







Languages
Pages
Legal


1
1
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
A.A. 2013-14
Chimica Fisica dei Materiali e laboratorio
Bartolomeo Civalleri Dip. Chimica IFM – Via P. Giuria 5 – 10125 Torino
Vibrazioni nei solidi
z
x
y
Libration (B1) Rz: 61 cm-1
2
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Atomic motion
• The crystal lattice is never rigid.
• Atoms actually move around their equilibrium positions inside the crystalline structure.
• Motions of atoms in solids provide the key to understand many physical phenomena mainly related to thermal effects, phase transitions, transport properties, and so forth.
• Theoretical calculation of atom vibrations then gives access to a number of properties (see next slides)

2
3
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Molecular dynamics Fourier transformation of the atomic
velocity autocorrelation function
Atomic trajectories
Disordered systems, high atomic
mobility
Better at high temperature
Include anharmonic effects
Accuracy depends on simulation
time (supercells)
Lattice dynamics Taylor expansion of the potential
energy surface harmonic approx.
Dynamical matrix
Crystalline systems
Better at low temperature
Anharmonic corrections (quasi-
harmonic approximation)
Thermodynamics through statistical
mechanics (supercells)
The two approaches provide complementary information
Vibrations in solids: computational tools
4
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
LD and Potential Energy Surface
Taylor expansion of the potential energy around the equilibrium configuration:
0
1 1 1...
2 3! 4!ij i j ijk i j k ijkl i j k l
ij ijk ijkl
E E H u u H u u u H u u u u
For an equilibrium structure first-derivatives are zero (stationary point)
Index i labels the triplet (G,t,a) with G as a translation vector of the primitive
lattice, t as an atom within the primitive unit cell and a as the Cartesian
coordinate of the atomic displacement u.
Usually, truncated at the second order terms harmonic approximation
Hij, Hijk and Hijkl are derivatives of the energy with respect to atomic
displacements. They are the harmonic, cubic and quartic force constants,
respectively

3
5
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Dynamical Matrix
In lattice dynamics the central role is played by the Dynamical Matrix
Where:
Mi is the mass of the atom associated to the i-th coordinate;
ui is the cartesian atomic displacements of the i-th coordinate;
R(G) = xi(0) - xj(G)
21
( ) exp ( )ij
i ji j
ED i
M M
0 GG
0
k k R Gu u
As for electronic energy levels, translation symmetry leads to a band structure
for vibrational energy levels (phonons). E.g. Silicon band structure and vDOSs
6
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Phonons: matter-radiation interaction
• Vibrational modes can be considered as being particle-like
(phonons)
• Phonons can interact with radiation and matter
• Phonon - Photon interaction
• Optic modes at k0
• Absorption: Infrared
• Scattering: Raman
• Acoustic modes at k0
• Scattering: Brillouin (elastic constants)
• Phonon-Neutron interaction
• Inelastic Neutron Scattering (INS)
Optic branch
Acoustic branch

4
7
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Dynamical Matrix and Phonon Dispersion
21
( ) exp ( )ij
SC i ji j
ED i
M M
0 G
G0
k k R Gu u
The dynamical matrix can be computed by using:
Linear response methods
Density Functional Perturbation Theory (S. Baroni, et al. Rev. Mod. Phys. (2001))
Finite displacements
Numerical derivatives (supercells must be used) (CRYSTAL)
A supercell calculation at G permits to map some k-points in the reciprocal space.
Number and kind of k-points depends on shape and size of the supercell
Covalent solids: reasonable approximation, fast decay of the 2nd derivatives, interpolation
schemes
Ionic and semi-ionic (polar) solids: slow decay, long-range contribution important, approximate
electrostatic models
Results can be compared with Inelastic Neutron Scattering (INS) experiments
8
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Frequencies Calculation in CRYSTAL - I
21( 0)ij
i ji j
ED
u uM M
0 0
0
k
CRYSTAL computes vibrational frequencies at G point (k=0)
The second-derivatives matrix is computed by numerical differentiation
of the analitical first-derivatives (gradients)
Special properties of the G point (k=0):
• D(0) is simple to calculate
• Three modes have zero frequency (acoustic branch – “translations”)
• D(0) possesses the point symmetry of the crystal (factorization)
• G point modes give rise to infrared and Raman spectra
LO/TO splitting, relevant to polar crystals, can be also computed by
using e and Z*.
2, ,,
0,..., ,...,0 0,..., ,...,0
2
i j i ji
i j j j
g u g ugE
u u u u
0 0
0 00
0 0 0 0
0 0

