







Languages
Pages
Legal


AMPA receptor activation induces association of G-betaprotein with the alpha subunit of the sodium channel inneurons
Philippe Marin,1 Laurent Fagni,2 Yvette Torrens,1 GiseÁ le Alcaraz,3 FrancËois Couraud,3 JoeÈ l Bockaert,2 JacquesGlowinski1 and JoeÈ l PreÂmont1¶1INSERM U114, ColleÁge de France, 11, Place Marcelin Berthelot, 75231 Paris Cedex 05, France2CNRS UPR 9023, CCIPE, 141, rue de la Cardonille, 34094 Montpellier Cedex 5, France3INSERM U464, Faculte de MeÂdecine Nord, Universite Aix-Marseille II, Boulevard Pierre Dramard, 13916 Marseille Cedex 20,
France
Keywords: AMPA receptor, Gb protein, mitochondrial calcium, mouse, sodium channel
Abstract
Glutamatergic transmission is mediated by ionotropic receptors that directly gate cationic channels and metabotropic receptors
that are coupled to second messenger generating systems and to ionic channels via heterotrimeric guanine-nucleotide binding-
(G) proteins. This distinction cannot be made for the ionotropic receptor subclass activated by a-amino-3-hydroxy-5-
methylisoxazole-4-propionic acid (AMPA), which has been shown to be physically associated with the a-subunit of Gi1 proteinand activates this G-protein. Here, we report that, in addition to a Ca2+ in¯ux, AMPA induces the mobilization of Ca2+ from the
mitochondrial pool by reversing the mitochondrial Na+/Ca2+ exchanger in mouse neurons in primary culture. Both processes
required the activation of tetrodotoxin-sensitive Na+ channels. AMPA receptor activation modi®ed the gating properties of theNa+ channel, independently of the AMPA current, suggesting a G-protein-mediated process. Indeed, co-immunoprecipitation
experiments indicated that AMPA receptor activation induced the association of Gb with the a-subunit of the Na+ channel. These
results suggest that, in addition to its ionic channel function, the AMPA receptor is coupled to Na+ channels through G-proteinsand that this novel metabotropic function is involved in the control of neuronal excitability.
Introduction
Neuronal voltage-gated Na+ channels [NaCh(s)] are heterotrimeric
complexes composed of a, b1 and b2 subunits (Catterall, 2000). Four
a-subunit genes (Nav1.1, Nav1.2, Nav1.3 and Nav1.6) are expressed
in the brain and the presence of RNA transcripts for 1.1, 1.2 and 1.3
genes have been evidenced in cultures of rat cortical neurons (Giraud
et al., 1998). Expression of the a-subunit alone is suf®cient to form
functional voltage-gated NaCh(s) (Noda et al., 1986; Goldin et al.,
1986; Smith & Goldin, 1998; Smith et al., 1998) and coexpression of
b1 and b2-subunits modulates channel expression and gating (Isom
et al., 1995a, b). It is now well established that ion channels including
NaCh can be regulated by a-subunits (Ga) and/or bg-subunits (Gbg)
of heterotrimeric guanine-nucleotide binding-(G) proteins. Activation
of brain NaCh is enhanced by G-proteins in hippocampal neurons and
CHO cells expressing type IIA a-subunit of the Na+ channel (aNaCh
IIA) (Ma et al., 1994). Moreover, in non-neuronal cells coexpressing
aNaCh IIA and different Gbg subunit combinations, a direct
association of Gbg to the C-terminal domain of the channel prolongs
inactivation of the Na+ current (Ma et al., 1997).
Fast excitatory synaptic transmission is mainly mediated by the
a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)
subtype of excitatory amino acid receptors at most synapses of the
central nervous system. One important consequence of AMPA
receptor activation is a marked increase in cytosolic free Ca2+
concentration, a necessary step in several physiological processes
including synaptic plasticity and many neuropathological disorders
such as neurotoxicity associated with cerebral ischemia, in which
AMPA receptors are implicated (Gill, 1994; Zamanillo et al., 1999).
Ca2+ entry in neurons exposed to AMPA can be mediated directly by
Ca2+-permeable AMPA receptor subtypes or indirectly by depolar-
ization-induced activation of voltage-gated Ca2+ channels (Murphy &
Miller, 1989; Weiss et al., 1990; Brorson et al., 1992).
Several studies suggest that, in addition to their ionic channel
function, AMPA receptors transduce some of their effects through
interaction with heterotrimeric G-proteins. Exposing rat cortical
neurons to AMPA induces the dissociation of the a-subunit from the
Gai1bg heterotrimeric complex, its association with the GluR1
AMPA receptor subunit, as well as the inhibition of adenylyl cyclase
activity (Wang et al., 1997). Moreover, AMPA receptors activate a
G-protein that suppresses a cGMP-gated current in retinal ganglion
cells (Kawai & Sterling, 1999).
In an attempt to identify other metabotropic functions of AMPA
receptors, we found that AMPA induced the mobilization of Ca2+
from the mitochondrial pool in cultured neurons. This effect occurred
through a mechanism involving tetrodotoxin- (TTX) sensitive
voltage-gated NaChs and the reverse function of the mitochondrial
Na+/Ca2+ exchanger. This process was accompanied by a prolonged
modi®cation of the gating properties of the TTX-sensitive NaChs that
Correspondence: Dr JoeÈl PreÂmont, 1INSERM U114, as above.E-mail: [email protected]
Received 6 August 2001, accepted 29 October 2001
European Journal of Neuroscience, Vol. 14, pp. 1953±1960, 2001 ã Federation of European Neuroscience Societies

was independent of the AMPA current. Additional experiments
indicated that AMPA receptor activation increased association of Gbproteins with aNaCh in neurons.
