments alsosuggest that R-loop association rates are affectedprimarily by the PAM, whereas R-loop stabilityis influenced mainly by protospacer elementsdistal to the PAM (82). Together with single-molecule and bulk biochemical experimentsusing mutated target DNAs, a mechanism canbe proposed whereby target DNA melting startsat the level of PAM recognition, resulting in di-rectional R-loop formation expanding towardthe distal protospacer end and concomitant RNAstrand invasion and RNA-DNA hybrid forma-tion (80–82).To assess the target-binding behavior of Cas9
in cells, researchers used chromatin immuno-precipitation and high-throughput sequencing(ChIP-seq) to determine the numbers and typesof Cas9 binding sites on the chromosome. Re-sults showed that in both human embryonic kid-ney (HEK293) cells (83) and mouse embryonicstem cells (mESCs) (84), a catalytically inactiveversion of Cas9 bound to many more sites thanthose matching the sequence of the sgRNA usedin each case. Such off-target interactions withDNA, typically at sites bearing a PAM and par-tially complementary to the guide RNA se-quence, are consistent with established modes
of DNA interrogation by Cas9 (80). Active Cas9rarely cleaves the DNA at off-target binding sites,implying decoupled binding and cleavage eventsin which nearly perfect complementarity betweenthe guide RNA and the target site are necessaryfor efficient DNA cleavage. These observations areconsistent with results obtained for Cas9–guideRNA complexes in single-molecule experiments(80). Furthermore,Cas9binding events occurmoredensely in areas of open chromatin as comparedto regions of compact, transcriptionally inactivechromatin. However, because the method involvescross-linking cells for ~10 min before quenchingthe reaction, transient and long-lived binding in-teractions cannot be distinguished. It is possiblethat many of the apparent off-target DNA in-teractions in fact reflect brief encounters thatwould not normally trigger strand invasion bythe guide RNA.
Engineering cells and model organisms
Following the 2012 publication of Jinek et al.(64), three studies in January 2013 demonstratedthat CRISPR-Cas9 represents an efficient tool toedit the genomes of human cells (75, 85, 86).The “humanized” versions of S. pyogenes Cas9(75, 85, 86) and S. thermophilus Cas9 (75) werecoexpressed with custom-designed sgRNAs(75, 85, 86) or with tracrRNA coexpressed withcustom-designed crRNAs (75) in human embry-onic kidney, chronic myelogenous leukemia, orinduced pluripotent stem cells (75, 85, 86) as wellas in mouse cells (75). The expected alterationsin the target DNA were observed, indicatingthat site-specific DSBs by RNA-guided Cas9 hadstimulated gene editing by nonhomologous endjoining repair or gene replacement by homology-directed repair (Fig. 4). Targeting with multiplesgRNAs—referred to as multiplexing—was alsosuccessfully achieved (75, 86). RNA-programmableS. pyogenes Cas9-mediated editing has now beenapplied to various human cells and embryonicstem cells [(87–90); for reviews, see (91–93)]. Al-though direct comparisons can be difficult toassess because of differences in target sites andprotein expression levels, some analyses showthat CRISPR-Cas9–mediated editing efficienciescan reach 80% or more depending on the target,
which is as high as or higher than levels observedusing ZFNs or TALENs (89, 94).These initial studies were only the beginning
of what has become an incredibly fast-paced fieldin which laboratories around the world have usedCRISPR-Cas9 to edit genomes of a wide range ofcell types and organisms (summarized in Fig. 5). Asof this writing, more than 1000 papers have beenpublished that include the CRISPR acronym inthe title or abstract, with the majority of thesepublished since the beginning of 2013. Many ofthese applications have been discussed in re-cent reviews (91–93). Here we highlight a fewexamples that illustrate the power of the tech-nology (Fig. 6). The first example is the precisereproduction of tumor-associated chromosomaltranslocations, which come about during carcino-genesis through illegitimate nonhomologousjoiningof two chromosomes. The ability of CRISPR-Cas9 to introduce DSBs at defined positions hasmade it possible to generate human cell lines andprimary cells bearing chromosomal translationsresembling those described in cancers such aslung cancer (95), acute myeloid leukemia, andEwing’s sarcoma (96, 97). An improved methodto generate liver cancer or myeloid malignancymodels in mice facilitated by CRISPR-Cas9 wasrecently reported (98, 99). CRISPR-Cas9 thus pro-vides a robust technology for studying genomicrearrangements and the development and pro-gression of cancers or other diseases.A second example is the systematic analysis of
gene functions in mammalian cells. A genome-scale lentiviral sgRNA library was developedto generate a pooled loss-of-function geneticscreening approach suitable for both positiveand negative selection (100, 101). This approachwas also used to identify genes essential forcell viability in cancer and pluripotent stem cells(102). Although such studies have been attemptedusing RNA interference (RNAi) to reduce theexpression of genes, this strategy does not allowthe generation of gene knockouts and can suf-fer from substantial off-target effects. The useof CRISPR-Cas9 for genome-wide studies willenable large-scale screening for drug targets andother phenotypes and thus will expand the natureand utility of genetic screens in human and other
nonmodel cell types and organisms.Other pertinent examples of
CRISPR-Cas9 applications with rel-evance to human health includethe ability to correct genetic muta-tions responsible for inherited dis-orders. A dominant mutation inthe Crygc gene responsible forcataracts was successfully correctedin mice (103). Using cultured pri-mary adult intestinal stem cellsderived from cystic fibrosis patients,the CFTR locus responsible forcystic fibrosis was corrected byhomologous recombination, result-ing in the clonal expansion ofminiature organlike cell cultures(organoids) harboring the desired,exact genetic change (104). These
1258096-4 28 NOVEMBER 2014 • VOL 346 ISSUE 6213 sciencemag.org SCIENCE
Fig. 3. Evolution and structure of Cas9.The structure of S. pyogenes Cas9 in the unliganded and RNA-DNA–boundforms [from (77, 81)].
RESEARCH | REVIEW
on
May
20,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
sgRNA (single guide RNA)
(Jinek et al., Science 2012 Doudna & CharpenGer, Science 2014)

What can you do with double-‐stranded DNA breaks?
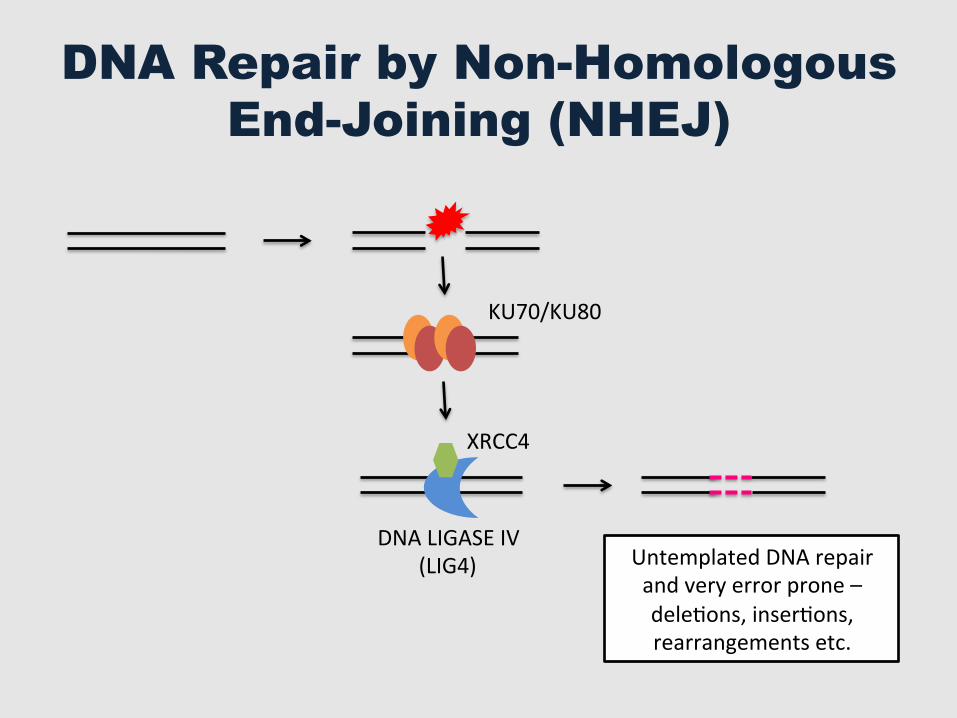
DNA Repair by Non-Homologous End-Joining (NHEJ)
DNA LIGASE IV (LIG4)
XRCC4
KU70/KU80
Untemplated DNA repair and very error prone – deleGons, inserGons, rearrangements etc.