5
9
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Frequencies Calculation in CRYSTAL - II
LO-TO splitting is computed by including a non-analytical term which
depends on the electronic dielectric tensor e and on the Born effective
charge tensor associated to each atom.
4( 0)
i jna
ijDV
k Z k Zk
k ε kWhere:
V is the volume of the unit cell;
Z* is the Born effective-charge tensor (analogous to the molecular GAPT
charges);
e is the electronic dielectric-constant tensor (CPHF/KS, see Bernasconi’s lecture)
All those quantities can be computed by CRYSTAL
In polar crystals, long range Coulomb effects give rise to macroscopic electric
fields for longitudinal optic modes (LO) at k0 (LO-TO splitting):
( 0) ( 0) ( 0)an na
ij ij ijD D Dk k k
10
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
The atomic Born tensors are key quantities for :
calculation of the IR intensities
calculation of the static (low-frequency) dielectric tensor, e0
calculation of the Longitudinal Optical (LO) modes
They are defined, in the cartesian basis, as (for atom a):
a
a a
*
ij i
j i j
EZ
u u
*i=component of an applied external field
**μ=cell dipole moment (polarization per unit cell)
The Born tensor

6
11
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
The IR intensity of the p-th mode:
2
p p
p
A dQ
a
a a
*
ij i
j i j
EZ
u uThe Born charge tensor:
,
2
p jp pA d Z
*dp=degeneracy of the p-th mode
The IR intensity - II
12
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
ε0 → static (low-frequency) dielectric constant
ε → electronic (high-frequency) dielectric constant
ωp → p-th frequency eigenvalue
e e
, ,0 4 p i p j
ij ij
p p
Z Z
Ω → unit cell volume
Ionic contribution
Only one component for each Zp is
non null
e e
2
,0 4 p i
ii ii
p p
Z
The static dielectric constant

7
, ,
2
4 p i p j
ij ij
p p p
Z Z
i
e e
2
1
1R
e
e
Spessartine …… Rcalc
___ Rexp
Reflectance Spectrum
Spessartine (garnet)
Mn3Al2 (SiO4)3
14
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Applications of Lattice Dynamics - I
Interpretation of vibrational spectra analysis of the normal modes assignment visualization/animations
symmetry analysis IR/Raman active/inactive
isotopic substitution
Thermodynamics calculation of thermodynamic functions phonon density of state
pressure- and temperature-dependent properties free energy
harmonic approximation, quasi-harmonic approximation simple models
Equations of state (p-V-T) phase diagrams phase stability
phase transitions pt
solid-state reactions
kinetics of transformation simple models

8
15
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Applications of Lattice Dynamics - II
Calculation of the Atomic Displacement Parameters (ADPs) computed from the eigeinvectors of the dynamical matrix
anisotropic thermal ellipsoids diffraction data
thermal motion corrections e.g. bond distances
Debye-Waller thermal factors dynamic X-ray structure factors
related to the intensity of Inelastic Neutron Scattering measurements
Characterization of the PES structure stability no imaginary frequencies
characterization of minima, transition states, higher-order saddle points
Isotopic equilibria isotope enrichment in minerals
Ab-initio derived semiempirical interatomic potentials basic information: E, X, g, i, Cij, B, ... construction (benchmark) of new (existing) interatomic potentials
transfer from the electronic to the atomic scale better transferability (?)
16
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Thermodynamics
, lnstG T E pV RT Z exp( 2 )
exp( ) 1i B
i
i i i B
h k TZ Z
h k T
, ln lnV
S T R Z RT Z T
Vibrational partition function
Phonon density of state
2
22
ln,
1V
V
R ZC T
T T
i i pi ipBZ
g d d k k k k 1g d
max
0
,V VC T C T g d
max
0
,f T f T g d
e.g.
;

