







Languages
Pages
Legal


Common InheritedCoagulation Disorders
Bob Miller, PA
2017
Inherited Coagulation Disorders
• Brief review of coagulation
• Disorders related to platelet dysfunction
(not the acquired thrombocytopenias)
• Disorders related to coagulation factor deficiencies
• Focus: Presenting features and initial lab tests
• Basics of treatment approaches
All coagulation factors and platelets in circulation(unactivated)
vWF+VIII
P
PP
P
P
PP
P PP
PvWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
VWF has two jobs ...Binds and protects FVIII & tethers to site of injury

Site of injury with exposure to subendothelium with platelet activation
vWF+VIII
P
PP
P
P
P
P PPvWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
VIII
vWF+VIII
P PP
Contact with “tissue factor” other subendothelial tissues
VWF “tethers” to exposed endothelium at site of injury
vWF+VIII
P
PP
P
P
P
P PP
VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
VIII
vWF+VIII
vWF vWF
ShearvWF
pP
Platelets are activated and “adhere” to VWF and then “aggregate” to form “platelet plug”
vWF+VIII
P
PP
P
P
P
P PP
VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
VIII
vWF+VIII
vWF vFW vWFP PP
PP
P

Activated coagulation proteins form fibrin strands
vWF+VIII
P
PP
P
P
P
P PP
X
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF+VIII
vWF vWF vWFP
PP
PP
PIX XIIIVII V
IIIVIII
Fibrin Strands
Fibrin Clot

Reproduced with permission from: Rao AK. Am J Med Sci.1998 316 69 76
A representation of normal platelet responses and the congenital disorders of
Bernard Soulier Syndrome
vWD Glanzmann’s Thrombasthenia
Deficiency of Platelet Coagulant Activities
Fibrinogen
GP IIb/III
a
Afibrinogenemia

Copyright © 2011 American Society of Hematology. Copyright restrictions may apply.
Peter Maslak, ASH Image Bank 2011; 2011-3689
Petechiae
Patterns of Bleeding
Platelet type …
• mucous membrane
• epistaxis / gums
• petechiae
• menorrhagia
Coagulation type ...
• bruising
• soft tissue
• muscles
• joints
If there is platelet pattern bleeding ...
• CBC gives you the platelet count
• And also the H&H may reflect the amount of bleeding possibly leading to anemia
• If the count is normal, look at platelet function

Platelet Function Evaluation• IVY bleeding time
• Plt Aggregation to : ADP
Epinephrine
Collagen
Ristocetin
Arachidonic acid
• PFA - Platelet function analyzer
• Consider VWD as cause of platelet pattern bleeding
Inherited Disorders Affecting Platelets• Low von Willebrand factor protein (very common)
von Willebrand Disease (VWD)
• Functional defects of the platelet (rare)
Glannsman’s thrombasthenia
Bernard-Soulier syndrome
• Vessel wall abnormalities (rare)
Connective tissue disorders
Reproduced with permission from: Rao AK. Am J Med Sci. 1998;316:69–76.
A representation of normal platelet responses and the congenital disorders of platelet function
Bernard Soulier Syndrome
vWD Glanzmann’s Thrombasthenia
Deficiency of Platelet Coagulant Activities
Fibrinogen
GP IIb/III
a
Afibrinogenemia

VWD….patterns of bleeding
VWD• Mucous membranes
• Epistaxis
• Gum bleeding
• Menorrhagia
• Superficial (petechiae)
Hemophilia• Deep bruising
• Hematomas
• Joints
• Muscles
von Willebrand Disease
• Estimated 1% of population (autosomal)
• Type 1 mild / most common
• Type 2 mild to moderate
• Type 3 severe
von Willebrand Disease
• Platelet count is normal (except in rare VWD 2B)
• The platelets are normal but have reduced adhesion to site of injury if inadequate VWF (the “glue”)
• The FVIII is normal but reduced amount because lack of VWF to carry and protect FVIII