Experimental procedures
Primary cultures
Pregnant mice were killed by prolonged exposure to a rising
concentration of CO2 and embryos (15 days old) were rapidly
removed from the uteri and placed into a phosphate-buffered saline
supplemented with glucose (33 mM). Primary cultures of mouse
cortical or cerebellar granule neurons were prepared as previously
described (Van Vliet et al., 1989; Marin et al., 1997). Cells were
maintained at 37 °C in a humidi®ed atmosphere containing 8% CO2
for 10±12 days without medium change. Under these conditions,
cultures were shown by immunocytochemistry experiments, using an
anti-Microtubule-Associated Protein 2 monoclonal antibody (Clone
HM2, Sigma, France), to be highly enriched in neurons and less than
7% of the cells exhibited immunoreactivity with a rabbit antibody
raised against glial ®brillary acid protein (Dakopatts, Glostrup,
Denmark) (data not shown).
Cytosolic Ca2+ measurement
Cytosolic free Ca2+ was monitored in cortical neurons cultured on
glass slides by quantitative ratio imaging of the ¯uorescent Ca2+
probe INDO-1 (Molecular Probes, Eugene, OR), as described
previously (Murphy et al., 1994). Cells were loaded for 60 min
with 12 mM of INDO-1 acetoxymethylester in HEPES buffer. After
loading, the glass slide was placed in a perfusion chamber where cells
were exposed to tested substances using a multichannel superfusion
device allowing the delivery of drugs to cells in less than 100 ms.
Cells were excited with a 75-W Xenon light, ®ltered at 340 nm with a
10-nm wide interferential ®lter. Excitation and emission spectra were
separated by a 380-nm dichroic long pass ®lter and the emission
spectra were divided in two halves by a 455-nm dichroic long pass
®lter (opticals were obtained from Nikon and Hamamatsu, Japan).
Two discriminant bands were selected by interferential ®lters from
the two halves at 400±410 nm and 470±480 nm and both ¯uorescent
images were digitized (eight video frames per digitized image,
permitting the recording of one image per second). The camera dark
noise was subtracted from the recorded crude image (camera and
digitizing system were from Hamamatsu). Results were expressed
as the ratio between ¯uorescence measured at 405 and 480 nm
(F405/F480).
Whole-cell patch-clamp recordings
Whole-cell, patch-clamp currents were recorded at room temperature
in cortical and cerebellar granule neurons cultured in 35-mm culture
dishes. The bathing medium contained (in mM): Na-gluconate, 140;
CaCl2, 2; KCl, 3; MgCl2, 2; HEPES, 10; D-glucose, 10; pH adjusted
to 7.4 with NaOH and to 330 mOsm with Na-gluconate. Drug
solutions were prepared in this bathing medium and the pH was
readjusted to 7.4. Patch pipettes were made from borosilicate glass,
coated with Sylgard, and the tip ®re polished. Pipettes had resistances
of 3±5 MW when ®lled with the following internal solution
(containing, in mM): Cs-acetate, 100; MgCl2, 2; HEPES, 10; glucose,
15, CsCl, 20; EGTA, 20 mM; Na2ATP, 2 mM and cAMP, 1 mM,
adjusted to pH 7.2 and to 300 mOsm with CsOH. Drugs were applied
by means of a gravity-driven, fast perfusion system as previously
described (Manzoni et al., 1992). Na+ currents were evoked by
voltage-clamp depolarizing pulses of 15 ms duration, from a holding
potential of ±60 mV. Voltage pulses were applied at a rate of 0.1 Hz.
Current signals were recorded with an Axopatch 200 ampli®er,
®ltered at 1 kHz with an 8-pole Bessel ®lter and sampled at 3 kHz on
a Pentium II PC computer. Analyses were performed using the
pClamp6 program of Axon Instruments and Na+ currents were
measured at their peak amplitude. Capacitance of the cells was
measured by delivering square wave hyperpolarizing pulses of 5 mV
amplitude, 10 ms duration, at a frequency of 2 Hz. Average
capacitance, input resistance and resting potential of the cells
were of 7.2 6 0.3 pF (n = 33), 11.2 6 0.9 MW (n = 33) and
±60 6 11 mV (n = 33), respectively.
Co-immunoprecipitation experiments
Cortical neurons, grown in 100-mm culture dishes were washed twice
in HEPES buffer (containing, in mM: HEPES, 20; glucose, 5.5; NaCl,
120; KCl, 5.5; MgCl2, 1.2; CaCl2, 1.2; pH 7.4) and then incubated in
the same medium for 5 min (unless otherwise indicated) in the
presence of drugs. Cells were scraped off in 5 mL of homogenization
buffer (sucrose 0.32 M, Tris-HCl 10 mM, pH 7.4, containing a
protease inhibitor cocktail (Roche, France), centrifuged for 5 min at
100 g and homogenized in 1 mL of the same buffer. Samples were
centrifuged for 10 min at 1000 g and the postnuclear fraction was
centrifuged for 30 min at 100 000 g. Immunoprecipitations were
conducted on solubilized membrane fractions as described previously
(Alcaraz et al., 1997). The membrane fraction (pellet) was incubated
for 30 min in 0.5 mL ice-cold solubilization buffer containing
100 mM NaCl, 50 mM Tris-HCl pH 7.4, 5 mM EDTA, 1% (wt/v)
Triton X-100 and the protease inhibitor cocktail, and centrifuged for
30 min at 100 000 g. Supernatant proteins (300 mg) were immuno-
precipitated with an antibody directed against a sequence identical to
that of the previously described SP20 antibody, which has been
largely used to detect NaChs in various tissues (Black et al., 1995a,
b). This antibody was obtained by immunization of rabbits with a
synthetic peptide derived from a conserved sequence in the second
intracellular loop of the a-subunit (amino acids 1111±1121, subtype
aII numbering: GESDFENLNTE). This serum was shown previously
to speci®cally immunoprecipitate aNaCh (Alcaraz et al., 1997).