Usefulness of DNA Repair by NHEJ
§ An effecGve form of mutagenesis § Diversity of breakpoints repaired by NHEJ = mulGple alleles are generated instantly
§ Make two breaks for large deleGons § Make mulGple breaks for translocaGons, desired rearrangements, cut and paste modificaGons
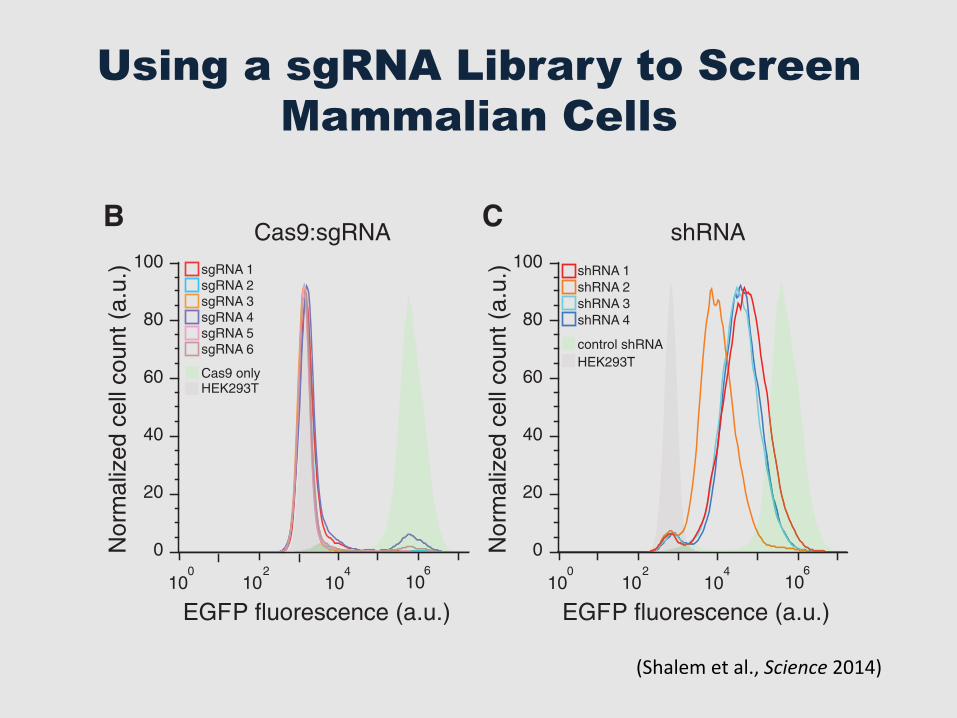
Using a sgRNA Library to Screen Mammalian Cells
(DSBs) at specific genomic loci (8, 9) through asynthetic single-guide RNA (sgRNA) (10), whichwhen targeted to coding regions of genes can cre-ate frame shift insertion/deletion (indel) mutationsthat result in a loss-of-function allele. Because thetargeting specificity of Cas9 is conferred by shortguide sequences, which can be easily generated atlarge scale by array-based oligonucleotide librarysynthesis (11), we sought to explore the potential ofCas9 for pooled genome-scale functional screening.
Lentiviral vectors are commonly used fordelivery of pooled short-hairpin RNAs (shRNAs)in RNAi because they can be easily titrated tocontrol transgene copy number and are stablymaintained as genomic integrants during subse-quent cell replication (2, 12, 13). Therefore, wedesigned a single lentiviral vector to deliver Cas9,a sgRNA, and a puromycin selection marker intotarget cells (lentiCRISPR) (Fig. 1A). The ability tosimultaneously deliver Cas9 and sgRNA through
a single vector enables application to any cell typeof interest, without the need to first generate celllines that express Cas9.
To determine the efficacy of gene knockout bylentiCRISPR transduction, we tested six sgRNAstargeting enhanced green fluorescent protein (EGFP)in a human embryonic kidney (HEK) 293T cellline containing a single copy of EGFP (fig. S1).After transduction at a lowmultiplicity of infection(MOI = 0.3) followed by selection with puromy-cin, lentiCRISPRs abolished EGFP fluorescencein 93 T 8% (mean T SD) of cells after 11 days(Fig. 1B). Deep sequencing of the EGFP locusrevealed a 92 T 9% indel frequency (n ! 104
sequencing reads per condition) (fig. S2). Incontrast, transduction of cells with lentiviralvectors expressing EGFP-targeting shRNA ledto incomplete knockdown of EGFP fluorescence(Fig. 1C).
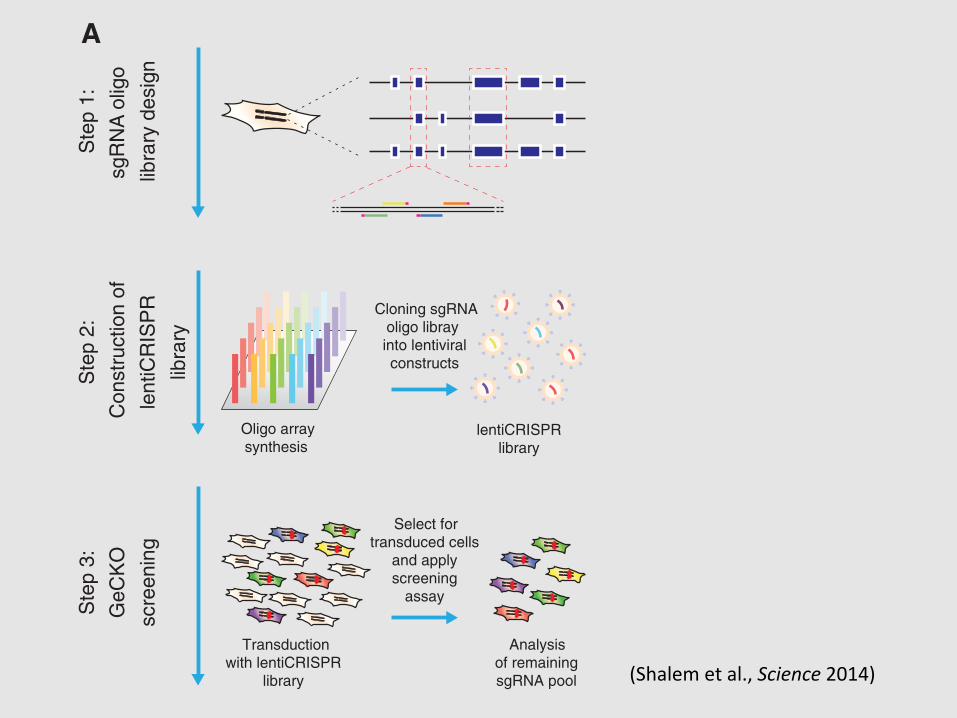
Given the high efficacy of gene knockoutby lentiCRISPR, we tested the feasibility of con-ducting genome-scale CRISPR-Cas9 knockout(GeCKO) screening with a pooled lentiCRISPRlibrary. We designed a library of sgRNAs target-ing 5! constitutive exons (Fig. 2A) of 18,080 genesin the human genome with an average coverageof 3 to 4 sgRNAs per gene (table S1), and eachtarget site was selected tominimize off-target mod-ification (14) (see supplementary text).
To test the efficacy of the full GeCKO libraryat achieving knockout of endogenous gene tar-gets, we conducted a negative selection screen byprofiling the depletion of sgRNAs targeting essen-tial survival genes (Fig. 2A). We transduced thehuman melanoma cell line A375 and the human
Fig. 1. Lentiviral deliv-ery of Cas9 and sgRNAprovidesefficientdeple-tion of target genes. (A)Lentiviral expression vec-tor for Cas9 and sgRNA(lentiCRISPR). puro, puro-mycin selection marker;psi+, psi packaging signal;RRE, rev response element;cPPT,centralpolypurinetract;EFS, elongation factor-1ashort promoter; P2A, 2Aself-cleavingpeptide;WPRE,posttranscriptional regu-latory element. (B) Distri-bution of fluorescence from293T-EGFP cells transducedby EGFP-targeting lenti-CRISPR (sgRNAs 1 to 6,outlined peaks) and Cas9-only (green-shaded peak)vectors, and nonfluorescent 293T cells (gray shaded peak). (C) Distribution of fluorescence from 293T-EGFP cellstransduced by EGFP-targeting shRNA (shRNAs 1 to 4, outlined peaks) and control shRNA (green-shaded peak)vectors, and nonfluorescent 293T cells (gray shaded peak).
A
B C
sgRNAU6 EFS SpCas9 WPREP2A Puro
cPPT
RREpsi+
lentiCRISPR
EGFP fluorescence (a.u.)
Nor
mal
ized
cel
l cou
nt (
a.u.
)
Nor
mal
ized
cel
l cou
nt (
a.u.