VWD….diagnosis
Test for these assays (levels)
FVIII (FVIII function)
VWF:Ag (presence of Ag)
Ristocetin cof (VWF function)
These are the basic initial lab tests to order to look for VWD
( some would add “VWF multimers” )
von Willebrand Disease
Type 1
• Reduced quantity of VWF (quantitative)
• VWF normal, amount is reduced
• Mild and most common type
• ~ 80% of all VWD
VWD….diagnosis
FVIII
VWF:Ag
RCof
Type 1All three tests partially decreased
to similar levels
Typically 20 to 50% (50-150)

Basic Bleeding Work-upCBC w/ platelet ct WNL
PT 11.2 (10-12)
PTT 46 (31-43)
Extra coag tube
FVIII 38 (50-150)
VWF Antigen 42 (50-150)
Ristocetin (RCof.) 35 (50-150)
Basic Bleeding Work-upCBC w/ platelet ct WNL
PT 10.9 (10-12)
PTT 46 (31-43)
Extra coag tube
FVIII 38 (50-150)
VWF Antigen 12 (50-150)
Ristocetin (RCof.) 5 (50-150)(discordant)
von Willebrand Disease
Type 2 “Qualitative” defects
Type 2A
Lacks HMW multimers
Type 2B
“Gain of function”
Increased platelet binding = low platelet ct

von Willebrand Disease
Type 2 “Qualitative” defects
Type 2M
Decreased binding to GP1b
Type 2N (Normandy)
Normal amount of VWF (Ag & Rcof normal)
Decreased binding to FVIII = low FVIII
(? Misdiagnosed as hemophilia A)
VWF Multimers
von Willebrand Disease
Type 3
< 5% of VWD
VWF very low or absent (quantitative)
Severe clinical features

Basic Bleeding Work-upCBC w/ platelet ct NL
PT 11.0 (10-12)
PTT, 1:1 mix 79 (31-43)
Extra coag tube
FVIII 6 (50-150)
VWF Antigen 3 (50-150)
Ristocetin RCof. 0 (50-150)
All 3 low
VWD ... lab tests to fine tune DX
• VIII
• VWF:Ag
• RCof
• Bleeding time
• VWF multimers
• Blood group
• RIPA
• Platelet count
• Genetic
von Willebrand Disease
Diagnosis
• Repeated testing may be needed if borderline values
• Bleeding history important
• Family history / inheritance
• Autosomal dominant / recessive

Basic Bleeding Work-upCBC w/ platelet ct NL
PT 11.1 (10-12)
PTT, 1:1 mix 43 (31-43)
Extra coag tube
FVIII c 60 (50-150)
VWF Antigen 41 (50-150)
Ristocetin (R cof.) 52 (50-150)
von Willebrand Disease Treatment
• DDAVP (desmopressin) intranasal (Stimate) or IV
Causes release of storage pool FVIII and VWF
Most Type 1 respond, some Type 2, no Type 3
Contraindicated in Type 2B
• Factor VIII concentrates containing VWF needed for cases unresponsive to DDAVP. (new rVWF available)
Inherited Defects of Coagulation Factors

Coagulation Testing (oversimplified) PTT XII
XI
IX PTVIII VII
X
V
Prothrombin
Thrombin
Fibrin
Fibrinogen clot XIII
(not tested by PT/PTT)
Coagulation Testing (oversimplified) PTT XII
XI
IX PTVIII VII
X
V
Prothrombin
Thrombin
Fibrin
Fibrinogen clot XIII
(not tested by PT/PTT)
Normal PT with abnormal PTTisolates the problem to these four factors
Thromboelastograph (TEG)
PT & PTT measures clot initiation
90% of clot dynamics occur after clot initiation

Queen Victoria
Hemophilias
“Classic” hemophilia A
Factor VIII deficiency
Hemophilia B
Factor IX deficiency
“Christmas disease”
Hemophilia Severity
Factor VIII or IX level
Normal = 50 –150%
•Severe < 1 %
•Moderate 1 - 5 %
•Mild 6 - 50 %