Immunocomplexes were resolved on either 10% polyacrylamide gels,
to detect Gb, or 4% polyacrylamide gels to detect aNaCh. The
amount of immunoprecipitated Gb was estimated by Western blotting
using a monoclonal antibody directed against Gb (clone 3, 1 : 1000
dilution, Transduction Laboratories, Lexington, KY). Immuno-
precipitated aNaCh was detected using a protein A-puri®ed Ig
fraction from an antiserum directed against a glutathione S-
transferase fusion protein containing the entire sequence of the
third intracellular loop of the a subunit (1.5±2 mg/mL). In Western
blots of rat brain membranes, this antibody recognizes a polypeptide
around 260 kDa that displays the smear characteristic of the highly
glycosylated aNaCh. The total amount of Gb was also estimated by
Western blotting of an aliquot of crude neuronal membranes (10 mg
protein) following each treatment.
ADP-ribosylation of Gai/o proteins by PTX
Cortical neurons grown in 100-mm culture dishes were washed twice
in HEPES buffer and incubated in the same medium for 5 min in the
presence of drugs (unless otherwise indicated). After two washes,
cells were scraped off and centrifuged for 10 min at 100 g. The cell
pellet was resuspended and homogenized in an ice-cold lysing buffer
containing 50 mM Tris-HCl (pH 7.5), 3 mM EDTA and protease
inhibitors, and centrifuged for 30 min at 100 000 g. Particulate
fraction (50 mg protein) was incubated for 1 h at 30 °C in 40 mL
ADP-ribosylation medium containing 100 mM Tris-HCl (pH 8),
1954 P. Marin et al.
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960

1 mM EDTA, 10 mM thymidine, 2 mM MgCl2, 1 mM ATP, 0.1 mM
GTP, 10 mM nicotinamide, 10 mg/mL Bordetella pertussis toxin
(PTX, preactivated with 1 mM dithiotreitol for 30 min at 37 °C), and
[a-32P]NAD+ (0.5 mM, 1 mCi, NEN-Dupont, France). The reaction
was stopped by the addition of 20 mL SDS (2%). Samples were
resolved on 10% polyacrylamide gels and subjected to autoradio-
graphy.
Results
Role of TTX-sensitive Na+ channels in AMPA-induced Ca2+
increase in neurons
As observed previously in several neuronal types (Murphy & Miller,
1989; Brorson et al., 1992), exposing mouse cortical neurons to a
maximally effective concentration of AMPA (30 mM) (Williams et al.,
1995) induced a prolonged increase in cytosolic Ca2+ in the presence
of extracellular Ca2+ (Fig. 1A). This effect was blocked by 6,7-
dinitroquinoxaline-2,3-dione (DNQX, 100 mM), an antagonist of
AMPA receptors (not shown). The AMPA-induced elevation of
cytosolic Ca2+ was also entirely suppressed by the coapplication of
TTX (1 mM, Fig. 1B).
Interestingly, AMPA also induced a transient (less than 1 min)
elevation of cytosolic Ca2+ in the absence of extracellular Ca2+
(Fig. 1C) and this response could be reliably observed following
successive applications of the agonist (Fig. 1D). This mobilization of
intracellular Ca2+ was totally suppressed by DNQX (not shown). It is
worth noting that washout of AMPA in a Ca2+-containing medium
was accompanied by a delayed Ca2+ increase (Fig. 1D). This Ca2+
response was probably mediated by activated voltage-gated Ca2+
channels, as it was suppressed by nifedipine, whereas, the transient
Ca2+ elevation detected in the absence of extracellular Ca2+ was not
altered (not illustrated).
Several pieces of evidence suggest that the mobilization of
intracellular Ca2+ induced by AMPA results from the reverse
function of the Na+/Ca2+ mitochondrial exchanger following the
presumed Na+ in¯ux through TTX-sensitive NaCh. The Ca2+
response was suppressed by TTX (Fig. 1E) and strongly reduced
when extracellular NaCl was replaced by choline chloride (120 mM)
or N-methyl-D-glucamine (240 mM) (84 6 11% and 81 6 13% of
decrease of the Ca2+ pike, compared with that measured in the
presence of extracellular Na+, n = 35, respectively); it was unaltered
by pretreating the neurons with thapsigargin (10 mM), which specif-
ically depletes endoplasmic reticulum Ca2+ stores (Fig. 1F); it was
suppressed in the presence of CGP 37 157 (25 mM), a speci®c
inhibitor of the mitochondrial Na+/Ca2+ exchanger (Fig. 1G) (Cox
et al., 1993; White & Reynolds, 1997). Finally, AMPA-induced
mitochondrial Ca2+ mobilization was mimicked by direct activation
of voltage-gated NaCh. Indeed, exposure of neurons to veratridine
(10 mM, in the absence of extracellular Ca2+), induced a transient
elevation of cytosolic Ca2+ (Fig. 1H), which was also suppressed by
CGP 37 157 (not shown).
Altogether, these results indicate that both Ca2+ in¯ux and
mobilization of mitochondrial Ca2+ induced by AMPA are strictly
dependent on the activation of TTX-sensitive NaCh.