)100
80
60
40
20
0
100 2 4 6
10 10 10
Cas9 onlyHEK293T
sgRNA 1sgRNA 2sgRNA 3sgRNA 4sgRNA 5sgRNA 6
Cas9:sgRNA100
80
60
40
20
0
100 2 4 6
10 10 10
control shRNAHEK293T
shRNA 1shRNA 2shRNA 3shRNA 4
EGFP fluorescence (a.u.)
shRNA
A B
D
Ste
p 1:
sgR
NA
olig
olib
rary
des
ign
Ste
p 2:
Con
stru
ctio
n of
lent
iCR
ISP
Rsc
reen
ing
libra
ry
Oligo arraysynthesis
lentiCRISPRlibrary
Cloning sgRNAoligo libray into lentiviralconstructs
Ste
p 3:
GeC
KO
Transductionwith lentiCRISPR
library
Select fortransduced cells
and applyscreening
assay
Analysisof remainingsgRNA pool
A375
12
Day 14
Day 3
Log2 normalized gene counts
Cum
ulat
ive
freq
uenc
y
0
0.2
0.4
0.6
0.8
1
2 4 6 8 10
HUES62
12
Day 14
Day 3
Log2 normalized gene counts
Cum
ulat
ive
freq
uenc
y
0
0.2
0.4
0.6
0.8
1
2 4 6 8 10
C
ERNA processing
Structural constituentof ribosome
Ribonucleoprotein complex (RNPC)
RNPC biogenesisand assembly
RNA binding
Gene rank2000 6000 10000 14000 18000
Translation
Kinetochore
RNA processing
Chromosome part
Depleted Enriched
Gene rank2000 6000 10000 14000 18000
Depleted Enriched
Structural constituentof ribosome
Fig. 2. GeCKO library design and application for genome-scale negativeselection screening. (A) Design of sgRNA library for genome-scale knockout ofcoding sequences in human cells (see supplementary text). (B and C) Cumulativefrequency of sgRNAs 3 and 14 days after transduction in A375 and human em-
bryonic stem cells, respectively. Shift in the 14-day curve represents the depletion in asubset of sgRNAs. (D and E) The five most significantly depleted gene sets in A375cells [nominal P< 10!5, false discovery rate (FDR)–corrected q< 10!5] and HUES62cells (nominal P < 10!5, FDR-corrected q < 10!3) identified by GSEA (15).
www.sciencemag.org SCIENCE VOL 343 3 JANUARY 2014 85
REPORTS
on
May
27,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
(Shalem et al., Science 2014)
(DSBs) at specific genomic loci (8, 9) through asynthetic single-guide RNA (sgRNA) (10), whichwhen targeted to coding regions of genes can cre-ate frame shift insertion/deletion (indel) mutationsthat result in a loss-of-function allele. Because thetargeting specificity of Cas9 is conferred by shortguide sequences, which can be easily generated atlarge scale by array-based oligonucleotide librarysynthesis (11), we sought to explore the potential ofCas9 for pooled genome-scale functional screening.
Lentiviral vectors are commonly used fordelivery of pooled short-hairpin RNAs (shRNAs)in RNAi because they can be easily titrated tocontrol transgene copy number and are stablymaintained as genomic integrants during subse-quent cell replication (2, 12, 13). Therefore, wedesigned a single lentiviral vector to deliver Cas9,a sgRNA, and a puromycin selection marker intotarget cells (lentiCRISPR) (Fig. 1A). The ability tosimultaneously deliver Cas9 and sgRNA through
a single vector enables application to any cell typeof interest, without the need to first generate celllines that express Cas9.
To determine the efficacy of gene knockout bylentiCRISPR transduction, we tested six sgRNAstargeting enhanced green fluorescent protein (EGFP)in a human embryonic kidney (HEK) 293T cellline containing a single copy of EGFP (fig. S1).After transduction at a lowmultiplicity of infection(MOI = 0.3) followed by selection with puromy-cin, lentiCRISPRs abolished EGFP fluorescencein 93 T 8% (mean T SD) of cells after 11 days(Fig. 1B). Deep sequencing of the EGFP locusrevealed a 92 T 9% indel frequency (n ! 104
sequencing reads per condition) (fig. S2). Incontrast, transduction of cells with lentiviralvectors expressing EGFP-targeting shRNA ledto incomplete knockdown of EGFP fluorescence(Fig. 1C).
Given the high efficacy of gene knockoutby lentiCRISPR, we tested the feasibility of con-ducting genome-scale CRISPR-Cas9 knockout(GeCKO) screening with a pooled lentiCRISPRlibrary. We designed a library of sgRNAs target-ing 5! constitutive exons (Fig. 2A) of 18,080 genesin the human genome with an average coverageof 3 to 4 sgRNAs per gene (table S1), and eachtarget site was selected tominimize off-target mod-ification (14) (see supplementary text).
To test the efficacy of the full GeCKO libraryat achieving knockout of endogenous gene tar-gets, we conducted a negative selection screen byprofiling the depletion of sgRNAs targeting essen-tial survival genes (Fig. 2A). We transduced thehuman melanoma cell line A375 and the human
Fig. 1. Lentiviral deliv-ery of Cas9 and sgRNAprovidesefficientdeple-tion of target genes. (A)Lentiviral expression vec-tor for Cas9 and sgRNA(lentiCRISPR). puro, puro-mycin selection marker;psi+, psi packaging signal;RRE, rev response element;cPPT,centralpolypurinetract;EFS, elongation factor-1ashort promoter; P2A, 2Aself-cleavingpeptide;WPRE,posttranscriptional regu-latory element. (B) Distri-bution of fluorescence from293T-EGFP cells transducedby EGFP-targeting lenti-CRISPR (sgRNAs 1 to 6,outlined peaks) and Cas9-only (green-shaded peak)vectors, and nonfluorescent 293T cells (gray shaded peak). (C) Distribution of fluorescence from 293T-EGFP cellstransduced by EGFP-targeting shRNA (shRNAs 1 to 4, outlined peaks) and control shRNA (green-shaded peak)vectors, and nonfluorescent 293T cells (gray shaded peak).
A
B C
sgRNAU6 EFS SpCas9 WPREP2A Puro
cPPT
RREpsi+
lentiCRISPR
EGFP fluorescence (a.u.)
Nor
mal
ized
cel
l cou
nt (
a.u.
)
Nor
mal
ized
cel
l cou
nt (
a.u.
)100
80
60
40
20
0
100 2 4 6
10 10 10
Cas9 onlyHEK293T
sgRNA 1sgRNA 2sgRNA 3sgRNA 4sgRNA 5sgRNA 6
Cas9:sgRNA100
80
60
40
20
0
100 2 4 6
10 10 10
control shRNAHEK293T
shRNA 1shRNA 2shRNA 3shRNA 4
EGFP fluorescence (a.u.)
shRNA
A B
D
Ste
p 1:
sgR
NA
olig
olib
rary
des
ign
Ste
p 2:
Con
stru
ctio
n of
lent
iCR
ISP
Rsc
reen
ing
libra
ry
Oligo arraysynthesis
lentiCRISPRlibrary
Cloning sgRNAoligo libray into lentiviralconstructs
Ste
p 3:
GeC
KO
Transductionwith lentiCRISPR
library
Select fortransduced cells
and applyscreening
assay
Analysisof remainingsgRNA pool
A375
12
Day 14
Day 3
Log2 normalized gene counts
Cum
ulat
ive
freq
uenc
y
0
0.2
0.4
0.6
0.8
1
2 4 6 8 10
HUES62
12
Day 14
Day 3
Log2 normalized gene counts
Cum
ulat
ive
freq
uenc
y
0
0.2
0.4
0.6
0.8
1
2 4 6 8 10
C
ERNA processing
Structural constituentof ribosome
Ribonucleoprotein complex (RNPC)
RNPC biogenesisand assembly
RNA binding
Gene rank2000 6000 10000 14000 18000
Translation
Kinetochore
RNA processing
Chromosome part
Depleted Enriched
Gene rank2000 6000 10000 14000 18000
Depleted Enriched
Structural constituentof ribosome
Fig. 2. GeCKO library design and application for genome-scale negativeselection screening. (A) Design of sgRNA library for genome-scale knockout ofcoding sequences in human cells (see supplementary text). (B and C) Cumulativefrequency of sgRNAs 3 and 14 days after transduction in A375 and human em-
bryonic stem cells, respectively. Shift in the 14-day curve represents the depletion in asubset of sgRNAs. (D and E) The five most significantly depleted gene sets in A375cells [nominal P< 10!5, false discovery rate (FDR)–corrected q< 10!5] and HUES62cells (nominal P < 10!5, FDR-corrected q < 10!3) identified by GSEA (15).