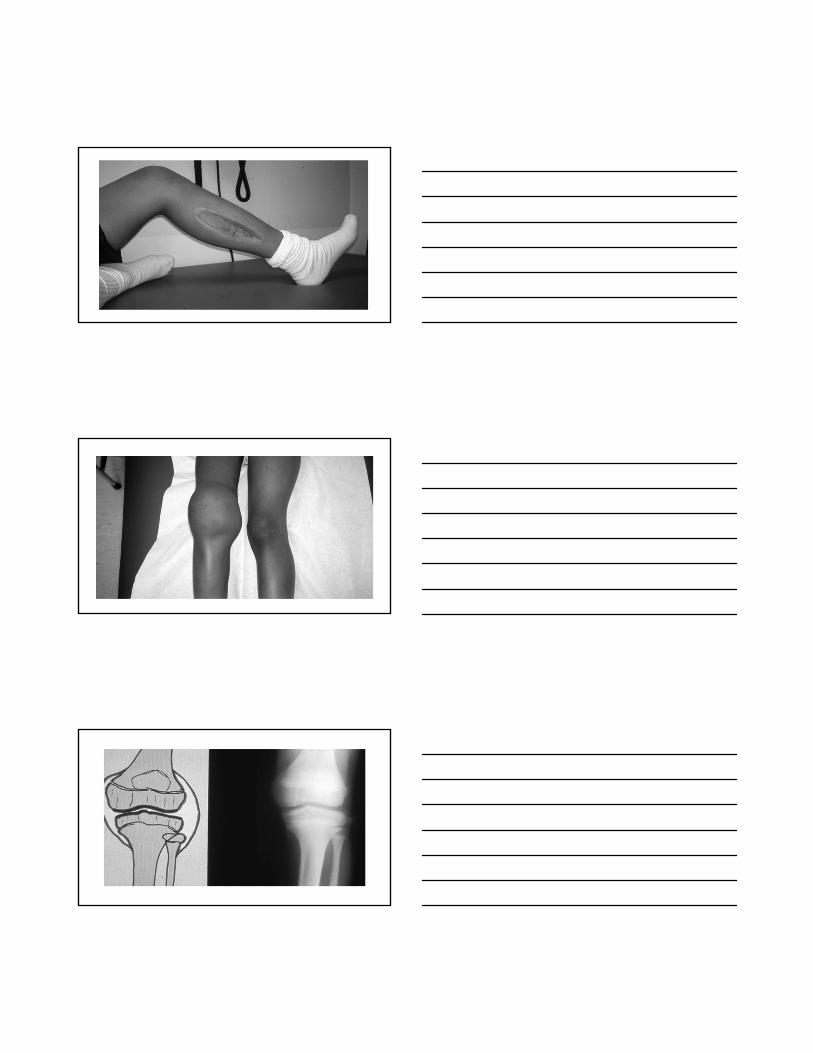
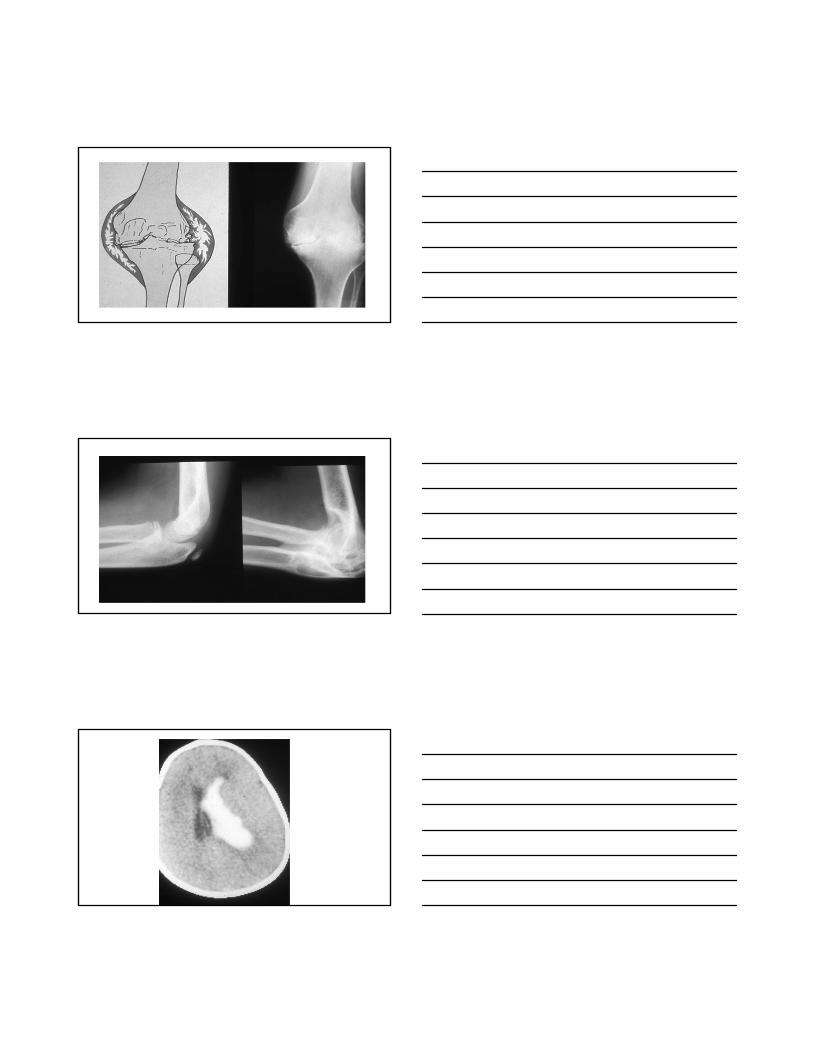