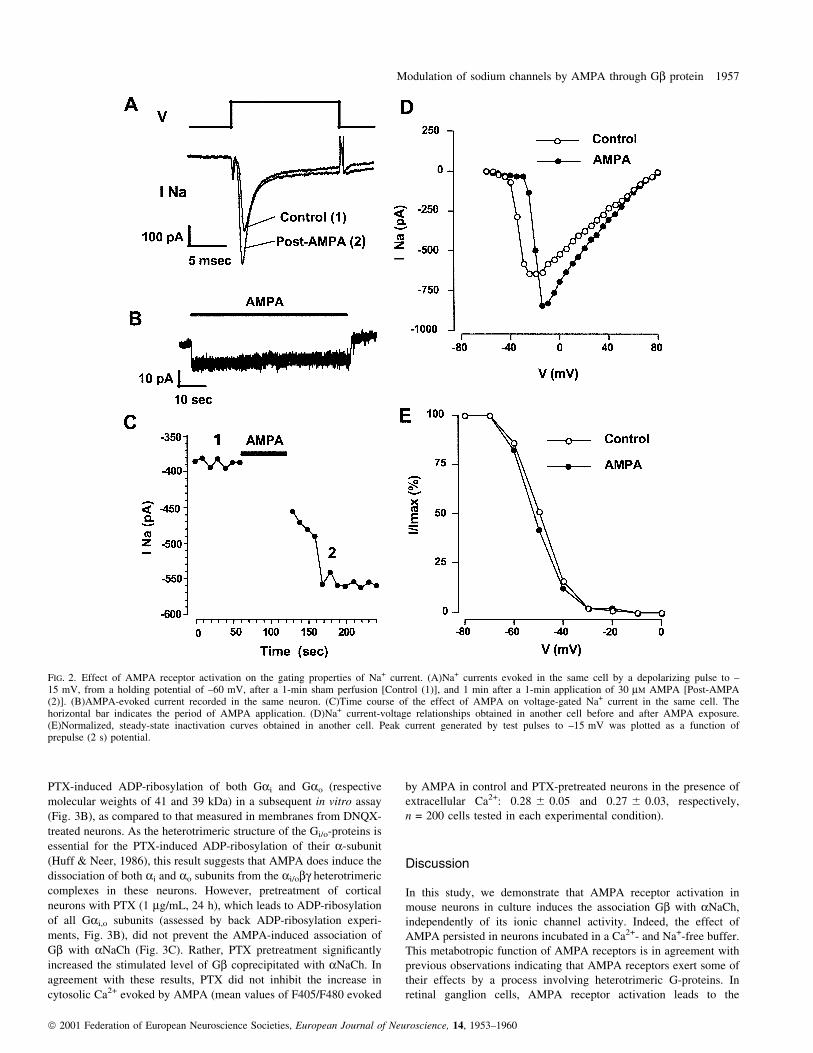
Effects of AMPA on the gating properties of the Na+ channel
Additional experiments were performed to examine the effect of
AMPA receptor activation on the properties of the voltage-gated
NaChs. Neurons exhibited stable Na+ currents (±145 6 10 pA/pF at
±15 mV, n = 11) that activated rapidly in response to depolarization
and then inactivated rapidly and virtually completely in less than
5 ms (Fig. 2A). The Na+ current was entirely suppressed by TTX
(1 mM, not shown). Exposing the neurons to AMPA (30 mM) induced
an inward current (2.4 6 0.6 pA/pF at ±60 mV membrane potential,
n = 11) in about 80% of the cells tested (n = 14), which rapidly
decayed after drug washout (Fig. 2B). Na+ currents were not recorded
during the application of AMPA because of the shunting effect of
AMPA-operated channels. The amplitude of the Na+ current was thus
compared in the same cell before and after a 1-min application of
AMPA followed by washout of the agonist (Fig. 2A and C).
Treatment with AMPA increased the amplitude of the Na+ current
(mean increase at ±15 mV, 31 6 6%, n = 9) in 80% of the neurons
examined (n = 11, Fig. 2A). This effect was rapid (less than 1 min)
and lasted for several minutes (Fig. 2C). Current±voltage relation-
ships of the Na+ currents obtained after the AMPA treatment
(Fig. 2D) shifted toward positive membrane potentials, showing that
the increase in current amplitude occurred only at potentials above ±
25 mV. Neither the time course of inactivation (Fig. 2A), nor the
steady-state inactivation curve (Fig. 2E) of the Na+ current were
signi®cantly affected by the AMPA treatment.
AMPA increases the association of Gb with aNaCh
A previous study demonstrated that heterotrimeric G-protein acti-
vation induced an increase in Na+ currents recorded in acutely
dissociated rat hippocampal neurons (Ma et al., 1994). Subsequent
experiments performed in non-neuronal cells coexpressing rat brain
aNaCh IIA and different Gbg subunit combinations indicated that the
binding of Gbg subunits to aNaCh IIA is responsible for the increase
of a component of the Na+ current displaying normal voltage-
dependence of activation but with a strongly slowed and incomplete
inactivation (Ma et al., 1997). Thus, we have examined whether
AMPA receptor activation stimulates the association of Gbg with
aNaCh in cortical neurons.
Co-immunoprecipitation experiments demonstrated that an anti-Gbantibody was able to detect a variable amount of Gb in
immunocomplexes obtained with an antibody directed against the
aNaCh in membranes from cortical neurons (Fig. 3A). Treating the
neurons with AMPA (30 mM) for 5 min increased the amount of Gbpresent in the immunoprecipitate, compared to that detected in both
untreated neurons and neurons exposed to DNQX (100 mM, Fig. 3A
and C). As expected, the effect of AMPA was totally suppressed by
DNQX (data not illustrated). In most experiments, exposing the
neurons to DNQX decreased the amount of Gb immunoprecipitated
with the anti-aNaCh antibody relative to untreated neurons (Fig. 3A).
This suggests that the association of Gb with aNaCh in untreated
neurons resulted from activation of AMPA receptors by ambient
glutamate. Thus, in subsequent experiments, the basal level of
immunoprecipitated Gb was determined in the presence of DNQX.
The observed changes in the amount of Gb associated to aNaCh were
not related to an altered association of Gb to neuronal membranes
after the treatments, as an equal amount of protein was found in crude
neuronal membranes (Fig. 3A, bottom).
In the presence of TTX (1 mM), the AMPA receptor-mediated
coprecipitation of Gb with aNaCh was signi®cantly increased
(Fig. 3C). This increase may be the consequence of a conformational
change of aNaCh induced by TTX (Tejedor et al., 1988). This result
also indicates that the association of Gb with aNaCh is not the result
of Na+ ¯ux through activated NaCh following AMPA receptor-
mediated depolarization. Further supporting this hypothesis, the
depolarization of neurons with KCl (50 mM) did not induce
association of Gb with aNaCh (Fig. 3C). Moreover, this treatment
did not further increase the level of Gb bound to aNaCh in AMPA-
exposed neurons (Fig. 3C). The association of Gb with aNaCh was
Modulation of sodium channels by AMPA through Gb protein 1955
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960

also independent of AMPA receptor ionic channel activity. Indeed,
the effect of AMPA persisted when neurons were incubated in a
Ca2+- and Na+-free buffer (Fig. 3C).