www.sciencemag.org SCIENCE VOL 343 3 JANUARY 2014 85
REPORTS
on
May
27,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
(Shalem et al., Science 2014)
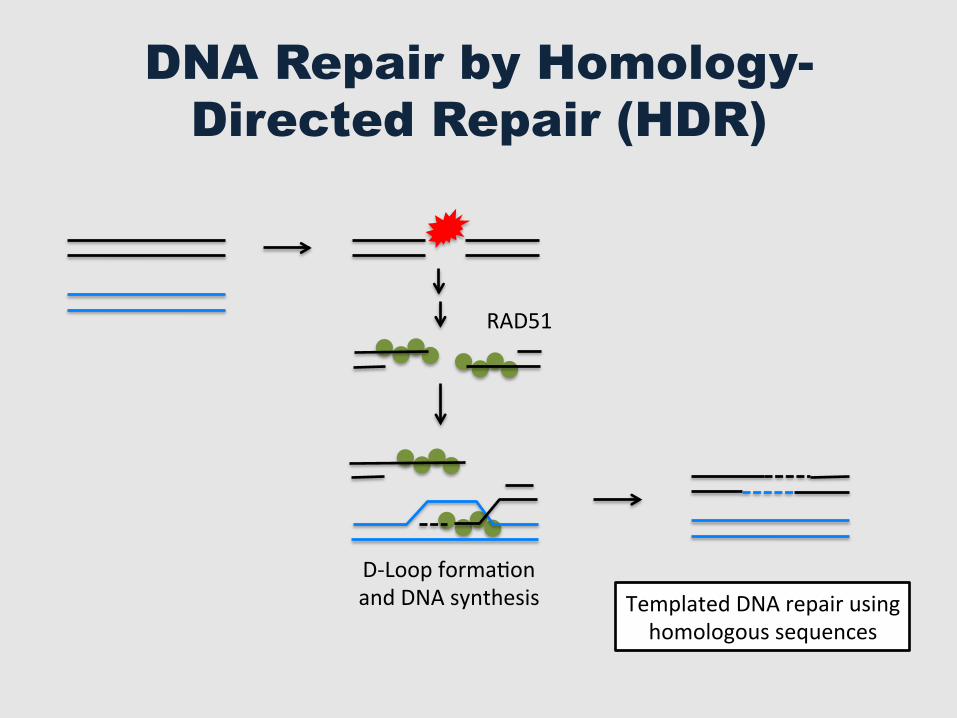
DNA Repair by Homology-Directed Repair (HDR)
RAD51
Templated DNA repair using homologous sequences
D-‐Loop formaGon and DNA synthesis

Usefulness of DNA Repair by HDR
§ Gene Therapy § Gene Replacement § Genome Engineering § SyntheGc Biology § Gene Drive – Control vectors such as mosquitoes
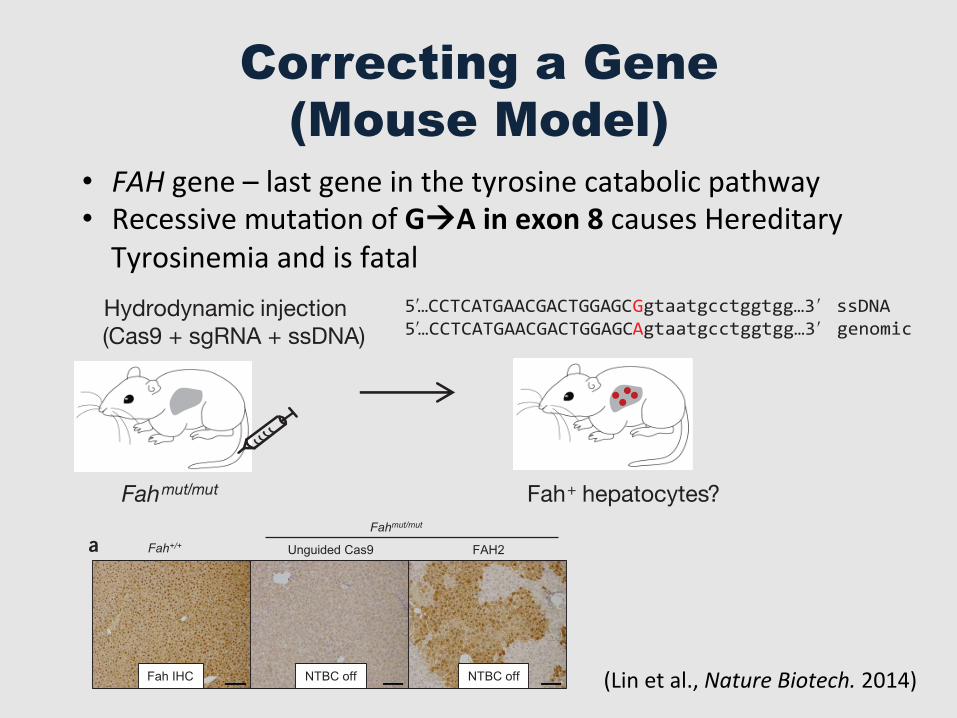
Correcting a Gene (Mouse Model)
• FAH gene – last gene in the tyrosine catabolic pathway • Recessive mutaGon of GàA in exon 8 causes Hereditary Tyrosinemia and is fatal
552 VOLUME 32 NUMBER 6 JUNE 2014 NATURE BIOTECHNOLOGY
(Fig. 2c). Quantitative RT-PCR using primers spanning exons 8 to 9 on liver samples from CRISPR-Cas9–treated mice showed they had Fah mRNA at 8–36% of levels in wild-type mice at the end of NTBC water withdrawal (Fig. 2d). These levels are consistent with the proportion of Fah+ hepatocytes detected by immunohistochemistry (Fig. 2a and Supplementary Fig. 3a) and the AAG correction rate (~9%, n = 2) detected by deep sequencing in FAH2-treated livers at 30 d off NTBC (Supplementary Fig. 3b–d and Supplementary Table 3). We note that an indel rate of ~26% was also detected by sequencing of these samples (Supplementary Fig. 3b–d and Supplementary Table 3); further work will be needed to assess the initial rate of indel formation compared to gene correction.
To examine potential off-target effects, we used a published predic-tion tool2 to identify potential off-target sites in the mouse genome for FAH1, 2 and 3 (Supplementary Figs. 4–6). In mouse 3T3 cells trans-fected with FAH 1, 2, 3 or control (unguided Cas9), the editing at the Fah locus and three or four potential off-target sites was measured using the mismatch-specific Surveyor nuclease assay1. Cleavage was detected at Fah in 3T3 cells, indicating that the one nucleotide mis-match between FAH1, 2 and 3 and the wild-type Fah gene does not prevent Cas9-mediated editing (Supplementary Figs. 4–6). Cleavage was not detected at the assayed three to four top-ranking off-target sites
Fahmut/mut mice had widespread patches of Fah+ hepatocytes (33.5% ± 3.3%, n = 3 mice) (Fig. 2a and Supplementary Fig. 3a). To measure the initial Fah gene repair frequency, we treated Fahmut/mut mice with FAH2 and kept them on NTBC water (to prevent positive selection of corrected cells) for 6 d before euthanizing them. As shown by immunohistochemical stain-ing of Fah+ cells, the initial repair frequency was 0.40 ± 0.12% (n = 3 mice) for mice treated with FAH2 compared to 0.01 ± 0.02% for those with unguided Cas9 (Fig. 2a). We also performed deep sequenc-ing to examine the initial repair rate; however, due to the error rate of sequencing, the mixture of nonparenchymal cells and polyploidy of hepatocytes, this approach could not detect low-frequency single-nucleotide polymorphisms in hepatocytes11.
We also carried out RT-PCR using primers spanning Fah exons 5–9 to determine whether the Fah splicing mutation was corrected in the liver. We found that wild-type mice had a 405-bp PCR band containing exon 8, Fahmut/mut mice had a 305-bp PCR band corresponding to the truncated Fah mRNA lacking exon 8 and Fahmut/mut mice injected with FAH1, 2 or 3 had both the 305- and 405-bp PCR bands, indicating that the exon 8 to exon 9 splicing is restored in a subset of hepatocytes (Fig. 2b). Sequencing of the 405-bp bands in CRISPR-Cas9 treated mice con-firmed that the corrected G nucleotide is included in the PCR product
Figure 1 Hydrodynamic injection of CRISPR components rescues lethal phenotype of Fah-deficient mice. (a) Experimental design. Fahmut/mut mice harbor a homozygous GAA point mutation at the last nucleotide of exon 8 (red), causing skipping of exon 8 during splicing. pX330 plasmids expressing Cas9 and a sgRNA targeting the Fah locus are delivered to the liver by hydrodynamic tail vein injection. A ssDNA oligo with the correct fragment of Fah sequence (i.e., the G allele) is co-injected to serve as a donor template to repair the ‘A’ mutation. Exon and intron sequences are in upper and lower cases, respectively. (b) Fahmut/mut mice were injected with saline only, ssDNA oligo only, ssDNA oligo plus pX330 (unguided Cas9), or ssDNA oligo plus pX330 expressing Cas9 and one of the three Fah sgRNAs (FAH1, FAH2 and FAH3). Body weight was monitored over time and normalized to pre-injection weight. Arrow indicates withdrawal of NTBC water (defined as day 0, which is 3 d after injection). (c) Mice injected with FAH1 or FAH3 in b were put back on NTBC water for 7 d and then again withdrawn from NTBC for 28 d. (d) H&E staining of liver sections from wild-type (Fah+/+) or Fahmut/mut mice injected with unguided Cas9 or Cas9 with the FAH2 sgRNA and kept off NTBC water. The FAH2 sample is from a mouse 30 d after NTBC withdrawal. Scale bars, 100 +m for upper panels, 20 +m for lower panels. (e–g) Liver damage markers (aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin) were measured in peripheral blood from Fahmut/mut mice injected with saline or ssDNA oligo only or unguided Cas9 (NTBC off) or FAH2 (NTBC off + FAH2, day 30). Fahmut/mut mice on NTBC water (NTBC on) served as a control. * P < 0.01 (n � 3 mice) using one-way ANOVA. Error bars, mean ( s.e.m.