Hemophilia Treatment
• Prevent bleeding !
• Consider prophylactic FVIII or IX (2-3 x wk)
• Treat bleeding early - replace the missing factor VIII or IX with concentrates given IV
• Monitor for complications such as orthopedic, viral and inhibitors
Hemophilia Inhibitors
• Antibodies “inhibitors” develop in ~ 20% of persons with severe hemophilia A
• Antibodies neutralize the infused coagulation factor
• May require an “activated” concentrate to control bleeding

Hemophilia Treatment
• Donor derived factor concentrates in the 70’s and 80’s led to viral complications
• HBV, HCV, HIV
MMWR
July 16, 1982Epidemiologic Notes and
Reports Pneumocystis carinii Pneumonia among
Persons with Hemophilia A
Hemophilia Treatment
• Safer plasma derived concentrates are now used
• Newer products using recombinant technology
• New products with a longer half-life
• DDAVP used in mild hemophilia A
A Recombinant Technology
MammalianCELLS
produce aprotein
Protein ispurified
Lyophilized
product
• Mammalian cells are provided with genetic information to produce a target protein.
• Cell lines may include CHO, BHK, HEK

CRISPR – Gene EditingClustered Regularly Interspaced Short Palindromic Repeats
(CRISPR)
• Genome can be cut to remove or add genes
• Palindrome is set of characters reading the same forward & backwards (madam / racecar) helps to localize and identify gene sequences
• ? Hemophilia, Thalassemia, SSA, CF, Others
CRISPR (Cut ... or Cut & Paste)
[ ]DNA target sequence
[ ] RNA guide
[ ]Cas9 Enzyme cuts both DNA strands
[x]Targeted gene is removed (silenced) orreplaced (improved)
Federal Regional Hemostasis & Thrombosis Centers (HTCs)
(Hemophilia Treatment Centers)

Captain Morgan, The Rescue Dog
Coagulation Testing (oversimplified) PTT XII
XI
IX PTVIII VII
X
V
Prothrombin
Thrombin
Fibrin
Fibrinogen clot XIII
(not tested by PT/PTT)
Normal PTT with abnormal PTisolates the problem to factor VII
Rare Coagulation Factor Deficiencies
Factor VII deficiency
• Autosomal
• Rare 1:500,000
• Bleeding variable
• Bleeding does not correlate with level
• Treat with rFVIIa
Factor XI deficiency
• Autosomal
• Rare > 1:100,000
• Ashkenazi Jews (8%)
• Bleeding variable
• Treat with FFP or rVIIa
(No FXI available)

Rare Coagulation Factor Deficiencies
Factor XII deficiency
• Autosomal
• Rare
• Prolongs the PTT
but does not result in
clinical bleeding
Surgery is OK
Factor XIII deficiency
• Autosomal
• Rare
• PT / PTT normal
• Excess bleeding from umbilical stump
Coagulation Testing (oversimplified) PTT XII
XI
IX PTVIII VII
X
V
Prothrombin
Thrombin
Fibrin
Fibrinogen clot XIII
(not tested by PT/PTT)
Top Related