Part of Gbg that associates with aNaCh upon AMPA receptor
stimulation might arise from PTX-sensitive G-proteins (Gi/Go).
Indeed, treating mouse cortical neurons with AMPA reduced the
FIG. 1. Role of TTX-sensitive Na+ channels in the AMPA-induced increase in cytosolic Ca2+ Variations in cytosolic Ca2+ concentration (ratio between the¯uorescence measured at 405 and 480 nm (F405/F480) were measured in INDO-1-loaded neurons. Horizontal black bars represent the periods of superfusionof drugs, performed in the presence (+ Ca2+) or absence (± Ca2+) of Ca2+. Between these periods, extracellular Ca2+ was present throughout the recordings.The following concentrations were used: AMPA, 30 mM; TTX, 1 mM; thapsigargin, 10 mM; CGP 37 157 (generously provided by Novartis, France), 25 mM;veratridine, 10 mM. The coapplication of CGP 37 157 with AMPA did not yield a signi®cant decrease of the AMPA-evoked response, a 30- s pre-exposure ofthe cells to the drug being required for full inhibition. The AMPA-evoked response was fully restored only 1 min after the removal of CGP 37 157. Tracesrepresent the mean of those obtained in at least 35 neurons originating from two sets of cultured neurons.
1956 P. Marin et al.
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960
PTX-induced ADP-ribosylation of both Gai and Gao (respective
molecular weights of 41 and 39 kDa) in a subsequent in vitro assay
(Fig. 3B), as compared to that measured in membranes from DNQX-
treated neurons. As the heterotrimeric structure of the Gi/o-proteins is
essential for the PTX-induced ADP-ribosylation of their a-subunit
(Huff & Neer, 1986), this result suggests that AMPA does induce the
dissociation of both ai and ao subunits from the ai/obg heterotrimeric
complexes in these neurons. However, pretreatment of cortical
neurons with PTX (1 mg/mL, 24 h), which leads to ADP-ribosylation
of all Gai,o subunits (assessed by back ADP-ribosylation experi-
ments, Fig. 3B), did not prevent the AMPA-induced association of
Gb with aNaCh (Fig. 3C). Rather, PTX pretreatment signi®cantly
increased the stimulated level of Gb coprecipitated with aNaCh. In
agreement with these results, PTX did not inhibit the increase in
cytosolic Ca2+ evoked by AMPA (mean values of F405/F480 evoked
by AMPA in control and PTX-pretreated neurons in the presence of
extracellular Ca2+: 0.28 6 0.05 and 0.27 6 0.03, respectively,
n = 200 cells tested in each experimental condition).
Discussion
In this study, we demonstrate that AMPA receptor activation in
mouse neurons in culture induces the association Gb with aNaCh,
independently of its ionic channel activity. Indeed, the effect of
AMPA persisted in neurons incubated in a Ca2+- and Na+-free buffer.
This metabotropic function of AMPA receptors is in agreement with
previous observations indicating that AMPA receptors exert some of
their effects by a process involving heterotrimeric G-proteins. In
retinal ganglion cells, AMPA receptor activation leads to the
FIG. 2. Effect of AMPA receptor activation on the gating properties of Na+ current. (A)Na+ currents evoked in the same cell by a depolarizing pulse to ±15 mV, from a holding potential of ±60 mV, after a 1-min sham perfusion [Control (1)], and 1 min after a 1-min application of 30 mM AMPA [Post-AMPA(2)]. (B)AMPA-evoked current recorded in the same neuron. (C)Time course of the effect of AMPA on voltage-gated Na+ current in the same cell. Thehorizontal bar indicates the period of AMPA application. (D)Na+ current-voltage relationships obtained in another cell before and after AMPA exposure.(E)Normalized, steady-state inactivation curves obtained in another cell. Peak current generated by test pulses to ±15 mV was plotted as a function ofprepulse (2 s) potential.
Modulation of sodium channels by AMPA through Gb protein 1957
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960

inhibition of cationic channels gated by cGMP through PTX-sensitive
G-proteins (Kawai & Sterling, 1999). In these neurons, AMPA
receptor activation opposes its cationic channel activity by inhibiting
the opening of another cationic channel through heterotrimeric G-
proteins. In rat cortical neurons, AMPA induces the dissociation of
the ai1 subunit of Gi-protein from the abg heterotrimeric complex
and increases its association with the AMPA receptor subunit GluR1.
This process is probably involved in the inhibition of adenylyl
cyclase activity (Wang et al., 1997). Another subclass of ionotropic
glutamate receptors, preferentially activated by kainate, appears to be
similarly coupled to heterotrimeric G-proteins and, thus, to display a
metabotropic function. Pharmacological manipulations that uncouple
G-proteins from their receptors have been shown to decrease the
binding of kainate receptor-selective agonists to hippocampal mem-
branes, as observed for agonists of classical metabotropic receptors
(Cunha et al., 1999). Moreover, activation of kainate receptors
decreases the release of g-aminobutyric acid in the hippocampus by a
mechanism sensitive to PTX and inhibitors of protein kinase C
(Rodriguez-Moreno & Lerma, 1998)´
The decreased ADP-ribosylation of Gai/o proteins by PTX,
following AMPA treatment, indicates that AMPA receptors are
capable of activating these G-protein subclasses in mouse cortical
neurons. In contrast, the association of Gb with aNaCh was totally
insensitive to the toxin. One possible explanation may be that, unlike
classical metabotropic receptors, the interaction of AMPA receptors
with the Ga subunit is not impaired by its ADP-ribosylation.
However, several observations demonstrate that PTX prevents the
activation of different biochemical cascades mediated by AMPA or
kainate (Wang & Durkin, 1995; Wang et al., 1997; Rodriguez-
Moreno & Lerma, 1998; Kawai & Sterling, 1999). This suggests that
Gbg subunits that bind aNaCh upon AMPA receptor activation in
cortical neurons mainly arise from PTX-insensitive G-proteins.