Hydrodynamic injection (Cas9 + sgRNA + ssDNA)
NTBC withdrawn
Fahmut/mut Fah+ hepatocytes?
5…CCTCATGAACGACTGGAGCGgtaatgcctggtgg…3 ssDNA 5…CCTCATGAACGACTGGAGCAgtaatgcctggtgg…3 genomic
7 8 9 7 8 9 A G
a
Fahmut/mut
f e
b
d
g
c
Unguided Cas9 Fah+/+ FAH2
H&E
H&E
!!
!!
1.1
Injec
tion d
ay (–
3) 7
Before
NTBC
NTB
C with
drawal
(0) 2 5 10 12 14 16 17 19 20 21 22 23 24 25 26 27 28
8 11 13 14 15 16 20 21 22 23 24 25 26 27 28 29 30
Day 0Time (days)
Time (days)
1.0
1.2
0.8
Wei
ght r
atio
Wei
ght r
atio
FAH1FAH2
FAH1FAH3
FAH3
SalinessDNA oligo
1,000
800
600
400
200
800 8
6
4
2
0
600
400
200
00
AS
T (IU
/L)
NTBC on
NTBC of
f
NTBC of
f + FA
H2
NTBC on
NTBC of
f
NTBC of
f + FA
H2
NTBC on
NTBC of
f
NTBC of
f + FA
H2
ALT
(IU
/L)
Tota
l bili
rubi
n (m
g/dl
)
Unguided Cas9
0.9
0.8
BR IEF COMMUNICAT IONS
NATURE BIOTECHNOLOGY VOLUME 32 NUMBER 6 JUNE 2014 553
for each sgRNA (Supplementary Figs. 4–6). The PCR products from three off-target sites of FAH2 were sequenced, and <0.3% indels were detected (Supplementary Fig. 5c and Supplementary Table 4). Wild-type FVB mice injected with Cas9 plus sgRNA showed no body weight loss, no signs of hyperplasia and extremely low Cas9 expression after 3 months (Supplementary Figs. 7 and 8), suggesting that hydrodynamic injection of these components was well-tolerated.
In summary, these data demonstrate the potential to correct dis-ease genes in vivo in adult mouse liver using a CRISPR-Cas9 system. Transient expression of Cas9, sgRNA and a co-injected ssDNA by non-viral hydrodynamic injection is sufficient to restore the weight loss of a mouse model of HTI. The strong positive selection and expansion of Fah+ hepatocytes in the Fahmut/mut liver may have contributed to the correction of the disease phenotype8, given the observed initial genetic correction rate of ~1/250 cells. We note that the initial efficiency of repair using zinc finger nucleases was also low and was substantially improved by subsequent work to correct hemophilia in mice12. We also note that hydrodynamic injection is unlikely to be used for clinical implemen-tation. Therefore, improvements to CRISPR delivery methods and repair efficiency will be required for its broad therapeutic application (Supplementary Discussion), in particular to increase the rate of gene correction and to target other tissues. Further studies will be required to evaluate the extent of off-target effects13, particularly in vivo, and strategies to reduce off-target effects (such as Cas9 nickases14) may help.
This proof-of-principle study indicates that correction of genetic disease in vivo with CRISPR-Cas9 may be possible, and we believe that recent advances in the delivery of nucleic acid therapeutics provide hope that CRISPR-Cas9–mediated correction may be translatable to humans15.
Accession codes. BioProject: PRJNA242331.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
ACKNOWLEDGMENTSWe thank I. Zhuang and W. Cai for technical assistance, F. Zhang for sharing pX330 CRISPR vectors, and D. Crowley and K. Cormier for histology. This work was supported in part by grants 2-PO1-CA42063 to P.A.S. and T.J. and core grant P30-CA14051 from the National Cancer Institute. This work was supported in part by National Institutes of Health (NIH) Grant R01-CA133404 and the Marie-D. & Pierre Casimir-Lambert Fund to P.A.S. T.J. is a Howard Hughes Investigator, the David H. Koch Professor of Biology and a Daniel K. Ludwig Scholar. H.Y. and S.C. are supported by 5-U54-CA151884-04 NIH Centers for Cancer Nanotechnology Excellence and the Harvard-MIT Center of Cancer Nanotechnology Excellence. W.X. is supported by grant 1K99CA169512. S.C. is a Damon Runyon Fellow (DRG-2117-12). The authors acknowledge the service of the late Sean Collier to the MIT community. We thank the Swanson Biotechnology Center for technical support.
AUTHOR CONTRIBUTIONSH.Y., W.X. and D.G.A. designed the study. H.Y., W.X., S.C., R.L.B. and E.B. performed experiments and analyzed data. M.G., V.K. and P.A.S. provided reagents and conceptual advice. H.Y., W.X., T.J. and D.G.A. wrote the manuscript.
COMPETING FINANCIAL INTERESTSThe authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
1. Cong, L. et al. Science 339, 819–823 (2013). 2. Hsu, P.D. et al. Nat. Biotechnol. 31, 827–832 (2013). 3. Mali, P. et al. Science 339, 823–826 (2013). 4. Cho, S.W., Kim, S., Kim, J.M. & Kim, J.S. Nat. Biotechnol. 31, 230–232 (2013). 5. Wu, Y. et al. Cell Stem Cell 13, 659–662 (2013). 6. Schwank, G. et al. Cell Stem Cell 13, 653–658 (2013). 7. Azuma, H. et al. Nat. Biotechnol. 25, 903–910 (2007). 8. Paulk, N.K. et al. Hepatology 51, 1200–1208 (2010). 9. Aponte, J.L. et al. Proc. Natl. Acad. Sci. USA 98, 641–645 (2001). 10. Liu, F., Song, Y. & Liu, D. Gene Ther. 6, 1258–1266 (1999). 11. Nielsen, R., Korneliussen, T., Albrechtsen, A., Li, Y. & Wang, J. PLoS ONE 7, e37558
(2012). 12. Li, H. et al. Nature 475, 217–221 (2011). 13. Fu, Y. et al. Nat. Biotechnol. 31, 822–826 (2013). 14. Ran, F.A. et al. Cell 154, 1380–1389 (2013). 15. Kanasty, R., Dorkin, J.R., Vegas, A. & Anderson, D. Nat. Mater. 12, 967–977 (2013).
d
b
c
CCTCATGAACGACTGGAGCGCACGAGACATCCAGCAA Exon 8 Exon 9
5 6 7 9
5 6 7 8 9
405 bp
305 bp
Actin
WT Fahmut/mut
1,000
500
bp
Fah
a
Fah IHC
Fahmut/mut
Unguided Cas9 Fah+/+ FAH2
NTBC on day 6 NTBC on day 6
NTBC off NTBC off
Fah exons
Rel
ativ
e m
RN
A e
xpre
ssio
n le
vel
(fold
)
Figure 2 CRISPR-Cas9–mediated editing corrects Fah splicing mutation in the liver. (a) Fah immunohistochemistry (IHC) of Fahmut/mut mice injected with unguided Cas9 or Cas9 plus the FAH2 sgRNA. Upper panel: FAH2 mice were off NTBC water for 30 d as in Figure 1d. There are 33.5% ( 3.3% Fah+ cells (n = 3 mice). Lower panel: mice were kept on NTBC water and euthanized at day 6 to estimate initial repair rate. Fah+ cell counts were 0.40 ( 0.12% for FAH2 and 0.01 ( 0.02% for unguided Cas9. P < 0.01 (n = 3 mice) using an unpaired t-test. Fah+/+ mice are shown as a control. Scale bars, 100 +m. (b) RT-PCR in liver RNA from wild-type (Fah+/+), Fahmut/mut and Fahmut/mut mice injected with FAH1, 2 or 3, using primers spanning exons 5–9 to amplify wild-type Fah (405 bp) and mutant Fah (305 bp, lacking exon 8). FAH-treated mice were harvested at the endpoints of NTBC withdrawal. (c) Representative sequence of the 405-bp bands in FAH2-treated mice. The corrected G nucleotide is highlighted in gray. (d) Quantitative RT-PCR measurement of wild-type Fah mRNA expression using primers spanning exons 8 and 9. Error bars, s.d. from three technical replicates for each individual mouse.
BR IEF COMMUNICAT IONS
(Lin et al., Nature Biotech. 2014)
552 VOLUME 32 NUMBER 6 JUNE 2014 NATURE BIOTECHNOLOGY
(Fig. 2c). Quantitative RT-PCR using primers spanning exons 8 to 9 on liver samples from CRISPR-Cas9–treated mice showed they had Fah mRNA at 8–36% of levels in wild-type mice at the end of NTBC water withdrawal (Fig. 2d). These levels are consistent with the proportion of Fah+ hepatocytes detected by immunohistochemistry (Fig. 2a and Supplementary Fig. 3a) and the AAG correction rate (~9%, n = 2) detected by deep sequencing in FAH2-treated livers at 30 d off NTBC (Supplementary Fig. 3b–d and Supplementary Table 3). We note that an indel rate of ~26% was also detected by sequencing of these samples (Supplementary Fig. 3b–d and Supplementary Table 3); further work will be needed to assess the initial rate of indel formation compared to gene correction.