9
17
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Thermodynamic functions: Pyrope
Specific Heat CV Entropy
M. Catti, F. Pascale and R. Dovesi, unpublished
18
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
From lattice dynamics to MSDs and ADPs
• e(q|j,k) corresponds to the atomic displacement (eigenvector component) of the atom q in the mode j along the wavevector k.
2
1( ) | |
Tj
atom
jq j
EB q q j q j
M
k
ke k e k
k
B
1 1
2 exp /k 1j j
j
ET
k kk
Atomic Anisotropic Displacement Parameters (ADPs, U(q)) can be
readily obtained from Batom(q) tensors
They can be compared with ADPs from X-ray or neutron diffraction
Visualized in terms of thermal ellipsoids
• Ej(k) is the energy of the vibrational mode
The atomic Mean Square Displacement (MSD) tensors (symmetric 3x3
tensor) can be computed as

10
19
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
From internal to external modes: supercell approach
B3LYP/6-31G(d,p)
ADPs strongly depend on the supercell size
A supercell of 2x2x2 size gives a reasonable agreement with experiment
ADPs of N atom show a great variability, with a large contribution from low frequency modes
123 K 0.13
0.39 0.98 3.36 0.96
Equal-probability ellipsoids (50%) Wavenumbers (cm-1)
Isotr
opic
MSD
(Å
2)
20
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
0.83
0.43
0.56
0.53
0.53 0.68
ADPs of Benzene, Urotropine and L-Alanine
B3LYP/6-31G(d,p) [2x2x2]
Equal-probability ellipsoids (50%) with similarity index
0.38
1.38
15 K 15 K
Equal-probability ellipsoids (75%)
23 K
0.24
0.06
0.17
0.46 0.37
0.31 0.22

11
21
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
0
2
4
6
8
10
12
14
16
18
MA
D (
cm-1
)
Accuracy: DFT methods vs experiment
Less than 20 cm-1 • Four different
systems: pyrope,
forsterite, quartz and
alumina
• Dataset of 134
vibrational frequencies
(IR and Raman data)
• 11 DFT methods:
LDA, GGA (standard
and for solids), hybrids
• Hybrid methods, in
particular, B3LYP and
WC1LYP give the
lowest MAD
Demichelis, Civalleri, Ferrabone, Dovesi, IJQC (2010)
22
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Interpretation of vibrational spectra
How to do that?
Scaling factors: Comparison between computed and experimental frequencies
Symmetry analysis IR/Raman active/inactive
Direction of transition moment vectors (TMV) (IR active modes)
Analysis of the normal modes assignment visualization/animations
Isotopic substitution
Known problems:
Anharmonicity (in particular: H-X vibrations and low-frequency modes)
Combination of modes: overtones and Fermi mixing
Approximations in the structural model
Deficiencies of the adopted level of theory