AMPA receptors may thus be coupled to different G-protein
subclasses, sensitive and insensitive to PTX.
Evidence for membrane depolarization-induced activation of Go-
protein has been reported previously in rat brain synaptoneurosomes
(Anis et al., 1999). In this model, voltage-dependent activation of
NaCh appears to be essential for the activation of Go, regardless of
Na+ current (Anis et al., 1999). In our experiments performed on
living cortical neurons, KCl-induced cell depolarization was, alone,
unable to stimulate the binding of Gb to NaCh and did not further
increase the level of Gb bound to aNaCh in AMPA-treated neurons.
This indicates that the association of Gb with aNaCh is not the
consequence of the activation of a G-protein following Na+ in¯ux
initiated by AMPA receptor activation and prolonged by the opening
of voltage-gated NaCh.
Parallel to this metabotropic effect of AMPA, we provide
electrophysiological experiments showing that AMPA induces a
shift in the voltage-dependence of activation of TTX-sensitive NaChs
and an increase in the Na+ current amplitude, which persisted after
agonist removal. A previous study has shown that brain NaCh activity
can be increased by a G-protein-dependent mechanism (Ma et al.,
1994). Further experiments performed in non-neuronal cells trans-
fected with aNaCh IIA and different Gbg subunit combinations,
indicated that direct association of Gbg with aNaCh IIA enhanced a
late component of the Na+ current with a normal voltage-dependence
of activation but slower and incomplete inactivation (Ma et al., 1997).
We propose that the modi®cations of electrophysiological properties
of NaCh following AMPA exposure in cortical neurons may be the
consequence of Gb association with aNaCh. Supporting the role of
the binding of Gb to aNaCh in the facilitation of Na+ current by
AMPA, both phenomena persisted in the absence of AMPA. Indeed,
the binding of Gb to aNaCh was observed in neuronal membranes
that were prepared in the absence of AMPA and the facilitation of
Na+ current also lasted several minutes after the AMPA withdrawal.
Contrasting with the results obtained in transfected non-neuronal
cells, we did not observe any persistent Na+ current nor any shift in
steady-state inactivation curve of Na+ current in neurons exposed to
AMPA. These discrepancies may result from the expression of an
excess of Gbg-subunits over free Ga-subunits or from the absence of
regulatory proteins associated with NaCh in the non-neuronal cells
used for tranfection experiments (Ma et al., 1997).
Our study highlights the importance of TTX-sensitive NaChs in the
regulation of the intracellular Ca2+ concentration in central neurons.
FIG. 3. AMPA receptor activation induces the association of Gb withaNaCh. (A) Neurons were exposed to sham treatment (None), DNQX100 mM or AMPA (30 mM) for 5 min and membrane proteins, solubilizedwith 1% Triton X-100, were immunoprecipitated with a polyclonal antibodyrecognizing aNaCh. Immunoprecipitated Gb and aNaCh were detected byWestern blotting. The total amount of Gb was determined in an aliquot ofthe crude membrane fraction in each experimental condition (bottom). Theillustrated experiment is representative of three independent experimentsperformed on different sets of cultured neurons. (B) Membrane proteins ofneurons challenged for 24 h with PTX (1 mg/mL) or for 5 min with eitherDNQX or AMPA were subjected to in vitro PTX-induced ADP-ribosylation.Data are representative of three experiments performed independently. (C)Neurons were incubated for 5 min in the absence or presence of eitherDNQX, AMPA, TTX (1 mM) or KCl (50 mM). In the experiments usingPTX, neurons were pretreated for 24 h with the toxin. For Na+- and Ca2+-free condition, cells were washed and incubated for 5 min in HEPES bufferwithout Ca2+ and containing choline chloride instead of NaCl. The amountof Gb co-immunoprecipitated with the anti-aNaCh antibody was determinedby densitometric analysis. Data are the mean 6 SEM of values obtained inthree experiments performed on different sets of cultured neurons and havebeen calculated as a percentage of immunoprecipitated Gb in neuronsexposed to AMPA in each experiment (hatched line). *P < 0.01 vs. DNQXin the corresponding condition. §P < 0.05 vs. AMPA alone (ANOVA
followed by Student's Newman±Keuls test). The above treatments did notchange the amount of Gb in crude membranes (data not illustrated).
1958 P. Marin et al.
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960

Indeed, we show that AMPA receptor stimulation induces a Ca2+
in¯ux and the mobilization of mitochondrial Ca2+ stores through the
reverse function of mitochondrial Na+/Ca2+ exchanger, both being
governed by a Na+ in¯ux through voltage-gated NaChs. The role of
Gb association with aNaCh in AMPA-induced Ca2+ elevation in
neurons remains to be elucidated. As the mobilization of mitochon-
drial Ca2+ is entirely dependent on the activity of NaCh, binding of
Gb to aNaCh may contribute, at least in part, to this AMPA-evoked
Ca2+ response. Experiments performed with TTX suggest that Na+
in¯ux through NaCh but not AMPA receptors is suf®cient to alter the
Na+/Ca2+ exchanger. This result is consistent with our electrophy-
siological recordings showing that the amplitude of steady-state
AMPA currents measured at ±60 mV was 50±60-fold smaller than
that of Na+ currents evoked by a depolarizing pulse. The AMPA
current was, however, suf®cient to activate voltage-gated NaChs
through membrane depolarization. In fact, Na+ concentration may
only reach a level capable of altering Na+/Ca2+ exchanger mode at
the immediate submembrane space following activation of NaCh.
Because of the limited intracellular volume of neuronal cells, in
particular in processes, one can suppose that peri-mitochondrial Na+
concentration also reaches a level that reverses the Na+/Ca2+
exchanger function. This hypothesis is supported by a recent study
showing that the increase in cytosolic Ca2+ in hippocampal neurons,
during in vitro ischemia, is mediated by a sequence of events
involving Na+ entry through voltage-gated NaCh and activation of
mitochondrial Na+/Ca2+ exchanger (Zhang & Lipton, 1999).