To examine potential off-target effects, we used a published predic-tion tool2 to identify potential off-target sites in the mouse genome for FAH1, 2 and 3 (Supplementary Figs. 4–6). In mouse 3T3 cells trans-fected with FAH 1, 2, 3 or control (unguided Cas9), the editing at the Fah locus and three or four potential off-target sites was measured using the mismatch-specific Surveyor nuclease assay1. Cleavage was detected at Fah in 3T3 cells, indicating that the one nucleotide mis-match between FAH1, 2 and 3 and the wild-type Fah gene does not prevent Cas9-mediated editing (Supplementary Figs. 4–6). Cleavage was not detected at the assayed three to four top-ranking off-target sites
Fahmut/mut mice had widespread patches of Fah+ hepatocytes (33.5% ± 3.3%, n = 3 mice) (Fig. 2a and Supplementary Fig. 3a). To measure the initial Fah gene repair frequency, we treated Fahmut/mut mice with FAH2 and kept them on NTBC water (to prevent positive selection of corrected cells) for 6 d before euthanizing them. As shown by immunohistochemical stain-ing of Fah+ cells, the initial repair frequency was 0.40 ± 0.12% (n = 3 mice) for mice treated with FAH2 compared to 0.01 ± 0.02% for those with unguided Cas9 (Fig. 2a). We also performed deep sequenc-ing to examine the initial repair rate; however, due to the error rate of sequencing, the mixture of nonparenchymal cells and polyploidy of hepatocytes, this approach could not detect low-frequency single-nucleotide polymorphisms in hepatocytes11.
We also carried out RT-PCR using primers spanning Fah exons 5–9 to determine whether the Fah splicing mutation was corrected in the liver. We found that wild-type mice had a 405-bp PCR band containing exon 8, Fahmut/mut mice had a 305-bp PCR band corresponding to the truncated Fah mRNA lacking exon 8 and Fahmut/mut mice injected with FAH1, 2 or 3 had both the 305- and 405-bp PCR bands, indicating that the exon 8 to exon 9 splicing is restored in a subset of hepatocytes (Fig. 2b). Sequencing of the 405-bp bands in CRISPR-Cas9 treated mice con-firmed that the corrected G nucleotide is included in the PCR product
Figure 1 Hydrodynamic injection of CRISPR components rescues lethal phenotype of Fah-deficient mice. (a) Experimental design. Fahmut/mut mice harbor a homozygous GAA point mutation at the last nucleotide of exon 8 (red), causing skipping of exon 8 during splicing. pX330 plasmids expressing Cas9 and a sgRNA targeting the Fah locus are delivered to the liver by hydrodynamic tail vein injection. A ssDNA oligo with the correct fragment of Fah sequence (i.e., the G allele) is co-injected to serve as a donor template to repair the ‘A’ mutation. Exon and intron sequences are in upper and lower cases, respectively. (b) Fahmut/mut mice were injected with saline only, ssDNA oligo only, ssDNA oligo plus pX330 (unguided Cas9), or ssDNA oligo plus pX330 expressing Cas9 and one of the three Fah sgRNAs (FAH1, FAH2 and FAH3). Body weight was monitored over time and normalized to pre-injection weight. Arrow indicates withdrawal of NTBC water (defined as day 0, which is 3 d after injection). (c) Mice injected with FAH1 or FAH3 in b were put back on NTBC water for 7 d and then again withdrawn from NTBC for 28 d. (d) H&E staining of liver sections from wild-type (Fah+/+) or Fahmut/mut mice injected with unguided Cas9 or Cas9 with the FAH2 sgRNA and kept off NTBC water. The FAH2 sample is from a mouse 30 d after NTBC withdrawal. Scale bars, 100 +m for upper panels, 20 +m for lower panels. (e–g) Liver damage markers (aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin) were measured in peripheral blood from Fahmut/mut mice injected with saline or ssDNA oligo only or unguided Cas9 (NTBC off) or FAH2 (NTBC off + FAH2, day 30). Fahmut/mut mice on NTBC water (NTBC on) served as a control. * P < 0.01 (n � 3 mice) using one-way ANOVA. Error bars, mean ( s.e.m.
Hydrodynamic injection (Cas9 + sgRNA + ssDNA)
NTBC withdrawn
Fahmut/mut Fah+ hepatocytes?
5…CCTCATGAACGACTGGAGCGgtaatgcctggtgg…3 ssDNA 5…CCTCATGAACGACTGGAGCAgtaatgcctggtgg…3 genomic
7 8 9 7 8 9 A G
a
Fahmut/mut
f e
b
d
g
c
Unguided Cas9 Fah+/+ FAH2
H&E
H&E
!!
!!
1.1
Injec
tion d
ay (–
3) 7
Before
NTBC
NTB
C with
drawal
(0) 2 5 10 12 14 16 17 19 20 21 22 23 24 25 26 27 28
8 11 13 14 15 16 20 21 22 23 24 25 26 27 28 29 30
Day 0Time (days)
Time (days)
1.0
1.2
0.8
Wei
ght r
atio
Wei
ght r
atio
FAH1FAH2
FAH1FAH3
FAH3
SalinessDNA oligo
1,000
800
600
400
200
800 8
6
4
2
0
600
400
200
00
AS
T (IU
/L)
NTBC on
NTBC of
f
NTBC of
f + FA
H2
NTBC on
NTBC of
f
NTBC of
f + FA
H2
NTBC on
NTBC of
f
NTBC of
f + FA
H2
ALT
(IU
/L)
Tota
l bili
rubi
n (m
g/dl
)
Unguided Cas9
0.9
0.8
BR IEF COMMUNICAT IONS

Outline
§ Genome EdiGng
§ The CRISPR-‐Cas9 System
§ The many flavors of Cas9
§ Genome EdiGng in the lab and beyond
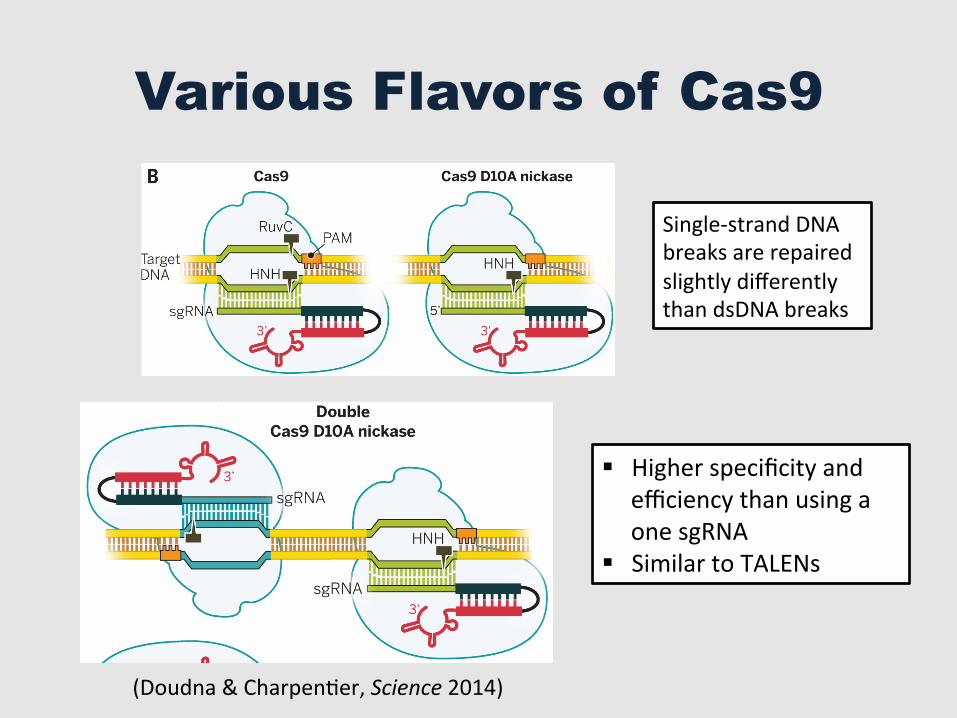
Various Flavors of Cas9
studies underscore the potential for this technol-ogy to be used for human gene therapy to treatgenetic disorders.A last example of CRISPR-Cas9 as a genome
engineering technology is its application to plantsand fungi. Since its demonstration as a genomeediting tool in Arabidopsis thaliana and Nicoti-ana benthamiana (105, 106), editing has beendemonstrated in crop plants including rice, wheat,and sorghumaswell as sweet orange and liverwort
(107–111). This technology promises to changethe pace and course of agricultural research. Forexample, a recent study in rice found that targetgeneswere edited innearly 50%of the embryogeniccells that received the Cas9–guide RNA constructs,and editing occurred before the first cell division(112). Furthermore, these genetic changes werepassed to the next generation of plants withoutnew mutation or reversion, and whole-genomesequencing did not reveal substantial off-target
editing. Such findings suggest that modificationof plant genomes to provide protection fromdisease and resistance to pestsmay bemuch easierthan has been the case with other technologies.The regulatory implications of CRISPR-Cas9 tech-nology for use in plants are not yet clear and willcertainly depend on the type of mutation(s) to beintroduced.In general, the lack of efficient, inexpensive,
fast-to-design, and easy-to-use precision genetic
SCIENCE sciencemag.org 28 NOVEMBER 2014 • VOL 346 ISSUE 6213 1258096-5
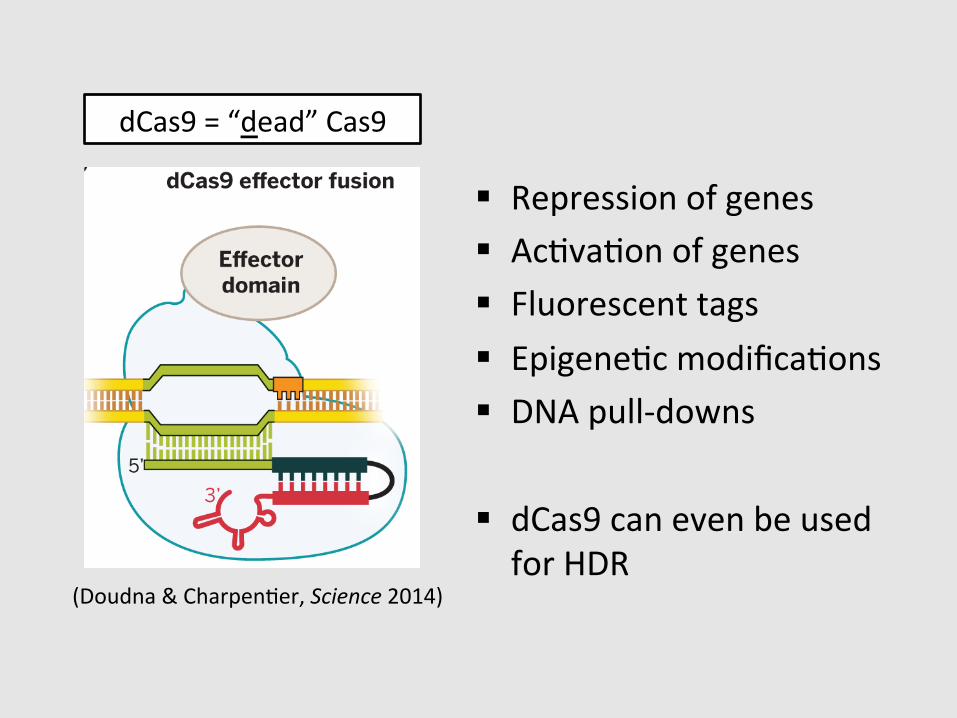