12
23
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
F. J. Torres, B. Civalleri, C. Pisani, P. Musto, A. R. Albunia, G. Guerra, J. Phys. Chem. B 111 (2007) 6327
Polystyrene: trans-Planar and s(2/1)2 Helix
F. J. Torres, B. Civalleri, A. Meyer, P. Musto, A. R. Albunia, P. Rizzo, G. Guerra, J. Phys. Chem. B 113 (2007) 5059
A. R. Albunia, P. Rizzo, G. Guerra, J. Torres, B. Civalleri, C. M. Zicovich-Wilson, Macromolecules 40 (2007) 3895
Trans-planar s(2/1)2 Helix
Pm2a (C2v)
P2122 (D2h)
24
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
3000 1500 1000 500
am
a
a
Ab
so
rba
nce
Wavenumber (cm-1)
am
calc
12201260130013401380
840880920
14521379
906
840
1440
IR spectrum of trans-planar sPS: spectrum
B3LYP/6-31G(d,p) scaled frequencies (scale factor: 0.9614)
Calc.
Exp.
IR intensities as % fraction of the max. computed intensity of 89 km/mol ( = 681 cm−1).
Lorentzian profile was used with a FWMH of 10 cm-1

13
25
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Trans-planar polystyrene: normal modes animation
B3LYP/6-31G(d,p) scaled frequencies (scale factor: 0.9614)
Animations of the normal modes:
http://www.crystal.unito.it/vibs/alpha-ps/
Main spectral regions:
3200 - 2800 cm-1
n(C-H) aromatic and alkyl
groups
1600 - 1350 cm-1
phenyl and alkyl groups:
n(CC) (1575 cm-1) and (CH)
1350 - 1000 cm-1
phenyl (CH) and alkyl
(CH) (1329 cm-1)
1000 - 500 cm-1
deformation aromatic rings
and CH groups (976 cm-1)
below 500 cm-1
collective vibrations
(torsions) (70 cm-1)
26
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Trans-planar sPS: exp. vs calc.
Full assignment of 50 IR and Raman frequencies
14
27
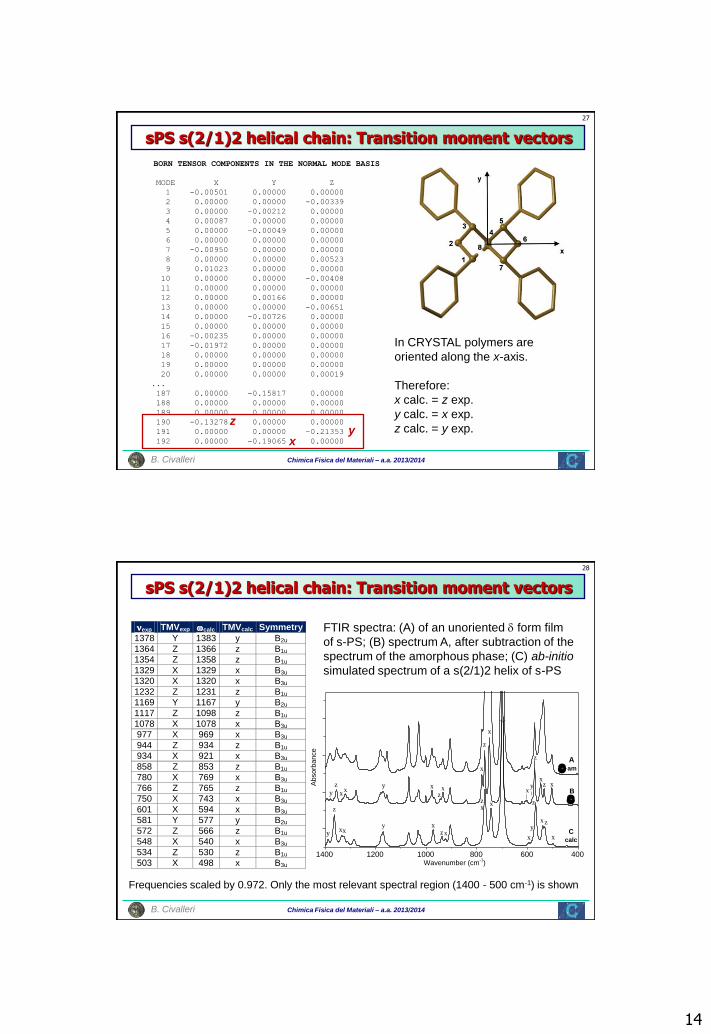
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
sPS s(2/1)2 helical chain: Transition moment vectors
In CRYSTAL polymers are