The physiological consequences of the mobilization of mitochon-
drial Ca2+ remain to be fully elucidated. It has been shown that the
uptake of Ca2+ into mitochondria plays a predominant role in the
buffering of intracellular Ca2+ during glutamate challenge (White &
Reynolds, 1997). This process is critically regulated by the rate of
Ca2+ release from mitochondria via the Na+/Ca2+ exchanger, which is
dictated by intracellular Na+ concentration. Indeed, the increase in
recovery time-course of cytosolic Ca2+ with the intensity and duration
of glutamate stimulation is due to an enhanced ef¯ux of mitochon-
drial Ca2+ (White & Reynolds, 1997). AMPA receptor stimulation
and Na+ entry-mediated activation of mitochondrial Na+/Ca2+
exchanger also plays a major role in the sustained increase in
neuronal cytosolic Ca2+ during in vitro ischemia in the hippocampus
(Zhang & Lipton, 1999). This observation, together with the
protective effects of both AMPA receptor antagonists and Na+
channel blockers, including TTX, in cerebral ischemia (Weber &
Taylor, 1994; Yatsugi et al., 1996; Turski et al., 1998) suggests that
the enhanced ef¯ux of Ca2+ from mitochondria following a robust
Na+ entry may be a key determinant in ischemic neuronal damage.
Physical coupling of AMPA receptors with PTX-sensitive G-
proteins as well as a direct interaction of bg subunits of G-
proteins with aNaCh had already been described (Ma et al., 1994;
Wang et al., 1997). These ®ndings are extended by the present
study, which suggests a functional coupling between AMPA
receptors and voltage-gated NaCh through the binding of Gb to
aNaCh, a process that may play an important role in the control
of neuronal excitability and contribute to neuronal death following
cerebral ischemia.
Acknowledgements
This work was supported by grants of Institut National de la Sante et de laRecherche MeÂdicale (INSERM), Centre National de la Recherche Scienti®que(CNRS) and ColleÁge de France.
Abbreviations
AMPA, a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; DNQX, 6,7-dinitroquinoxaline-2,3-dione; G-protein, guanine-nucleotide binding- protein;NaCh, Na+ channel; PTX, Bordetella pertussis toxin; TTX, tetrodotoxin.
References
Alcaraz, G., Sampo, B., Tricaud, N., Giraud, P., Martin-Eauclaire, M.F.,Couraud, F. & Dargent, B. (1997) Down-regulation of voltage-dependentsodium channels coincides with a low expression of alphabeta1 subunitcomplexes. Brain Res. Mol. Brain Res., 51, 143±513.
Anis, Y., Nurnberg, B., Visochek, L., Reiss, N., Naor, Z. & Cohen-Armon, M.(1999) Activation of Go-proteins by membrane depolarization traced by insitu photoaf®nity labeling of Gao-proteins with [a32P]GTP-azidoanilide. J.Biol. Chem., 274, 7431±7440.
Black, J.A., Westenbroek, R.E., Catterall. W.A. & Waxman, S.G. (1995a)Type II brain sodium channel expression in non-neuronal cells: embryonicrat osteoblasts. Brain Res. Mol. Brain Res., 34, 89±98.
Black, J.A., Westenbroek, R., Minturn, J.E., Ransom, B.R., Catterall, W.A. &Waxman, S.G. (1995b) Isoform-speci®c expression of sodium channels inastrocytes in vitro: immunocytochemical observations. Glia, 14, 133±144.
Brorson, J.R., Bleakman, D., Chard, P.S. & Miller. R.J. (1992) Calciumdirectly permeates kainate/alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in cultured cerebellar Purkinje neurons.Mol. Pharmacol., 41, 603±608.
Catterall, W.A. (2000) From ionic currents to molecular mechanisms: Thestructure and function of voltage-gated sodium channels. Neuron, 26, 13±25.
Cox, D.A., Conforti, L., Sperelakis, N. & Matlib, M.A. (1993) Selectivity ofinhibition of Na(+) -Ca2+ exchange of heart mitochondria bybenzothiazepine CGP-37157. J. Cardiovasc. Pharmacol., 21, 595±599.
Cunha, R.A., Malva, J.O. & Ribeiro, J.A. (1999) Kainate receptors coupled toG(i) /G(o) proteins in the rat hippocampus. Mol. Pharmacol., 56, 429±433.
Gill, R. (1994) The pharmacology of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) /kainate antagonists and their role in cerebralischaemia. Cerebrovasc. Brain Metab. Rev., 6, 225±256.
Giraud, P., Alcaraz, G., Jullien, F., Sampo, B., Jover, E., Couraud, F. &Dargent, B. (1998) Multiple pathways regulate the expression of genesencoding sodium channel subunits in developing neurons. Brain Res. Mol.Brain Res., 56, 238±255.
Goldin, A.L., Snutch, T., Lubbert, H., Dowsett, A., Marshall, J., Auld, V.,Downey, W., Fritz, L.C., Lester, H.A., Dunn, R. & Catterall, W.A. (1986)Davidson N Messenger RNA coding for only the alpha subunit of rat brainNa channel is suf®cient for expression of functional channels in Xenopusoocytes. Proc. Natl. Acad. Sci. USA, 83, 7503±7507.
Huff, R.M. & Neer, E.J. (1986) Subunit interactions of native and ADP-ribosylated alpha 39 and alpha 41, two guanine nucleotide-binding proteinsfrom bovine cerebral cortex. J. Biol. Chem., 261, 1105±1110.
Isom, L.L., Ragsdale, D.S., De Jongh, K.S., Westenbroek, R.E., Reber, B.F.,Scheuer, T. & Catterall, W.A. (1995a) Structure and function of the beta 2subunit of brain sodium channels, a transmembrane glycoprotein with aCAM motif. Cell, 83, 433±442.
Isom, L.L., Scheuer, T., Brownstein, A.B., Ragsdale, D.S., Murphy, B.J. &Catterall, W.A. (1995b) Functional co-expression of the beta 1 and type IIAalpha subunits of sodium channels in a mammalian cell line. J. Biol. Chem.,270, 3306±3310.