Fig. 4. CRISPR-Cas9 as agenome engineering tool. (A)Different strategies for intro-ducing blunt double-strandedDNA breaks into genomic loci,which become substrates forendogenous cellular DNA repairmachinery that catalyze non-homologous end joining (NHEJ)or homology-directed repair(HDR). (B) Cas9 can function asa nickase (nCas9) when engi-neered to contain an inactivat-ing mutation in either the HNHdomain or RuvC domain activesites. When nCas9 is used withtwo sgRNAs that recognizeoffset target sites in DNA, astaggered double-strand breakis created. (C) Cas9 functionsas an RNA-guided DNA bindingprotein when engineered tocontain inactivating mutationsin both of its active sites. Thiscatalytically inactive or deadCas9 (dCas9) can mediatetranscriptional down-regulationor activation, particularlywhen fused to activator orrepressor domains. In addition,dCas9 can be fused to fluores-cent domains, such as greenfluorescent protein (GFP), forlive-cell imaging of chromo-somal loci. Other dCas9 fusions,such as those including chro-matin or DNA modificationdomains, may enable targetedepigenetic changes togenomic DNA.
RESEARCH | REVIEW
on
May
20,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
studies underscore the potential for this technol-ogy to be used for human gene therapy to treatgenetic disorders.A last example of CRISPR-Cas9 as a genome
engineering technology is its application to plantsand fungi. Since its demonstration as a genomeediting tool in Arabidopsis thaliana and Nicoti-ana benthamiana (105, 106), editing has beendemonstrated in crop plants including rice, wheat,and sorghumaswell as sweet orange and liverwort
(107–111). This technology promises to changethe pace and course of agricultural research. Forexample, a recent study in rice found that targetgeneswere edited innearly 50%of the embryogeniccells that received the Cas9–guide RNA constructs,and editing occurred before the first cell division(112). Furthermore, these genetic changes werepassed to the next generation of plants withoutnew mutation or reversion, and whole-genomesequencing did not reveal substantial off-target
editing. Such findings suggest that modificationof plant genomes to provide protection fromdisease and resistance to pestsmay bemuch easierthan has been the case with other technologies.The regulatory implications of CRISPR-Cas9 tech-nology for use in plants are not yet clear and willcertainly depend on the type of mutation(s) to beintroduced.In general, the lack of efficient, inexpensive,
fast-to-design, and easy-to-use precision genetic
SCIENCE sciencemag.org 28 NOVEMBER 2014 • VOL 346 ISSUE 6213 1258096-5
Fig. 4. CRISPR-Cas9 as agenome engineering tool. (A)Different strategies for intro-ducing blunt double-strandedDNA breaks into genomic loci,which become substrates forendogenous cellular DNA repairmachinery that catalyze non-homologous end joining (NHEJ)or homology-directed repair(HDR). (B) Cas9 can function asa nickase (nCas9) when engi-neered to contain an inactivat-ing mutation in either the HNHdomain or RuvC domain activesites. When nCas9 is used withtwo sgRNAs that recognizeoffset target sites in DNA, astaggered double-strand breakis created. (C) Cas9 functionsas an RNA-guided DNA bindingprotein when engineered tocontain inactivating mutationsin both of its active sites. Thiscatalytically inactive or deadCas9 (dCas9) can mediatetranscriptional down-regulationor activation, particularlywhen fused to activator orrepressor domains. In addition,dCas9 can be fused to fluores-cent domains, such as greenfluorescent protein (GFP), forlive-cell imaging of chromo-somal loci. Other dCas9 fusions,such as those including chro-matin or DNA modificationdomains, may enable targetedepigenetic changes togenomic DNA.
RESEARCH | REVIEW
on
May
20,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
§ Higher specificity and efficiency than using a one sgRNA
§ Similar to TALENs
Single-‐strand DNA breaks are repaired slightly differently than dsDNA breaks
(Doudna & CharpenGer, Science 2014)
studies underscore the potential for this technol-ogy to be used for human gene therapy to treatgenetic disorders.A last example of CRISPR-Cas9 as a genome
engineering technology is its application to plantsand fungi. Since its demonstration as a genomeediting tool in Arabidopsis thaliana and Nicoti-ana benthamiana (105, 106), editing has beendemonstrated in crop plants including rice, wheat,and sorghumaswell as sweet orange and liverwort
(107–111). This technology promises to changethe pace and course of agricultural research. Forexample, a recent study in rice found that targetgeneswere edited innearly 50%of the embryogeniccells that received the Cas9–guide RNA constructs,and editing occurred before the first cell division(112). Furthermore, these genetic changes werepassed to the next generation of plants withoutnew mutation or reversion, and whole-genomesequencing did not reveal substantial off-target
editing. Such findings suggest that modificationof plant genomes to provide protection fromdisease and resistance to pestsmay bemuch easierthan has been the case with other technologies.The regulatory implications of CRISPR-Cas9 tech-nology for use in plants are not yet clear and willcertainly depend on the type of mutation(s) to beintroduced.In general, the lack of efficient, inexpensive,
fast-to-design, and easy-to-use precision genetic
SCIENCE sciencemag.org 28 NOVEMBER 2014 • VOL 346 ISSUE 6213 1258096-5
Fig. 4. CRISPR-Cas9 as agenome engineering tool. (A)Different strategies for intro-ducing blunt double-strandedDNA breaks into genomic loci,which become substrates forendogenous cellular DNA repairmachinery that catalyze non-homologous end joining (NHEJ)or homology-directed repair(HDR). (B) Cas9 can function asa nickase (nCas9) when engi-neered to contain an inactivat-ing mutation in either the HNHdomain or RuvC domain activesites. When nCas9 is used withtwo sgRNAs that recognizeoffset target sites in DNA, astaggered double-strand breakis created. (C) Cas9 functionsas an RNA-guided DNA bindingprotein when engineered tocontain inactivating mutationsin both of its active sites. Thiscatalytically inactive or deadCas9 (dCas9) can mediatetranscriptional down-regulationor activation, particularlywhen fused to activator orrepressor domains. In addition,dCas9 can be fused to fluores-cent domains, such as greenfluorescent protein (GFP), forlive-cell imaging of chromo-somal loci. Other dCas9 fusions,such as those including chro-matin or DNA modificationdomains, may enable targetedepigenetic changes togenomic DNA.