oriented along the x-axis.
Therefore:
x calc. = z exp.
y calc. = x exp.
z calc. = y exp.
BORN TENSOR COMPONENTS IN THE NORMAL MODE BASIS
MODE X Y Z
1 -0.00501 0.00000 0.00000
2 0.00000 0.00000 -0.00339
3 0.00000 -0.00212 0.00000
4 0.00087 0.00000 0.00000
5 0.00000 -0.00049 0.00000
6 0.00000 0.00000 0.00000
7 -0.00950 0.00000 0.00000
8 0.00000 0.00000 0.00523
9 0.01023 0.00000 0.00000
10 0.00000 0.00000 -0.00408
11 0.00000 0.00000 0.00000
12 0.00000 0.00166 0.00000
13 0.00000 0.00000 -0.00651
14 0.00000 -0.00726 0.00000
15 0.00000 0.00000 0.00000
16 -0.00235 0.00000 0.00000
17 -0.01972 0.00000 0.00000
18 0.00000 0.00000 0.00000
19 0.00000 0.00000 0.00000
20 0.00000 0.00000 0.00019
...
187 0.00000 -0.15817 0.00000
188 0.00000 0.00000 0.00000
189 0.00000 0.00000 0.00000
190 -0.13278 0.00000 0.00000
191 0.00000 0.00000 -0.21353
192 0.00000 -0.19065 0.00000 x y
z
28
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
sPS s(2/1)2 helical chain: Transition moment vectors
nexp TMVexp calc TMVcalc Symmetry
1378 Y 1383 y B2u
1364 Z 1366 z B1u
1354 Z 1358 z B1u
1329 X 1329 x B3u
1320 X 1320 x B3u
1232 Z 1231 z B1u
1169 Y 1167 y B2u
1117 Z 1098 z B1u
1078 X 1078 x B3u
977 X 969 x B3u
944 Z 934 z B1u
934 X 921 x B3u
858 Z 853 z B1u
780 X 769 x B3u
766 Z 765 z B1u
750 X 743 x B3u
601 X 594 x B3u
581 Y 577 y B2u
572 Z 566 z B1u
548 X 540 x B3u
534 Z 530 z B1u
503 X 498 x B3u
1400 1200 1000 800 600 400
z
z
x
xx
x
x
x
z
z
z
z
z
z
z y
y
x
y
y
y
y
xx
x
x
x
xx
xxx
C
calc
B
A+am
Ab
sorb
an
ce
Wavenumber (cm-1)
z
x
FTIR spectra: (A) of an unoriented form film
of s-PS; (B) spectrum A, after subtraction of the
spectrum of the amorphous phase; (C) ab-initio
simulated spectrum of a s(2/1)2 helix of s-PS
Frequencies scaled by 0.972. Only the most relevant spectral region (1400 - 500 cm-1) is shown