Kawai, F. & Sterling, P. (1999) AMPA receptor activates a G-protein thatsuppresses a cGMP-gated current. J. Neurosci., 19, 2954±2959.
Ma, J.Y., Catterall, W.A. & Scheuer, T. (1997) Persistent sodium currentsthrough brain sodium channels induced by G protein betagamma subunits.Neuron, 19, 443±452.
Ma, J.Y., Li, M., Catterall, W.A. & Scheuer, T. (1994) Modulation of brainNa+ channels by a G-protein-coupled pathway. Proc. Natl. Acad. Sci. USA,91, 12351±12355.
Manzoni, O., Prezeau, L., Marin, P., Desagher, S., Bockaert, J. & Fagni, L.(1992) Nitric oxide-induced blockade of NMDA receptors. Neuron, 8, 653±662.
Marin, P., Nastiuk, K.L., Daniel, N., Girault, J.A., Czernik, A.J., Glowinski, J.,Nairn, A.C. & Premont, J. (1997) Glutamate-dependent phosphorylation ofelongation factor-2 and inhibition of protein synthesis in neurons. J.Neurosci., 17, 3445±3454.
Murphy, N.P., Cordier, J., Glowinski, J. & Premont, J. (1994) Is protein kinase
Modulation of sodium channels by AMPA through Gb protein 1959
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960

C activity required for the N-methyl-D-aspartate-evoked rise in cytosolicCa2+ in mouse striatal neurons? Eur. J. Neurosci., 6, 854±860.
Murphy, S.N. & Miller, R.J. (1989) Two distinct quisqualate receptorsregulate Ca2+ homeostasis in hippocampal neurons in vitro. Mol.Pharmacol., 35, 671±680.
Noda, M., Ikeda, T., Suzuki, H., Takeshima, H., Takahashi, T., Kuno, M. &Numa, S. (1986) Expression of functional sodium channels from clonedcDNA. Nature, 322, 826±828.
Rodriguez-Moreno, A. & Lerma, J. (1998) Kainate receptor modulation ofGABA release involves a metabotropic function. Neuron, 20, 1211±1218.
Smith, R.D. & Goldin, A.L. (1998) Functional analysis of the rat I sodiumchannel in Xenopus oocytes. J. Neurosci., 18, 811±820.
Smith, M.R., Smith, R.D., Plummer, N.W., Meisler, M.H. & Goldin, A.L.(1998) Functional analysis of the mouse Scn8a sodium channel. J.Neurosci., 18, 6093±6102.
Tejedor, F.J., McHugh, E. & Catterall, W.A. (1988) Stabilization of a sodiumchannel state with high af®nity for saxitoxin by intramolecular cross-linking. Evidence for allosteric effects on saxitoxin binding. Biochemistry,27, 2389±2397.
Turski, L., Huth, A., Sheardown, M., McDonald, F., Neuhaus, R., Schneider,H.H., Dirnagl, U., Wiegand, F., Jacobsen, P. & Ottow, E. (1998) ZK200775:a phosphonate quinoxalinedione AMPA antagonist for neuroprotection instroke and trauma. Proc. Natl. Acad. Sci. USA, 95, 10960±10965.
Van Vliet, B.J., Sebben, M., Dumuis, A., Gabrion, J., Bockaert, J. & Pin, J.P.(1989) Endogenous amino acid release from cultured cerebellar neuronalcells: effect of tetanus toxin on glutamate release. J. Neurochem., 52, 1229±1239.
Wang, Y. & Durkin, J.P. (1995) alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, but not N-methyl-D-aspartate, activates mitogen-
activated protein kinase through G-protein beta gamma subunits in ratcortical neurons. J. Biol. Chem., 270, 22783±22787.
Wang, Y., Small, D.L., Stanimirovic, D.B., Morley, P. & Durkin, J.P. (1997)AMPA receptor-mediated regulation of a Gi-protein in cortical neurons.Nature, 389, 502±504.
Weber, M.L. & Taylor, C.P. (1994) Damage from oxygen and glucosedeprivation in hippocampal slices is prevented by tetrodotoxin, lidocaineand phenytoin without blockade of action potentials. Brain Res., 664, 167±177.
Weiss, J.H., Hartley, D.M., Koh, J. & Choi, D.W. (1990) The calcium channelblocker nifedipine attenuates slow excitatory amino acid neurotoxicity.Science, 247, 1474±1477.
White, R.J. & Reynolds, I.J. (1997) Mitochondria accumulate Ca2+ followingintense glutamate stimulation of cultured rat forebrain neurones. J. Physiol.(Lond.), 498, 31±47.
Williams, R.J., Murphy, N., Glowinski, J. & Premont, J. (1995) Glucoseregulates glutamate-evoked arachidonic acid release from cultured striatalneurons. J. Neurochem., 65, 241±249.
Yatsugi, S., Takahashi, M., Kawasaki-Yatsugi, S., Koshiya, K., Sakamoto, S.,Uematsu, D. & Shimizu-Sasamata, M. (1996) Neuroprotective effect ofYM90K, a novel AMPA/kainate receptor antagonist, in focal cerebralischemia in cats. J. Cereb. Blood. Flow Metab., 16, 959±966.
Zamanillo, D., Sprengel, R., Hvalby, O., Jensen, V., Burnashev, N., Rozov, A.,Kaiser, K.M., Koster, H.J., Borchardt, T., Worley, P., Lubke, J., Frotscher,M., Kelly, P.H., Sommer, B., Andersen, P., Seeburg, P.H. & Sakmann, B.(1999) Importance of AMPA receptors for hippocampal synaptic plasticitybut not for spatial learning. Science, 284, 1805±1811.
Zhang, Y. & Lipton, P. (1999) Cytosolic Ca2+ changes during in vitroischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J. Neurosci., 19, 3307±3315.
1960 P. Marin et al.
ã 2001 Federation of European Neuroscience Societies, European Journal of Neuroscience, 14, 1953±1960
Top Related