RESEARCH | REVIEW
on
May
20,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
§ Repression of genes § AcGvaGon of genes § Fluorescent tags § EpigeneGc modificaGons § DNA pull-‐downs
§ dCas9 can even be used for HDR
dCas9 = “dead” Cas9
(Doudna & CharpenGer, Science 2014)

Outline
§ Genome EdiGng
§ The CRISPR-‐Cas9 System
§ The many flavors of Cas9
§ Genome EdiGng in the lab and beyond
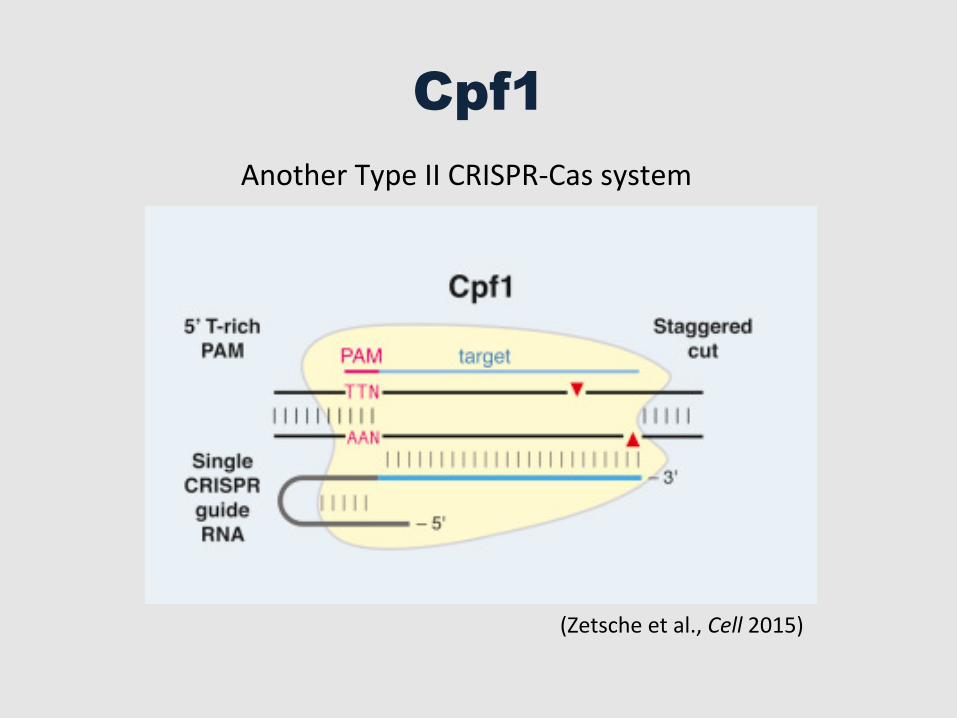
Cpf1
(Zetsche et al., Cell 2015)
Another Type II CRISPR-‐Cas system
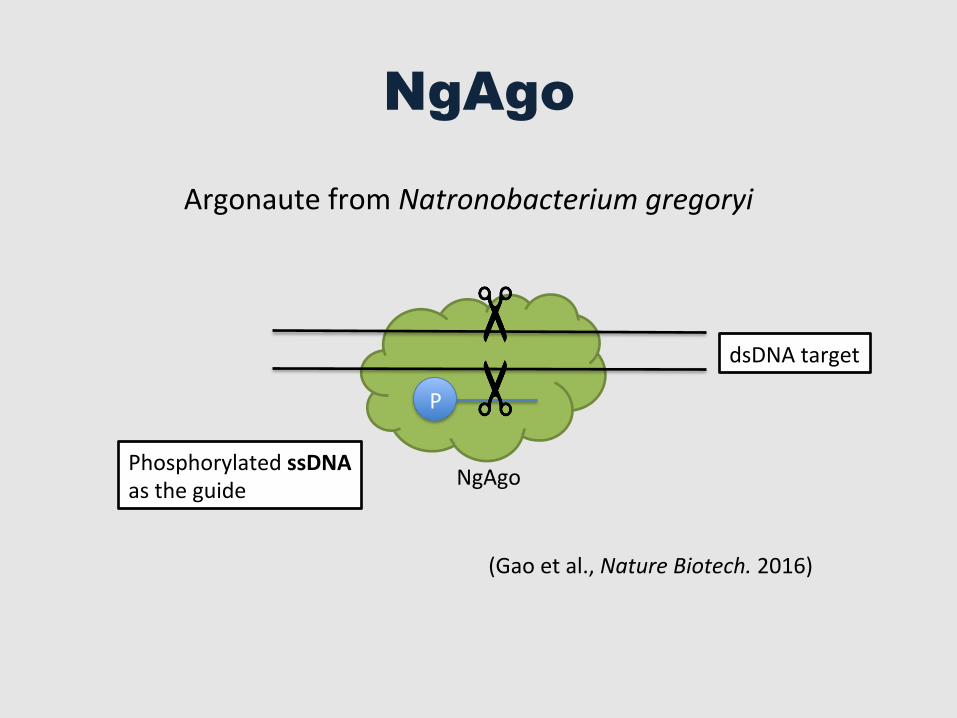
NgAgo
P
Phosphorylated ssDNA as the guide
(Gao et al., Nature Biotech. 2016)
Argonaute from Natronobacterium gregoryi
NgAgo
dsDNA target
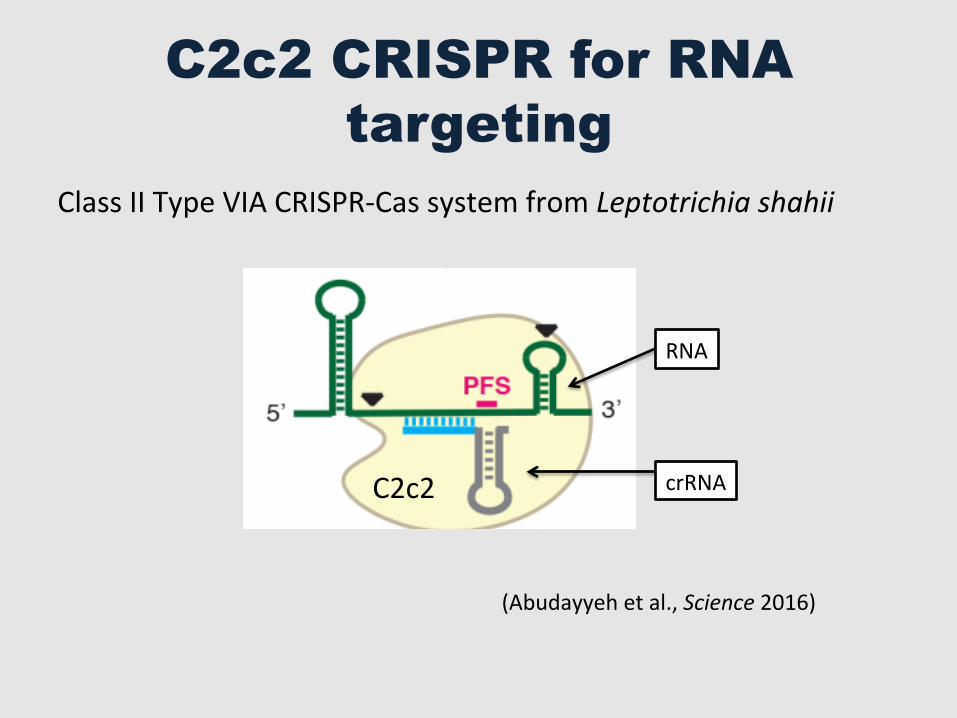
C2c2 CRISPR for RNA targeting
(Abudayyeh et al., Science 2016)
Fig. 7. C2c2 as a putative RNA-targeting prokaryotic immune system. The C2c2-crRNA complex recognizes target RNA via base pairing with the cognate protospacer and cleaves the target RNA. In addition, binding of the target RNA by C2c2-crRNA activates a non-specific RNase activity which may lead to promiscuous cleavage of RNAs without complementarity to the crRNA guide sequence. Through this non-specific RNase activity, C2c2 may also cause abortive infection via programmed cell death or dormancy induction.
First release: 2 June 2016 www.sciencemag.org (Page numbers not final at time of first release) 16
on
June
17,
201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from
C2c2 crRNA
RNA
Class II Type VIA CRISPR-‐Cas system from Leptotrichia shahii

Take Home
§ Genome ediGng is a powerful tool that is under constant development – CRISPR-‐Cas systems in parGcular
§ The advantages of CRISPR-‐Cas9 are its ease of use and its efficacy in almost any organism that has been tested so far
§ Roadblock to genome engineering has been removed with CRISPR-‐Cas9 technology
§ The potenGal impacts that genome engineering can have in our lives is just emerging – stay tuned!
Top Related