15
29
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
• As a tool for the assignment of the modes and for the
interpretation of the spectrum
• One atom at a time (e.g. 29Al for 27Al)
(experimental data available for comparison)
• In some cases also infinite mass:
Advantages with respect to subunits investigated with clusters
a) the atoms move in the field created by the infinite system.
b) and in the presence of the other atoms
c) and the hessian matrix is the correct one
Isotopic substitution and isotopic shift
21( 0)ij
i ji j
ED
u uM M
0 GG
0
k
30
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
B3LYP
Isotopic
substitution 62
Dn(exp.t)
13C 18O
12÷8 28÷27
9
15 38
16
37
Exp. Data: P. Gillet, et al. Geochim. Cosmochim. Acta 60 (1996) 3471; M.E. Böttcher, et al. Solid State Ion. 101-103 (1997) 1379
Vibrational frequencies of Calcite (CaCO3)

16
31
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
E2
E1
E0
02
01
exe=(2 01- 02) / 2
X-H stretching fully decoupled
from any other normal modes
A wide range (0.5 Å) of X-H
distances must be explored to
properly evaluate E1 and E2
Direct comparison with
experiment for fundamental
frequency, first overtone and
anharmonicity constant
(ANHARM)
X-H stretching modes are highly anharmonic. How to deal with that?
Anharmonicity: the problem of X-H modes
32
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
Isolated OH groups in crystals: model structures/1
M O
H
M=Mg Brucite M=Ca Portlandite
Edingtonite surface
Chabazite
All calculations with 6-31G(d,p) basis set
17
33
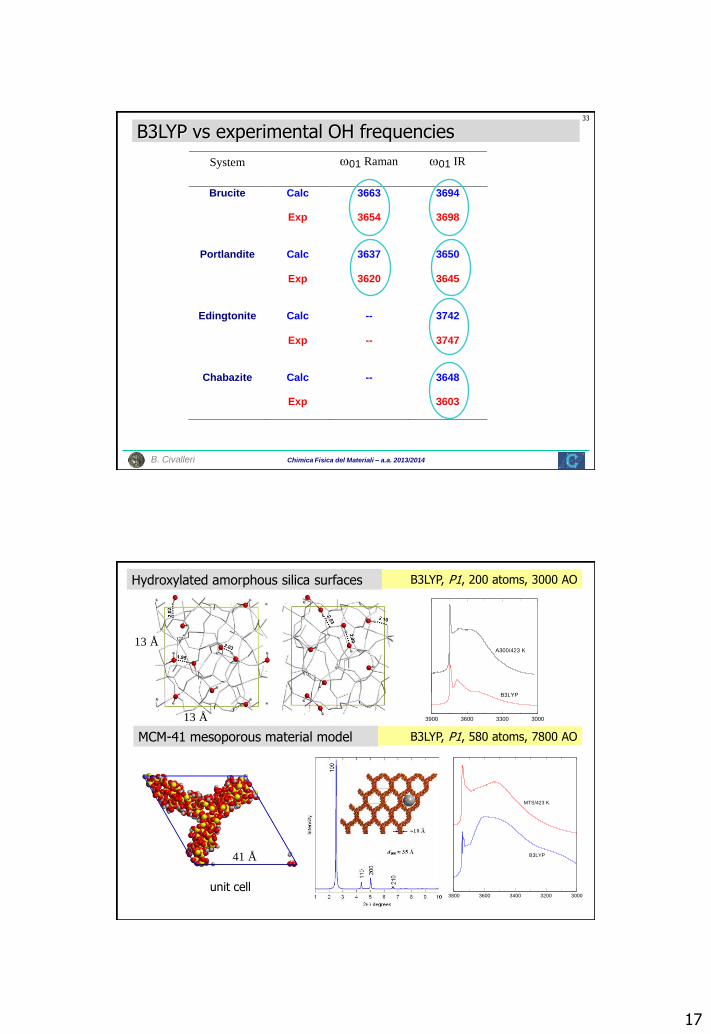
Chimica Fisica del Materiali – a.a. 2013/2014 B. Civalleri
B3LYP vs experimental OH frequencies
System 01 Raman 01 IR
Brucite Calc 3663 3694
Exp 3654 3698
Portlandite Calc 3637 3650
Exp 3620 3645
Edingtonite Calc -- 3742
Exp -- 3747
Chabazite Calc -- 3648
Exp 3603
Hydroxylated amorphous silica surfaces
MCM-41 mesoporous material model
3000330036003900
A300/423 K
B3LYP
30003200340036003800
MTS/423 K
B3LYP
B3LYP, P1, 200 atoms, 3000 AO
B3LYP, P1, 580 atoms, 7800 AO
unit cell
13 Å
13 Å
41 Å
Top Related